

Rotavirus capping enzyme VP3 is a stable tetramer with unique RNA binding, RTPase, and RNA helicase activities.
Abstract
In many viruses, including rotavirus (RV), the major pathogen of infantile gastroenteritis, capping of viral messenger RNAs is a pivotal step for efficient translation of the viral genome. In RV, VP3 caps the nascent transcripts synthesized from the genomic dsRNA segments by the RV polymerase VP1 within the particle core. Here, from cryo–electron microscopy, x-ray crystallography, and biochemical analyses, we show that VP3 forms a stable tetrameric assembly with each subunit having a modular domain organization, which uniquely integrates five distinct enzymatic steps required for capping the transcripts. In addition to the previously known guanylyl- and methyltransferase activities, we show that VP3 exhibits hitherto unsuspected RNA triphosphatase activity necessary for initiating transcript capping and RNA helicase activity likely required for separating the RNA duplex formed transiently during endogenous transcription. From our studies, we propose a new mechanism for how VP3 inside the virion core caps the nascent transcripts exiting from the polymerase.
INTRODUCTION
The 5′-end capping of nascent pre-mRNA is an evolutionarily conserved process in eukaryotes and viruses. The capping of the pre-mRNA involves three sequential enzymatic steps. First, the removal of terminal phosphate from 5′-pppNpRNA through RNA triphosphatase (RTPase) activity; second, covalent addition of guanosine monophosphate (GMP) to the RNA to form GpppNpRNA involving guanylyltransferase (GTase) activity; and last, a two-step methyltransferase (MTase) activity, using S-adenosyl-l-methionine (SAM) as the methyl donor, leading to the formation of cap-0 (m7GpppRNA) and cap-1 (m7GpppNm2pRNA). Viruses implement various strategies to perform capping of nascent viral transcripts to mimic host mRNA capping, which allows utilization of the host machinery for efficient translation of viral mRNA as well as evasion of the host antiviral innate immune response (1, 2). In viruses from the Reoviridae family such as rotavirus (RV), reovirus, and bluetongue virus (BTV), which have multiple genomic double-stranded RNA (dsRNA) segments, the 5′-end cap on the positive-sense RNA strand is essential not only for efficient translation in host cells but also to initiate endogenous transcription from the genomic dsRNA by the viral RNA-dependent RNA polymerase (RdRp), a process that occurs within the constrained environment of the capsid interior (3–5).
In RV, which is the major causative agent of life-threatening diarrhea among children (6), VP3 mediates capping of the nascent transcripts synthesized by the viral RdRp, VP1, during endogenous transcription. Both VP3 and VP1 together with the 11 dsRNA segments are encapsidated in the core of the RV particle that is surrounded by three concentric icosahedral capsid layers formed by four structural proteins (VP2, VP6, VP7, and VP4) (7). Although the structures of the RV proteins that constitute the multilayer capsid and that of VP1 are well defined, the structure of VP3 has remained elusive. In addition, how it caps the nascent transcripts synthesized by VP1, within the capsid interior, has been a mystery. Previous biochemical studies showed that VP3 exhibits RNA binding, GTase and MTase activities required for capping (8–10), but it has been unclear whether VP3 exhibits RTPase activity, the first step in mRNA capping, or whether it is assisted by another viral protein. If VP3 has RTPase activity, then a relevant question is how does it bind to nascent transcripts and which domain is involved in this process. During endogenous transcription, as suggested by Jenni et al. (4), the nascent transcripts synthesized by VP1 transiently form a duplex with the negative-strand template of the dsRNA, which needs to be separated before the capping of the transcripts. As VP1 is not known to exhibit helicase activity, it remains unknown how strand separation occurs before capping and whether VP3 is involved in this process. In addition to its function as a capping enzyme, VP3 also plays an important role during RV replication as an antagonist of the antiviral 2′-5′-oligoadenylate synthetase/ribonuclease L (RNase L) pathway with its phosphodiesterase (PDE) activity accomplished through its C-terminal PDE domain (11, 12). To gain mechanistic insights into the multiple functions of this enigmatic protein, we have determined its full-length structure and analyzed its biochemical activities.
RESULTS
Recombinant VP3 exhibits GTase and MTase activities
Following successful expression of the full-length RV VP3 (amino acid residues 1 to 835; ~98 kDa) using the baculovirus expression system, the purified recombinant VP3 was used to determine the cryo–electron microscopy (cryo-EM) structure to a resolution of ~2.7 Å (Fig. 1, A to C) and further functional characterization. Before structural analysis, we confirmed that the recombinant VP3 exhibits the previously characterized GTase and MTase activities required for mRNA capping (9, 10). In addition to autoguanylylation, as previously shown to infer GTase activity, we demonstrated a direct transfer of guanylyl group from guanosine triphosphate (GTP) and similarly of methyl group from SAM onto the rotaviral transcript (segment 10) (fig. S1, A to D). An unexpected finding from this experiment is that the GMP transfer is more efficient in the presence of SAM, suggesting the N7-methylation of the GMP takes place before GMP transfer onto the 5′-end of the transcript (fig. S1B). The sequential steps of RNA capping in RV appear to follow a similar pattern as in alphaviruses, wherein GMP is methylated before the transfer of the guanylyl group onto the mRNA (13). This finding also strengthens previous reports that show that the addition of SAM is critical for in vitro endogenous transcription of full-length transcripts by double-layered particles (DLPs) (14).
Fig. 1. Cryo-EM single-particle reconstruction and atomic model of VP3.
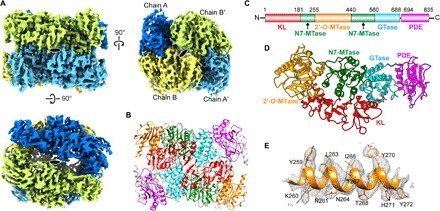
(A) The cryo-EM structure of the VP3 tetramer [Electron Microscopy Data Bank (EMDB) ID: EMD-0632) is shown along the twofold axes of the D2 symmetry. Individual subunits chain A (aqua blue) and chain B (lemon) and their twofold related symmetry pairs chain A′ (sky blue) and chain B′ (honey dew) are shown in shades of blue and yellow. The top panel shows a dimeric subunit with one twofold axis, and the bottom panel shows second twofold axis. (B) Ribbon representation of the atomic model of VP3 tetramer relative to the orientation in the top panel of (A). Each individual dimer is formed by two antiparallel monomers. Individual domains are shown in separate colors: kinase-like (KL) domain (red), guanine-N7 MTase (N7-MTase) domain (green), 2′-O-MTase domain (orange), GTase domain (cyan), and PDE domain (magenta). The same coloring scheme is used in other figures. (C) Domain demarcation along the VP3 peptide chain sequence based on the structural annotation. (D) Modular domain organization of VP3 monomer structure. (E) A representative sample of the 2.7 Å cryo-EM map showing side-chain densities (from the 2′-O-MTase domain) along with the modeled structure. See movies S1 to S3.
VP3 forms a stable tetramer
Our biochemical analysis of recombinant VP3 using size-exclusion chromatography and analytical ultracentrifugation (AUC) analysis showed that VP3 formed a tetramer (fig. S1, E and F). The VP3 tetramer remained stable in a wide range of salt concentrations (50 mM to 1 M NaCl) and pH (5.5 to 9.0), further indicating that VP3 intrinsically favors this oligomeric state (fig. S1G). The cryo-EM structure of VP3 was determined from the images of a frozen-hydrated sample using a well-established single-particle reconstruction workflow (fig. S2, A to E). The cryo-EM reconstruction showed a tetrameric assembly, as expected from the biochemical analysis, with a D2 symmetry formed by two antiparallel dimers (Fig. 1, A and B, and movie S1). The cryo-EM map was of sufficient quality, with the majority of the side-chain densities and the loop regions well defined, for ab initio model building (Fig. 1E, fig. S3A, and movie S2). The previously determined crystal structure of the C-terminal VP3-PDE domain (residues 694 to 835) was unambiguously identified in the model (12). Other domains were identified on the basis of similarities with the domains in the structure of BTV VP4 (15), the closest structural homolog of VP3 (16), and other structural homologs as revealed by the Dali search (17). In addition, the cryo-EM model of the monomer allowed phasing of the ~3.5 Å x-ray diffraction data of the native VP3 crystal, which otherwise could not be phased. The x-ray structure of VP3 showed a similar tetrameric association of VP3 monomeric subunits, as observed by cryo-EM (fig. S3, B and C), and allowed recognition of other subtle features as noted below. The subunits in the tetramer interact extensively with a total calculated buried surface area of ~17,080 Å2, further strengthening the notion that the tetrameric assembly is the most favored state for VP3.
VP3 exhibits a modular domain organization for enzymatic activities
The VP3 monomer exhibits a modular organization with five distinct domains: N-terminal kinase-like (KL) domain (residues 1 to 181), guanine-N7 MTase (N7-MTase) domain (residues 182 to 255 and 441 to 560) interspersed around the ribose 2′-O-MTase domain (residues 256 to 440), and GTase domain (residues 561 to 688) connected to the C-terminal PDE domain (residues 694 to 835) through a short linker (Fig. 1, B to D, and movie S3). Although the overall domain organization in the VP3 monomer is similar to BTV VP4, with the catalytic residues in the GTase and MTase domains locationally conserved, there are notable differences in the relative orientations of the domains resulting in an overall root mean square deviation of 5.4 Å for the matching Cα atoms when the two structures were superimposed (fig. S4A). A distinctive difference between the RV VP3 and the BTV VP4 is the absence of the PDE domain in the latter and comparatively longer N-terminal KL domain (fig. S4, A and B).
The GTase domain in VP3 shows the expected features consistent with its enzymatic activity of adding a GMP to the nascent RNA (Fig. 2A). In both the cryo-EM and x-ray maps of VP3, an additional density is located at the GTase catalytic center that was modeled as GMP (Fig. 2B and fig. S5A). This modeling was confirmed by simulated annealing difference maps using x-ray diffraction data. The GMP is coordinated with the conserved residues Ser570, His574, Arg577, Tyr588, Arg591, His612, His650, and Glu654 (Fig. 2B and figs. S5A and S7). As the GMP was not added during either the purification or the crystallization procedures, it is likely that it copurified with VP3. The location of the GMP and the interacting residues are consistent with previous mutational studies, which indicated that mutating Arg591, His612, or His650 abrogates autoguanylylation activity (16). The x-ray map also showed a bound sulfate interacting with Arg571, a conserved residue in group A RVs (figs. S5B and S7). A similarly bound sulfate is also observed in the structure of the human GTase domain (18) and is suggested to be analogous to the phosphate released from the GTP during the GTase reaction. The GTase domain also participates in the VP3 dimer formation through interactions with the N-terminal KL domain of the neighboring monomer subunit (Fig. 2A).
Fig. 2. VP3 domain organization and structural comparisons.
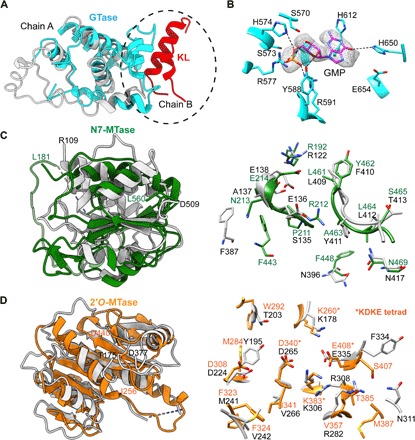
(A) Structural comparison of the GTase domains in RV VP3 (cyan) and BTV VP4 (light gray) showing similar structural folds. GTase domain of VP3 additionally participates in the interaction with the KL domain (helices 165 to 178, shown in red) from the neighboring subunit at the dimeric interface (circled). (B) Electron density for the GMP observed at the catalytic center of GTase domain in the x-ray structure. Conserved catalytic residues interacting with the GMP are denoted. (C) Structural comparison of the guanine N7-MTase (green) and (D) 2′-O-MTase (orange) domains in RV VP3 with those in BTV VP4 (light gray). Note that the substrate binding region shows ~50% conservation in amino acid sequences. The 2′-O-MTase domain also contains the well-characterized catalytic KDKE tetrad motif (in asterisks), which is a conserved feature of MTase domains of other capping enzymes. N- and C-terminal residues of the MTase domains in RV VP3 (L181 to L560 for N7-MTase and I256 to D440 for 2′-O-MTase) and BTV VP4 (R109 to D509 for N7-MTase and T175 to D377 for 2′-O-MTase) are indicated (domain colors as in previous figures).
The sequential addition of cap-0 and cap-1 to the 5′-end of the nascent RNA is performed by the N7-MTase and 2′-O-MTase domains, respectively. Despite low sequence similarity, the MTase domains in RV VP3 are structurally similar to the MTase domains in BTV VP4. Both 2′-O-MTase and N7-MTase domains contain the expected class I SAM-dependent MTase fold with six β-strands surrounded by three helices. While the 2′-O-MTase domain has the characteristic Lys260-Asp340-Lys383-Glu408 (KDKE) catalytic tetrad, this motif is not present in the N7-MTase domain (Fig. 2, C and D). Both of the MTase domains are extensively involved in interactions with the PDE and GTase domains at the dimer interface with twofold related subunits (fig. S4, C and D). The N7-MTase domain contains a positively charged cavity beneath a loop region that is likely the site for nucleotide binding (fig. S5C). The corresponding cavity in the BTV VP4 N7-MTase domain structure contains a bound S-adenosyl-l-homocysteine (SAH) albeit it is negatively charged (fig. S5D). Although the MTase domains in VP3 exhibit overall structural similarities with the corresponding domains in BTV VP4, the residues implicated in SAM/SAH and RNA substrate binding in the BTV VP4 structure are not completely conserved in VP3, suggesting a slightly altered mechanism for substrate recognition and binding in RV VP3 (Fig. 2, C and D, and table S3).
The PDE domain of VP3 exhibits a novel RTPase activity
Before the GTase and MTase activities, an obligatory step in 5′-mRNA capping is the removal of the terminal γ-phosphate (RTPase activity) from the nascent transcript (1). Previous studies suggested that the GTase and MTase activities are associated with VP3 (9, 10), yet the RTPase activity was not demonstrated. Among the RV proteins, nonstructural protein NSP2 is the only protein known to exhibit RTPase/NTPase (nucleoside triphosphatase) activity (19). The lack of NSP2 inside the core particle raises the question of how mRNA capping takes place in RV during endogenous transcription. With this question in mind, we investigated whether purified VP3 exhibits RTPase activity. Our in vitro assays clearly demonstrated that VP3 exhibits RTPase activity (Fig. 3A). We further investigated which region or domain of the VP3 could be responsible for the RTPase activity. We hypothesized that the distal and exposed PDE domain might be an appropriate candidate for the RTPase activity. Our in vitro biochemical assay confirmed that the PDE domain alone exhibits RTPase activity in a dose-dependent manner (Fig. 3A).
Fig. 3. VP3 exhibits RTPase and RNA helicase activity.
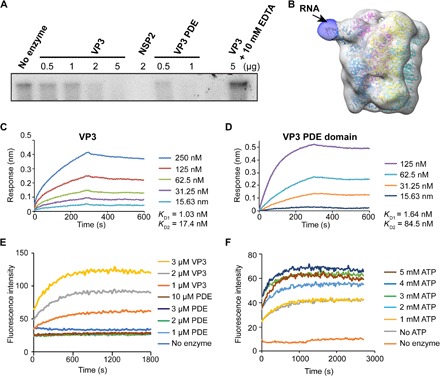
(A) An autoradiograph showing a VP3 or PDE domain concentration-dependent decrease in the 5′-end [γ-32P] GTP-labeled RNA band intensity confirms that VP3 exhibits RTPase activity and that this activity is associated with the PDE domain. RV NSP2, which was previously shown to have RTPase activity, is used as a positive control. The RTPase activity of VP3 is inhibited in the presence of EDTA indicating that the RTPase activity of VP3 is metal dependent. (B) Low-resolution cryo-EM single-particle reconstruction of VP3 in complex with 8-mer single-stranded RNA (ssRNA), suggests that the RNA binds near a cleft in the PDE domain (black arrow). The difference map was obtained using low-resolution cryo-EM map from apo-VP3 and cryo-EM map from VP3-ssRNA complex in Chimera. (C and D) Biolayer interferometry (BLI) analysis of RV VP3 and PDE domain binding with ssRNA (8-mer) shows that both full-length VP3 and PDE domain bind with ssRNA with similar nanomolar affinity. The two–binding site model fits the data with excellent R2 (~0.99) and χ2 (~0) values. (E and F) Fluorescence-based helicase assay. The 6-fluorescin amidite (6-FAM)–labeled RNA strand is quenched by a complementary quencher [Black Hole Quencher (BHQ)–labeled] strand in dsRNA. Increased fluorescence intensity with increasing concentrations of VP3 indicates the strand separation confirming the dsRNA helicase activity of VP3. Helicase activity requires the full-length VP3; the PDE domain alone does not show any significant increase in fluorescence intensity. (F) The fluorescence intensity is also modulated by adenosine triphosphate (ATP) concentrations.
To perform RTPase activity, the PDE domain should first bind to the single-stranded RNA (ssRNA) substrate, and biolayer interferometry (BLI) experiments showed that both full-length VP3 and the PDE domain bind to 8-mer biotinylated ssRNA (Fig. 3, C and D) with nanomolar affinity. The best fit of the binding sensograms in each case was with 2 (RNA):1 (VP3 or PDE) (R2 ~ 0.99, χ2 ~ 0), suggesting the possibility of two simultaneous RNA binding modes of similar affinity. The structure of the PDE domain clearly shows a surface-exposed cleft with an effective radius of 4.69 Å lined predominantly by positively charged residues (fig. S6, A to C), which is likely the site for ssRNA binding. RNA binding to the PDE domain of VP3 is further supported by our low-resolution (~12 Å) cryo-EM reconstruction map of VP3-ssRNA complex (unmodified 8-mer) in which we could unambiguously locate 8-mer ssRNA bound to the cleft in the PDE domain (Fig. 3B and fig. S8). The reconstruction shows that RNA binds to only one of the four PDE domains in the tetramer. In the cryo-EM images, we observed that several VP3 tetramers are decorated with more than one RNA; however, the majority of the VP3 tetramers in these images show binding with only one RNA molecule, and these particles were the ones selected for reconstruction (fig. S8). There are several factors such as binding kinetics, incubation time, RNA concentration, and nature of RNA used in our experiments, which might have affected the simultaneous binding of RNA to all four PDE domains in the tetramer. Nonetheless, our results clearly demonstrate the ability of VP3 to bind RNA through its PDE domain.
Association of RTPase activity with the PDE domain is indeed unique. The fold of the PDE domain is highly conserved among the proteins that belong to the 2H PDE superfamily, which all share two similarly spaced H-X-T-X motifs (20). This family of proteins includes several cellular proteins involved in a variety of signaling processes (20). To our knowledge, RTPase activity has not been shown to be associated with any of these proteins. Whether VP3 is a singular example or whether other cellular proteins with this fold also exhibit RTPase activity remains an open question.
VP3 exhibits RNA helicase activity
A recent report by Jenni et al. (4) showed that the nascent transcript and the negative-strand template form a transient duplex inside the catalytic cavity of VP1 during endogenous transcription. Considering that there is no reported helicase activity for VP1 and that the RNA duplex has to be separated for the nascent transcript to interact with VP3 for capping, we investigated whether VP3 exhibits a helicase-like activity. Our fluorescence-based assay showed that VP3 indeed exhibits helicase activity and that this activity is modulated by the addition of adenosine triphosphate (ATP) and MgCl2 (Fig. 3, E and F, and fig. S6D). VP3 exhibits basal helicase activity even in the absence of ATP (Fig. 3F). However, depletion of MgCl2, with the addition of EDTA, abolishes helicase activity, suggesting that divalent metal ions are critical for VP3-associated helicase activity (fig. S6D). We have also observed a decrease in the helicase activity at higher ATP (>4 mM) and MgCl2 (>3 mM) concentrations, suggesting that free ATP and free MgCl2 hinders the helicase activity, similar to what was previously observed in the case of the NS3 helicase of hepatitis C virus (21). We further tested whether this activity is preferentially affected by a particular nucleoside triphosphate (NTP) and found that ATP and UTP (uridine triphosphate) are preferred over GTP and CTP (cytidine triphosphate) (fig. S6E).
On the basis of the structural features of VP3, it is unlike any known RNA helicase. It is unclear as to the precise mechanism by which VP3 unwinds the RNA duplex and which specific domain(s) in VP3 are involved in this process. It is likely that the PDE domain, because of its distal location and the associated RNA binding and RTPase activities, and the GTase domain, because of its inherent NTPase activity, act in a concerted manner to unwind the RNA duplex. Our conjecture is that while the PDE domain binds to one strand of the RNA duplex, an additional site is created as a result of possible conformational changes due to NTP hydrolysis, which also might fuel the unwinding process that will engage the other strand. Because of the inherent nature of VP3 to form a highly stable tetramer, it is also likely that such an oligomeric assembly is critical for this function. Our attempts to destabilize the tetramer into any smaller oligomeric states to examine whether the tetrameric assembly is crucial for the helicase activity were not successful.
DISCUSSION
Here, by using a combination of cryo-EM, x-ray crystallography, and biochemical assays, we have provided a detailed picture of the structure-activity relationship in the RV VP3. In addition to determining the atomic structure, we have discovered several previously uncharacterized features associated with this multifunctional protein. Our results show that recombinant VP3 not only exhibits GTase and MTase activities but also exhibits previously undiscovered RTPase and RNA binding activities, indicating that VP3 has all the necessary properties required for 5′-RNA capping. Another fascinating finding is that VP3 exhibits a novel RNA helicase activity that can be modulated by ATP and the divalent metal ion MgCl2. Such a confluence of various enzymatic activities including helicase activity embedded in a tetrameric assembly is indeed unique and unprecedented. One of the unexpected results from our studies is that the recombinant VP3 forms a stable tetramer and that it maintains its tetrameric assembly under a variety of conditions. This raises several intriguing questions such as whether VP3 is incorporated into the particle as a tetramer and if so, how such an oligomeric state facilitates capping of nascent transcripts within the confines of the RV capsid interior.
Transcription of the dsRNA by VP1 followed by capping of the nascent transcript occurs within the intact DLP, which is formed by the removal of the outer capsid layer during the process of virus entry into host cells (7, 22). Cryo-EM reconstructions of actively transcribing DLPs have shown that the capped mRNA exits through the aqueous channels at the fivefold axes of the DLP (4). Previous in vitro biochemical studies on transcribing particles show that transcripts are capped by the time the sixth or seventh nucleotide is synthesized by VP1, suggesting that VP3 is in close proximity to VP1 during endogenous transcription (14). Although asymmetric reconstructions of the DLPs have unequivocally demonstrated that VP1 is attached to the innermost layer of the core formed by VP2 at the fivefold axes (4, 23), VP3 was not visualized in these reconstructions, and thus, how VP3 coordinates capping of the nascent mRNA exiting from VP1 is unclear. The absence of VP3 in the asymmetric reconstructions perhaps suggests that VP3 is not firmly anchored to VP1 (or VP2) and that its spatial arrangement inside the particle interior is dynamic.
On the basis of our studies, we envision a plausible functional model for endogenous transcription that incorporates the various novel observations that we have made for VP3. Although the oligomeric state of VP3 inside the RV particle is yet unknown, on the basis of our observations that VP3 forms highly stable tetramers, we hypothesize that VP3 is incorporated into the particles as tetramers. In our model, a VP3 tetramer is associated with each of the nonredundant pairs of neighboring VP1 molecules inside the core, noting that the distance between the RNA exit channels of the neighboring VP1 (~130 Å) matches well with the length of the VP3 tetramer (~126 Å) (Fig. 4A). The distance between the exit channel in the VP1 and the nearest PDE domain in VP3 would approximate to a distance that is spanned by ~5 nucleotides, which is consistent with the biochemical studies that suggest that transcripts are capped by the time the sixth or seventh nucleotide is synthesized by VP1 (14). During endogenous transcription, the duplex formed by the template strand and the nascent transcript that exits from VP1 is separated by the RNA helicase activity associated with VP3. While the transcript continues through the process of capping, the template is returned to form a duplex with the partner positive strand to go through the iterative process of transcription. In this model, following the removal of the γ-phosphate by the PDE domain, the emerging transcript transits through the GTase domain and then through the MTase domains of the twofold related subunit for subsequent steps of capping. The capped transcript then exits via the closest channel at the fivefold axis in the VP2 layer (Fig. 4B). A similar path for a second transcript is envisioned for the other end of the VP3 tetramer that is closest to the neighboring VP1. Similar association of VP3 tetramers with other nonredundant VP1 pairs will ensure independent and simultaneous production of capped transcripts during endogenous transcription. The proposed transcript path and the location of the VP3 parallel to the inner surface of VP2 layer are consistent with a previous proposal by Estrozi et al. (24). Note that this model also works with an antiparallel dimer with the PDE domain of each subunit facing the adjacent VP1. However, the redundancy of the VP3 tetrameric assembly in the dynamic environment of the DLP during endogenous transcription may be advantageous to allow efficient capture the nascent transcripts exiting from the VP1.
Fig. 4. Proposed model for mRNA capping during endogenous transcription.
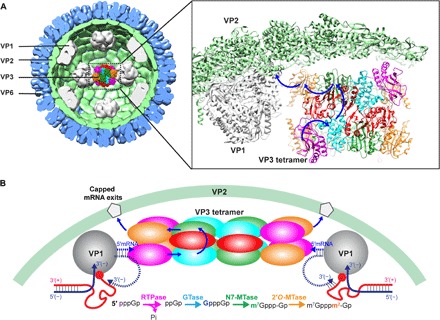
(A) The VP3 tetramer is positioned inside the core particle between two VP1 molecules such that the PDE domains on either side of the VP3 tetramer face the transcript exit channels in the two neighboring VP1 molecules at each fivefold axis. Note that the length of the VP3 tetramer (~126 Å) matches the distance between the two VP1 exit channels (~130 Å). Similar placement of VP3 tetramers between the other five nonredundant VP1 pairs inside the core particle is envisioned. The VP6 and VP2 layers of the DLP are shown in blue and green shades, respectively, along with VP1 molecules (in gray) attached to VP2 at the fivefold axes. In this arrangement of VP3, two simultaneous capping activities can take place using the VP3 oligomeric assembly. The blue arrows indicate the possible path for the nascent transcript during the capping process. (B) Cartoon representation of a possible path for the nascent transcript emerging from VP1 through the PDE and GTase domains of one VP3 subunit and the MTase domains in the twofold related VP3 subunit, followed by its exit through the closest exit channel at the fivefold axes (denoted by a pentagon) of the DLP. Before capping, the RNA duplex formed by the nascent transcript and the template strand needs to be separated. The dsRNA helicase activity exhibited by VP3 suggests such a role for VP3 in RNA duplex separation with subsequent 5′-end capping.
Considering that the entire process of simultaneous transcription of the genomic dsRNA segments within the capsid interior resulting in the extrusion of the individual capped transcripts is necessarily a highly dynamic process, significant conformational rearrangements in VP3 have to be anticipated. Previous studies have indeed shown that VP1 undergo noticeable conformational changes during endogenous transcription (4, 23). Although the precise mechanism of RNA unwinding by the VP3 tetramer is yet to be determined, it is likely that the process of capping, which involves the passage of the nascent transcripts through the various catalytic domains, leads to significant conformational rearrangements within each VP3 subunit and also between the subunits in the tetramer to facilitate strand separation. Elucidation of the precise details of such a fascinating mechanism of how VP3 assists in strand separation and subsequent capping inside the constrained particle interior requires further structural and biochemical studies.
In conclusion, with the elucidation of the VP3 structure, we now have atomic structures of all six structural proteins that define the RV capsid. Our studies have revealed a number of hitherto unsuspected novel aspects of VP3 and provided mechanistic insight into how this protein with its modular domain organization carries out multiple enzymatic activities essential for RV replication. Given its critical role during RV replication, VP3 is a potential target for designing antivirals to counter RV infection that is responsible for the deaths of ~200,000 children annually worldwide, and for such an endeavor, the VP3 structure provides a rational platform.
MATERIALS AND METHODS
Full-length VP3 and PDE cloning, expression, and purification
RV A VP3 sequence (accession number: ABC66299) was codon-optimized for the baculovirus expression system and cloned by Epoch Life Science into the pVL1392 vector using restriction sites (Not I/Eco RI) with a C-terminal 6xHis tag. Recombinant baculoviruses were generated by the Protein and Monoclonal Antibody Production Core at Baylor College of Medicine. Viral plaques were screened for expression and an optimally expressing clone was scaled up for expression in Sf9 cells at a multiplicity of infection of 3 for 96 hours in the presence of protease inhibitor cocktail tablets (Thermo Fisher Scientific) at 24, 48, and 72 hours. Cells were washed with phosphate-buffered saline and pelleted down at 5000 rpm for 10 min at 4°C. Sf9 cells were suspended in lysis buffer A [50 mM tris-HCl (pH 8.0), 500 mM NaCl, 2 mM [Tris(2-carboxyethyl)phosphine hydrochloride (TCEP)], 10% glycerol, 1 mM phenylmethylsulfonyl fluoride, 0.1% Triton X-100, 1× EDTA-free protease inhibitor cocktail, and 500 U of benzonase] and lysed using a homogenizer (KIMBLE Dounce tissue grinder set 15 ml, Millipore Sigma). The cell lysate was cleared by centrifugation at 27,216g (rotor: Beckman Coulter JA-25.50 fixed-angle aluminum rotor) for 60 min at 4°C, and the cleared supernatant was incubated with Ni-NTA (nitrilotriacetic acid) agarose resin (QIAGEN) preequilibrated with lysis buffer. Ni-NTA resin was washed with 15 to 20 column volumes of wash buffer B [50 mM tris-HCl (pH 8.0), 500 mM NaCl, 30 mM imidazole, and 5% glycerol], followed by elution with elution buffer C [50 mM tris-HCl (pH 8.0), 500 mM NaCl, 5 mM TCEP, 10% glycerol, and 300 mM imidazole]. Eluted protein was further purified on a gel filtration column (Superdex 200 Increase 10/300 GL, GE Healthcare) preequilibrated with gel filtration buffer D [50 mM tris-HCl (pH 8.0), 200 mM NaCl, 5 mM dithiothreitol (DTT), 10% glycerol, and 2 mM MgCl2]. Purified protein was concentrated using Amicon Ultra-15 Centrifugal Filter Units (Millipore Sigma) to ~7 mg/ml and used for further studies. The VP3-PDE domain was purified using a previously established protocol (12).
Cryo-EM sample preparation, data collection, and single-particle reconstruction
For cryo-EM sample preparation, VP3 was diluted to ~0.5 mg/ml in a nonglycerol buffer [50 mM tris-HCl (pH 8.0), 200 mM NaCl, 5 mM DTT, and 2 mM MgCl2]. The sample was kept on ice before vitrification (25) on the grid. A 3.0-μl aliquot of VP3 was applied onto a 200-mesh R1.2/1.3 Quantifoil holey carbon grid covered with a graphene oxide support film. After applying the sample, the grid was blotted and rapidly frozen in liquid ethane using a Vitrobot IV (FEI), with constant temperature and humidity during the process of blotting. The grid was stored in liquid nitrogen before imaging. Movie stacks (1690) were collected at 300 kV on a JEM-3200FSC electron microscope (JEOL) with an in-column energy filter (30-eV width) equipped with a direct electron detector K2 Summit camera (Gatan). Images were collected semiautomatically by SerialEM (26) in dose fractionation super-resolution counting mode at 40,000× magnification, corresponding to a calibrated physical pixel size of 0.837 Å. The images were collected with a defocus range from −1.2 to −2.4 μm. The total exposure time for the dataset was 10 s, leading to a total accumulated dose of 53 electrons Å−2 on the specimen. Each image stack was fractionated into 50 subframes, each with an accumulation time of 0.2 s per frame. The final frame average was computed from averages of every three consecutive frames to correct beam-induced motion during exposure by MotionCor2 (27). The image in each frame was weighted according to radiation damage (28). CTF (Contrast Transfer Function) parameters of the particles in each frame average were determined by program e2ctf.py in EMAN2.2 (29) and Gctf 0.50 (30). In total, 133,712 particle images were automatically boxed out by e2boxer.py in EMAN 2.2 (31) with a box size of 300 × 300 pixels using the averaged sum of 50 raw frames per specimen area. Two-dimensional (2D) reference-free class averages were computed using RELION 2.1 (32). Initial models for every reconstruction were generated from scratch by e2initialmodel.py program (29) using selected good quality 2D averages with D2 symmetry based on the 2D averages results. This initial model was low-pass–filtered to 60 Å, and further refinements were carried out using RELION 2.1 (32), with a search angle of 7.5° for 11 iterations and subsequently with an angular sampling of 0.9375° for 14 iterations. Particle images (70,892) were applied for a final reconstruction yielding a resolution of 3.4 Å at 0.143 of the Fourier shell correlation and 2.7 Å after applying postprocessing. For cryo-EM data collection of the VP3-RNA complex, 8-mer ssRNA (5′-rG.rG.rC.rU.rU.rU.rU.rA-3′; Dharmacon Inc.) at a stoichiometric ratio of 1:1000 was incubated with VP3 for 30 min at room temperature and purified by gel filtration (Superdex 200 Increase 10/300 GL, GE Healthcare) column preequilibrated with nonglycerol buffer [50 mM tris-HCl (pH 8.0), 200 mM NaCl, 5 mM DTT, and 2 mM MgCl2]. We followed a similar protocol for VP3-RNA complex cryo-EM single-particle reconstruction. In total, 210,818 particles are boxed out, and 40,694 particles are used for final reconstruction of VP3-RNA density map at 11.8 Å resolution. The cryo-EM data acquisition, image processing, model building, and refinement statistics for both datasets are given in table S1.
Cryo-EM model building and refinement
The full-length VP3 model was built from a postprocessed density map at 2.7 Å resolution. We started de novo model building for full-length VP3 by docking the C-terminal PDE domain [Protein Data Bank (PDB) ID: 4RPT] (12) and segmenting monomer map (segment map feature in Chimera) (33) from the tetramer cryo-EM map. The monomer model was constructed manually in Coot (34) using the C-terminal PDE domain as a starting point toward the N-terminal domain using secondary structure prediction to trace the chain direction. The monomer model was docked into the cryo-EM map in Chimera (35) to generate a tetramer model and refined using Rosetta density refinement (36) and phenix.real_space_refine (37) with secondary structure restraints. The model quality was validated using MolProbity (38) online server and EMRinger tool from the Phenix suite (39). Local resolution was estimated using ResMap (40). Detailed statistics for model building and refinements are given in table S1. The final maps and models were submitted to the Electron Microscopy Data Bank (EMDB) [ID: EMD-0632 (native VP3) and EMD-20159 (VP3-RNA)] and PDB (ID: 6O6B).
Full-length VP3 crystallization, data collection, data processing, and structure determination
Crystallization of full-length VP3 (concentration, ~7 mg/ml) was set up using the hanging drop vapor diffusion method at 20°C. The native crystals were obtained in a crystallization condition containing 15% ethanol, 0.1 M sodium citrate (pH 5.5), and 0.2 M Li2SO4. Selenomethionine-derived crystals were also obtained for use in SAD (single-wavelength anomalous diffraction) phasing. However, selenomethionine-derived crystals diffracted to very low resolution and could not be used for phasing and structure determination. The native VP3 crystals were harvested with 20% ethylene glycol as a cryoprotectant and flash-frozen in liquid nitrogen. The diffraction data were collected at Advanced Light Source, Lawrence Berkeley National Laboratory beamline 5.0.2 at a temperature of 100 K. The native crystal diffracted at 3.5 Å resolution and was further processed and scaled by HKL2000 (41). Because of the lack of a good structural model, including the BTV VP4 structure, phasing by molecular replacement (MR) was not successful. Subsequently, once the cryo-EM model of VP3 was available, MR using the monomer subunit in the phaser module of Phenix suite (42) provided a unique MR solution. The model was further refined using Phenix Refine (43) and validated using MolProbity server (PDB accession code: 6O3V) (38). The ligand density was confirmed with the polder map module in Phenix suite (44). The data processing and model statistics are shown in table S2. The buried surface area was analyzed using PISA server (45). The electrostatic surface potential was rendered in PyMOL using APBS plugin (46, 47). The figures were prepared in Chimera (35), ChimeraX (48), and PyMOL (46). The PDE domain cavity radius was determined by 3V server (49). Multiple sequence alignment was performed by T-Coffee multiple sequence alignment online server (50), and structure-based sequence alignment was performed by ESPript 3.0 server (51).
Enzymatic characterization
VP3 autoguanylylation activity was performed with purified VP3 by incubating increasing concentrations of VP3 with 1 μl of [α-32P] GTP in GTase buffer [20 mM tris-HCl (pH 7.4), 200 mM NaCl, 5 mM DTT, 2 mM MgCl2, and 5% glycerol] in a 20-μl reaction mixture at room temperature for 1 hour as previously described (9). The reaction mixture was resolved by SDS–polyacrylamide gel electrophoresis, followed by transfer onto a polyvinylidene difluoride (PVDF) membrane. The autoguanylylation was observed by autoradiography of the PVDF membrane. RV NSP2 was used as a negative control, and the vaccinia virus capping system (New England Biolabs Inc.) was used as a positive control. For showing direct transfer of GMP on transcript (GTase assay), we followed a previously established protocol with slight modification (52). For this purpose, first we synthesized RV gene 10 transcript using an in vitro transcription kit (Ambion T7 MEGAscript). GTase reaction mixture (20 μl) was prepared by adding 2 μl of IVT (in vitro transcription) synthesized ssRNA (200 ng/μl) with increasing concentrations of VP3 protein in GTase buffer [20 mM Hepes (pH 8.0), 150 mM NaCl, 5 mM DTT, 5 mM MgCl2, and 5% glycerol], 2 μl of [α-32P] GTP, and 1 μl of murine RNase inhibitor (New England Biolabs Inc.) with or without SAM (2 mM), followed by incubation at 30°C for 3 hours. The reaction was stopped by incubating the reaction mixture at 70°C for 5 min and adding 2× RNA gel loading buffer II (Thermo Fisher Scientific). The reaction mixture was resolved on a 6% tris-borate-EDTA [Tris-borate-EDTA (TBE)]–urea gel (Thermo Fisher Scientific). After completely vacuum drying the gel, GTase activity was confirmed by autoradiography. The vaccinia capping system (New England Biolabs Inc.) was used as a positive control. We also used this reaction mixture to show transfer of a methyl group onto the RNA transcript. For that purpose, we purified the RNA from the above reaction mixture using the Zymo RNA clean and concentrator-5 kit (Zymo Research) and eluted the RNA in 7 μl of RNase-free water. The eluted RNA was digested with P1 Nuclease in a 7-μl reaction mixture: 5 μl of purified RNA from the previous step, 0.6 μl of RNase-free water, 0.7 μl of P1 Nuclease buffer [500 mM sodium acetate (pH 5.2)], and 0.7 μl of P1 Nuclease (0.625 U/μl). The reaction mixture was gently mixed and incubated at 37°C for 30 min. Thin-layer chromatography (TLC) was run on a Macherey-Nagel Cellulose MN 300 PEI TLC plate (Fisher Scientific; prerun in RNase-free water and air-dried before loading the samples) in a 0.4 M ammonium sulfate mobile phase. The TLC plate was completely air-dried, and the film was developed for autoradiography. Nonmethylated GpppG and methylated mGpppG (New England Biolabs Inc.) cap standards were used to mark the spots for the capped products. We also performed a colorimetric MTase assay with a commercially available MTase colorimetric assay kit (Cayman Chemical). The SAM-dependent MTase uses SAM as a methyl donor to transfer the methyl group on the substrate. The transfer of the methyl group from SAM (AdoMet) generates SAH (AdoHcy). The MTase assay kit detects the level of free SAH by a colorimetric reaction. Briefly, a 20-μl reaction mixture was prepared with increasing amounts of VP3, 2.5 mM nonmethylated cap analog (GpppG; New England Biolabs Inc.), and 1 mM SAM in MTase buffer [100 mM tris-HCl (pH 8.0), 150 mM NaCl, 2 mM MgCl2, and 5% glycerol]. The reaction mixture was incubated at 37°C for 1 hour. Furthermore, we added 100 μl of the MTase colorimetric assay kit master mix with 15 μl of the reaction mixture, followed by measuring the absorbance at 510 nm for 30 min on a FlexStation 3 (Molecular Devices). RV VP8* was used as a negative control. For RTPase activity, RV gene 10 transcript was synthesized using an in vitro transcription kit (Ambion T7 MEGAshortscript T7 transcription kit) in the presence of 5 μl of [γ-32P]-GTP, followed by 4 hours of incubation at 37°C. RTPase reaction mixture (20 μl) was prepared by adding 2 μl of [γ-32P]-GTP 5′-end labeled gene 10 RNA with increasing concentrations of VP3 and VP3-PDE domain proteins in RTPase buffer [20 mM Hepes (pH 8.0), 150 mM NaCl, 5 mM DTT, 5 mM MgCl2, and 10% glycerol] and 1 μl of murine RNase inhibitor (Invitrogen). The reaction mixture was incubated at room temperature for 1 hour. The previously established RV NSP2 RTPase was used as a positive control (19). The reaction was stopped by adding 10 mM EDTA (pH 8) and 2× RNA gel loading buffer II (Thermo Fisher Scientific), followed by heating at 95°C for 5 min. The reaction mixture was resolved on a 6% TBE-urea gel (Thermo Fisher Scientific). After completely vacuum drying the gel, RTPase activity was confirmed by autoradiography. For RNA helicase activity, a fluorescence resonance energy transfer–based assay was performed using a previously established protocol (53). Briefly, a dsRNA strand was prepared by mixing 6-fluorescin amidite (6-FAM)–labeled RNA strand (6-FAM-5′-UAGUACCGCCACCCUCAGAACCUUUUUUUUUUUUUU-3′; Sigma-Aldrich) and a Black Hole Quencher (BHQ)–labeled quencher strand (5′-GGUUCUGAGGGUGGCCCUACUA-3′-BHQ; Sigma-Aldrich) at 2:1 molar ratio. RNA duplex formation reaction was set up in 25 mM Mops-NaOH (pH 6.5) and RiboLock (Invitrogen; 10 U) buffer by heating at 99°C for 5 min, followed by incubation at 65°C for 30 min and then overnight incubation at 25°C. The RNA helicase assay was performed with increasing concentrations of VP3 in helicase buffer [25 mM Mops-NaOH (pH 6.5), 2.5 mM MgCl2, and 2 mM DTT], 4 U of RiboLock (Invitrogen), 50 nM dsRNA substrate, 5 mM ATP, and 100 nM DNA capture strand (5′-TAGTACCGCCACCCTCAGAACC-3′; IDT) in 50 μl of reaction volume. Fluorescence intensity was measured on a FlexStation 3 (Molecular Devices; maximum excitation/emission, 494/516) at 37°C for 45 min at 20-s intervals. For ATP- and MgCl2-modulated helicase activity, the assay was performed with constant VP3 concentration (3 μM) and varying concentrations of ATP or MgCl2 (1 to 5 mM).
Kinetics of RNA-VP3 and RNA-PDE binding using BLI
Octet RED96 instrument (ForteBio) was used to carry out BLI. Biotinylated 8-mer ssRNA (5′-rG.rG.rC.rU.rU.rU.rU.rA-3′; Dharmacon Inc.) was loaded onto streptavidin-coated biosensors (ForteBio) at a concentration of 0.02 μM in BLI running buffer [50 mM Hepes (pH 8.0), 200 mM NaCl, 2 mM DTT, and 2 mM MgCl2] for 600 s, resulting in capture levels of 0.4 to 0.6 nm within a row of eight tips. RNA-VP3 or RNA-PDE association and dissociation curves were obtained through twofold serial dilutions of VP3 (250, 125, 62.5, 31.25, and 15.625 nM) or PDE domain (125, 62.5, 31.25, and 15.625 nM), respectively, with buffer blanks by using the Octet acquisition software. The binding data acquired were fitted to 2:1 binding model, respectively, using the Octet analysis software based on their R2 values (~0.99) and χ2/degrees of freedom values (~0).
AUC of VP3
To determine the oligomeric state of VP3, AUC experiments were performed using a Beckman Coulter XL-A analytical ultracentrifuge equipped with a TiAn50 eight-hole rotor. Sedimentation velocity experiments were conducted using two-channel epon centerpiece (12 mm) and a quartz window at 3.5 μM concentration. Sample (VP3; 420 μl) and reference solution buffer [50 mM Hepes (pH 8.0), 200 mM NaCl, 2 mM DTT, 2 mM MgCl2, and 5% glycerol; 440 μl] were loaded into the cell. Before starting the experiment, the rotor was equilibrated at 25°C in the vacuum chamber for 1 to 2 hours. Absorbance (optical density at 280 nm) scans were collected at 1-min intervals during sedimentation at 40,000 rpm. The resulting scans were analyzed using the continuous distribution [c(s)] analysis module in the program SEDFIT (54). Partial specific volume of the proteins, solvent density (ρ), and solvent viscosity (η) were calculated from standard tables using the program SEDNTERP (55).
Supplementary Material
Acknowledgments
Funding: We acknowledge support from NIH grant AI36040 (to B.V.V.P.) and the Robert Welch Foundation (Q1279) to B.V.V.P. and (Q1967) to Z.W. The Berkeley Center for Structural Biology is supported, in part, by the NIH, National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences of the U.S. Department of Energy under contract no. DE-AC02-05CH11231. The ALS-ENABLE beamlines are supported in part by the National Institutes of Health, National Institute of General Medical Sciences, grant P30 GM124169. The CryoEM is supported by Advanced Technology Cores (ATC) CryoEM/ET core at Baylor College of Medicine. This project was supported by the Protein and Monoclonal Antibody Production Shared Resource at Baylor College of Medicine with funding from NIH Cancer Center Support Grant P30 CA125123. Author contributions: D.K., X.Y., S.E.C., R.M., R.A., S.K., J.J., and L.H. performed the experiments, data analysis, and interpretation. B.S. assisted with synchrotron data collection. M.K.E., Z.W., and B.V.V.P. contributed to experimental design, analysis, and interpretation. B.V.V.P. contributed to the overall design of the project and wrote the manuscript with D.K. and significant input from all the authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Structural data (EM map, PDB files, and structure factor) are uploaded onto the RCSB PDB and EMDB databases. Raw cryo-EM data would be provided upon request. RVA VP3 sequence accession number: ABC66299. The final cryo-EM maps and model have been submitted to EMDB (ID: EMD-0632 for native VP3 and EMD-20159 for VP3-RNA) and RCSB PDB (ID: 6O6B). The crystal structure has been submitted to the RCSB database (PDB ID: 6O3V).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/16/eaay6410/DC1
REFERENCES AND NOTES
- 1.Decroly E., Ferron F., Lescar J., Canard B., Conventional and unconventional mechanisms for capping viral mRNA. Nat. Rev. Microbiol. 10, 51–65 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramanathan A., Robb G. B., Chan S. H., mRNA capping: Biological functions and applications. Nucleic Acids Res. 44, 7511–7526 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tao Y., Farsetta D. L., Nibert M. L., Harrison S. C., RNA synthesis in a cage - Structural studies of reovirus polymerase λ3. Cell 111, 733–745 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Jenni S., Salgado E. N., Herrmann T., Li Z., Grant T., Grigorieff N., Trapani S., Estrozi L. F., Harrison S. C., In situ structure of rotavirus VP1 RNA-dependent RNA polymerase. J. Mol. Biol. 431, 3124–3138 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He Y., Shivakoti S., Ding K., Cui Y., Roy P., Zhou Z. H., In situ structures of RNA-dependent RNA polymerase inside bluetongue virus before and after uncoating. Proc. Natl. Acad. Sci. U.S.A. 116, 16535–16540 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Troeger C., Khalil I. A., Rao P. C., Cao S., Blacker B. F., Ahmed T., Armah G., Bines J. E., Brewer T. G., Colombara D. V., Kang G., Kirkpatrick B. D., Kirkwood C. D., Mwenda J. M., Parashar U. D., Petri W. A. Jr., Riddle M. S., Steele A. D., Thompson R. L., Walson J. L., Sanders J. W., Mokdad A. H., Murray C. J. L., Hay S. I., Reiner R. C. Jr., Rotavirus vaccination and the global burden of rotavirus diarrhea among children younger than 5 years. JAMA Pediatr. 172, 958–965 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trask S. D., McDonald S. M., Patton J. T., Structural insights into the coupling of virion assembly and rotavirus replication. Nat. Rev. Microbiol. 10, 165–177 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patton J. T., Chen D., RNA-binding and capping activities of proteins in rotavirus open cores. J. Virol. 73, 1382–1391 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu M., Mattion N. M., Estes M. K., Rotavirus VP3 expressed in insect cells possesses guanylyltransferase activity. Virology 188, 77–84 (1992). [DOI] [PubMed] [Google Scholar]
- 10.Chen D., Luongo C. L., Nibert M. L., Patton J. T., Rotavirus open cores catalyze 5′-capping and methylation of exogenous RNA: Evidence that VP3 is a methyltransferase. Virology 265, 120–130 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Zhang R., Jha B. K., Ogden K. M., Dong B., Zhao L., Elliott R., Patton J. T., Silverman R. H., Weiss S. R., Homologous 2',5'-phosphodiesterases from disparate RNA viruses antagonize antiviral innate immunity. Proc. Natl. Acad. Sci. U.S.A. 110, 13114–13119 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ogden K. M., Hu L., Jha B. K., Sankaran B., Weiss S. R., Silverman R. H., Patton J. T., Prasad B. V. V., Structural basis for 2′-5′-oligoadenylate binding and enzyme activity of a viral RNase L antagonist. J. Virol. 89, 6633–6645 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahola T., Kääriäinen L., Reaction in alphavirus mRNA capping: Formation of a covalent complex of nonstructural protein nsP1 with 7-methyl-GMP. Proc. Natl. Acad. Sci. U.S.A. 92, 507–511 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lawton J. A., Estes M. K., Prasad B. V. V., Identification and characterization of a transcription pause site in rotavirus. J. Virol. 75, 1632–1642 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sutton G., Grimes J. M., Stuart D. I., Roy P., Bluetongue virus VP4 is an RNA-capping assembly line. Nat. Struct. Mol. Biol. 14, 449–451 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Ogden K. M., Snyder M. J., Dennis A. F., Patton J. T., Predicted structure and domain organization of rotavirus capping enzyme and innate immune antagonist VP3. J. Virol. 88, 9072–9085 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holm L., Laakso L. M., Dali server update. Nucleic Acids Res. 44, W351–W355 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chu C., Das K., Tyminski J. R., Bauman J. D., Guan R., Qiu W., Montelione G. T., Arnold E., Shatkin A. J., Structure of the guanylyltransferase domain of human mRNA capping enzyme. Proc. Natl. Acad. Sci. U.S.A. 108, 10104–10108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vasquez-Del Carpio R., Gonzalez-Nilo F. D., Riadi G., Taraporewala Z. F., Patton J. T., Histidine triad-like motif of the rotavirus NSP2 octamer mediates both RTPase and NTPase activities. J. Mol. Biol. 362, 539–554 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mazumder R., Iyer L. M., Vasudevan S., Aravind L., Detection of novel members, structure-function analysis and evolutionary classification of the 2H phosphoesterase superfamily. Nucleic Acids Res. 30, 5229–5243 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frick D. N., Banik S., Rypma R. S., Role of divalent metal cations in ATP hydrolysis catalyzed by the hepatitis C virus NS3 helicase: Magnesium provides a bridge for ATP to fuel unwinding. J. Mol. Biol. 365, 1017–1032 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lawton J. A., Estes M. K., Prasad B. V., Mechanism of genome transcription in segmented dsRNA viruses. Adv. Virus Res. 55, 185–229 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding K., Celma C. C., Zhang X., Chang T., Shen W., Atanasov I., Roy P., Zhou Z. H., In situ structures of rotavirus polymerase in action and mechanism of mRNA transcription and release. Nat. Commun. 10, 2216 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Estrozi L. F., Settembre E. C., Goret G., McClain B., Zhang X., Chen J. Z., Grigorieff N., Harrison S. C., Location of the dsRNA-dependent polymerase, VP1, in rotavirus particles. J. Mol. Biol. 425, 124–132 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dubochet J., Adrian M., Chang J. J., Homo J. C., Lepault J., McDowall A. W., Schultz P., Cryo-electron microscopy of vitrified specimens. Q. Rev. Biophys. 21, 129–228 (1988). [DOI] [PubMed] [Google Scholar]
- 26.Mastronarde D. N., Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Zheng S. Q., Palovcak E., Armache J.-P., Verba K. A., Cheng Y., Agard D. A., MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z., Hryc C. F., Bammes B., Afonine P. V., Jakana J., Chen D.-H., Liu X., Baker M. L., Kao C., Ludtke S. J., Schmid M. F., Adams P. D., Chiu W., An atomic model of brome mosaic virus using direct electron detection and real-space optimization. Nat. Commun. 5, 4808 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., Ludtke S. J., EMAN2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 1–296 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Zhang K., Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bell J. M., Chen M., Baldwin P. R., Ludtke S. J., High resolution single particle refinement in EMAN2.1. Methods 100, 25–34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scheres S. H. W., RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pintilie G. D., Zhang J., Goddard T. D., Chiu W., Gossard D. C., Quantitative analysis of cryo-EM density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to regions. J. Struct. Biol. 170, 427–438 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 36.DiMaio F., Song Y., Li X., Brunner M. J., Xu C., Conticello V., Egelman E., Marlovits T., Cheng Y., Baker D., Atomic-accuracy models from 4.5-Å cryo-electron microscopy data with density-guided iterative local refinement. Nat. Methods 12, 361–365 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Afonine P. V., Poon B. K., Read R. J., Sobolev O. V., Terwilliger T. C., Urzhumtsev A., Adams P. D., Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. Sect. D Struct. Biol. 74, 531–544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen V. B., Arendall W. B. III, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barad B. A., Echols N., Wang R. Y.-R., Cheng Y., DiMaio F., Adams P. D., Fraser J. S., EMRinger: Side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kucukelbir A., Sigworth F. J., Tagare H. D., Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Otwinowski Z., Minor W., [20] Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 42.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Headd J. J., Echols N., Afonine P. V., Grosse-Kunstleve R. W., Chen V. B., Moriarty N. W., Richardson D. C., Richardson J. S., Adams P. D., Use of knowledge-based restraints in phenix.refine to improve macromolecular refinement at low resolution. Acta Crystallogr. Sect. D Biol. Crystallogr. 68, 381–390 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liebschner D., Afonine P. V., Moriarty N. W., Poon B. K., Sobolev O. V., Terwilliger T. C., Adams P. D., Polder maps: Improving OMIT maps by excluding bulk solvent. Acta Crystallogr. Sect. D Struct. Biol. 73, 148–157 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krissinel E., Henrick K., Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007). [DOI] [PubMed] [Google Scholar]
- 46.L. Schrödinger, The PyMOL molecular graphics system, version 1.8. (2015); https://www.pymol.org/citing.
- 47.Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A., Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goddard T. D., Huang C. C., Meng E. C., Pettersen E. F., Couch G. S., Morris J. H., Ferrin T. E., UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Voss N. R., Gerstein M., 3V: Cavity, channel and cleft volume calculator and extractor. Nucleic Acids Res. 38, W555–W562 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Di Tommaso P., Moretti S., Xenarios I., Orobitg M., Montanyola A., Chang J.-M., Taly J.-F., Notredame C., T-Coffee: A web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39, W13–W17 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robert X., Gouet P., Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trotman J. B., Schoenberg D. R., RNA Cap methyltransferase activity assay. Bio Protoc. 8, e2767 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tani H., Fujita O., Furuta A., Matsuda Y., Miyata R., Akimitsu N., Tanaka J., Tsuneda S., Sekiguchi Y., Noda N., Real-time monitoring of RNA helicase activity using fluorescence resonance energy transfer in vitro. Biochem. Biophys. Res. Commun. 393, 131–136 (2010). [DOI] [PubMed] [Google Scholar]
- 54.Schuck P., Perugini M. A., Gonzales N. R., Hewlett G. J., Schubert D., Size-distribution analysis of proteins by analytical ultracentrifugation: Strategies and application to model systems. Biophys. J. 82, 1096–1111 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.T. M. Laue, B. D. Shah, T. M. Ridgeway, S. L. Pelletier. Computer-aided interpretation of analytical sedimentation data for proteins, in Analitical Ultracentrifugation in Biochemistry and Polymer Science, J. C. H. S. E. Harding, A. J. Rowe Eds. (Royal Society of Chemistry, 1992), pp. 90–125. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/16/eaay6410/DC1