

Abstract
Background:
In the United States, high-risk medical devices must be cleared through the premarket approval (PMA) pathway, which requires clinical evidence ensuring safety and efficacy. Approved devices can be modified and reintroduced to market without additional study through the PMA supplemental review track. This study characterizes the changes of high-risk plastic surgery devices once they undergo initial clearance.
Methods:
A retrospective, cross-sectional analysis of the Food and Drug Administration (FDA) PMA database. The following data were extracted from the PMA database (January 1, 1980 to December 31, 2018): initial clearance date, device type, the number and type of supplement, supplement reason, and product withdrawal date. Data from the FDA medical device recall database were also extracted and reported. The median number of device modifications and median lifetime of device-years were calculated.
Results:
There have been 39 original plastic surgery devices approved by the FDA. There was no significant change with respect to initial clearance dates for original devices over time (r = 0.28; P = 0.084). PMA supplement usage has significantly increased with time (rs = 0.9174, P = 0.000). Overall, approved plastic surgery devices have undergone a median of 11 changes (IQR, 3–35). Breast implant devices collectively underwent the most modifications with a median of 28 modifications per device (IQR, 20.25–33.25).
Conclusions:
Over the past 2 decades, plastic surgery device manufacturers have significantly increased the use of supplement track review. High-risk plastic surgery devices may undergo frequent minor changes without clinical evidence to support the safety and efficacy of modified versions.
INTRODUCTION
The rapidly growing medical device industry is projected to be valued at 800 billion dollars by the year 2030.1 The label “medical device” can be applied to a wide range of products, from gauze to synthetic skin substitutes.
The Food and Drug Administration (FDA) is the professional body responsible for the regulation and safety of medical devices in the United States.2 They define a medical device as: “an instrument, apparatus, implement, machine, […] intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease […].”2 The FDA originally began regulating medical devices under the Medical Device Regulation Act (MDRA) in 1976 following the adverse events and deaths related to intrauterine devices, which harmed an estimated 200,000 women and their families.3
The MDRA was passed in 1976 to establish risk-based classifications for the evaluation of medical devices. The MDRA framework included 3 categories (Class I–III), in order of increasing potential risk (Table 1).2 Low-risk devices (Class I) do not require FDA clearance before marketing (eg, tongue depressors). Moderate-risk devices (Class II) require a 501k Premarket Notification, a less rigorous pathway to market. Class III devices are considered to have the highest potential risk and undergo the most stringent requirements before being able to be marketed to the public.2 A high-risk device is that which “supports or sustains human life, is of substantial importance in preventing impairment of human health, or presents a potential, unreasonable risk of illness or injury.”2 Class III devices must submit an application for review by an advisory committee that is responsible for confirming the validity of the evidence and ensuring the device is safe and effective. Examples of Class III devices include breast implants, vascular sutures, and dermal implant devices.
Table 1.
FDA Medical Device Classification
| FDA Device Class | Risk | Examples | Regulation |
|---|---|---|---|
| Class I | Low risk | Tourniquet sterile dressing | General |
| Class II | Moderate risk | Negative pressure wound therapy system | 510(k) Premarket notification |
| Class III | High risk | Integra – bilayer wound matrix | PMA approval |
The application for plastic surgery devices is reviewed by the General and Plastic Surgery Advisory Committee, a panel responsible for assessing device safety and effectiveness.3 Plastic surgery devices approved by the FDA can be identified by filtering the database for devices indexed as approved by the General and Plastic Surgery Advisory Committee.
After the first device is approved, manufacturers may apply to make postmarket changes. Depending on the modification type, manufacturer’s may apply to different premarket approval (PMA) supplement tracks, which include: panel track, 180-day track, real-time, special (immediate) track, 30-day notice, and 135-day track2 (Table 2). Modifications can range from changing the manufacturing process, device indications, and device design. In the majority of tracks, the clinical data supporting the original device are considered sufficient enough to inform decision making regarding approval of the device modification and new clinical data are often not required.
Table 2.
FDA Supplement Review Track Pathways
| FDA Supplement Tracks | Panel Track | Special (Immediate) Track | 180-day Track | Real-time Track | 30-day Notice | 135-day Supplement |
|---|---|---|---|---|---|---|
| Primary indication | Labeling change to expand indication, or major design change | Labeling change to improve safety* | Significant design change** | Minor design change** | Manufacturing change | Manufacturing change |
| Year of track introduction | 1990 | 1986 | 1986 | 1997 | 1997 | 1997 |
| Supporting evidence | Clinical study | Requires no specific new data | Preclinical, confirmatory clinical data in some cases | Preclinical only | No specific requirement | No specific requirement |
| Fee in US dollars (2019) | $241,610 | NA | $48,322 | $22,550 | $5,154 | NA |
| Reviewer | Panel of subject matter experts and FDA staff | FDA staff | FDA staff | FDA staff | FDA staff | FDA staff |
*May include process changes.
**May include labeling changes.
Devices that undergo many supplements (ie, modifications) may experience “design drift.”4 This phenomenon occurs when a modified medical device is found to be substantially different from the original device. These cumulative changes may significantly transform the original device and undermine previous safety and efficacy testing.
Modification trends remain undescribed in the plastic surgery literature. The primary objective of this study was to highlight the number and quality of device changes over the lifespan of a high-risk plastic surgery device. Secondary objectives included describing the types of Class III plastic surgery devices receiving FDA approval, the types of PMA supplement tracks used, and the number of device recalls and withdrawals. Previous studies in orthopedics,4 dermatology,5 otolaryngology,6 and cardiology7 have identified substantial design changes in high-risk devices approved through the PMA pathway.
METHODS
Search Strategy and Eligibility
A retrospective, cross-sectional analysis was conducted on all high-risk plastic surgery devices receiving US marketing approval through the FDA PMA pathway by the General and Plastic Surgery Advisory Committee. The publicly accessible PMA database was searched for all original PMAs, device recalls, and device withdrawal records reported through December 31, 2018.
The devices included in this analysis met the following inclusion criteria: devices manufactured primarily for the use by plastic surgeons; devices that are not used commonly in fields outside of plastic surgery; and medical devices included may overlap with other specialties but should reflect plastic surgery specifically as opposed to other areas of medicine. Two authors (O.R.O. and N.S.) screened each approved original PMA device against the inclusion criteria established a priori for eligibility to be included in the analysis. If consensus could not be reached, disagreements were resolved by a third blinded senior reviewer (M.H.M.) to determine final eligibility.
Extraction of Data
Two authors (O.R.O and D.O) collaborated in the extraction of device data from the PMA database to characterize each original high-risk plastic surgery device and related device supplements. The following data were extracted: initial clearance date, device type, product classification code, implantable status, the number of supplements, type of modification (supplement track), supplement reason, and product withdrawal dates. The product code for each original device was searched in the FDA medical device recall database and the recall information was also extracted. This database was first implemented on November 1, 2002, and contained all recalls issued thereafter.
To best reflect incremental changes in high-risk devices, only labeling, design, and production changes were included in our analysis. FDA applications regarding the change of a manufacturing location or postapproval study design were excluded.
Statistical Analysis
The interobserver agreement for the inclusion of plastic surgery devices was calculated using Cohen kappa coefficient. The kappa coefficient was interpreted according to the Landis and Koch (1977) guidelines and was categorized a priori as k = 0.81–1.00 as almost perfect agreement, k = 0.61–0.8 as substantial agreement, k = 0.41–0.6 as moderate agreement, and k = 0.21–0.40 as fair agreement.8 Pearson correlation and Spearman’s rank were used to measure the strength and direction of association between different variables.
Descriptive statistics such as median and measures of variance (eg, interquartile range [IQR] and SDs, 95% confidence interval [CI]) are presented where applicable. Descriptive statistics were used to characterize the median number of devices approved per year and the median number of modifications per year. The number of PMA-approved Class III plastic surgery devices were summed and reported. The number of devices in each category was classified using product codes. The number of original devices and supplement applications approved per year were also calculated and reported. Linear regression was used to determine the change in the original annual PMA approvals and approved supplements over time.
Stata version 12.0 (StataCorp, LP, College Station, TX, USA) was used to perform all statistical analysis. Two-tailed statistical tests were used and a probability of less than 0.05 was considered statistically significant. Google Sheets (Google, California, USA) was utilized for the development of extraction forms and figures.
RESULTS
The FDA has cleared 39 original high-risk plastic surgery devices via the PMA pathway which has served as the basis for a collection of modified devices (Table 3). The independent duplicate screening demonstrated almost perfect inter-rater agreement with a kappa value of k = 0.86 (95% [CI] 0.61–0.90) for device inclusion. The type of devices included breast implants, dermal implants, lasers, sutures, vascular sealants, wound dressings, and an electrical impedance spectrometer.
Table 3.
Summary of High-risk (Class III) Plastic Surgery Devices
| PMA Number | Manufacturer | Device | Approval Year | Product Codes | Type of Device | No. Supplements | FDA Recall Class (Posting Date) | Withdrawal Date | Device-years |
|---|---|---|---|---|---|---|---|---|---|
| P800022 | Allergan | Zyderm collagen implant(Zyderm CI) | 1981 | LMH | Dermal implant | 35 | 10/25/2011 | 30 | |
| P850053 | Mentor Corp. | Fibrel | 1988 | LMH | Dermal implant | 4 | 02/28/2008 | 20 | |
| P870069 | UDL Laboratories, Inc. | Biobrane(R) II | 1989 | FRO | Wound device | 1 | 01/19/2010 | 21 | |
| P890002 | Alcon Laboratories | Polypropylene Surgical Suture | 1989 | GAW | Suture | 0 | 29 | ||
| P900033 | Integra LifeSciences Corp. | Integra Dermal Regeneration Template | 1996 | MGR | Wound device | 64 | 2(2009) 3(2008) | 22 | |
| P960007 | Shire Regenerative Medicine | Transcyte Human Fibroblast-Derived Temporary Skin Substitute | 1997 | MGR | Wound device | 12 | 21 | ||
| P950032 | Organogenesis, Inc. | Apligraf (Graftskin) | 1998 | MGR | Wound device | 63 | 2(2011) 2(2011) 2(2010) 2(2009) 2(2008) 3(2006) | 20 | |
| P990004 | Ferrosan Medical Devices A/S | Surgifoam Absorbable Gelatin Sponge, USP | 1999 | LMF | Hemostasis adjunct | 0 | 1(2012) 1(2012) 2(2012) | 19 | |
| P990019 | Dusa Pharmaceuticals, Inc. | Blu -U Blue Light Photodynamic Therapy Illuminator | 1999 | MVF | Laser | 5 | 19 | ||
| P990021 | Concordia Laboratories, Inc | Diomed 630 PDT Laser | 2000 | MVF | Laser | 3 | 18 | ||
| P990049 | Lumenis | Coherent Opal Photoactivator Laser System | 2000 | MVF | Laser | 1 | 09/10/2010 | 10 | |
| P990074 | Allergan | Natrelle Saline Breast Implants | 2000 | FWM | Breast implant | 34 | 3(2005) 3(2005) | 18 | |
| P990075 | Mentor Worldwide LLC | Mentor Corporation Saline-Filled And Spectrum (R) Mammary Prostheses | 2000 | FWM | Breast implant | 41 | 2(2017) | 18 | |
| P000036 | Shire Regenerative Medicine | Dermagraft | 2001 | MGR | Wound device | 13 | 2(2003) | 17 | |
| P010016 | Forticell Bioscience | Orcel Bilayered Cellular Matrix | 2001 | MGR | Wound device | 2 | 17 | ||
| P020023 | Q-Med AB | Restylane Injectable Gel | 2003 | LMH | Dermal implant | 12 | 15 | ||
| P060028 | Mentor Worldwide LLC | Mentor Memoryshape Breast Implants | 2003 | FTR | Breast implant | 24 | 15 | ||
| P010061 | Photo Cure Asa | Curelight Broadband (Model Curelight 01) | 2004 | MYH | Laser | 0 | 08/28/2008 | 4 | |
| P030032 | Genzyme Biosurgery | Hylaform (Hylan B Gel) | 2004 | LMH | Dermal implant | 12 | 02/10/2016 | 12 | |
| P030050 | Q-Med AB | Sculptra And Sculptra Aesthetic | 2004 | LMH | Dermal implant | 19 | 14 | ||
| P040024 | Q-Med AB | Restylane Injectable Gel | 2005 | LMH | Dermal implant | 92 | 13 | ||
| P020012 | Suneva Medical, Inc. | Artefill, Bellafill PMMA Collagen Permanent Dermal Filler | 2006 | LMH | Dermal implant | 15 | 12 | ||
| P020056 | Allergan | Natrelle Silicone-Filled Breast Implants | 2006 | FTR | Breast implant | 33 | 12 | ||
| P030053 | Mentor Corp. | Memorygel Silicone Gel-Filled Breast Implants | 2006 | FTR | Breast implant | 31 | 2(2016) | 12 | |
| P050033 | Anika Therapeutics, Inc. | Hydrelle | 2006 | LMH | Dermal implant | 17 | 12 | ||
| P050037 | Merz North America, Inc | Radiesse 1.3CC And 0.3CC | 2006 | LMH | Dermal implant | 85 | 2(2011) 3(2011) | 12 | |
| P050047 | Allergan | Juvederm 24HV, Juvederm 30 And Juvederm 30HV Gel Implants | 2006 | LMH | Dermal implant | 61 | 12 | ||
| P050052 | Merz North America, Inc | Radiesse Injectable Implant | 2006 | LMH | Dermal implant | 93 | 2(2016) 2(2015) 2(2011) 3(2011) | 12 | |
| P070013 | Colbar Lifescience Ltd. | Evolence Collagen Filler | 2008 | LMH | Dermal implant | 3 | 12/03/2010 | 2 | |
| P060029 | Ethicon, Inc. | Ethicon Omnex Surgical Sealant | 2010 | NBE | Vascular reconstruction adhesive | 3 | 8 | ||
| P090016 | Merz North America, Inc | Belotero Balance | 2011 | LMH | Dermal implant | 23 | 7 | ||
| P070004 | Sientra, Inc | Sientra Silicone Gel Breast Implants | 2012 | FTR | Breast implant | 8 | 6 | ||
| P120011 | Idealimplant | Ideal Implant Saline-Filled Breast Implant | 2012 | FWM | Breast implant | 12 | 6 | ||
| P040046 | Allergan | Natrelle Highly Cohesive Silicone-Filled Breast Implants | 2013 | FTR | Breast implant | 23 | 5 | ||
| P110033 | Allergan | Juvederm Voluma XC | 2013 | LMH | Dermal implant | 37 | 5 | ||
| P140029 | Q-Med AB | Restylane Refyne, Restylane Defyne | 2016 | LMH | Dermal implant | 12 | 2 | ||
| P150046 | Scibase AB | Nevisense | 2017 | ONV | Electrical impedance spectrometer | 2 | 1 | ||
| P160042 | Prollenium Medical Technologies Inc. | Revanesse Ultra | 2017 | LMH | Dermal implant | 2 | 1 | ||
| P170002 | Teoxane S.A. | RHA 2, RHA 3, RHA 4 | 2017 | LMH | Dermal implant | 0 | 1 |
Pearson’s correlation failed to demonstrate a significant association between original high-risk devices release over time (r = 0.28; P = 0.084) (Fig. 1a). The relationship between PMA supplement usage over time demonstrated an exponential relationship. As such, Spearman’s correlation was used to assess the relationship between supplement usage and time (Fig. 1b). There was a strong positive correlation between supplement approval over time, which was statistically significant (rs = 0.9174, P = 0.000). The FDA cleared 897 total incremental changes for 39 original devices through December 31, 2018 (Table 4). There were 161 modifications which did not represent device or process changes and were excluded from our analysis (eg, location change or postapproval study protocol change).
Fig. 1.
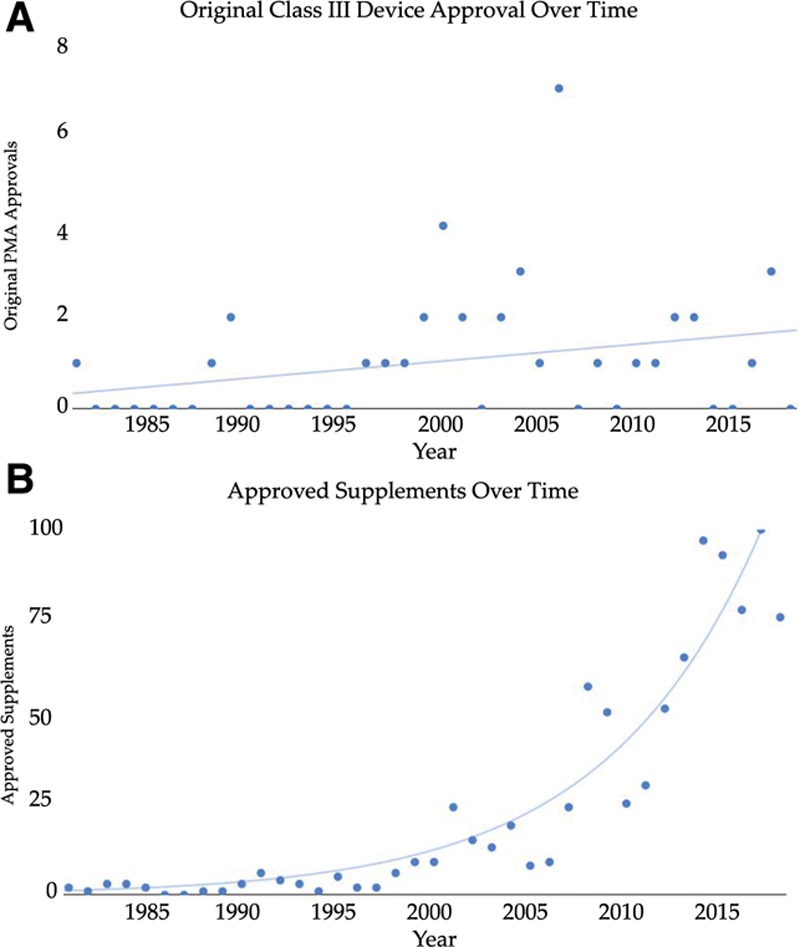
A, Changes in original devices between 1980 and 2018. B, PMA supplements approved between 1980 and 2018.
Table 4.
Characteristics of Included Approved High-risk Plastic Surgery Devices
| Devices (n = 39) | |
|---|---|
| Initial clearance date | n (%) |
| 1980–1989 | 4 (10.0%) |
| 1990–1999 | 5 (13.0%) |
| 2000–2009 | 20 (51.0%) |
| 2010–present | 10 (26.0%) |
| Implantable | |
| Yes | 26 (67.0%) |
| No | 13 (33.0%) |
| Device type | |
| Wound device | 6 (15.0%) |
| Breast implant | 8 (20.5%) |
| Dermal implant | 17 (43.6%) |
| Laser | 4 (10.0%) |
| Hemostasis adjunct | 1 (2.50%) |
| Vascular reconstruction adhesive | 1 (2.5%) |
| Electrical impedance spectrometer | 1 (2.5%) |
| Suture | 1 (2.5%) |
| Total | 39 |
| No. supplements | |
| Breast implant | 206 (23.0%) |
| Dermal implant | 522 (58.2%) |
| Electrical impedance spectrometer | 2 (0.2%) |
| Laser | 9 (1.0%) |
| Suture | 0 (0.0%) |
| Vascular reconstruction adhesive | 3 (0.3%) |
| Wound device | 155 (17.4%) |
| Total | 897 |
Table 5.
Total Approved Supplements by Supplement Track
| Supplement Track | No. Supplements |
|---|---|
| 135 Review track for 30-day notice | 108 |
| 30-day notice | 487 |
| Normal 180-day track | 125 |
| Not reported | 11 |
| Panel track | 17 |
| Real-time process | 102 |
| Special (immediate track) | 47 |
| Total | 897 |
Breast and dermal implants represented 64.1% of the high-risk devices that have come to market via the PMA approval pathway and comprised 81.2% of all modifications to devices which come to market. Radiesse injectable implant underwent the greatest number of changes with 93 supplements since 2006. Of the changes, 57 (62%) were 30-day notice, 6 (6.5%) panel track, 8 (8.7%) special track, 2 (2.2%) real-time track, and 8 (8.7%) normal 180-day track changes. Furthermore, the dermal implant underwent the highest rate of postmarket device changes per device-year, with 7.75 modifications approved per active device-year over a 6-year lifespan. Restylane underwent the second greatest number of changes with 92 modifications since 2005. Dermal medical devices represent 43.6% of the original devices, yet undergo 58% of the device changes, and have a median of 17 changes per device (IQR, 12–38; range, 0–93). Although dermal implants had the greatest number of modifications to an individual device, breast implant devices had the greatest median number of changes. Breast implant devices underwent a median of 27.5 modifications per device (IQR, 20.25–33.25; range, 8–41). Four original devices did not undergo any supplement changes (Table 3).
Recall and Withdrawal
Following initial FDA clearance, 9 (23%) original devices contributed to 22 recalls (Table 3). There was a moderate positive correlation between the number of supplements and the number of device recalls (r = 0.5413, P = 0.0004, R2 = 0.239). Only 2 of the recalls were classified as a Class I recall, which is a circumstance where a product is likely to cause serious adverse health consequences or death.9 Apligraf, skin substitute, required the most device recalls with 6 recalls which occurred over 5 years. There were 7 devices withdrawn from the market by manufacturers. FDA databases do not provide a reason for withdrawal.
Types of Postmarket Modifications and Device Lifespan
The median lifespan of high-risk therapeutic plastic surgery devices was 12 years (IQR, 6.5–18; range, 1–30 years) (Table 3). There was an approved total of 620 process changes, 124 design changes, and 64 labeling changes (Table 6).
Table 6.
Types of Postmarket Modifications Approved Changes to Devices
| Type of Modification | Count |
|---|---|
| Labeling change – indications/instructions/shelf life/trade name | 139 |
| Change design/components/specifications/material | 123 |
| Other report | 3 |
| Process change – manufacturer/sterilizer/packager/supplier | 620 |
| Special report | 1 |
| Before PMA pathway | 11 |
| Total | 897 |
Refer PMA Database PMA Number (P800022-S035, P950032-S002, P950032-S028) for details.
After the 30-day notice and 135-day track review were formally introduced in 1997, the supplement per active device increased from 0.389 supplements per active device (1990–1999) to 0.630 supplements per active device in the next decade (2000–2009) and to 2.44 supplements per active device from 2010 to 2018.
PMA Supplemental Review Tracks
The use of different types of PMA supplement review tracks has changed over time (Fig. 2). The 30-day notice and 135-day review track are the most common type of supplemental review. Approximately 54% (n = 487) of approved supplements were through the 30-day notice track and 12% (n = 108) of the 30-day notice applications result in the FDA requesting for more information via the 135-day review pathway. Real-time review track which requires preclinical data and is intended for minor design changes reviews represented 11% (n = 102) of supplement applications. Special (immediate) track intended for labeling changes meant to enhance device safety represented 5.2% (n = 47) of the supplements. Fourteen percent (n = 125) of supplements were normal 180-day track changes, which are intended for major design changes; 1.9% (n = 17) plastic surgery devices were approved through panel track review, which requires substantial new clinical data and is used to expand use or remove contraindications.
Fig. 2.
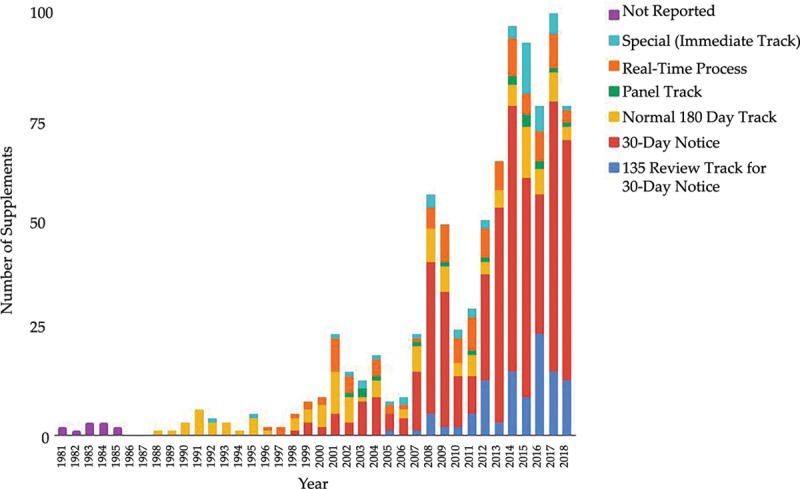
Variations in PMA supplement utilization over time.
Changes by Supplement Review Track
There was congruence between the published supplement reason and the supplement type (Fig. 3). All changes made through the 30-day (n = 487, 100%) and the 135 review track (n = 108, 100%) supplements were related to production changes. Labeling changes is a broad classification by the FDA, which may include changes to indications, instructions, shelf life, and trade name.10
Fig. 3.
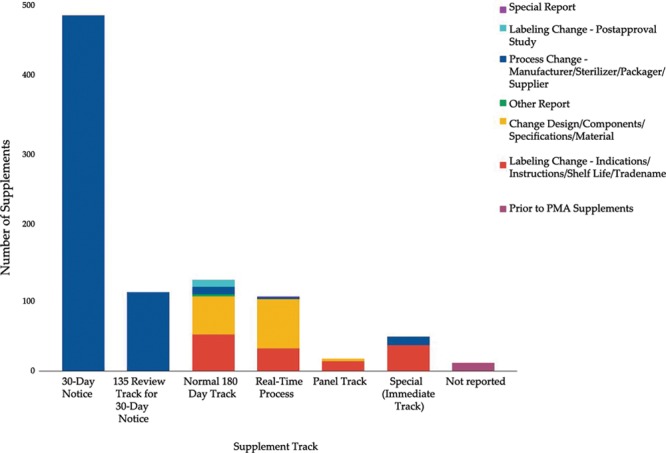
Types of changes in plastic surgery devices by supplement track.
There were 60 (47.8%) labeling modifications which were approved through the 180-day track and 31 (30.4%) labeling modifications through real-time supplement track. Special (immediate) track was used primarily for labeling changes (n = 35, 74.5%) and process changes (n = 12, 25.5%). Panel track review was used to approve 13 labeling changes (76.5%) and 4 design changes (23.5%).
DISCUSSION
This cross-sectional analysis of the FDA PMA database included 39 high-risk therapeutic plastic surgery devices which were approved through the original PMA pathway. These devices were found to undergo a median of 12 changes related to design and labeling over a 12-year median device lifespan. The number of changes a medical device underwent was device specific. Breast and dermal implants composed 81% of all postmarket device modifications but represented 63% of the included plastic surgery devices. Breast implant devices underwent the greatest number of changes with a median of 27.5 changes over a median of 12 active device-years. This may be due to the relatively large commercial market relative to other devices.
The most utilized pathway, 30-day notice, requires no specific additional study or investigation and is primarily used for minor production changes such as switching suppliers for a component.10 Hauser and Maron11 describe a 21-year-old patient who was unknowingly implanted with a defective implantable cardioverter defibrillator and subsequently died. Before the incident, the manufacturer identified the defect and applied for production changes but left the previous iterations on the market. There are other many well-documented cases of medical devices being approved through the PMA pathway, undergoing several approved changes without clinical study and then contributing to preventable harm to patients.11 While such occurrences have not been reported in the plastic surgery literature, it is important to highlight that newer devices not only lack evidence on long-term safety, as this is ascertained in postmarket evaluation, but also undergo several changes with variable amounts of evidence in the premarket stages.
The issues highlighted in this paper remain at the policy level, with the role of surgeons being quite limited. In fact, manufactures are not required to report which supplements apply to a specific model on the packaging and surgeons are often unfamiliar of which specific iteration they are using.12 Nonetheless, the deficiencies of the PMA process highlight the importance of reporting clinical outcomes and adverse events in national registries which enable accurate device assessment in postapproval studies. The high rate of device turnover raises a new challenge of tracking long-term evidence on the safety of devices, especially on rare events.
Previous studies have characterized the types of postmarket device changes in cardiology,7 dermatology,5 otolaryngology,6 and orthopedics.4 High-risk plastic surgery devices undergo lower rates of changes related to design (32.6%) compared to high-risk otolaryngology (52%) and cardiology devices (37%), but more modifications compared to orthopedic devices (22.5%). To the author’s knowledge, this is the first study to characterize postmarket modification trends in high-risk plastic surgery devices.
There were a significant number of labeling changes approved through pathways primarily intended for design (Fig. 3). Labeling modifications may include changes to indications, instructions, shelf life, and trade name.10 Changes to device indications should be made exclusively through the panel review track, and other labeling changes can be made through 180-day or real-time review supplement tracks.10 Though a formal audit of the approved supplements was beyond the scope of this analysis, the authors identified a 180-day supplement used to expand the indications of a device, which should exclusively undergo panel track review.10,13 Another observation was that real-time and 180-day tracks were used to approve identical design changes in the style and sizes of breast implants.14,15 This was observed across different devices, where similar products may undergo identical changes but require different levels of review before being brought onto the market. The difference between the real-time and 180-day tracks are the depth of review and the amount of evidence used to support the device change.10 While the FDA has the ultimate authority on what devices are approved through each pathway, without consistency, there is a risk for devices being approved via less rigorous pathways. Future studies should assess the congruency between the published supplement track and the approval order statement.
In an effort to promote transparency, the FDA has developed a pilot program releasing select PMA summary review memos for select 180-day design changes and total product life cycle reports, which provide information regarding product clearances, approvals, adverse events, and recalls.16 These initiatives are encouraged as they enhance transparency and should be expanded to include memos on other review tracks. The FDA device user experience database should be indexed by type of incident reported, which may prove useful for research and clinical decision-making purposes.
In plastic surgery, breast implant-associated anaplastic large cell lymphoma (BIA-ALCL) and other safety concerns have led to profound developments by the FDA in adverse event reporting and device monitoring. In early 2019, the FDA announced the termination of the alternative summary reporting program, a program intended for internal review but concealed reports regarding unusual, unique, or uncommon adverse events, such as breast implant-associated anaplastic large cell lymphoma and breast implant illness.17 Additionally, the FDA has partnered with 2 registries: Patient Registry and Outcomes for Breast Implants and Anaplastic Large Cell Lymphoma Etiology and Epidemiology and National Breast Implant Registry and is taking further steps to enhance the rigour of postapproval studies.17 This is important as these studies will be fundamental in providing long-term safety data. Evidently, there are rapid advancements being made to protect the public and improve the device approval process.
Study Limitations
There are several limitations of this study. First, we restricted our analysis to devices published in the PMA database and then selected devices for inclusion. Although reviewers were blinded and agreed with high confidence, there is a risk of selection bias regarding which devices were included and excluded.
Utilizing the PMA database limits our analysis to only high-risk devices approved in the PMA pathway. Evidence suggests that high-risk devices are also inappropriately approved through the less stringent 510(k) pathways.9 Another limitation is that device market withdrawal information is largely dictated by the manufacturer and there is no information regarding the rationale posted on the withdrawal database. Additionally, modifications approved before 1986 remain unclassified. Nonetheless, this is only a small proportion of the included plastic surgery device supplements. Consistent with the nature of any cross-sectional analysis, our study only describes a snapshot in time. The observations made in this analysis were quantitative in nature, and the impact of these modifications on safety and function requires further study.
CONCLUSIONS
Medical devices are essential tools used in the field of plastic surgery, and surgeons should be knowledgeable of how these devices come to market. This analysis of the PMA database included 39 original plastic surgery devices which have underwent a total of 897 modifications. The most frequently used supplements do not require additional clinical testing, which may contribute to substantial design drift in select devices.
Footnotes
Published online 19 February 2020.
Disclosure: The authors have no financial interest in any of the products, devices, or drugs mentioned in this article.
REFERENCES
- 1.Medical devices 2030. https://advisory.kpmg.us/articles/2018/medical-devices-2030.html. Accessed March 29, 2019.
- 2.U.S. Department of Health and Human Services. Regulated Products – Medical Device Overview. https://www.fda.gov/forindustry/importprogram/importbasics/regulatedproducts/ucm510630.htm. Accessed April 14, 2019.
- 3.Faris O, Shuren J. An FDA viewpoint on unique considerations for medical-device clinical trials. N Engl J Med. 2017;376:1350–1357. [DOI] [PubMed] [Google Scholar]
- 4.Samuel AM, Rathi VK, Grauer JN, et al. How do orthopaedic devices change after their initial FDA premarket approval? Clin Orthop Relat Res. 2016;474:1053–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ezaldein HH, Scott JF, Yin ES, et al. Transparency and dermatologic device approval by the US Food and Drug Administration. JAMA Dermatol. 2018;154:273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rathi VK, Ross JS, Samuel AM, et al. Postmarket modifications of high-risk therapeutic devices in otolaryngology cleared by the US Food and Drug Administration. Otolaryngol Head Neck Surg. 2015;153:400–408. [DOI] [PubMed] [Google Scholar]
- 7.Rome BN, Kramer DB, Kesselheim AS. FDA approval of cardiac implantable electronic devices via original and supplement premarket approval pathways, 1979-2012. JAMA. 2014;311:385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Landis JR, Koch GG. The measurement of observer agreement for categorical data. Biometrics. 1977;33:159. [PubMed] [Google Scholar]
- 9.U.S Food and Drug Administration. Recalls, Corrections and Removals (Devices). https://www.fda.gov/medical-devices/postmarket-requirements-devices/recalls-corrections-and-removals-devices. Accessed April 28, 2019.
- 10.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Devices and Radiological Health. Guidance for industry and FDA staff modifications to devices subject to preparket approval (PMA) -- The PMA supplement decision-making process. 2007. http://www.fda.gov/cdrh/ode/guidance/1584.pdf. Accessed April 14, 2019.
- 11.Hauser RG, Maron BJ. Lessons from the failure and recall of an implantable cardioverter-defibrillator. Circulation. 2005;112:2040–2042 [DOI] [PubMed] [Google Scholar]
- 12.Zheng SY, Redberg RF. Premarket approval supplement pathway: do we know what we are getting? Ann Intern Med. 2014;160:798–799. [DOI] [PubMed] [Google Scholar]
- 13.U.S Food and Drug Administration. Premarket Approval (PMA). https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P040024S039. Accessed April 28, 2019.
- 14.U.S Food and Drug Administration. Premarket Approval (PMA). https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P040024S039. Accessed May 18, 2019.
- 15.U.S Food and Drug Administration. Premarket Approval (PMA). https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P020056S036. Accessed May 18, 2019.
- 16.U.S Food and Drug Administration. Premarket Approval (PMA) Summary Review Memos for 180-Day Design Changes. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pmamemos.cfm. Accessed April 28, 2019.
- 17.U.S. Food and Drug Administration. Statement from FDA Principal Deputy Commissioner Amy Abernethy, M.D., Ph.D., and Jeff Shuren, M.D., J.D., director of the FDA’s Center for Devices and Radiological Health on FDA’s new efforts to protect women’s health and help to ensure thesafety of breast implants. https://www.fda.gov/news-events/press-announcements/statement-fda-principal-deputy-commissioner-amy-abernethy-md-phd-and-jeff-shuren-md-jd-director-fdas. Accessed May 15, 2019.