

Dominance of dissolved extracellular enzymes indicates that deep-sea prokaryotes are associated mainly with particulate matter.
Abstract
Heterotrophic prokaryotes express extracellular hydrolytic enzymes to cleave large organic molecules before taking up the hydrolyzed products. According to foraging theory, extracellular enzymes should be cell associated in dilute systems such as deep sea habitats, but secreted into the surrounding medium in diffusion-limited systems. However, extracellular enzymes in the deep sea are found mainly dissolved in ambient water rather than cell associated. In order to resolve this paradox, we conducted a global survey of peptidases and carbohydrate-active enzymes (CAZymes), two key enzyme groups initiating organic matter assimilation, in an integrated metagenomics, metatranscriptomics, and metaproteomics approach. The abundance, percentage, and diversity of genes encoding secretory processes, i.e., dissolved enzymes, consistently increased from epipelagic to bathypelagic waters, indicating that organic matter cleavage, and hence prokaryotic metabolism, is mediated mainly by particle-associated prokaryotes releasing their extracellular enzymes into diffusion-limited particles in the bathypelagic realm.
INTRODUCTION
Marine dissolved organic carbon (DOC) is one of the largest actively cycling carbon reservoirs, similar in magnitude to atmospheric CO2 (1). The concentration and bioavailability of DOC decrease with water column depth (2, 3), and the apparent age of DOC is about 5000 to 6000 years, indicating rather slow remineralization rates (1). However, the cell-specific respiration rates of deep-sea prokaryotes are similar to that of surface water prokaryotes, despite the fact that their abundance is about two orders of magnitude lower than in surface waters; hence, particulate organic carbon (POC) has been suggested as the trophic basis of the deep-sea prokaryotic community (4, 5). In general, DOC remineralization is largely controlled by heterotrophic prokaryotes using extracellular enzymes to hydrolyze POC and high–molecular weight DOC into low–molecular weight compounds before the incorporation into the cells (6). Thus, extracellular enzymatic activities (EEAs) are referred to as the rate-limiting step in the marine carbon cycle (6, 7).
The extracellular enzymes can either be associated with the periplasmic space and/or to the cell wall of prokaryotes (i.e., cell-attached EEA) or alternatively released into the ambient water (i.e., dissolved or secretory EEA). As predicted by the foraging model, free-living aquatic prokaryotic extracellular enzymes should be associated with the cell, leading to a tight hydrolysis-uptake coupling. In contrast, in diffusion-limited systems such as particles, foraging theory predicts that secretory enzymes, released into the diffusion-limited environment such as in marine snow, are advantageous, where the rate of substrate solubilization is much higher than the growth requirement of the microorganism indicating a loose hydrolysis-uptake coupling (8, 9).
Heterotrophic prokaryotic production in the deep ocean is generally limited by the availability of organic carbon. Consequently, according to foraging theory, one would assume that extracellular enzymes are cell associated in this environment where DOC is refractory and the diversity of the organic molecules is high (2). In the deep ocean, however, EEA is almost exclusively found in the dissolved phase rather than cell associated (10–12), which apparently contradicts the foraging model. Using a combined -omics approach, we addressed this enigma of the prevalence of dissolved or secretory extracellular enzymes in the deep ocean, which is considered as a dilute trophic environment with a plethora of organic solutes at low concentrations (13, 14).
Also, despite the central role of EEAs, little is known about the functional diversity of these enzymes, the factors determining their diversity, and how their functional diversity relates to the phylogenetic composition of microbes throughout the oceanic water column. This lack of understanding is due in part to the large contribution of dissolved material to the total EEA, which could generate a disconnect between the prokaryotes producing these extracellular enzymes and the EEAs, i.e., a decoupling of in situ hydrolysis rates from actual prokaryotic dynamics (12). However, using a multi-omics approach, it is possible to directly link different extracellular enzymes to their potential prokaryotic producers. Moreover, as the exoproteome is a mixture of secretory enzymes released by the living cells and nonsecretory enzymes from dead cells, we examined the signal peptide presence of all predicted carbohydrate-active enzyme (CAZyme) and peptidase sequences. Signal peptides direct the corresponding enzyme to the secretory pathway for transmembrane translocation (15). Thus, by mapping the identified protein spectra derived from the exoproteome data to the signal-peptide-containing sequences, we can distinguish between secretory and nonsecretory enzymes (e.g., between cell-free and cell-associated enzymes).
RESULTS AND DISCUSSION
We examined the abundance, diversity, functional classification, phylogenetic affiliation, and metabolic expression of genes encoding CAZymes and peptidases at a global scale using 345 metagenomes, 52 metatranscriptomes, and 42 metaproteomes covering epipelagic (0 to 200 m), mesopelagic (200 to 1000 m), and bathypelagic (>1000 m) water masses including oxygen minimum zones (OMZ) (fig. S1A and table S1). We focused on CAZymes and peptidases because they are key enzymes involved in the degradation of carbohydrates and proteins (6), respectively, which are the major macromolecules in organisms including prokaryotes inhabiting marine detrital organic matter such as marine snow.
Our metagenomic analyses resulted in 7,065,591 bacterial and 291,414 archaeal CAZyme sequences and 14,180,885 bacterial and 719,134 archaeal peptidase sequences identified after assembly. Because CAZymes and peptidases can be secretory (16), and a large fraction of the total marine EEA is performed by secreted enzymes (10), the quantification and characterization of the gene diversity of the extra- and intracellular enzyme pools provide novel and valuable insights into the ecology of marine prokaryotes and organic matter cycling. Consequently, we differentiated between the secretory and cytoplasmic enzymes, the former characterized by the presence of signal peptides. This approach revealed 263,440 bacterial and 24,158 archaeal CAZyme sequences and 575,553 bacterial and 64,398 archaeal peptidase sequences with secretory potential (see Materials and Methods). Furthermore, to examine the level of agreement between the genomic potential and metabolic expression, metatranscriptomes from the bathypelagic realm were analyzed. Metaproteome analyses were performed to further distinguish between cell-associated proteins (i.e., endoproteome, >0.22-μm fraction) and cell-free proteins (i.e., exoproteome, 0.22-μm to 5000-Da fraction).
The large number (345) of metagenomes analyzed in this study allowed for a complete representation of functions of our target enzymes, as confirmed by a permutation analysis of detected enzyme families (fig. S1B). A principal coordinate analysis against Bray-Curtis dissimilarity, based on the gene abundance of CAZyme and peptidase families, revealed genomic differences in the composition of enzyme families between the epipelagic, mesopelagic, and bathypelagic realm (fig. S1, C and D). This suggests the potential for a depth-stratified clustering of dissolved organic matter (DOM) utilization. The predicted CAZymes and peptidases in the OMZ originally clustered with mesopelagic and/or bathypelagic CAZymes and peptidases in the PCoA (principal coordinate analysis) analyses (fig. S1, C and D). However, a more specific, supervised learning approach (17) teased apart the OMZ cluster from the mesopelagic and bathypelagic clusters (fig. S1, E and F). This indicates that the OMZ selects for enzymes specific for low-oxygen environments, indicating different organic matter utilization pathways between aerobic and anaerobic or microaerobic heterotrophic prokaryotes. Constrained analysis on the principal coordinates suggested that depth was the major factor driving the clustering pattern, and in the surface layer, oxygen, temperature, and salinity also explained the distribution of predicted enzyme composition (fig. S2).
The gene abundance of the predicted total (intracellular and secretory) CAZymes and peptidases was similar in the epipelagic and mesopelagic but significantly decreased in the bathypelagic realm (fig. S3, A and C). In contrast, the abundance of genes encoding secretory CAZymes and peptidases increased with depth from the epipelagic to the bathypelagic realm (fig. S3, B and D). This increase in the percentage of predicted secretory relative to total CAZyme and peptidase genes with depth (Fig. 1A) is consistent with the reported increase in cell-specific total EEA (glucosidase, protease, and phosphatase) and in the proportion of dissolved to total EEA with depth using fluorescent substrate analogs (10). Because the prokaryotic extracellular enzymes are defined as the enzymes outside of the periplasmatic membrane, the relative increase in genes encoding secretory enzymes might indicate that most of the secretory enzymes encoded by these genes are actually released into the environment and, hence, can be considered dissolved or cell-free enzymes (18, 19). A focused analysis on the abundance of genes encoding the four major CAZyme classes (i.e., targeting animal-derived carbohydrates, plant cell walls, peptidoglycans, and fungal carbohydrates) (20) also revealed an increase in the percentage of secretory CAZyme genes with depth in all classes (fig. S3, E to H). This indicates a widespread genomic adaptation of mesopelagic and bathypelagic prokaryotes toward secretory enzymes.
Fig. 1. Increasing contribution of genes tentatively encoding secretory CAZymes and peptidases with depth.

Percent distribution (A), Shannon index (B), and Bray-Curtis dissimilarity–based β-diversity (C) for CAZyme and peptidase encoding genes. Box shows median and interquartile range (IQR); whiskers show 1.5 × IQR of the lower and upper quartiles or range; and outliers extend to the data range. Statistics are based on Wilcoxon test. Letters are used to indicate statistical significance (P < 0.05); a shared letter means no significant difference. Epi, epipelagic (n = 216); Meso, mesopelagic (n = 68); Bathy, bathypelagic (n = 54); OMZ (n = 7).
The α-diversity (Shannon index) of the enzyme-encoding genes was higher for the total than for the secretory enzymes (Fig. 1B), but still, 79% (441 of 553) of total CAZyme families and 47% (992 of 2091) of total protease families belonged to the secretory enzyme gene pool. In addition, a higher variability was found in the secretory relative to the total enzyme gene pool (Fig. 1C), indicative of a more dynamic nature of the secretory enzymes. Overall, the α-diversity of genes encoding enzymes (both total and secretory) was generally higher in the bathypelagic than in the epipelagic waters (Fig. 1B). This higher diversity of genes encoding enzymes in the deep ocean is in agreement with the hypothesis that the low reactivity and the refractory nature of the deep-sea DOM are due to the low concentration of a plethora of diverse organic compounds (2, 3). This increase in the refractory nature of the DOM with depth has also been linked to higher cell-specific prokaryotic EEA in the bathypelagic than in the epipelagic waters (10, 19). However, the vast majority of the deep-sea DOM is in the low–molecular weight size fraction (<1000 Da) (2, 21). These small molecules do not need to be cleaved by extracellular enzymes before prokaryotic uptake. Hence, it is more likely that the deep-sea heterotrophic prokaryotes are associated with the marine snow-type particle pool ubiquitous in mesopelagic and bathypelagic waters (22, 23). Recent metaproteomic studies on organic matter transporters of deep-sea prokaryotes revealed their fairly constant distribution throughout the water column. This also supports the idea that deep-sea microbes are responding to a limited range of labile organic matter substrates, likely cleaved from the particulate organic matter (POM), rather than to the more diverse but recalcitrant deep-sea DOM (24).
A pronounced depth stratification of the phylogenetic composition of the bacterial community was found for genes encoding CAZymes and peptidases (Fig. 2, A and C). In the epipelagic realm, the main groups harboring total CAZyme and peptidase encoding genes were Alphaproteobacteria, Gammaproteobacteria, Bacteroidetes, and Cyanobacteria. The relative contribution of Alphaproteobacteria, Bacteroidetes, and Cyanobacteria decreased with depth, while that of a group of unclassified Bacteria and Gammaproteobacteria increased with depth (Fig. 2, A and C, left). The major contributors to the genes encoding secretory enzymes were Gammaproteobacteria throughout the water column, Bacteroidetes in the epipelagic zone, and the unclassified Bacteria in mesopelagic waters (Fig. 2, A and C, right). In contrast to the high abundance of Cyanobacteria-affiliated genes encoding total CAZymes and peptidases, cyanobacterial sequences of genes encoding secretory enzymes were rarely detected (Fig. 2, A and C). This suggests that photoautotrophic marine microbes such as Cyanobacteria are only minor contributors to the secretory pool of enzymes in the ocean. The decreasing proportion of Bacteroidetes with depth in the gene pool might be linked to the characteristic decrease in high–molecular weight DOM and sinking POM with depth, because Bacteroidetes is preferentially associated with high–molecular weight DOM and particles (25). In the bathypelagic realm, however, the metatranscriptomic data indicated that the contribution of Bacteroidetes to the secretory CAZyme and peptidase pool (20 and 10%, respectively) is comparable to the contribution of Gammaproteobacteria (Fig. 3, A and C), tentatively indicating that in bathypelagic waters POM, like fast-sinking particles or nonsinking marine snow-type particles, is a suitable habitat for Bacteroidetes.
Fig. 2. Phylogenetic affiliation and functional classification of genes encoding bacterial CAZymes and peptidases throughout the water column.
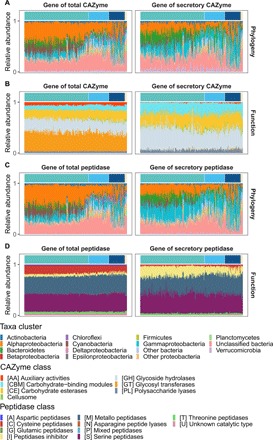
Taxonomic variability at the phylum level (class level for Proteobacteria) of genes encoding CAZymes (A) and peptidases (C); functional composition of genes encoding CAZymes (B) and peptidases (D). Color bars along x axes indicate samples from different depth: green, epipelagic; light blue, mesopelagic; dark blue, bathypelagic; sandy yellow, OMZ.
Fig. 3. Phylogenetic affiliation and functional classification of transcripts for gene encoding bacterial CAZymes and peptidases in the bathypelagic ocean.
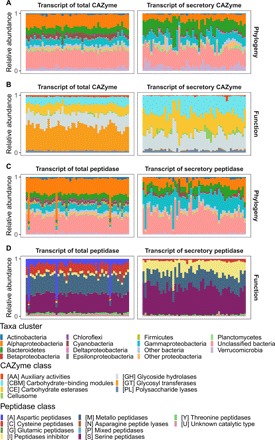
Taxonomic variability at the phylum level (class level for Proteobacteria) of transcripts for genes encoding CAZymes (A) and peptidases (C); functional composition of transcripts for genes encoding CAZymes (B) and peptidases (D).
The metaproteome (endoproteome and exoproteome) analysis showed no clear depth stratification pattern (Fig. 4, A and C) and, similar to the metagenomic data, also identified Alphaproteobacteria, Gammaproteobacteria, and the unclassified bacterial group as the main contributors to the CAZyme and peptidase pool in the endoproteome (Fig. 4, A and C, left). However, Gammaproteobacteria accounted for ca. 75% of the secretory CAZyme and peptidase pool in the exoproteome (Fig. 4, A and C, right, and fig. S4). Bacteroidetes-affiliated CAZymes and peptidases (total and secretory) were also present throughout the water column, consistent with the metatranscriptome data. The high contribution of Gammaproteobacteria and the presence of Bacteroidetes-derived secretory enzymes in the exoproteome (Fig. 4, A and C, right) might indicate preferential utilization of POM by the bacterial community in the deep ocean (26, 27). As the concentration of sinking POM decreases exponentially toward bathypelagic waters, the expression of genes and the presence of proteins related to organic matter hydrolysis might indicate an autochthonous supply of POM in bathypelagic waters either from the nonsinking particle pool, chemoautotrophically produced organic matter, or injection of POM via the physical injection pump (4, 28, 29). Actually, it has been shown that there is a stable background concentration of neutrally buoyant or slow-sinking POM throughout the dark ocean’s water column (4, 22, 23).
Fig. 4. Phylogenetic affiliation and functional classification of bacterial CAZymes and peptidases throughout water column.
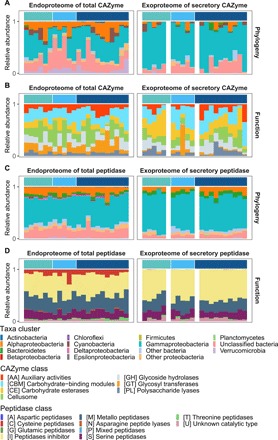
Taxonomic variability at the phylum level (class level for Proteobacteria) of CAZymes (A) and peptidases (C); functional composition of CAZymes (B) and peptidases (D). Color bars along x axes indicate samples from different depth: green, epipelagic; light blue, mesopelagic; dark blue, bathypelagic; sandy yellow, OMZ. Missing data are in white gap.
The high contribution of Gammaproteobacteria (ca. 75%) to the secretory CAZyme and peptidase pool in the exoproteome did not significantly change with depth (fig. S4). This, together with the longer lifetime of extracellular enzymes in the deep versus surface waters, would imply an accumulation of cell-free enzymes in deep waters, consistent with the increase in the proportion of dissolved to total EEA with depth (10, 11). In contrast to the depth-stratified phylogenetic composition of the prokaryotic community, the composition of genes encoding specific enzymatic functions (both total and secretory) was remarkably stable throughout the water column (Fig. 2, B and D). This functional redundancy of microbes in terms of expressed total and secretory enzymes was also detected in the metaproteomic data (Fig. 4, B and D). It is also consistent with the conclusions obtained in a recent metaproteomic study on membrane-associated transporters of deep-sea prokaryotic communities (28).
Despite this apparent stability in the functional diversity of enzymes with depth, there were fundamental differences in the genes tentatively encoding the prevalent enzyme classes of total versus secretory enzymes. In the total CAZyme gene pool, the dominant CAZyme classes were glycosyl transferases (GTs; 37.3 ± 4.3%), glycoside hydrolases (GHs; 24.9 ± 2.0%), carbohydrate esterases (CEs; 18.2 ± 2.2%), carbohydrate-binding modules (CBMs; 9.3 ± 1.7%), and auxiliary activities (AAs; 5.9 ± 1.2%) (Fig. 2B, left). In the secretory CAZyme fraction, however, GTs (0.7 ± 1.0%), which catalyze the sugar bond formation of glycoside inside the cell, were almost absent and the AAs (1.1% ± 0.9%) low in abundance, while the degradative CAZymes, GHs (41.7 ± 8.2%), CEs (26.3 ± 4.8%) (i.e., the families GH23 and GH103, lytic transglucosylase; GH74, xyloglucanase; CE1, acetyl-xylan esterase; CE10, carboxyl esterase) and polysaccharide lyases (PLs; 6.5 ± 2.2%) were highly abundant (Fig. 2B, right, and table S2).
Among the genes encoding peptidases, the contribution of the cysteine peptidase (C) class was substantially lower in the secretory (3.4 ± 2.0%) than in the total fraction (16.0 ± 2.0%). In contrast, a subset of metallo (M) and serine (S) peptidase-like M38 (dipeptidase) and S09 (oligopeptidase) as well as the peptidase inhibitor (I) class (I39) dominated the secretory peptidase gene pool throughout the water column (M: 34.6 ± 5.4%; S: 38.9 ± 4.9%; I: 18.7 ± 4.7%; Fig. 2D and table S2). These functional differences between the total and secretory enzyme gene pool were also detected in both the metatranscriptome and metaproteome (Figs. 3, B and D, and 4, B and D, and tables S4 and S6). In the exoproteome, the degradative CAZymes (i.e., GHs, 7.1 ± 6.6%; CEs, 11.2 ± 6.3%; PLs, 4.7 ± 4.4%) and the CBMs (14.1 ± 10.5%) dominated the secretory CAZyme pool, while GTs were barely detected.
The fluctuation in the composition of CAZymes in both endoproteome and exoproteome throughout the water column might indicate metabolic adaptation to organic matter utilization. The proportion of GHs in the exoproteome (7.1 ± 6.6%; Fig. 4B) is much lower than in the secretory enzyme genes encoded in the metagenome (41.7 ± 8.2%; Fig. 2B). This decrease in the relative proportions from the secreting enzymes encoding genes to the secreted proteins of GHs was more pronounced than that of CEs (from 26.3 ± 4.8% to 11.2 ± 6.3%; Figs. 2B and 4B) and PLs (6.5 ± 2.2% to 4.7 ± 4.4%; Figs. 2B and 4B), leading to a higher relative contribution of CEs and PLs to the degradative CAZyme pool in the exoproteome (Fig. 4B). This suggests different lifetimes of cell-free enzymes in the water column (12). As the balance between peptidases and their inhibitors is critical to the hydrolysis, the higher relative abundance of peptidase inhibitors in the secretory peptidase pool than in the pool of genes encoding secretory enzymes (Fig. 4D) reflects the intensive regulation of proteolytic activities (30).
Archaea also contributed to the prokaryotic gene pool encoding CAZymes and peptidases, although to a minor extent (mean ± SD; 4.8 ± 4.0% of total CAZymes, 6.3 ± 5.0% of secretory CAZymes; 7.1 ± 6.7% of total peptidases, 10.7 ± 8.6% of secretory peptidases; n = 345; table S3). Metatranscriptomic and metaproteomic analyses revealed that mainly Euryarchaeota contributed to the archaeal CAZyme and peptidase pool, and Euryarchaeota contributed only 2 to 3% to the secretory CAZyme and peptidase gene transcripts. In the exoproteome, the archaeal CAZymes and peptidases were barely detected (figs. S5 and S6, and tables S5 and S7).
The repertoire of genes encoding peptidases and CAZymes of the two major bacterial groups, i.e., Alphaproteobacteria and Gammaproteobacteria, was further analyzed (Fig. 5). While the abundance of gammaproteobacterial genes encoding secretory enzymes increased with depth (fig. S7, B and D), the number of alphaproteobacterial genes encoding secretory enzymes decreased from the epipelagic to the mesopelagic layer and increased again in the bathypelagic realm (fig. S7, A and C). Although the proportion of secretory to total enzymes was higher in Gammaproteobacteria than in Alphaproteobacteria, this proportion increased in both bacterial groups (at the community level) with depth (Fig. 5). A detailed gene analysis of the functional diversity of the enzyme classes of different bacterial taxa with depth revealed different levels of variability among phylogenetic groups (fig. S8, B and D). Specifically, Alphaproteobacteria exhibited a higher variability in the relative abundance of genes encoding secretory CAZymes and peptidases with depth. This was in contrast to the rather stable abundance of genes encoding CAZymes and peptidases in other groups such as Gammaproteobacteria (fig. S8, F and H).
Fig. 5. Percentage of genes encoding secretory CAZymes and peptidases of Alphaproteobacteria and Gammaproteobacteria increasing with depth.
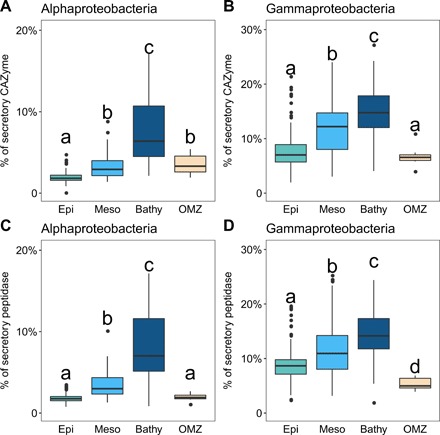
(A and C), Alphaproteobacteria; (B and D), Gammaproteobacteria. Box shows median and IQR; whiskers show 1.5 × IQR of the lower and upper quartiles or range; outliers extend to the data range. Statistics are based on Wilcoxon test, and letters are used to show statistical significance (P < 0.05); a shared letter means no significant difference. Epipelagic (n = 216); mesopelagic (n = 68); bathypelagic (n = 54); OMZ (n = 7).
A further phylogenetic analysis of total and secretory CAZymes and peptidases at the order level revealed a strong shift in alphaproteobacterial genes, with Pelagibacterales dominating in surface waters and Sphingomonadales and Rhodobacterales dominating in bathypelagic waters (fig. S8, A and C). This strong shift in the dominating phylogenetic groups in Alphaproteobacteria contrasts with the more gradual changes observed in gammaproteobacterial enzymes, where the contribution of Alteromonadales and Oceanospirillales gradually increased with depth (fig. S8, E and G). The metatranscriptomic data are in agreement with the metagenomic results indicating that Sphingomonadales, Rhodobacterales, Alteromonadales, and Oceanospirillales were dominating organic matter utilization in the bathypelagic realm (fig. S9). While Alteromonadales secreted both CAZymes and peptidases into the ambient water as indicated by the proteome analyses, Sphingomonadales and Rhodobacterales exhibited preferential expression of secretory peptidases only (fig. S10). Roseobacter, a member of the Rhodobacterales, is about equally important as SAR11 in amino acid assimilation in coastal environments (31). In contrast to Gammaproteobacteria, Alphaproteobacteria showed distinct patterns in the CAZyme and peptidase gene expression, indicating that the phylogenetic difference in community composition was detectable not only on the 16S ribosomal RNA gene level but also on the metabolic level (figs. S9 and S10).
The relative increase in secreted enzymes in Alphaproteobacteria from the epipelagic to the bathypelagic waters might be due to the decline of Pelagibacterales and the increase of Sphingomonadales and Rhodobacterales with depth (fig. S8). Pelagibacterales accounted for the majority of Alphaproteobacteria in epipelagic waters and are typical oligotrophic organisms with a streamlined genome (32). Hence, it is possible that the small genome of Pelagibacterales reduces the possibility of harboring genes encoding for secreted enzymes. In contrast, the relative increase in genes encoding secretory CAZyme and peptidase originating from Rhodobacterales in the bathypelagic waters reflects the characteristic metabolic flexibility in energy acquisition and carbon utilization of the Roseobacter clade (33). The increase in the number of gammaproteobacterial genes encoding secreted enzymes with depth is caused by Alteromonas, the most abundant Gammaproteobacterium in bathypelagic waters, harboring the widespread type II secretion (T2S) system and mediating enzyme secretion (34). Sphingomonadales and Gammaproteobacteria, such as Alteromonadales and Oceanospirillales, use TonB-dependent transporters for DOM uptake (35). Isolates of deep-sea ecotypes of Sphingomonadales, Alteromonadales, and Oceanospirillales produce exopolysaccharides (36–38), which can trap nutrients and retain extracellular enzymes (39). This indicates that deep-ocean heterotrophic prokaryotes, generally considered substrate limited, might benefit from producing exopolymeric material to facilitate scavenging of organics from the environment in combination with the use of TonB transporters and secreting enzymes (24, 40). This strategy might allow for an increased efficiency in the acquisition and processing of organic matter in a diluted environment such as the deep ocean.
The Alteromonadales are widespread in the marine environment, especially in the deep sea (41, 42). Also, Alteromonadales respond rapidly to changes in DOM (43). Given the refractory nature of deep-sea DOM, chemolithotroph production of POM or DOM originating from chemolithotrophs may support gammaproteobacterial growth in deep ocean waters. Although multilevel physiological regulation and adaptations are needed (44), our data indicate that the capacity of secreting extracellular enzymes targeting both carbohydrates and proteins might be one of the complex functional properties. Such strategy could also be used by other phylotypes like Rhodobacterales, Sphingomonadales, and Oceanospirillales and help the establishment of bacterial generalists occupying a wide ecological niche throughout the water column despite the heterogeneous composition of organic matter supply (45).
These integrated meta-omics (genomic, transcriptomic, and proteomic) analyses, focusing on the repertoire of prokaryotic functional genes tentatively encoding CAZymes and peptidases and on the respective enzyme distribution as revealed by the proteomics approach, indicate a high level of functional redundancy among oceanic prokaryotes throughout the water column. While on a phylogenetic level the prokaryotic community composition was depth-stratified, the functional classes of CAZyme- and peptidase-encoding genes remained fairly constant. A similar conclusion has been reached by a recent metaproteomic study on the distribution of transporter proteins responsible for DOM uptake by prokaryotes (24). Nonetheless, this meta-omics approach is inherently biased to the reference database and availability of samples. Compared to the large number of metagenomic datasets, the availability of metaproteomic databases is limited. Furthermore, only limited data are available from the vast regions of the Arctic and Southern Ocean.
Together, our data provide an unprecedented view of the link between two major categories of extracellular enzymes and specific prokaryotic taxa throughout the oceanic water column. Moreover, our analyses suggest genomic and functional adaptation of dark ocean prokaryotes in response to the low concentration and diverse nature of the deep-sea DOM pool. The main adaptation is the increasing proportion of genes encoding secreted enzymes with depth. This conclusion is supported by the increase in cell-specific total EEA and by the proportion of dissolved versus total EEA with depth measured with substrate analogs (10, 19). The higher relative proportion of secreted enzymes of deep-ocean prokaryotes might be related to their preferential particle-associated lifestyle (10, 23, 26) because particle-associated prokaryotes tend to show a loose hydrolysis-uptake coupling mediated by the release of extracellular enzymes into the environment (9). Our results also indicate that the documented changes in organic matter composition with depth are reflected by the genes encoding enzymes for organic matter cleavage as the capability of prokaryotes such as Alteromonadales to secrete enzymes into the environment increases with depth. According to foraging theory, releasing enzymes into the environment is advantageous only if the environment is diffusion limited. Hence, we conclude that prokaryotes and prokaryotic activity are mainly associated with particles in the deep sea where neutrally buoyant particles are abundant (22) and prokaryotic activity is related to particle concentration (23). This conclusion is also supported by the findings of Follett et al. (46) reporting that about 30% of the bathypelagic DOC is of recent origin, while the remaining DOC is more than 30,000 years old, and with the notion that the POC flux supports the bulk (ca. 90%) of the respiratory carbon demand in the dark ocean (5). We present compelling evidence that, on the basis of the increasing contribution of genes encoding secretory enzymes, deep-sea prokaryotes and their metabolism are likely associated with particles rather than on the utilization of ambient-water DOC. This finding has important implications for the oceanic organic carbon inventory, as a potential explanation for the paradoxical discrepancy between the high metabolic activity detected in deep-sea prokaryotes on a single-cell level and the century- to millnium-long turnover times of deep-sea DOC indicative of a high degree of metabolic recalcitrance.
MATERIALS AND METHODS
Metagenomic and meta (endo- and exo-)proteomic sampling
Two hundred to 400 liters of seawater were filtered through 0.22-μm pore-size polycarbonate membranes (142-mm diameter) at 22 stations from the Pacific, Atlantic, and Southern Ocean (table S1). Using tangential flow filtration with a molecular weight cutoff of 5000 Da, the filtrates were further concentrated to a final volume of 50 ml following the method of Wang et al. (47) for exoproteomic analyses as described below. The filters and the concentrated filtrates were immediately frozen in liquid nitrogen and stored at −80°C until extraction. The filters were used for both DNA (metagenomics) and protein extraction (endoproteomics). The DNA was extracted from one-quarter of each filter using a standard phenol extraction protocol (48) and sequenced by Microsynth AG, Switzerland.
For endoproteome analyses, protein extraction was performed according to Dong et al. (49). Briefly, the filter sections were cut into smaller pieces and resuspended in lysis buffer containing 7 M urea, 2 M thiourea, 1% dithiothreitol, 2% CHAPS, and protease inhibitor cocktail. The mixture was homogenized with bead beating and thereafter sonicated at high power with 10-s pulses for 10 min. The supernatant from the slurry and the concentrated filtrates were further concentrated with a 3000-Da Amicon Ultra-15 Centrifugal Filter Unit (Millipore). The protein fraction was precipitated with cold ethanol overnight at −20°C and resuspended with 7 M urea and 2 M thiourea. The protein concentration was quantified with a bicinchoninic acid assay (Thermo Fisher Scientific). Ten to 30 μg of protein of each sample were used for in-solution trypsin digestion (1:100, w/w). The peptides were sequenced on a Q Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Scientific) after desalting. In total, 8 metagenomes, 22 endoproteomes, and 20 exoproteomes were generated (table S1).
Acquisition of global ocean metagenomes and metatranscriptomes
Besides the metagenomes and metaproteomes generated, 337 marine metagenomes and 52 marine metatranscriptomes were downloaded from the National Center for Biotechnology Information website (table S1). The corresponding metadata (coordinates, depth, and environmental parameters) were retrieved from the original publications and websites, if available.
Metagenomic and metatranscriptomic analysis
Reads from the metagenomic dataset were assembled individually using Megahit (1.1.2) (50) with default settings. Putative genes were then predicted on contigs longer than 200 base pairs using Prodigal (2.6.3) (51) under metagenome mode (-p meta). The abundance of each predicted gene was evaluated by mapping reads back with Burrows-Wheeler Aligner (BWA) algorithm (0.7.16a) (52) and then normalized with the following equation: RPM = 1M × (mapped reads/gene length)/(sum of mapped reads/gene length). For all the predicted genes, CAZymes were annotated using hmmsearch against the dbCAN database (53) (e value <1 × 10−10; coverage, >0.3). The domain with the highest coverage was selected for sequences overlapping multiple CAZyme domains. Peptidases were annotated using DIAMOND (0.8.36) BLASTp (54) searches against the MEROPS database (55) using cutoffs of e value <1 × 10−10. The phylogenetic affiliation of CAZyme and peptidase sequences was determined using the lowest common ancestor algorithm adapted from DIAMOND (0.8.36) (54) blast by searching against the nonredundant database. The top 10% hits with an e value <1 × 10−5 were used for phylogeny determination (--top 10). SignalP (4.0) (56) was used to detect the presence of signal peptides for bacterial sequences. Actinobacteria- and Firmicutes-affiliated sequences were predicted under Gram-positive mode, while other bacterial sequences were predicted under Gram-negative mode. For archaeal sequences, PSORTb (3.0.2) (57) was used to predict the subcellular location, because SignalP (4.0) (56) does not support sequences of Archaea. To evaluate the gene expression in the metatranscriptomic data, reads from each metatranscriptome were mapped to the CAZyme and peptidase gene category from the metagenomic assembly with BWA algorithm (0.7.16a) (52) and normalized as follows: TPM = 1M × (mapped transcripts/gene length)/(sum of mapped reads/gene length).
Proteomic annotation and analysis
The tandem mass spectrometry spectra from each proteomic sample were searched using SEQUEST-HT(12) engines against a curated protein database of metagenomic CAZyme and peptidase sequences, which were clustered at 90% similarity (-c 0.9 -G 0 -aS 0.9) using CD-HIT(4.6.8) (58) and validated with Percolator in Proteome Discoverer 2.1 (Thermo Fisher Scientific). To reduce the probability of false peptide identification, the target-decoy approach (59) was used, and results <1% false discovery rate at the peptide level were kept. A minimum of two peptides and one unique peptide was required for protein identification. Protein quantification was conducted with a chromatographic peak area-based label-free quantitative method (60).
Statistical analysis and visualization
All the statistics and visualization were performed using specific packages in R (www.r-project.org/). Vegan, rtk, and ggplot2 were used for ordination, diversity calculation, and visualization, respectively.
Supplementary Material
Acknowledgments
We thank T. Reinthaler and G. Dangl for the help with sampling. We also thank M. Guerreiro for bioinformatics input. Funding: The study was supported by the Austrian Science Fund (FWF) project ARTEMIS (project number P28781-B21) to G.J.H. This work is in partial fulfillment of the requirements for a PhD degree from the University of Vienna to Z.Z. Author contributions: Z.Z. and G.J.H. conceived the project. Z.Z. collected the samples, did the experiments, and performed the analysis. Z.Z., F.B., and G.J.H. interpreted the data and wrote the paper. Competing interest: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/16/eaaz4354/DC1
REFERENCES AND NOTES
- 1.Hansell D. A., Recalcitrant dissolved organic carbon fractions. Ann. Rev. Mar. Sci. 5, 421–445 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Arrieta J. M., Mayol E., Hansman R. L., Herndl G. J., Dittmar T., Duarte C. M., Ocean chemistry. Dilution limits dissolved organic carbon utilization in the deep ocean. Science 348, 331–333 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Jiao N., Herndl G. J., Hansell D. A., Benner R., Kattner G., Wilhelm S. W., Kirchman D. L., Weinbauer M. G., Luo T., Chen F., Azam F., Microbial production of recalcitrant dissolved organic matter: Long-term carbon storage in the global ocean. Nat. Rev. Microbiol. 8, 593–599 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Herndl G. J., Reinthaler T., Microbial control of the dark end of the biological pump. Nat. Geosci. 6, 718–724 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aristegui J., Duarte C. M., Agustí S., Doval M., Álvarez-Salgado X. A., Hansell D. A., Dissolved organic carbon support of respiration in the dark ocean. Science 298, 1967 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Arnosti C., Microbial extracellular enzymes and the marine carbon cycle. Ann. Rev. Mar. Sci. 3, 401–425 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Azam F., Malfatti F., Microbial structuring of marine ecosystems. Nat. Rev. Microbiol. 5, 782–791 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Vetter Y. A., Deming J. W., Jumars P. A., Krieger-Brockett B. B., A predictive model of bacterial foraging by means of freely released extracellular enzymes. Microb. Ecol. 36, 75–92 (1998). [DOI] [PubMed] [Google Scholar]
- 9.Smith D. C., Simon M., Alldredge A. L., Azam F., Intense hydrolytic enzyme-activity on marine aggregates and implications for rapid particle dissolution. Nature 359, 139–142 (1992). [Google Scholar]
- 10.Baltar F., Arístegui J., Gasol J. M., Sintes E., van Aken H., Herndl G. J., High dissolved extracellular enzymatic activity in the deep central Atlantic Ocean. Aquat. Microb. Ecol. 58, 287–302 (2010). [Google Scholar]
- 11.Baltar F., Aristegui J., Gasol J. M., Yokokawa T., Herndl G. J., Bacterial versus archaeal origin of extracellular enzymatic activity in the Northeast Atlantic deep waters. Microb. Ecol. 65, 277–288 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Baltar F., Watch out for the "living dead": Cell-free enzymes and their fate. Front. Microbiol. 8, 2438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arrieta J. M., Mayol E., Hansman R. L., Herndl G. J., Dittmar T., Duarte C. M., Dilution limits dissolved organic carbon utilization in the deep ocean. Science 348, 331–333 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Shen Y., Benner R., Molecular properties are a primary control on the microbial utilization of dissolved organic matter in the ocean. Limnol. Oceanogr. 9999, 1–11 (2019). [Google Scholar]
- 15.K. Kapp, S. Schrempf, M. K. Lemberg, B. Dobberstein, Madame Curie Bioscience Database [Internet] (Landes Bioscience, 2013). [Google Scholar]
- 16.M. R. Pollock, Exoenzymes. in The bacteria, E. C. Gunsalus, R. Y. Stanier, Eds. (Academic Press, 1962), vol. 4, pp. 121–178. [Google Scholar]
- 17.J. Ye, R. Janardan, Q. Li, Two-dimensional linear discriminant analysis, in Advances in Neural Information Processing Systems 17 - Proceedings of the 2004 Conference, NIPS 2004 (Advances in Neural Information Processing Systems), (Neural Information Processing Systems Foundation, 2005), pp. 1569–1576. [Google Scholar]
- 18.Karner M., Herndl G. J., Extracellular enzymatic-activity and secondary production in free-living and marine-snow-associated bacteria. Mar. Biol. 113, 341–347 (1992). [Google Scholar]
- 19.Baltar F., Arístegui J., Sintes E., van Aken H., Gasol J. M., Herndl G. J., Prokaryotic extracellular enzymatic activity in relation to biomass production and respiration in the meso- and bathypelagic waters of the (sub)tropical Atlantic. Environ. Microbiol. 11, 1998–2014 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Cantarel B. L., Lombard V., Henrissat B., Complex carbohydrate utilization by the healthy human microbiome. PLOS ONE 7, e28742 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benner R., Pakulski J. D., McCarthy M., Hedges J. I., Hatcher P. G., Bulk chemical characteristics of dissolved organic matter in the ocean. Science 255, 1561–1564 (1992). [DOI] [PubMed] [Google Scholar]
- 22.Bochdansky A. B., van Aken H. M., Herndl G. J., Role of macroscopic particles in deep-sea oxygen consumption. Proc. Natl. Acad. Sci. U.S.A. 107, 8287–8291 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baltar F., Aristegui J., Gasol J. M., Sintes E., Herndl G. J., Evidence of prokaryotic metabolism on suspended particulate organic matter in the dark waters of the subtropical North Atlantic. Limnol. Oceanogr. 54, 182–193 (2009). [Google Scholar]
- 24.Bergauer K., Fernandez-Guerra A., Garcia J. A. L., Sprenger R. R., Stepanauskas R., Pachiadaki M. G., Jensen O. N., Herndl G. J., Organic matter processing by microbial communities throughout the Atlantic water column as revealed by metaproteomics. Proc. Natl. Acad. Sci. U.S.A. 115, E400–E408 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kirchman D. L., The ecology of Cytophaga-Flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 39, 91–100 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Aristegui J., Gasol J. M., Duarte C. M., Herndl G. J., Microbial oceanography of the dark ocean's pelagic realm. Limnol. Oceanogr. 54, 1501–1529 (2009). [Google Scholar]
- 27.Benner R., Amon R. M., The size-reactivity continuum of major bioelements in the ocean. Ann. Rev. Mar. Sci. 7, 185–205 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Boyd P. W., Claustre H., Levy M., Siegel D. A., Weber T., Multi-faceted particle pumps drive carbon sequestration in the ocean. Nature 568, 327–335 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Baltar F., Arístegui J., Sintes E., Gasol J. M., Reinthaler T., Herndl G. J., Significance of non-sinking particulate organic carbon and dark CO2 fixation to heterotrophic carbon demand in the mesopelagic northeast Atlantic. Geophys. Res. Lett. 37, (2010). [Google Scholar]
- 30.Bode W., Huber R., Natural protein proteinase inhibitors and their interaction with proteinases. Eur. J. Biochem. 204, 433–451 (1992). [DOI] [PubMed] [Google Scholar]
- 31.Kujawinski E. B., The impact of microbial metabolism on marine dissolved organic matter. Ann. Rev. Mar. Sci. 3, 567–599 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Giovannoni S. J., SAR11 bacteria: The most abundant plankton in the oceans. Ann. Rev. Mar. Sci. 9, 231–255 (2017). [DOI] [PubMed] [Google Scholar]
- 33.A. Buchan, J. M. González, in Handbook of Hydrocarbon and Lipid Microbiology, K. N. Timmis, Ed. (Springer Berlin Heidelberg, Berlin, Heidelberg, 2010), pp. 1335–1343. [Google Scholar]
- 34.Evans F. F., Egan S., Kjelleberg S., Ecology of type II secretion in marine gammaproteobacteria. Environ. Microbiol. 10, 1101–1107 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Tang K., Jiao N., Liu K., Zhang Y., Li S., Distribution and functions of TonB-dependent transporters in marine bacteria and environments: Implications for dissolved organic matter utilization. PLOS ONE 7, e41204 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar A. S., Mody K., Jha B., Bacterial exopolysaccharides—A perception. J. Basic Microbiol. 47, 103–117 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Gutierrez T., Berry D., Yang T., Mishamandani S., McKay L., Teske A., Aitken M. D., Role of bacterial exopolysaccharides (eps) in the fate of the oil released during the deepwater horizon oil spill. PLOS ONE 8, e67717 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Le Costaouëc T., Cérantola S., Ropartz D., Ratiskol J., Sinquin C., Colliec-Jouault S., Boisset C., Structural data on a bacterial exopolysaccharide produced by a deep-sea Alteromonas macleodii strain. Carbohydr. Polym. 90, 49–59 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Decho A. W., Microbial exopolymer secretions in ocean environments: Their role(s) in food webs and marine processes. Oceanogr. Mar. Biol. 28, 73–154 (1990). [Google Scholar]
- 40.Heissenberger A., Leppard G. G., Herndl G. J., Relationship between the intracellular integrity and the morphology of the capsular envelope in attached and free-living marine bacteria. Appl. Environ. Microbiol. 62, 4521–4528 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DeLong E. F., Preston C. M., Mincer T., Rich V., Hallam S. J., Frigaard N.-U., Martinez A., Sullivan M. B., Edwards R., Brito B. R., Chisholm S. W., Karl D. M., Community genomics among stratified microbial assemblages in the ocean's interior. Science 311, 496–503 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Agogué H., Lamy D., Neal P. R., Sogin M. L., Herndl G. J., Water mass-specificity of bacterial communities in the North Atlantic revealed by massively parallel sequencing. Mol. Ecol. 20, 258–274 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi Y., Tyson G. W., Eppley J. M., DeLong E. F., Integrated metatranscriptomic and metagenomic analyses of stratified microbial assemblages in the open ocean. ISME J. 5, 999–1013 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boeuf D., Edwards B. R., Eppley J. M., Hu S. K., Poff K. E., Romano A. E., Caron D. A., Karl D. M., DeLong E., Biological composition and microbial dynamics of sinking particulate organic matter at abyssal depths in the oligotrophic open ocean. Proc. Natl. Acad. Sci. U.S.A. 116, 11824–11832 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mou X., Sun S., Edwards R. A., Hodson R. E., Moran M. A., Bacterial carbon processing by generalist species in the coastal ocean. Nature 451, 708–711 (2008). [DOI] [PubMed] [Google Scholar]
- 46.Follett C. L., Repeta D. J., Rothman D. H., Xu L., Santinelli C., Hidden cycle of dissolved organic carbon in the deep ocean. Proc. Natl. Acad. Sci. U.S.A. 111, 16706–16711 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang D.-Z., Dong H.-P., Xie Z.-X., Dai M.-H., Hong H.-S., Metaproteomic characterization of dissolved organic matter in the water column of the South China Sea. Limnol. Oceanogr. 56, 1641–1652 (2011). [Google Scholar]
- 48.Paul J. H., Section 1 update: Extraction of microbial DNA from aquatic sources: Marine environments. Mol. Microbial Ecol. Manual 1, 1931–1942 (1996). [Google Scholar]
- 49.Dong H.-P., Wang D.-Z., Dai M., Chan L. L., Hong H.-S., Shotgun proteomics: Tools for analysis of marine particulate proteins. Limnol. Oceanogr.-Meth. 7, 865–874 (2009). [Google Scholar]
- 50.Li D., Liu C.-M., Luo R., Sadakane K., Lam T.-W., MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Hyatt D., Chen G.-L., LoCascio P. F., Land M. L., Larimer F. W., Hauser L. J., Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li H., Durbin R., Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang H., Yohe T., Huang L., Entwistle S., Wu P., Yang Z., Busk P. K., Xu Y., Yin Y., dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46, W95–W101 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buchfink B., Xie C., Huson D. H., Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Rawlings N. D., Barrett A. J., Thomas P. D., Huang X., Bateman A., Finn R. D., The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 46, D624–D632 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Petersen T. N., Brunak S., von Heijne G., Nielsen H., SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 (2011). [DOI] [PubMed] [Google Scholar]
- 57.Yu N. Y., Wagner J. R., Laird M. R., Melli G., Rey S., Lo R., Dao P., Sahinalp S. C., Ester M., Foster L. J., Brinkman F. S. L., PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26, 1608–1615 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li W., Godzik A., Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Elias J. E., Gygi S. P., Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Zhang Y., Wen Z., Washburn M. P., Florens L., Improving label-free quantitative proteomics strategies by distributing shared peptides and stabilizing variance. Anal. Chem. 87, 4749–4756 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/16/eaaz4354/DC1