

Cryo-EM structures of the b0,+AT-rBAT complex are revealed, providing clues for the substrate recognition and transport mechanism.
Abstract
Heteromeric amino acid transporters (HATs) catalyze the transmembrane movement of amino acids, comprising two subunits, a heavy chain and a light chain, linked by a disulfide bridge. The b0,+AT (SLC7A9) is a representative light chain of HATs, forming heterodimer with rBAT, a heavy chain which mediates the membrane trafficking of b0,+AT. The b0,+AT-rBAT complex is an obligatory exchanger, which mediates the influx of cystine and cationic amino acids and the efflux of neutral amino acids in kidney and small intestine. Here, we report the cryo-EM structure of the human b0,+AT-rBAT complex alone and in complex with arginine substrate at resolution of 2.7 and 2.3 Å, respectively. The overall structure of b0,+AT-rBAT exists as a dimer of heterodimer consistent with the previous study. A ligand molecule is bound to the substrate binding pocket, near which an occluded pocket is identified, to which we found that it is important for substrate transport.
INTRODUCTION
Amino acids are essential for living cells as necessary nutrients, metabolite precursors, and building blocks for proteins. Heteromeric amino acid transporters (HATs) are a unique kind of amino acid transporters that comprise a heavy chain and a light chain linked by a conserved disulfide bridge (1–3). Two homologous heavy chains, rBAT (related to b0,+ amino acid transporter) (SLC3A1) and 4F2hc (SLC3A2), which belong to the solute carrier family 3, have been identified (2). The light chains of HATs are members of the solute carrier family 7, which belongs to the amino acids, polyamines, and organocations transporter superfamily. 4F2hc heterodimerizes with six light chains (2, 4). The rBAT protein is expressed mainly in the apical membrane of epithelial cells of kidney and small intestine (5, 6), forming a heterodimer with b0,+AT (SLC7A9) and AGT1 (SLC7A13) (7, 8). The HAT formed by b0,+AT and rBAT will be referred as the b0,+AT-rBAT complex hereafter for clarity. The b0,+AT-rBAT heterodimer mediates the Na+-independent electrogenic obligatory exchange of extracellular cationic amino acids and cystine with intracellular neutral amino acids (9–11). The b0,+AT-rBAT complex exhibits a high affinity for cationic amino acids and cystine on the extracellular side and a relative lower affinity for neutral amino acids on the intracellular side (12). Two b0,+AT-rBAT heterodimers have been found to form a noncovalent heterotetramer through rBAT (13). Heterodimerization with b0,+AT prevents degradation of unassembled rBAT in endoplasmic reticulum (14), whereas the stability of b0,+AT seems independent of rBAT (12, 15). rBAT has a longer sequence than 4F2hc, with ~27% amino acid sequence identity between them. Both proteins are type II membrane glycoproteins, with an intracellular N terminus and a bulky extracellular C terminus. The sequence of the ectodomains of rBAT (rBAT-ECD) and 4F2hc (4F2hc-ECD) shows homology with insect maltases and bacterial α-amylases (16, 17). The crystal structure of the human 4F2hc-ECD shows domain A and domain C, which contain a triose phosphate isomerase barrel [(α/β)8] and eight antiparallel β-strands, respectively (16). Sequence homology suggests that rBAT-ECD has also domain B in addition to domains A and C (18). It is not known whether rBAT has glucosidase-like activity.
Mutations in b0,+AT or rBAT cause cystinuria, an inherited autosomal recessive disease characterized by hyperexcretion of cystine in urine (19, 20). Cystinuria is classified into type I and non-type I. All mutations in rBAT and some cases of mutations in b0,+AT cause type I cystinuria, in which heterozygotes have normal urine amino acids (21, 22), whereas other mutations in b0,+AT cause non-type I cystinuria, in which heterozygotes have moderate-to-high amino acid-uria (23, 24).
The structural studies related with HATs include the crystal structure of the ectodomain of human 4F2hc (16), the low-resolution negatively staining electron microscopy (EM) structures of the large neutral amino acid transporter small subunit 2 (LAT2)–4F2hc complex (25, 26), the crystal structures of the bacterial homologs (27–31), and the cryo-EM structures of the LAT1/2-4F2hc complex (32–34). However, there is no structural study directly related to rBAT HATs. Here, we report the cryo-EM structures of the human b0,+AT-rBAT complex, which exists as a dimer of heterodimer in inward-open conformation, solved at 2.3-Å resolution for the overall structure and at 2.7-Å resolution for the transmembrane (TM) domains. A ligand molecule is bound to an inward-open binding pocket in the middle of the transporter, which was confirmed by comparing with the apo structure. Several key residues in the pockets, and the residues which mutations are correlated with cystinuria, were surveyed biochemically. These results provide new insights into the working mechanism of the b0, +AT-rBAT complex.
RESULTS
Biochemical characterizations of the human b0,+AT-rBAT complex
The purified b0,+AT-rBAT complex was examined by SDS–polyacrylamide gel electrophoresis (PAGE) gels under different redox conditions. The proteins migrated as separated bands or as a single band in reducing or oxidizing environment, respectively (Fig. 1A). To test whether the purified complex was functional, we reconstituted the complex into liposome and performed a counterflow assay, in which the uptake of the substrate [14C]-cystine into the liposome was monitored. Time-course analysis of [14C]-cystine uptake showed that the amount of accumulated substrate increased during the first 10 min and gradually decreased after that, a typical profile for counterflow assays (Fig. 1B). To investigate the substrate selectivity, we also set up substrate competition experiments in the [14C]-cystine counterflow assay. Results showed that the transport activity was inhibited notably by cationic amino acids Arg and Lys and neutral amino acids Met, Leu, Phe, Tyr, and Ala. Cys, which might be oxidized into cystine in the air, has the strongest inhibition effects (Fig. 1C).
Fig. 1. In vitro characterization of the transport activity of the b0,+AT-rBAT complex.
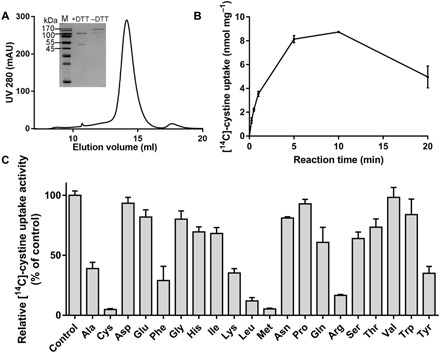
(A) Representative SEC purification profile of the b0,+AT-rBAT complex in the presence of GDN detergent. Inset, SDS-PAGE under reducing [+DTT (dithiothreitol)] or oxidizing (−DTT) conditions, visualized by Coomassie blue staining. M, molecular mass markers. (B) Liposome-based counterflow assay for the purified WT b0,+AT-rBAT complex. A typical time course of [14C]-cystine and leucine exchange. (C) Substrate competition assays for cystine transport by the b0,+AT-rBAT complex. UV, ultraviolet.
Structural determination of the b0,+AT-rBAT complex
To solve the structure of the b0,+AT-rBAT complex, we collected a total of 3453 micrographs of the b0,+AT-rBAT complex incubated with 10 mM Arg, and 798 micrographs were from the b0,+AT-rBAT complex with no ligands. For clarity, these two samples are referred to as b0,+AT-rBAT+Arg and apo-b0,+AT-rBAT, respectively. Details of cryo-EM sample preparation, data acquisition, and processing can be found in Materials and Methods and figs. S1 and S2. The two-dimensional (2D) class average reveals that the b0,+AT-rBAT heterodimer forms a dimer (fig. S1A). The cryo-EM maps for the b0,+AT-rBAT+Arg complex were determined at 2.3-Å resolution for overall structure and the soluble domains and at 2.6- and 2.9-Å resolution for TM domains with the b0,+AT-rBAT+Arg dataset and the apo-b0,+AT-rBAT dataset, respectively (figs. S1, S3, and S4). The TM domains of b0,+AT-rBAT+Arg and apo-b0,+AT-rBAT are nearly identical (fig. S1H). Nearly all of sequences of both rBAT and b0,+AT were clearly resolved (figs. S3 and S4), except the N-terminal dozens of residues, the density of which was invisible in the cryo-EM map. We also built >100 water molecules in each heterodimer that benefited from the high resolution (Fig. 2A).
Fig. 2. Structural determination of the b0,+AT-rBAT complex.
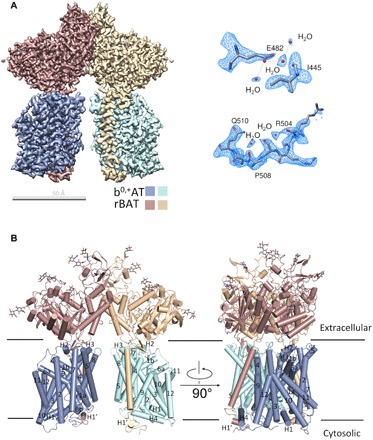
(A) Cryo-EM map of the b0,+AT-rBAT complex. The map is generated by merging the focused refined maps. Right: The water molecules are resolved in the cryo-EM map. (B) Cartoon representation of the atomic model of the b0,+AT-rBAT complex. The glycosylation moieties are shown as sticks.
The extracellular domain (ECD, residues 111 to 685) of rBAT, which has five glycosylation sites, is located above b0,+AT and is connected to the TM helix with a much shorter linker than that in LAT1-4F2hc complex (Fig. 2B and fig. S5A). The alignment of b0,+AT and LAT1 shows that the structures of these two transporters are very similar (fig. S5B). The TM domains of rBAT and b0,+AT is not so tilted as the LAT1-4F2hc complex on the whole view (fig. S5C). The first 10 TM helices of b0,+AT are arranged in a canonical LeuT fold with two inverted repeats, namely, TM1-5 and TM6-10, related to each other via a pseudo twofold symmetry axis parallel to the membrane. Similar to other LeuT-fold transporters, TM1 and TM6 of b0,+AT are disrupted by a short loop in the middle. The half helices hence were referred to as TM1a/1b and TM6a/6b.
The rBAT interacts with b0,+AT through multiple interfaces similar to the LAT1-4F2hc complex (fig. S6). Besides the disulfide bond between rBAT-Cys114 and b0,+AT-Cys144, there are many polar interactions at the extracellular side, intracellular side, and two extra lipid molecules bound to pockets on the intracellular side of the membrane, which are quite similar to that in the LAT1-4F2hc complex (32).
rBAT mediates the dimerization of HATs
The b0,+AT-rBAT complex dimerizes via multiple interfaces in rBAT, which contain numerous hydrophobic and polar interactions and can be divided into three patches (Fig. 3A). The upper patch includes His324, Arg326, Ile329, Asp349, and Tyr347 (Fig. 3B). Arg326 is stacked between cis-His324 and trans-Tyr347 via cation-π interactions, meanwhile, hydrogen bonded with trans-Asp349. In the middle patch (Fig. 3C), Lys323 is hydrogen bonded with trans-Asp391. The lower patch (Fig. 3D) contains a hydrogen bond network formed by Asp359 and Arg362 from both two protomers. There are also numerous hydrophobic interactions in all of three dimerization patches, including Ile329 in the upper patch, Val355 and Met395 in the middle patch, and Phe401 and Ile402 in the lower patch. Most of the amino acid residues involved in dimerization mentioned above are located in the extra loops of rBAT than in the 4F2hc (Fig. 3E and fig. S7), explaining why the rBAT HATs exist as dimer of heterodimer, while the 4F2hc HATs exist as heterodimer.
Fig. 3. Dimerization interface of rBAT.
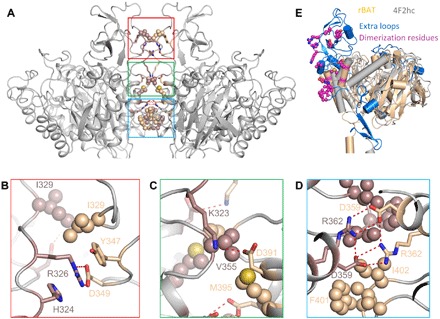
(A) rBAT dimerizes via several patches. The details of these patches are shown in (B) to (D) boxed with corresponding colors. (E) The extra inserted loops of rBAT than 4F2hc contain the most residues involved in dimerization.
The interactions mentioned above make the b0,+AT-rBAT dimer quite stable. We tried to disrupt the dimer of the b0,+AT-rBAT complex by introducing mutations to the dimer interfaces. However, we failed to get the b0,+AT-rBAT heterodimer, with some mutations affecting the expression of the complex. We also tested whether the b0,+AT-rBAT complex has the α-glucosidase activity. The results showed that the complex has no α-glucosidase activity when compared with the positive control isomaltase of Saccharomyces cerevisiae (fig. S8).
Mapping of cystinuria-related mutations
Cystinuria is caused by mutations in rBAT and b0,+AT, whereas how these mutations affect the transport activity of b0,+AT-rBAT complex remains unknown. The high-resolution structure of b0,+AT-rBAT complex provides basis for mechanistic interpretation of representative cystinuria-related mutations (Fig. 4A) (35). To investigate the effects of these mutations, we cloned all these mutations and purified proteins (Fig. 4B). All the rBAT mutants, V183A, T216M, and M467T, and the b0,+AT mutants, P52L and G259R, are not stable and cannot be purified homogeneously, indicating that these residues are essential for the stability of the complex. The mutation A70V, T123M, and A182T in b0,+AT showed partial activity (48 to 60%), which are similar to previous cell-based measurement (35). For other mutants with mutation in b0,+AT, W230R and A354T showed almost no transport activity, and V170M, R333W, A382T, and P482L only retained 5 to 10% activity compared with wild-type (WT) complex. On the basis of these results, these mutations can be divided into three classes. Class I is required for the stability of the complex, including V183A, T216M, and M467T in rBAT and P52L and G259R in b0,+AT. Class II is responsible for substrate binding or lining the transport path, including T123M, W230R, and A382T in b0,+AT. Class III comprises other mutations, including A70V, V170M, A182T, R333W, A354T, and P482L, which might upset the TM rotations when mutated into larger or different charged residues. Besides, some mutations might affect the stability of mRNA and protein in vivo (35).
Fig. 4. Cystinuria-related mutations mapping in the b0,+AT-rBAT complex and putative working model.
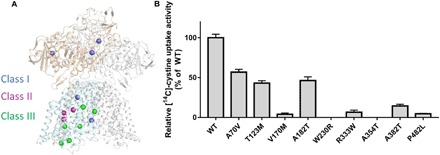
(A) Cystinuria-related mutations mapping in the complex, which can be classified into classes I, II, and III that are colored in blue, purple, and green, respectively. (B) The transport activity of disease-related mutations.
Putative working mechanism of b0,+AT-rBAT complex
The cryo-EM structure of the b0,+AT-rBAT complex adopted an inward-open conformation similar to the LAT1-4F2hc complex (Fig. 5A). However, in contrast to the LAT1-4F2hc complex, the b0,+AT-rBAT complex has an occluded pocket near the open intracellular pocket (Fig. 5A). These two pockets are named as pockets 2 and 1 hereafter for clarity. Extra density was found in each of these two pockets in the cryo-EM map of the b0,+AT-rBAT+Arg complex (fig. S4, C and D). We built an arginine in pocket 1 according to the experimental condition and the cryo-EM map (Fig. 5, A and B, and fig. S4C), which will be referred to as Arg 1 hereafter. The extra density in pocket 2 has a strip-like shape. We compared the cryo-EM map with the apo-b0,+AT-rBAT complex and found that it also contains similar extra density in pocket 2 (fig. S4D), so we excluded the possibility of amino acid molecules and built four water molecules in pocket 2 (Fig. 5C and fig. S4D).
Fig. 5. The ligand binding site and pockets.
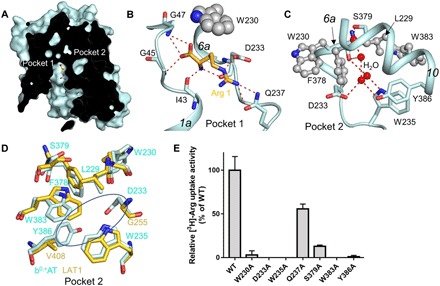
(A) b0,+AT adopts an inward-open conformation with a ligand bound to pocket 1, near which is pocket 2. (B) The ligand-binding site in pocket 1. (C) The pocket 2 contains several water molecules, which are surrounded by a ring of hydrophobic residues and hydrogen bonded with residues at the upper and lower ends. (D) Comparison of b0,+AT with LAT1 in pocket 2 region. (E) The effects of the alanine mutation of the residues involved in pockets 2 and 1 on transport activity measured by [3H]-arginine uptake.
The Arg 1, which is located in the bottom of pocket 1, is hydrogen bonded with Ile43, Gly45, and Gly47 on TM1 and Trp230, Asp233, and Gln237 on TM6 (Fig. 5B), via a traditional mechanism, which is similar to the LAT1-4F2hc complex and also the prokaryotic homologs BasC and GkApcT, with a little difference that the side chain of Arg substrate mainly interacted with residues in TM6b (fig. S9) (30, 31). A water molecule is found near pocket 1, which hydrogen bonds with Ser46, Ser131, and Trp230, thus connecting TM1, TM3, and TM6 (fig. S10). The pocket 2 is surrounded by residues from TM6 and TM10. Four water molecules are hydrogen bonded to each other and packed in pocket 2. These water molecules are also hydrogen bonded to Ser379 on TM10 at the upper end of pocket 2, and to Asp233 and Trp235 on TM6 and Tyr386 on TM10 at the lower end of pocket 2. The middle region of pocket 2 is lined by several hydrophobic residues, including Leu229 and Trp230 from TM6 and Phe378 and Trp383 from TM10. We aligned the pocket 2 in the structures of the b0,+AT-rBAT complex with the corresponding residues in LAT1-4F2hc complex (Fig. 5D). Results showed that the most notable differences happened to Asp233 and Tyr386 of b0,+AT, the corresponding residues of which in LAT1 are Gly255 and Val408. Sequence alignment shows that the residues on these two sites are not conserved in light chains of HAT (fig. S11).
To further investigate the roles of the residues in pocket 2, we aligned the sequences of b0,+AT with that of other species, and results show that these residues are quite conserved among these species (fig. S12). We designed the alanine mutations of Trp230, Asp233, Trp235, Gln237, Ser379, Trp383, and Tyr386 and purified protein for all variants. Results show that Q237A and S379A remain about 50 and 20% of transport activity, respectively, compared with WT complex in the [3H]-arginine counterflow assay (Fig. 5E). The W230A, D233A, W235A, W383A, and Y386A show almost no transport activity (Fig. 5E), indicating their critical roles for transport activity. We also measured the uptake of [14C]-cystine with these mutants; results show similar transport activity compared with the [3H]-arginine counterflow assay, except Q237A and S379A remain about 30 and 50% transport activity, respectively (fig. S13). Together, these results show that the residues in pocket 2 are important for the transport activity of b0,+AT-rBAT complex.
Molecular dynamics (MD) simulation might provide clues for transport mechanism. We first checked the water molecules in pocket 2 and run a MD simulation with pocket 2 initially filled with four water molecules and an arginine molecule in pocket 1. We analyzed the distribution of the water molecules in pocket 2, which showed that these water molecules formed several clusters, according to which we built five water molecules, four of which well coincided with the water molecules we built in pocket 2 initially (fig. S14). We also observed longer average residence time compared with other structural water molecules in b0,+AT-rBAT complex. The hydrogen bond network of these waters is facilitated by characteristic residues of pocket 2 including Trp235, Asp233, Ser379, and Tyr386. These results indicate that the density in pocket 2 corresponds to water than other molecules.
To further investigate the role that the two pockets play in the transport cycle, we performed MD simulations with different system setups. MD simulations were carried out with an arginine molecule built into pocket 2 only or simultaneously with a leucine molecule built into pocket 1. We found that the arginine in pocket 2 adopts a stable binding pose in the time scale of 100 ns. In this pose, the backbone of arginine forms hydrogen bonds with Trp230, Ser379, and Asn227, while the guanidine group forms salt bridge with Asp233 (see movie S1). Similar simulations carried out with other amino acids (Phe, Tyr, Leu, and Met) in pocket 2 indicated that these substrates can fit into this occluded pocket with the same backbone interaction pattern but different side-chain recognitions (see movies S2 to S5). An open movement between TM6a and TM10 is observed coupling with the binding of pocket 2 with amino acid molecule (fig. S15).
DISCUSSION
The b0,+AT-rBAT complex mediates the transport of cationic amino acids and cystine across the cell membrane, some mutations in which are associated with cystinuria diseases. In this work, we solved the high-resolution structures of the b0,+AT-rBAT complex in apo or Arg-bound status with single-particle cryo-EM. In these two structures, b0,+AT exhibits inward-open conformation with similar topology and structure to LAT1 and its prokaryotic homologs. There is an additional pocket 2 near the classical substrate binding pocket 1. The pocket 2 has two polar patches on its ends and is surrounded by hydrophobic residues in its middle region. We speculate that the lower end of pocket 2, which comprises Asp233, Trp235, and Tyr386, might accommodate the polar moiety of the side chain of the dibasic amino acids and one of two ends of cystine. The corresponding residues in LAT1 for Asp233 and Tyr386 are Gly255 and Val408, which are small residues and unable to recognize dibasic amino acids or cystine. These analyses might provide clues into explaining why b0,+AT transports the cationic amino acids and cystine, while LAT1 transports large neutral amino acids.
We tried to propose a working model for the b0,+AT-rBAT complex based on these results (Fig. 6). Two pockets exist in b0,+AT and are separated by a conserved gating residue Trp230, the conformation of which can change markedly, just like the corresponding residue Trp202 in AdiC shows (29). The pockets 1 and 2 might merge together when the transporter experiences conformational change. Actually, LAT1 contains a merged pocket instead of two separated pockets (fig. S16) (32, 33), which is proposed to be able to accommodate bulky hydrophobic side chains (33). The substrates of transporter might be exchanged by each other in the binding pocket, resulting in the transport cycle according to the alternating access model. Together, the results of the structure determination and analysis and the transport activity assays in this work are a step toward a detailed, mechanistic understanding of HAT antiporters and related diseases.
Fig. 6. A proposed model for substrate transport mediated by b0,+AT-rBAT complex.
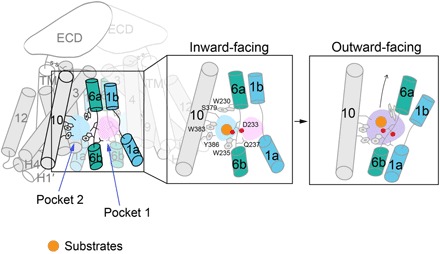
In this model, the pockets 1 and 2 might merge together when the transporter experiences conformational change. The substrates of transporter might be exchanged by each other in the binding pocket, resulting in the transport cycle according to the alternating access model.
MATERIALS AND METHODS
Protein preparation
The full-length human complementary DNAs of b0,+AT (accession number: NM_001126335.1) and rBAT (accession number: NM_000341.4) were subcloned into pCAG with N-terminal FLAG tag and pCAG with N-terminal 10× His tag, respectively. A standard two-step polymerase chain reaction was used to generate the mutations.
For protein expression, human embryonic kidney 293F cells (Invitrogen) were cultured in SMM 293T-I medium (Sino Biological Inc.) at 37°C under 5% CO2 in a Multitron-Pro shaker (130 rpm, Infors). For transfection, 3 mg of polyethylenimines (Polysciences), 0.75 mg of the b0,+AT plasmid, and 0.75 mg of the rBAT plasmid were preincubated for 15 min with fresh medium in a final volume of 50 ml and was added into 1 liter of cell culture whose cell density was ~2.0 × 106/ml. The transfected cells were harvested 48 hours after transfection.
For purification of the b0,+AT-rBAT complex, the cells were collected by centrifugation at 3800g for 10 min and resuspended in a buffer containing 25 mM tris (pH 8.0), 150 mM NaCl, and three protease inhibitors, aprotinin (0.2 μM, AMRESCO), pepstatin (1 μM, AMRESCO), and leupeptin (10.1 μM, AMRESCO). The membrane fraction was solubilized at 4°C for 2 hours with 2% (w/v) n-dodecyl-β-d-maltoside (Anatrace). Cell debris was removed by centrifugation at 18,700g for 45 min, and the supernatant was loaded onto anti-FLAG M2 affinity resin (Sigma-Aldrich). After the resin was rinsed with the wash buffer containing 25 mM tris (pH 8.0), 150 mM NaCl, and 0.05% glyco diosgenin (GDN) (w/v) (Anatrace), the protein was eluted with wash buffer plus FLAG peptide (0.2 mg/ml). Then, the eluent was loaded onto nickel resin (Ni-NTA, Qiagen) and washed with wash buffer plus 10 mM imidazole. The protein complex was eluted from the nickel resin with wash buffer plus 300 mM imidazole, following by concentrating and subjecting to size exclusion chromatography (SEC; Superose 6 Increase 10/300 GL, GE Healthcare) in buffer containing 25 mM tris (pH 8.0), 150 mM NaCl, and 0.02% GDN. The peak fractions were collected and concentrated for EM analysis and in vitro liposome-based transport assay.
For α-glucosidase activity measurement, the transfected cells were resuspended in a buffer containing 25 mM Hepes (pH 7.0), 150 mM NaCl, and the three protease inhibitors mentioned above. The buffer used for affinity chromatography and gel filtration was almost the same as that mentioned above except tris (pH 8.0) was replaced by Hepes (pH 7.0).
α-Glucosidase activity measurement
The open reading frame of IMA1 (accession number: NM_001181416.3) from S. cerevisiae was subcloned into pET15b and expressed in Escherichia coli BL21 (DE3). For protein expression and purification, cells were induced with 0.2 mM isopropyl-β-d-thiogalactopyranoside at an optical density at 600 nm (OD600nm) of 1.2 for 12 hours. Cells were harvested, homogenized in a buffer containing 25 mM Hepes (pH 7.0) and 150 mM NaCl, and sonicated for disruption. After centrifugation at 18,700g for 45 min, the supernatant was loaded onto nickel resin (Ni-NTA, Qiagen), and the nickel resin was washed by the wash buffer containing 25 mM Hepes (pH 7.0), 150 mM NaCl, and 20 mM imidazole. The protein was eluted by wash buffer plus 280 mM imidazole and subjected into gel filtration (Superose 6 Increase 10/300 GL, GE Healthcare). Peak fractions were collected and used to measure α-glucosidase activity. α-Glucosidase activity measurement was performed by α-Glucosidase Activity Colorimetric Assay Kit (Solarbio) and was referred to its description.
For standard curve measurement, ρ-nitrophenol standards (5 μmol/ml) were diluted five times with reagent III, which was then diluted with double-distilled H2O (ddH2O) to indicated concentrations (100, 50, 25, 12.5, and 6.25 nmol/ml). OD400nm of ρ-nitrophenol at concentrations mentioned above and ddH2O was measured.
For α-glucosidase activity measurement, 400 μl of reagent I, 500 μl of reagent II, and 100 μl (0.73 nmol) of b0,+AT-rBAT complex, isomaltase (1.46 nmol), or buffer containing 25 mM Hepes (pH 7.0), 150 mM NaCl, and 0.02% (w/v) GDN (Anatrace) were mixed and reacted at 37°C for 30 min. Then, reaction was stopped by incubating at 100°C for 5 min. When cooled to room temperature, the mixture was centrifuged at 8000g at 4°C. Two minutes after mixing 500 μl of supernatant and 1000 μl of reagent III, OD400nm was measured. Empty control was performed almost the same as that was described above, except 400 μl of reagent I was added into the reaction system after reaction was stopped instead of before. α-Glucosidase activity unit means 1 nmol of ρ-nitrophenol is produced by 1 nmol of protein at 1 ml of reaction system every hour. The data for α-glucosidase activity measurement were processed by GraphPad Prism software.
In vitro transport activity assay
Liposomes and proteoliposomes were prepared as described previously with a slight modification (32). The reaction buffer contains 20 mM potassium phosphate (pH 6.5), 150 mM KCl, and 10 mM l-leucine (Sigma-Aldrich) inside liposomes and proteoliposomes. All transport activity assays were performed at room temperature. The in vitro transport reaction was initiated by adding 100 μl of reaction buffer containing 20 mM potassium phosphate (pH 6.5), 150 mM KCl, and 5 μM (0.1 μCi) l-[14C] cystine [0.184 μM (1 μCi) L-[3H] arginine] (PerkinElmer Life Sciences) to 4 μl of proteoliposome. 14C-labeled cystine uptake or 3H-labeled arginine was terminated after 1 min by rapidly filtering the reaction solution through a 0.22-μm GSTF filter (Millipore) and washed with 2 ml of ice-cold wash buffer [20 mM potassium phosphate (pH 6.5) and 150 mM KCl]. The filter was then used for liquid scintillation counting.
The substrate competition assay for the b0,+AT-rBAT complex was initiated by adding reaction buffer plus 1 mM unlabeled amino acids into 4 μl of proteoliposome (50 mg/ml). Liposome containing no protein was introduced as empty control in each measurement. GraphPad Prism software was used for data processing.
Cryo-EM sample preparation and data acquisition
The purified b0,+AT-rBAT complex was concentrated to ~10 mg/ml and incubated with 10 mM Arg for 2 hours if necessary before being applied to the grids. For clarity, the samples added no substrate or Arg are referred to as apo b0,+AT-rBAT and b0,+AT-rBAT+Arg herein. Aliquots (4 μl) of the protein complex were placed on glow-discharged holey carbon grids (Quantifoil Cu R1.2/1.3). The grids were blotted for 3.5 s and flash-frozen in liquid ethane cooled by liquid nitrogen with Vitrobot (Mark IV, Thermo Fisher Scientific). The prepared grids were transferred to a Titan Krios operating at 300 kV equipped with Cs corrector, Gatan K2 Summit detector, and GIF Quantum energy filter. A total of 798 and 3453 movie stacks were automatically collected using AutoEMation (36) for apo b0,+AT-rBAT and b0,+AT-rBAT+Arg, respectively, with a slit width of 20 eV on the energy filter and a preset defocus range from −1.2 to −2.2 μm in super-resolution mode at a nominal magnification of ×105,000. Each stack was exposed for 5.6 s with an exposure time of 0.175 s per frame, resulting in a total of 32 frames per stack. The total dose rate was approximately 48 e−/Å2 for each stack. The stacks were motion-corrected with MotionCor2 (37) and binned twofold, resulting in a pixel size of 1.091 Å per pixel. Meanwhile, dose weighting was performed (38). The defocus values were estimated with Gctf (39).
Data processing
A total of 1,677,374 or 313,238 particles were automatically picked from 3086 or 677 manually selected micrographs using Relion 3 (40–44) for the b0,+AT-rBAT+Arg and apo b0,+AT-rBAT complex, respectively. After 2D classification, a total of 1,172,618 or 254,699 particles were selected for the b0,+AT-rBAT+Arg and apo b0,+AT-rBAT complex, respectively. The selected particles were subjected to global angular searching 3D classification against an initial model generated with Relion (45) with C2 symmetry. For each of the last several iterations of the global angular searching 3D classification, a local angular searching 3D classification was performed, during which the particles were classified into four classes. A total of 1,030,894 or 175,370 nonredundant particles were selected from the local angular searching 3D classification for the b0,+AT-rBAT+Arg and apo b0,+AT-rBAT complex, respectively. Then, these selected particles were subjected to multireference 3D classification and contrast transfer function refinement (44). The overall resolutions of the 3D auto-refinement after postprocessing were 2.3 or 2.7 Å, with a particle number of 665,827 or 127,377, for the b0,+AT-rBAT+Arg and apo b0,+AT-rBAT complex, respectively. To further improve the map quality, the soluble domains and the TM domains were focused refined with adapted masks separately. The applied symmetry was C2 and C1 for the soluble domains and the TM domains, respectively. The dataset was symmetry-expanded to C1 before the focused refinement for the TM domains, resulting in doubled particle number. The resolutions of the soluble domain–focused refinement were 2.3 and 2.5 Å for the b0,+AT-rBAT+Arg and apo b0,+AT-rBAT complex, respectively. The resolutions of the TM domain-focused refinement were 2.6 and 2.9 Å for the b0,+AT-rBAT+Arg and apo b0,+AT-rBAT complex, respectively.
The 2D classification, 3D classification, and auto-refinement were performed with Relion 3. The resolution was estimated with the gold-standard Fourier shell correlation 0.143 criterion (46, 47) with high-resolution noise substitution (48).
Model building and structure refinement
Model building of the b0,+AT-rBAT complex was based on the focused refined cryo-EM maps. The structure of the extracellular soluble domain of rBAT was ab initio built with Coot (49). The TM domains of the b0,+AT-rBAT complex was built using the structure of the TM domains of the LAT1-4F2hc complex [Protein Data Bank (PDB) ID: 6IRT] as a starting template. The subsequent modeling was performed in Coot with aromatic residues as land markers, as most of these residues were clearly visible in our cryo-EM map. Each residue was manually checked with the chemical properties considered during model building.
A total of 2162 amino acid residues were constructed for the dimer of the b0,+AT-rBAT heterodimer complex. The N-terminal sequences of both rBAT and b0,+AT were not modeled because of the invisibility of the corresponding density in the map. Ten sugar moieties, one Ca2+ ion, two lipid moieties, one ligand amino acid molecules, and more than 100 water molecules were assigned for each b0,+AT-rBAT heterodimer according to the cryo-EM map. The model composition of apo b0,+AT-rBAT is similar to b0,+AT-rBAT +Arg, except that there is no ligand amino acid.
Structure refinement was performed with Phenix (50) with secondary structure and geometry restraints to prevent structure overfitting. To monitor the overfitting of the model, the model was refined against one of the two independent half maps from the gold-standard 3D refinement approach. Then, the refined model was tested against the other map (51). Statistics associated with data collection, 3D reconstruction, and model refinement can be found in table S1.
MD simulations
All-atom MD simulations of b0,+AT-rBAT in the context of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipid bilayers were carried out using CHARMM (52) and OpenMM (53), with proteins described by the CHARMM36m force field (54) and lipids by the CHARMM36 force field (55). Simulation systems contain the TM domain of rBAT, with its extracellular part truncated to reduce the simulation system size. The membrane protein systems then solvated in water box, reaching to the size of 108 Å by 108 Å by 100 Å, with Na+ and Cl− ions added to neutralize the system and maintain ion concentration. The total number of atoms in the simulation system is about 94,000. The system was minimized for 5000 steps, and then six rounds of 100 ps simulations were carried out with gradual decrease of constraints on heavy atoms, and then, the production simulations (60 to 600 ns) were performed with all atoms free under the NPT (constant Number of atoms, constant Temperature and constant Pressure ensemble) ensemble in periodic boundary conditions. The temperature was controlled at 310 K with Andersen thermostat (56), and the pressure was maintained at 1 atm with the Monte Carlo Anisotropic Barostat algorithm. Particle mesh Ewald summation for electrostatic calculation and the 12-Å cutoff for nonbond interactions were used throughout the whole simulations. The integration time step is 2 fs, and covalent bonds including hydrogen were kept rigid with SHAKE. The obtained trajectory was analyzed with VMD (57) and MDTraj (58). To study the water molecule distribution in pocket, 2600 ns MD simulation was performed with pocket 2 initially filled with four water molecules. Frames were saved every 100 ps, and water coordinates were plotted together after correction with respect to the coordinates of the transporter at each frame, which exhibits several clusters, according to which water molecules were built.
To test whether the pocket 2 can accommodate an amino acid ligand, we built different amino acids at the pocket 2 in the context of both the pocket 1 site occupied with amino acid or not. On the basis of the observation of amino acid binding poses in pocket 2 from a series of simulations, we determine the initial conformation to validate the stability of amino acid binding in the pocket 2.
Supplementary Material
Acknowledgments
We thank the cryo-EM facility and the Bio-Computing Platform of the Tsinghua University Branch of China National Center for Protein Sciences (Beijing) for providing the cryo-EM computation support, respectively. We thank X. Li at the cryo-EM facility for technical support. Funding: This work was funded by the National Natural Science Foundation of China (projects 31971123, 81920108015, and 31930059), the Key R&D Program of Zhejiang Province (2020C04001), and Zhejiang Provincial Natural Science Foundation of China (LR19B030001). Author contributions: Q.Z., J.H., and R.Y. conceived the project. Q.Z., R.Y., and Y.S. designed the experiments. R.Y., Y.L., Y.S., J.Z., and Q.Z. did the experiments. J.L. facilitated data collection. All authors contributed to data analysis. Q.Z., R.Y., and J.H. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Atomic coordinates and cryo-EM maps for the b0,+AT-rBAT+Arg complex [PDB: 6LI9; EMDB: EMD-0903 (whole map), EMD-0907 (soluble domain–focused refined), and EMD-0908 (TM domain-focused refined)] and the apo b0,+AT-rBAT complex [PDB: 6LID; EMDB: EMD-0904 (whole map), EMD-0905 (soluble domain–focused refined), and EMD-0906 (TM domain-focused refined)] have been deposited in the Protein Data Bank (www.rcsb.org) and the Electron Microscopy Data Bank (www.ebi.ac.uk/pdbe/emdb/). Additional data related to this paper may be requested from the corresponding authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/16/eaay6379/DC1
REFERENCES AND NOTES
- 1.Pfeiffer R., Spindler B., Loffing J., Skelly P. J., Shoemaker C. B., Verrey F., Functional heterodimeric amino acid transporters lacking cysteine residues involved in disulfide bond. FEBS Lett. 439, 157–162 (1998). [DOI] [PubMed] [Google Scholar]
- 2.Fotiadis D., Kanai Y., Palacín M., The SLC3 and SLC7 families of amino acid transporters. Mol. Aspects Med. 34, 139–158 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Mastroberardino L., Spindler B., Pfeiffer R., Skelly P. J., Loffing J., Shoemaker C. B., Verrey F., Amino-acid transport by heterodimers of 4F2hc/CD98 and members of a permease family. Nature 395, 288–291 (1998). [DOI] [PubMed] [Google Scholar]
- 4.Palacín M., Errasti-Murugarren E., Rosell A., Heteromeric amino acid transporters. In search of the molecular bases of transport cycle mechanisms. Biochem. Soc. Trans. 44, 745–752 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Furriols M., Chillarón J., Mora C., Castelló A., Bertran J., Camps M., Testar X., Vilaró S., Zorzano A., Palacín M., rBAT, related to L-cysteine transport, is localized to the microvilli of proximal straight tubules, and its expression is regulated in kidney by development. J. Biol. Chem. 268, 27060–27068 (1993). [PubMed] [Google Scholar]
- 6.Pickel V. M., Nirenberg M. J., Chan J., Mosckovitz R., Udenfriend S., Tate S. S., Ultrastructural localization of a neutral and basic amino acid transporter in rat kidney and intestine. Proc. Natl. Acad. Sci. U.S.A. 90, 7779–7783 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feliubadaló L., Font M., Purroy J., Rousaud F., Estivill X., Nunes V., Golomb E., Centola M., Aksentijevich I., Kreiss Y., Goldman B., Pras M., Kastner D. L., Pras E., Gasparini P., Bisceglia L., Beccia E., Gallucci M., de Sanctis L., Ponzone A., Rizzoni G. F., Zelante L., Bassi M. T., George A. L. Jr., Manzoni M., De Grandi A., Riboni M., Endsley J. K., Ballabio A., Borsani G., Reig N., Fernández E., Estévez R., Pineda M., Torrents D., Camps M., Lloberas J., Zorzano A., Palacín M., Non-type I cystinuria caused by mutations in SLC7A9, encoding a subunit (bo,+AT) of rBAT. Nat. Genet. 23, 52–57 (1999). [DOI] [PubMed] [Google Scholar]
- 8.Nagamori S., Wiriyasermkul P., Guarch M. E., Okuyama H., Nakagomi S., Tadagaki K., Nishinaka Y., Bodoy S., Takafuji K., Okuda S., Kurokawa J., Ohgaki R., Nunes V., Palacín M., Kanai Y., Novel cystine transporter in renal proximal tubule identified as a missing partner of cystinuria-related plasma membrane protein rBAT/SLC3A1. Proc. Natl. Acad. Sci. U.S.A. 113, 775–780 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Busch A. E., Herzer T., Waldegger S., Schmidt F., Palacin M., Biber J., Markovich D., Murer H., Lang F., Opposite directed currents induced by the transport of dibasic and neutral amino acids in xenopus oocytes expressing the protein rBAT. J. Biol. Chem. 269, 25581–25586 (1994). [PubMed] [Google Scholar]
- 10.Chillarón J., Estévez R., Mora C., Wagner C. A., Suessbrich H., Lang F., Gelpí J. L., Testar X., Busch A. E., Zorzano A., Palacín M., Obligatory amino acid exchange via systems bo,+-like and y+L-like. A tertiary active transport mechanism for renal reabsorption of cystine and dibasic amino acids. J. Biol. Chem. 271, 17761–17770 (1996). [DOI] [PubMed] [Google Scholar]
- 11.Pfeiffer R., Loffing J., Rossier G., Bauch C., Meier C., Eggermann T., Loffing-Cueni D., Kühn L. C., Verrey F., Luminal heterodimeric amino acid transporter defective in cystinuria. Mol. Biol. Cell 10, 4135–4147 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reig N., Chillarón J., Bartoccioni P., Fernández E., Bendahan A., Zorzano A., Kanner B., Palacín M., Bertran J., The light subunit of system bo,+ is fully functional in the absence of the heavy subunit. EMBO J. 21, 4906–4914 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernandez E., Jiménez-Vidal M., Calvo M., Zorzano A., Tebar F., Palacín M., Chillarón J., The structural and functional units of heteromeric amino acid transporters. The heavy subunit rBAT dictates oligomerization of the heteromeric amino acid transporters. J. Biol. Chem. 281, 26552–26561 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Rius M., Chillarón J., Carrier subunit of plasma membrane transporter is required for oxidative folding of its helper subunit. J. Biol. Chem. 287, 18190–18200 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bauch C., Verrey F., Apical heterodimeric cystine and cationic amino acid transporter expressed in MDCK cells. Am. J. Physiol. Renal Physiol. 283, F181–F189 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Fort J., de la Ballina L. R., Burghardt H. E., Ferrer-Costa C., Turnay J., Ferrer-Orta C., Usón I., Zorzano A., Fernández-Recio J., Orozco M., Lizarbe M. A., Fita I., Palacín M., The structure of human 4F2hc ectodomain provides a model for homodimerization and electrostatic interaction with plasma membrane. J. Biol. Chem. 282, 31444–31452 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Gabriško M., Janeček Š., Looking for the ancestry of the heavy-chain subunits of heteromeric amino acid transporters rBAT and 4F2hc within the GH13 α-amylase family. FEBS J. 276, 7265–7278 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Chillarón J., Font-Llitjós M., Fort J., Zorzano A., Goldfarb D. S., Nunes V., Palacín M., Pathophysiology and treatment of cystinuria. Nat. Rev. Nephrol. 6, 424–434 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Bröer S., Palacín M., The role of amino acid transporters in inherited and acquired diseases. Biochem. J. 436, 193–211 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Brons A.-K., Henthorn P. S., Raj K., Fitzgerald C. A., Liu J., Sewell A. C., Giger U., SLC3A1 and SLC7A9 mutations in autosomal recessive or dominant canine cystinuria: A new classification system. J. Vet. Intern. Med. 27, 1400–1408 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calonge M. J., Gasparini P., Chillarón J., Chillón M., Gallucci M., Rousaud F., Zelante L., Testar X., Dallapiccola B., Di Silverio F., Barceló P., Estivill X., Zorzano A., Nunes V., Palacín M., Cystinuria caused by mutations in rBAT, a gene involved in the transport of cystine. Nat. Genet. 6, 420–425 (1994). [DOI] [PubMed] [Google Scholar]
- 22.Calonge M. J., Volpini V., Bisceglia L., Rousaud F., de Sanctis L., Beccia E., Zelante L., Testar X., Zorzano A., Estivill X., Genetic heterogeneity in cystinuria: The SLC3A1 gene is linked to type I but not to type III cystinuria. Proc. Natl. Acad. Sci. U.S.A. 92, 9667–9671 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dello Strologo L., Pras E., Pontesilli C., Beccia E., Ricci-Barbini V., de Sanctis L., Ponzone A., Gallucci M., Bisceglia L., Zelante L., Jimenez-Vidal M., Font M., Zorzano A., Rousaud F., Nunes V., Gasparini P., Palacín M., Rizzoni G., Comparison between SLC3A1 and SLC7A9 cystinuria patients and carriers: A need for a new classification. J. Am. Soc. Nephrol. 13, 2547–2553 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Leclerc D., Boutros M., Suh D., Wu Q., Palacin M., Ellis J. R., Goodyer P., Rozen R., SLC7A9 mutations in all three cystinuria subtypes. Kidney Int. 62, 1550–1559 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Meury M., Costa M., Harder D., Stauffer M., Jeckelmann J.-M., Brühlmann B., Rosell A., Ilgü H., Kovar K., Palacín M., Fotiadis D., Detergent-induced stabilization and improved 3D map of the human heteromeric amino acid transporter 4F2hc-LAT2. PLOS ONE 9, e109882 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosell A., Meury M., Álvarez-Marimon E., Costa M., Pérez-Cano L., Zorzano A., Fernández-Recio J., Palacín M., Fotiadis D., Structural bases for the interaction and stabilization of the human amino acid transporter LAT2 with its ancillary protein 4F2hc. Proc. Natl. Acad. Sci. U.S.A. 111, 2966–2971 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fang Y., Jayaram H., Shane T., Kolmakova-Partensky L., Wu F., Williams C., Xiong Y., Miller C., Structure of a prokaryotic virtual proton pump at 3.2 Å resolution. Nature 460, 1040–1043 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao X., Lu F., Zhou L., Dang S., Sun L., Li X., Wang J., Shi Y., Structure and mechanism of an amino acid antiporter. Science 324, 1565–1568 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Gao X., Zhou L., Jiao X., Lu F., Yan C., Zeng X., Wang J., Shi Y., Mechanism of substrate recognition and transport by an amino acid antiporter. Nature 463, 828–832 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Jungnickel K. E. J., Parker J. L., Newstead S., Structural basis for amino acid transport by the CAT family of SLC7 transporters. Nat. Commun. 9, 550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Errasti-Murugarren E., Fort J., Bartoccioni P., Díaz L., Pardon E., Carpena X., Espino-Guarch M., Zorzano A., Ziegler C., Steyaert J., Fernández-Recio J., Fita I., Palacín M., L amino acid transporter structure and molecular bases for the asymmetry of substrate interaction. Nat. Commun. 10, 1807 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yan R., Zhao X., Lei J., Zhou Q., Structure of the human LAT1-4F2hc heteromeric amino acid transporter complex. Nature 568, 127–130 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Lee Y., Wiriyasermkul P., Jin C., Quan L., Ohgaki R., Okuda S., Kusakizako T., Nishizawa T., Oda K., Ishitani R., Yokoyama T., Nakane T., Shirouzu M., Endou H., Nagamori S., Kanai Y., Nureki O., Cryo-EM structure of the human L-type amino acid transporter 1 in complex with glycoprotein CD98hc. Nat. Struct. Mol. Biol. 26, 510–517 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Jeckelmann J.-M., Fotiadis D., Volta phase plate cryo-EM structure of the human heterodimeric amino acid transporter 4F2hc-LAT2. Int. J. Mol. Sci. 20, E931 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Font M. A., Feliubadaló L., Estivill X., Nunes V., Golomb E., Kreiss Y., Pras E., Bisceglia L., d’Adamo A. P., Zelante L., Gasparini P., Bassi M. T., George A. L. Jr., Manzoni M., Riboni M., Ballabio A., Borsani G., Reig N., Fernández E., Zorzano A., Bertran J., Palacín M., Functional analysis of mutations in SLC7A9, and genotype–Phenotype correlation in non-type I cystinuria. Hum. Mol. Genet. 10, 305–316 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Lei J., Frank J., Automated acquisition of cryo-electron micrographs for single particle reconstruction on an FEI Tecnai electron microscope. J. Struct. Biol. 150, 69–80 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Zheng S. Q., Palovcak E., Armache J.-P., Verba K. A., Cheng Y., Agard D. A., MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grant T., Grigorieff N., Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. eLife 4, e06980 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang K., Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scheres S. H., A Bayesian view on cryo-EM structure determination. J. Mol. Biol. 415, 406–418 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scheres S. H., RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scheres S. H., Semi-automated selection of cryo-EM particles in RELION-1.3. J. Struct. Biol. 189, 114–122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kimanius D., Forsberg B. O., Scheres S. H. W., Lindahl E., Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. eLife 5, e18722 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zivanov J., Nakane T., Forsberg B. O., Kimanius D., Hagen W. J. H., Lindahl E., Scheres S. H. W., New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Punjani A., Rubinstein J. L., Fleet D. J., Brubaker M. A., cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Rosenthal P. B., Henderson R., Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333, 721–745 (2003). [DOI] [PubMed] [Google Scholar]
- 47.S. H. Scheres, S. Chen, in Nature Methods (United States, 2012), vol. 9, pp. 853–854.
- 48.Chen S., McMullan G., Faruqi A. R., Murshudov G. N., Short J. M., Scheres S. H. W., Henderson R., High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 135, 24–35 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Amunts A., Brown A., Bai X.-c., Llácer J. L., Hussain T., Emsley P., Long F., Murshudov G., Scheres S. H. W., Ramakrishnan V., Structure of the yeast mitochondrial large ribosomal subunit. Science 343, 1485–1489 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brooks B. R., Brooks C. L. III, Mackerell A. D. Jr., Nilsson L., Petrella R. J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A. R., Feig M., Fischer S., Gao J., Hodoscek M., Im W., Kuczera K., Lazaridis T., Ma J., Ovchinnikov V., Paci E., Pastor R. W., Post C. B., Pu J. Z., Schaefer M., Tidor B., Venable R. M., Woodcock H. L., Wu X., Yang W., York D. M., Karplus M., CHARMM: The biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eastman P., Swails J., Chodera J. D., Mc Gibbon R. T., Zhao Y., Beauchamp K. A., Wang L.-P., Simmonett A. C., Harrigan M. P., Stern C. D., Wiewiora R. P., Brooks B. R., Pande V. S., OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLOS Comput. Biol. 13, e1005659 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., de Groot B. L., Grubmüller H., MacKerell A. D. Jr., CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pastor R. W., MacKerell A. D. Jr., Development of the CHARMM force field for lipids. J. Phys. Chem. Lett. 2, 1526–1532 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Andersen H. C., Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. 72, 2384–2393 (1980). [Google Scholar]
- 57.Humphrey W., Dalke A., Schulten K., VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
- 58.McGibbon R. T., Beauchamp K. A., Harrigan M. P., Klein C., Swails J. M., Hernández C. X., Schwantes C. R., Wang L.-P., Lane T. J., Pande V. S., MDTraj: A modern open library for the analysis of molecular dynamics trajectories. Biophys. J. 109, 1528–1532 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/16/eaay6379/DC1