

Signaling from the mitochondrial respiratory chain is essential for acute vascular oxygen sensing in the lung.
Abstract
Mitochondria play an important role in sensing both acute and chronic hypoxia in the pulmonary vasculature, but their primary oxygen-sensing mechanism and contribution to stabilization of the hypoxia-inducible factor (HIF) remains elusive. Alteration of the mitochondrial electron flux and increased superoxide release from complex III has been proposed as an essential trigger for hypoxic pulmonary vasoconstriction (HPV). We used mice expressing a tunicate alternative oxidase, AOX, which maintains electron flux when respiratory complexes III and/or IV are inhibited. Respiratory restoration by AOX prevented acute HPV and hypoxic responses of pulmonary arterial smooth muscle cells (PASMC), acute hypoxia-induced redox changes of NADH and cytochrome c, and superoxide production. In contrast, AOX did not affect the development of chronic hypoxia-induced pulmonary hypertension and HIF-1α stabilization. These results indicate that distal inhibition of the mitochondrial electron transport chain in PASMC is an essential initial step for acute but not chronic oxygen sensing.
INTRODUCTION
Hypoxic pulmonary vasoconstriction (HPV) is a vital response mechanism that diverts pulmonary blood flow away from poorly ventilated to well-ventilated lung alveoli, thereby optimizing arterial oxygenation under conditions of local alveolar hypoxia (1, 2). Disturbed or inappropriate HPV may cause life-threatening oxygen deprivation, for instance during anesthesia, pneumonia, adult respiratory distress syndrome, septic events, or liver failure. It also contributes to the development of high-altitude pulmonary edema. Under global alveolar hypoxia, HPV can also cause an increase in pulmonary vascular pressure. This occurs naturally in the hypoxic lungs of the unborn fetus, and inappropriate continuation of HPV after birth causes persistent pulmonary hypertension (PH) of the newborn, which is associated with significant mortality and morbidity (3). HPV also contributes to the development of PH caused by chronic hypoxia-induced pulmonary vascular remodeling, which can eventually lead to right heart failure (4).
Recently, we provided evidence that acute HPV may be regulated by a central oxygen sensor within the mitochondrial respiratory chain, which depends on the presence of the mitochondrial complex IV [cIV; cytochrome c (Cyt c) oxidase] subunit 4 isoform 2 (Cox4i2) (5). We were able to demonstrate that acute hypoxia induces Cox4i2-dependent mitochondrial hyperpolarization, leading to increased mitochondrial superoxide release from the respiratory complex III (cIII; ubiquinol:Cyt c oxidoreductase) and/or complex I [cI; reduced form of nicotinamide adenine dinucleotide (NADH):ubiquinone oxidoreductase] with subsequent inhibition of cellular potassium channels (KV), cellular membrane depolarization, and activation of voltage-gated calcium channels, resulting in intracellular calcium increase and HPV. Similarly, we found that chronic hypoxia results in mitochondrial membrane hyperpolarization (6) and KV channel inhibition (7), leading to a proliferative and antiapoptotic phenotype of pulmonary arterial smooth muscle cells (PASMC). However, the underlying mechanism for these alterations remains elusive. In particular, it remains unclear whether inhibition of mitochondrial respiration by oxygen deprivation at cIV could act as the actual trigger for HPV under acute hypoxia, as HPV is caused already by mild hypoxia at oxygen levels at which cIV oxygen affinity should overcome inhibitory consequences. Moreover, the downstream signaling consequences are still under debate and decreased reactive oxygen species (ROS) release from cI (8) during acute hypoxia contrasts to the concept of increased ROS from cIII being the major cause (9). Conversely, there is broad consensus that chronic hypoxia inhibits mitochondrial respiration regulated by hypoxia-inducible factor 1α (HIF-1α)–dependent gene expression. HIF-1α stabilization, in turn, has been suggested to depend on mitochondrial ROS release. We, thus, investigated (i) whether HPV and chronic hypoxia-induced PH and HIF-1α stabilization are prompted by hypoxia-induced inhibition of mitochondrial respiration, (ii) the mechanisms of downstream signaling of the mitochondrial respiratory chain, and (iii) whether partial restoration of mitochondrial respiration by expression of alternative oxidase (AOX) could prevent HPV and hypoxia-induced PH.
To this end, we made use of a mouse model ubiquitously expressing Ciona intestinalis AOX (10), hereafter referred to as AOX mouse. Many organisms, including plants and some metazoans, but not insects and mammals, have AOX, which branches the mitochondrial respiratory chain when electron flux through cIII and cIV is impaired and the quinone pool is highly reduced (11, 12). Under such conditions, AOX accepts electrons from ubiquinol and directly reduces oxygen to water in a thermogenic reaction. AOX thus prevents overreduction of the mitochondrial quinone pool and the resulting excess ROS production and maintains the activity of the Krebs cycle. Despite its enzymatic function, AOX expression alone does not disturb normal mouse physiology (10, 13). However, it is able to correct pathological states associated with respiratory inhibition, notably those affecting cIII and/or cIV (10, 13–16). This makes AOX a valuable tool to study the involvement of mitochondria and their hierarchy in physiological and pathophysiological processes affecting health and disease (17–19).
RESULTS
AOX is expressed in the pulmonary vasculature and inhibits HPV under acute hypoxia
We first verified widespread AOX expression in the lung, including the vasculature and airways (Fig. 1A), and then tested the effects of AOX on cyanide- (fig. S1) and hypoxia-induced vasoconstriction (Fig. 1B). Vasoconstriction was quantified as the increase in pulmonary arterial pressure (∆PAP) in isolated, ventilated lungs perfused with buffer at constant flow. In wild-type (WT) lungs, ∆PAP reached a maximum value after 10 min of hypoxic ventilation (acute HPV), transiently decreasing before rising again under prolonged hypoxia of up to 3 hours (sustained HPV) (Fig. 1B). AOX-expressing lungs showed inhibition of HPV under acute hypoxia (Fig. 1B) and a diminished PAP response in the presence of cyanide (fig. S1). HPV inhibition by AOX was confirmed using the AOX-specific inhibitor n-propyl gallate (nPG). The presence of nPG during 10 min of hypoxic ventilation abrogated the AOX effect and restored HPV (Fig. 1C). To verify that the effect of AOX was not due to nonspecific lung damage and/or impaired calcium signaling, we stimulated isolated lungs with the hypoxia-independent vasoconstrictor U46619, a thromboxane mimetic. WT and AOX lungs responded similarly to U46619 (Fig. 1D), with an increase in PAP indicating intact pulmonary vasculature physiology, showing the AOX effect to be specific to HPV and, thus, oxygen-dependent vasoconstriction.
Fig. 1. Acute HPV is absent in AOX-expressing isolated murine lungs.
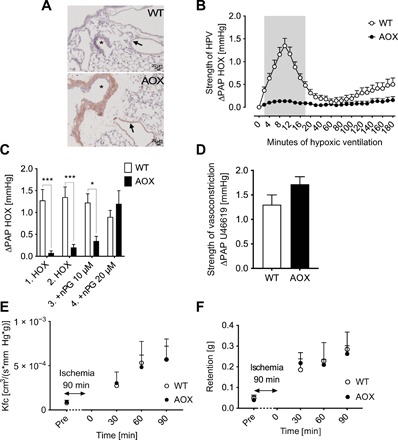
(A) AOX protein expression detected as brownish color in bronchial walls (*) and pulmonary arteries (arrows). (B) PAP response of isolated, buffer-perfused WT and AOX murine lungs ventilated with 1% O2 for time as indicated. Data are shown as means ± SEM of n = 9 experiments. Gray area indicates significant difference with P < 0.05 tested by multiple t tests. (C) PAP response to hypoxic (HOX; 1% O2) challenge with and without AOX inhibitor nPG applied 5 min before sequential HOX. Data are shown as means ± SEM of n = 4 experiments. *P < 0.05, ***P < 0.001 for comparison as indicated, analyzed by two-way analysis of variance (ANOVA) and Sidak’s multiple comparisons test. (D) PAP response to pulmonary artery infusion of the thromboxane mimetic U46619. Data are shown as means ± SEM of n = 6 experiments. (E) Kfc after 90 min of ischemia. Data are shown as means ± SEM of n = 3 experiments. (F) Lung weight gain (retention) during reperfusion after 90 min of ischemia. Data are shown as means ± SEM of n = 3 experiments.
Moreover, there was no difference between isolated WT and AOX-overexpressing lungs with regard to postischemic endothelial damage during ischemia-reperfusion, measured as the increase in capillary filtration rate (Kfc; Fig. 1E) and gain of lung weight (Fig. 1F). These results support the conclusion that AOX expression in murine lungs specifically inhibited the response of the pulmonary vasculature to acute and sustained hypoxia and, notably, implicate different underlying mechanisms in ischemia-reperfusion injury.
AOX decreases hypoxia-induced cellular membrane depolarization in PASMC
We next investigated the hypoxia response in isolated PASMC. Since cellular membrane depolarization is an essential step in HPV signaling, upstream of cytoplasmic calcium increase but downstream of superoxide release, we measured cellular membrane potential by patch clamp analysis. In WT PASMC, cellular membrane potential was increased upon exposure to hypoxia (Fig. 2, A and C). By contrast, the hypoxic response in AOX PASMC was blunted (Fig. 2, B and D), with the membrane potential reaching a lower plateau level than in WT (Fig. 2E). AOX inhibition by nPG renormalized membrane depolarization (Fig. 2, B, D, and E), while the basal membrane potential did not differ between WT and AOX PASMC (Fig. 2, C and D). Again, this indicates that AOX, by accepting electrons from ubiquinol, specifically interferes with the hypoxic signal originating from mitochondria, leaving general cellular physiology unaffected.
Fig. 2. Hypoxia-induced cellular membrane depolarization is decreased in AOX-expressing PASMC.
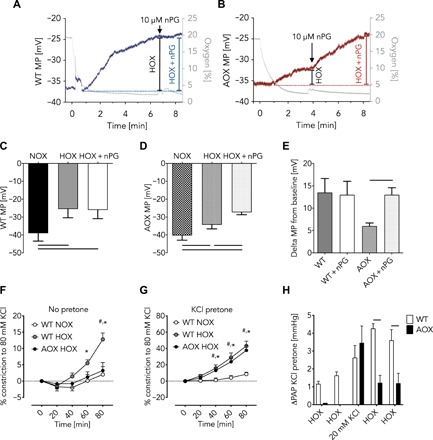
(A and B) Representative traces of patch clamp measurements to determine cellular membrane potential (MP) during acute HOX (1% O2) in mouse WT (A) and AOX (B) PASMC. Gray traces depict oxygen concentration in %; blue (WT) and red (AOX) traces indicate MP in millivolts. Addition of AOX inhibitor nPG as indicated. Cellular MP in mouse WT (C) and AOX (D) PASMC during normoxia (NOX) and acute HOX or acute HOX plus nPG. (E) Change of cellular MP compared with NOX in the absence and presence of nPG as indicated. Data of (C) to (E) shown as means ± SEM of n = 6 experiments. Horizontal bars indicate significant difference with P < 0.05 analyzed by repeated-measures one-way ANOVA and Tukey’s multiple comparisons test. (F) Vasoconstriction of isolated pulmonary arteries during superfusion with hypoxic (1% O2) or normoxic KCl-free buffer shown as % of response to 80 mM KCl. Data are shown as means ± SEM of n = 8 experiments. *P < 0.05 WT NOX versus WT HOX; #P < 0.05 WT HOX versus AOX HOX analyzed by two-way ANOVA and Tukey’s multiple comparisons test. (G) Vasoconstriction as in (F) but in the presence of ~20 mM KCl. Data are shown as means ± SEM of n = 8 experiments. *P < 0.05 WT NOX versus WT HOX; #P < 0.05 WT NOX versus AOX HOX analyzed by two-way ANOVA and Tukey’s multiple comparisons test. (H) PAP response of isolated WT and AOX lungs during HOX (10% O2) ventilation before and after infusion of 20 mM KCl. Data are shown as means ± SEM of n = 3 experiments. Horizontal bars indicated significant difference with P < 0.05 analyzed by two-way ANOVA and Sidak’s multiple comparisons test.
To test this assumption in a more physiological context, we measured hypoxia-induced vasoconstriction of pulmonary arterial vessels, which was again prevented by AOX (Fig. 2F). However, vasoconstriction was restored in isolated vessels expressing AOX when perfused with potassium chloride (Fig. 2, F and G). Moreover, we observed that addition of potassium chloride to the perfusate in isolated AOX lungs also restored HPV (Fig. 2H). This indicates that mitochondria, and specifically the electron flux through the respiratory chain, lie upstream of potassium channel inhibition in the hierarchy of acute HPV signaling, and reveals that other minor hypoxia-dependent processes must contribute to reaching the threshold of depolarization necessary to trigger HPV.
AOX inhibits hypoxia-induced mitochondrial superoxide release and membrane hyperpolarization in PASMC
Since a hallmark of impaired electron flux through the respiratory chain is the production of ROS and reactive nitrogen species (RNS), we tested whether ROS/RNS are the mitochondrial signals that trigger HPV. We measured ROS/RNS release in hypoxia-exposed snap-frozen mouse PASMC using the spin probe CMH (1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine) with electron spin resonance (ESR) spectroscopy (Fig. 3A). To determine the proportion of superoxide within the total ROS/RNS load, we incubated parallel samples with polyethylene glycol–conjugated superoxide dismutase (pSOD) (Fig. 3, A and B). Superoxide levels increased under hypoxic conditions in WT but not in AOX PASMC. To validate this finding, we expressed either native AOX or a catalytically inactive mutant AOX (20) in WT mouse PASMC. Only the inactive mutant AOX showed a hypoxia-dependent increase in superoxide (Fig. 3C). To investigate the mechanism behind increased superoxide production, we measured the mitochondrial membrane potential (∆𝜓), which was increased in WT but not in AOX PASMC during superfusion with hypoxic medium (Fig. 3, D and E). We also assayed the oxygen dependence of mitochondrial respiration, in this case using transfected rat PASMC (rPASMC) to obtain sufficient material. rPASMC transfected with an AOX expression plasmid or with empty vector both showed decreased respiration under oxygen deprivation. However, at low oxygen concentrations, AOX-expressing rPASMC had slightly, but significantly, higher oxygen consumption than the control (Fig. 3F). These data confirm that AOX is catalytically engaged during acute hypoxic inhibition of the respiratory chain, thereby affecting, besides ROS production, the mitochondrial redox state and accumulation of Krebs cycle intermediates.
Fig. 3. AOX inhibits hypoxia-induced superoxide release and mitochondrial membrane hyperpolarization in PASMC and affects the redox state of mitochondrial biomarkers.
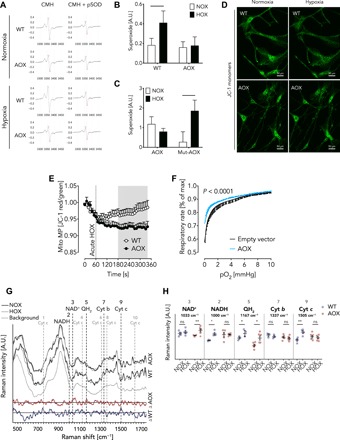
(A) Representative ESR spectra from mouse WT and AOX PASMC using the probe CMH and pSOD for control. (B) Superoxide production in mouse WT and AOX PASMC during exposure to normoxia (NOX) and hypoxia (HOX, 1% O2) for 5 min. A.U., arbitrary units. Data are shown as means ± SEM of n = 4 experiments. The horizontal bar indicates significant difference with P < 0.05 analyzed by two-way ANOVA and Sidak’s multiple comparisons test. (C) Superoxide production in WT mouse PASMC transfected with plasmids encoding native Ciona AOX or a catalytically inactive mutant AOX exposed to NOX and HOX. Data are shown as means ± SEM of n = 12 experiments. The horizontal bar indicates significant difference with P < 0.05 analyzed by two-way ANOVA and Sidak’s multiple comparisons test. No significant differences were detected between the genotypes. Data in (B) and (C) are depicted as the portion of CMH signal inhibited by pSOD. (D) Representative mouse WT and AOX PASMC stained with JC-1 under normoxia and hypoxia (1% O2) as indicated. (E) Mitochondrial MP (Mito MP) determined as JC-1 red/green ratio under acute HOX as indicated. Data are shown as means ± SEM of n = 10 (WT) and n = 12 (AOX) experiments. Gray area depicts significant difference with P < 0.05 analyzed by two-way ANOVA and uncorrected Fisher’s least significant difference (LSD). (F) Oxygen-dependent respiratory rate of 100,000 to 300,000 intact rPASMC per experiment transfected with an empty vector or AOX encoding plasmid. Data are shown as means ± SEM in % of rate at normoxia (max) with P < 0.0001 indicating significant difference between WT and AOX analyzed by paired t test. (G) Normalized fluorescence-corrected Savitzky-Golay reconstructed Raman spectra taken from WT (n = 4) and AOX (n = 6) PASMC before (black line = NOX) and after (gray line) exposure to acute hypoxia (HOX, 5% O2). Background spectrum from microfluidic system and buffer solution is shown as dashed line (n = 25). Differences between NOX and HOX are highlighted with a difference spectrum for each corresponding sample type (WT, blue; AOX, red). Peak locations for mitochondrial biomarkers NAD+ (3: 1033 cm−1), NADH (2: 1000 cm−1), ubiquinol (QH2; 5: 1167 cm−1), and cytochrome b (Cyt b; 7: 1337 cm−1) as well as peaks associated with reduced (1: 750 cm−1; 4: 1127 cm−1; 6: 1313 cm−1; 9: 1505 cm−1) and oxidized forms of Cyt c (8: 1369 cm−1; 10: 1638 cm−1) are shown as vertical dashed lines. (H) Raman intensity for individual biomarkers are shown as means ± SEM of n ≥ 3 experiments. ns, not significant. *P < 0.05; **P < 0.01; ***P < 0.001 analyzed by two-way ANOVA and uncorrected Fisher’s LSD.
AOX affects the mitochondrial redox state
In respiring mitochondria, the redox state of the individual chain complexes is highly dynamic and depends particularly on oxygen supply. Accordingly, hypoxic inhibition at cIV should impair electron flux in WT PASMC, thereby reducing the individual mitochondrial complexes and ultimately resulting in an increased cellular NADH/NAD+ ratio. Since AOX branches the mitochondrial chain when electron flux through cIII and cIV is impaired (14), we hypothesized that both the redox state of mitochondrial complexes downstream of AOX and the NADH/NAD+ ratio would be unaffected by hypoxic challenge in AOX PASMC. To test this hypothesis, we performed Raman spectroscopy, which allows the redox state of individual mitochondrial complexes and NADH/NAD+ ratios to be simultaneously studied in single living PASMC (21–25). As expected, acute hypoxia shifted NADH, ubiquinol (QH2), and Cyt c in WT PASMC toward a more reduced state (Fig. 3, G and H, and fig. S2, A and B). The redox state of cytochrome b (Cyt b) was unaltered, which is indicative for an electron leak at cIII. In accordance with our hypothesis, no change in redox state at cIII (Cyt b) and cIV (Cyt c) was observed in AOX PASMC (Fig. 3H and fig. S2B), consistent with a bypass of electrons from reduced ubiquinol to AOX. Furthermore, NAD+, but not NADH levels, increased upon hypoxia (Fig. 3H). These findings further support the concept of AOX being catalytically engaged in acute hypoxic inhibition and suggest that NADH is the respective electron donor for mitochondrial respiration through AOX.
AOX does not affect adaptation processes upon chronic hypoxia
Notably, mitochondrial impairment has also been identified as an underlying mechanism for chronic hypoxia-induced PH (26), a life-threatening disease occurring, for example, at high altitude. To test whether AOX expression can also prevent the adverse effects of generalized chronic hypoxia, we exposed WT and AOX mice to 4 weeks of hypoxia (10% O2). In contrast to expectations, both WT and AOX mice developed PH to a similar degree, with similar alterations in right ventricular systolic pressure (Fig. 4A), right ventricular hypertrophy (Fulton index; Fig. 4B), cardiac output (Fig. 4C), and pulmonary vascular muscularization (Fig. 4D). Also, we failed to find an advantage of expressing AOX in other cardiac and/or systemic parameters characterizing the response to hypoxia (figs. S3 and S4). HIF-1α expression was similarly increased in PASMC of both strains after 3 days of hypoxia (1% O2; Fig. 4, E and F), indicating that, at least under these conditions, mitochondrial ROS are not a relevant trigger for HIF-1α stabilization. By contrast, PASMC proliferation was induced by hypoxia in both strains, but its level was generally lower in the presence of AOX (Fig. 4G).
Fig. 4. Adaptation processes upon chronic hypoxia in WT and AOX transgenic mice.
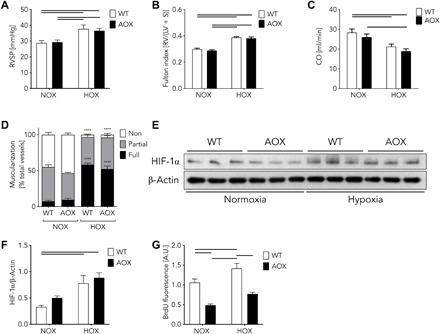
(A) Right ventricular systolic pressure (RVSP) after normoxia (NOX) or hypoxia (HOX, 10% O2) for 28 days. Data are shown as means ± SEM of n ≥ 9 experiments. Horizontal bars indicate significant difference with P < 0.05 analyzed by two-way ANOVA and Tukey’s multiple comparisons test. (B) Ratios of right ventricle (RV) and left ventricle (LV) plus septum (S) (Fulton index). Data are shown as means ± SEM of n = 10 experiments. Horizontal bars indicate significant difference with P < 0.05 analyzed by two-way ANOVA and Tukey’s multiple comparisons test. (C) Cardiac output (CO) measured by echocardiography. Data are shown as means ± SEM of n = 10 experiments. Horizontal bars indicate significant difference with P < 0.05 analyzed by two-way ANOVA and Tukey’s multiple comparisons test. (D) Vascular remodeling quantified as degree of muscularization of small (20- to 70-μm diameter) pulmonary arterial vessels. Vessel muscularization categorized as non, partial, or full after immunostaining against α-smooth muscle actin as marker for PASMC and von Willebrand factor to discard endothelium. Data are shown as means ± SEM of n ≥ 4 experiments. ****P < 0.0001 NOX versus HOX analyzed by two-way ANOVA and Tukey’s multiple comparisons test. Note, no difference observed between WT versus AOX. (E) Western blots showing HIF-1α and β-actin expression in WT and AOX transgenic PASMC under normoxia and hypoxia. (F) Quantification of protein expression shown in (E). Data are shown as means ± SEM of n = 3 experiments. Horizontal bars indicate significant difference with P < 0.05 analyzed by two-way ANOVA and Sidak’s multiple comparisons test. (G) Proliferation assay of WT and AOX PASMC cultured under normoxia (NOX) or hypoxia (HOX, 1% O2) for 48 hours. Data are shown as means ± SEM of n = 9 experiments. Horizontal bars indicate significant difference with P < 0.05 analyzed by two-way ANOVA and Tukey’s multiple comparisons test. BrdU, bromodeoxyuridine.
DISCUSSION
Our data substantiate the concept that an increase in superoxide production at cIII is an essential trigger for the HPV signaling cascade, as bypassing cIII during acute hypoxia by AOX inhibits HPV and hypoxia-induced responses in PASMC (summarized in Fig. 5). Going beyond, we demonstrate that inhibition of the electron flux distal from the quinone pool and, most likely, at the level of cIV results in electron accumulation at the electron transport chain, which may promote superoxide release from cIII. Restoration of even a small amount of electron flux by bypassing cIII and cIV with AOX is sufficient to prevent HPV. These findings indicate that the mitochondrial respiratory chain can be the primary step in acute hypoxia signaling in the lung, resulting in HPV. Moreover, bypassing the Cox4i2 isoform of cIV as the oxygen-sensitive mitochondrial component at the head of the pathway, which we previously showed to be essential for hypoxia-induced ROS release and mitochondrial hyperpolarization (5), may inhibit hypoxic signaling. Last, our data show that distinct mechanisms underlie acute and chronic oxygen sensing and signaling in the pulmonary vasculature.
Fig. 5. Integration of present findings into current concepts of oxygen sensing and HPV in the murine lung.
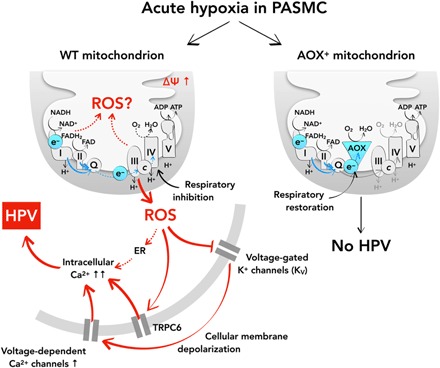
Acute HPV depends on a central oxygen sensor within the mitochondrial respiratory chain. Hypoxia induces mitochondrial membrane hyperpolarization, an increase in mitochondrial superoxide release, and subsequent inhibition of cellular potassium channels (KV), which leads to cellular membrane depolarization and activation of voltage-gated calcium channels. This sequence of events results in intracellular calcium increase and HPV. AOX prevents HPV in mouse PASMC by preventing mitochondrial ROS production and release. This places mitochondria and electron flux through the mitochondrial respiratory chain at the top level in the hierarchy of oxygen sensing and signaling controlling HPV. ADP, adenosine diphosphate; FAD, flavin adenine dinucleotide.
It has been suggested that ROS release from cIII into the intermembrane space could be the essential step in HPV (27, 28). Our previous findings implicating a cIV subunit in hypoxia signaling and cellular membrane depolarization (5) were consistent with this hypothesis, but the initial triggering mechanism of acute hypoxia remained unclear. In the present study, we found that AOX, which is catalytically engaged only when the quinone pool is sufficiently reduced (11), can prevent mitochondrial membrane hyperpolarization, increased superoxide production, and consequent hypoxic signaling. Thus, distal mitochondrial respiratory inhibition and electron flux stalling may act as the initial step in HPV, resulting either in electron accumulation at the quinone pool or the Krebs cycle. As AOX supports electron flux in PASMC under low-oxygen conditions that are sufficient to trigger HPV, accumulation of electrons (and thus the hypoxic signaling) is inhibited. A limitation of the current approach using AOX is that one cannot conclude from its effect alone whether the exact source of the excess ROS is cI, cIII, or both. However, we are not aware of any study proposing an increase in ROS formation from cI as a trigger of HPV. Moreover, we cannot exclude that a hypoxia-induced increase or decrease in Krebs cycle intermediates also contributes to the inhibition of hypoxic signaling by AOX. Furthermore, Cox4i2-dependent mitochondrial hyperpolarization may be inhibited as a result of bypassing cIII/cIV. Although the underlying mechanism is not fully elucidated, mitochondrial hyperpolarization may be downstream of ROS, or more distant mechanisms such as glycolytic adenosine triphosphate (ATP) production may contribute to hyperpolarization. Consistent with our study, AOX has previously been shown to decrease mitochondrial ROS production in mouse tissues and cells under various conditions (10, 13, 15, 16). Despite the possibility of multiple trigger mechanisms, the prominent role of hypoxia-induced superoxide release from cIII has been demonstrated previously using the specific cIII inhibitor S3QEL2 (29), which was shown to prevent HPV (5). This finding is now substantiated by our investigation, using AOX. Moreover, knockdown of subunits of cIII was demonstrated to inhibit acute hypoxia-induced responses of the pulmonary vasculature (27).
Hypoxia-dependent PASMC membrane depolarization was only partially prevented by AOX, and hypoxia-induced vasoconstriction could be restored by experimentally shifting the cellular membrane potential to higher levels. These findings suggest that multiple sensing mechanisms may contribute to reaching a threshold of cellular membrane depolarization necessary for the final execution of HPV (30). Cellular membrane depolarization might be achieved by a change in cellular redox state or activation of ROS-dependent signaling pathways that influence cell membrane ion channels (1), such as the hydrogen peroxide–mediated inhibition of KV channels (5). Accordingly, KV channel inhibition causes cell membrane depolarization and subsequent activation of L- and T-type calcium channels, increasing intracellular calcium levels and triggering HPV (31, 32). The threshold for activation of voltage-dependent calcium channels has been shown to be at a membrane potential between −30 and − 20 mV in mouse PASMC (33). Thus, while WT PASMC can reach this threshold in hypoxia, the presence of AOX prevents membrane depolarization from reaching the critical threshold. The mechanism behind the partial depolarization remains to be identified. A plethora of additional oxygen-sensitive components in PASMC have been suggested (1), including hypoxia-sensitive potassium channels (34, 35). Substituting the hypoxic mitochondrial signal by shifting the cellular membrane potential to a critical threshold, as in our experiments by the addition of potassium chloride, may therefore bypass the mitochondrial signaling and thus restore hypoxia-induced cellular membrane depolarization to a level that can trigger calcium increase and HPV. This indicates that mitochondria must be acting far upstream in the signaling hierarchy, and thus, our results demonstrate the essential role of the mitochondrial respiratory chain in acute oxygen sensing and, in addition, provide an explanation for previous, seemingly contradictory, findings with regard to oxygen-sensitive components in PASMC.
The acute and chronic responses of the pulmonary vasculature to hypoxia have hitherto been assumed to involve similar primary mechanisms and to share regulatory components. However, our data point toward different signaling pathways operating in the two conditions and thus support previous studies that demonstrated a different mechanism of acute HPV and chronic hypoxia-induced PH (5, 30, 36). Confusingly, previous studies have implicated directionally opposite changes in mitochondrial ROS in chronic hypoxia showing both an increase (28) and a decrease in mitochondrial ROS (37) affecting PASMC proliferation and pulmonary vascular remodeling (38, 39). There are also contradictory reports on the effects of mitochondrial ROS on HIF-1α, which is a major driver for the development of chronic hypoxia-induced PH (39), with some authors finding evidence of its stabilization by ROS (28, 40), while others found no such effect (41). Recently, we found evidence for a decrease in PASMC ROS levels in chronic as opposed to acute hypoxia (36). The fact that AOX did not inhibit chronic hypoxia-induced PH, nor interfere with hypoxia-induced stabilization of HIF-1α, may help resolve these discordant findings, recasting the acute and chronic effects of hypoxia in lungs as two fundamentally different processes. The discrepancy to other studies may be due to cell type–specific mechanisms of HIF-1α stabilization, or time- and oxygen concentration–dependent differences. For example, nonmitochondrial ROS sources, such as NADPH (reduced form of nicotinamide adenine dinucleotide phosphate) oxidases, may be of greater importance in the pulmonary vasculature under chronic hypoxic conditions. Although under chronic hypoxia AOX did not influence general physiological parameters, we did record a lower proliferation capacity in PASMC expressing AOX. However, this may be a cell culture effect arising from partial activation of AOX under high glycolytic conditions. It should be emphasized, however, that in vitro effects of mitochondrial superoxide release with regard to HIF-1α stabilization and PASMC proliferation may differ from the in vivo situation.
In summary, our data indicate that respiratory inhibition is an upstream event in HPV signaling, substantiate that mitochondrial superoxide derived from cIII is essential for acute HPV, and provide evidence for the multifactorial nature of HPV with several hypoxia-sensitive components contributing to hypoxia-induced cellular membrane depolarization in PASMC (Fig. 5). Conversely, oxygen sensing under chronic hypoxia in pulmonary cells operates by distinct mechanisms. The differential effect of AOX on acute and chronic hypoxia in the lung illustrates its value as a tool to study disease etiologies, and eventually opens the possibility of a novel therapeutic approach.
MATERIALS AND METHODS
Reagents and resources
A list of reagents and resources is given in Table 1.
Table 1. List of reagents and resources.
| Reagents | Source | Identifier |
| Antibodies | ||
| AOX antiserum (custom raised in rabbit) | 21st Century Biochemicals, Marlborough, MA, USA | (42) |
| HIF-1α (C-Term) polyclonal antibody | Cayman Chemical | 10006421 |
| Anti-beta actin antibody [mAbcam 8226]—loading control (HRP) |
Abcam | ab20272 |
| Anti-rabbit IgG (H + L), HRP conjugate | Promega | W4011 |
| Anti-mouse IgG (H + L), HRP conjugate | Promega | W4021 |
| Lentiviral particles | ||
| Lentiviral Ciona intestinalis AOX | This paper | N/A |
| Lentiviral catalytic inactive (mutant) AOX | This paper | (20) |
| Chemicals | ||
| n-Propyl gallate (nPG, AOX inhibitor) | Sigma-Aldrich | P3130 |
| U46619 (prostaglandin H2/thromboxane A2 receptor agonist) |
Enzo Life Sciences, Exeter, UK | PG-023 |
| Sodium chloride (NaCl) | Sigma-Aldrich | 31434 |
| Potassium chloride (KCl) | Carl Roth, Karlsruhe, Germany | 5346.1 |
| Calcium chloride (CaCl2) | Carl Roth | CN93.1 |
| Magnesium chloride (MgCl2) | Carl Roth | KK36.1 |
| Sodium dihydrogen phosphate dihydrate (NaH2PO4·2H2O) |
Merck Millipore, Darmstadt, Germany | 1063421000 |
| Sodium hydrogen carbonate (NaHCO3) | Carl Roth | 8551 |
| d(+)-Glucose | Carl Roth | X997.1 |
| l-Aspartic acid potassium salt (l-aspartate) | Sigma-Aldrich | A6558 |
| Adenosine 5′-triphosphate magnesium salt, ATP | Sigma-Aldrich | A9187 |
| Ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′- tetraacetic acid (EGTA) |
Sigma-Aldrich | 03777 |
| Hepes (PUFFERAN®) | Carl Roth | 9105.3 |
| Potassium hydroxide (KOH) | Carl Roth | 6751 |
| 1-Hydroxy-3-methoxycarbonyl −2,2,5,5-tetramethylpyrrolidine (CMH spin probe) |
Noxygen, Elzach, Germany | NOX-02.2 |
| Superoxide dismutase–polyethylene glycol (pSOD, PEG-SOD) |
Sigma-Aldrich | S9549 |
| 5,5′,6,6’-Tetrachloro-1,1′,3,3′-tetraethyl- imidacarbocyanine iodide (JC-1) |
ThermoFisher Scientific | T3168 |
| Penicillin-streptomycin | Gibco, ThermoFisher Scientific | 1507–063 |
| Normocin—antimicrobial reagent | InvivoGen, San Diego, USA | ant-nr-1 |
| Assays | ||
| ZytoChem Plus (HRP) Polymer Bulk Kit | Zytomed Systems GmbH, Berlin, Germany | POLHRP-100 |
| VECTOR NovaRED Peroxidase (HRP) Substrate Kit | Vector Laboratories, Burlingame, USA | SK-4800 |
| Enhanced chemiluminescence ECL Prime Western Blotting System |
GE Healthcare | RPN2232 |
| Cell Proliferation ELISA, BrdU (colorimetric) assay | Sigma-Aldrich | 11647229001 |
| Primary cells | ||
| Mouse PASMC from precapillary pulmonary arterial vessels |
(30) | N/A |
| Rat PASMC from precapillary pulmonary arterial vessels |
(45) | N/A |
| Rodent models | ||
| Mouse: Rosa26-Aox (aka AoxRosa26) | (10) | N/A |
| Mouse: C57BL/6J | Charles River, Sulzfeld, Germany | Strain Code 632 |
| Rat: Sprague-Dawley (Crl:SD) | Charles River | Strain Code 400 |
| Plasmid and vectors | ||
| pWPXL (constitutive lentiviral plasmid) | Addgene, Boston, MA, USA | Plasmid #12257 |
| pMD2.G (envelope vector) | Addgene | Plasmid #12259 |
| psPAX2 (packing vector) | Addgene | Plasmid #12260 |
| Software and algorithms | ||
| Patchmaster software | HEKA, Lambrecht, Germany | v2x90 |
| IGOR Pro software | Wavemetrics, Lake Oswego, OR, USA | version 6.37 |
| DatLab software | Oroboros Instruments, Innsbruck, Austria | DatLab 5 |
| Prism software | GraphPad Software; San Diego, CA, USA | version 8.2.0 |
| Other | ||
| Multi Wire Myograph System | Danish Myo Technology A/S, Aarhus, Denmark | 620 M |
| N2-balanced gas (21% O2, 5% CO2) | Praxair, Danbury, CT, USA | 592091 (custom made) |
| N2-balanced gas (1% O2, 5% CO2) | Praxair | 650329 (custom made) |
| Optical Oxygen Meter (FireStingO2) | Pyro Science, Aachen, Germany | FSO2–2 |
| Culture dishes | Greiner Bio-One, Frickenhausen, Germany | 627860 |
| Multi-line in-line solution heater | Warner Instruments, Hamden, CT, USA | SHM-8 |
| Borosilicate glass capillary tubes | Sutter Instrument, Novato, CA, USA | BF150–86-10HP |
| DMZ universal electrode puller | Zeitz, Martinsried, Germany | N/A |
| EPC 10 USB patch clamp amplifier | HEKA | 895000 |
| EMXmicro ESR spectrometer | Bruker Biospin GmbH, Rheinstetten, Germany | N/A |
| OROBOROS-2k oxygraph | Oroboros Instruments | N/A |
| Media 199—Mammalian Cell Culture | Gibco, ThermoFisher Scientific | 31150–022 |
| Vevo2100 high-resolution imaging system equipped with a 40-MHz transducer |
VisualSonics, Toronto, Canada | N/A |
| Raman spectrometer Shamrock 303i | Andor, Technology, Belfast, UK | SR-303i-A |
| Laser | Altechna, Vilnius, Lithuania | DPSS 532 |
| Raman filter cube with dichroic mirror | Semrock, Rochester, NY, USA | zt532/640rpc |
| 532-nm EdgeBasic best-value long-pass edge filter | Semrock | BLP01-532R-25 |
| ×40 magnification objective | Olympus, Tokyo, Japan | LUCPLFLN |
Animal housing and experiments
All animals were maintained and treated according to the regulations of the animal welfare agencies, and all of the experimental protocols were approved by the local “Animal Investigation Care and Use Committee” and the “Regierungspräsidium Darmstadt” (B2/292). Mice and rats were housed in a humidity- and temperature-regulated animal facility with food and water provided ad libitum. A general description and characterization of the AOX-expressing strain have been published elsewhere (10). C57BL/6J mice and Sprague-Dawley rats were obtained from Charles River (Sulzfeld, Germany) (Table 1). A list of reagents and resources is given in Table 1 (see below).
Immunohistochemistry
Formalin-fixed paraffin-embedded mouse lung tissue was used for staining with AOX antiserum (1:2000; custom raised in rabbit; 21st Century Biochemicals, Marlborough, MA, USA) (42), visualized using the ZytoChem Plus (HRP) Polymer Bulk Kit (Zytomed Systems GmbH, Berlin, Germany) and VECTOR NovaRED Peroxidase (HRP) Substrate Kit (Vector Laboratories, Burlingame, USA).
Hemodynamic measurements in isolated, perfused, and ventilated mouse lungs
Lungs of WT or AOX littermates were isolated, perfused, and ventilated as described elsewhere (30, 43). The AOX inhibitor nPG (Sigma-Aldrich) was titrated to confirm AOX dependence (10 to 20 μM). The thromboxane analog U46619 (Enzo Life Sciences, Exeter, UK) was used to test the intactness of vasoconstriction (1 ml of bolus with a concentration of 66 ng/ml).
Intrapulmonary artery isolation and tension measurements
Mouse intrapulmonary arteries were isolated and mounted in wire myograph chambers (Danish Myo Technology A/S, Aarhus, Denmark) as described (44). Each experiment was started by 3 × 3–min exposures to 80 mM KCl containing physiological saline solution. To induce hypoxia upon reaching a stable vascular tone, the N2-balanced gassing was switched from 21% O2, 5% CO2 (Praxair, Danbury, CT, USA) to 1% O2, 5% CO2 (Praxair). The partial pressure of O2 during the hypoxic challenge was 15 to 22 mmHg tested by a FireStingO2 oxygen meter (PyroScience, Aachen, Germany). Changes in isometric tension from the baseline values were expressed as percentages of the maximum constriction induced by the final exposure to 80 mM KCl. In some experiments, 15 to 20 mM KCl was applied before a hypoxic challenge to achieve preconstriction that was ~20% of the maximal constriction induced by 80 mM KCl.
Isolation of mouse and rPASMC and mouse pulmonary arterial vessels
PASMC and pulmonary arterial vessels were isolated as described previously (30, 45).
Estimation of the cellular membrane potential
The cellular membrane potential was measured using the patch clamp technique. Mouse PASMC (passage 0), grown on culture dishes (Greiner Bio-One, Frickenhausen, Germany), were continuously perfused with extracellular analogous bath solution [126.7 mM NaCl (Sigma-Aldrich), 5.4 mM KCl (Carl Roth, Karlsruhe, Germany), 1.8 mM CaCl2 (Carl Roth), 1.05 mM MgCl2 (Carl Roth), 0.42 mM NaH2PO4 (Merck Millipore, Darmstadt, Germany), 22 mM NaHCO3 (Carl Roth), 10 mM glucose (Carl Roth), pH7.4]. Bath solution was gassed—normoxic (21% O2, 5.3% CO2, rest N2) or hypoxic (1% O2, 5.3% CO2, rest N2)—and preheated to 37°C via an in-line solution heater (Warner Instruments, Hamden, CT, USA). Acute hypoxia was applied by switching from a normoxic to a hypoxic bath solution, and the pO2 near the cell was recorded by an optical needle-type FireStingO2 oxygen meter (PyroScience, Aachen, Germany).
Fire-polished patch pipettes with a tip resistance of 3 to 5 megohms were pulled from borosilicate glass capillary tubes (Sutter Instrument, Novato, CA, USA) using a DMZ universal electrode puller (Zeitz, Martinsried, Germany) and filled with intracellular analogous solution containing 105 mM l-aspartate (Sigma-Aldrich), 25 mM KCl (Carl Roth), 4 mM NaCl (Sigma-Aldrich), 1 mM MgCl2 (Carl Roth), 4 mM ATP (Sigma-Aldrich), 10 mM EGTA (Sigma-Aldrich), and 10 mM Hepes (Carl Roth), pH 7.2 adjusted with 1 M KOH (Carl Roth). Experimentally determined liquid junction potential (+12.3 mV) was corrected electronically. Cells used for experiments were either relaxed or partially contracted and exhibited spindle-shaped morphology (round and fully contracted cells were discarded). The membrane potential was recorded under current clamp mode (I = 0) in whole-cell configuration using an EPC 10 USB patch clamp amplifier (HEKA, Lambrecht, Germany). nPG (Sigma-Aldrich) was added to confirm AOX dependence (10 μM). Data were filtered at 2.9 kHz and sampled at 50 Hz using Patchmaster software (HEKA). Data were analyzed using IGOR Pro software (Wavemetrics, Lake Oswego, OR, USA).
Quantification of superoxide levels by ESR spectroscopy
Intracellular and extracellular ROS concentration was measured using an EMXmicro ESR spectrometer (Bruker Biospin GmbH, Rheinstetten, Germany) using 0.5 mM of the spin probe CMH (Noxygen, Elzach, Germany). The superoxide portion of ROS was determined by subtracting the ESR signal of the sample with pSOD (Sigma-Aldrich) from the sample incubated for 90 min without pSOD (45 U/ml) in ESR-Krebs Hepes buffer (46, 47). Hypoxia was applied by incubating the cells in a hypoxic chamber (1% O2 for 5 min).
Lentivirus production and mouse PASMC transduction
Native or mutated AOX were subcloned into the pWPXL plasmid and packed in a second-generation lentivirus transduction system with pMD2.G as the envelope and psPAX2 as a packing vector (all lentiviral plasmids from Addgene, Boston, MA, USA). Lentiviral transduction was performed with a titer of at least 1 × 107 particles according to established protocols (see http://tronolab.epfl.ch/ for more details). After 3 days of transduction, PASMC were used for further experiments.
Measuring mitochondrial membrane potential
The mitochondrial membrane potential was studied by fluorescence microscopy using 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide (JC-1) as recommended by the manufacturer (Thermo Fisher Scientific, Waltham, MA, USA). Acute hypoxia was induced by switching from a normoxic perfusion buffer to a hypoxic buffer (1% O2).
High-resolution respirometry
Oxygen consumption was determined at 37°C using an OROBOROS-2 k oxygraph (Oroboros Instruments, Innsbruck, Austria) as described previously (5). Briefly, primary rPASMC were trypsinized, washed two times, and resuspended in M199 medium (Gibco, Thermo Fisher Scientific, Hampton, NH, USA) containing 1% penicillin-streptomycin (Gibco, Thermo Fisher Scientific) and 10% Hepes (Carl Roth). PASMC transfected with an empty or AOX-containing vector were measured in parallel. Oxygen consumption was recorded continuously until the oxygen was depleted. DatLab software (Oroboros Instruments) was used to calculate oxygen consumption as the time derivative of the oxygen content, standardized to cell number.
Raman spectroscopy on PASMC
Raman spectra of mouse WT and AOX PASMC were measured using a home-built microscope equipped with a ×40 magnification microscope objective (Olympus, Tokyo, Japan), a Raman spectrometer (Andor Technology, Belfast, UK), and a laser with an excitation wavelength of 532 nm (Altechna, Vilnius, Lithuania). The Raman signal was filtered out from the laser light using a filter cube containing a dichroic mirror (Semrock, Rochester, NY, USA) and a 532-nm edge filter (Semrock). An in-house–designed gas-tight microfluidic system was used to register Raman spectra on the cultured PASMC at different oxidation states, i.e., normoxic and hypoxic conditions (48). The sample was illuminated with a laser output power of 3.1 mW with an integration time of 60 s. Acquired Raman data were processed in two steps. First, Cosmic rays were removed using the 2D second difference (49). Second, an optimal reconstruction was done using a Savitzky-Golay filter (50). To confirm the Raman signal from the single live cells, background spectra of the microfluidic system and the buffer were taken.
Right ventricular hemodynamics and morphometry and vascular remodeling
Mice were exposed to normobaric hypoxia (10% O2) for 4 weeks. Quantification of PH was performed as described (30). Measurement of transthoracic echocardiography was performed in the mice before measurement of invasive hemodynamics using a Vevo2100 high-resolution imaging system equipped with a 40-MHz transducer (VisualSonics, Toronto, Canada) as described previously (5, 36).
Western blot analysis
Hypoxic exposure of PASMC, protein extraction, and Western blot analysis were performed as described (5). Primary antibodies used were anti–HIF-1α (1:1000 dilution; Cayman Chemical) and anti–β-actin (1:50,000 dilution; Abcam). Specific immune-reactive signals were detected using enhanced chemiluminescence ECL Prime Western Blotting System (GE Healthcare) using a proprietary secondary antibody coupled to horseradish peroxidase (1:5000 dilution; Promega).
Bromodeoxyuridine proliferation assay
For assessment of proliferation, mouse PASMC (5000 cells per well) from passage 1 were seeded in 24-well plates in smooth muscle cell basal medium containing 1% normocin (InvivoGen, San Diego, USA). After 24 hours of starvation, cells were exposed to normoxia (21% O2, 5% CO2) or hypoxia (1% O2, 5% CO2) at 37°C in water-saturated incubators for 72 hours. Bromodeoxyuridine (BrdU) labeling reagent was added 18 hours before the end of the incubation period. The proliferation assay was performed according to the manufacturer’s instructions [Cell Proliferation ELISA, BrdU (colorimetric), Sigma-Aldrich]. The absorbance of the substrate reaction was measured after 15 min of incubation at 370 nm (reference wavelength, 492 nm).
Statistical analysis
All data depicted are shown as means ± SEM. The exact group size is stated in each figure legend. If not stated otherwise, statistical analysis was done using Prism (GraphPad Software). Differences were considered statistically significant at a P value <0.05.
Supplementary Material
Acknowledgments
We thank K. Homberger, E. Kappes, I. Breitenborn-Mueller, and M. Partanen for technical assistance. Funding: This work was supported by the European Research Council (Advanced Grant 232738), Academy of Finland (Centre of Excellence grant 272376 and Academy Professorship grant 256615), Tampere University Hospital Medical Research Fund and Sigrid Juselius Foundation (to H.T.J.), the Swedish Research Council (grant 2016-04220 to K.R.), and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, project no. 268555672, SFB 1213, A06 to N.W. and N.S.). Author contributions: All authors performed research or compiled and analyzed data. N.S., N.W., H.T.J., and M.S. designed and supervised the research and drafted the manuscript, which was then finalized by all authors. Competing interests: H.T.J. is an inventor on a PATENT related to this work filed by Inst Nat Sante Rech Med (no. 7572616, published 10/09/2008). M.S. declares himself a shareholder in a startup company to develop AOX therapeutics. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The mice xenotopically expressing Ciona intestinalis alternative oxidase (AOX) can be provided by the Faculty of Medicine and Health Technology, Tampere University, FI-33520 Tampere, Finland, pending scientific review and a completed material transfer agreement. Requests for the mice should be submitted to Prof. Howard T. Jacobs (howard.jacobs@tuni.fi).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/16/eaba0694/DC1
REFERENCES AND NOTES
- 1.Sommer N., Strielkov I., Pak O., Weissmann N., Oxygen sensing and signal transduction in hypoxic pulmonary vasoconstriction. Eur. Respir. J. 47, 288–303 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Sylvester J. T., Shimoda L. A., Aaronson P. I., Ward J. P. T., Hypoxic pulmonary vasoconstriction. Physiol. Rev. 92, 367–520 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mathew B., Lakshminrusimha S., Persistent pulmonary hypertension in the newborn. Children 4, 63 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grimminger J., Richter M., Tello K., Sommer N., Gall H., Ghofrani H. A., Thin air resulting in high pressure: Mountain sickness and hypoxia-induced pulmonary hypertension. Can. Respir. J. 2017, 8381653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sommer N., Hüttemann M., Pak O., Scheibe S., Knoepp F., Sinkler C., Malczyk M., Gierhardt M., Esfandiary A., Kraut S., Jonas F., Veith C., Aras S., Sydykov A., Alebrahimdehkordi N., Giehl K., Hecker M., Brandes R. P., Seeger W., Grimminger F., Ghofrani H. A., Schermuly R. T., Grossman L. I., Weissmann N., Mitochondrial complex IV subunit 4 isoform 2 is essential for acute pulmonary oxygen sensing. Circ. Res. 121, 424–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pak O., Sommer N., Hoeres T., Bakr A., Waisbrod S., Sydykov A., Haag D., Esfandiary A., Kojonazarov B., Veit F., Fuchs B., Weisel F. C., Hecker M., Schermuly R. T., Grimminger F., Ghofrani H. A., Seeger W., Weissmann N., Mitochondrial hyperpolarization in pulmonary vascular remodeling. Mitochondrial uncoupling protein deficiency as disease model. Am. J. Respir. Cell Mol. Biol. 49, 358–367 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Mittal M., Gu X. Q., Pak O., Pamenter M. E., Haag D., Fuchs D. B., Schermuly R. T., Ghofrani H. A., Brandes R. P., Seeger W., Grimminger F., Haddad G. G., Weissmann N., Hypoxia induces Kv channel current inhibition by increased NADPH oxidase-derived reactive oxygen species. Free Radic. Biol. Med. 52, 1033–1042 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Dunham-Snary K. J., Wu D., Potus F., Sykes E. A., Mewburn J. D., Charles R. L., Eaton P., Sultanian R. A., Archer S. L., Ndufs2, a core subunit of mitochondrial complex I, is essential for acute oxygen-sensing and hypoxic pulmonary vasoconstriction. Circ. Res. 124, 1727–1746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waypa G. B., Smith K. A., Schumacker P. T., O2 sensing, mitochondria and ROS signaling: The fog is lifting. Mol. Aspects Med. 47-48, 76–89 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Szibor M., Dhandapani P. K., Dufour E., Holmström K. M., Zhuang Y., Salwig I., Wittig I., Heidler J., Gizatullina Z., Gainutdinov T.; German Mouse Clinic Consortium, Fuchs H., Gailus-Durner V., de Angelis M. H., Nandania J., Velagapudi V., Wietelmann A., Rustin P., Gellerich F. N., Jacobs H. T., Braun T., Broad AOX expression in a genetically tractable mouse model does not disturb normal physiology. Dis. Model. Mech. 10, 163–171 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dry I. B., Moore A. L., Day D. A., Wiskich J. T., Regulation of alternative pathway activity in plant mitochondria: Nonlinear relationship between electron flux and the redox poise of the quinone pool. Arch. Biochem. Biophys. 273, 148–157 (1989). [DOI] [PubMed] [Google Scholar]
- 12.Robb E. L., Hall A. R., Prime T. A., Eaton S., Szibor M., Viscomi C., James A. M., Murphy M. P., Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem. 293, 9869–9879 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.El-Khoury R., Dufour E., Rak M., Ramanantsoa N., Grandchamp N., Csaba Z., Duvillié B., Bénit P., Gallego J., Gressens P., Sarkis C., Jacobs H. T., Rustin P., Alternative oxidase expression in the mouse enables bypassing cytochrome c oxidase blockade and limits mitochondrial ROS overproduction. PLOS Genet. 9, e1003182 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szibor M., Gainutdinov T., Fernández-Vizarra E., Dufour E., Gizatullina Z., Debska-Vielhaber G., Heidler J., Wittig I., Viscomi C., Gellerich F., Moore A. L., Bioenergetic consequences from xenotopic expression of a tunicate AOX in mouse mitochondria: Switch from RET and ROS to FET. Biochim. Biophys. Acta Bioenerg. 1861, 148137 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Mills E. L., Kelly B., Logan A., Costa A. S. H., Varma M., Bryant C. E., Tourlomousis P., Däbritz J. H. M., Gottlieb E., Latorre I., Corr S. C., McManus G., Ryan D., Jacobs H. T., Szibor M., Xavier R. J., Braun T., Frezza C., Murphy M. P., O’Neill L. A., Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167, 457–470.e13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giordano L., Farnham A., Dhandapani P. K., Salminen L., Bhaskaran J., Voswinckel R., Rauschkolb P., Scheibe S., Sommer N., Beisswenger C., Weissmann N., Braun T., Jacobs H. T., Bals R., Herr C., Szibor M., Alternative oxidase attenuates cigarette smoke-induced lung dysfunction and tissue damage. Am. J. Respir. Cell Mol. Biol. 60, 515–522 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El-Khoury R., Kemppainen K. K., Dufour E., Szibor M., Jacobs H. T., Rustin P., Engineering the alternative oxidase gene to better understand and counteract mitochondrial defects: State of the art and perspectives. Br. J. Pharmacol. 171, 2243–2249 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dassa E. P., Dufour E., Goncalves S., Jacobs H. T., Rustin P., The alternative oxidase, a tool for compensating cytochrome c oxidase deficiency in human cells. Physiol. Plant. 137, 427–434 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Rustin P., Jacobs H. T., Respiratory chain alternative enzymes as tools to better understand and counteract respiratory chain deficiencies in human cells and animals. Physiol. Plant. 137, 362–370 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Andjelković A., Oliveira M. T., Cannino G., Yalgin C., Dhandapani P. K., Dufour E., Rustin P., Szibor M., Jacobs H. T., Diiron centre mutations in ciona intestinalis alternative oxidase abolish enzymatic activity and prevent rescue of cytochrome oxidase deficiency in flies. Sci. Rep. 5, 18295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brazhe N. A., Treiman M., Faricelli B., Vestergaard J. H., Sosnovtseva O., In situ Raman study of redox state changes of mitochondrial cytochromes in a perfused rat heart. PLOS ONE 8, e70488 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teng H., Lv M., Liu L., Zhang X., Zhao Y., Wu Z., Xu H., Quantitative detection of NADH using a novel enzyme-assisted method based on surface-enhanced raman scattering. Sensors 17, E788 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spiro T. G., Strekas T. C., Resonance Raman spectra of hemoglobin and cytochrome c: Inverse polarization and vibronic scattering. Proc. Natl. Acad. Sci. U.S.A. 69, 2622–2626 (1972). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erjavec N., Pinato G., Ramser K., Raman spectroscopy as a tool for detecting mitochondrial fitness. J. Raman Spectrosc. 47, 933–939 (2016). [Google Scholar]
- 25.Choi S., Lee J. J., Wei Y. H., Spiro T. G., Resonance Raman and electronic spectra of heme a complexes and cytochrome oxidase. J. Am. Chem. Soc. 105, 3692–3707 (1983). [Google Scholar]
- 26.Marshall J. D., Bazan I., Zhang Y., Fares W. H., Lee P. J., Mitochondrial dysfunction and pulmonary hypertension: Cause, effect, or both. Am. J. Physiol. Lung Cell. Mol. Physiol. 314, L782–L796 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waypa G. B., Marks J. D., Guzy R. D., Mungai P. T., Schriewer J. M., Dokic D., Ball M. K., Schumacker P. T., Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. Am. J. Respir. Crit. Care Med. 187, 424–432 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schumacker P. T., Lung cell hypoxia: Role of mitochondrial reactive oxygen species signaling in triggering responses. Proc. Am. Thorac. Soc. 8, 477–484 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Orr A. L., Vargas L., Turk C. N., Baaten J. E., Matzen J. T., Dardov V. J., Attle S. J., Li J., Quackenbush D. C., Goncalves R. L. S., Perevoshchikova I. V., Petrassi H. M., Meeusen S. L., Ainscow E. K., Brand M. D., Suppressors of superoxide production from mitochondrial complex III. Nat. Chem. Biol. 11, 834–836 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weissmann N., Dietrich A., Fuchs B., Kalwa H., Ay M., Dumitrascu R., Olschewski A., Storch U., Mederos y Schnitzler M., Ghofrani H. A., Schermuly R. T., Pinkenburg O., Seeger W., Grimminger F., Gudermann T., Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc. Natl. Acad. Sci. U.S.A. 103, 19093–19098 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Veit F., Pak O., Brandes R. P., Weissmann N., Hypoxia-dependent reactive oxygen species signaling in the pulmonary circulation: Focus on ion channels. Antioxid. Redox Signal. 22, 537–552 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Firth A. L., Remillard C. V., Platoshyn O., Fantozzi I., Ko E. A., Yuan J. X.-J., Functional ion channels in human pulmonary artery smooth muscle cells: Voltage-dependent cation channels. Pulm. Circ. 1, 48–71 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ko E. A., Wan J., Yamamura A., Zimnicka A. M., Yamamura H., Yoo H. Y., Tang H., Smith K. A., Sundivakkam P. C., Zeifman A., Ayon R. J., Makino A., Yuan J. X.-J., Functional characterization of voltage-dependent Ca2+ channels in mouse pulmonary arterial smooth muscle cells: Divergent effect of ROS. Am. J. Physiol. Cell Physiol. 304, C1042–C1052 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olschewski A., Veale E. L., Nagy B. M., Nagaraj C., Kwapiszewska G., Antigny F., Lambert M., Humbert M., Czirják G., Enyedi P., Mathie A., TASK-1 (KCNK3) channels in the lung: From cell biology to clinical implications. Eur. Respir. J. 50, 1700754 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Weir E. K., Olschewski A., Role of ion channels in acute and chronic responses of the pulmonary vasculature to hypoxia. Cardiovasc. Res. 71, 630–641 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Pak O., Scheibe S., Esfandiary A., Gierhardt M., Sydykov A., Logan A., Fysikopoulos A., Veit F., Hecker M., Kroschel F., Quanz K., Erb A., Schäfer K., Fassbinder M., Alebrahimdehkordi N., Ghofrani H. A., Schermuly R. T., Brandes R. P., Seeger W., Murphy M. P., Weissmann N., Sommer N., Impact of the mitochondria-targeted antioxidant MitoQ on hypoxia-induced pulmonary hypertension. Eur. Respir. J. 51, 1701024 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Dromparis P., Sutendra G., Michelakis E. D., The role of mitochondria in pulmonary vascular remodeling. J. Mol. Med. 88, 1003–1010 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Adesina S. E., Kang B.-Y., Bijli K. M., Ma J., Cheng J., Murphy T. C., Michael Hart C., Sutliff R. L., Targeting mitochondrial reactive oxygen species to modulate hypoxia-induced pulmonary hypertension. Free Radic. Biol. Med. 87, 36–47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ball M. K., Waypa G. B., Mungai P. T., Nielsen J. M., Czech L., Dudley V. J., Beussink L., Dettman R. W., Berkelhamer S. K., Steinhorn R. H., Shah S. J., Schumacker P. T., Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1α. Am. J. Respir. Crit. Care Med. 189, 314–324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martínez-Reyes I., Diebold L. P., Kong H., Schieber M., Huang H., Hensley C. T., Mehta M. M., Wang T., Santos J. H., Woychik R., Dufour E., Spelbrink J. N., Weinberg S. E., Zhao Y., DeBerardinis R. J., Chandel N. S., TCA cycle and mitochondrial membrane potential are necessary for diverse biological functions. Mol. Cell 61, 199–209 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chua Y. L., Dufour E., Dassa E. P., Rustin P., Jacobs H. T., Taylor C. T., Hagen T., Stabilization of hypoxia-inducible factor-1α protein in hypoxia occurs independently of mitochondrial reactive oxygen species production. J. Biol. Chem. 285, 31277–31284 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dassa E. P., Dufour E., Gonçalves S., Paupe V., Hakkaart G. A. J., Jacobs H. T., Rustin P., Expression of the alternative oxidase complements cytochrome c oxidase deficiency in human cells. EMBO Mol. Med. 1, 30–36 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weissmann N., Sydykov A., Kalwa H., Storch U., Fuchs B., Mederos y Schnitzler M., Brandes R. P., Grimminger F., Meissner M., Freichel M., Offermanns S., Veit F., Pak O., Krause K.-H., Schermuly R. T., Brewer A. C., Schmidt H. H. H. W., Seeger W., Shah A. M., Gudermann T., Ghofrani H. A., Dietrich A., Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nat. Commun. 3, 649 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Strielkov I., Krause N. C., Sommer N., Schermuly R. T., Ghofrani H. A., Grimminger F., Gudermann T., Dietrich A., Weissmann N., Hypoxic pulmonary vasoconstriction in isolated mouse pulmonary arterial vessels. Exp. Physiol. 103, 1185–1191 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Veit F., Pak O., Egemnazarov B., Roth M., Kosanovic D., Seimetz M., Sommer N., Ghofrani H. A., Seeger W., Grimminger F., Brandes R. P., Schermuly R. T., Weissmann N., Function of NADPH oxidase 1 in pulmonary arterial smooth muscle cells after monocrotaline-induced pulmonary vascular remodeling. Antioxid. Redox Signal. 19, 2213–2231 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Weissmann N., Kuzkaya N., Fuchs B., Tiyerili V., Schäfer R. U., Schütte H., Ghofrani H. A., Schermuly R. T., Schudt C., Sydykov A., Egemnazarow B., Seeger W., Grimminger F., Detection of reactive oxygen species in isolated, perfused lungs by electron spin resonance spectroscopy. Respir. Res. 6, 86 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dikalov S. I., Li W., Mehranpour P., Wang S. S., Zafari A. M., Production of extracellular superoxide by human lymphoblast cell lines: Comparison of electron spin resonance techniques and cytochrome C reduction assay. Biochem. Pharmacol. 73, 972–980 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Knoepp F., Wahl J., Andersson A., Borg J., Weissmann N., Ramser K., Development of a gas-tight microfluidic system for Raman sensing of single pulmonary arterial smooth muscle cells under Normoxic/Hypoxic conditions. Sensors 18, E3238 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schulze H. G., Turner R. F. B., A two-dimensionally coincident second difference cosmic ray spike removal method for the fully automated processing of Raman spectra. Appl. Spectrosc. 68, 185–191 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Savitzky A., Golay M. J. E., Smoothing and differentiation of data by simplified least squares procedures. Anal. Chem. 36, 1627–1639 (1964). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/16/eaba0694/DC1