

Abstract
Pathogen‐selective labeling was achieved by using the novel gemcitabine metabolite analogue 2′‐deoxy‐2′,2′‐difluoro‐5‐ethynyluridine (dF‐EdU) and click chemistry. Cells infected with Herpes Simplex Virus‐1 (HSV‐1), but not uninfected cells, exhibit nuclear staining upon the addition of dF‐EdU and a fluorescent azide. The incorporation of the dF‐EdU into DNA depends on its phosphorylation by a herpes virus thymidine kinase (TK). Crystallographic analyses revealed how dF‐EdU is well accommodated in the active site of HSV‐1 TK, but steric clashes prevent dF‐EdU from binding human TK. These results provide the first example of pathogen‐enzyme‐dependent incorporation and labeling of bioorthogonal functional groups in human cells.
Keywords: Bioorthogonale Chemie, Chemische Reporter, Klick‐Chemie, Nukleoside, Viren
Abstract
Pathogeninduzierte Metabolitenmarkierung: Mit dem Herpes‐simplex‐Virus 1 (HSV‐1) infizierte Zellen wurden selektiv mit dem Gemcitabin‐Metabolitenanalogon dF‐EdU und einem azidkonjugierten Fluorophor markiert. Die Selektivität dieses Prozesses stützt sich auf die Phosphorylierung von dF‐EdU durch virale, jedoch nicht durch humane Thymidin‐Kinasen. Somit steht ein neuer pathogenspezifischer bioorthogonaler chemischer Markierungsansatz offen.

Bioorthogonal chemical reporters selectively modify biomolecules in their native context.1 In this approach, endogenous biosynthetic pathways metabolically incorporate a bioorthogonal functional group (e.g., azide, alkyne, alkene) into a biological macromolecule. Subsequent click reactions such as [2+3] alkyne–azide and [2+4] alkene–tetrazine cycloadditions can be used to visualize and/or capture the labeled biomolecules.2, 3 Labeling is usually selective with respect to macromolecule type, but nonselective with respect to cell type. This imposes severe limitations on the study of specific pathways in whole systems.4 Advances in cell‐type‐selective labeling have recently been published,5 but the detection of a pathogenic organism in human cells by using a bioorthogonal chemical reporter has not so far been demonstrated. Based on the fundamental principles of medicinal chemistry,6 we anticipated that pathogen‐specific reporters could be developed from “clickable” biomolecular building blocks that are metabolized by pathogen‐infected cells but rejected by healthy ones. To explore this concept, we selected Herpes Simplex Virus‐1 (HSV‐1) as a model system.
Recent studies have demonstrated that modified nucleosides containing a terminal alkyne can be metabolically incorporated into the genomes of adenovirus, herpes virus, vaccinia virus, and papillomavirus.7 After screening a collection of ethynyl nucleosides including 5‐ethynyl‐2′‐deoxyuridine (EdU),8 (2′S)‐2‐deoxy‐2′‐fluoro‐5‐ethynyluridine (F‐ara‐EdU),9 5‐ethynyl‐2′‐deoxycytidine (EdC),10 and 7‐deaza‐7‐ethynyl‐2′‐deoxyadenosine (EdA),11 we discovered that ethynyl‐modified viral genomes could be produced without negatively impacting viral infectivity or egress.7 All of these nucleosides, however, were also incorporated into cellular genomes.8–11 In samples in which the cells and viruses were simultaneously replicating, viral DNA could not be detected over the large background of cellular DNA.7
Herein, we report a strategy for pathogen‐selective labeling that utilizes the relaxed fidelity of pathogen‐encoded enzymes.6 The new gemcitabine metabolite analogue 2′‐deoxy‐2′,2′‐difluoro‐5‐ethynyluridine (dF‐EdU) is selectively metabolized in HSV‐1 infected cells owing to the expression of a viral thymidine kinase (TK).12 Subsequent treatment with CuI and an azide‐conjugated fluorophore stains cells harboring an HSV‐1 infection but not uninfected cells. In addition to the large number of viruses that encode nucleoside kinases,13a this general approach should be applicable to viruses that encode error‐prone polymerases (HIV, influenza virus, rhinovirus, poliovirus, coronavirus, etc.),13b,c as well as parasitic mycoplasma that encode unique purine salvage enzymes.13d
The chemotherapeutic drug gemcitabine (dFdC, 1) is deaminated in vivo14 to give 2′‐deoxy‐2′,2′‐difluorouridine (dFdU, 2; Scheme 1). Both dFdC and dFdU are phosphorylated and incorporated into nucleic acids by endogenous human enzymes.15 dFdU is significantly less toxic than dFdC16 and was therefore selected for further development as a metabolic label. dFdU derivatives containing substituents at the 5‐position, such as 2′‐deoxy‐2′,2′‐difluoro‐5‐bromouridine (BrdFdU), are nontoxic to mammalian cell cultures but they inhibit herpes virus replication.17 These results suggest the presence of BrdFdU‐selective metabolism by one or more virus‐encoded enzymes, including herpes virus TK. We therefore selected the 5‐position of dFdU as the attachment site for a bioorthogonal functional group. While a variety of bioorthogonal functional groups, including methylazide,2g vinyl,3e and ethynyl8–10 groups can be utilized at this position, an ethynyl group was selected for its robust biocompatibility with DNA synthesis. According to these design criteria, dF‐EdU (3) was synthesized from gemcitabine over four steps with a total yield of isolated product of 42 % (Scheme S1 in the Supporting Information).
Scheme 1.
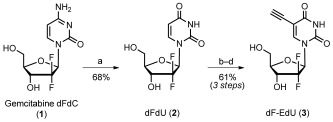
Synthesis of 2′‐deoxy‐2′‐difluoro‐5‐ethynyluridine (dF‐EdU, 3). a) Isopentyl nitrite, 0.1 N HCl, 70 °C, overnight; b) Ce(NH4)2(NO3)6, I2, HOAc, 80 °C, 2 h; c) ethynyltrimethylsilane, Pd(PPh3)2Cl2, CuI, Et3N, DMF, RT, 60 min; d) NaOH/H2O/MeOH, rt, 15 min. DMF=dimethylformamide.
To evaluate the phosphorylation of dF‐EdU in vitro, HSV‐1 thymidine kinase (HSV1‐TK) or human thymidine kinase 1 (hTK) were incubated with dF‐EdU in solutions containing ATP. Reversed‐phase ion‐pair chromatography was used to quantify the conversion of ATP into ADP, as well as the conversion of dF‐EdU into dF‐EdU‐5′‐monophosphate (dF‐EdUMP). As a positive control, hTK was incubated with 2′‐deoxythymidine (dT; Figure 1 a). According to this assay, hTK was unable to catalyze the phosphorylation of dF‐EdU (Figure 1 b). In contrast, when HSV1‐TK was used in place of hTK, ATP was converted into ADP and dF‐EdU was converted into dF‐EdUMP (Figure 1 c, d and Figure S1 in the Supporting Information).
Figure 1.
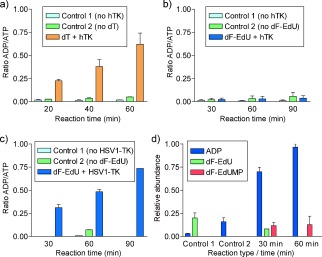
a, b) In vitro phosphorylation of dT (a) or dF‐EdU (b) by hTK. c, d) Phosphorylation of dF‐EdU by HSV1‐TK according to the relative quantities of ADP, ATP, dF‐EdU, and dF‐EdUMP. In all panels, Control 1 lacks the kinase and Control 2 lacks the nucleoside.
To evaluate the structural basis of the TK selectivity of dF‐EdU, a comparative crystallographic analysis was conducted. Crystals of HSV1‐TK were soaked in solutions containing dF‐EdU and the X‐ray data solved at 2.1 Å resolution by molecular replacement. The overall α/β architecture of the TK homodimer was similar to previous HSV1‐TK structures.18, 19 The cocrystal structure was refined to an R factor of 17.8 % and R free of 22.2 % (Table S1 in the Supporting Information) to provide the first reported structure of a TK bound to an ethnyl nucleoside. The (F obs−F calc) electron density map contoured at 2.5 σ was clearly interpretable, with dF‐EdU in the active site, along with a sulfate ion in the ATP binding pocket (Figure 2 a). The 5‐ethynyl group of dF‐EdU is accommodated in a hydrophobic pocket defined by the side chains of Trp 88, Tyr 132, Ala 167, and Ala 168. The sugar moiety of dF‐EdU adopts a 2′‐endo conformation, forming hydrogen bonds with Glu 83, Arg 163, and Glu 225 in an analogous fashion to that of dT itself (Figure S2).20 These observations are consistent with the ability of dF‐EdU to be phosphorylated by HSV1‐TK.
Figure 2.
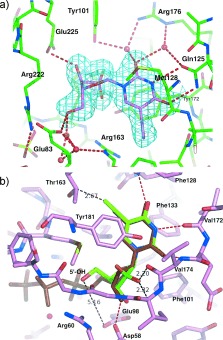
a) Cocrystal structure of dF‐EdU (violet) bound in the active site of HSV1‐TK (green, PDB entry code 4OQL). The electron density map (2F obs−F calc) of dF‐EdU with a resolution of 2.1 Å is contoured at 1 σ and shown in cyan. Water molecules are depicted as red spheres and hydrogen bonds as dashed red lines. b) Superposition of dF‐EdU (green) onto dTTP (brown) within the active site of hTK (pink, PDB entry code 1W4R).22
To evaluate why dF‐EdU is rejected by hTK, a superimposition model of dF‐EdU onto a dTTP‐hTK cocrystal was generated by using the “Superpose ligand” function in the molecular graphic program COOT (Figure 2 b).21 According to this model, the terminal ethynyl carbon of dF‐EdU is only 2.67 Å from the Cβ of Thr 163, a position known to mediate steric control over substrate specificity.22 This potential steric clash is insufficient for the rejection of dF‐EdU because 5‐ethynyl‐2′‐deoxyuridine (EdU) is phosphorylated by hTK (Figure S3). Likewise, 2′‐deoxy‐2′,2′‐difluorouridine (dFdU, Figure 1) is also a substrate for hTK.15, 16 The combined interplay of steric constraints from both the 5‐ethnyl group and the 2′‐fluorine atoms is therefore responsible for the inability of hTK to phosphorylate dF‐EdU. Consistent with this conclusion, the superimposition model places the (2′R) fluorine atom of dF‐EdU 2.20–2.32 Å from Val 174/Ile 175, and its terminal ethynyl carbon atom 2.67 Å from Thr 163 (Figure 2 b).
The ability of dF‐EdU to be phosphorylated by HSV1‐TK and rejected by hTK suggested that dF‐EdU might be selectively phosphorylated by HSV1‐infected cells and incorporated into DNA. To evaluate this possibility, Vero cells were infected by HSV‐1 in the presence of 10 μm of dF‐EdU for eight hours. Samples were then fixed and stained with an azide‐conjugated fluorophore (green, Figure 3) and the DNA stain DAPI (blue, Figure 3). dF‐EdU click labeling was apparent in infected (Figure 3 a) but not in uninfected (Figure 3 b) cells. dF‐EdU staining of HSV‐1 viral replication compartments was apparent in the DAPI‐excluded areas of the nuclei (circle in Figure 3 a, and Figure 3 c). These morphologically distinct regions were confirmed as HSV‐1 replication compartments by immunostaining of ICP8 as an HSV‐specific marker.7 Cellular nucleic acids were also stained in some cells (rectangle in Figure 3 a, and Figure 3 d), thus suggesting that both viral and cellular polymerases were capable of utilizing dF‐EdU triphosphate as a substrate.
Figure 3.
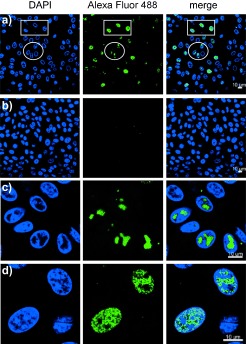
a) Vero cells infected with HSV‐1 (multiplicity of infection (MOI)=10) were incubated with dF‐EdU (10 μm) for 8 h, fixed, and stained with azide‐conjugated Alexa Fluor 488 in the presence of CuI. b) Control samples were treated identically but received no HSV‐1. c) High‐magnification images illustrating labeling of the viral replication compartments. d) High‐magnification images illustrating labeling of cellular nucleic acids. Scale bars: 10 μm.
To evaluate wheher viral TK expression is required for the incorporation of dF‐EdU into cellular and viral nucleic acids, HeLa cells were transduced with a lentiviral vector encoding HSV1‐TK and green fluorescent protein (GFP), and selected by fluorescence‐activated cell sorting (FACS) to generate the cell line HeLa‐HSVtk(+). These cells were subjected to a second round of transduction and more stringent FACS selection to generate the cell line HeLa‐HSVtk(++). Cell cultures were then treated with dF‐EdU, fixed, stained, and characterized by fluorescence microscopy and flow cytometry (Figure 4). Wild‐type HeLa cells exhibited little or no fluorescent staining of dF‐EdU when dF‐EdU was applied at 1–100 μm (Figure 4 a). In contrast, HeLa‐HSVtk(+) and HeLa‐HSVtk(++) cells exhibited strong labeling by 0.1–1.0 μm of dF‐EdU. The intensity of dF‐EdU staining was approximately 10‐fold higher in HeLa‐HSVtk(++) cells than HeLa‐HSVtk(+) cells (Figure 4 b, c). DNA was the primary target of dF‐EdU incorporation, as revealed by the selective staining of metaphase chromosomes (Figure S4). dF‐EdU was essentially nontoxic to all three cell lines tested (Figure S5), thus indicating a high degree of compatibility with DNA synthesis.12 Taken together, these results demonstrate that the metabolic incorporation of dF‐EdU into the DNA of HSV‐1‐infected cells (Figure 3) is dependent upon HSV1‐TK expression (Figure 4).
Figure 4.
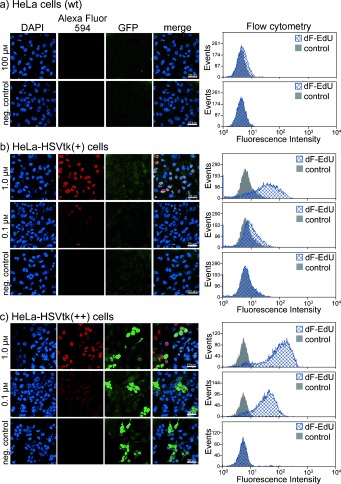
Metabolic incorporation and staining of dF‐EdU in HeLa, HeLa‐HSVtk(+), or HeLa‐HSVtk(++) cells. Variable concentrations of dF‐EdU were incubated with wild‐type HeLa cells (a), HeLa‐HSVtk(+) cells (b), or HeLa‐HSVtk(++) cells (c) for 24 h. The cells were then fixed and stained with 10 μm of azide‐conjugated Alexa Fluor dye (Alexa Fluor 594 for microscopy samples; Alexa Fluor 647 for flow cytometry samples) in the presence of CuI. To maintain the fluorescence signal from GFP, aminoguanidinium HCl and the CuI ligand THPTA25 were included (Figure S6). Negative controls did not receive any nucleoside analogue but were otherwise treated identically. Scale bars: 50 μm.
The tendency of pathogen‐encoded enzymes to exhibit relaxed substrate specificity has been widely exploited in medicinal chemistry6 but it has not previously been utilized for the pathogen‐dependent incorporation of bioorthogonal functional groups into mammalian cells. Herein, we report the clickable nontoxic nucleoside analogue dF‐EdU, which is selectively metabolized by virus‐infected cells as a result of their expression of a low‐fidelity thymidine kinase. As a reporter molecule, dF‐EdU is compatible with chain elongation following its incorporation into DNA, thus allowing highly sensitive and selective click staining of infected cells. This same type of approach should also be applicable to pathogens that encode unique polynucleotide polymerases and/or nucleoside salvage enzymes.13 Nucleoside derivatives containing bioorthogonal functional groups will therefore enable a wide variety of diagnostic and therapeutic applications,23 including cell‐fate control and suicide‐gene therapies.24
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
We thank Dr. Anna Paula de Oliveira, Prof. Cornel Fraefel, and the X06DA beam‐line scientists for technical assistance. We thank the Swiss National Science Foundation (146754) and the University of Zürich Forschungskredit for financial support. L.P. was a recipient of a grant from the Novartis Consumer Health Foundation.
References
- 1. Prescher J. A., Bertozzi C. R., Nat. Chem. Biol. 2005, 1, 13–21. [DOI] [PubMed] [Google Scholar]
- 2.Azide–alkyne cycloaddition:
- 2a. Wang Q., Chan T. R., Hilgraf R., Fokin V. V., Sharpless K. B., Finn M. G., J. Am. Chem. Soc. 2003, 125, 3192–3193; [DOI] [PubMed] [Google Scholar]
- 2b. Link A. J., Tirrell D. A., J. Am. Chem. Soc. 2003, 125, 11164–11165; [DOI] [PubMed] [Google Scholar]
- 2c. Sawa M., Hsu T.‐L., Itoh T., Sugiyama M., Hanson S. R., Vogt P. K., Wong C.‐H., Proc. Natl. Acad. Sci. USA 2006, 103, 12371–12376; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2d. Baskin J. M., Prescher J. A., Laughlin S. T., Agard N. J., Chang P. V., Miller I. A., Lo A., Codelli J. A., Bertozzi C. R., Proc. Natl. Acad. Sci. USA 2007, 104, 16793–16797; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2e. Ning X., Guo J., Wolfert M. A., Boons G.‐J., Angew. Chem. Int. Ed. 2008, 47, 2253–2255; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 2285–2287; [Google Scholar]
- 2f. Dommerholt J., Schmidt S., Temming R., Hendriks L. J. A., Rutjes F. P. J. T., van Hest J. C. M., Lefeber D. J., Friedl P., van Delft F. L., Angew. Chem. Int. Ed. 2010, 49, 9422–9425; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 9612–9615; [Google Scholar]
- 2g. Neef A. B., Luedtke N. W., ChemBioChem 2014, 15, 789–793. [DOI] [PubMed] [Google Scholar]
- 3.Tetrazine–alkene cycloaddition:
- 3a. Blackman M. L., Royzen M., Fox J. M., J. Am. Chem. Soc. 2008, 130, 13518–13519; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Devaraj N. K., Weissleder R., Hilderbrand S. A., Bioconjugate Chem. 2008, 19, 2297–2299; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Lang K., Davis L., Torres‐Kolbus J., Chou C., Deiters A., Chin J. W., Nat. Chem. 2012, 4, 298–304; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Kaya E., Vrabel M., Deiml C., Prill S., Fluxa V. S., Carell T., Angew. Chem. Int. Ed. 2012, 51, 4466–4469; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 4542–4545; [Google Scholar]
- 3e. Rieder U., Luedtke N. W., Angew. Chem. Int. Ed. 2014, 53, 9168–9172; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9322–9326. [Google Scholar]
- 4.Recent reviews:
- 4a. Grammel M., Hang H. C., Nat. Chem. Biol. 2013, 9, 475–784; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Xie R., Hong S., Chen X., Curr. Opin. Chem. Biol. 2013, 17, 747–752; [DOI] [PubMed] [Google Scholar]
- 4c. Patterson D. M., Nazarova L. A., Prescher J. A., ACS Chem. Biol. 2014, 9, 592–605. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Ngo J. T., Champion J. A., Mahdavi A., Tanrikulu I. C., Beatty K. E., Connor R. E., Yoo T. H., Dieterich D. C., Schuman E. M., Tirrell D. A., Nat. Chem. Biol. 2009, 5, 715–717; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Chang P. V., Dube D. H., Sletten E. M., Bertozzi C. R., J. Am. Chem. Soc. 2010, 132, 9516–9518; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Truong F., Yoo T. H., Lampo T. J., Tirrell D. A., J. Am. Chem. Soc. 2012, 134, 8551–8556; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d. Grammel M., Dossa P. D., Taylor‐Salmon E., Hang H. C., Chem. Commun. 2012, 48, 1473–1474; [DOI] [PubMed] [Google Scholar]
- 5e. Xie R., Hong S., Feng L., Rong J., Chen X., J. Am. Chem. Soc. 2012, 134, 9914–9917; [DOI] [PubMed] [Google Scholar]
- 5f. Ngo J. T., Schuman E. M., Tirrell D. A., Proc. Natl. Acad. Sci. USA 2013, 110, 4992–4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Garfield E., Essays of an informed scientist, vol. 12, ISI Press, Philadelphia, 1989, pp. 201–211; [Google Scholar]
- 6b. De Clercq E., Field H. J., Br. J. Pharmacol. 2006, 147, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang I. H., Suomalainen M., Andriasyan V., Kilcher S., Mercer J., Neef A., Luedtke N. W., Greber U. F., Cell Host Microbe 2013, 14, 468–480. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Gierlich J., Burley G. A., Gramlich P. M., Hammond D. M., Carell T., Org. Lett. 2006, 8, 3639–3642; [DOI] [PubMed] [Google Scholar]
- 8b. Salic A., Mitchison T. J., Proc. Natl. Acad. Sci. USA 2008, 105, 2415–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Neef A. B., Luedtke N. W., Proc. Natl. Acad. Sci. USA 2011, 108, 20404–20409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Qu D., Wang G., Wang Z., Zhou L., Chi W., Cong S., Ren X., Liang P., Zhang B., Anal. Biochem. 2011, 417, 112–121; [DOI] [PubMed] [Google Scholar]
- 10b. Guan L., van der Heijden G. W., Bortvin A., Greenberg M. M., ChemBioChem 2011, 12, 2184–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Neef A. B., Samain F., Luedtke N. W., ChemBioChem 2012, 13, 1750–1753. [DOI] [PubMed] [Google Scholar]
- 12.Antiviral prodrugs like acyclovir are selectively phosphorylated by herpes TK but their incoporation into DNA is self‐limiting owing to subsequent inhibition of DNA synthesis.[6]
- 13.
- 13a. Brown J. C., Newcomb W. W., Curr. Opin. Virol. 2011, 1, 142–149; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Berdis A. J., Biochemistry 2008, 47, 8253–8260; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13c. Tsai C. H., Lee P. Y., Stollar V., Li M. L., Curr. Pharm. Des. 2006, 12, 1339–1355; [DOI] [PubMed] [Google Scholar]
- 13d. Wang L., Westberg J., Bölske G., Eriksson S., Mol. Microbiol. 2001, 42, 1065–1073. [DOI] [PubMed] [Google Scholar]
- 14. Giusti G., Mangoni C., De Petrocellis B., Scarano E., Enzymol. Biol. Clin. 1970, 11, 375–383. [DOI] [PubMed] [Google Scholar]
- 15. Veltkamp S. A., Pluim D., van Eijndhoven M. A., Bolijn M. J., Ong F. H., Govindarajan R., Unadkat J. D., Beijnen J. H., Schellens J. H., Mol. Cancer. Ther. 2008, 7, 2415–2425. [DOI] [PubMed] [Google Scholar]
- 16. Benyumov A., Gurvich V. J., Lis L. G., Williams B. W., Kirstein M. N., ChemMedChem 2011, 6, 457–464. [DOI] [PubMed] [Google Scholar]
- 17. Quintiliani M., Persoons L., Solaroli N., Karlsson A., Andrei G., Snoeck R., Balzarini J., McGuigan C., Bioorg. Med. Chem. 2011, 19, 4338–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vogt J., Perozzo R., Pautsch A., Prota A., Schelling P., Pilger B., Folkers G., Scapozza L., Schulz G. E., Proteins 2000, 41, 545–553. [DOI] [PubMed] [Google Scholar]
- 19. Martić M., Pernot L., Westermaier Y., Perozzo R., Kraljevic T. G., Kristafor S., Raic‐Malic S., Scapozza L., Ametamey S., Nucleosides Nucleotides Nucleic Acids 2011, 30, 293–315. [DOI] [PubMed] [Google Scholar]
- 20. Champness J. N., Bennett M. S., Wien F., Visse R., Summers W. C., Herdewijn P., de Clerq E., Ostrowski T., Jarvest R. L., Sanderson M. R., Proteins 1998, 32, 350–361. [DOI] [PubMed] [Google Scholar]
- 21. Emsley P., Cowtan K., Acta Crystallogr. Sect. D 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 22. Birringer M. S., Claus M. T., Folkers G., Kloer D. P., Schulz G. E., Scapozza L., FEBS Lett. 2005, 579, 1376–1382. [DOI] [PubMed] [Google Scholar]
- 23. Knight J. C., Cornelissen B., Am. J. Nucl. Med. Mol. Imaging 2014, 4, 96–113. [PMC free article] [PubMed] [Google Scholar]
- 24.
- 24a. Wang H. E., Yu H. M., Liu R. S., Lin M., Gelovani J. G., Hwang J. J., Wei H. J., Deng W. P., J. Nucl. Med. 2006, 47, 1161–1171; [PubMed] [Google Scholar]
- 24b. Shao D., Zeng Q., Fan Z., Li J., Zhang M., Zhang Y., Li O., Chen L., Kong X., Zhang H., Biomaterials 2012, 33, 4336–4344; [DOI] [PubMed] [Google Scholar]
- 24c. Sato T., Neschadim A., Lavie A., Yanagisawa T., Medin J. A., PLoS One 2013, 8, e78711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hong V., Presolski S. I., Ma C., Finn M. G., Angew. Chem. Int. Ed. 2009, 48, 9879–9883; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 10063–10067. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information