

Abstract
Cyclic peptides are found in a diverse range of organisms and are characterized by their stability and role in defense. Why is only one class of cyclic peptides found in mammals? Possibly we have not looked hard enough for them, or the technologies needed to identify them are not fully developed. We also do not yet understand their intriguing biosynthesis from two separate gene products. Addressing these challenges will require the application of chemical tools and insights from other classes of cyclic peptides. Herein, we highlight recent developments in the characterization of theta defensins and describe the important role that chemistry has played in delineating their modes of action. Furthermore, we emphasize the potential of theta defensins as antimicrobial agents and scaffolds for peptide drug design.
Keywords: antimicrobial compounds, cyclic peptides, drug design, host defense, peptides
Climbing up the ladder: Theta defensins are the only known cyclic backbone peptides from mammals and are characterized by the cyclic cystine ladder motif. They have promising applications as antimicrobials and peptide drug scaffolds, but little is known about their distribution in primate species or biosynthesis from two gene products. The Review discusses the progress in understanding the chemistry and biology of theta defensins and highlights remaining challenges and questions.
1. Naturally Occurring Cyclic Peptides
1.1. Introduction
Ribosomally synthesized cyclic peptides have now been discovered in bacteria, fungi, plants, and animals,1–4 and have a wide range of sizes, structures, and biological properties (Figure 1).5–8 As the pharmaceutical industry has gained interest in peptide drugs, cyclic peptides have shown great potential because of their stability to degradation.9–11 Theta defensins (θ‐defensins) are the only known class of cyclic peptides from mammals and the focus of this review, but they need to be understood in the broader context of cyclic peptides as they share common features and challenges. We describe how some of these challenges have been addressed for other classes of cyclic peptides and how they might add to our understanding of θ‐defensins.
Figure 1.

Diversity of cyclic peptides. a) Cyclic peptides occur in bacteria, plants, animals, and fungi. b) They vary in their sequences, size, and disulfide connectivities. Amino acid sequences of AS‐48, kalata B1, RTD‐1, and α‐amanitin with disulfide bonds as grey lines and post‐translationally modified residues marked with an asterisk. c) Three‐dimensional structures of AS‐48 (PDB 1E68),5 kalata B1 (PDB 1NB1),6 RTD‐1 (PDB 2LYF),7 and α‐amanitin (PDB 1K83)8 shown as cartoons.
The cyclic peptides described herein are all direct gene products, but we note that there are many cyclic peptides synthesized nonribosomally.12–13 An example is cyclosporin, a fungal peptide that is used as an immune suppressant.14 There is also a growing number of bioactive peptides that have been artificially cyclized to take advantage of the increased stability afforded by a cyclic peptide backbone. An example is the cyclized cone snail venom peptide Vc1.1, which showed increased stability and potency in an animal pain model.15
Cyclic peptide discovery is challenging because the lack of termini makes cyclic peptides resistant to traditional sequencing methods. Furthermore, the unknown position of the “termini” and small size of the genes hamper the search for cyclic peptides at the nucleic acid level. However, new search methods are being developed, particularly using mass spectrometry,16 and data on cyclic peptides that have been discovered are collected in Cybase.17 We anticipate that as new discovery methods are developed, other classes of cyclic peptides will emerge, with novel bioactivities and applications.
1.2. Bacterial Cyclic Peptides
Gene‐encoded cyclic peptides produced by bacteria4 typically comprise 30–70 amino acids and were the first reported class of ribosomal cyclic peptides; the cyclic backbone of AS‐48, a bacteriocin, was elucidated in 1994.18 Cyclic bacteriocins are produced by bacteria in the phylum Firmicutes and have globular structures comprising α‐helices.4 Cyanobacteria produce the cyanobactins, which contain posttranslational modifications in addition to cyclization.19 Both cyclic bacteriocins and cyanobactins seem to be produced for defense against competing bacteria and have potential applications as antimicrobial agents. In contrast, the cyclic pilins have a structural role and are used to transfer genetic material.20 The biosynthetic mechanisms of bacterial cyclic peptides are better understood than those of their eukaryotic counterparts because the genes that encode them are often clustered with genes encoding their processing enzymes.19, 21 Understanding these mechanisms in bacteria will enable recombinant cyclic peptide production, thus facilitating the development of cyclic peptide therapeutics.
1.3. Fungal Cyclic Peptides
Fungi in the Amanitaceae family produce two classes of cyclic peptides, the amatoxins and phallocidins, both of which contain unusual posttranslational modifications.2, 22 While phallocidins comprise seven residues with a Cys‐Trp sulfide link, amatoxins comprise eight residues with a Cys‐Trp sulfoxide link and are hydroxylated.23 Fungal cyclic peptides are flanked by Pro residues at their N‐ and C‐terminal processing sites and are thought to be cyclized by a prolyl oligopeptidase.22, 24 Both the amatoxins and phallocidins are highly toxic to humans; amatoxins inhibit transcription by binding to RNA polymerase II and phallocidins bind to F‐actin, which stabilizes filament structures within the cell.23, 25
1.4. Plant Cyclic Peptides
Plant cyclic peptides are grouped into three families, cyclotides,26 sunflower trypsin inhibitors,27 and orbitides.28 Cyclotides comprise approximately 30 residues and are characterized by the cyclic cystine knot motif.26, 29 Following discovery of the prototypic cyclotide kalata B1,29, 30 cyclotides have been found in the Violaceae, Rubiaceae, Fabaceae, Solanaceae, and Cucurbitaceae families, and their structures, activities, and applications have been extensively reviewed.2, 31 Cyclotides are divided into the bracelet, Möbius, and trypsin inhibitor subfamilies, based on their sequences and activities.26, 32 Although the natural function of cyclotides is thought to be as plant defense molecules against insect pests,33 several unrelated pharmacological activities have been reported: anti‐HIV,34 antibacterial,35 antitumor,36 hemolytic,37 and uterotonic38 activities. Membrane binding appears to be a mechanism common to these activities.39 The exact mechanism of cyclotide biosynthesis is still not clear; however, it is thought that the cyclotide precursor is cleaved at the N terminus and an asparaginyl endopeptidase (AEP) carries out a transpeptidation reaction to form the cyclic backbone.40 In addition to their obvious application in agriculture for plant protection, cyclotides have shown potential as a stable scaffold for the design of peptide drugs41, 42 and possible developments include their production in “plant factories”.43
The sunflower trypsin inhibitors are smaller than cyclotides, comprising only 14 residues and one disulfide bond.27 The nucleic acid sequences that encode the sunflower family of cyclic peptides are buried within genes encoding albumin seed storage proteins and are cyclized by an AEP.44 The potent trypsin inhibitory activity of sunflower trypsin inhibitor 1 (SFTI‐1) has been exploited for the design of potential anticancer compounds; chemically synthesized analogues of SFTI‐1 inhibit matriptase45 and kallikrein‐related peptidase‐4,46 proteases involved in breast and prostate cancer, respectively.
A recent report recommended the name orbitides for small cyclic plant peptides, earlier known as cyclolinopeptides or caryophyllaceae‐type peptides.47 Orbitides comprise five to 12 amino acids and, in contrast to cyclotides, have a high proportion of hydrophobic residues and lack disulfide bonds.28, 48 More than 100 orbitides have been discovered in ten plant families and, as cyclotides, their natural function in plants is thought to be as antiherbivore or antibacterial agents, although they have many different bioactivities in vivo.28, 47 DNA precursors of orbitides were identified in Saponaria vaccaria and their biosynthesis appears to involve cleavage of the precursor by an oligopeptidase followed by cyclization mediated by a serine protease.49, 50
1.5. Mammalian Cyclic Peptides
Since their discovery in 1999,51 significant progress has been made in understanding the antimicrobial activities of θ‐defensins, but many questions remain, especially with regard to their biosynthesis. Recent progress in the synthesis and characterization of θ‐defensins should facilitate efforts to address these unsolved problems and identify new applications for θ‐defensins. We focus on these challenges and opportunities in the remainder of this article.
2. Mammalian Defensins
2.1. α‐, β‐, and θ‐Defensins
Defensins are disulfide‐rich peptides that form part of the mammalian innate immune system, providing defense against microbial pathogens and regulating the immune response.52–54 α‐Defensins comprise 29–35 amino acids and contain three disulfide bonds in a I–VI, II–IV, III–V arrangement (Figure 2).55, 56 Six α‐defensins are found in humans: the human neutrophil peptides (HNP1‐4) and human defensins (HD5 and HD6), which are expressed in the Paneth cells of the intestine.57 β‐Defensins are larger than α‐defensins, comprising approximately 45 amino acids and three disulfide bonds in a I–V, II–IV, III–VI arrangement.55, 58 More than 30 β‐defensins are found in humans, predominantly in epithelial cells and the male reproductive tract.59 The third class of mammalian defensins is the θ‐defensins (so named because of their structural similarity to the Greek letter theta), which comprise 18 residues and three disulfide bonds in a I–VI, II–V, III–IV arrangement.51, 60
Figure 2.
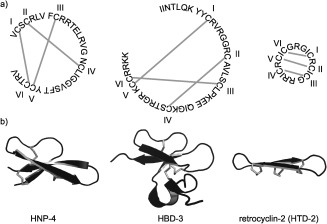
Mammalian defensins. a) Mammalian defensins are divided into three classes, α‐defensins (e.g. HNP‐4), β‐defensins (e.g. HBD‐3), and θ‐defensins (e.g. HTD‐2). Amino acids are represented by their one‐letter codes, grey lines represent disulfide bonds and disulfide connectivities are shown by Roman numerals. b) Three‐dimensional structures of HNP‐4 (PDB 1ZMM),56 HBD‐3 (PDB 1KJ6),58 and retrocyclin‐2 (PDB 2ATG)60 are shown in cartoon representation with disulfide bonds in grey.
2.2. Discovery of θ‐Defensins
The first cyclic mammalian peptide, rhesus θ‐defensin 1 (RTD‐1), was discovered during a study of defensin expression in rhesus macaque (Macacca mulata) leukocytes.51 Fractionation and screening of a leukocyte extract resulted in the isolation of an 18‐residue peptide with potent antibacterial activity, and sequencing of overlapping fragments led to the elucidation of the cyclic backbone. A series of thermolysin digestions revealed three parallel disulfide bonds, which are now termed the cyclic cystine ladder.7, 51, 60 A search for the gene that encodes RTD‐1 surprisingly revealed two genes (GenBank AF191102 and AF191103), each encoding a nine‐residue fragment of RTD‐1 (demidefensins RTD1a and RTD1b), followed by three residues and a stop codon (Figure 3 a).51 θ‐Defensin genes and pseudogenes are named DEFT (defensin theta) and ψDEFT genes, respectively. The genes appear to be truncated α‐defensin genes,51 and the two encoded demidefensins61 are spliced together to form the cyclic 18‐residue θ‐defensin. To date, θ‐defensins are the only cyclic peptides to be biosynthesized from two separate gene products.3
Figure 3.
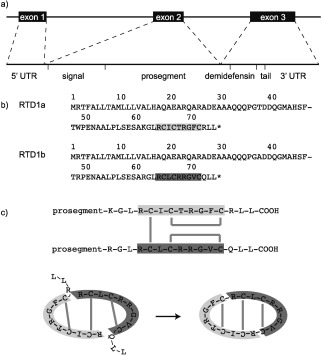
θ‐Defensins are synthesized by binary ligation of two nine‐residue demidefensins.51, 53 a) Demidefensin genes comprise three exons and two introns. b) Demidefensin precursors comprise a signal sequence, prosegment, nine‐residue demidefensin (RTD‐1a, light grey and RTD1b, dark grey), and a three‐residue tail. Amino acids are represented by one‐letter codes and asterisks represent stop codons. c) In a proposed biosynthetic mechanism, one intermolecular and two intramolecular disulfide bonds (grey lines) join the two demidefensins. The N‐terminal prosegment is cleaved and two new peptide bonds ligate the demidefensins, with concomitant loss of the tail residues to form the cyclic peptide.
An unsolved question is why heterodimeric θ‐defensins are more abundant than homodimeric θ‐defensins. Isolation of the homodimers of RTD1a and RTD1b (RTD‐2 and RTD‐3) illustrated that n demidefensin genes can give rise to (n/2)(n+1) θ‐defensins.61, 62 A third demidefensin, RTD1c, was later identified, and the six corresponding θ‐defensins RTD‐1 to RTD‐6 were isolated from macaque neutrophils.61, 63 Although the amount of θ‐defensin peptide varies by as much as a factor of three between individuals, RTD‐1 accounts for around 50 % of the total θ‐defensin content.63 The preference for heterodimeric species over homodimeric species and the high proportion of RTD‐1 compared to the other five variants might be a result of different gene copy numbers or different expression levels, but these questions remain to be addressed.
Another major challenge is to understand why θ‐defensin genes in humans exist as pseudogenes and so do not give rise to θ‐defensin peptides. Selsted and co‐workers noted the sequence similarity of the genes encoding RTD‐1 to the gene encoding a human α‐defensin‐related pseudogene (GenBank U10267),51 which contains a stop codon in the signal sequence. Lehrer and co‐workers synthesized the θ‐defensin that would be produced from two transcripts of the pseudogene in the absence of the premature stop codon.64 The peptide was named retrocyclin and showed potent anti‐HIV activity and low cytotoxicity, adding an ironic twist to the question of why humans have not inherited intact θ‐defensin genes.64
3. Distribution and Diversity
3.1. Sequence Diversity
Less is known about the distribution and diversity of θ‐defensins than other cyclic peptides; at the time of writing this Review, 532 cyclotides had been isolated from 55 species of plants17 since the elucidation of their cyclic structure in 1995,29 but only 11 different θ‐defensins had been isolated from three species of primates since their discovery in 1999.51 This contrast might reflect the relative difficulties in obtaining primate tissue samples compared to plants, or a lack of efficient discovery tools. In addition to the six θ‐defensins isolated from rhesus macaques (Macacca mulata), four θ‐defensin cDNAs were identified in olive baboon (Papio anubis) leukocytes, suggesting that ten θ‐defensins could be synthesized; however, only five were isolated.65 The two peptides isolated from hamadryas baboon (Papio hamadryas) leukocytes, PhTD‐1 and PhTD‐3, were sequenced by MALDI‐MS and had identical sequences to BTD‐1 and BTD‐3, respectively.66 To date, θ‐defensins isolated at the peptide level are named according to the first letter of the species of primate in which they are found. However, as θ‐defensins are discovered in new species, a systematic nomenclature will need to be developed to avoid ambiguities.
A missing piece in our understanding of the biosynthesis of θ‐defensins is a lack of knowledge about their sequence diversity. The sequences of all known demidefensins encoded by genes and pseudogenes are shown in Table 1. All θ‐defensins isolated at the peptide level contain six cysteine residues. However, it is interesting that the Pan troglodytes and Pan paniscus pseudogenes only encode four cysteines. The greatest sequence variation occurs in the four residues of the β‐turn and the number of arginine residues, which result in charges from +2 to +6.67 In contrast to cyclotides, no acyclic θ‐defensin analogues have been reported, an observation that might reflect a different biosynthetic mechanism to cyclotides.
Table 1.
Sequences of demidefensins encoded by θ‐defensin (DEFT) genes and pseudogenes (ψDEFT).
|
Species |
Demidefensin |
Sequence[a] |
|---|---|---|
|
DEFT2 (RTD‐1a) |
RCICTRGFCRLL |
|
|
DEFT1 (RTD‐1b) |
RCLCRRGVCQLL |
|
|
DEFT3 (RTD‐1c) |
RCICVLGICRLL |
|
|
DEFT4 |
RCICTRGVCQLL |
|
|
ψDEFT1,2,3,5,6 |
RCICGRGICRLL |
|
|
ψDEFT4 |
RCICGRRICRLL |
|
|
Pongo Pygmaeus abelii 67 (Sumatran orangutan) |
ψDEFT1 |
RCICRRGVCRFL |
|
DEFT1,2,4 |
RCICRRGVCRLL |
|
|
DEFT3 |
RCICGRGVCRLL |
|
|
Gorilla gorilla 67 (lowland gorilla) |
ψDEFT1 |
RCICGRGICRLL |
|
Pan troglodytes 67 (common chimpanzee) |
ψDEFT1 |
RCIGGRGICGLL |
|
Pan paniscus 67 (bonobo chimpanzee) |
ψDEFT1 |
RCIGGRGICGLL |
|
Hylobates syndactylus 67 (siamang) |
DEFT1 |
RCICGRGVCRLL |
|
Macaca nemestrina 67 (pig‐tailed macaque) |
DEFT1 |
RCICRRGVCQLL |
|
Colobus guereza kikutensis [67] (kikuyu colobus) |
DEFT1 |
RCVCTRGFCHLL |
|
Papio anubis 65 (olive baboon)/ Papio hamadryas 66 (hamadryas baboon) |
BTD‐a |
RCVCTRGFCRLL |
|
BTD‐b |
RCVCRRGVCQLL |
|
|
BTD‐c |
RCICLLGICRLL |
|
|
BTD‐d |
RCFCRRGVCQLL |
[a] Demidefensin sequence with cysteines in bold and the three‐residue “tail” in grey.
3.2. Distribution of θ‐Defensins in Primates
The distribution of θ‐defensins in primate species and the origin of the stop codon in ψDEFT genes are as yet not understood. A phylogenetic study of 21 species of old and new world primates identified new DEFT and ψDEFT genes (Table 1)67 and concluded that the silencing mutation in ψDEFT genes occurred in a common ancestor of humans, chimpanzees, and gorillas around 7.5–10 million years ago when the orangutan lineage diverged.67 Orangutans (Pongo Pygmaeus abelii) have both intact and silenced copies of DEFT genes.68 Furthermore, the stop codon appears to have arisen by a series of stepwise mutations in codon 17; CAC/T (histidine) in old world monkeys, CAG (glutamine) in orangutans, and TAG (stop) in new world monkeys.59, 67
In accordance with their role in innate immunity, θ‐defensins are produced mainly in the bone marrow.51 They are expressed in the phagocytic cells, neutrophils and monocytes, but not lymphocytes or eosinophils, and do not appear to be secreted but are localized in azurophil granules.51, 54 However, expression of RTD‐1 by Paneth cells in crypts of the small intestine of rhesus macaques was reported recently.69 In humans, expression of ψDEFT genes is highest in bone marrow but is also observed in skeletal muscle, spleen, thymus, and testis.67
3.3. Human Retrocyclins
Does our inability to inately produce θ‐defensins make humans more susceptible to HIV? This burning question will be difficult to answer conclusively. No θ‐defensin peptides have been found in humans, but there are six ψDEFT genes in the human genome: five (ψDEFT1‐5) on chromosome 8p23 (gi: 501091),70 upstream of the HNP‐1 gene, and one (ψDEFT6) on chromosome 1q41.59, 67 Although retrocyclin‐1 inhibits two Thai HIV‐1 subtypes, sequencing of DEFT genes from persistently HIV‐1 seronegative female sex‐workers from Thailand showed that they all had ψDEFT genes.71 These results led to the conclusion that the HIV‐1 resistance of these women was not due to θ‐defensin production. Nevertheless, the possibility remains that some isolated populations might have retained intact DEFT genes, and hence have intrinsic protection from HIV.71
4. Gene Structure and Biosynthesis
4.1. Structure of DEFT Genes
DEFT genes comprise three exons and two introns (Figure 3 a). The exons encode a 76‐residue propeptide comprising a 20‐residue signal peptide, 44‐residue prosegment, nine‐residue demidefensin, and three‐residue tail (Figure 3 b).51 Although the structures and disulfide connectivities of α‐ and θ‐defensins differ, common features in their precursor peptides suggest that similar processing mechanisms might be used.53
4.2. Biosynthesis of Cyclic Peptides
Understanding the biosynthetic mechanism is a challenge for all classes of cyclic peptides, but much less is known about the biosynthesis of θ‐defensins than of other classes. Cyclization is thought to have arisen separately in the different kingdoms, although similar features, such as a protease‐mediated transpeptidation, are present.1, 72 In cyclotides, conserved Gly‐Leu‐Pro sequences at the N termini of both the cyclotide and propeptide are recognized by the AEP protease. The C‐terminal propeptide is thought to be cleaved, allowing the N terminus of the cyclotide to replace it in the recognition site. The N‐terminal Gly then acts as a nucleophile, cyclizing the peptide chain.40 Similarly, the TrbC pilin in bacteria is cleaved and cyclized by the TraF protease.20, 53 θ‐Defensin biosynthesis also probably involves a protease; however, two new peptide bonds need to be formed.
4.3. θ‐Defensins: Putting the Pieces Together
How do two peptide and three disulfide bonds form between two demidefensins to build a θ‐defensin? A likely explanation is that two disulfide‐stabilized β‐hairpins are formed and held together by the interchain disulfide for ligation (Figure 3 c).51 Structural properties of the precursors might also direct association of demidefensins for ligation.62 Possible developments arising from the elucidation of θ‐defensin biosynthesis are the identification of new cyclic peptides and insights into how peptide cyclization has arisen in mammals.
Evidence for whether human cells have retained an ability to process demidefensin transcripts is inconclusive. In two separate studies, human promyelocytic HL‐60 cells, which are able to process α‐defensins, were transfected with pro‐θ‐defensin cDNA. In one study, neither mature θ‐defensins nor θ‐defensin precursors were detected,53 but in the other study, the presence of cyclic retrocyclins that had anti‐HIV activity was reported.73 Furthermore, in the latter study, aminoglycosides were used to read through the stop codon in the human ψDEFT gene, restoring endogenous expression of retrocyclin.73 A yeast two‐hybrid screen was used to identify proteins that interact with defensin precursors and might be involved in their processing. Stromal cell derived factor‐2‐like protein 1 (SDF2 L1), which is part of the endoplasmic reticulum chaperone complex, interacted with α‐, β‐, and θ‐defensins, thus suggesting that it might have a role in the biosynthesis or packaging.74
5. Structure and Stability
5.1. NMR Structures of θ‐Defensins in Solution
Structures of naturally occurring and modified θ‐defensins have been determined from NMR data, giving insights into their stability and mechanism of action. The β‐sheet structure cross‐braced by three disulfide bonds was predicted by a molecular dynamics model51 and later confirmed by the NMR structure of RTD‐1 (PDB 1HVZ) in solution.75 Broadened NMR signals and few nuclear Overhauser enhancement (NOE) interactions indicated that that RTD‐1 is flexible (Figure 4 a).75 The structure of synthetic retrocyclin (HTD‐2, PDB 2ATG) was then determined in the presence of sodium dodecyl sulphate (SDS) micelles, which might stabilize the extended conformation of the peptide.60 More recently, the development of NMR methods facilitated the determination of well‐defined structures of three θ‐defensins and the characterization of the cyclic cystine ladder (Figure 4 b): RTD‐1 (PDB 2LYF); HTD‐2 (PDB 2LZI); and the first symmetrical θ‐defensin BTD‐2 (PDB 2LYE).7 The structures comprise two highly constrained β‐strands joined by β1′ turns, and heteronuclear NOEs and predicted order parameters suggested that θ‐defensins are more rigid than previously thought.7 Several structures of θ‐defensins are now available in the Protein Data Bank for modeling and peptide design applications.76, 77
Figure 4.
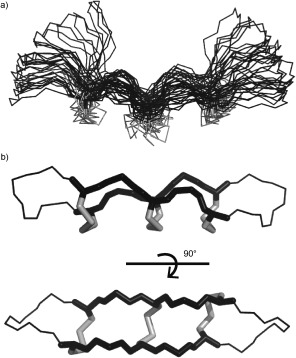
NMR structures of θ‐defensins. a) Ensemble of the 20 lowest‐energy structures of RTD‐1 (PDB 1HVZ)75 with disulfide bonds in grey. This was the first θ‐defensin structure published and suggested flexibility of the turn region. b) The symmetrical θ‐defensin BTD‐2 (PDB 2LYE)7 with the cyclic cystine ladder motif shown as sticks.
The possible role of self‐association in the mechanism of action of θ‐defensins is still unclear. Density ultracentrifugation and NMR diffusion measurements suggested that retrocyclin forms trimers at high concentrations. A model was proposed in which the peptides self‐associate along their longitudinal axes with the disulfide bonds facing inwards, based on selective broadening of the cysteine signals.60 NMR diffusion measurements on RTD‐1, BTD‐2, and HTD‐2 supported this hypothesis and extended the concentration range, suggesting that other aggregation states might be formed.7
The disulfide connectivities in the cyclic cystine ladder are well established and have been confirmed by several methods: a computational method that predicts disulfide bond connectivities in peptides;78 comparison of the energy‐minimized structures of all 15 possible disulfide connectivities; calculation of the probabilities of each possible disulfide connectivity using PADLOC79 (Pattern of disulfides from local constraints); and measurement of cysteine–cysteine distances in a structure calculated without disulfide bond restraints.76
5.2. Thermal and Proteolytic Stability of θ‐Defensins
The constrained structure of θ‐defensins gives them remarkable stability, consistent with resistance to high concentrations of proteases in inflamed tissue.51, 53 Synthetic θ‐defensin analogues with varying numbers and arrangements of disulfide bonds revealed the dependence of the structure and stability on the cyclic cystine ladder.77 Although analogues with three, two, or one central disulfide bond were structured and stable, those lacking disulfide bonds or a cyclic backbone were unstructured and rapidly degraded in serum. NMR studies showed that θ‐defensin analogues with two or three disulfide bonds retained their structure at temperatures above 80 °C.77 In contrast, the disulfide bonds were not necessary for antibacterial or membrane‐binding properties; however, the cyclic cystine ladder might have a role in molecular recognition and antibacterial activity at physiological salt concentrations.53 Acyclic θ‐defensin analogues have weaker antibacterial activity than cyclic θ‐defensins, suggesting that the cyclic backbone is important for activity.75, 77 A d‐retrocyclin analogue synthesized for even greater stability showed more potent anti‐HIV activity than l‐retrocyclin.80
6. Synthesis
6.1. Chemical Synthesis
The accessibility of θ‐defensins by chemical synthesis (Figure 5) has facilitated studies of their activities and mechanisms. Early syntheses employed fluorenylmethoxycarbonyl (Fmoc) chemistry for the assembly of the peptide backbone by solid‐phase peptide synthesis (SPPS), followed by oxidation to bring the termini (residues 1 and 18) into proximity for ligation using 1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide (EDC) and hydroxybenzotriazole (HOBt) as coupling reagents.51, 61 Although this strategy was successful, racemization of the cysteine residues led to poor yields.53 In a later study, three methods were investigated for cyclization of the oxidized RTD‐1 precursor: 1) O‐benzotriazole‐N,N,N′,N′‐tetramethyl‐uronium‐hexafluoro‐phosphate (HBTU) and N,N‐diisopropylethylamine (DIPEA) in dimethylformamide (DMF); 2) (benzotriazol‐1‐yloxy)tris(dimethyl amino)phosphonium hexafluorophosphate (BOP) and DIPEA in DMF; and 3) HOBt and EDC in dimethylsulfoxide (DMSO).75 While a mixture of products was obtained with HBTU owing to side reactions, both BOP and EDC/HOBt gave the desired major product. The EDC/HOBt method was recommended for ease of purification.75
Figure 5.
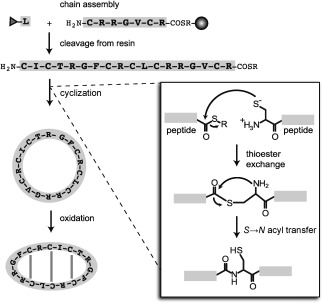
Chemical synthesis of θ‐defensins involves three stages: assembly of the peptide chain on resin; cleavage from the resin; and cyclization and oxidation to form the circular backbone and three disulfide bonds. A grey sphere represents the resin bead and amino acids are represented by one‐letter codes. The peptide chain is assembled from C to N terminus by coupling N‐terminal protected (grey triangle) amino acids to the free N terminus of the peptide chain. Inset: cyclization by native chemical ligation. The reaction involves thioester exchange between a thioester linker (COSR) at the C terminus of the peptide with the free thiol of an N‐terminal cysteine. A spontaneous S→N acyl transfer then releases the free cysteine thiol and forms a peptide bond.
The challenge of θ‐defensin cyclization has now been largely overcome by the use of native chemical ligation. Native chemical ligation81 was originally developed for the ligation of two peptide chains, but is also ideal for the synthesis of cyclic peptides, especially those containing multiple cysteine residues.82 The reaction involves thioester exchange between a C‐terminal thioester linker and the free thiol of an N‐terminal cysteine residue. Formation of the peptide bond is achieved by an irreversible S→N acyl transfer, which regenerates the free cysteine thiol (Figure 5 inset).81 Several native θ‐defensins and analogues have been synthesized using Boc SPPS followed by concurrent cyclization by native chemical ligation and oxidation.7, 77 Fmoc‐compatible strategies are also becoming more available, and recently a ‘one‐pot’ cyclization and oxidation of a linear θ‐defensin precursor was reported.83
6.2. Semirecombinant Synthesis
A lack of efficient in vivo cyclization methods has hindered the recombinant production of θ‐defensins, but several semirecombinant approaches have been reported. RTD‐1 and an analogue containing three N‐methylated residues were synthesized using codon reprograming in a cell‐free expression system.84 The peptide precursors contained a C‐terminal Cys‐Pro‐glycolic acid sequence that self‐rearranges to form a diketopiperadine thioester, which then rearranges to cyclize the peptide.84 An advantage of this approach is that nonstandard or N‐methylated amino acids can be included. A bacterial expression system has also been used to produce RTD‐intein precursors, which are then cyclized either in vitro or in vivo by a modified intein‐splicing mechanism.85 This strategy opens up the possibility of producing cyclic peptide libraries or isotope‐labeled peptides recombinantly; however, the yields need to be improved for large‐scale production.85
6.3. Chemically Modified θ‐Defensins
Many θ‐defensin analogues have been synthesized chemically to study the their mechanism of action or to optimize antibacterial or antiviral properties. Examples include pseudogene products, Lys mutants to enable labeling with chromophores, Tyr mutants, and retro‐ and enantioanalogues for anti‐HIV and carbohydrate‐binding studies.86, 87 Chemical synthesis has also been used to produce simplified retrocyclin analogues,88 disulfide‐bond analogues, and θ‐defensin analogues containing non‐native bioactive epitopes.76, 77 Oxidation of unprotected cysteine residues appears to give the native I–VI, II–V, III–IV (laddered) disulfide connectivity.76 Nevertheless, in one study, RTD‐1 was synthesized using orthogonally protected cysteines, and it was noted that correct disulfide pairing was assisted by the formation of β sheets.89 RTD‐1 has also been used to illustrate a method for synthesizing peptides with multiple disulfides in complex arrangements without the need for orthogonal protection; the disulfide bonds in RTD‐1 were replaced with Watson–Crick base pairs.90 The resulting RTD‐1 analogue had a similar twisted β‐sheet structure to native RTD‐1, low hemolytic activity, and slightly higher antibacterial activity than native RTD‐1.
7. Antimicrobial Activity
7.1. Antibacterial and Membrane‐Disruptive Activity
The antimicrobial activity of θ‐defensins led to their initial discovery and is thought to be their natural function.51 While α‐, β‐, and θ‐defensins are all antimicrobial at 0.5–5 μm, θ‐defensins have the advantage of a lower sensitivity to physiological salt concentrations.54 The cyclic backbone might have a role in this salt insensitivity; the acyclic analogue oRTD‐1 is three times less active than cyclic RTD‐1 against E. coli and S. aureus.51 In contrast, the disulfide bonds are not essential for antibacterial activity.77 θ‐Defensins also bind to and neutralize bacterial toxins. Retrocyclin‐1 and its analogues are active against anthrax bacilli and spores, and bind to anthrax lethal toxin.91 However, acyclic or reduced and alkylated analogues of retrocyclin lack activity, suggesting that the cyclic backbone and disulfide bonds have a role in toxin binding.91 Furthermore, retrocyclin‐1 is active against L. monocytogenes and inhibits listeriolysin O, a pore‐forming toxin that enables bacteria to escape from phagosomes.92 Antifungal activities against C. albicans, C. neoformans, V. dahliae, and F. oxysporum have also been reported.51, 65, 93
Understanding the mechanism of antimicrobial activity in the complex inflammatory environment where numerous inhibitors and enhancers are present is extremely challenging.54 The antibacterial activity of θ‐defensins is thought to be caused by their interaction with membranes or glycoproteins, as both enantiomers have similar antibacterial activity.80 As illustrated by the activity of RTD‐1 against clinical isolates of antibiotic‐resistant S. aureus and P. aeruginosa, θ‐defensins might be effective against resistant bacteria because they target the cell membrane rather than specific enzymes.94 Differential scanning calorimetry and X‐ray scattering measurements showed that θ‐defensins are selective for anionic membranes over zwitterionic membranes and indicated that the positively charged face of θ‐defensins interacts with anionic phospholipid headgroups.95 Surface plasmon resonance (SPR) measurements also showed that BTD‐2 has higher affinity for anionic membranes than zwitterionic membranes and the number of disulfide bonds did not affect membrane‐binding affinity.77 Measurements of membrane curvature by small‐angle X‐ray scattering showed that θ‐defensins generate negative saddle‐splay curvature in bacterial membrane models, indicating a potential for membrane disruption dependent on the composition of cationic amino acids.96 Oriented circular dichroism and X‐ray diffraction of RTD‐1 in lipid bilayers showed both a parallel membrane‐bound state and a transmembrane pore‐forming state of the peptide. However, the transition between these two states and the exact mechanism was unclear.97 Solid‐state NMR measurements indicated that 15N‐labeled retrocyclin‐2 adopted a transmembrane orientation in dilauroylphosphatidylcholine (DLPC) bilayers, but a more in‐plane (≈65°) orientation in the thicker 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphatidylcholine (POPC) bilayer.98
7.2. Antiviral Activity
The potent anti‐HIV properties of retrocyclin were first reported by Cole and co‐workers,64 and their therapeutic potential has generated much interest.68 Retrocyclin inhibits HIV‐1 replication at low micromolar concentrations but only when added to the cells before infection, as it does not directly inactivate the virus, but prevents its entry by blocking the fusion of the viral envelope and cell membrane (Figure 6).64, 86, 99 Inhibition of the viral entry might be mediated by the ability of retrocyclin to recognize and bind sugar molecules on cell surfaces; a fluorescently labeled retrocyclin analogue formed patches on the surface of CD4+ cells.64, 87 Cell‐fusion assays and SPR showed that retrocyclin‐1 binds to the C‐heptad repeat of gp41, preventing the formation of a six‐helix bundle (Figure 6).100 Docking studies revealed glutamic acid residues in the C‐heptad repeat‐2 segments of gp41 to which arginine residues of retrocyclin might bind. However, mutation of these residues did not lead to resistance to retrocyclin because the mutations rendered the virus less infective.101 In a recent study, RC‐101 was effective against HIV strains that were resistant to the fusion inhibitor enfuvirtide, suggesting that the two antivirals bind to different sites on the heptad repeat‐2 domain.102 Furthermore, the effects of compensatory mutations in viral strains resistant to RC‐101 were studied, to help guide the development of fusion inhibitors based on retrocyclin.102
Figure 6.
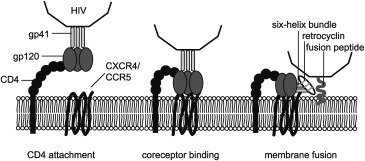
Simplified mechanism of HIV virus entry into a host cell. The HIV virus attaches to the host cell membrane by binding of the viral gp120 protein with a CD4 receptor on the host cell surface. Changes in the conformation of gp120 then allow it to bind to the coreceptor, either chemokine receptor CXCR4 or CCR5. Coreceptor binding exposes the hydrophobic gp41 fusion peptide, which inserts into the host cell membrane. Each subunit of the trimeric gp41 protein then folds in half to form the six‐helix bundle, drawing the viral and host cell membranes together and resulting in membrane fusion. Retrocyclin is thought to inhibit HIV entry by binding to the six‐helix bundle of gp41.
The important role that chemistry has had in elucidating the structure–activity relationships of θ‐defenins is illustrated by the variety of θ‐defensin analogues that have been synthesized. For example, a more active retrocyclin analogue (retrocyclin‐101) was developed by replacing Arg9 with Lys.103 In another study, the retro‐analogue of retrocyclin was actually found to increase HIV infection.104 The ability of retrocyclin to inhibit HIV entry is dependent on the HIV subtype, possibly because of different glycoproteins displayed.103 HIV‐infected human cells grown in the presence of retrocyclin‐101 to select for resistance showed only a five‐fold decrease in HIV‐1 susceptibility. Three mutations were identified in the resistant viruses, all of which were in the envelope glycoproteins gp120 and gp41 and were replacements of negative or neutral residues with positively charged residues.105
θ‐Defensins also have anti‐influenza and antiherpes activities, in which their carbohydrate‐binding “lectin” properties might have a role.106 Retrocyclin is active against the influenza A virus, binding to surfactant protein D107 and inhibiting viral entry by cross‐linking and immobilizing membrane glycoproteins that have a role in membrane fusion.108 Cells and chicken embryos transfected with retrocyclin‐2 showed reduced infection with H5N1 avian influenza virus, but it was not clear whether the cells expressed cyclic retrocyclin.109 Chemically synthesized truncated θ‐defensin analogues have also shown anti‐influenza A activity.88, 110 Furthermore, a series of θ‐defensins and analogues inhibited the entry of herpes simplex virus into cells by binding surface glycoproteins111 and retrocyclin‐2 was effective as a prophylaxis against herpes simplex virus type 1 in a keratitis mouse model.112
8. Applications
The most obvious application of θ‐defensins is their use as antimicrobials. Although their antibacterial activities are comparable to those of other antimicrobial peptides, the potent anti‐HIV activity of θ‐defensins has garnered the most interest. Several reports have proposed the use of retrocyclins in topical microbicides for reducing HIV‐1 transmission.113, 114 A dissolving polyvinyl alcohol film formulation of RC‐101 has been tested for in vitro and ex vivo safety and efficacy as a vaginal microbicide, but long‐term stability and bioavailability testing still need to be carried out.115
In a recent report it was suggested that θ‐defensins could also be used as immunomodulatory agents.116, 117 While α‐ and β‐defensins stimulate the adaptive immune response, θ‐defensins suppress the production of proinflammatory cytokines, modulating the immune response.55, 117 In a polymicrobial sepsis model, mice dosed with RTD‐1 had lower levels of tumor necrosis factor and interleukins. Furthermore, systemically administered θ‐defensins were nontoxic, stable in plasma, and did not stimulate an immune response in adult chimpanzees.117 In an earlier study, prophylactic treatment with RTD‐1 prevented lethal infection of mice with a mouse‐adapted strain of severe acute respiratory syndrome coronavirus.118 However, θ‐defensins are neither virus‐ nor lipopolysaccharide (LPS)‐neutralizing, suggesting that their immunomodulatory activity and ability to bind to carbohydrate‐containing cell‐surface receptors helped prevent viral infection.117, 118 Furthermore, binding to cell‐surface receptors or plasma proteins might concentrate θ‐defensins at infection sites.59
θ‐Defensins are attractive scaffolds for peptide drug design because of their constrained structure, stability, low cytotoxicity, and inherent bioactivities.77 The concept of a ‘prototypic design template’ has been applied to tachyplesin analogues, which have similar rigid structures to θ‐defensins, but do not have the additional advantage of a cyclic backbone.89 As a proof‐of‐concept that θ‐defensins can be used as scaffolds for peptide drugs, the cyclic cystine ladder of RTD‐1 was used to constrain and stabilize the integrin‐binding Arg‐Gly‐Asp (RGD) sequence in either one or both loops.76 The RGD‐containing θ‐defensins showed potent and selective integrin‐binding activity, illustrating that the θ‐defensin scaffold can acquire a non‐native bioactivity. Furthermore, higher integrin‐binding activity of analogues containing RGD in both loops suggested that the natural symmetry of θ‐defensins might be exploited in the design of bifunctional peptide therapeutics.76 Development of chemical synthesis strategies for θ‐defensin analogues will help to drive their application as drug scaffolds by providing access to novel molecules containing unnatural amino acids, labels, mutations, and chemical modifications.
9. Summary and Outlook
The 1990s saw the emergence of the new field of cyclic peptides, as the structures and ribosomal origins of cyclic peptides in bacteria, fungi, and plants were elucidated. The discovery of θ‐defensins in 1999 revealed that mammals also produce cyclic peptides. Since 2000, chemical tools for discovery, sequencing, synthesis, and structure determination have facilitated investigations into the structures and mechanisms of action of cyclic peptides. While interest in the antimicrobial properties of θ‐defensins has led to mechanistic insights, the discovery of new θ‐defensins has lagged considerably behind other cyclic peptides. Furthermore, the biosynthetic mechanism of θ‐defensins from two separate gene products remains one of the most intriguing yet elusive questions in the field. The growing interest in peptide‐based drugs in the pharmaceutical industry, as well as the increasing number of validated targets for drugs that disrupt protein–protein interactions, provides many opportunities for applications of θ‐defensins. Further studies on the structures, mechanisms of action, and synthesis of θ‐defensins will help to promote their development as therapeutic agents. We anticipate that the next few years will see the discovery of new θ‐defensins and the development of novel applications based on their favorable chemical properties and biological activities.
Biographical Information
Anne Conibear completed her B.Sc.(Hons) and M.Sc. in Chemistry (2010) at Rhodes University, South Africa. She is currently in the final stages of her Ph.D. studies under the supervision of David Craik at the Institute for Molecular Bioscience at The University of Queensland, Australia. Her Ph.D. research focuses on the characterization and applications of θ‐defensins, and she is also interested in the application of NMR techniques in chemistry and biology.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Biographical Information
David Craik received his Ph.D. from La Trobe University, Australia, in 1981 and undertook postdoctoral research at Florida State and Syracuse Universities, USA. In 1988, he was appointed as Head of the School of Pharmaceutical Chemistry at the Victorian College of Pharmacy in Melbourne, Australia. In 1995 he took up a research appointment at the Institute for Molecular Bioscience at The University of Queensland, where his research focuses on applications of NMR spectroscopy in drug design and development.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Acknowledgements
Work in our laboratory on cyclic peptides is funded by the Australian Research Council (grant ID DP0984390) and the National Health and Medical Research Council (grant ID APP1047857). D.J.C. is a NHMRC Professorial Fellow (grant ID APPP1026501). A.C.C. is supported by a University of Queensland International PhD Scholarship.
References
- 1. Craik D. J., Science 2006, 311, 1563–1564. [DOI] [PubMed] [Google Scholar]
- 2. Göransson U., Burman R., Gunasekera S., Strömstedt A. A., Rosengren K. J., J. Biol. Chem. 2012, 287, 27001–27006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lehrer R. I., Cole A. M., Selsted M. E., J. Biol. Chem. 2012, 287, 27014–27019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Montalbán‐López M., Sánchez‐Hidalgo M., Cebrián R., Maqueda M., J. Biol. Chem. 2012, 287, 27007–27013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gonzalez C., Langdon G. M., Bruix M., Galvez A., Valdivia E., Maqueda M., Rico M., Proc. Natl. Acad. Sci. USA 2000, 97, 11221–11226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rosengren K. J., Daly N. L., Plan M. R., Waine C., Craik D. J., J. Biol. Chem. 2003, 278, 8606–8616. [DOI] [PubMed] [Google Scholar]
- 7. Conibear A. C., Rosengren K. J., Harvey P. J., Craik D. J., Biochemistry 2012, 51, 9718–9726. [DOI] [PubMed] [Google Scholar]
- 8. Bushnell D. A., Cramer P., Kornberg R. D., Proc. Natl. Acad. Sci. USA 2002, 99, 1218–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bockus A. T., McEwen C. M., Lokey R. S., Curr. Top. Med. Chem. 2013, 13, 821–836. [DOI] [PubMed] [Google Scholar]
- 10. Craik D. J., Fairlie D. P., Liras S., Price D., Chem. Biol. Drug Des. 2013, 81, 136–147. [DOI] [PubMed] [Google Scholar]
- 11. Giordanetto F., Kihlberg J., J. Med. Chem. 2014, 57, 278–295. [DOI] [PubMed] [Google Scholar]
- 12. Sieber S. A., Marahiel M. A., J. Bacteriol. 2003, 185, 7036–7043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Walsh C. T., Science 2004, 303, 1805–1810. [DOI] [PubMed] [Google Scholar]
- 14. Borel J. F., Feurer C., Gubler H. U., Stahelin H., Agents Actions 1976, 6, 468–475. [DOI] [PubMed] [Google Scholar]
- 15. Clark R. J., Jensen J., Nevin S. T., Callaghan B. P., Adams D. J., Craik D. J., Angew. Chem. 2010, 122, 6695–6698; [Google Scholar]; Angew. Chem. Int. Ed. 2010, 49, 6545–6548. [DOI] [PubMed] [Google Scholar]
- 16. Mohimani H., Liu W.‐T., Mylne J. S., Poth A. G., Colgrave M. L., Tran D., Selsted M. E., Dorrestein P. C., Pevzner P. A., J. Proteome Res. 2011, 10, 4505–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang C. K., Kaas Q., Chiche L., Craik D. J., Nucleic Acids Res. 2008, 36, D206–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Samyn B., Martinez‐Bueno M., Devreese B., Maqueda M., Galvez A., Valdivia E., Coyette J., Van Beeumen J., FEBS Lett. 1994, 352, 87–90. [DOI] [PubMed] [Google Scholar]
- 19. Sivonen K., Leikoski N., Fewer D., Jokela J., Appl. Microbiol. Biotechnol. 2010, 86, 1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kalkum M., Eisenbrandt R., Lanka E., Curr. Protein Pept. Sci. 2004, 5, 417–424. [DOI] [PubMed] [Google Scholar]
- 21. Maqueda M., Sanchez‐Hidalgo M., Fernandez M., Montalban‐Lopez M., Valdivia E., Martinez‐Bueno M., FEMS Microbiol. Rev. 2008, 32, 2–22. [DOI] [PubMed] [Google Scholar]
- 22. Hallen H. E., Luo H., Scott‐Craig J. S., Walton J. D., Proc. Natl. Acad. Sci. USA 2007, 104, 19097–19101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vetter J., Toxicon 1998, 36, 13–24. [DOI] [PubMed] [Google Scholar]
- 24. Luo H., Hallen‐Adams H. E., Walton J. D., J. Biol. Chem. 2009, 284, 18070–18077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaplan C. D., Larsson K.‐M., Kornberg R. D., Mol. Cell 2008, 30, 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Craik D. J., Daly N. L., Bond T., Waine C., J. Mol. Biol. 1999, 294, 1327–1336. [DOI] [PubMed] [Google Scholar]
- 27. Luckett S., Garcia R. S., Barker J. J., Konarev A. V., Shewry P. R., Clarke A. R., Brady R. L., J. Mol. Biol. 1999, 290, 525–533. [DOI] [PubMed] [Google Scholar]
- 28. Tan N.‐H., Zhou J., Chem. Rev. 2006, 106, 840–895. [DOI] [PubMed] [Google Scholar]
- 29. Saether O., Craik D. J., Campbell I. D., Sletten K., Juul J., Norman D. G., Biochemistry 1995, 34, 4147–4158. [DOI] [PubMed] [Google Scholar]
- 30. Gran L., Medd. Nor. Farm. Selsk. 1970, 12, 173–180. [Google Scholar]
- 31. Cemazar M., Kwon S., Mahatmanto T., Ravipati A. S., Craik D. J., Curr. Top. Med. Chem. 2012, 12, 1534–1545. [DOI] [PubMed] [Google Scholar]
- 32. Chiche L., Heitz A., Gelly J. C., Gracy J., Chau P. T., Ha P. T., Hernandez J. F., Le‐Nguyen D., Curr. Protein Pept. Sci. 2004, 5, 341–349. [DOI] [PubMed] [Google Scholar]
- 33. Jennings C., West J., Waine C., Craik D., Anderson M., Proc. Natl. Acad. Sci. USA 2001, 98, 10614–10619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gustafson K. R., McKee T. C., Bokesch H. R., Curr. Protein Pept. Sci. 2004, 5, 331–340. [DOI] [PubMed] [Google Scholar]
- 35. Tam J. P., Lu Y. A., Yang J. L., Chiu K. W., Proc. Natl. Acad. Sci. USA 1999, 96, 8913–8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Svangård E., Göransson U., Hocaoglu Z., Gullbo J., Larsson R., Claeson P., Bohlin L., J. Nat. Prod. 2004, 67, 144–147. [DOI] [PubMed] [Google Scholar]
- 37. Barry D. G., Daly N. L., Clark R. J., Sando L., Craik D. J., Biochemistry 2003, 42, 6688–6695. [DOI] [PubMed] [Google Scholar]
- 38. Gran L., Sandberg F., Sletten K., J. Ethnopharmacol. 2000, 70, 197–203. [DOI] [PubMed] [Google Scholar]
- 39. Henriques S. T., Huang Y.‐H., Castanho M. A. R. B., Bagatolli L. A., Sonza S., Tachedjian G., Daly N. L., Craik D. J., J. Biol. Chem. 2012, 287, 33629–33643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Craik D. J., Malik U., Curr. Opin. Chem. Biol. 2013, 17, 546–554. [DOI] [PubMed] [Google Scholar]
- 41. Craik D. J., Cemazar M., Daly N. L., Curr. Opin. Drug Discov. Devel. 2006, 9, 251–260. [PubMed] [Google Scholar]
- 42. Jagadish K., Camarero J. A., Biopolymers 2010, 94, 611–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Craik D. J., Mylne J. S., Daly N. L., Cell. Mol. Life Sci. 2010, 67, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mylne J. S. D., Colgrave N. L., Daly N. L., Chanson M. L., Elliot A., McCallum A. G., Jones E. J., Craik A., Nat. Chem. Biol. 2011, 7, 257–259. [DOI] [PubMed] [Google Scholar]
- 45. Li P., Jiang S., Lee S.‐L., Lin C. Y., Johnson M. D., Dickson R. B., Michejda C. J., Roller P. P., J. Med. Chem. 2007, 50, 5976–5983. [DOI] [PubMed] [Google Scholar]
- 46. Swedberg J. E., et al., Chem. Biol. 2009, 16, 633–643. [DOI] [PubMed] [Google Scholar]
- 47. Arnison P. G., et al., Nat. Prod. Rep. 2013, 30, 108–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Morita H., Takeya K., Heterocycles 2010, 80, 739–764. [Google Scholar]
- 49. Condie J. A., Nowak G., Reed D. W., Balsevich J. J., Reaney M. J. T., Arnison P. G., Covello P. S., Plant J. 2011, 67, 682–690. [DOI] [PubMed] [Google Scholar]
- 50. Barber C. J. S., Pujara P. T., Reed D. W., Chiwocha S., Zhang H. X., Covello P. S., J. Biol. Chem. 2013, 288, 12500–12510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tang Y.‐Q., Yuan J., Ösapay G., Ösapay K., Tran D., Miller C. J., Ouellette A. J., Selsted M. E., Science 1999, 286, 498–502. [DOI] [PubMed] [Google Scholar]
- 52. Lehrer R. I., Nat. Rev. Microbiol. 2004, 2, 727–738. [DOI] [PubMed] [Google Scholar]
- 53. Selsted M. E., Curr. Protein Pept. Sci. 2004, 5, 365–371. [DOI] [PubMed] [Google Scholar]
- 54. Selsted M. E., Ouellette A. J., Nat. Immunol. 2005, 6, 551–557. [DOI] [PubMed] [Google Scholar]
- 55. Lehrer R. I., Ganz T., Curr. Opin. Immunol. 2002, 14, 96–102. [DOI] [PubMed] [Google Scholar]
- 56. Szyk A., Wu Z. B., Tucker K., Yang D., Lu W. Y., Lubkowski J., Protein Sci. 2006, 15, 2749–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ouellette A. J., Bevins C. L., Inflamm. Bowel. Dis. 2001, 7, 43–50. [DOI] [PubMed] [Google Scholar]
- 58. Schibli D. J., Hunter H. N., Aseyev V., Starner T. D., Wiencek J. M., McCray P. B., Tack B. F., Vogel H. J., J. Biol. Chem. 2002, 277, 8279–8289. [DOI] [PubMed] [Google Scholar]
- 59. Cole A. M., Wang W., Waring A. J., Lehrer R. I., Curr. Protein Pept. Sci. 2004, 5, 373–381. [DOI] [PubMed] [Google Scholar]
- 60. Daly N. L., Chen Y. K., Rosengren K. J., Marx U. C., Phillips M. L., Waring A. J., Wang W., Lehrer R. I., Craik D. J., Biochemistry 2007, 46, 9920–9928. [DOI] [PubMed] [Google Scholar]
- 61. Leonova L., Kokryakov V. N., Aleshina G., Hong T., Nguyen T., Zhao C., Waring A. J., Lehrer R. I., J. Leukocyte Biol. 2001, 70, 461–464. [PubMed] [Google Scholar]
- 62. Tran D., Tran P. A., Tang Y. Q., Yuan J., Cole T., Selsted M. E., J. Biol. Chem. 2002, 277, 3079–3084. [DOI] [PubMed] [Google Scholar]
- 63. Tongaonkar P., Tran P., Roberts K., Schaal J., Ösapay G., Tran D., Ouellette A. J., Selsted M. E., J. Leukocyte Biol. 2011, 89, 283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cole A. M., et al., Proc. Natl. Acad. Sci. USA 2002, 99, 1813–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Garcia A. E., Ösapay G., Tran P. A., Yuan J., Selsted M. E., Infect. Immun. 2008, 76, 5883–5891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stegemann C., Tsvetkova E. V., Aleshina G. M., Lehrer R. I., Kokryakov V. N., Hoffmann R., Rapid Commun. Mass Spectrom. 2010, 24, 599–604. [DOI] [PubMed] [Google Scholar]
- 67. Nguyen T. X., Cole A. M., Lehrer R. I., Peptides 2003, 24, 1647–1654. [DOI] [PubMed] [Google Scholar]
- 68. Penberthy W. T., Chari S., Cole A. L., Cole A. M., Cell. Mol. Life Sci. 2011, 68, 2231–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lucero C. M., et al., Clin. Vacc. Immunol. 2013, 20, 1320–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Palfree R. G. E., Sadro L. C., Solomon S., Mol. Endocrinol. 1993, 7, 199–205. [DOI] [PubMed] [Google Scholar]
- 71. Yang C., Boone L., Nguyen T. X., Rudolph D., Limpakarnjanarat K., Mastro T. D., Tappero J., Cole A. M., Lal R. B., Infect. Genet. Evol. 2005, 5, 11–15. [DOI] [PubMed] [Google Scholar]
- 72. Conlan B. F., Anderson M. A., Curr. Pharm. Des. 2011, 17, 4318–4328. [DOI] [PubMed] [Google Scholar]
- 73. Venkataraman N., Cole A. L., Ruchala P., Waring A. J., Lehrer R. I., Stuchlik O., Pohl J., Cole A. M., PLoS Biol. 2009, 7, e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tongaonkar P., Selsted M. E., J. Biol. Chem. 2009, 284, 5602–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Trabi M., Schirra H. J., Craik D. J., Biochemistry 2001, 40, 4211–4221. [DOI] [PubMed] [Google Scholar]
- 76. Conibear A. C., Bochen A., Rosengren K. J., Stupar P., Wang C., Kessler H., Craik D. J., ChemBioChem 2014, 15, 451–459. [DOI] [PubMed] [Google Scholar]
- 77. Conibear A. C., Rosengren K. J., Daly N. L., Henriques S. T., Craik D. J., J. Biol. Chem. 2013, 288, 10830–10840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Shehu A., Kavraki L. E., Clementi C., Protein Sci. 2008, 17, 482–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Poppe L., Hui J. O., Ligutti J., Murray J. K., Schnier P. D., Anal. Chem. 2012, 84, 262–266. [DOI] [PubMed] [Google Scholar]
- 80. Owen S. M., Rudolph D., Wang W., Cole A. M., Sherman M. A., Waring A. J., Lehrer R. I., Lal R. B., J. Pept. Res. 2004, 63, 469–476. [DOI] [PubMed] [Google Scholar]
- 81. Dawson P. E., Muir T. W., Clark‐Lewis I., Kent S. B., Science 1994, 266, 776–779. [DOI] [PubMed] [Google Scholar]
- 82. Camarero J. A., Muir T. W., Chem. Commun. 1997, 1369–1370. [Google Scholar]
- 83. Aboye T. L., Li Y. L., Majumder S., Hao J. F., Shekhtman A., Camarero J. A., Bioorg. Med. Chem. Lett. 2012, 22, 2823–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kawakami T., Ohta A., Ohuchi M., Ashigai H., Murakami H., Suga H., Nat. Chem. Biol. 2009, 5, 888–890. [DOI] [PubMed] [Google Scholar]
- 85. Gould A., Li Y. L., Majumder S., Garcia A. E., Carlsson P., Shekhtman A., Camarero J. A., Mol. Biosyst. 2012, 8, 1359–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Münk C., Wei G., Yang O. O., Waring A. J., Wang W., Hong T., Lehrer R. I., Landau N. R., Cole A. M., AIDS Res. Hum. Retroviruses 2003, 19, 875–881. [DOI] [PubMed] [Google Scholar]
- 87. Wang W., Cole A. M., Hong T., Waring A. J., Lehrer R. I., J. Immunol. 2003, 170, 4708–4716. [DOI] [PubMed] [Google Scholar]
- 88. Ruchala P., et al., Int. J. Pept. Res. Ther. 2011, 17, 325–336. [Google Scholar]
- 89. Tam J. P., Lu Y. A., Yang J. L., Biochem. Biophys. Res. Commun. 2000, 267, 783–790. [DOI] [PubMed] [Google Scholar]
- 90. Rapireddy S., Nhon L., Meehan R. E., Franks J., Stolz D. B., Tran D., Selsted M. E., Ly D. H., J. Am. Chem. Soc. 2012, 134, 4041–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wang W., et al., J. Biol. Chem. 2006, 281, 32755–32764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Arnett E., Lehrer R. I., Pratikhya P., Lu W., Seveau S., Cell. Microbiol. 2011, 13, 635–651. [DOI] [PubMed] [Google Scholar]
- 93. Ni M., Zhao Y. J., Bibi N., Shao M. Y., Yuan S. N., Fan K., Zhang G. X., Li F., Wang X. D., Appl. Microbiol. Biotechnol. 2013, 97, 2043–2052. [DOI] [PubMed] [Google Scholar]
- 94.K. P. Tai, K. Kamdar, J. Yamaki, V. V. Le, D. Tran, P. Tran, M. E. Selsted, A. J. Ouellette, A. Wong‐Beringer, Innate Immun. 2013, DOI: 10.1177/1753425913514784 [DOI] [PMC free article] [PubMed]
- 95. Abuja P. M., Zenz A., Trabi M., Craik D. J., Lohner K., FEBS Lett. 2004, 566, 301–306. [DOI] [PubMed] [Google Scholar]
- 96. Schmidt N. W., et al., J. Am. Chem. Soc. 2011, 133, 6720–6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Weiss T. M., Yang L., Ding L., Waring A. J., Lehrer R. I., Huang H. W., Biochemistry 2002, 41, 10070–10076. [DOI] [PubMed] [Google Scholar]
- 98. Tang M., Waring A. J., Lehrer R. I., Hong M., Biophys. J. 2006, 90, 3616–3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Seidel A., Ye Y., de Armas L. R., Soto M., Yarosh W., Marcsisin R. A., Tran D., Selsted M. E., Camerini D., PLoS ONE 2010, 5, e9737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Gallo S. A., et al., J. Biol. Chem. 2006, 281, 18787–18792. [DOI] [PubMed] [Google Scholar]
- 101. Fuhrman C. A., Warren A. D., Waring A. J., Dutz S. M., Sharma S., Lehrer R. I., Cole A. L., Cole A. M., FEBS J. 2007, 274, 6477–6487. [DOI] [PubMed] [Google Scholar]
- 102. Wood M. P., Cole A. L., Ruchala P., Waring A. J., Rohan L. C., Marx P., Tarwater P. M., Gupta P., Cole A. M., PLoS ONE 2013, 8, e55478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Owen S. M., Rudolph D. L., Wang W., Cole A. M., Waring A. J., Lal R. B., Lehrer R. I., AIDS Res. Hum. Retroviruses 2004, 20, 1157–1165. [DOI] [PubMed] [Google Scholar]
- 104. Wang Q. W., et al., AIDS Res. Hum. Retroviruses 2007, 23, 508–514. [DOI] [PubMed] [Google Scholar]
- 105. Cole A. L., Yang O. O., Warren A. D., Waring A. J., Lehrer R. I., Cole A. M., J. Immunol. 2006, 176, 6900–6905. [DOI] [PubMed] [Google Scholar]
- 106. Wang W., Owen S. M., Rudolph D. L., Cole A. M., Hong T., Waring A. J., Lal R. B., Lehrer R. I., J. Immunol. 2004, 173, 515–520. [DOI] [PubMed] [Google Scholar]
- 107. Doss M., et al., J. Immunol. 2009, 182, 7878–7887. [DOI] [PubMed] [Google Scholar]
- 108. Leikina E., et al., Nat. Immunol. 2005, 6, 995–1001. [DOI] [PubMed] [Google Scholar]
- 109. Liang Q.‐l., Zhou K., He H.‐x., Biotechnol. Lett. 2010, 32, 387–392. [DOI] [PubMed] [Google Scholar]
- 110. Doss M., et al., J. Immunol. 2012, 188, 2759–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Yasin B., Wang W., Pang M., Cheshenko N., Hong T., Waring A. J., Herold B. C., Wagar E. A., Lehrer R. I., J. Virol. 2004, 78, 5147–5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Brandt C. R., Akkarawongsa R., Altmann S., Jose G., Kolb A. W., Waring A. J., Lehrer R. I., Invest. Ophthalmol. Visual Sci. 2007, 48, 5118–5124. [DOI] [PubMed] [Google Scholar]
- 113. Cole A. M., Lehrer R. I., Curr. Pharm. Des. 2003, 9, 1463–1473. [DOI] [PubMed] [Google Scholar]
- 114. Cole A. M., Cole A. L., Am. J. Reprod. Immunol. 2008, 59, 27–34. [DOI] [PubMed] [Google Scholar]
- 115. Sassi A. B., Cost M. R., Cole A. L., Cole A. M., Patton D. L., Gupta P., Rohan L. C., Antimicrob. Agents Chemother. 2011, 55, 2282–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Ding J., Chou Y.‐Y., Chang T. L., J. Innate Immun. 2009, 1, 413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Schaal J. B., et al., PLoS ONE 2012, 7, e51337. [Google Scholar]
- 118. Wohlford‐Lenane C. L., Meyerholz D. K., Perlman S., Zhou H., Tran D., Selsted M. E., P. B. McCray, Jr. , J. Virol. 2009, 83, 11385–11390. [DOI] [PMC free article] [PubMed] [Google Scholar]