

Abstract
T cells recognize and respond to self antigens in both cancer and autoimmunity. One strategy to influence this response is to incorporate amino acid substitutions into these T cell-specific epitopes. This strategy is being reconsidered now with the goal of increasing time to regression with checkpoint blockade therapies in cancer and antigen-specific immunotherapies in autoimmunity. We discuss how these amino acid substitutions change the interactions with the MHC class I or II molecule and the responding T cell repertoire. Amino acid substitutions in epitopes that are the most effective in therapies bind more strongly to T cell receptor and/or MHC molecules and cross-react with the same repertoire of T cells as the natural antigen.
Keywords: Peptide mimotopes, Peptide analogues, Peptide variants, Peptide agonists, Heteroclitic peptides, Altered peptide ligands, TCR-peptide-MHC interactions, Molecular mimicry, Tumor antigens, Autoimmunity antigens
1. Introduction
The relative risk of developing cancer increases with autoimmunity, and autoimmunity increases the risk of cancer (reviewed in [1]), although both disease categories involve diverse cell types and mechanisms. One similarity in both conditions, however, is that T cells interact with self antigens. Specifically, the antigen receptor on T cells (TCRs) interacts directly with peptides derived from self proteins presented in the groove of MHC molecules. One case study involving a melanoma patient suggests that T cells and antigens in both diseases could be the same. The patient’s melanoma was treated with expanded autologous tumor infiltrating lymphocytes (TILs) resulting in durable and complete remission of the cancer. This patient also developed autoimmunity resembling Vogt-Koyanagi-Harada disease (VKH) [2]. VKH is an autoimmune disease targeting organs that contain melanocytes, such as eye, ear, skin, and meninges.
Most T cells that effectively react to self antigens and attack self tissues, so called antitumor T cells in cancer or pathogenic T cells (Tpaths) in autoimmunity, are culled in the thymus during T cell development. Regulatory T cells (Tregs) promote tolerance to self antigens and stop the function of the other T cells to avoid autoimmunity. Interventions to activate T cells against self/tumor antigens for immunotherapies of cancer or to impair effector T cells (Tpath) cells against self antigens in autoimmunity therapies require innovative methods. To achieve such goals, here we discuss the use of mimotopes, mimics of epitopes, which substitute amino acids in the antigenic peptide with either natural or artificial amino acids. Mimotopes have also been tested in HIV and other infectious diseases, but are beyond our scope and have recently been reviewed [3].
Cross-reactivity of TCRs
For T cell immunity to be successful in modulating immune responses, TCRs must efficiently bind to more than one peptide-MHC (pMHC) molecule [4]. Each αβ T cell expresses one TCR generated by somatic recombination of gene fragments and random addition/subtraction of nucleotides at the gene fragment junctions (reviewed in [5]). This process results in vast TCR diversity making it possible for TCRs to interact with pMHC, and contributes to antigen binding specificity of the T cell. However, there are not enough T cells in our bodies for one T cell clone to interact with one peptide [eg, assuming 20 amino acids and 10 amino acid long peptides, there are 2010 (~1 × 1013) possible peptides, and there is the possibility of even more because peptides come in different lengths, may receive post-translational modifications, and there are multiple MHC-restricting molecules,). In addition, there is no mechanism for a single cognate antigen to find a single T cell clone during an infection. There are a number of hints that suggest TCRs interact with multiple peptide antigens. First, T cells are exposed to at least two pMHC molecules: one during thymic development and another as they function in the periphery. Another observation that suggests significant polyspecificity of T cells was the identification of diverse peptide ligands using synthetic combinatorial peptide libraries [6–8]. Using these libraries, it was estimated that between two thousand and two million stimulatory peptides contribute to each T cell activation. Another example of cross-reactivity was in a clinical trial for melanoma: patients received affinity-enhanced T cells that were meant to target and kill MAGE-3A expressing tumor cells, but those T cells also reacted with Titin-expressing cardiac cells [9–11]. The two proteins had only 55% sequence overlap. To directly address the extent of cross-reactivity, Wooldridge and colleagues used experiments and modeling to estimate that a single TCR can respond to more than one million 10-mer peptides, making TCRs the most degenerate receptors known [12].
The cross-reactivity of TCRs to multiple pMHCs has inspired researchers to alter antigen-specific T cell function in tumor immunity and autoimmunity using native peptides with substituted amino acids or new post-translational modifications. These substituted peptides have been given many names: peptide mimotopes, heteroclitic peptides, peptide analogues, altered peptide ligands, peptide variants, and peptide agonists. We have chosen “mimotopes”, mimics of epitopes, for this review. Substituting amino acids in peptides is not the only method to change TCR specificity. There are numerous ways that peptides can be modified naturally or synthetically and remain recognized by TCRs. The concept of molecular mimicry also requires TCRs to be cross-reactive: pathogens and self-antigens share sequence or structural similarities (reviewed in [13]). The antigen from a pathogen infection, raises T cells that crossreact with self antigens and may result in autoimmunity. Using this framework, the pathogen is a mimotope for the T cells that cross-react with self. Structural studies [14] and computational molecular dynamics simulations [15] propose that major features of the pMHC structure for the myelin basic protein and peptides from Herpes simplex virus and Pseudomonas aeruginosa bind to a cross-reactive TCR from a multiple sclerosis patient in a similar manner.
Three issues need consideration to develop ideal mimotopes that can robustly modulate disease activity. First, peptides must stimulate efficient TCR signaling. Such peptides need to be efficiently presented by MHC molecules, and pMHCs need to be efficiently recognized by TCRs. Both of these interactions are most likely achieved with peptides that bind to the MHC molecule and the pMHC complex has relatively strong affinity for the TCR. Second, the TCR repertoire that reacts with an original epitope must be similar to the repertoire reacting with the mimotopes so that the mimotopes modulate T cells which make a difference in the disease (Fig 1). Third, in autoimmunity elimination or modification of the entire Tpath repertoire is necessary to stop the autoimmune process, whereas targeting a portion of the tumor-specific or autoimmune Treg repertoire may be sufficient to fight the disease (Fig 2). With these requirements for the development of effective mimotopes in mind, here we discuss past accomplishments and future directions in identification effective mimotopes.
Figure 1. Venn diagrams demonstrate commonalities between antigen-specific T cell repertoires.
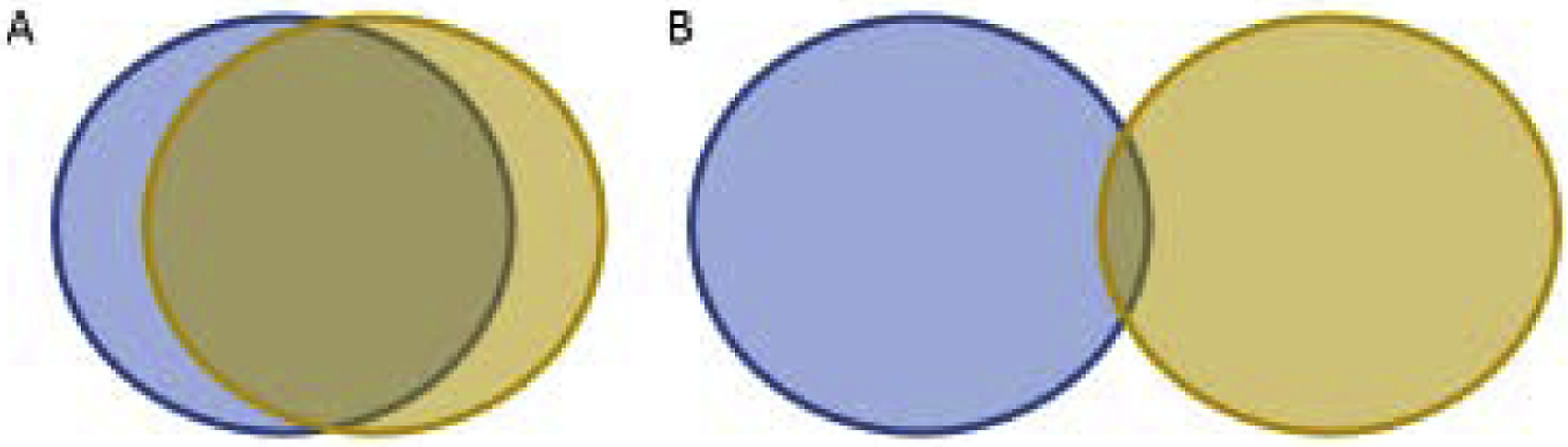
The T cell repertoires that respond to the wild type antigen are in blue (left) and the mimotope antigen is in brown (right). When targeting effector T cells for antitumor immunity or regulatory T cells for autoimmunity, we predict from the literature reviewed here that more overlap between T cell repertoires that respond to the wild type and mimotope antigens result in more robust T cell responses (A). We predict that peptide-MHC and mimotope-MHC with similar structures will stimulate similar T cell repertoires. Substitutions in amino acids that contact the TCR and primary and secondary MHC anchor residues can change the structure recognized by the TCR. If the repertoires are disparate (B), non-specific T cells will be recruited. Methods of focusing the small over-lapping repertoire to the wild type T cells are discussed in Part 5.
Figure 2. Venn diagrams demonstrate how disease-associated T cell repertoires differ in cancer and autoimmunity.
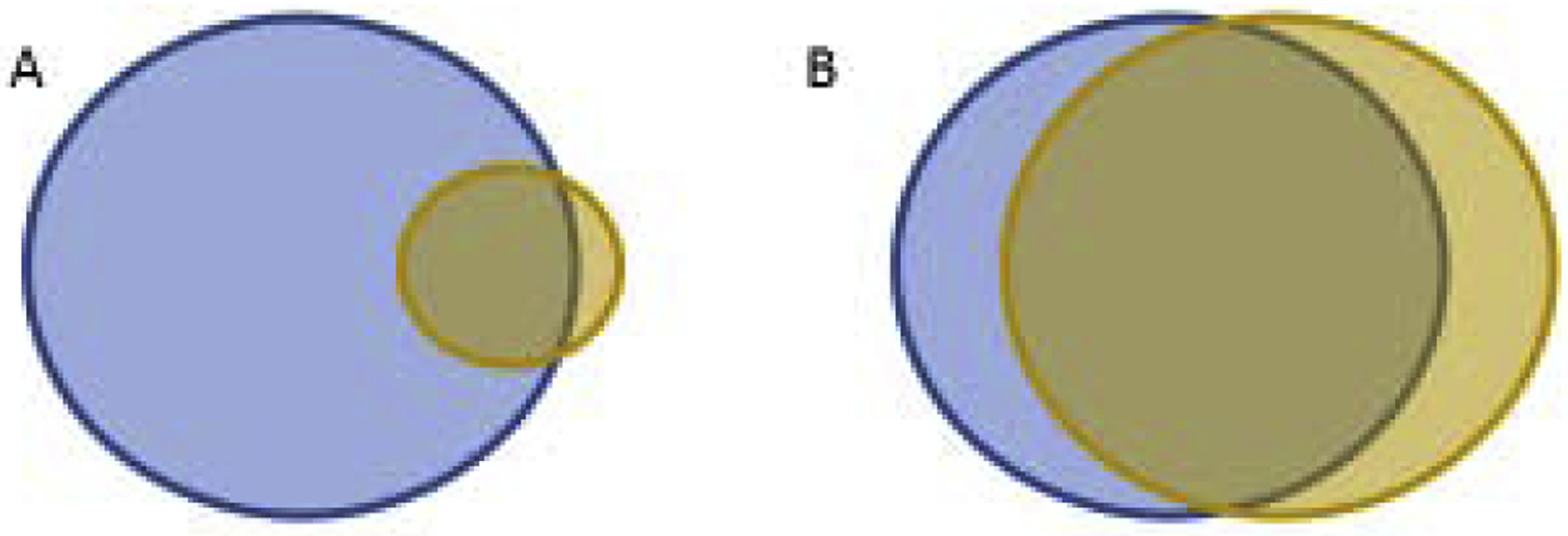
The T cell repertoires that respond to disease-associated antigen(s) are in blue (left) and the mimotope are in brown (right). Assuming that all tumor cells express the mimotope-targeted antigen, activating a fraction of the T cells as noted in A, will recognize all of the tumor cells (overlap). This scenario may also be true in autoimmunity if the mimotope targets Tregs or converts pathogenic T cells to Tregs. Alternatively, in autoimmunity if the mimotope only targets a fraction of the pathogenic T cells, the remaining pathogenic T cells in blue will still be involved in the autoimmune process (A). If most/all of the pathogenic T cells are targeted in autoimmunity as in B, then further autoimmune processes will be halted. Unless the antigen repertoire of the autoimmune disease is narrow, the possibility of discovering such a mimotope for autoimmunity is unlikely, especially in the progressive stages of disease.
2. Substitutions in the MHC anchor amino acids
MHC class I (MHCI) and MHC class II (MHCII) molecules bound to peptide are a classic example of how form reflects function. The peptide binding grooves of the MHC molecules face away from the antigen presenting cell, so that the peptide can interact with the TCR (reviewed in [16, 17] and by many others). The groove is bordered by two alpha helices and the floor of the groove is made of a beta-pleated sheet. The peptide lies in the groove in an extended state allowing the amino acid side chains to point in optimal directions for interactions with TCR. In MHCI molecules, depending on the allele and the polymorphisms, the floor of the groove has a series of 6 pockets, A through F, that interact with the peptide [18, 19]. The amino terminus of the peptide binds directly to the A pocket in a side-chain-independent manner. The residue of the last amino acid is buried in the F pocket locking the peptide into the groove, elucidating one position of the MHCI allele’s peptide binding motif. Since the ends of the peptide are “attached” and the ends of the MHCI grooves are closed, the peptide can bulge in the middle allowing for conformational changes upon TCR binding [20]. The size, shape, and electronic charge of the pockets determine which peptides interact with that MHC allele and with what binding affinity. Many mimotopes for human antigens are designed and tested for presentation by the MHCI HLA-A*0201 allele (HLA-A2) because, relative to other HLA alleles, HLA-A2 is frequently identified in many human populations and there are many reagents (such as antibodies, antigen databases, atomic structures, and transgenic animals) that facilitate its study.
Peptide binding to MHCII molecules differ from MHCI in that the TCR binding surface is made of two proteins, alpha and beta, and the peptide extends beyond the open ends of the groove [21, 22]. Hydrogen bonds between the peptide backbone and the MHCII molecules are generally conserved and sequence-independent. The peptide binds in a more extended form than MHCI, with more hydrogen bonds and lacks the central bulge sometimes found in MHCI. However, like MHCI, the amino acid side chains of MHCII contribute to individual pockets which interact specifically with peptide amino acids. These pockets are named after the position of the peptide side chains that they accept. The most frequently used pockets are P1 and P9 near the ends of the groove, followed by the more central P4 and P6. Thus, the peptide binding motifs found in MHCII peptides are usually, but not always, in positions 1, 9, 4, and 6.
As the MHC molecules have been identified and analyzed, consensus peptide binding motifs have been determined for many alleles. These motifs are used by the ever improving algorithms to predict which peptides effectively bind to MHC molecules (reviewed in [23]). To date, researchers most frequently substitute amino acids in peptide antigens that interact directly with the MHC molecule, referred to as MHC anchor-residue modifications (partial list: [22, 24–32]). The general rationale for making these substitutions is if a peptide does not bind well to the MHC molecule, the stability of the peptide-MHC interaction and presentation of the peptide antigen to TCR will be weak, reducing immunogenicity. Numerous studies have introduced MHC anchor-residue modifications with the goal of leading to stronger or modulated antigen-specific T cell responses in immunotherapies of cancer and autoimmunity. Interestingly, in T cell fingerprinting analyses, mutations in the anchor amino acids of MHCI-restricted peptides did not hinder TCR interactions as predicted [33]. We do not include a comprehensive list here, but discuss studies that justify how these mimotopes performed in vivo.
2.1. In cancer
Early clinical trials, targeting antigens shared by melanoma and differentiating melanocytes, with substitutions in the MHC anchor residues, such as Melan-A/MART-126–35 A27L, showed little efficacy (WT EAAGIGILTV, A27L ELAGIGILTV, the substituted amino acid is bolded) [34]. Later trials were more promising. In a prime/boost study, melanoma patients were first vaccinated with a plasmid encoding Melan-A/MART-126–35 A27L, followed by a boost with peptides [35]. Another trial with the MART-1 mimotope compared the responses to peptide encapsulated in noninfectious virus-like particles with and without LAG-3Ig designed to engage the LAG-3 checkpoint protein. One patient of 12 had a partial response in this trial [36].
Cole et al. examined differences between the Melan-A/MART-126–35 wild type and mimotope peptides [37]. Using two different T cell clones, they showed that the peptides bind to the MHC and TCR molecules with different binding properties and stimulated different responses in vitro. They also stimulated peripheral blood mononuclear cells (PBMCs) from HLA-A2 donors with these peptides followed by sequencing of the complementarity-determining region 3 of the TCR’s beta chain (CDR3P) regions. They identified only 15 of 101 unique clones elicited by both peptides, suggesting that different T cell repertoires respond to both peptides. Structural studies of this tumor antigen and mimotope peptide also indicate differences in the surfaces identified by the TCR, likely resulting in different TCR repertoires of responding T cells, see Figure 1A [38, 39]. In another study, Speiser et al. vaccinated human melanoma patients with these peptides and a strong adjuvant (CpG oligodeoxynucleotides) [40]. Although there were more T cells responding to the modified peptide vaccines, the quality and function of the T cells that cross reacted with the natural peptide was weaker than those responding to the wild type antigen vaccine. In the same vein, there were a number of examples when the natural epitopes make higher affinity interactions with TCRs than the mimotope resulting in better T cell responses [40].
A clinical trial using four well-characterized tumor antigen peptides with amino acid substitutions that increased their binding to HLA-A2 was performed in patients with early stage melanoma [41]. The patients were vaccinated with mimotope peptides and adjuvant then analyzed for responses. High frequencies of mimotope-specific effector-memory T cells were identified in the PBMCs and the tumor lesions; however, there was no improvement in diseasefree survival or overall survival in the patients. When the responding T cells were further analyzed, there were stronger responses to the mimotope peptide that did not cross-react with the wild type tumor antigen. The peptide sequences were Melan-A/MART-1 WT and A27L see above, gp100209-217 WT ITDQVPFSV, gp100 2M IMDQVPFSV, NY-ESO-1157–165 WT SLLMWITQC, NY-ESO-1 C165V SLLMWITQV, and Survivin WT ELTLGEFLKL, Survivin T97M ELMLGEFLKL.
A randomized phase 3 trial, which enrolled 185 patients at 21 centers, was conducted in patients with advanced melanoma [42]. Patients were given IL-2 with or without the gp100209–217 2M peptide in the adjuvant Montanide ISA-51. The patients who received the mimotope vaccine did significantly better: overall clinical response was higher with the 2M peptide, 16% vs. 6%, and patients had longer progression-free survival, 2.2 months vs. 1.6 months. Consistent with clinical outcomes, different conclusions were made when analyzing this peptide than the anchor modified mimotopes discussed above. The 2M mimotope bound HLA-A2 with 9-fold stronger affinity than the gp100209–217 wild type peptide [43]. In addition, crystal structures of peptide bound to the HLA-A2 molecule were very similar and ELISpot assays showed that the frequency of IFNγ producing T cells in response to the wild type and mimotope peptides were similar too. Antigen-specific memory T cells were identified in vaccinated individuals, and these T cells responded to melanoma cell lines suggesting that both peptides stimulated similar repertoires (Fig 1B) [44].
There have also been a number of clinical trials evaluating T cell responses in breast and ovarian cancer patients with HER2/neu-positive tumors vaccinated the HER2/neu peptide GP2 which binds to HLA-A2 molecules (reviewed in [45]). One caveat to these trials is that GP2 does not bind well to HLA-A2 (WT IISAVVGIL). Thus, there have been a number of attempts at altering a primary MHC anchor in the HER2/neu tumor antigen with the hopes of improving the peptide binding to MHC and the immunogenicity. Crystal studies of these substitutions showed substantial changes in the T cell binding surface suggesting that few T cells that cross-react with WT peptide would be raised by this vaccine (I2L/V5L ILSALVGIL) [46].
2.2. In autoimmunity
In autoimmunity, delivery of high affinity signals into T cells using mimotopes can improve therapeutic efficacy through a number of mechanisms. Induction of stronger antigen-specific responses by Tregs, modulation of functions and phenotypes of Tpaths, and deleting Tpaths all assist identifying and targeting self antigen-reactive T cells in an efficient manner. An important aspect of the latter two strategies that aim to kill or modify response of Tpaths is that targeting T cells specific for a single epitope may not be sufficient to stop autoimmune responses (Fig 2); antigen-spreading of therapy effects to non-mimotope-specific Tpaths may also be necessary to halt autoimmunity.
Genetic risks of a number of autoimmune diseases are detected within the HLA loci [47, 48]. Specific HLA alleles and haplotypes provide susceptibility or protection from the disease, such as susceptibility with DQ2 and DQ8 in celiac disease [49–51], DR3-DQ2/DR4-DQ8 in type 1 diabetes [52–54], DQB1*0602 in narcolepsy [55, 56], and DR15-DQ6 in multiple sclerosis [57, 58]. In contrast, DQB1*0602 provides dominant protection from the development of type 1 diabetes [53, 54]. These strong HLA associations with autoimmune diseases suggest that T cells reactive to epitopes that are specifically presented by risk HLA molecules play a critical role in the development of these autoimmune diseases.
In type 1 diabetes, which is a T cell-mediated autoimmune disease against pancreatic beta cells, the lack of an acidic amino acid residue (i.e. aspartic acid and glutamic acid) at position 57 of the DQ8 beta chain (β57) is associated with increased risk of developing this disease [52, 59]. As shown in studies analyzing peptide repertoires presented by both human DQ8 and the mouse ortholog I-Ag7, which is the MHCII molecule in NOD mice, a non-obese model of type 1 diabetes, without this acidic residue the peptide repertoire in these alleles are typically negatively charged [60, 61]. Mimotopes with amino acid substitution with acidic residues at P9 have been tested as superagonists that are capable of efficiently detecting self-reactive T cells [62–64] and potentially suppressing diabetes development more effectively than natural epitopes [65]. Teyton and his colleagues demonstrated that lack of an acidic amino acid at β57 in I-Ag7 or DQ8 results in TCR repertoires with negatively charged amino acids in the CDR3P region [66, 67]. Thus, TCR repertoires will change when the mimotope has alterations in the pockets of the MHC molecule.
Celiac disease, which is distinguished by an intensive immune response to dietary gluten, is another autoimmune disease with a strong genetic association to HLA [49–51]. The majority of patients have DQ2 (DQA1*05:01-DQB1*02:01, so called DQ2.5) and/or DQ8 (DQA1*03:01-DQB1*03:02) HLA class II haplotypes. Intestinal T cells of patients recognize peptides derived from gliadins, a protein component of gluten [68], and the majority of patients develop antibodies directed to transglutaminase 2, which deamidates glutamine to glutamic acid. It is well appreciated that DQ2 prefers acidic amino acid residues at P4 and proline at P6, whereas DQ8 has strong preference of acidic residues at P9 [70] as mentioned above. Gliadins are extremely rich in glutamine and proline, and gliadin peptides modified by transglutaminase 2 elicit responses by intestinal T cells of patients much more vigorously than unmodified gliadin peptides. Thus, deamidation of gliadin peptides so that they preferentially bind to DQ2 or DQ8 results in “naturally produced mimotopes” that induce robust T cell responses.
Citrullination of arginine is a well appreciated post-translational modification that generates antigens for rheumatoid arthritis [71, 72]. HLA-DRB1 alleles having particular sequences, the so called “HLA shared epitope alleles,” that are associated with a risk of rheumatoid arthritis developing anti-citrullinated protein antibodies. The HLA shared epitope sequences, amino acids 70–74 of DRB1, shape P4 of the peptide binding groove in HLA-DR molecules, and peptides with citrulline rather than arginine at P4 bind to shared epitope HLAs more efficiently [73, 74]. These peptides provide another example of naturally produced mimotopes that bind to MHC with high affinity, and elicit robust T cell responses. A major benefit of using such naturally produced mimotopes for therapies is that TCR repertoires targeting these mimotopes are all disease-associated, whereas experimentally designed mimotopes are likely to be recognized by only a portion of TCR repertoires targeting cognate disease associated antigens. Unlike for enhancing antitumor responses, all of the antigen-specific repertoire must be targeted in autoimmunity (Fig 2); otherwise, remaining Tpaths may continue to contribute to autoimmune progression.
3. Substitutions in secondary MHC anchor amino acids
One strategy to improve binding of peptides to MHC molecules is to make substitutions in secondary anchor amino acids. These amino acids are unique from the dominant consensus anchor amino acids discussed above, but also point into the MHC groove and contribute to stabilizing the pMHC interaction [75, 76]. One testable assumption made with these substitutions is that the interaction between the peptide and MHC molecule may change, but with the right substitution the surface that interacts with the TCR may not change and a similar repertoire of T cells may respond to the mimotope as the wild type peptide. The majority of studies of peptides with altered secondary anchor residues are in MHCI-restricted peptides [7, 77–81], although some mimotope substitutions, synthesized or natural, in secondary anchor residues for MHCII-restricted antigens have been characterized [82, 83]. Peptides that bind to MHCII are more heterogeneous in length and more degenerate in MHC binding specificity than those that bind to MHCI. In addition, substitutions in peptides that bind weakly to MHCII molecules, might change the register of the peptide in the MHCII molecule since the ends of the peptide binding groove are open [84, 85].
3.1. In cancer
Since the HLA-DRB1*0401 (DR4) allele is well characterized, Chen et al examined DR4-restricted mimotopes from the gp10044–59 peptide and their potential to contribute to melanoma therapies [83]. In this study, they solved the crystal structure of DR4 complexed with the self/tumor antigen and determined that the secondary anchor residues are in positions P4, P7, and P9. They made substitutions in secondary anchor amino acids, which bound with stronger affinity that the wild type peptide; however, different clones responded differently to the mimotopes. Some of them responded less than the wild type leading them to the conclusion that this method of identifying mimotopes does not provide predictable results. Potential factors that may have contributed to this conclusion are that changes in the secondary anchor amino acids may have changed the surface recognized by T cells or the mimotopes may have shifted the binding register in DR4. Alternatively, the test clones may not represent the antigen-specific repertoire.
One early report that aimed to break immune tolerance to the differentiation antigen gp100, expressed in human melanomas and the mouse B16 model for melanoma, showed that substituting amino acids other than those in the primary anchor motifs may be effective. This study showed that xenoimmunization of mice with human gp10025–33 (KVPRNQDWL) elicited T cells that cross-reacted with mouse gp10025–33 (EGSRNQDWL) [86]. Although the study did not show tumor protection, the T cells from mice vaccinated with the human antigen produced more IFNγ in response to the mouse peptide than T cells from the mouse vaccinated with the mouse peptide. The MHC binding motif for H-2Db has an asparagine at P5 and a leucine at P9 and the amino acids at P2 and P3 are buried deep within the Db groove [87]. Thus, the human gp10025–33 peptide may represent a mimotope with advantageous secondary anchor substitutions for the mouse.
Many preclinical and clinical studies on the immune response to common epithelial tumors has focused on HLA-A2-restricted antigens from the carcinoembryonic antigen (CEA) and Muc-1 proteins. Both proteins are frequently expressed by many tumors, such as breast, ovary, bladder, lung, colon, stomach, and thyroid tumors. Schlom’s team identified the CAP-1 peptide, which encodes consensus anchor amino acids for HLA-A2. To improve binding, they tested a panel of single amino acid substitutions against a T cell line made from a patient immunized with recombinant vaccinia virus expressing CEA. They found that a mimotope with an aspartic acid in the secondary anchor at P6 improved immunogenicity and presentation (CAP-1 WT YLSGANLNL, CAP-6D YLSGADLNL) [88]. The same team analyzed the Muc-1 tumor antigen, which is found on the same subsets of tumors. They identified an HLA-A2-restricted antigen outside the immunogenic variable number of tandem repeat region, and included a substitution in the primary anchor residue (Muc-192–101 WT ATWGQDVTSV, Muc-1 93L ALWGQDVTSV) [89].
In a randomized phase 2 study of metastatic breast cancer, patients were treated with the CEA and Muc-1 mimotopes, in addition to chemotherapy docetaxel and many other immune stimulatory molecules, resulting in an unprecedented doubling of progression-free survival [90]. The other immune stimulatory molecules cytokines and recombinant viruses for T cell costimulatory. Interestingly, the CAP-6D peptide expanded different T cells than the CAP-1 peptides; those expanded by CAP-6D had a lower functional avidity for the WT antigen (less sensitive) [91]. However, in the presence of this therapeutic immunostimulatory vaccine, T cells with low functional avidity may have been activated and/or the T cells in the tumor may have already been of high functional avidity, and the mimotopes may have offered specific T cells a functional boost.
Sharma and colleagues aimed to enhance the stability of the GP2 peptide from the HER-2/neu protein to the HLA-A2 molecule to improve antitumor responses [46]. They started with a version of GP2 that substituted both MHC anchor amino acids to encode the consensus motifs and then made a third substitution by changing valine to leucine in a central amino acid with the hope to further stabilize peptide binding (ILSALVGIV). They found that this change did the opposite: the peptide did not bind as well to the MHC molecule. To determine why, they crystalized the complexes, and showed that the amino acids which would be pointing toward the TCR had significantly changed. Another study of the GP2 peptide binding HLA-A2 showed that a substitution in a secondary anchor residue, phenylalanine in P7, improves peptide binding [78]. Joseph et al showed when vaccinating HLA-A2 transgenic mice with either the G7F mimotope or wild type GP2 peptide, T cells produced more IFNγ in response to wild type GP2 suggesting that this substitution may improve antitumor T cell responses.
We used an immunogenic mouse model to better understand the mechanism of action of these mimotopes. The immunodominant CD8 T cell response to the mouse CT26 colon cancer cell line is to GP70423–431, also known as AH1, bound to the MHCI molecule H-2Ld (Ld)[92]. The AH1 peptide encodes both consensus anchor residues for binding to Ld (proline in P2 and phenylalanine/leucine in P9) and it binds Ld with high affinity (ca 300–400 nM range) [93]. Thus, we made other substitutions in the antigenic peptide to identify potential mimotope peptides to improve the antitumor response. Initial experiments using a tumor antigen-specific T cell clone, antigen presenting cells, and a panel of peptides that replaced each amino acid in AH1 with an alanine, suggested that changing an amino acid in P5 from valine to alanine, A5, would increase the affinity to the T cell clone and enhance the antitumor response (AH1 WT SPSYVYHQF, A5 SPSYAYHQF) [93]. Using a strong adjuvant [94], the A5 peptide protected all vaccinated mice from developing a tumor, whereas the wild type AH1 peptide protected none of the mice [95]. We also identified the valine to alanine substitution using a combinatorial peptide library [6]. Although most of the peptides that we identified using this library also had the anchor amino acids, one peptide that protected 60% of mice from developing tumors, had a substitution in one of the Ld anchor residues (proline in P2 was substituted with asparagine) and six of nine amino acid substitutions (PS39, MNKYAYHML). Notably, all of the peptides identified from the combinatorial peptide library harbored the alanine substitution in P5 and, like the wild type peptide, tyrosines in positions 4 and 6 predicted (and later shown) to point toward the TCR.
To understand why mimotopes such as PS39 and A5 protect from tumor growth and other peptides do not, we used MHC tetramers that were labeled with different fluorophores and loaded with different peptides [95]. We used peptides that were covalently linked to the β2m molecule of the tetramer so that peptides could not reveal inaccurate results by loading MHC molecules in other tetramer complexes during the staining process. We simultaneously stained cells from mimotope-vaccinated mice with mimotope- and AH1-loaded tetramers. All mimotope vaccines immunized equally well, as mimotope-vaccinated mice had similar frequencies of mimotope-specific T cells, but not all of the mimotope vaccines elicited similar frequencies of T cells that also cross-reacted with the AH1-loaded tetramers. The more T cells that double stained with the AH1- and mimotope-loaded tetramers, the better that mimotope protected from tumor development. Protection also correlated with IFNγ production in response to ex vivo stimulation with the AH1 peptide. We concluded that the frequency of T cells that cross-reacted with the wild type antigen and the sensitivity of those T cells to make IFNγ was critical for optimal performance of the mimotope vaccine. More results using the immunogenic CT26 system are included in the next section, since although the amino acid in P5 points into the MHC molecule, it significantly changed TCR binding.
4. Amino acid substitutions that improve T cell responses
Research using surface plasmon resonance to study binding affinity and kinetics of the monomeric TCR-pMHC interaction suggest that the physiologic affinity range is 100 to 1 micromolar (μM) [96]. However, a recent study by Zhang et al, which examined Hepatitis C-specific T cells using an in situ TCR affinity and sequence assay, found 1000-fold range in affinity in specific CD8+ T cells [97]. Since TCRs are the only antigen-specific molecules on the surface of T cells, a simple assumption is that the affinity of the TCR for pMHC correlates with the strength of the T cell response, although there are a number of noted exceptions (reviewed in [98]). The monomeric affinity between TCR and pMHC molecule is weak relative to other receptor-ligand interactions and T cells have on average 105 TCR molecules on their surfaces [99]. In addition, many other receptor-ligand interactions take place after TCR-pMHC binding resulting in a stronger avidity [100]. Krogsgaard and colleagues showed the threshold affinity is 10 μM—stronger binding does not further increase the avidity of the interaction [101]. The overall function of mimotopes in therapies of cancer and autoimmunity is to increase the biding of this interaction.
4.1. In cancer
One of the most highly studied tumor antigens with potential for adding specificity to tumor vaccines is the cancer testes antigen NY-ESO-1. NY-ESO-1 is immunogenic in a wide range of malignancies and is presented by many class I and class II HLAs [102]. As mentioned above, peptide substitutions have been made in NY-ESO-1157–165 to improve binding to the HLA-A2 molecule (WT SLLMWITQC). Studies have also been performed targeting residues that interact with the TCR. With structure trials and binding assays, Webb et al showed that substitution of the non-consensus amino acid in P9 cysteine, an HLA-A2 anchor position, with a non-natural epitope, 2-aminoisobutyric acid (Abu), did not result in stronger HLA binding, but did bind TCRs from T cell clones with higher affinity [103]. Of note, this study considered potential oxidative damage that occurs with peptides harboring cysteine, which can be applied to other peptide vaccines. Testing the hypothesis that peptides substituted with conserved TCR binding amino acids improve T cell responses, Shang et al used a molecular simulation strategy in positions 4–8 in the HLA-A2-restricted NY-ESO-1 peptide. When the tryptophan in position 5 was substituted with a phenylalanine, the peptide continued to bind to HLA-A2, and was more immunogenic in a number of assays. Specifically, a cross-reactive repertoire was identified in HLA-A2-expressing PBMCs from healthy individuals, IFNγ production was detected in response to the wild type peptide, and T cells raised to the mimotope with phenylalanine substitution killed target cells loaded with the wild type peptide [104].
Using high throughput sequencing and the mouse CT26 tumor model discussed above, we investigated the sequences of the cross-reactive TCRs from T cells responding to mimotope vaccines and the tumor [105]. We asked whether the protective mimotopes elicited a de novo subset of T cells or T cells that were already activated by the tumor antigen. We found that most of the tumor-specific T cells were similar to those expanded by the tumor (Fig 1B) and that many of the mimotope-elicited T cells that cross-reacted with the AH1 peptide shared a specific motif in their CDR3P sequences. Over 8% of tumor infiltrating T cells had a CDR3P motif. Binding assays showed that TCRs with this motif had weaker binding affinity (≥100 μM) than the original sub-cloned TCR (5 μM) [106].
To determine if TCRs encoding the motif identified more therapeutic mimotopes than the higher affinity TCR, we screened peptide libraries with both TCRs [107]. Almost half of the peptides identified with both TCRs had the beneficial alanine at P5 (mimotope A5). We used pools of mimotopes identified by both TCRs in tumor protection assays; most mice grew tumors. However, if we followed the initial immunization with a boost with the AH1 peptide which does not protect on its own, we observed significantly more protection with the peptides identified with the common TCR than the high affinity, 70% vs. 5%, respectively. Thus, it was more important for the TCR used to identify mimotopes to be representative of the population to be immunized than to be of high affinity.
Finally, we performed structural studies to understand why the mimotope A5 protects from tumor formation and the wild type peptide AH1 does not [106]. The structure of the peptides bound to MHC look very similar, the only difference is that the bulge in the center of the A5 peptide is shifted toward the MHC molecule about 1Å, explaining the overlapping repertoires. We showed that the monomeric affinity of the A5 peptide in the TCR-pMHC interaction was significantly stronger than the wild type peptide, ~11 μM versus >100 μM, respectively. Interestingly, binding of the A5 complex displayed second order kinetics, suggesting that there was a conformational change in the complex occurring after initial binding of the TCR. Further structural studies of the T cell receptor binding to A5-MHC showed this conformational change in the tyrosines quite dramatically—one tyrosine rotated 135° which results in numerous interactions between the identified motif in the CDR3β and the peptide, explaining the increase in affinity and protection.
4.2. In autoimmunity
Many mimotopes have been designed for autoimmunity based on the responsiveness of T cells isolated from patients or animal models. T cell clones, derived from targeted organs or PBMCs cells, are tested to identify amino acid substitutions of peptides that reduce pathogenic responses or elicit strong regulatory responses. Some of the mimotopes identified have been tested for therapeutic purposes in clinical trials. Examples of such clinical trials to preserve pancreatic beta cell function in diabetes patients include a mimotope modified from the human insulin B chain peptide 9–23 restricted by HLA-DQ8 (WT SHLVEALYLVCGERG, NBI-6024 SHLVEALALVAGERG) [108]. The substitutions in NBI-6024 are known to be important in the diabetes-prone NOD mouse model [109–111]. Importantly, we have recently shown that CD4 T cells reactive to the wild type peptide are present in the islets of organ donors with type 1 diabetes [112]. However, the decline of C-peptide concentrations, a measure of pancreatic beta cell function, were not improved by subcutaneous administration of the mimotopes over two years compared to placebo [108].
Mimotopes of myelin basic protein were also tested in clinical trials for multiple sclerosis. Amino acid substitutions were made in the DR15-restricted myelin basic protein peptide 83–99 targeted by CD4 T cells in the central nervous system of multiple sclerosis patients (WT ENPVVHFFKNIVTPRTP). Two amino acids at positions 89 and 91 have been shown to be important as TCR contact residues. Subcutaneous administration of mimotope NBI-5788 was effective in inducing mimotope-reactive CD4 T cells with a Th2-like phenotype with no progression of disease (NBI-5788 AKPVVHLFANIVTPRTP). Unfortunately, there was no improvement in patients receiving a lower dose of the mimotope and many of those who received a higher dose suffered from hypersensitivity reactions, so the trial was discontinued [113–115]. Another mimotope, CGP77116 which also has amino acid substitutions at positions 89 and 91, was tested in patients with relapsing-remitting multiple sclerosis. In this trial some of the patients receiving high doses of the peptides (50 mg weekly) showed exacerbation of disease [113]. Although unsuccessful, these studies showed a trend that efficacy of the treatment depends on peptide dose and schedule [115]. Also, one explanation for poor trial outcomes may be due to the diversity of peptides administered. Antigens targeted by T cells involved in disease progression vary unless therapies are given at the initial stage, thereby anticipating the need for eliminating or modifying phenotypes of Tpaths that react with a variety of antigens. Indeed, a recent clinical trial using four peptides derived from myelin basic protein (ATX MS-1467) showed a preferable outcome with expansion of Tregs [116, 117] consistent with studies performed in humanized (DR2 × Ob1)F1 mice [118]. Thus, whether or not peptides are natural or altered, it is important to consider as many disease-associated T cells as possible early in disease progression.
Antigen repertoires that are targeted by T cells in the intestines of celiac disease patients target are considered relatively narrow. Therefore, rationally designed epitope-specific immunotherapies for celiac disease are a logical solution. As mentioned above, celiac disease-associated T cells preferably react with “naturally produced mimotopes” that result from deamidation of gliadin-derived peptides. Clinical trials using the combination of three gluten-derived peptides, which contain at least five gliadin-specific T cell epitopes presented by HLA-DQ2.5 (Nexvax2), were conducted with HLA-DQ2.5-positive celiac disease patients [119]. While the Phase 1 studies showed preferable outcomes in terms of safety and tolerability, it was recently announced that the Nexvax 2 phase-2 trial was discontinued due to lack of protection by the therapy from gluten challenge [120]. While the negative outcome was unfortunate, this well-designed study leaves us with important suggestions for future trials. Analyses of antigen specificity of T cells associated with alternation of phenotypes between groups receiving different doses and schedules of therapy will also be beneficial in understanding needs for future therapies.
5. Future mimotopes
The concept that adding antigen-specific T cell responses to immunotherapies has reemerged as patients are relapsing after checkpoint therapies for cancer and antigens are being discovered in autoimmunities. Methods to improve mimotopes that enhance binding of peptide to MHC or pMHC to T cell repertoire are evolving, and technologies that predict epitopes and mimotopes recognized by particular TCRs are being developed [33, 121–123]. Chemically modified antigens or antigens with unnatural amino acids, such as d-amino acids, may help to implement subtle changes in antigens; those that improve binding but do not change the antigen surface [124, 125].
In the search for the perfect antitumor mimotope, we identified many that were suboptimal and did not protect from tumor growth. Most of these mimotopes raised T cell repertoires that were not cross-reactive with the wild type antigen. However, we determined that we could improve these suboptimal mimotopes by boosting with the wild type antigen [126, 127]. The first immunization with the mimotope would stimulate a small fraction of the crossreactive T cells with higher affinity than the wild type antigen did, then the booster immunization with the wild type antigen did not have to be as strong to expand the T cells differentiating into cytotoxic T lymphocytes as these T cells have a much lower threshold for stimulation. For this reason, the order of the vaccination could not be reversed.
Unlike in tumor immunity, in autoimmunity some mimotopes have ultimately been found to bind with lower affinity than the wild type peptide. Some self antigens originally thought to weakly interact with their cognate T cell repertoire, but were actually mis-identified antigens. For example, a panel of T cell clones made by the Haskins’ group (including BDC-2.5 and BDC-10.1), were isolated from the spleen of diabetes-prone NOD mice. These TCRs induce diabetes in adoptive transfer experiments and transgenic mouse models (summarized in [128]). Until recently antigens targeted by these TCRs were unknown. A number of mimotopes that stimulate these T cell clones were identified during the past two decades [[129] and other refs], and finally Haskins and colleagues discovered that a fusion peptide composed of preproinsulin and chromogranin A is a target that can stimulate BDC-2.5 and other several T cell clones a magnitude of order more strongly than the natural chromogranin A peptide [130]. Importantly, T cells recognizing this fusion peptide, 2.5HIP (hybrid insulin peptide), are the most frequent in the islets of NOD mice among T cells recognizing mimotopes that can stimulate the BDC-2.5 and other T cell clones [130], suggesting that only a portion of TCR repertoires recognizing the mimotopes overlaps with repertoires of the wild type antigen.
Reactivity of tumor-specific and autoreactive T cells is typically lower than pathogen-reactive T cells, and indeed atypical binding between autoreactive TCRs and peptide-MHC complexes has been structurally demonstrated [131–134]. Therefore, mimotopes that have high affinity to MHC or to TCR and are recognized by T cells more efficiently have been explored for antigen-specific therapies. However, the studies pursuing antigen specificity for BDC-2.5 and other diabetogenic T cell clones raise an important possibility that antigens truly targeted by disease-involving T cells may react to the antigen as strongly as pathogen-reactive T cells and may be more appropriate to be utilized for therapies than artificially-designed mimotopes as mimotopes could influence only a portion of T cell repertoires. Thus, identifying antigens that are truly targeted by disease-associated T cells may be critical for the development of robust antigen-specific immunotherapies.
Another important concept required to develop ideal mimotopes for autoimmune diseases is to distinguish antigens that are preferably recognized by Tregs from those for Tpaths. For example, the majority of HIP2.5-specific T cells in the islets of NOD mice have a pathogenic phenotype, whereas T cells specific for an insulin peptide contain a higher proportion of FoxP3-positive Tregs [135]. Depending on the use of mimotopes for therapies (i.e. delete Tpaths or elicit Tregs), mimotopes that are preferably recognized by either Tregs or Tpaths need to be identified to be used for therapies (Fig 2).
In summary, structural, functional, and repertoire studies show that amino acid substitutions must be examined for each antigen. To date there is not a formulae that will work to identify efficacious mimotopes without significant investigation, although the decades of study have established what needs to be addressed for each antigen. These studies, in addition to the adjuvants required to get a significant response, the dosing regimen, the time during disease progression, and the antigen itself all contribute to making these antigen-specific therapies successful.
Abbreviations
- APC
antigen presenting cell
- CDR3
complementarity-determining region 3
- CEA
carcinoembryonic antigen
- ELISpot
enzyme-linked immunospot
- HIP
Hybrid insulin peptide
- HLA
human leukocyte antigen
- IFNγ
interferon-gamma
- MHC
major histocompatibility complex
- MHCI
MHC class I
- MHCII
MHC class II
- μM
micromolar
- P
amino acid position (in a peptide)
- PBMC
peripheral blood mononuclear cell
- pMHC
peptide-MHC complex
- TCR
T cell receptor
- TIL
tumor infiltrating lymphocytes
- Tpath
pathogenic T cell
- Treg
regulatory T cell
- VKH
Vogt-Koyanagi-Harada disease
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Franks AL, Slansky JE, Multiple associations between a broad spectrum of autoimmune diseases, chronic inflammatory diseases and cancer, Anticancer Res 32(4) (2012) 1119–36. [PMC free article] [PubMed] [Google Scholar]
- [2].Yeh S, Karne NK, Kerkar SP, Heller CK, Palmer DC, Johnson LA, Li Z, Bishop RJ, Wong WT, Sherry RM, Yang JC, Dudley ME, Restifo NP, Rosenberg SA, Nussenblatt RB, Ocular and systemic autoimmunity after successful tumor-infiltrating lymphocyte immunotherapy for recurrent, metastatic melanoma, Ophthalmology 116(5) (2009) 981–989 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Adegoke AO, Grant MD, Enhancing Human Immunodeficiency Virus-Specific CD8(+) T Cell Responses with Heteroclitic Peptides, Front Immunol 6 (2015) 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mason D, A very high level of crossreactivity is an essential feature of the T-cell receptor, Immunol Today 19(9) (1998) 395–404. [DOI] [PubMed] [Google Scholar]
- [5].Abbey JL, O’Neill HC, Expression of T-cell receptor genes during early T-cell development, Immunol Cell Biol 86(2) (2008) 166–74. [DOI] [PubMed] [Google Scholar]
- [6].McMahan RH, McWilliams JA, Jordan KR, Dow SW, Wilson DB, Slansky JE, Relating TCR-peptide-MHC affinity to immunogenicity for the design of tumor vaccines, J Clin Invest 116(9) (2006) 2543–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sidney J, Assarsson E, Moore C, Ngo S, Pinilla C, Sette A, Peters B, Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries, Immunome Res 4 (2008) 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wucherpfennig KW, Allen PM, Celada F, Cohen IR, De Boer R, Garcia KC, Goldstein B, Greenspan R, Hafler D, Hodgkin P, Huseby ES, Krakauer DC, Nemazee D, Perelson AS, Pinilla C, Strong RK, Sercarz EE, Polyspecificity of T cell and B cell receptor recognition, Semin Immunol 19(4) (2007) 216–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, Grand F, Brewer JE, Gupta M, Plesa G, Bossi G, Vuidepot A, Powlesland AS, Legg A, Adams KJ, Bennett AD, Pumphrey NJ, Williams DD, Binder-Scholl G, Kulikovskaya I, Levine BL, Riley JL, Varela-Rohena A, Stadtmauer EA, Rapoport AP, Linette GP, June CH, Hassan NJ, Kalos M, Jakobsen BK, Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells, Sci Transl Med 5(197) (2013) 197ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, Binder-Scholl GK, Smethurst DP, Gerry AB, Pumphrey NJ, Bennett AD, Brewer JE, Dukes J, Harper J, Tayton-Martin HK, Jakobsen BK, Hassan NJ, Kalos M, June CH, Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma, Blood 122(6) (2013) 863–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM, Phan GQ, Hughes MS, Kammula US, Miller AD, Hessman CJ, Stewart AA, Restifo NP, Quezado MM, Alimchandani M, Rosenberg AZ, Nath A, Wang T, Bielekova B, Wuest SC, Akula N, McMahon FJ, Wilde S, Mosetter B, Schendel DJ, Laurencot CM, Rosenberg SA, Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy, J Immunother 36(2) (2013) 133–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wooldridge L, Ekeruche-Makinde J, van den Berg HA, Skowera A, Miles JJ, Tan MP, Dolton G, Clement M, Llewellyn-Lacey S, Price DA, Peakman M, Sewell AK, A single autoimmune T cell receptor recognizes more than a million different peptides, J Biol Chem 287(2) (2012) 1168–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cusick MF, Libbey JE, Fujinami RS, Molecular mimicry as a mechanism of autoimmune disease, Clin Rev Allergy Immunol 42(1) (2012) 102–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sethi DK, Gordo S, Schubert DA, Wucherpfennig KW, Crossreactivity of a human autoimmune TCR is dominated by a single TCR loop, Nat Commun 4 (2013) 2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kumar A, Delogu F, Dynamical footprint of cross-reactivity in a human autoimmune T-cell receptor, Sci Rep 7 (2017) 42496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Natarajan K, Li H, Mariuzza RA, Margulies DH, MHC class I molecules, structure and function, Rev Immunogenet 1(1) (1999) 32–46. [PubMed] [Google Scholar]
- [17].Nelson CA, Fremont DH, Structural principles of MHC class II antigen presentation, Rev Immunogenet 1(1) (1999) 47–59. [PubMed] [Google Scholar]
- [18].Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC, Structure of the human class I histocompatibility antigen, HLA-A2, Nature 329(6139) (1987) 506–12. [DOI] [PubMed] [Google Scholar]
- [19].Li H, Natarajan K, Malchiodi EL, Margulies DH, Mariuzza RA, Three-dimensional structure of H-2Dd complexed with an immunodominant peptide from human immunodeficiency virus envelope glycoprotein 120, J Mol Biol 283(1) (1998) 179–91. [DOI] [PubMed] [Google Scholar]
- [20].Wieczorek M, Abualrous ET, Sticht J, Alvaro-Benito M, Stolzenberg S, Noe F, Freund C, Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation, Front Immunol 8 (2017) 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dessen A, Lawrence CM, Cupo S, Zaller DM, Wiley DC, X-ray crystal structure of HLA-DR4 (DRA*0101, DRB1*0401) complexed with a peptide from human collagen II, Immunity 7(4) (1997) 473–81. [DOI] [PubMed] [Google Scholar]
- [22].Wang Y, Sosinowski T, Novikov A, Crawford F, Neau DB, Yang J, Kwok WW, Marrack P, Kappler JW, Dai S, C-terminal modification of the insulin B:11–23 peptide creates superagonists in mouse and human type 1 diabetes, Proc Natl Acad Sci U S A 115(1) (2018) 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liao WW, Arthur JW, Predicting peptide binding to Major Histocompatibility Complex molecules, Autoimmun Rev 10(8) (2011) 469–73. [DOI] [PubMed] [Google Scholar]
- [24].Bakker AB, van der Burg SH, Huijbens RJ, Drijfhout JW, Melief CJ, Adema GJ, Figdor CG, Analogues of CTL epitopes with improved MHC class-I binding capacity elicit anti-melanoma CTL recognizing the wild-type epitope, Int J Cancer 70(3) (1997) 302–9. [DOI] [PubMed] [Google Scholar]
- [25].Bownds S, Tong-On P, Rosenberg SA, Parkhurst M, Induction of tumor-reactive cytotoxic T-lymphocytes using a peptide from NY-ESO-1 modified at the carboxy-terminus to enhance HLA-A2.1 binding affinity and stability in solution, J Immunother 24(1) (2001) 1–9. [DOI] [PubMed] [Google Scholar]
- [26].Dyall R, Bowne WB, Weber LW, LeMaoult J, Szabo P, Moroi Y, Piskun G, Lewis JJ, Houghton AN, Nikolic-Zugic J, Heteroclitic immunization induces tumor immunity, J Exp Med 188(9) (1998) 1553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Harig S, Witzens M, Krackhardt AM, Trojan A, Barrett P, Broderick R, Zauls AJ, Gribben JG, Induction of cytotoxic T-cell responses against immunoglobulin V region-derived peptides modified at human leukocyte antigen-A2 binding residues, Blood 98(10) (2001) 2999–3005. [DOI] [PubMed] [Google Scholar]
- [28].Mimura K, Kono K, Southwood S, Fikes J, Takahashi A, Miyagawa N, Sugai H, Fujii H, Substitution analog peptide derived from HER-2 can efficiently induce HER-2-specific, HLA-A24 restricted CTLs, Cancer Immunol Immunother 55(11) (2006) 1358–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Parkhurst MR, Salgaller ML, Southwood S, Robbins PF, Sette A, Rosenberg SA, Kawakami Y, Improved induction of melanoma-reactive CTL with peptides from the melanoma antigen gp100 modified at HLA-A*0201-binding residues, J Immunol 157(6) (1996) 2539–48. [PubMed] [Google Scholar]
- [30].Pinilla-Ibarz J, May RJ, Korontsvit T, Gomez M, Kappel B, Zakhaleva V, Zhang RH, Scheinberg DA, Improved human T-cell responses against synthetic HLA-0201 analog peptides derived from the WT1 oncoprotein, Leukemia 20(11) (2006) 2025–33. [DOI] [PubMed] [Google Scholar]
- [31].Valmori D, Pittet MJ, Vonarbourg C, Rimoldi D, Lienard D, Speiser D, Dunbar R, Cerundolo V, Cerottini JC, Romero P, Analysis of the cytolytic T lymphocyte response of melanoma patients to the naturally HLA-A*0201-associated tyrosinase peptide 368–376, Cancer Res 59(16) (1999) 4050–5. [PubMed] [Google Scholar]
- [32].Wang Y, Sosinowski T, Novikov A, Crawford F, White J, Jin N, Liu Z, Zou J, Neau D, Davidson GW, Nakayama M, Kwok WW, Gapin L, Marrack P, Kappler JW, Dai S, How C-terminal additions to insulin B-chain fragments create superagonists for T cells in mouse and human type 1 diabetes, Sci Immunol 4(34) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bentzen AK, Such L, Jensen KK, Marquard AM, Jessen LE, Miller NJ, Church CD, Lyngaa R, Koelle DM, Becker JC, Linnemann C, Schumacher TNM, Marcatili P, Nghiem P, Nielsen M, Hadrup SR, T cell receptor fingerprinting enables in-depth characterization of the interactions governing recognition of peptide-MHC complexes, Nat Biotechnol (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Rosenberg SA, Yang JC, Restifo NP, Cancer immunotherapy: moving beyond current vaccines, Nat Med 10(9) (2004) 909–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ribas A, Weber JS, Chmielowski B, Comin-Anduix B, Lu D, Douek M, Ragavendra N, Raman S, Seja E, Rosario D, Miles S, Diamond DC, Qiu Z, Obrocea M, Bot A, Intra-lymph node prime-boost vaccination against Melan A and tyrosinase for the treatment of metastatic melanoma: results of a phase 1 clinical trial, Clin Cancer Res 17(9) (2011) 2987–96. [DOI] [PubMed] [Google Scholar]
- [36].Romano E, Michielin O, Voelter V, Laurent J, Bichat H, Stravodimou A, Romero P, Speiser DE, Triebel F, Leyvraz S, Harari A, MART-1 peptide vaccination plus IMP321 (LAG-3Ig fusion protein) in patients receiving autologous PBMCs after lymphodepletion: results of a Phase I trial, J Transl Med 12 (2014) 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cole DK, Edwards ES, Wynn KK, Clement M, Miles JJ, Ladell K, Ekeruche J, Gostick E, Adams KJ, Skowera A, Peakman M, Wooldridge L, Price DA, Sewell AK, Modification of MHC anchor residues generates heteroclitic peptides that alter TCR binding and T cell recognition, J Immunol 185(4) (2010) 2600–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Borbulevych OY, Insaidoo FK, Baxter TK, Powell DJ Jr., Johnson LA, Restifo NP, Baker BM, Structures of MART-126/27–35 Peptide/HLA-A2 complexes reveal a remarkable disconnect between antigen structural homology and T cell recognition, J Mol Biol 372(5) (2007) 1123–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Madura F, Rizkallah PJ, Holland CJ, Fuller A, Bulek A, Godkin AJ, Schauenburg AJ, Cole DK, Sewell AK, Structural basis for ineffective T-cell responses to MHC anchor residue-improved “heteroclitic” peptides, Eur J Immunol 45(2) (2015) 584–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Speiser DE, Baumgaertner P, Voelter V, Devevre E, Barbey C, Rufer N, Romero P, Unmodified self antigen triggers human CD8 T cells with stronger tumor reactivity than altered antigen, Proc Natl Acad Sci U S A 105(10) (2008) 3849–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Filipazzi P, Pilla L, Mariani L, Patuzzo R, Castelli C, Camisaschi C, Maurichi A, Cova A, Rigamonti G, Giardino F, Di Florio A, Asioli M, Frati P, Sovena G, Squarcina P, Maio M, Danielli R, Chiarion-Sileni V, Villa A, Lombardo C, Tragni G, Santinami M, Parmiani G, Rivoltini L, Limited induction of tumor cross-reactive T cells without a measurable clinical benefit in early melanoma patients vaccinated with human leukocyte antigen class I-modified peptides, Clin Cancer Res 18(23) (2012) 6485–96. [DOI] [PubMed] [Google Scholar]
- [42].Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, Gailani F, Riley L, Conlon K, Pockaj B, Kendra KL, White RL, Gonzalez R, Kuzel TM, Curti B, Leming PD, Whitman ED, Balkissoon J, Reintgen DS, Kaufman H, Marincola FM, Merino MJ, Rosenberg SA, Choyke P, Vena D, Hwu P, gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma, N Engl J Med 364(22) (2011) 2119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Borbulevych OY, Baxter TK, Yu Z, Restifo NP, Baker BM, Increased immunogenicity of an anchor-modified tumor-associated antigen is due to the enhanced stability of the peptide/MHC complex: implications for vaccine design, J Immunol 174(8) (2005) 4812–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Walker EB, Miller W, Haley D, Floyd K, Curti B, Urba WJ, Characterization of the class I-restricted gp100 melanoma peptide-stimulated primary immune response in tumor-free vaccine-draining lymph nodes and peripheral blood, Clin Cancer Res 15(7) (2009) 2541–51. [DOI] [PubMed] [Google Scholar]
- [45].Clive KS, Tyler JA, Clifton GT, Holmes JP, Ponniah S, Peoples GE, Mittendorf EA, The GP2 peptide: a HER2/neu-based breast cancer vaccine, J Surg Oncol 105(5) (2012) 452–8. [DOI] [PubMed] [Google Scholar]
- [46].Sharma AK, Kuhns JJ, Yan S, Friedline RH, Long B, Tisch R, Collins EJ, Class I major histocompatibility complex anchor substitutions alter the conformation of T cell receptor contacts, J Biol Chem 276(24) (2001) 21443–9. [DOI] [PubMed] [Google Scholar]
- [47].Matzaraki V, Kumar V, Wijmenga C, Zhernakova A, The MHC locus and genetic susceptibility to autoimmune and infectious diseases, Genome Biol 18(1) (2017) 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Thorsby E, Lie BA, HLA associated genetic predisposition to autoimmune diseases: Genes involved and possible mechanisms, Transpl Immunol 14(3–4) (2005) 175–82. [DOI] [PubMed] [Google Scholar]
- [49].Hunt KA, Zhernakova A, Turner G, Heap GA, Franke L, Bruinenberg M, Romanos J, Dinesen LC, Ryan AW, Panesar D, Gwilliam R, Takeuchi F, McLaren WM, Holmes GK, Howdle PD, Walters JR, Sanders DS, Playford RJ, Trynka G, Mulder CJ, Mearin ML, Verbeek WH, Trimble V, Stevens FM, O’Morain C, Kennedy NP, Kelleher D, Pennington DJ, Strachan DP, McArdle WL, Mein CA, Wapenaar MC, Deloukas P, McGinnis R, McManus R, Wijmenga C, van Heel DA, Newly identified genetic risk variants for celiac disease related to the immune response, Nat Genet 40(4) (2008) 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Liu E, Lee HS, Aronsson CA, Hagopian WA, Koletzko S, Rewers MJ, Eisenbarth GS, Bingley PJ, Bonifacio E, Simell V, Agardh D, Group TS, Risk of pediatric celiac disease according to HLA haplotype and country, N Engl J Med 371(1) (2014) 42–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sollid LM, The roles of MHC class II genes and post-translational modification in celiac disease, Immunogenetics 69(8–9) (2017) 605–616. [DOI] [PubMed] [Google Scholar]
- [52].Todd A, Bell JI, McDevitt HO, HLA-DQ beta gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus, Nature 329(6140) (1987) 599–604. [DOI] [PubMed] [Google Scholar]
- [53].Steck AK, Rewers MJ, Genetics of type 1 diabetes, Clin Chem 57(2) (2011) 176–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Concannon P, Rich SS, Nepom GT, Genetics of type 1A diabetes, N Engl J Med 360(16) (2009) 1646–54. [DOI] [PubMed] [Google Scholar]
- [55].Mignot E, Hayduk R, Black J, Grumet FC, Guilleminault C, HLA DQB1*0602 is associated with cataplexy in 509 narcoleptic patients, Sleep 20(11) (1997) 1012–20. [PubMed] [Google Scholar]
- [56].Lin L, Hungs M, Mignot E, Narcolepsy and the HLA region, J Neuroimmunol 117(1–2) (2001) 9–20. [DOI] [PubMed] [Google Scholar]
- [57].Olerup O, Hillert J, HLA class II-associated genetic susceptibility in multiple sclerosis: a critical evaluation, Tissue Antigens 38(1) (1991) 1–15. [DOI] [PubMed] [Google Scholar]
- [58].Schmidt H, Williamson D, Ashley-Koch A, HLA-DR15 haplotype and multiple sclerosis: a HuGE review, Am J Epidemiol 165(10) (2007) 1097–109. [DOI] [PubMed] [Google Scholar]
- [59].Hu X, Deutsch AJ, Lenz TL, Onengut-Gumuscu S, Han B, Chen WM, Howson JM, Todd JA, de Bakker PI, Rich SS, Raychaudhuri S, Additive and interaction effects at three amino acid positions in HLA-DQ and HLA-DR molecules drive type 1 diabetes risk, Nat Genet 47(8) (2015) 898–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Suri A, Walters JJ, Gross ML, Unanue ER, Natural peptides selected by diabetogenic DQ8 and murine I-A(g7) molecules show common sequence specificity, J Clin Invest 115(8) (2005) 2268–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].van Lummel M, van Veelen PA, Zaldumbide A, de Ru A, Janssen GM, Moustakas AK, Papadopoulos GK, Drijfhout JW, Roep BO, Koning F, Type 1 diabetes-associated HLA-DQ8 transdimer accommodates a unique peptide repertoire, J Biol Chem 287(12) (2012) 9514–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Yang J, Chow IT, Sosinowski T, Torres-Chinn N, Greenbaum CJ, James EA, Kappler JW, Davidson HW, Kwok WW, Autoreactive T cells specific for insulin B:11–23 recognize a low-affinity peptide register in human subjects with autoimmune diabetes, Proc Natl Acad Sci U S A 111(41) (2014) 14840–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Nakayama M, McDaniel K, Fitzgerald-Miller L, Kiekhaefer C, Snell-Bergeon JK, Davidson HW, Rewers M, Yu L, Gottlieb P, Kappler JW, Michels A, Regulatory vs. inflammatory cytokine T-cell responses to mutated insulin peptides in healthy and type 1 diabetic subjects, Proc Natl Acad Sci U S A 112(14) (2015) 4429–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Spanier JA, Sahli NL, Wilson JC, Martinov T, Dileepan T, Burrack AL, Finger EB, Blazar BR, Michels AW, Moran A, Jenkins MK, Fife BT, Increased Effector Memory Insulin-Specific CD4(+) T Cells Correlate With Insulin Autoantibodies in Patients With Recent-Onset Type 1 Diabetes, Diabetes 66(12) (2017) 3051–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zhang L, Crawford F, Yu L, Michels A, Nakayama M, Davidson HW, Kappler JW, Eisenbarth GS, Monoclonal antibody blocking the recognition of an insulin peptide-MHC complex modulates type 1 diabetes, Proc Natl Acad Sci U S A 111(7) (2014) 2656–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Yoshida K, Corper AL, Herro R, Jabri B, Wilson IA, Teyton L, The diabetogenic mouse MHC class II molecule I-Ag7 is endowed with a switch that modulates TCR affinity, J Clin Invest 120(5) (2010) 1578–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Gioia L, Holt M, Costanzo A, Sharma S, Abe B, Kain L, Nakayama M, Wan X, Su A, Mathews C, Chen YG, Unanue E, Teyton L, Position beta57 of I-A(g7) controls early antiinsulin responses in NOD mice, linking an MHC susceptibility allele to type 1 diabetes onset, Sci Immunol 4(38) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Sollid LM, Qiao SW, Anderson RP, Gianfrani C, Koning F, Nomenclature and listing of celiac disease relevant gluten T-cell epitopes restricted by HLA-DQ molecules, Immunogenetics 64(6) (2012) 455–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Stepniak D, Wiesner M, de Ru AH, Moustakas AK, Drijfhout JW, Papadopoulos GK, van Veelen PA, Koning F, Large-scale characterization of natural ligands explains the unique gluten-binding properties of HLA-DQ2, J Immunol 180(5) (2008) 3268–78. [DOI] [PubMed] [Google Scholar]
- [70].Hovhannisyan Z, Weiss A, Martin A, Wiesner M, Tollefsen S, Yoshida K, Ciszewski C, Curran SA, Murray JA, David CS, Sollid LM, Koning F, Teyton L, Jabri B, The role of HLA-DQ8 beta57 polymorphism in the anti-gluten T-cell response in coeliac disease, Nature 456(7221) (2008) 534–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Valesini G, Gerardi MC, Iannuccelli C, Pacucci VA, Pendolino M, Shoenfeld Y, Citrullination and autoimmunity, Autoimmun Rev 14(6) (2015) 490–7. [DOI] [PubMed] [Google Scholar]
- [72].Kampstra ASB, Toes REM, HLA class II and rheumatoid arthritis: the bumpy road of revelation, Immunogenetics 69(8–9) (2017) 597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].James EA, Moustakas AK, Bui J, Papadopoulos GK, Bondinas G, Buckner JH, Kwok WW, HLA-DR1001 presents “altered-self” peptides derived from joint-associated proteins by accepting citrulline in three of its binding pockets, Arthritis Rheum 62(10) (2010) 2909–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Hill JA, Southwood S, Sette A, Jevnikar AM, Bell DA, Cairns E, Cutting edge: the conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC class II molecule, J Immunol 171(2) (2003) 538–41. [DOI] [PubMed] [Google Scholar]
- [75].Kersh GJ, Miley MJ, Nelson CA, Grakoui A, Horvath S, Donermeyer DL, Kappler J, Allen PM, Fremont DH, Structural and functional consequences of altering a peptide MHC anchor residue, J Immunol 166(5) (2001) 3345–54. [DOI] [PubMed] [Google Scholar]
- [76].Ruppert J, Sidney J, Celis E, Kubo RT, Grey HM, Sette A, Prominent role of secondary anchor residues in peptide binding to HLA-A2.1 molecules, Cell 74(5) (1993) 929–37. [DOI] [PubMed] [Google Scholar]
- [77].Gianfrani C, Troncone R, Mugione P, Cosentini E, De Pascale M, Faruolo C, Senger S, Terrazzano G, Southwood S, Auricchio S, Sette A, Celiac disease association with CD8+ T cell responses: identification of a novel gliadin-derived HLA-A2-restricted epitope, J Immunol 170(5) (2003) 2719–26. [DOI] [PubMed] [Google Scholar]
- [78].Joseph MA, Mitchell ML, Evanseck JD, Kovacs JR, Jia L, Shen H, Meng WS, Secondary anchor substitutions in an HLA-A*0201-restricted T-cell epitope derived from Her-2/neu, Mol Immunol 44(4) (2007) 322–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Nicholls S, Piper KP, Mohammed F, Dafforn TR, Tenzer S, Salim M, Mahendra P, Craddock C, van Endert P, Schild H, Cobbold M, Engelhard VH, Moss PA, Willcox BE, Secondary anchor polymorphism in the HA-1 minor histocompatibility antigen critically affects MHC stability and TCR recognition, Proc Natl Acad Sci U S A 106(10) (2009) 3889–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].McMahon RM, Friis L, Siebold C, Friese MA, Fugger L, Jones EY, Structure of HLA-A*0301 in complex with a peptide of proteolipid protein: insights into the role of HLA-A alleles in susceptibility to multiple sclerosis, Acta Crystallogr D Biol Crystallogr 67(Pt 5) (2011) 447–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Lasso P, Cardenas C, Guzman F, Rosas F, Thomas MC, Lopez MC, Gonzalez JM, Cuellar A, Campanera JM, Luque FJ, Puerta CJ, Effect of secondary anchor amino acid substitutions on the immunogenic properties of an HLA-A*0201-restricted T cell epitope derived from the Trypanosoma cruzi KMP-11 protein, Peptides 78 (2016) 68–76. [DOI] [PubMed] [Google Scholar]
- [82].Deng L, Langley RJ, Brown PH, Xu G, Teng L, Wang Q, Gonzales MI, Callender GG, Nishimura MI, Topalian SL, Mariuzza RA, Structural basis for the recognition of mutant self by a tumor-specific, MHC class II-restricted T cell receptor, Nat Immunol 8(4) (2007) 398–408. [DOI] [PubMed] [Google Scholar]
- [83].Chen S, Li Y, Depontieu FR, McMiller TL, English AM, Shabanowitz J, Kos F, Sidney J, Sette A, Rosenberg SA, Hunt DF, Mariuzza RA, Topalian SL, Structure-based design of altered MHC class II-restricted peptide ligands with heterogeneous immunogenicity, J Immunol 191(10) (2013) 5097–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Stadinski BD, Zhang L, Crawford F, Marrack P, Eisenbarth GS, Kappler JW, Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register, Proc Natl Acad Sci U S A 107(24) (2010) 10978–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Mohan JF, Petzold SJ, Unanue ER, Register shifting of an insulin peptide-MHC complex allows diabetogenic T cells to escape thymic deletion, J Exp Med 208(12) (2011) 2375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Overwijk WW, Tsung A, Irvine KR, Parkhurst MR, Goletz TJ, Tsung K, Carroll MW, Liu C, Moss B, Rosenberg SA, Restifo NP, gp100/pmel 17 is a murine tumor rejection antigen: induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand, J Exp Med 188(2) (1998) 277–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Glithero A, Tormo J, Haurum JS, Arsequell G, Valencia G, Edwards J, Springer S, Townsend A, Pao YL, Wormald M, Dwek RA, Jones EY, Elliott T, Crystal structures of two H-2Db/glycopeptide complexes suggest a molecular basis for CTL cross-reactivity, Immunity 10(1) (1999) 63–74. [DOI] [PubMed] [Google Scholar]
- [88].Zaremba S, Barzaga E, Zhu M, Soares N, Tsang KY, Schlom J, Identification of an enhancer agonist cytotoxic T lymphocyte peptide from human carcinoembryonic antigen, Cancer Res 57(20) (1997) 4570–7. [PubMed] [Google Scholar]
- [89].Tsang KY, Palena C, Gulley J, Arlen P, Schlom J, A human cytotoxic T-lymphocyte epitope and its agonist epitope from the nonvariable number of tandem repeat sequence of MUC-1, Clin Cancer Res 10(6) (2004) 2139–49. [DOI] [PubMed] [Google Scholar]
- [90].Heery CR, Ibrahim NK, Arlen PM, Mohebtash M, Murray JL, Koenig K, Madan RA, McMahon S, Marte JL, Steinberg SM, Donahue RN, Grenga I, Jochems C, Farsaci B, Folio LR, Schlom J, Gulley JL, Docetaxel Alone or in Combination With a Therapeutic Cancer Vaccine (PANVAC) in Patients With Metastatic Breast Cancer: A Randomized Clinical Trial, JAMA Oncol 1(8) (2015) 1087–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Hou Y, Kavanagh B, Fong L, Distinct CD8+ T cell repertoires primed with agonist and native peptides derived from a tumor-associated antigen, J Immunol 180(3) (2008) 1526–34. [DOI] [PubMed] [Google Scholar]
- [92].Huang AY, Gulden PH, Woods AS, Thomas MC, Tong CD, Wang W, Engelhard VH, Pasternack G, Cotter R, Hunt D, Pardoll DM, Jaffee EM, The immunodominant major histocompatibility complex class I-restricted antigen of a murine colon tumor derives from an endogenous retroviral gene product, Proc Natl Acad Sci U S A 93(18) (1996) 9730–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Slansky JE, Rattis FM, Boyd LF, Fahmy T, Jaffee EM, Schneck JP, Margulies DH, Pardoll DM, Enhanced antigen-specific antitumor immunity with altered peptide ligands that stabilize the MHC-peptide-TCR complex, Immunity 13(4) (2000) 529–38. [DOI] [PubMed] [Google Scholar]
- [94].Jordan KR, McMahan RH, Oh JZ, Pipeling MR, Pardoll DM, Kedl RM, Kappler JW, Slansky JE, Baculovirus-infected insect cells expressing peptide-MHC complexes elicit protective antitumor immunity, J Immunol 180(1) (2008) 188–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Jordan KR, McMahan RH, Kemmler CB, Kappler JW, Slansky JE, Peptide vaccines prevent tumor growth by activating T cells that respond to native tumor antigens, Proc Natl Acad Sci U S A 107(10) (2010) 4652–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Stone JD, Chervin AS, Kranz DM, T-cell receptor binding affinities and kinetics: impact on T-cell activity and specificity, Immunology 126(2) (2009) 165–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Zhang SQ, Parker P, Ma KY, He C, Shi Q, Cui Z, Williams CM, Wendel BS, Meriwether AI, Salazar MA, Jiang N, Direct measurement of T cell receptor affinity and sequence from naive antiviral T cells, Sci Transl Med 8(341) (2016) 341ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Hebeisen M, Allard M, Gannon PO, Schmidt J, Speiser DE, Rufer N, Identifying Individual T Cell Receptors of Optimal Avidity for Tumor Antigens, Front Immunol 6 (2015) 582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Schodin BA, Tsomides TJ, Kranz DM, Correlation between the number of T cell receptors required for T cell activation and TCR-ligand affinity, Immunity 5(2) (1996) 137–46. [DOI] [PubMed] [Google Scholar]
- [100].Pardoll DM, The blockade of immune checkpoints in cancer immunotherapy, Nat Rev Cancer 12(4) (2012) 252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Zhong S, Malecek K, Johnson LA, Yu Z, Vega-Saenz de Miera E, Darvishian F, McGary K, Huang K, Boyer J, Corse E, Shao Y, Rosenberg SA, Restifo NP, Osman I, Krogsgaard M, T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy, Proc Natl Acad Sci U S A 110(17) (2013) 6973–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Thomas R, Al-Khadairi G, Roelands J, Hendrickx W, Dermime S, Bedognetti D, Decock J, NY-ESO-1 Based Immunotherapy of Cancer: Current Perspectives, Front Immunol 9 (2018) 947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Webb AI, Dunstone MA, Chen W, Aguilar MI, Chen Q, Jackson H, Chang L, Kjer-Nielsen L, Beddoe T, McCluskey J, Rossjohn J, Purcell AW, Functional and structural characteristics of NY-ESO-1-related HLA A2-restricted epitopes and the design of a novel immunogenic analogue, J Biol Chem 279(22) (2004) 23438–46. [DOI] [PubMed] [Google Scholar]
- [104].Shang X, Wang L, Niu W, Meng G, Fu X, Ni B, Lin Z, Yang Z, Chen X, Wu Y, Rational optimization of tumor epitopes using in silico analysis-assisted substitution of TCR contact residues, Eur J Immunol 39(8) (2009) 2248–58. [DOI] [PubMed] [Google Scholar]
- [105].Jordan KR, Buhrman JD, Sprague J, Moore BL, Gao D, Kappler JW, Slansky JE, TCR hypervariable regions expressed by T cells that respond to effective tumor vaccines, Cancer Immunol Immunother 61(10) (2012) 1627–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Wei P, Jordan KR, Buhrman JD, Lei J, Deng H, Marrack P, Kappler JW, Slansky JE, Yin L, Structures reveal a strategy for converting weak self-peptides into superagonists for CD8 T cells in cancer, (2019). [DOI] [PMC free article] [PubMed]
- [107].Buhrman JD, Jordan KR, Munson DJ, Moore BL, Kappler JW, Slansky JE, Improving antigenic peptide vaccines for cancer immunotherapy using a dominant tumor-specific T cell receptor, J Biol Chem 288(46) (2013) 33213–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Walter M, Philotheou A, Bonnici F, Ziegler AG, Jimenez R, Group NBIS, No effect of the altered peptide ligand NBI-6024 on beta-cell residual function and insulin needs in newonset type 1 diabetes, Diabetes Care 32(11) (2009) 2036–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, Yu L, Wegmann DR, Hutton JC, Elliott JF, Eisenbarth GS, Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice, Nature 435(7039) (2005) 220–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Nakayama M, Beilke JN, Jasinski JM, Kobayashi M, Miao D, Li M, Coulombe MG, Liu E, Elliott JF, Gill RG, Eisenbarth GS, Priming and effector dependence on insulin B:9–23 peptide in NOD islet autoimmunity, J Clin Invest 117(7) (2007) 1835–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Crawford F, Stadinski B, Jin N, Michels A, Nakayama M, Pratt P, Marrack P, Eisenbarth G, Kappler JW, Specificity and detection of insulin-reactive CD4+ T cells in type 1 diabetes in the nonobese diabetic (NOD) mouse, Proc Natl Acad Sci U S A 108(40) (2011) 16729–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Michels AW, Landry LG, McDaniel KA, Yu L, Campbell-Thompson M, Kwok WW, Jones KL, Gottlieb PA, Kappler JW, Tang Q, Roep BO, Atkinson MA, Mathews CE, Nakayama M, Islet-Derived CD4 T Cells Targeting Proinsulin in Human Autoimmune Diabetes, Diabetes 66(3) (2017) 722–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Kappos L, Comi G, Panitch H, Oger J, Antel J, Conlon P, Steinman L, Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. The Altered Peptide Ligand in Relapsing MS Study Group, Nat Med 6(10) (2000) 1176–82. [DOI] [PubMed] [Google Scholar]
- [114].Crowe PD, Qin Y, Conlon PJ, Antel JP, NBI-5788, an altered MBP83–99 peptide, induces a T-helper 2-like immune response in multiple sclerosis patients, Ann Neurol 48(5) (2000) 758–65. [PubMed] [Google Scholar]
- [115].Steinman L, Antigen-specific therapy of multiple sclerosis: the long-sought magic bullet, Neurotherapeutics 4(4) (2007) 661–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Streeter HB, Rigden R, Martin KF, Scolding NJ, Wraith DC, Preclinical development and first-in-human study of ATX-MS-1467 for immunotherapy of MS, Neurol Neuroimmunol Neuroinflamm 2(3) (2015) e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Chataway J, Martin K, Barrell K, Sharrack B, Stolt P, Wraith DC, Group A-MS, Effects of ATX-MS-1467 immunotherapy over 16 weeks in relapsing multiple sclerosis, Neurology 90(11) (2018) e955–e962. [DOI] [PubMed] [Google Scholar]
- [118].De Souza ALS, Rudin S, Chang R, Mitchell K, Crandall T, Huang S, Choi JK, Okitsu SL, Graham DL, Tomkinson B, Dellovade T, ATX-MS-1467 Induces Long-Term Tolerance to Myelin Basic Protein in (DR2 x Ob1)F1 Mice by Induction of IL-10-Secreting iTregs, Neurol Ther 7(1) (2018) 103–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Goel G, King T, Daveson AJ, Andrews JM, Krishnarajah J, Krause R, Brown GJE, Fogel R, Barish CF, Epstein R, Kinney TP, Miner PB Jr., Tye-Din JA, Girardin A, Taavela J, Popp A, Sidney J, Maki M, Goldstein KE, Griffin PH, Wang S, Dzuris JL, Williams LJ, Sette A, Xavier RJ, Sollid LM, Jabri B, Anderson RP, Epitope-specific immunotherapy targeting CD4-positive T cells in coeliac disease: two randomised, double-blind, placebo-controlled phase 1 studies, Lancet Gastroenterol Hepatol 2(7) (2017) 479–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Australia C, ImmusanT Discontinues Phase 2 Clinical Trials for Nexvax2®, 2019. https://www.coeliac.org.au/news-stories/immusant-discontinues-phase-2-clinical-trials-for-nexvax2/.
- [121].Boggiano C, Moya R, Pinilla C, Bihl F, Brander C, Sidney J, Sette A, Blondelle SE, Discovery and characterization of highly immunogenic and broadly recognized mimics of the HIV-1 CTL epitope Gag77–85, Eur J Immunol 35(5) (2005) 1428–37. [DOI] [PubMed] [Google Scholar]
- [122].Crawford F, Jordan KR, Stadinski B, Wang Y, Huseby E, Marrack P, Slansky JE, Kappler JW, Use of baculovirus MHC/peptide display libraries to characterize T-cell receptor ligands, Immunol Rev 210 (2006) 156–70. [DOI] [PubMed] [Google Scholar]
- [123].Birnbaum ME, Mendoza JL, Sethi DK, Dong S, Glanville J, Dobbins J, Ozkan E, Davis MM, Wucherpfennig KW, Garcia KC, Deconstructing the peptide-MHC specificity of T cell recognition, Cell 157(5) (2014) 1073–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Hoppes R, Oostvogels R, Luimstra JJ, Wals K, Toebes M, Bies L, Ekkebus R, Rijal P, Celie PH, Huang JH, Emmelot ME, Spaapen RM, Lokhorst H, Schumacher TN, Mutis T, Rodenko B, Ovaa H, Altered peptide ligands revisited: vaccine design through chemically modified HLA-A2-restricted T cell epitopes, J Immunol 193(10) (2014) 4803–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Meister D, Taimoory SM, Trant JF, Unnatural amino acids improve affinity and modulateimmunogenicity: Developing peptides to treat MHCtype II autoimmune disorders, Pep Sci 111:e24058 (2018) 1–21. [Google Scholar]
- [126].Buhrman JD, Jordan KR, U’Ren L, Sprague J, Kemmler CB, Slansky JE, Augmenting antitumor T-cell responses to mimotope vaccination by boosting with native tumor antigens, Cancer Res 73(1) (2013) 74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Buhrman JD, Slansky JE, Mimotope vaccine efficacy gets a “boost” from native tumor antigens, Oncoimmunology 2(4) (2013) e23492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Haskins K, Pathogenic T-cell clones in autoimmune diabetes: more lessons from the NOD mouse, Adv Immunol 87 (2005) 123–62. [DOI] [PubMed] [Google Scholar]
- [129].Yoshida K, Martin T, Yamamoto K, Dobbs C, Munz C, Kamikawaji N, Nakano N, Rammensee HG, Sasazuki T, Haskins K, Kikutani H, Evidence for shared recognition of a peptide ligand by a diverse panel of non-obese diabetic mice-derived, islet-specific, diabetogenic T cell clones, Int Immunol 14(12) (2002) 1439–47. [DOI] [PubMed] [Google Scholar]
- [130].Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, Armstrong M, Powell RL, Reisdorph N, Kumar N, Elso CM, DeNicola M, Bottino R, Powers AC, Harlan DM, Kent SC, Mannering SI, Haskins K, Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion, Science 351(6274) (2016) 711–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Hahn M, Nicholson MJ, Pyrdol J, Wucherpfennig KW, Unconventional topology of self peptide-major histocompatibility complex binding by a human autoimmune T cell receptor, Nat Immunol 6(5) (2005) 490–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Maynard J, Petersson K, Wilson DH, Adams EJ, Blondelle SE, Boulanger MJ, Wilson DB, Garcia KC, Structure of an autoimmune T cell receptor complexed with class II peptide-MHC: insights into MHC bias and antigen specificity, Immunity 22(1) (2005) 81–92. [DOI] [PubMed] [Google Scholar]
- [133].Li Y, Huang Y, Lue J, Quandt JA, Martin R, Mariuzza RA, Structure of a human autoimmune TCR bound to a myelin basic protein self-peptide and a multiple sclerosis associated MHC class II molecule, EMBO J 24(17) (2005) 2968–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Wucherpfennig KW, Call MJ, Deng L, Mariuzza R, Structural alterations in peptide-MHC recognition by self-reactive T cell receptors, Curr Opin Immunol 21(6) (2009) 590–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Baker RL, Jamison BL, Wiles TA, Lindsay RS, Barbour G, Bradley B, Delong T, Friedman RS, Nakayama M, Haskins K, CD4 T Cells Reactive to Hybrid Insulin Peptides Are Indicators of Disease Activity in the NOD Mouse, Diabetes 67(9) (2018) 1836–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]