

Abstract
Objective
To compare the genetic regulation of collagen‐induced arthritis (CIA) with that of pristane‐induced arthritis (PIA) in rats.
Methods
A genome‐wide linkage analysis of an (E3 × DA)DA backcross of rats with CIA (n = 364 male rats; the same strain combinations as previously used to determine the genetic control of PIA) was performed. The strongest loci in both CIA and PIA (i.e., Cia12/Pia4 and Cia13/Pia7) were isolated in congenic strains. Susceptibility in both congenic strains was tested in rats with CIA and in rats with PIA.
Results
We found a striking, although not complete, similarity of the arthritis‐controlling loci in CIA and in PIA, as well as the previously defined loci associated with cartilage destruction, antibody production, and the acute‐phase response. All major PIA quantitative trait loci (QTLs) identified in early severe arthritis were also strong regulators of CIA. The 2 strongest QTLs, Cia12/Pia4 on chromosome 12 and Cia13/Pia7 on chromosome 4, were also analyzed in congenic strains with DA or E3 as the background genome. Consistent with the results of linkage analysis, the congenic strain experiments showed that the chromosome 4 locus was more penetrant in CIA than in PIA, while the chromosome 12 locus almost completely dominated the control of PIA severity.
Conclusion
The underlying genetic control of CIA was found to have many, but not all, pathogenic mechanisms in common with PIA, despite the use of a cartilage‐specific antigen (type II collagen) to induce CIA but not PIA.
Rheumatoid arthritis (RA) is a polygenic disease that primarily affects peripheral joints, with chronic inflammation, cartilage and bone destruction, and ultimately, joint deformation. The pathogenesis of RA is poorly understood, and the diagnosis is based on clinical descriptions rather than an understanding of the disease mechanisms (1). RA is believed to be the result of environmental factors in combination with a genetic predisposition to the disease. Several genetic studies of families, twins, and siblings showing familial aggregation and twin and sibling concordance have demonstrated a clear genetic component of arthritis (2, 3). Inheritance of RA has been shown to depend upon the major histocompatibility complex (MHC) haplotype as well as other, hitherto‐unidentified genes with varying influences (4, 5). A recently published linkage analysis of human RA estimated the heritable component of RA to be as high as 60% (5).
Unfortunately, heterogeneity of the population hampers linkage analyses of RA in humans. Therefore, considerable effort has been invested in establishing animal models of disease that resemble RA in humans. These animal models, being inbred, have the advantage of identical genetic composition. Furthermore, since these animals can be studied under environmentally controlled conditions, inherited components of arthritis can be isolated.
There is a range of available rat models of arthritis that more or less resemble RA in humans. These models include cartilage‐restricted antigen‐induced arthritis, such as type II collagen–induced arthritis (CIA) (6, 7), adjuvant‐induced arthritis, such as the classic Mycobacterium‐induced arthritis (8), as well as oil‐induced arthritis (OIA) (9) and pristane‐induced arthritis (PIA) (10). In all these models, the induction protocols are different. In the CIA model, type II collagen (CII) emulsified in Freund's incomplete adjuvant (IFA) is injected. Immunization with the collagen emulsification induces an antibody response followed by clinical manifestations of arthritis (11). In PIA and OIA, adjuvants (pristane and IFA, respectively) are injected to induce arthritis. These nonimmunogenic, adjuvant‐induced diseases are believed to depend largely on the activation of T cells, as shown by α/β T cell depletion and T cell–transferable arthritis but no antibody response to any specific joint antigen (10, 12, 13, 14). Cartilage‐restricted antigen‐induced arthritis involves an antibody‐mediated component in the effector pathway (15).
We previously reported our genetic analyses of PIA in genetic segregating crosses between the arthritis‐susceptible DA rat and the arthritis‐resistant E3 rat (16). In those studies, we demonstrated strong linkage between arthritis phenotypes and several different chromosome regions. Eight quantitative trait loci (QTLs) (Pia1–Pia8), including the MHC region on chromosome 20, have been found to be associated with the clinical manifestations of PIA (10, 16, 17). Two QTLs that control the acute‐phase response (Apr1 and Apr2) have also been correlated with disease (18). Taken together, all these loci have major effects on the outcome of arthritis, reflecting the complexity of the disease even in the context of only 2 genomes.
With regard to CIA in rats, most linkage analyses have been performed in the setting of bovine CII–induced arthritis and in strain combinations other than E3 with DA (19, 20, 21, 22, 23). Therefore, in order to make a reasonable comparison of the genetic inheritance of CIA and PIA in rats, we performed linkage analysis of CIA in the same strain combination and induced by the same collagen source that was used for the PIA studies. Hence, arthritis was induced with homologous rat CII in IFA in a backcross of 364 male (E3 × DA)DA rats. The CIA QTLs obtained were then compared with previously identified PIA‐derived QTLs, thus matching the inheritance of these 2 rat models of arthritis in the same rat strains.
MATERIALS AND METHODS
Animals.
Rats of the E3 and DA strains (Zentralinstitut für Versuchstierzucht, Hannover, Germany) were maintained in our animal facilities in a climate‐controlled environment with 12‐hour cycles of light/dark. Rats were housed in polystyrene cages containing wood shavings and were fed standard rodent chow and water ad libitum. The rats were found to be free of common pathogens, including Sendai virus, Hantaan virus, coronavirus, reovirus, cytomegalovirus, and Mycoplasma pulmonis. Breeding to produce (E3 × DA)DA offspring and arthritis experiments were performed in the same pathogen‐free animal facilities.
Female E3 rats were intercrossed with male DA rats to produce (E3 × DA)F1 offspring that were further backcrossed with DA rats to produce the 364 male (E3 × DA)DA rats that were used in the linkage analysis. DA.Pia4 (D12Rat28 to D12Mgh3; N10) and DA.Pia7 (D4Mit16 to D4Mgh11; N7) congenic rats were obtained through conventional backcross breeding to parental DA rats, with negative selection for all known QTLs and positive selection for microsatellite markers on chromosome 12 or chromosome 4, respectively. The same conventional backcross breeding procedure was used to develop the E3.Pia4 and E3.Pia7 congenic strains (N9). For the arthritis experiments, double‐heterozygous congenic strains with the E3 background were intercrossed. The experimental animals were stratified for Cia12/Pia4 and Cia13/Pia7 genotypes in the analyses.
Induction and evaluation of arthritis.
Lathyritic rat CII was purified from Swarm rat chondrosarcoma grown in male rats that were receiving β‐aminopropionitrile monofumaratic salt in their drinking water during the tumor‐growing period, as previously described (24, 25). CIA was induced by an intradermal injection at the base of the tail with 150 μg of lathyritic rat CII dissolved in 75 μl of 0.1M acetic acid and emulsified in 75 μl of IFA; for the DA.Pia7 congenic strain experiment, 75 μg of lathyritic rat CII was used. PIA was induced by an intradermal injection at the base of the tail with 150 μl of pristane (2,6,10,14‐tetramethylpentadecane; Aldrich, Milwaukee, WI). Rats were ages 8–12 weeks at the time of arthritis induction.
Arthritis development was monitored in all 4 paws and scored according to macroscopic appearance. For each limb, 1 point was given for each swollen or red toe, 1 point for each swollen midfoot, digit, or knuckle, and 5 points for a swollen ankle (maximum score per limb 15). The scores of the 4 limbs were added to yield a total score for each rat (maximum total score per rat 60). Rats were examined 1–4 times each week for 5 months after arthritis induction. Blood was collected (by cutting the tip of the tail) on days 21 and 49 after arthritis induction. To prevent coagulation, 10 μl of heparin (5,000 units/ml; Lövens Läkemedel, Malmö, Sweden) was mixed with 500–1,000 μl of blood. Plasma was separated from blood cells by centrifugation, removed, and stored at −70°C until assayed.
Determination of plasma protein concentrations.
Levels of α1‐acid glycoprotein were measured with a soluble competitive radioimmunoassay (26). Rat α1‐acid glycoprotein (Zivic‐Miller Laboratories, Zelienpole, PA) and a polyclonal rabbit antibody against α1‐acid glycoprotein (Agrisera, Vännäs, Sweden) were used.
Plasma concentrations of cartilage oligomeric matrix protein (COMP) were determined by a competitive enzyme‐linked immunosorbent assay (ELISA), using similar conditions as described elsewhere for determining human COMP concentrations (27), with modifications. Rat COMP was used to coat the microtiter plates and to prepare the standard curve included in each plate, and a polyclonal antiserum raised against rat COMP was used as the capture antibody.
Determination of antibodies.
Antibodies against rat cartilage in plasma were analyzed by ELISA. Briefly, 96‐well plates (Costar, Cambridge, MA) were coated overnight at 4°C with 50 μl/well of phosphate buffered saline (PBS) containing 10 μg/ml of rat CII. All washings were performed with Tris buffered saline (NaCl 1.3M, Tris 0.1M, pH 7.4) containing 0.1% Tween 20. Plasma was diluted in PBS–0.1% Tween 20 and analyzed in duplicate. Levels of bound IgG antibody were estimated after incubation with a donkey anti‐rat IgG coupled to alkaline phosphatase (Jackson ImmunoResearch, West Grove, PA). Paranitrophenol was used as a chromogenic substrate, and the absorbance was determined with a SpectraMax instrument (Molecular Devices, Sunnyvale, CA). The relative amount of plasma antibody was determined by comparison against an anti‐CII–positive control serum. Rheumatoid factor (RF) concentrations were determined in the same manner, except that the plates were coated with 8 μg/ml of rabbit IgG (Sigma‐Aldrich, St. Louis, MO).
Genotyping and linkage analysis.
DNA was prepared from toe biopsy tissues by heating the sample in 1 ml of 50 mM NaOH for 1 hour. The DNA solution was neutralized with 100 μl of 1M Tris buffer and used directly in the polymerase chain reaction (PCR) (28). Primer sequences for rat microsatellite markers defined as DxMity, DxMghy, DxRaty, and DxGoty were obtained from Research Genetics (Huntsville, AL), and those for markers defined as DxWoxy were obtained from the Wellcome Institute for Human Genetics (Oxford, UK). All markers were assayed by PCR on a PTC‐200 Thermal Cycler (MJ Research, Waltham, MA) according to standard protocol. The resulting PCR products were run on an ABI 377 DNA sequencer (Perkin Elmer, Emeryville, CA) or a MegaBACE 1000 sequencer (Amersham Pharmacia Biotech, Uppsala, Sweden), and data were analyzed with the software packages GeneScan 3.1 and either Genotyper 2.1 (Perkin Elmer) or Genetic Profiler 1.1 through comparison with amplified samples from parental strain rats.
To produce linkage maps covering the complete genome, all 364 backcross progeny were genotyped using 238 markers. More than 94% of the rat genome was within 10 cM of 1 microsatellite marker (maximum intermarker distance 28 cM). An improved linkage map based on several crosses involving E3 and DA can be found at our Internet site at http://net.inflam.lu.se.
Map Manager QTX13 software (29) was used to perform the QTL analysis and permutation tests. Chromosomal QTL maps showing the logarithm of the likelihood that a given QTL controlled arthritis or the arthritis‐regulated blood phenotypes were drawn using Qgene 3.06v software (30). Threshold values for significance obtained from the permutation tests were determined by randomizing the phenotypes against the genotypes 500 times in order to calculate relevant significance levels, since permutation calculations based on the features under investigation provide a more accurate estimation of significance levels (31). For a claim of significant linkage, we used a threshold of logarithm of odds (LOD) values ≥2.8, as determined by permutation analysis of the respective traits used for the analysis of the backcross. All phenotype traits were transformed by natural logarithm in order to normalize the distribution of the analyzed data. Quantitative data for congenic strains are expressed as the mean ± SEM. Significance analysis was performed using the nonparametric Mann‐Whitney U test.
RESULTS
Linkage analysis of CIA.
Previously published reports regarding linkages in crosses of the arthritis‐susceptible DA rat with the arthritis‐resistant E3 rat have focused on PIA. Linkages identified in PIA involve regulation of the onset (Pia2 and Pia3), severity (Pia4, Pia7, and Pia8), and chronicity (Pia1, Pia5, and Pia6) of arthritis, as well as the acute inflammatory response, analyzed as plasma concentrations of acute‐phase proteins (Apr1 and Apr2) (10, 16, 17, 18).
The clinical manifestations of CIA are similar to those of PIA, although the genetic susceptibility and pathogenesis are slightly different (32). E3 rats are resistant to both PIA and CIA. DA rats are 100% susceptible to PIA and are slightly less susceptible to CIA. Lower degrees of arthritis severity and later onset of arthritis are also observed in CIA compared with PIA. F1 offspring show complete resistance to CIA, whereas >50% of F1 offspring are affected by PIA, but with late onset and milder arthritis (16, 18) (Table 1).
Table 1.
Clinical arthritis in parental rat strains and segregating crosses of rats with CIA and PIA*
| No. of rats | Day of arthritis onset, mean ± SEM | Maximum clinical score, mean ± SEM | Incidence, % | |
|---|---|---|---|---|
| Rats with CIA | ||||
| E3 | 9 | – | – | 0 |
| DA | 18 | 22 ± 5 | 19 ± 1 | 78 |
| (E3 × DA)F1 | 18 | – | – | 0 |
| (E3 × DA)DA | 364 | 26 ± 1 | 8 ± 1 | 34 |
| Rats with PIA | ||||
| E3 | 10 | – | – | 0 |
| DA | 104 | 12 ± 0.2 | 32 ± 0.7 | 100 |
| (E3 × DA)F1 | 82 | 23 ± 1.0 | 9 ± 1.1 | 59 |
| (E3 × DA)DA | 650 | 16 ± 0.2 | 14 ± 0.6 | 70 |
Values are the mean ± SEM. CIA = type II collagen–induced arthritis; PIA = pristane‐induced arthritis.
We used the CIA model to identify new QTLs inherited from E3 and DA rats and to verify QTLs that correlate between CIA and PIA. In this CIA linkage analysis, 364 male (E3 × DA)DA rats were genotyped using 238 markers, and linkages between genotype and clinical arthritis phenotypes (onset of disease and maximum arthritis severity) or plasma phenotypes (COMP, α1‐acid glycoprotein, and antibody levels) were determined.
In the present study, we identified significant linkage between a locus on chromosome 3 and clinical arthritis. This arthritis‐regulating locus overlaps with the previously reported Cia11 locus, which was identified in experiments using DA and BN rats (20). Other significant loci that regulate clinical arthritis have previously been reported, as follows: on chromosome 4, Cia3/Pia5 and Cia13/Pia7, on chromosome 6, Cia20/Pia3, and on chromosome 12, Cia12/Pia4 (Table 2 and Figure 1A). Four loci were identified as strong regulators of the acute‐phase response (detected as plasma levels of α1‐acid glycoprotein) and cartilage destruction (measured as plasma levels of COMP). The QTLs linked to α1‐acid glycoprotein and COMP were found on chromosomes 4 and 12, respectively, and were co‐inherited with clinical arthritis traits (Cia3/Pia5, Cia13/Pia7, and Cia12/Pia4/Apr1, respectively) (Table 2 and Figure 1B).
Table 2.
Linkages identified in the (E3 × DA)DA rat CIA experiment and previously identified PIA loci*
| Phenotype, SSLP marker | LOD | Inheritance† | CIA QTL‡ | PIA QTL‡ | Other QTLs | ||
|---|---|---|---|---|---|---|---|
| QTL | Ref. | QTL | Ref. | ||||
| Maximum arthritis score | |||||||
| D3mit6 | 3.5 | DA | Cia11 | 20 | – | – | – |
| D4rat109 | 4.7 | DA | Cia3 | 22 | Pia5 | 16 | Aia3, Eau1 |
| D4eno2 | 7.5 | DA | Cia13 | 20 | Pia7 | 17 | Oia2, Ciaa4 |
| D12rat72 | 6.6 | DA | Cia12 | 20 | Pia4 | 16 | Eae5, Eau2 |
| Arthritis onset | |||||||
| D3mit6 | 2.8 | DA | Cia11 | 20 | – | – | – |
| D4rat109 | 5.2 | DA | Cia3 | 22 | Pia5 | 16 | Aia3, Eau1 |
| D4eno2 | 10.8 | DA | Cia13 | 20 | Pia7 | 17 | Oia2, Ciaa4 |
| D6mgh10 | 2.9 | E3 | Cia20 | – | Pia3 | 16 | – |
| D12rat72 | 7.6 | DA | Cia12 | 20 | Pia4 | 16 | Eae5, Eau2 |
| COMP | |||||||
| D4eno2 | 4.0 | DA | Cia13 | 20 | Pia7 | 17 | Oia2, Ciaa4 |
| D12rat72 | 3.6 | DA | Cia12 | 20 | Pia4 | 16 | Eae5, Eau2, Apr1 |
| α1‐acid glycoprotein | |||||||
| D4eno2 | 5.1 | DA | Cia13 | 20 | Pia7 | 17 | Oia2, Ciaa4 |
| D12rat72 | 8.3 | DA | Cia12 | 20 | Pia4 | 16 | Eae5, Eau2, Apr1 |
| Anti‐CII IgG | |||||||
| D1mgh11 | 3.3 | DA | Ciaa6 | – | – | – | Eae6, Eae7 |
| D20mgh4 | 5.8 | E3 | Ciaa1 | 22 | Pia1 | 10 | Cia1, Eae1, Oia1 |
| IgG rheumatoid factors | |||||||
| D5rat37 | 3.0 | DA | Ciaa5 | 19 | – | – | Apr2 |
| D14wox12 | 3.2 | DA | Ciaa7 | – | Pia6 | 16 | Eae10 |
The maximum arthritis score represents the maximum clinical score obtained during the experiment. Arthritis onset represents the first day of visible signs of arthritis after induction of type II collagen–induced arthritis (CIA). Plasma levels of cartilage oligomeric matrix protein (COMP), α1‐acid glycoprotein, anti–type II collagen (anti‐CII) IgG (reactive with rat type II collagen), and IgG rheumatoid factors were measured as described in Materials and Methods. SSLP = simple sequence‐length polymorphism; PIA = pristane‐induced arthritis; LOD = logarithm of odds; QTL = quantitative trait locus.
Inheritance pattern determined as being DA or E3 promoting.
Figure 1.
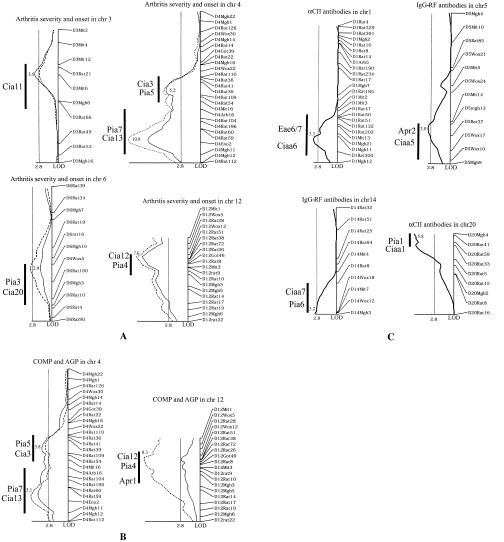
Linkage to clinical arthritis and plasma phenotypes in collagen‐induced arthritis (CIA). A, Logarithm of odds (LOD) score plots showing significant linkage of CIA phenotypes for clinical arthritis, determined as the maximum arthritis score (solid line) and the day of arthritis onset (broken line). B, LOD score plots of the plasma proteins cartilage oligomeric matrix protein (COMP; solid line) and α1‐acid glycoprotein (AGP; broken line) as correlated with arthritis. C, LOD score plots of anti–type II collagen (αCII) antibodies of IgG isotype and IgG rheumatic factors (IgG‐RF). All cosegregating traits are plotted in a single graph. An LOD score of 2.8 was designated as the level of significance, as determined by permutation analysis. Thick vertical bars indicate borders of previously identified quantitative trait loci (QTLs) in the same chromosome region. Individual LOD scores are indicated at maximum peaks. Plot scale represents 10 cM/cm and 2 LOD/cm. chr = chromosome.
Plasma concentrations of IgG‐RF and antibodies directed against CII were analyzed as a reflection of the B cell response to CIA. The production of RF was controlled by loci on chromosomes 5 (Ciaa5/Apr2) and 14 (Ciaa7/Pia6). Linkage to anti‐CII antibodies was identified on chromosome 1 (Ciaa6/Eae6/7 and acute‐phase response). Linkage with anti‐CII antibodies was also identified on chromosome 20 (Ciaa1/Pia1), which contains the MHC region (Table 2 and Figure 1C).
Comparison of linked QTLs between CIA and PIA.
We were unable to address female‐dependent inherited regulation (i.e., Pia2 and Pia8) in the present study since only male rats were used. The remaining published PIA QTLs (i.e., Pia1, Pia3, Pia4, Pia5, Pia6, and Pia7, as well as Apr1 and Apr2) were identified using the CIA model on the (E3 × DA)DA cross. Pia5 and Pia6 are both linked to chronic arthritis in PIA; however, in the CIA backcross analysis, they were associated with arthritis severity and RF production, respectively. Whether these loci were also associated with chronic CIA could not be determined, since the analysis was performed only during the acute phase of disease (<50 days). A schematic representation of the rat genome is depicted in Figure 2, with all linkages obtained in this CIA analysis indicated together with previously published loci from PIA linkage analyses using E3 and DA parental rats.
Figure 2.
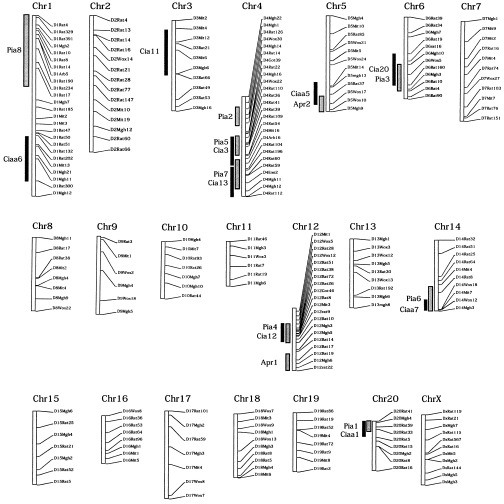
Schematic genome view of the inheritance of pristane‐induced arthritis (PIA) and collagen‐induced arthritis (CIA) in linkage analyses of E3 and DA rats. Results of linkage analyses from the present study of CIA are shown together with previously published linkage analyses of PIA, acute‐phase response, and antibody level. Thick black vertical bars show linkages obtained with the CIA model, using identical parental founder strains (E3 × DA); thick shaded vertical bars show the published quantitative trait locus regions of PIA linkage. Chr = chromosome.
Confirmation of the major loci in congenic strains.
Two of the major QTLs that regulate arthritis severity are the Cia12/Pia4 locus on chromosome 12 and the Cia13/Pia7 locus on chromosome 4 (16, 17). We produced congenic strains of the chromosome 12 region (DA.Pia4 and DA.Pia7) and the chromosome 4 region (DA.Pia4 and DA.Pia7) by transferring the Cia12/Pia4 or the Cia13/Pia7 locus, respectively, from the arthritis‐resistant E3 strain to the arthritis‐susceptible DA strain. By using both PIA and CIA, the genetic fragments in the Cia12/Pia4 (Figure 3) and the Cia13/Pia7 (Figure 4) regions were confirmed to have a strong ameliorating effect on arthritis. The ameliorating effect of Cia12/Pia4 was more pronounced in PIA than in CIA, whereas the opposite was observed in the Cia13/Pia7 congenic strain.
Figure 3.
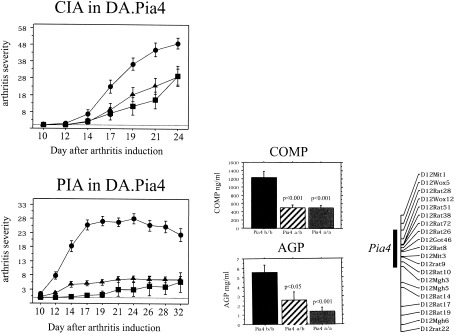
Clinical arthritis in collagen‐induced arthritis (CIA) and pristane‐induced arthritis (PIA) in DA.Pia4 and DA.Pia7 congenic rats. Arthritis severity was determined in DA rats (•; n = 13 for CIA and n = 16 for PIA), DA.Pia4 E3/DA rats (▴; n = 16 in each group), and DA.Pia4 E3/E3 rats (▪; n = 7 for CIA and n = 16 for PIA). There were significant differences between DA and DA.Pia4 and DA.Pia7 congenic rats (P < 0.01 for CIA, showing a significant difference beginning on day 17; P < 0.0001 for PIA, showing a significant difference beginning on day 10, as determined by Mann‐Whitney U test). Levels of cartilage oligomeric matrix protein (COMP) and α1‐acid glycoprotein (AGP) were significant in the DA.Pia4 a/a (E3/E3) and DA.Pia4 a/b (E3/DA) rats compared with the DA.Pia4 b/b (DA/DA) rats (a = E3 allele; b = DA allele). The size of the Cia12/Pia4 congenic fragment is depicted in a schematic representation of chromosome 12.
Figure 4.
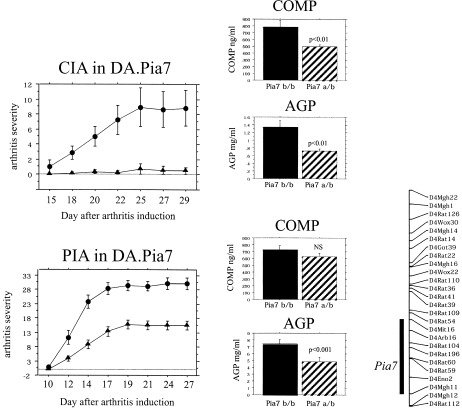
Clinical arthritis in collagen‐induced arthritis (CIA) and pristane‐induced arthritis (PIA) in DA.Pia4 and DA.Pia7 congenic rats. Arthritis severity was determined in DA rats (•; n = 26 for CIA and n = 21 for PIA) and DA.Pia7 E3/DA rats (▴; n = 21 for CIA and n = 29 for PIA). There were significant differences between the DA rats and the DA.Pia4 and DA.Pia7 congenic rats (P < 0.001 for CIA, showing a significant difference beginning on day 18; P < 0.001 for PIA, showing a significant difference beginning on day 12, as determined by Mann‐Whitney U test). Levels of cartilage oligomeric matrix protein (COMP) and α1‐acid glycoprotein (AGP) were significant in Pia7 a/b rats compared with the Pia7 b/b rats (a = E3 allele; b = DA allele). The size of the Cia13/Pia7 congenic fragment is depicted in a schematic representation of chromosome 12.
Since both these loci also were shown to regulate plasma levels of COMP (reflecting cartilage destruction) and α1‐acid glycoprotein (reflecting systemic inflammation), plasma concentrations of these proteins during the acute phase of the disease were evaluated. The DA.Pia4 congenic strain, which showed the greatest difference in the presence of PIA compared with CIA, showed significantly decreased COMP and α1‐acid glycoprotein levels. Unfortunately, no blood samples were obtained during the CIA experiment, and these blood proteins therefore could not be analyzed.
In the DA.Pia7 congenic strain, both COMP and α1‐acid glycoprotein were significantly decreased in rats with CIA, whereas in rats with PIA, only the α1‐acid glycoprotein level was decreased. To further analyze the different effects of these 2 loci on arthritis, reciprocal congenic strains were developed. Both PIA and CIA were tested in an intercross experiment between (E3.Pia4 × E3.Pia7)F1 congenic rats. We observed that in the presence of CIA, the Cia13/Pia7 locus had the most significant impact, whereas the Cia12/Pia4 locus had no significant effect in the E3 background (Figure 5). None of these congenic strains was susceptible to PIA.
Figure 5.
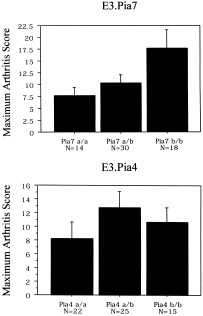
Maximum arthritis scores in (E3.Pia4 × E3.Pia7)F1 intercrossed rats with collagen‐induced arthritis, stratified for Pia7 and Pia4. Significantly more arthritis was induced through carrying 2 DA alleles (b/b) of Cia13/Pia7 (P < 0.05), whereas no effect DA.Cia12/Pia4 was observed. a = E3 allele.
DISCUSSION
There is a striking resemblance between the genetic control of PIA and the genetic control of CIA, as shown by linkage analysis and studies of congenic strains. The present study was designed to be highly discriminating. We used 364 male rats of the (E3 × DA)DA backcross and analyzed arthritis onset and severity as well as antibody levels, RF production, and acute‐phase response. Since this has also been done in E3 × DA crosses after injection with pristane (10, 16, 17, 18), a careful comparison between these arthritis models could be made. In the CIA linkage experiment, we were able to identify all published PIA loci except for those related strictly to female inheritance (Pia2 and Pia8) and to chronicity (Pia5 and Pia6), since the analysis was performed on male rats during the acute phase of arthritis. However, the relative contribution of the various loci to the traits we examined differed, and we found additional loci associated with arthritis and anti‐CII antibody production, reflecting the divergent pathogeneses of these diseases.
Arthritis is inducible in rats by the injection of CII emulsified in either IFA (mineral oil) or CFA, as in CIA, or by the injection of pure pristane oil, as in PIA. Therefore, the major difference between PIA and CIA is the use of CII in the immunization emulsion in CIA. CIA and PIA are clinically very similar, aside from the fact that CIA affects the phalangeal joints more profoundly than does PIA and that relapses are more prominent during chronic disease in PIA. PIA is a T cell–dependent disease, although the specificity of the autoimmune reaction is unknown (10). CIA is more complex, having the added influence of B cells due to the in vivo affinity of CII‐specific antibodies for cartilage (34). Since CIA shares T cell dependence with PIA but has the additional antibody‐dependent mechanism, it is expected that CIA would share some genetic linkages with PIA as well as demonstrate some specific genetic regulation. Loci identified in both CIA and PIA may therefore reflect genes involved in regulating the T cell response caused by the injection of adjuvant, while the CIA‐specific QTLs may be involved in the more antigen‐specific response directed against CII.
CIA loci that were found to regulate the onset and severity of arthritis (i.e., chromosomes 3, 4, 6, and 12, which are equivalent to Cia11 and Cia3/Pia5, Cia12/Pia7, Cia20/Pia3, and Cia12/Pia4, respectively) were identical in CIA and PIA, except for the Cia11 locus. These findings suggest that these loci harbor genes that control the general inflammatory response elicited by the adjuvant. The antibody QTLs in CIA found on chromosomes 20 and 14 are equivalent to Ciaa1/Pia1 and Ciaa7/Pia6, respectively. Both Pia1 and Pia6 are loci that regulate chronic arthritis. Hence, the identification of these loci with the use of anti‐CII antibodies or RFs in CIA possibly reflects an antibody‐regulated phase, which is observed in the chronic phase of PIA. In CIA, an immune response to joint‐specific antigen (CII) is triggered soon after immunization, which possibly accounts for the involvement of these loci in acute disease. However, in PIA, where both Pia1 and Pia6 play a role strictly during the chronic phase of disease, these loci may be triggered only after exposure of cartilage protein to the immune system and breakage of immune tolerance due to the ongoing joint erosion and inflammation.
In previous linkage analyses of PIA using genetic segregation between E3 and DA rats, the Cia12/Pia4 locus on chromosome 12 was found to be the strongest regulator of arthritis in these strains (16). This locus also showed a strong effect on disease, since the E3 allele of this locus conferred 90% protection against arthritis severity in PIA (Figure 3). Recently, the Cia12/Pia4 QTL was found to confer structural polymorphism in the Ncf1 gene (35). The effect of this genetic variation resulted in differential production of free radicals by the NADPH oxidase complex (36). We hypothesized that the production of oxygen radicals by the antigen‐presenting cells reduces the priming of autoreactive T cells. We found that in CIA, the effect of the Cia12/Pia4 locus was less potent, while Cia13/Pia7 had the greatest effect on clinical arthritis. The different effects of these 2 loci were clarified in isolated congenic strains of both DA and E3 backgrounds (Figures 4 and 5), where it was clear that the Cia13/Pia7 locus was of greatest importance in the regulation of CIA. This result is highly significant, since it suggests that the Cia13/Pia7 locus on chromosome 4 represents genes that are involved in the processing/presentation of, and response to, joint antigens. Furthermore, we showed the usefulness of different disease‐reflecting phenotypes, such as COMP and α1‐acid glycoprotein, in characterizing and evaluating the impact of QTLs, both in linkage studies and in studies of congenic strains.
There have been several reports of other arthritis models with a matching position of the loci of importance in controlling different types of arthritis. For example, both oil‐induced arthritis and squalene‐induced arthritis are regulated by a QTL on rat chromosome 10, which was successfully isolated in a congenic strain (37). Interestingly, Oia2, the locus corresponding to Cia13/Pia7, was also recently isolated and characterized in a congenic strain in which the disease‐protective chromosome 4 allele from PVG rats was transferred to the DA background. This congenic strain also showed significant protection against PIA, CIA, squalene‐induced arthritis, and adjuvant‐induced arthritis (38). Thus, it is plausible that the Cia13, Oia2, and Pia7 loci share a gene in E3 and PVG rats that also confers disease protection in the DA strain. Moreover, Mycobacterium butyricum adjuvant–induced arthritis has been found to be regulated by previously identified and isolated CIA loci, such as Cia5 (Aia5) (39). These findings stress the importance of using several models to study the regulation of arthritis by the QTLs of interest, since information concerning the function of the underlying genes may be identified. This would be especially important in studies of arthritis in rats, in which both antigen‐induced and adjuvant‐induced disease (PIA or oil‐induced arthritis) can be investigated (40).
In conclusion, PIA and CIA share most of the arthritis susceptibility loci. The majority are involved in T cell regulation induced by the adjuvant, with or without the addition of cartilage‐specific proteins; however, loci that regulate the B cell response (observed as antibody production) are also involved. Thus, it is possible that our results can be extended to all rat models of arthritis. It is therefore important that the arthritis‐regulating genes be identified and evaluated in as many rat models of arthritis as possible. The information gathered from such genetic analyses in rats will eventually help unravel the pathways involved in human RA and may provide novel therapeutic targets for the treatment of autoimmune disease.
REFERENCES
- 1. Feldmann M, Brennan FM, Maini RN. Rheumatoid arthritis. Cell 1996; 85: 307–10. [DOI] [PubMed] [Google Scholar]
- 2. Aho K, Koskenvuo M, Tuominen J, Kaprio J. Occurrence of rheumatoid arthritis in a nationwide series of twins. J Rheumatol 1986; 13: 899–902. [PubMed] [Google Scholar]
- 3. Silman AJ, MacGregor AJ, Thomson W, Holligan S, Carthy D, Farhan A, et al. Twin concordance rates for rheumatoid arthritis: results from a nationwide study. Br J Rheumatol 1993; 32: 903–7. [DOI] [PubMed] [Google Scholar]
- 4. Cornelis F, Faure S, Martinez M, Prud'homme JF, Fritz P, Dib C, et al. New susceptibility locus for rheumatoid arthritis suggested by a genome‐wide linkage study. Proc Natl Acad Sci U S A 1998; 95: 10746–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. MacGregor AJ, Snieder H, Rigby AS, Koskenvuo M, Kaprio J, Aho K, et al. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum 2000; 43: 30–7. [DOI] [PubMed] [Google Scholar]
- 6. Trentham DE, Townes AS, Kang AH. Autoimmunity to type II collagen: an experimental model of arthritis. J Exp Med 1977; 146: 857–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Griffiths MM, DeWitt CW. Genetic control of collagen‐induced arthritis in rats: the immune response to type II collagen among susceptible and resistant strains and evidence for multiple gene control. J Immunol 1984; 132: 2830–6. [PubMed] [Google Scholar]
- 8. Pearson CM. Development of arthritis, periarthritis and periostitis in rats given adjuvants. Proc Soc Exp Biol Med 1956; 91: 91–101. [DOI] [PubMed] [Google Scholar]
- 9. Kleinau S, Erlandsson H, Holmdahl R, Klareskog L. Adjuvant oils induce arthritis in the DA rat. I. Characterization of the disease and evidence for an immunological involvement. J Autoimmun 1991; 4: 871–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vingsbo C, Sahlstrand P, Brun JG, Jonsson R, Saxne T, Holmdahl R. Pristane‐induced arthritis in rats: a new model for rheumatoid arthritis with a chronic disease course influenced by both major histocompatibility complex and non‐major histocompatibility complex genes. Am J Pathol 1996; 149: 1675–83. [PMC free article] [PubMed] [Google Scholar]
- 11. Holmdahl R, Vingsbo C, Malmstrom V, Jansson L, Holmdahl M. Chronicity of arthritis induced with homologous type II collagen (CII) in rats is associated with anti‐CII B‐cell activation. J Autoimmun 1994; 7: 739–52. [DOI] [PubMed] [Google Scholar]
- 12. Carlson BC, Jansson AM, Larsson A, Bucht A, Lorentzen JC. The endogenous adjuvant squalene can induce a chronic T cell‐mediated arthritis in rats. Am J Pathol 2000; 156: 2057–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jansson AM, Lorentzen JC, Bucht A. CD8+ cells suppress oil‐induced arthritis. Clin Exp Immunol 2000; 120: 532–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kleinau S, Klareskog L. Oil‐induced arthritis in DA rats: passive transfer by T cells but not with serum. J Autoimmun 1993; 6: 449–58. [DOI] [PubMed] [Google Scholar]
- 15. Taurog JD, Kerwar SS, McReynolds RA, Sandberg GP, Leary SL, Mahowald ML. Synergy between adjuvant arthritis and collagen‐induced arthritis in rats. J Exp Med 1985; 162: 962–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vingsbo‐Lundberg C, Nordquist N, Olofsson P, Sundvall M, Saxne T, Pettersson U, et al. Genetic control of arthritis onset, severity and chronicity in a model for rheumatoid arthritis in rats. Nat Genet 1998; 20: 401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nordquist N, Olofsson P, Vingsbo‐Lundberg C, Pettersson U, Holmdahl R. Complex genetic control in a rat model for rheumatoid arthritis. J Autoimmun 2000; 15: 425–32. [DOI] [PubMed] [Google Scholar]
- 18. Olofsson P, Nordquist N, Vingsbo‐Lundberg C, Larsson A, Falkenberg C, Pettersson U, et al. Genetic links between the acute‐phase response and arthritis development in rats. Arthritis Rheum 2002; 46: 259–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Furuya T, Salstrom JL, McCall‐Vining S, Cannon GW, Joe B, Remmers EF, et al. Genetic dissection of a rat model for rheumatoid arthritis: significant gender influences on autosomal modifier loci. Hum Mol Genet 2000; 9: 2241–50. [DOI] [PubMed] [Google Scholar]
- 20. Griffiths MM, Wang J, Joe B, Dracheva S, Kawahito Y, Shepard JS, et al. Identification of four new quantitative trait loci regulating arthritis severity and one new quantitative trait locus regulating autoantibody production in rats with collagen‐induced arthritis. Arthritis Rheum 2000; 43: 1278–89. [DOI] [PubMed] [Google Scholar]
- 21. Gulko PS, Kawahito Y, Remmers EF, Reese VR, Wang J, Dracheva SV, et al. Identification of a new non–major histocompatibility complex genetic locus on chromosome 2 that controls disease severity in collagen‐induced arthritis in rats. Arthritis Rheum 1998; 41: 2122–31. [DOI] [PubMed] [Google Scholar]
- 22. Remmers EF, Longman RE, Du Y, O'Hare A, Cannon GW, Griffiths MM, et al. A genome scan localizes five non‐MHC loci controlling collagen‐induced arthritis in rats. Nat Genet 1996; 14: 82–5. [DOI] [PubMed] [Google Scholar]
- 23. Dracheva SV, Remmers EF, Gulko PS, Kawahito Y, Longman RE, Reese VR, et al. Identification of a new quantitative trait locus on chromosome 7 controlling disease severity of collagen‐induced arthritis in rats. Immunogenetics 1999; 49: 787–91. [DOI] [PubMed] [Google Scholar]
- 24. Smith BD, Martin GR, Miller EJ, Dorfman A, Swarm R. Nature of the collagen synthesized by a transplanted chondrosarcoma. Arch Biochem Biophys 1975; 166: 181–6. [DOI] [PubMed] [Google Scholar]
- 25. Miller EJ, Rhodes RK. Preparation and characterization of the different types of collagen. Methods Enzymol 1982; 82: 33–64. [DOI] [PubMed] [Google Scholar]
- 26. Åkerström B. Immunological analysis of α1‐microglobulin in different mammalian and chicken serum: α1‐microglobulin is 5–8 kilodaltons larger in primates. J Biol Chem 1985; 260: 4839–44. [PubMed] [Google Scholar]
- 27. Saxne T, Heinegård D. Cartilage oligomeric matrix protein: a novel marker of cartilage turnover detectable in synovial fluid and blood [published erratum appears in Br J Rheumatol 1993;32:247]. Br J Rheumatol 1992; 31: 583–91. [DOI] [PubMed] [Google Scholar]
- 28. Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, Warman ML. Preparation of PCR‐quality mouse genomic DNA with hot sodium hydroxide and Tris (HotSHOT). Biotechniques 2000; 29: 52–54. [DOI] [PubMed] [Google Scholar]
- 29. Manly KF, Cudmore RH Jr, Meer JM. Map Manager QTX, cross‐platform software for genetic mapping. Mamm Genome 2001; 12: 930–2. [DOI] [PubMed] [Google Scholar]
- 30. Nelson J. QGENE: software for marker‐based genomic analysis and breeding. Mol Breeding 1997; 239–45. [Google Scholar]
- 31. Churchill GA, Doerge RW. Empirical threshold values for quantitative trait mapping. Genetics 1994; 138: 963–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Holmdahl R, Lorentzen JC, Lu S, Olofsson P, Wester L, Holmberg J, et al. Arthritis induced in rats with nonimmunogenic adjuvants as models for rheumatoid arthritis. Immunol Rev 2001; 184: 184–202. [DOI] [PubMed] [Google Scholar]
- 33. Bergsteinsdottir K, Yang HT, Pettersson U, Holmdahl R. Evidence for common autoimmune disease genes controlling onset, severity, and chronicity based on experimental models for multiple sclerosis and rheumatoid arthritis. J Immunol 2000; 164: 1564–8. [DOI] [PubMed] [Google Scholar]
- 34. Kraetsch HG, Unger C, Wernhoff P, Schneider C, Kalden JR, Holmdahl R, et al. Cartilage‐specific autoimmunity in rheumatoid arthritis: characterization of a triple helical B cell epitope in the integrin‐binding‐domain of collagen type II. Eur J Immunol 2001; 31: 1666–73. [DOI] [PubMed] [Google Scholar]
- 35. Olofsson P, Holmberg J, Tordsson J, Lu S, Åkerström B, Holmdahl R. Positional identification of Ncf1 as a gene that regulates arthritis severity in rats. Nat Genet 2003; 33: 25–32. [DOI] [PubMed] [Google Scholar]
- 36. Babior BM. NADPH oxidase: an update. Blood 1999; 93: 1464–76. [PubMed] [Google Scholar]
- 37. Holm BC, Xu HW, Jacobsson L, Larsson A, Luthman H, Lorentzen JC. Rats made congenic for Oia3 on chromosome 10 become susceptible to squalene‐induced arthritis. Hum Mol Genet 2001; 10: 565–72. [DOI] [PubMed] [Google Scholar]
- 38. Bäckdahl L, Ribbhammar U, Lorentzen JC. Mapping and functional characterization of rat chromosome 4 regions that regulate arthritis models and phenotypes in congenic strains. Arthritis Rheum 2003; 48: 551–9. [DOI] [PubMed] [Google Scholar]
- 39. Joe B, Cannon GW, Griffiths MM, Dobbins DE, Gulko PS, Wilder RL, et al. Evaluation of quantitative trait loci regulating severity of mycobacterial adjuvant‐induced arthritis in monocongenic and polycongenic rats: identification of a new regulatory locus on rat chromosome 10 and evidence of overlap with rheumatoid arthritis susceptibility loci. Arthritis Rheum 2002; 46: 1075–85. [DOI] [PubMed] [Google Scholar]
- 40. Remmers EF, Joe B, Griffiths MM, Dobbins DE, Dracheva SV, Hashiramoto A, et al. Modulation of multiple experimental arthritis models by collagen‐induced arthritis quantitative trait loci isolated in congenic rat lines: different effects of non–major histocompatibility complex quantitative trait loci in males and females. Arthritis Rheum 2002; 46: 2225–34. [DOI] [PubMed] [Google Scholar]
- 41. Kawahito Y, Cannon GW, Gulko PS, Remmers EF, Longman RE, Reese VR, et al. Localization of quantitative trait loci regulating adjuvant‐induced arthritis in rats: evidence for genetic factors common to multiple autoimmune diseases. J Immunol 1998; 161: 4411–9. [PubMed] [Google Scholar]
- 42. Lorentzen JC, Glaser A, Jacobsson L, Galli J, Fakhrai‐rad H, Klareskog L, et al. Identification of rat susceptibility loci for adjuvant‐oil‐induced arthritis. Proc Natl Acad Sci U S A 1998; 95: 6383–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sun SH, Silver PB, Caspi RR, Du Y, Chan CC, Wilder RL, et al. Identification of genomic regions controlling experimental autoimmune uveoretinitis in rats. Int Immunol 1999; 11: 529–34. [DOI] [PubMed] [Google Scholar]