

Abstract
The UniProt Knowledgebase (UniProtKB) collects and centralises functional information on proteins across a wide range of species. In addition to the functional information added to all protein entries, for enzymes, which represent 20-40% of most proteomes, UniProtKB provides additional information about EC classification, catalytic activity, cofactors, enzyme regulation, kinetics and pathways, all based on critical assessment of published experimental data. Computer-based analysis and structural data are used to enrich the annotation of the sequence through the identification of active sites and binding sites.
While the annotation of enzymes is well defined, the curation of pseudoenzymes in UniProtKB has highlighted some challenges: how to identify them, how to assess their lack of catalytic activity, how to annotate their lack of catalytic activity in a consistent way and how much can be inferred and propagated from experimental data obtained from other species. Through various examples, we illustrate some of these issues and discuss some of the changes we propose to enhance the annotation and discovery of pseudoenzymes.
Ultimately, improving the curation of pseudoenzymes will provide the scientific community with a comprehensive resource for pseudoenzymes which will lead to a better understanding of the evolution of these molecules, the aetiology of related diseases and the development of drugs.
Keywords: protein database, curation, pseudoenzyme, UniProtKB
Graphical Abstract
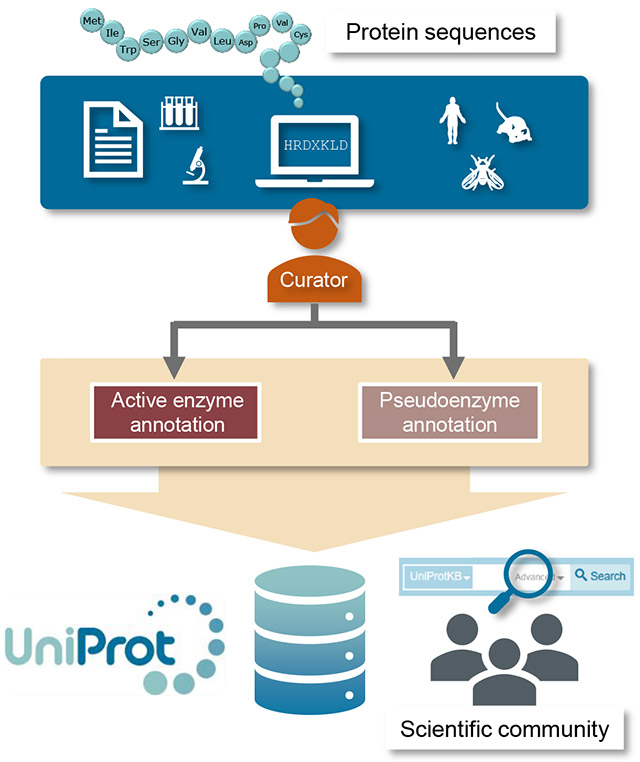
The UniProt Knowledgebase (UniProtKB) collects and centralises protein sequences and functional information across a wide range of species. This review summarizes the improvements made to the annotation of pseudoenzymes to facilitate their discovery and to provide a comprehensive resource for these proteins which will lead to a better understanding of their biological roles and evolution.
Introduction
During enzyme evolution, gene duplication and the accumulation of mutations affecting residues involved in catalysis have given rise to a group of enzyme-related proteins that have lost their capacity to catalyse biochemical reactions [1,2] (Thornton et al, this issue). Despite the loss of their original catalytic function, these proteins, known as pseudoenzymes, are remarkably well conserved. They are found in almost all enzyme families, where they represent between 10-15% of the members, and are distributed across the whole tree of life. Recent years have witnessed a surge in pseudoenzyme research uncovering their biological roles, particularly those belonging to the most abundant enzyme groups, namely kinases [3–5], phosphatases [4] and proteases [6,7]. These studies have revealed that, despite the lack of enzymatic activity, these proteins have evolved essential catalytic-independent functions, explaining why there has been a selective pressure to retain them. These roles, which are described in more detail in [8,9], include: (1) allosteric activation of an active enzyme; for example, myotubularin-related pseudophosphatase MTMR9 (UniProtKB Q96QG7) binds to MTMR6 and increases MTMR6 lipid phosphatase activity [10], (2) control of the localization and/or assembly of macromolecular complexes; for example, pseudophosphatase STYX (serine/threonine/tyrosine-interacting protein; UniProtKB Q8WUJ0) anchors the mitogen-activated protein kinases MAPK1 and MAPK3 in the nucleus [11], (3) assemblage of signalling cascades; for example, KSR1 (Kinase suppressor of Ras 1; UniProtKB Q8IVT5) recruits various components of the MAPK/Erk signalling cascade [12] and (4) competition for substrate binding or complex assembly; for example, C.elegans pseudophosphatase egg-4 (UniProtKB O01767) sequesters and inhibits phosphorylated kinase mbk-2 [13].
It has become apparent from these studies that some pseudoenzymes are also linked to diseases [4,14]. A well-characterized case is Charcot-Marie-Tooth disease, a neurodegenerative disorder caused by mutations affecting pseudophosphatases SBF2/MTMR13 (UniProtKB Q86WG5) and SBF1/MTMR5 (UniProtKB O95248) [15,16]. In part due to their capacity to regulate enzymes, pseudoenzymes have also attracted interest as potential targets for therapeutic treatments [14].
The growing interest in pseudoenzymes led to two successful international meetings in 2016 and 2018 where various topics were discussed, including how bioinformatics tools could advance pseudoenzyme study. Among these tools, protein databases play an instrumental role by providing repositories for protein-related data where functional information and protein sequences are brought together. For example, the Protein Kinase Ontology (ProKinO) resource [17] has established a list of all known and predicted pseudokinases across all kingdoms of life [18]. Similarly, the peptidase database MEROPS includes pseudoproteases where they are defined as non-peptidase homologues [19]. While these resources provide invaluable data, they focus only on one specific enzyme family.
The UniProt Knowledgebase (UniProtKB) provides the scientific community with free access to more than 150 million protein sequences (release 2019_05) annotated with high-quality functional information [20]. Reviewed entries (also known as UniProtKB/Swiss-Prot entries), have been enriched with information extracted from peer-reviewed literature by expert curators. Unreviewed entries (also known as UniProtKB/TrEMBL entries), have functional information added automatically by transferring annotation from well-studied, closely related orthologs.
UniProtKB records are regularly assessed and revised to integrate new advances in the protein biology field. This ensures that we provide users with accurate and up-to-date information. The recent advances made in the pseudoenzyme field prompted us to revisit those records in UniProtKB describing pseudoenzymes and update their content.
In this study, we present an outline of the process and the challenges faced in reviewing pseudoenzymes including how they are identified, how the information related to their loss of activity is captured and presented in a concise manner and finally, how we improve their discoverability. The ongoing improvements to pseudoenzyme annotation will provide the scientific community with a valuable resource to facilitate pseudoenzyme biology and the study of pseudoenzyme and enzyme evolution.
Identification of pseudoenzymes
Demonstrating unequivocally that an enzyme is catalytically inactive is notoriously challenging. In UniProtKB, curators use three main types of evidence: (a) evidence based on sequence analysis and/or structural data, (b) evidence based on experimental assays and (c) evidence based on sequence similarity or orthology. Each of these evidence types provides some information but also has its own caveats and may also conflict with other evidence types. All these evidences are combined and carefully assessed before a decision is made regarding the activity of the protein.
Sequence analysis-based evidence
Computer-based protein sequence analysis is commonly used to predict the lack of catalytic activity. The recent curation of the C.elegans kinome [21] and phosphatome (Zaru et al, manuscript in preparation) showed that, among the reviewed members that have been functionally characterized, >95% of the proteins identified as inactive are classified as pseudokinases or pseudophosphatases based on sequence analysis evidence only (Figure 1A–B). Usually, this method is based on the absence of essential residues that have been shown experimentally to be critical for the enzymatic reaction. For example, the myotubularin-related phosphatase family contains 5 members in C. elegans. They are involved in the dephosphorylation of the D3 position of phosphatidylinositol 3-phosphate and phosphatidylinositol 3,5-bisphosphate [22]. The reaction mechanism involves a highly conserved C-X5-R motif which containing the essential cysteine and arginine residues which stabilize the substrate by forming a thiol-phosphate intermediate [23]. An alignment of their sequences shows that two out its 5 members lacks the essential cysteine residue in the phosphatase domain and thus are predicted to be inactive (Figure 1C).
Fig. 1.

C.elegans kinome (A) and phosphatome (B). Percentage of reviewed kinase/phosphatase entries that are active or inactive. The distribution of pseudokinases/pseudophosphatases according to the type of evidence supporting the lack of catalytic activity is shown. Figures reproduced from [41]. (C) Clustal Omega alignment [46] of the catalytic site sequence of C.elegans myotubularins. The consensus sequence for the catalytic motif (C-X5-R; C:Cysteine, X: any amino acid, R:arginine) is highlighted and the cysteine intermediate is highlighted in orange in the active myotubularin-related (MTMR) proteins.
While this method is valuable in predicting the lack of catalytic activity, it has some limitations and can sometimes be misleading. Firstly, sequence analysis software relies on a good understanding of residues and/or motifs implicated in the reaction. Thus, for enzymes for which the residues involved in catalysis have not yet been identified, the capacity of sequence analysis methods to predict the lack of catalytic activity will be low.
Secondly, the catalytic mechanism may have evolved to result in the use of alternative residues. One of the best characterized examples of this is the serine/threonine-protein kinase WNK1 (Protein kinase with no lysine 1; UniProtKB Q9JIH7). Based on sequence analysis, WNK1 is predicted to be inactive as it lacks the catalytic lysine in the kinase subdomain II that is crucial for binding to ATP. However, in kinase assays, WNK1 was proven to be catalytically active due to an alternative lysine at position 233 in the kinase subdomain I becoming involved in ATP binding [24].
Thirdly, sequence analysis may mistakenly predict the lack of catalytic activity or predict the wrong enzymatic activity. This has been elegantly shown for two bacterial enzymes SelO [25] and SidJ [26]. Based on sequence analysis, these two proteins contain a domain that resembles the protein kinase domain. Only by combining experimental data with 3D structure analysis was their actual catalytic activity determined; SelO turns out to use ATP to AMPylate proteins and SidJ acts as a protein polyglutamase. These last examples illustrate the importance of combining sequence information with experimental data to assess the enzymatic activity of a protein.
Experimental evidence
The most convincing method for confirming the predicted lack of enzymatic activity is to test the protein in a biological assay, usually comparing the predicted pseudoenzyme with a closely related active enzyme. Often, site-directed mutagenesis of the missing catalytic site is used to restore activity. For example, mutating the glycine residue at position 120 in the C-X5-R motif of human pseudophosphatase STYX to the catalytic cysteine restores its phosphatase activity [11]. This method, although not without its own caveats, confirms that the lack of catalytic activity predicted by sequence analysis is due the replacement of the catalytic site residue. While having convincing experimental confirmation of the lack of catalytic activity is highly desirable, some caution is nonetheless required for the interpretation of the results as the results of enzymatic assay can be misleading. The lack of detectable activity can be the result of inappropriate experimental conditions. pH and temperature can affect the activity of an enzyme as demonstrated by lysosomal proteases which require an acidic environment for their activity whereas thermophilic DNA polymerases are active only at high temperature. Often, the physiological substrate is unknown or despite being closely related, two enzymes can have very different targets. For example, Nat8f2 (N-acetyltransferase family 8 member 2; UniProtKB Q8CHQ9) is predicted to be an acetyltransferase but, so far, no histone acetyltransferase activity has been detected, although histone proteins are well characterized substrates of other Camello family members [27].
Enzymes are rarely constitutively active and often require either post-translational modifications such as phosphorylation and/or binding to other protein partner(s) or small molecules. The possible contamination of the assay with an active enzyme can also result in the wrong attribution of activity, a common problem when the source of the pseudoenzyme is obtained via immunoprecipitation. This is an important issue to consider as pseudoenzymes often associate with and regulate the activity of their active counterparts. For example, the pseudophosphatase SBF2/MTMR13 binds to MTMR2 to promote MTMR2 phosphatidylinositol phosphatase activity [28]. Sometimes the activity detected is very low. In this circumstance, the pseudoenzyme designation is made on a case-by-case basis, considering all the available evidence. For example, in the kinase domain of KSR2 (UniProtKB Q6VAB6), the lysine residue in the VAIK motif is replaced by an arginine, suggesting that the protein is inactive but low protein kinase activity has been detected in vitro [29]. The interaction with BRAF is proposed to induce a conformation change that increases the low intrinsic kinase activity. In this specific case, KSR2 has been recorded as active in UniProtKB with a comment added to explain that KSR2 kinase activity is currently unsure.
Orthology-based evidence
UniProt makes use of orthology to allow the propagation of functional information between similar proteins in different species and to provide consistent information across orthologs. To identify putative orthologs, curators combine results from reciprocal Blast searches with data from other resources including scientific literature, sequence analysis tools, phylogenetic and comparative genomics databases, and other specialised databases such as species-specific collections.
In some cases, orthology and sequence analysis prediction give rise to apparent contradictory results. This is particularly true when, for example, orthologs use alternative residues instead of the canonical catalytic sites [30]. The proteolytic activity of serine proteases is based on a Ser/His/Asp triad where the serine residue acts as a nucleophile (Figure 2A). Among the 32 mammalian PRSS50/TSP50 (Testis-specific protease-like protein 50) protein entries in UniProtKB, 25% have a threonine instead of a serine residue, suggesting that they may be devoid of proteolytic activity (Figure 2B). However, it has been shown that the threonine can replace the serine residue in the reaction mechanism [31]. This case illustrates the ability of reaction mechanisms to evolve by exploiting closely related residue substitutions and the importance of experimental evidence to support sequence analysis.
Fig. 2.

Serine pseudopeptidases. (A) Reaction mechanism and canonical catalytic triad residues (Asp/His/Ser) for the peptidase S1 family members. (B) Sequence logos of catalytic triad residues for 32 mammalian PRSS50 proteins. The red arrows indicate the position of the three catalytic residues. In rat Prss50 (UniProtKB D4A1L9), a serine residue acts as nucleophile whereas in human PRSS50 (UniProtKB Q9UI38) this role is performed by a threonine residue. (C) Analysis of UniProtKB reviewed members of the peptidase S1 family. The percentage of active and inactive proteases is shown. The number of active and inactive proteases is indicated in brackets. (D) Characterisation of the reviewed S1 pseudoproteases. Taxonomy distribution (left), evidence for lack of catalytic activity (middle) and function (right) are shown.
Evolution can result in residue changes that lead to the loss of catalytic activity, a situation that becomes apparent when comparing distant homologs. For example, C.elegans ddr-1 (UniProtKB Q18163) and ddr-2 (UniProtKB Q95ZV7) are predicted homologs of human DDR1 (Discoidin domain receptor 1; UniProtKB Q08345) and DDR2 (UniProtKB Q16832). In human DDR1 and DDR2 and C. elegans ddr-2, the catalytic site is conserved whereas in C.elegans ddr-1, the aspartic acid residue has been replaced by a histidine suggesting that ddr-1 is inactive. These two examples illustrate the importance of combining various evidence when deciding if a protein has catalytic activity or not.
Specific annotation for pseudoenzymes
Once a protein sequence has been identified as a potential pseudoenzyme and all the available evidence has been assessed, the next step in the curation process is to translate this information into meaningful annotation. This annotation also needs to reflect the type of evidence used and enable pseudoenzyme discovery using the UniProtKB search engine. UniProtKB provides a wealth of protein-related information including function, subcellular location, expression, and interacting partners as well as key residues within the protein sequence such as those which are post-translationally modified. This concise summary uses a combination of controlled vocabularies and free text which facilitates the retrieval and discoverability of proteins matching specific criteria.
For active enzymes, which represent 45% of the reviewed entries, we add enzyme-specific information including the catalytic activity, the regulation mechanism, whether a cofactor is required and the positions of active site(s), cofactor and substrate binding sites (Figure 3A). While the annotation of enzymes is well-established [21], the current annotation of pseudoenzymes needed to be revised to integrate new advances in the field. The revision process involved addressing various challenges to make sure that the new annotation workflow was appropriate. For this task, we considered two perspectives, the user point-of-view and the curator point-of-view. To provide the best information to our users, the challenges were: (1) where and how to display the information about the lack of catalytic activity, (2) how to provide the user with an evidence-supported reason such as lack of sites important for catalysis or cofactor binding; in other words, how to convey that despite sharing a similar catalytic domain with active enzymes, the domain of the pseudoenzyme is not functional, (3) how to ensure consistency in the annotation of pseudoenzymes, (4) how to highlight the fact that, despite their lack of catalytic activity, they share sequence homology with their active counterparts and (5) how to ensure that the annotation is sufficiently unique to facilitate their discoverability? Curators needed to understand: (1) which criteria to use to identify bona fide pseudoenzymes, (2) how to evaluate the evidence available, (3) how to deal with conflicting results and (4) how to efficiently apply the revised annotation to “existing” reviewed pseudoenzyme entries. Ultimately, the re-evaluation of the existing pseudoenzyme annotation resulted in various improvements which are described in more detail below and highlighted in Figure 3B.
Fig. 3.

UniProtKB annotation for enzymes (A) and pseudoenzyme (B) entries. (A) For active enzymes (example: UniProtKB O43293), UniProt provides information about catalytic activity, cofactor, kinetics, the type and position of sites important for the catalysis, and enzyme-related keywords in dedicated fields. (B) For pseudoenzymes (example: UniProtKB Q9C0I1), the lack of catalytic activity is explained in a “Caution” comment and reflected in the protein name using “inactive”.
Protein name
A protein name is often what provides researchers with a first hint about a protein function. Usually, when authors name a protein, they devise a meaningful name that offers a first indication of the protein function. During the curation of a UniProtKB entry, an official recommended protein name based on the name(s) provided by the literature and/or nomenclature committees is added. When the protein is known by more than one name, these names are included as synonyms. By providing users with a comprehensive list of protein names, the mining of the scientific literature is thus facilitated.
Whilst the names given to active enzymes often reflect their catalytic activity, naming their inactive counterparts has proven to be more challenging and various approaches have been used. Some names reflect the non-catalytic function of the pseudoenzymes (for example, PPAF2 name is phenoloxidase-activating factor 2, UniProtKB Q9GRW0), while other use names that highlight their lack of enzymatic activity by including words such as “inactive”, ”-like” or “homologue” (for example, DPP10 name is inactive dipeptidyl peptidase 10, UniProtKB Q8N608). To standardise pseudoenzyme names and avoid ambiguities that ”-like” or “homologue” could cause, curators follow the International Protein Nomenclature Guidelines (https://www.uniprot.org/docs/International_Protein_Nomenclature_Guidelines.pdf) and now include the word “inactive” followed by the missing enzymatic activity in the official name or in a synonym (Figure 3B).
Caution
The basis for the lack of catalytic activity is reported in the “Function” section in a caution comment highlighted in yellow in the entry view on the UniProt website (Figure 3B). The comment describes the nature of the conserved active site residues which are changed, any experimental evidence of inactivity, if available, and conflicting results. Importantly, the evidence used to infer this information is provided (Figure 4 and below).
Fig. 4.

The “caution” comment provides evidence for the lack of catalytic activity. Examples of “caution” comments for the pseudokinase PEAK1 (UniProtKB Q9H792), the pseudophosphatase egg-4 (UniProtKB O01767) and the pseudoisomerase FKBP6 (UniProtKB O75344). The yellow labels provide access to the evidence supporting the annotation. The arrow head allows expansion of the label to see the evidence in more detail (insert).
Sequence features
UniProtKB indicates important residues and regions within the protein sequence such as catalytic sites, functional domains and post-translational modifications obtained from computer-based sequence analysis in combination with experimental evidence. For both enzymes and pseudoenzymes, the position of the catalytic domain is usually provided based on sequence analysis tools such as InterPro (www.ebi.ac.uk/interpro/). For active enzymes, the position of the active site(s) is annotated while, for pseudoenzymes, they are omitted even when the residue is conserved as illustrated by C.elegans pseudokinase kin-32 (UniProtKB Q95YD4) which has been experimentally proven to be inactive [32]. However, when residues involved in cofactor binding or substrate binding are conserved, these are indicated, especially when they are supported by experimental evidence such as a 3D structure, as they can be important to stabilize the structure or to enable to protein to perform its non-catalytic function. For example, for pseudokinases, ATP binding to the inactive kinase domain is essential in maintaining their correct folding or in promoting their binding to other proteins [33]. Similarly, the annotation of substrate-binding residues is important as one of the functions of pseudoenzymes is to sequester substrates as illustrated by C.elegans pseudophosphatase egg-4 mentioned previously.
Protein family
Although pseudoenzymes lack catalytic activity, they retain sequence similarities with active enzymes of the same protein family. For example, both active MTMR6 (UniProtKB Q9Y217) and inactive MTMR9 (UniProtKB Q96QG7) belong to the “protein-tyrosine phosphatase family, non-receptor class myotubularin subfamily”. To enable the identification of proteins with similar sequences, UniProtKB provides this information in the ‘Sequence similarities’ subsection of the ‘Family and domains’ section (Figure 3A–B). Proteins are assigned to families using a range of sources including protein family databases, sequence analysis tools, scientific literature and sequence similarity search tools.
Inactive isoforms
In some rare cases, alternative RNA splicing during expression of enzyme-coding genes can result in the production of inactive isoforms. For example, HDAC9 (Histone deacetylase 9; UniProtKB Q9UKV0) produces 11 isoforms. Isoform 1 displays histone deacetylase activity whereas isoform 3 is inactive due to the loss of the domain containing the catalytic site residue [34]. The lack of enzymatic activity is indicated in a note attached to the isoform sequence.
Protein with catalytic and non-catalytic domains
Interestingly, some proteins that contain multiple catalytic domains have one that is inactive. These domains appear to have conformational roles either in stabilizing the protein or by providing a mechanism to regulate the activity of the other domains. Such proteins are found in many enzyme families. For example, the receptor guanylate cyclases, members of both the guanylate cyclase and protein kinase families, contain one active guanylate cyclase domain and one inactive kinase domain [35]. These entries are annotated as active enzymes; however, curators also report the lack of catalytic activity of one of the catalytic domains in the caution comment together with an alternative name describing the lost enzymatic function (Figure 5).
Fig. 5.

Example of the annotation of a protein with two catalytic domains, one active and one inactive. PPIL2 (UniProtKB Q13356) has an active ubiquitin ligase domain while its peptidyl-prolyl cis-trans isomerase domain is not functional. For the ubiquitin ligase domain (U-box), whose positions are indicated in the “Family & Domains” section, the name, the EC number and catalytic activity provide information related to its active status. For the peptidyl-prolyl cis-trans isomerase domain (PPIase cyclophilin-type), whose positions are indicated in the “Family & Domains” section, its inactive status is reflected in the name (“Inactive..”) and in the caution comment where the evidence for the lack of activity is provided.
Conflicting results
To provide our users with an accurate identification of pseudoenzymes, curators ensure that the annotation reflects as much as possible the evidence available. This is particularly crucial when the various pieces of evidence described previously appear to contradict each other. The most common case is when a protein is predicted to be inactive based on sequence analysis but shows activity when tested experimentally, or vice versa. For example, C.elegans kinase drl-1 (UniProtKB Q86ME2) is predicted to be inactive as the catalytic site is not conserved. However, in an in vitro assay, some kinase activity has been detected [36]. After carefully assessing the evidence, drl-1 was annotated as inactive with a caution comment highlighting the discrepancy: “Although the residues involved in the catalytic activity are absent, suggesting that the kinase is inactive, some kinase activity has been detected.”
Similarly, the mannosidase activity of EDEM1 (ER degradation-enhancing alpha-mannosidase-like protein 1; UniProtKB Q92611) and EDEM2 (UniProtKB Q9BV94), which belong to the glycosyl hydrolase 47 family, is controversial [37]. In this case, they have been annotated as inactive, while mentioning that some mannosidase activity has been detected, until further evidence becomes available.
GO annotation
As part of the manual curation process, UniProtKB entries are enriched with Gene Ontology (GO) terms which describe gene products in terms of their associated biological processes, molecular functions and cellular components in a species-independent manner [38,39]. UniProtKB curators assign GO terms to all reviewed entries based on experimental data from the curated literature. The “molecular function” ontology contains GO terms for most of the Enzyme Commission (EC) numbers. There is no GO term as such to describe the lack of catalytic activity. Instead, the NOT qualifier is used in combination with the GO term corresponding to the expected specific enzymatic activity. For example, the GO annotation for inactive MTMR5 is NOT + GO term phosphatase activity (GO:0016791). Ideally, the annotation is supported by experimental manual evidence but, for most pseudoenzymes, an evidence code based on sequence analysis only (IKR, inferred from key residues) is used.
Data evidence
As demonstrated above, the evidence source is crucial to assess the strength of the information used to support the lack of catalytic activity. For each piece of information that we annotate, UniProtKB provides a direct link to its original source so that users can easily identify its origin and evaluate it. UniProtKB makes use of a subset of evidence codes from the Evidence and Conclusion Ontology (ECO) to indicate data origin [40]. These ECO codes are shown directly in the text version of the entries while, on the UniProtKB website, they are transformed into user-friendly, easy to understand labels (Figure 4) [21]. For instance, for information inferred from experimental data, we provide a link to the original paper. For information which has been transferred from a related experimentally characterized protein, the accession number of the characterized protein is indicated, providing a link to the entry with experimental evidence. Similarly, information based on computer-based sequence is indicated as such. An analysis of the serine endopeptidase (S1 protease) family showed that, out of the 874 reviewed UniProt entries, 74 are annotated as inactive (Figure 2C–D). Strikingly, only one of them has experimental evidence for the lack of catalytic activity whereas, for the active S1 proteases, more than 30% have experimental evidence to support their catalytic activity. This is in agreement with what we found for the C.elegans pseudokinome and pseudophosphatome described earlier where the predominant evidence for the loss of enzymatic activity comes from sequence analysis prediction.
Prediction and automatic annotation
The advances in sequencing techniques in the last decades have led to an explosion in the number of sequenced genomes. In 2018 alone, 29316 new proteomes were imported into UniProt and the flow of new sequenced genomes is not slowing down. In the first half of 2019, 28180 proteomes have already been integrated. These newly imported sequences are presented as unreviewed entries ( > 150 Mio entries in 2019_05 release) and, because of both the time required for manual curation and the lack of available experimental characterization, most of them will remain unreviewed. Yet, UniProt does provide functional information for these entries using rule-based systems to automatically annotate and classify them. Together with predictions from a suite of sequence analysis methods, they enrich the records with information describing protein names, function, catalytic activity, pathway and family memberships, and subcellular location, along with sequence-specific information. These rules are kept up-to-date and all predictions are refreshed with each UniProtKB release to ensure the latest state-of-knowledge is applied.
The Unified Rule system, or UniRule, contains rules designed and tested by curators using experimental data from manually reviewed entries [20]. These rules use the presence of specific protein signatures together with taxonomy to predict the biochemical features and biological role of a protein (Figure 6).
Fig. 6.

UniProtKB curation process for enzymes. Potential enzyme entries are selected, an expert curator assesses the evidence available (identification/curation step). Based on the evidence, an annotation specific for enzyme or pseudoenzyme is made (annotation step). The entries are integrated into the reviewed/Swiss-Prot section of the UniProtKB database. Template entries corresponding to well characterized enzymes are selected to create rules for automatic annotation which are then applied to the unreviewed entries. These entries can be retrieved using the UniProtKB search engine.
Out of the 7222 UniRules implemented in the UniProt automatic annotation pipeline, 2580 rules (36%) (release 2019_05) are specific for annotating enzymes. These rules provide annotation for the name, EC number, catalytic activity, active sites, cofactor, enzyme-related keywords and GO terms for more than 20 million unreviewed entries (13% of the total) covering the four superkingdoms (bacteria, eukaryotes, viruses, archaea). While enzyme and pseudoenzyme identification is linked, there is no rule yet for the annotation of pseudoenzymes as such. At present, if an entry does not meet the criteria for an active enzyme, i.e. the presence of critical residues such as active site(s) in a specific family, no annotation is made, and the following caution comment is usually added: “Lacks conserved residue(s) required for the propagation of feature annotation.”
Could rules be designed to automatically annotate pseudoenzymes? Or could the existing enzyme prediction rules be improved by including additional and/or more stringent criteria? Answering these questions is not an easy task as there are many challenges affecting the design of these rules that need to be considered, including (1) reliable criteria for prediction, (2) well characterized templates and (3) conservation of these criteria across species. For enzymes where the reaction mechanism is well known, such as protein kinases, it could be possible to update the existing rules adding new conditions which would enable the identification and labelling of potential pseudokinases. While addressing these challenges described above is still an ongoing project, in the end, these rules will provide an invaluable tool to expand the prediction and identification of potential pseudoenzymes in UniProtKB.
Searching for pseudoenzymes in UniProt
One important goal behind the revision of the pseudoenzyme annotation was to improve their discoverability. UniProtKB can be queried using the search box on the top of the website page either by typing terms directly into the box or by using the advanced search options. The advanced search allows our users to restrict search terms to specific fields in a UniProtKB entry and, if required, to combine multiple fields using Boolean logic. For example, using the search term “inactive” in the “Protein name” field allows the retrieval of pseudoenzymes. This search retrieved 455 reviewed entries (release 2019_05). Although this number is far from reflecting the total number of existing inactive enzymes, the analysis of these members offers a preliminary insight in terms of what type of information can be retrieved. As shown by previous studies, the majority of pseudoenzymes identified so far are from eukaryotic species but a substantial number are also found in bacteria and viruses. They belong to over 100 enzyme families confirming that inactive members are present in almost all families. 21 of them have an inactive domain combined with an active domain. So far, 269 have a caution comment providing an explanation for the lack of catalytic activity.
Discussion
In the era of high throughput experiments, databases play an instrumental role in the analysis of large datasets. They not only provide a tool for identification but are also often the initial source of functional information. In the protein field, UniProt is a unique resource that currently gives access to more than 150 million sequences belonging to over 800,000 species combined with functional information based on expert curation and automatic predicted annotation. To ensure that users are provided with the latest knowledge, the annotation is constantly revised and updated. This is made possible by keeping up to date with the latest advances in specific protein fields through the literature, conferences, workshops and, most importantly, through discussion with scientific experts. Curators play an active role in community workshops and are involved in activities such as writing nomenclature guidelines or classification systems which can then be adopted by the UniProt database [41,42]. One such collaboration has been particularly fruitful, leading to improvements in the curation of pseudoenzymes, their description and in enhancing their discoverability [41]. Researchers with specialist knowledge are actively encouraged to contribute to the manual curation process by highlighting key publications and critical information which should be included in specific entries. To this end, we provide mechanisms by which users can feedback on UniProtKB entries, for example enabling researchers to submit additional bibliography to UniProt entries, with ORCIDs used to both validate and credit contributions (http://insideuniprot.blogspot.com/2019/07/) and by providing direct feedback links from every protein record. To expand the information contained in a UniProt entry, we also integrate data from other specialized databases, including several enzyme resources. Thus, in each UniProt entries, users can find direct links to relevant external resources - UniProt release 2019_07 provides cross-references to 170 specialized external resources - which they can use to find further information on their protein of interest.
The criteria used to identify pseudoenzymes are intricately linked to how catalytic activity is assessed in their active counterparts. The capacity to predict accurately that a protein is devoid of catalytic activity correlates with how well the reaction mechanism, in terms of the residues involved, is known in the active members of the related family. Among the various methods used to identify pseudoenzymes, the most commonly used are, by far, based on sequence analysis prediction (up to 95%). This highlights a need to improve and extend our understanding of the molecular mechanism of active enzymes and manually curated repositories such the Mechanism and Catalytic Site Atlas (M-CSA) reaction database are instrumental in this [43]. Comparison of structural data between enzyme and pseudoenzyme has been instrumental in understanding the evolution of the catalytic domain, the reaction mechanism, in particular, which are the key residues and/or motifs and how pseudoenzymes achieved their catalytic independent functions. Importantly, the increasing number of experimentally solved 3D structures (>150000 in the Protein structure database PDB) together with structural protein domain evolution databases such as CATH/Gene3D [44,45] and the advances in 3D prediction model software will facilitate their study. A better understanding of the enzymatic reaction mechanism at the molecular level will contribute to the development of accurate prediction tools or rules to identify and automatically annotate putative pseudoenzymes.
While this review focuses mainly on how pseudoenzymes are identified and how the lack of catalytic activity is reported in UniProt, we obviously also annotate their catalytic independent roles which are described in the “Function” section of an entry. The effort invested in reporting a “non”-function may appear trivial. However, the molecular reasons behind the loss of catalytic activity often provide crucial clues to understand the actual functions of a pseudoenzyme.
UniProt provides researchers with a unique resource for the study of pseudoenzymes, providing a snapshot of the magnitude of the biological processes they are involved in and helping to explain why their catalytic domain is no longer functional. Importantly, these data will lead to a better understanding of the evolution of pseudoenzymes and their active counterparts and the aetiology of related diseases. It will also support the ongoing quest to target pseudoenzymes for therapeutic treatments and offer some insight into the expanding field of enzyme engineering.
Acknowledgements
The authors thank the participants of the Pseudoenzymes 2016 and 2018 conferences for helpful discussions. UniProt has been prepared by Alex Bateman, Michele Magrane, Maria Martin, Sandra Orchard, Emily Bowler, Ramona Britto, Hema Bye-A-Jee, Penelope Garmiri, George Georghiou, Emma Hatton-Ellis, Yvonne Lussi, Alistair MacDougall, Elena Speretta, Nidhi Tyagi, Kate Warner, Rossana Zaru, Shadab Ahmed, Emanuele Alpi, Borisas Bursteinas, Leonardo Gonzales, Alexandr Ignatchenko, Giuseppe Insana, Rizwan Ishtiaq, Vishal Joshi, Dushyanth Jyothi, Jie Luo, Mahdi Mahmoudy, Andrew Nightingale, Joseph Onwubiko, , Sangya Pundir, Guoying Qi, Daniel Rice, Rabie Saidi, Edward Turner, Preethi Vasudev, Vladimir Volynkin, Xavier Watkins and Hermann Zellner at the European Bioinformatics Institute; Alan Bridge, Lionel Breuza, Elisabeth Coudert, Damien Lieberherr, Ivo Pedruzzi, Sylvain Poux, Manuela Pruess, Nicole Redaschi, Lucila Aimo, Ghislaine Argoud-Puy, Andrea Auchincloss, Kristian Axelsen, Emmanuel Boutet, Cristina Casals-Casas, Anne Estreicher, Livia Famiglietti, Marc Feuermann, Arnaud Gos, Nadine Gruaz, Chantal Hulo, Nevila Hyka-Nouspikel, Florence Jungo, Philippe Lemercier, Patrick Masson, Anne Morgat, Sandrine Pilbout, Catherine Rivoire, Christian Sigrist, Shyamala Sundaram, Parit Bansal, Delphine Baratin, Teresa Batista Neto, Jerven Bolleman, Beatrice Cuche, Edouard De Castro, Elisabeth Gasteiger, Sebastien Gehant, Arnaud Kerhornou, Thierry Lombardot and Monica Pozzato at the SIB Swiss Institute of Bioinformatics; Cathy Wu, Cecilia Arighi, Hongzhan Huang, Peter McGarvey, Darren Natale, John S. Garavelli, Kati Laiho, Karen Ross, C. R. Vinayaka, Qinghua Wang, Lai-Su Yeh, Leslie Arminski, Chuming Chen, Yongxing Chen, Yuqi Wang and Jian Zhang at the Protein Information Resource.
This work was supported by the National Eye Institute (NEI), National Human Genome Research Institute (NHGRI), National Heart, Lung, and Blood Institute (NHLBI), National Institute of Allergy and Infectious Diseases (NIAID), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institute of General Medical Sciences (NIGMS), and National Institute of Mental Health (NIMH) of the National Institutes of Health under Award Number [U24HG007822] ; Swiss Federal Government through the State Secretariat for Education, Research and Innovation; European Molecular Biology Laboratory core funds.
Abbreviations
- MTMR
Myotubularin-related protein
- MAPK
Mitogen-activated protein kinase
- KSR
Kinase suppressor of Ras
- ECO
Evidence and Conclusion Ontology
- GO
Gene Ontology
- UniProtKB
Universal Protein Knowledgebase
Footnotes
Conflict of interest
The authors report no conflicts of interest.
References
- 1.Todd AE, Orengo CA & Thornton JM (2002) Sequence and structural differences between enzyme and nonenzyme homologs. Structure 10, 1435–51. [DOI] [PubMed] [Google Scholar]
- 2.Pils B & Schultz J (2004) Inactive enzyme-homologues find new function in regulatory processes. J. Mol. Biol 340, 399–404. [DOI] [PubMed] [Google Scholar]
- 3.Jacobsen A V & Murphy JM (2017) The secret life of kinases: insights into non-catalytic signalling functions from pseudokinases. Biochem. Soc. Trans 45, 665–681. [DOI] [PubMed] [Google Scholar]
- 4.Reiterer V, Eyers PA & Farhan H (2014) Day of the dead: pseudokinases and pseudophosphatases in physiology and disease. Trends Cell Biol. 24, 489–505. [DOI] [PubMed] [Google Scholar]
- 5.Boudeau J, Miranda-Saavedra D, Barton GJ & Alessi DR (2006) Emerging roles of pseudokinases. Trends Cell Biol. 16, 443–52. [DOI] [PubMed] [Google Scholar]
- 6.Reynolds SL & Fischer K (2015) Pseudoproteases: mechanisms and function. Biochem. J 468, 17–24. [DOI] [PubMed] [Google Scholar]
- 7.Lemberg MK & Adrain C (2016) Inactive rhomboid proteins: New mechanisms with implications in health and disease. Semin. Cell Dev. Biol 60, 29–37. [DOI] [PubMed] [Google Scholar]
- 8.Murphy JM, Mace PD & Eyers PA (2017) Live and let die: insights into pseudoenzyme mechanisms from structure. Curr. Opin. Struct. Biol 47, 95–104. [DOI] [PubMed] [Google Scholar]
- 9.Murphy JM, Farhan H & Eyers PA (2017) Bio-Zombie: the rise of pseudoenzymes in biology. Biochem. Soc. Trans 45, 537–544. [DOI] [PubMed] [Google Scholar]
- 10.Zou J, Chang S-C, Marjanovic J & Majerus PW (2009) MTMR9 increases MTMR6 enzyme activity, stability, and role in apoptosis. J. Biol. Chem 284, 2064–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reiterer V, Fey D, Kolch W, Kholodenko BN & Farhan H (2013) Pseudophosphatase STYX modulates cell-fate decisions and cell migration by spatiotemporal regulation of ERK1/2. Proc. Natl. Acad. Sci. U. S. A 110, E2934–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clapéron A & Therrien M (2007) KSR and CNK: two scaffolds regulating RAS-mediated RAF activation. Oncogene 26, 3143–58. [DOI] [PubMed] [Google Scholar]
- 13.Cheng KC-C, Klancer R, Singson A & Seydoux G (2009) Regulation of MBK-2/DYRK by CDK-1 and the pseudophosphatases EGG-4 and EGG-5 during the oocyte-to-embryo transition. Cell 139, 560–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kung JE & Jura N (2019) Prospects for pharmacological targeting of pseudokinases. Nat. Rev. Drug Discov [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Azzedine H, Bolino A, Taïeb T, Birouk N, Di Duca M, Bouhouche A, Benamou S, Mrabet A, Hammadouche T, Chkili T, Gouider R, Ravazzolo R, Brice A, Laporte J & LeGuern E (2003) Mutations in MTMR13, a new pseudophosphatase homologue of MTMR2 and Sbf1, in two families with an autosomal recessive demyelinating form of Charcot-Marie-Tooth disease associated with early-onset glaucoma. Am. J. Hum. Genet 72, 1141–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakhro K, Park J-M, Hong Y Bin, Park JH, Nam SH, Yoon BR, Yoo JH, Koo H, Jung S-C, Kim H-L, Kim JY, Choi K-G, Choi B-O & Chung KW (2013) SET binding factor 1 (SBF1) mutation causes Charcot-Marie-Tooth disease type 4B3. Neurology 81, 165–73. [DOI] [PubMed] [Google Scholar]
- 17.McSkimming DI, Dastgheib S, Talevich E, Narayanan A, Katiyar S, Taylor SS, Kochut K & Kannan N (2015) ProKinO: a unified resource for mining the cancer kinome. Hum. Mutat 36, 175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwon A, Scott S, Taujale R, Yeung W, Kochut KJ, Eyers PA & Kannan N (2019) Tracing the origin and evolution of pseudokinases across the tree of life. Sci. Signal 12, eaav3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rawlings ND, Barrett AJ, Thomas PD, Huang X, Bateman A & Finn RD (2018) The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 46, D624–D632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Consortium UniProt (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47, D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zaru R, Magrane M, O’Donovan C & UniProt Consortium (2017) From the research laboratory to the database: the Caenorhabditis elegans kinome in UniProtKB. Biochem. J 474, 493–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma J, Zeng F, Ho WT, Teng L, Li Q, Fu X & Zhao ZJ (2008) Characterization and functional studies of a FYVE domain-containing phosphatase in C. elegans. J. Cell. Biochem 104, 1843–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsu F & Mao Y (2015) The structure of phosphoinositide phosphatases: Insights into substrate specificity and catalysis. Biochim. Biophys. Acta 1851, 698–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu B, English JM, Wilsbacher JL, Stippec S, Goldsmith EJ & Cobb MH (2000) WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J. Biol. Chem 275, 16795–801. [DOI] [PubMed] [Google Scholar]
- 25.Sreelatha A, Yee SS, Lopez VA, Park BC, Kinch LN, Pilch S, Servage KA, Zhang J, Jiou J, Karasiewicz-Urbańska M, Łobocka M, Grishin N V, Orth K, Kucharczyk R, Pawłowski K, Tomchick DR & Tagliabracci VS (2018) Protein AMPylation by an Evolutionarily Conserved Pseudokinase. Cell 175, 809–821. e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Black MH, Osinski A, Gradowski M, Servage KA, Pawłowski K, Tomchick DR & Tagliabracci VS (2019) Bacterial pseudokinase catalyzes protein polyglutamylation to inhibit the SidE-family ubiquitin ligases. Science 364, 787–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karmodiya K, Anamika K, Muley V, Pradhan SJ, Bhide Y & Galande S (2014) Camello, a novel family of Histone Acetyltransferases that acetylate histone H4 and is essential for zebrafish development. Sci. Rep 4, 6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berger P, Berger I, Schaffitzel C, Tersar K, Volkmer B & Suter U (2006) Multi-level regulation of myotubularin-related protein-2 phosphatase activity by myotubularin-related protein-13/set-binding factor-2. Hum. Mol. Genet 15, 569–79. [DOI] [PubMed] [Google Scholar]
- 29.Brennan DF, Dar AC, Hertz NT, Chao WCH, Burlingame AL, Shokat KM & Barford D (2011) A Raf-induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature 472, 366–9. [DOI] [PubMed] [Google Scholar]
- 30.Ekici OD, Paetzel M & Dalbey RE (2008) Unconventional serine proteases: variations on the catalytic Ser/His/Asp triad configuration. Protein Sci. 17, 2023–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu H, Shan J, Jurukovski V, Yuan L, Li J & Tian K (2007) TSP50 encodes a testis-specific protease and is negatively regulated by p53. Cancer Res. 67, 1239–45. [DOI] [PubMed] [Google Scholar]
- 32.Cram EJ, Fontanez KM & Schwarzbauer JE (2008) Functional characterization of KIN-32, the Caenorhabditis elegans homolog of focal adhesion kinase. Dev. Dyn 237, 837–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeqiraj E, Filippi BM, Goldie S, Navratilova I, Boudeau J, Deak M, Alessi DR & van Aalten DMF (2009) ATP and MO25alpha regulate the conformational state of the STRADalpha pseudokinase and activation of the LKB1 tumour suppressor. PLoS Biol. 7, e1000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou X, Richon VM, Rifkind RA & Marks PA (2000) Identification of a transcriptional repressor related to the noncatalytic domain of histone deacetylases 4 and 5. Proc. Natl. Acad. Sci 97, 1056–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edmund AB, Walseth TF, Levinson NM & Potter LR (2019) The pseudokinase domains of guanylyl cyclase-A and -B allosterically increase the affinity of their catalytic domains for substrate. Sci. Signal 12, eaau5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chamoli M, Singh A, Malik Y & Mukhopadhyay A (2014) A novel kinase regulates dietary restriction-mediated longevity in Caenorhabditis elegans. Aging Cell 13, 641–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ninagawa S, Okada T, Sumitomo Y, Kamiya Y, Kato K, Horimoto S, Ishikawa T, Takeda S, Sakuma T, Yamamoto T & Mori K (2014) EDEM2 initiates mammalian glycoprotein ERAD by catalyzing the first mannose trimming step. J. Cell Biol 206, 347–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huntley RP, Sawford T, Mutowo-Meullenet P, Shypitsyna A, Bonilla C, Martin MJ & O’Donovan C (2015) The GOA database: gene Ontology annotation updates for 2015. Nucleic Acids Res 43, D1057–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.The Gene Ontology Consortium (2019) The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res 47, D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chibucos MC, Mungall CJ, Balakrishnan R, Christie KR, Huntley RP, White O, Blake JA, Lewis SE & Giglio M (2014) Standardized description of scientific evidence using the Evidence Ontology (ECO). Database (Oxford) 2014, bau075–bau075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ribeiro AJM, Das S, Dawson N, Zaru R, Orchard S, Thornton JM, Orengo C, Zeqiraj E, Murphy JM & Eyers PA (2019) Emerging concepts in pseudoenzyme classification, evolution, and signaling. Sci. Signal 12, eaat9797. [DOI] [PubMed] [Google Scholar]
- 42.Persson B, Kallberg Y, Bray JE, Bruford E, Dellaporta SL, Favia AD, Duarte RG, Jörnvall H, Kavanagh KL, Kedishvili N, Kisiela M, Maser E, Mindnich R, Orchard S, Penning TM, Thornton JM, Adamski J & Oppermann U (2009) The SDR (short-chain dehydrogenase/reductase and related enzymes) nomenclature initiative. Chem. Biol. Interact 178, 94–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ribeiro AJM, Holliday GL, Furnham N, Tyzack JD, Ferris K & Thornton JM (2018) Mechanism and Catalytic Site Atlas (M-CSA): a database of enzyme reaction mechanisms and active sites. Nucleic Acids Res. 46, D618–D623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dawson NL, Sillitoe I, Lees JG, Lam SD & Orengo CA (2017) CATH-Gene3D: Generation of the Resource and Its Use in Obtaining Structural and Functional Annotations for Protein Sequences. Methods Mol. Biol 1558, 79–110. [DOI] [PubMed] [Google Scholar]
- 45.Tyzack JD, Furnham N, Sillitoe I, Orengo CM & Thornton JM (2019) Exploring Enzyme Evolution from Changes in Sequence, Structure, and Function. Methods Mol. Biol 1851, 263–275. [DOI] [PubMed] [Google Scholar]
- 46.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD & Higgins DG (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]