

Introduction
Heart failure (HF) and renal dysfunction are common coexisting problems in clinical practice. There are bi-directional interrelationships between the heart and kidney, and it has been stated that the kidney is the most important organ related in acute HF. Acute or chronic dysfunction of one organ induces acute or chronic dysfunction of the other, described as “cardio-renal syndrome’ (CRS) (1, 2). In addition, renal dysfunction in HF may lead to reduced diuretic efficiency, diuretic resistance, worsening of congestion followed by further deteriorated renal function, which becomes a vicious cycle. Renal dysfunction is also a strong independent predictor for short- and long-term outcomes in patients with acute HF (3–9). Traditionally, the causes of CRS have been attributed to renal hypo-perfusion resulting from low cardiac output and over diuresis. However, in the past decades, data have increasingly demonstrated more of a correlation between venous congestion and CRS, rather than low cardiac output, linking the failing right heart to CRS. Indeed, right heart failure (RHF) and CRS both have complex and intertwining pathophysiologies that may involve various organs and systems beyond isolated dysfunction of the heart and/or kidneys. This review focuses on pathophysiology intersections between RHF and cardio-centric phenotypes of CRS (“Type 1” and “Type 2”) as well as considerations of therapeutic management.
Definition and Classification of Cardio-Renal Syndrome
The Working Group of the National Heart, Lung, and Blood Institute in 2004 first described CRS as the result of interaction between the kidney and other circulatory compartments that increase circulating volume and symptoms of HF and disease progression(10). The most extreme progression of cardio-renal dysregulation leads to the term “cardio-renal syndrome”, and therapy for congestive symptoms is limited by decline in renal function. This physiologic concept is in stark contrast with classification schemes proposed by Ronco et al (1) and the Acute Dialysis Quality Initiative (ADQI)(2) that described the primary organ dysfunction (heart or kidney) and time course (acute or chronic), with an additional subtype for a systemic condition affecting both organs simultaneously (Table 1). Although useful to clarify more precisely the clinical presentation of CRS, the overlap between the Ronco/ADQI subtypes and frequent evolution of one subtype to another could be challenging and has limited such a classification scheme to guide therapeutic management. Furthermore, there are limited quantifications of cardiac or renal functional assessments to determine these subtypes, rendering them largely descriptive and academic in clinical applications.
Table 1.
Types of Cardio-renal syndrome (CRS)
| Type of CRS | Definition | Conditions |
|---|---|---|
| Type 1: Acute CRS | Acute worsening of heart function leading to kidney injury and/or dysfunction | Acute HF, Acute cardiogenic shock |
| Type 2: Chronic CRS | Chronic abnormalities in heart function leading to progressive and permanent CKD | Chronic HF |
| Type 3: Acute Renocardiac syndrome | AKI causing acute heart dysfunction | Acute glomerunephritis/ AKI cause acute HF, |
| Type 4: Chronic Renocardiac syndrome | CKD leading to chronic heart disease and CKD progression | CKD cause cardiac hypertrophy, decreased cardiac function |
| Type 5: Secondary CRS | Systemic diseases leading to heart and kidney damage/dysfunction | Diabetes, sepsis, septic shock |
CRS: Cardio Renal Syndrome, HF: Heart failure, AKI: Acute Kidney Injury, CKD: Chronic Kidney damage
Traditional Concept of Impaired Forward Perfusion in Cardio-Renal Syndrome
Traditionally, “right heart failure” is often characterized by the inability of the right ventricle (RV) to generate enough stroke volume, thereby resulting in systemic venous congestion, under filling of the left ventricle, and in the most advanced cases, cardiogenic shock. Acute RHF can occur because of abruptly increased RV afterload (pulmonary embolus, hypoxia, and acidemia) or decreased RV contractility (RV ischemia, myocarditis, post cardiotomy shock). Each condition represents a unique hemodynamic challenge for the RV. In addition, the failing right heart can be a downstream manifestation of a primary insult (e.g., pulmonary hypertension, tricuspid valve dysfunction due to pacemaker lead interference), even though the downstream disturbances of the cardio-renal interactions are likely similar.
Historically, impairment in forward flow (cardiac output) leading to decreased renal arterial perfusion and neurohormonal activation has been considered as a key events in CRS (11, 12). It is therefore logical that the RV as the primary contributor to adequate preload directly contributes to CRS. Original descriptions of CRS postulated that renal blood flow could be preserved until the cardiac index fell below 1.5 L/min/m2(12), which is often described as “pre-renal.” Arterial under filling, secondary to low cardiac output and increased peripheral vascular resistance, may activate the renin-angiotensin-aldosterone system (RAAS), sympathetic nervous system (SNS) and release of arginine vasopressin resulting in water and sodium retention and worsening HF(11).
Clinical Evidence of Systemic Venous Congestion and Impaired Renal Function
Over the past decade, there is increasing evidence that worsening renal function (WRF) in the setting of CRS may not be adequately explained solely due to arterial under filling (4, 13–16). The ADHERE registry (Acute Decompensated Heart Failure National Registry) observed the same incidence rate of renal dysfunction in AHF with reduced and preserved systolic function (17), indicating that impairment of forward flow is not likely the primary culprit in the large majority of patients experiencing CRS. Meanwhile, WRF following treatment occurs more often in the setting of HF with preserved ejection fraction than those with severely reduced ejection fraction (18). It is therefore important to recognize that the occurrence of WRF in the setting of impaired perfusion (cardiogenic or distributive shock leading to intravascular depletion that is often a detrimental consequence) can be significantly different from that in the setting of systemic venous congestion. The latter often leads to increased venous pressure and increases the backward pressure into the intra-abdominal organs that can be reversed with effective decongestion. Indeed, signs and symptoms of congestion such as the presence of elevated jugular venous pressure (JVP), orthopnea, ascites and edema were independently related to the reduced estimated glomerular filtration rate (GFR) and were associated with increased mortality(19). In patients with predominant RHF, systemic venous congestion assessed by inferior vena cava (IVC) diameter was an independent determinant of GFR (20), and reduction of IVC size after treatment was associated with improvement of GFR. This is supported by the study of non-invasive and invasive measurements of venous congestion that showed correlation between high central venous pressure (CVP) with baseline renal impairment(4, 15, 16, 21) and WRF in acute HF (14), and WRF less frequently in patients with CVP < 8 mmHg after intensive medical therapy(14). Moreover, high CVP was also an independent predictor for cardiac rehospitalization (22) and all-cause mortality (4, 15). Finally, in patients who had congestion confirmed by echocardiography, relief of venous congestion showed significant renal function improvement in HF with RV dysfunction (23). Table 2 is a summary of studies that showed an association between venous congestion and renal dysfunction. It is important to also recognize that WRF in the setting of aggressive diuresis can also lead to hemoconcentration that is not necessarily “worsening” and in fact can indicate an effective therapeutic response with effective relief of systemic venous congestion.
Table 2.
Summary of studies linking venous congestion and renal dysfunction in heart failure
| Author, year (Reference) | Objectives | Study population and study design | Results | Conclusion |
|---|---|---|---|---|
| Nohria et al, 2008(4) | To evaluate correlation between hemodynamic parameters using pulmonary artery catheter and renal function | Prospective RCT of 433 patient with AHF | RAP correlated with serum Cr | Renal dysfunction or WRF does not related to poor forward flow alone |
| Mullens et al, 2009(14) | To determine whether venous congestion, rather than low CO is primarily associated with worsening renal function inADHF | Prospective observational study of 145 ADHF treated with PAC guided | WRF associated with high CVP with 75% of patients with baseline CVP > 24 immHg developed WRF | Venous congestion is more important factor driving WRF |
| Damman et al, 2009(15) | To investigate the relationship between increased CVP, renal function and mortality | Retrospective study of 2557 CVD patients with RHC | CVP>6 mmHg showed steep declined in eGFR | Increased CVP is associated with impaired renal function |
| Damman et al, 2010(19) | To investigate the relationship between signs and symptoms of congestion, renal impairment and outcomes | Double-blind RCT of 2647 HF NYHAclasslll/VI | Signs and symptoms of congestion were independently related to low eGFR and increase in mortality | Signs and symptoms of congestion are associated with renal impairment and independent determinants of prognosis |
| Guglin et al, 2011(16) | To assess correlation of congestion and renal function | Retrospective study of 178 patients with HF and underwent RHC | Serum Cr correlated with CVP, PCWP and renal perfusion pressure but not with Cl or LVEF | Renal dysfunction correlated with venous congestion and low renal perfusion |
| Testani et al, 2010(23) | To assess diuresis in RV dysfunction and resulting in reduced venous congestion and improving in renal function | Prospective study of 141 patients with HF with congestion assessed by echocardiography | RV dysfunction had more frequent venous congestion and lower incidence of worsening renal function | Relief of venous congestion in RV dysfunction likely leads to improved renal function |
Pathophysiology of Right Heart Failure and Cardio-Renal Syndrome
As previously mentioned, the overarching pathophysiology of CRS in RHF is complex through a variety of mechanisms due to the complex interrelationship between the heart and kidney as shown in Figure 1. Systemic venous congestion develops in the context of RHF - often the final pathway of many cardiovascular diseases – can be found in either isolated RV failure or biventricular failure. The consequences of venous congestion on various organs (backward congestion) play a pivotal role in the pathophysiology of CRS and have both local and systemic effects. Local venous congestion effects the kidneys and splanchnic organs and leads to renal and splanchnic congestion. Also, venous congestion produces systemic vascular congestion. These lead to mechanical, biological and immune responses which contribute to CRS development.
Figure 1: Summary of pathophysiology of RHF and CRS.
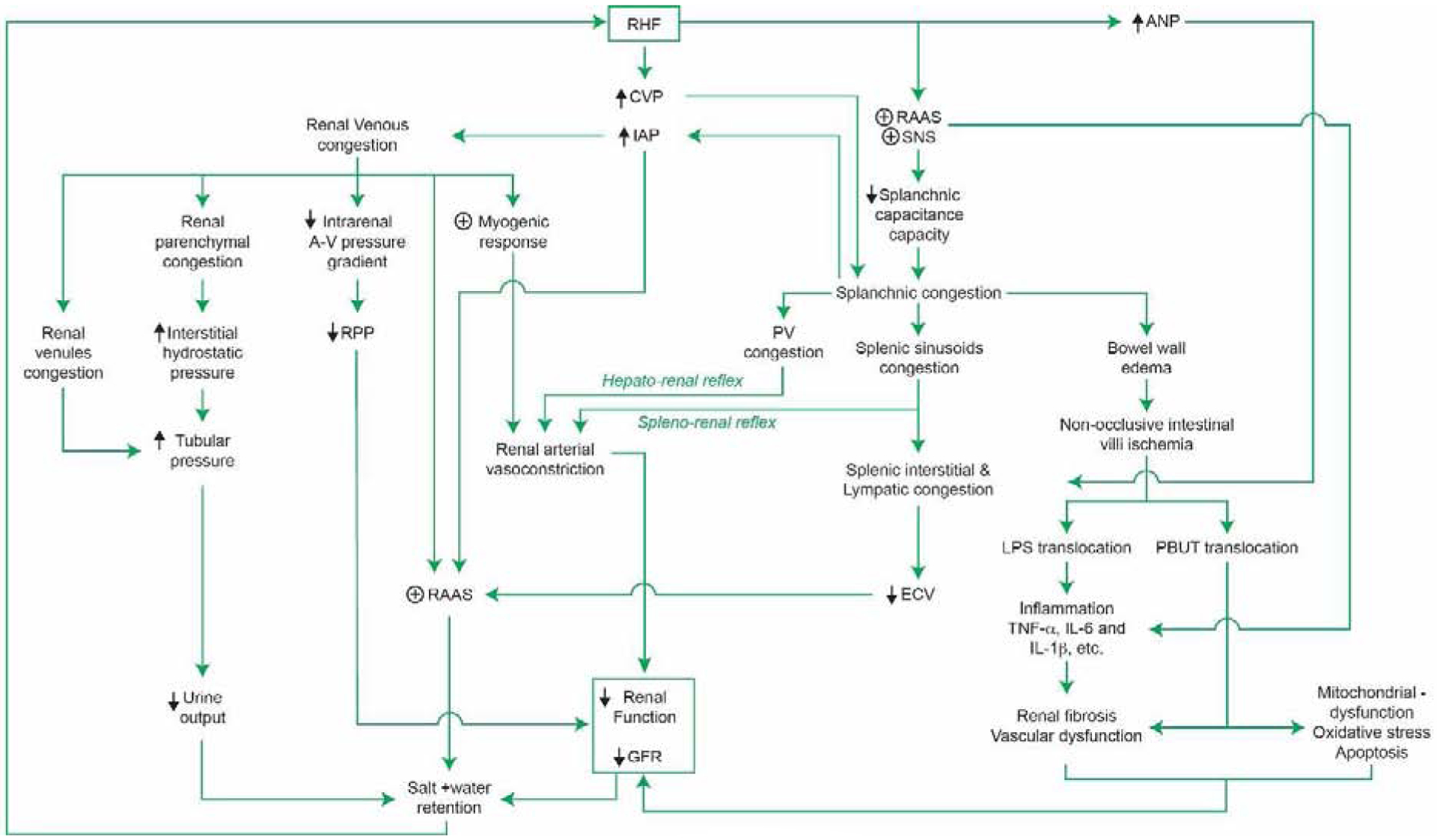
RHF: Right heart failure, CVP: Central venous pressure, IAP: Intra-abdominal pressure, ANP: Atrial natriuretic peptide, RAAS: Renin-angiotensin-aldosterone system, SNS: Sympathetic nervous system, A-V: Arterio-venous, RPP: Renal perfusion pressure, PV: Portal vein, ECV: Effective circulatory volume, LPS: Lipopolysaccharide, TNF- α: Tumor necrotic factor – α, IL-6: Interleukin-6, IL-1 β: Interleukin-1β, GFR: Glomerular filtration rate.
Several mechanisms have been postulated to explain the effect of venous congestion and renal dysfunction involving various organs, not only the heart and kidneys, which were originally called CRS. Elevated renal venous pressure obliterated renal tubules by distending renal venules (24), reduced renal perfusion pressure, increased renal interstitial pressure by fluid extravasation leading to a hypoxic state of renal parenchyma, tubular dysfunction. It also activated the RAAS (25–29) and SNS (30), activated vascular inflammation by endothelial cell dysfunction(31), and contributed to other abdominal organs (i.e. splanchnic venous and intestinal congestion) and lymphatic congestion (32). Clearly, these mechanisms are not mutually exclusive, and the large majority of them have been difficult to assess at the bedside. We will expand on these mechanisms further as they relate to the pathophysiology of RHF and CRS.
Renal Congestion
Effect of the pressure
Backward pressure from systemic venous into renal veins can generate increased renal venous pressure that can directly impair a wide range of kidney functions(33). Indeed, studies demonstrated that increased renal venous pressure showed a relationship with reduction in urine flow and alteration in glomerular and tubular function. Furthermore, renal blood flow decreased by increased venous pressure more than an equivalent decrease in arterial pressure(34). It was previously postulated that increased renal venous pressure leads to renal parenchymal congestion within the non-distensible renal capsule (so-called “renal tamponade”) resulting in increased interstitial pressure that affects the entire capillary bed and tubules (35, 36). Experimental models have demonstrated the effects of increased renal venous pressures on changes in filtration fraction, flow from baroreceptor, intrinsic vascular reflex response (myogenic response), tubuloglomerular feedback (TGF), RAAS and SNS (37).
Clinically, renal venous congestion secondary to increased systemic pressure, often is reflected by high CVP, may lead to decreased intra-renal arterio-venous gradient and, theoretically, decreased renal perfusion pressure (RPP). It is a gradient between aortic and renal venous pressure that is equal to mean arterial pressure (MAP) minus CVP. However, studies in acute HF patients showed incongruous results which demonstrated the similarity in renal perfusion pressure (estimated by MAP-CVP) in those with and without WRF during acute HF hospitalization(14). These observations may imply that mechanisms of WRF may be more directly related to venous congestion. Meanwhile, in animal experimental studies, an increased renal venous pressure retards urine flow equal to the decrease in arterial pressure (24, 38). Therefore, congested renal venules and increased interstitial hydrostatic pressure may compress renal tubules and obstruct the urine flow (24, 39). Moreover, increased renal interstitial pressure may decrease renal blood flow even in the presence of furosemide (to block tubuloglomerular feedback), renal decapsulation (local sympathetic inhibition) or systemic inhibition of SNS (using phentolamine)(40). In a human study, abdominal compression led to increased intra-abdominal and renal venous pressure and was associated with a fall in urine output(41). Regarding filtration function, stepwise increase in renal venous pressure particularly during volume expansion showed decrease of GFR(42–46) which is explained by increased renal venous pressure resulting in increased tubular pressure(43) which will oppose filtration and net ultrafiltration pressure(39). As early as 1913, the experimental study of chronic passive renal congestion in dogs created by selective banding of the unilateral renal vein showed effect on renal excretory function(47). Conversely, reduction in venous pressure demonstrated reversibility of impaired natriuresis/aquaresis and GFR (42, 45).
Neural reflex and Neurohormonal mechanism
Myogenic response is an autoregulatory mechanism in mammalian kidney which is the intrinsic capability of the renal vasculature, particularly in the small diameter vessels in response to an increase in wall tension which results in smooth muscle cell contraction by increased intracellular calcium and activation of myosin light chain kinase(48). Rise in renal venous pressure, by a partially obstructed renal vein resulted in vasoconstriction, has been studied in animal models and showed strong effect on arterial microcirculation by triggered sympathetic vasoconstrictive neural responses(49) that were both extrarenal and intrarenal mechanisms(50). Surgical or pharmacological sympathectomy partially prevent the effects of venous congestion(50). In addition, stimulation of mechanoreceptors in an intra-renal vein by venous pressure enhanced local sympathetic renal nerve activation, resulted in intra-renal arterial vasoconstriction and decreased GFR (51, 52).
Experimental studies have demonstrated both increase in intra-abdominal pressure and renal venous pressure affecting an increase in plasma renin activity and aldosterone level (53–55) even though they may not result in significant changes in hemodynamic parameters such as cardiac index or systemic blood pressure(46). An animal study as early as 1949 showed elevated renal venous pressure can significantly diminish sodium and water excretion, but with modest elevations in renal venous pressure and slight changes in renal plasma flow, GFR, and filtration fraction(45). These effects of congestion impinging on renal sodium excretion lead to a vicious cycle of salt and water retention and more renal congestion(56).
Splanchnic Congestion
Studies of the interaction between splanchnic congestion and CRS have demonstrated that they might be a strong contributor to development of renal dysfunction in HF. Indeed, the splanchnic veins have a function as a blood reservoir and actively function in regulation for cardiac preload during changes in volume status (57) which is regulated by passive (transmural pressure changes) or active mechanisms (SNS regulation) (57–59). Furthermore, splanchnic veins have high capability to pool additional blood volume which contributed up to 66% of total additional volume(58). However, maladaptation of splanchnic capacitance function in HF has been demonstrated (60) which raised speculation that incremental capacity of splanchnic capacitance is limited, and thereby could be the result of long standing venous congestion and neurohormonal activation in advanced HF(61) and might be a significant factor in CRS development.
Liver and spleen are crucial visceral organs in the splanchnic circulation and contain approximately a quarter of total blood volume in the human body (58, 62). Studies in splanchnic compartment and CRS showed correlation between major visceral organs and kidney by local reflex systems. First, hepatorenal reflex, which is regulated by receptors in intrahepatic circulation, which result in the kidney being neurally-mediated (63, 64). This occurs through (i) chronic splanchnic congestion resulting in portal vein distension and stretch of the venous wall which leads to renal vasoconstriction(65) and (ii) increased intrahepatic adenosine by portal vasoconstriction activated by SNS mediated α-adrenergic receptor and release to circulation. This results in renal vasoconstriction and sodium retention by activation TGF(66, 67). Second, splenorenal reflex can lead to increased intrasplenic venous pressure and neutrally-mediated renal vasoconstriction (68, 69). Conversely, interruption of either afferent or efferent reflex pathway resultsin elimination of both hepato- and spleno-renal reflexes (64, 69). Furthermore, splenic congestion leads to interstitial edema by intrasplenic fluid extravasation from splenic sinusoids, which are freely permeable for plasma protein. Thus intravascular hydrostatic pressure becomes a main determinant for fluid transport. Fluid extravasation from splenic sinusoids to lymphatic circulation and interstitial tissue, respectively, lead to perception of decreased effective circulatory volume and exacerbate neurohormonal activation, creating a vicious cycle of sodium and water retention(32). Atrial natriuretic peptide (ANP) might be another contributor to promote reduction in central plasma volume. Infusion of ANP in rats resulted in hemoconcentration and reduction in plasma volume which are not entirely accounted for by excretion of urine(70). This effect was explained as ANP having effects on splanchnic hemodynamics including increased intrasplenic pressure and increased intrasplenic fluid extravasation(71).
Dysfunction of intestinal endothelial cells secondary to bowel edema due to backward congestion and consequences in alteration of gut microbiota have been demonstrated and speculated to be a contributor to development of CRS in RHF. Intestinal villi have a unique microcirculation, and the tips of intestinal villi are the most susceptible to anoxic damage (72). Splanchnic congestion, co-existing with low perfusion and sympathetic activation, leads to increased risk of non-occlusive ischemia of intestinal villi(73). In addition, increased intestinal wall thickness and edema have been observed in patients with HF and have been shown to have a direct correlation with increased paracellular intestinal permeability (74) which is secondary to hypoxia, hypoperfusion and endotoxin production by intestinal gram-negative bacteria (74, 75). These structural and functional alterations of gut barrier result in translocation of gut microbiota and their components into host circulation including lipopolysaccharides (LPS) and protein-bound uremic toxins produced by gut microbiota which contribute to renal dysfunction (76–78). LPS, found in the outer membrane of gram-negative bacteria, further promote mucosal barrier function deterioration and systemic inflammatory processes(79). LPS and pro-inflammatory cytokines levels were found to be higher in edematous chronic HF than non-edematous and healthy volunteers. Also, reduction could be demonstrated after diuretic treatment (80). Inflammation activation is further discussed in the section on Inflammation and CRS.
Although, evaluation of splanchnic congestion is not clinically available unless there are detectable ascites, increased intra-abdominal pressure (IAP) might be used and reflects splanchnic congestion. As reported in the study, increased IAP (≥ 8 mmHg) was found in 60% of patients admitted with advanced HF(81), and ascites were found in a small number which suggested splanchnic congestion could contribute to increased IAP(32). Furthermore, increased IAP was associated with impaired renal function, and changes in IAP were strong predictors of change in renal function compared to hemodynamic parameters (41, 81). IAP may contribute to renal dysfunction by increased renal parenchyma, renal venous and intraglomerular pressures and decreased renal perfusion reflected by reduced GFR (41, 46, 55, 81–83). Moreover, increased IAP was also associated with RAAS activation, which showed increased plasma renin activity and aldosterone level(55). Reduction in IAP by ultrafiltration or paracentesis has corresponded with improvement in renal function in some patients (84).
Inflammation and Cardio-Renal Syndrome
There are increasing data that inflammation could be related to pathogenesis of CRS. Production of pro-inflammatory cytokines as a consequence of HF could be from neurohormonal activation, venous congestion including either local congestion, such as splanchnic congestion and intra-renal venous congestion, or systemic venous congestion. Elevated cytokines in HF have been demonstrated, such as tumor necrotic factor-α (TNF-α), interleukin-6 (IL-6) and interleukin-1 (IL-1) (85, 86) and correlate with poor clinical outcomes (87–89). These cytokines have direct biological effects to both structural and functional damage to various end-organs including the heart, vasculature and kidney(90). Studies have shown that inflammation results in depressed cardiac function, vascular dysfunction, renal fibrosis and progressive renal dysfunction (91–97). Finally, inflammation may increase vascular permeability and promote absorption of pro-inflammatory endotoxins from the bowel(90). Hence, a vicious cycle develops leading to increased congestion due to end-organ damage and more inflammation activation.
Inflammation consequences of neurohormonal activation
Increased activity of RAAS and SNS in HF leading to chronic inflammation have been demonstrated. Angiotensin II (AII) increases TNF-α biosynthesis in myocardium which is mediated through the angiotensin 1 receptor (AT1R) (98). Similarly, in animal studies, AII treated rats showed increased renal tissue expression of TNF-α (glomeruli, mainly at endothelium of tubules and vasculature), activated nuclear factor-kappa B (NF-kappa B) and systemic infusion of AII induced renal synthesis of IL-6, monocyte chemoattractant protein-1 (MCP-1) co-existing with glomerular and interstitial inflammatory cells in kidney(99, 100). Chronic blockage of AT1R supports the role of AII inflammatory responses which showed reduction in circulating pro-inflammatory cytokines such as TNF-α (101). SNS also promoted inflammatory responses which was demonstrated in the animal study when isoproterenol infusion increased expression of TNF-α, IL-6 and IL-1β in myocardium cells and cardiac blood vessels(102) and beta-adrenergic blocker administration decreases those effects(102).
Inflammatory Consequences of Venous Congestion
Venous and tissue congestion may promote inflammatory responses by various mechanisms(90). Mesenteric venous congestion leads to bowel wall edema and increased vascular permeability, gram-negative bacterial translocation through the endothelial cells of the intestinal villi, and thereby endotoxin release has been proposed(103). Endotoxins, such as LPS, which is elevated in HF(104), promotes the secretion of pro-inflammatory cytokines through the effect on human monocyte and macrophage function such as TNF-α, the IL-1 family, IL-6, IL-8, the IL-10 family, the IL-12 family, IL-15 and transforming growth factor beta (TGF-β)(105). LPS and cytokine levels have shown to be increased in edematous chronic HF (80). According to a cohort study in chronic HF, more elevated LPS levels in patients with peripheral edema were demonstrated, and these levels showed a reduction after acute diuretic treatment (80). In addition, supporting data showed higher endotoxin levels in the hepatic vein compared to the left ventricle in acute HF which suggests bacterial or endotoxin translocation from bowel to the blood stream(106).
Beside bacterial and endotoxin translocation, hemodynamic stimulation by intravascular volume expansion can promote vascular inflammation and endothelial cell activation through cytokine secretion itself (31, 90) and alter other bioactive molecules such as nitric oxide (NO) and prostacyclin function(107). Regarding inflammation, both in vitro and in vivo studies have shown evidence of activation of vascular endothelial cells and increased production of a variety of vasoactive meditators, including inflammatory cytokines after biomechanical stress including TNF-α, IL-6 (108–110). A specific study of fluid load in normal dogs created venous congestion accompanied by vascular endothelial activation of inflammation and oxidative stress (111). Moreover, in a human study, peripheral congestion was created through applied pressure using a tourniquet on the arms of healthy subjects which caused release of inflammatory markers, IL-6 and endothelin-1 (ET-1)(112).
Hence, evidence of systemic inflammation in HF has been increasing and is postulated to contribute to CRS development which could be secondary to neurohormonal and sympathetic activation, and vascular and tissue congestion. Inflammation leads to end-organ damage, causing progressive fluid accumulation thereby further inflammatory activation occurs.
Effects of Right Ventricular Volume Overload
Right ventricular dysfunction and dilation in RHF secondary to increased RV filling pressure leads to ventricular interdependence which result in leftward shift of interventricular septum and altered left ventricle (LV) geometry(113). Hence, reducing LV distensibility, preload, reducing cardiac output and thereby reducing renal arterial pressure which might be another contributing factor in CRS(15, 113, 114). This is often seen in isolated RHF as in advanced pulmonary arterial hypertension.
Novel Diagnostic Strategies for Congestion and Cardio-Renal Syndrome
Serum and Urine Biomarkers
Biomarkers provide a wide spectrum of prevention, early diagnosis, treatment and outcomes of organ injury including in the heart and kidney (115). There are various biomarkers for renal and cardiac injury which provide different roles in diagnosis and prognosis in acute kidney injury (AKI), HF and CRS (116, 117). B-type natriuretic peptide (BNP), a cardiac biomarker, is a marker of myocardial stretch, has both diagnostic and prognostic roles in HF and CRS(116). In HF with impaired renal function, including CRS, there are higher BNP levels compared with patients with normal renal function which could be attributed to impaired renal excretion, volume overload by impaired renal function and cardiomyopathy associated with renal dysfunction(118–120). In CRS, there is elevation of other cardiac biomarkers such as cardiac troponin (120, 121) and galactin-3(122). High levels of glactin-3 and cardiac troponin were associated with higher mortality in HF (121, 122).
Beside cardiac biomarkers which are directly associated with congestion, renal biomarkers also have value for both diagnosis and prognosis in CRS. Neutrophil gelatinase-associated lipocalin (NGAL) is a large lysosomal enzyme originating in the proximal tubular cell; detection of urine NGAL indicates proximal tubular injury (123). Elevated NGAL was observed in HF with renal dysfunction(120), and elevated serum and urine levels of NGAL could be a useful predictor for dialysis and death in AKI including CRS(124). Serial measurement of NGAL in AHF is an accurate predictor of WRF (125), although the majority of patients with CRS have relatively low urine NGAL levels as significant intrinsic kidney injuries are relatively uncommon. AKI biomarkers such as NGAL are not readily available for clinical use.
Cystatin C (CysC) is a renal biomarker present in all nucleated cells with a constant production rate. It is freely filtrated, completely reabsorbed and not secreted by renal tubules(117). Cystatin C is dramatically better than creatinine for measuring GFR, because CysC is not primarily determined by muscle mass (126). The studied use of CysC calculation for GFR and reclassified CKD stage showed stronger correlation and more linear correlation to death along with all eGFR (127). Combined CysC and others biomarkers, such as N-terminal pro-BNP (NT pro-BNP) and cardiac troponin T, had additive prognostic value for adverse events in AHF (128). Albuminuria in HF without concomitant comorbidity such as renal dysfunction, hypertension and diabetes might provide greater diagnostic power for CRS than by using eGFR alone (129, 130). Albuminuria also had a prognostic value in HF and is associated with increased mortality and increased admissions for HF independent of eGFR, diabetes, and hypertension (129, 131).
It is important to recognize that few cardiac or renal biomarkers are specific to RHF or CRS. Biomarker-guided strategies have also failed to uniformly provide incremental treatment benefits over standard of care, largely because the above-mentioned biomarkers have not distinguished RHF as a unique contributor to CRS, and the majority of biomarkers have been developed with diagnosing a clinical condition in mind (e.g., AKI or HF).
Renal Ultrasonography
Renal vein flow pattern using Doppler ultrasound is a non-invasive tool which has been studied in several situations including HF with renal venous congestion (132–137). In HF, intrarenal venous flow patterns depend on right atrial pressure (RAP) and are strongly correlated with clinical outcomes (136). Patterns of renal venous vein flow were stratified by RAP. Continuous renal vein flow patterns were correlated with normal RAP (RAP <8 mmHg). A discontinuous renal flow pattern was associated with increased RAP, and a monophasic pattern had the highest RAP and poorest prognosis (<40% survival at 1 year) (136). The discontinuous pattern is explained by transmission of backward pressure from increased RAP to the renal vein which results in increased pulsatility of flow pattern and reflects the response of renal vessels in increased intrarenal pressure within the encapsulated capsule(132) (Figure 2). Moreover, renal flow pattern, rather than renal resistive index (RI), which is calculated from renal arterial waveform, was shown to have incremental prognostic value (136) that could reflect the more important role of renal venous congestion rather than renal hypoperfusion. However, reversibility of renal flow pattern has not been demonstrated by any current therapeutic options; hence, we lack data to support the use of renal venous flow pattern to make the decision for decongestive therapeutic strategies. In addition, this tool requires expertise to perform, technical feasibility and needs validation for consistency of Doppler waveform sampling by operators and in a diverse group of patients.
Figure 2. Ultrasound Profiles Across the Spectrum of Elevated Right Atrial Pressure As Indication for Cardio-Renal Syndrome in Right Heart Failure.
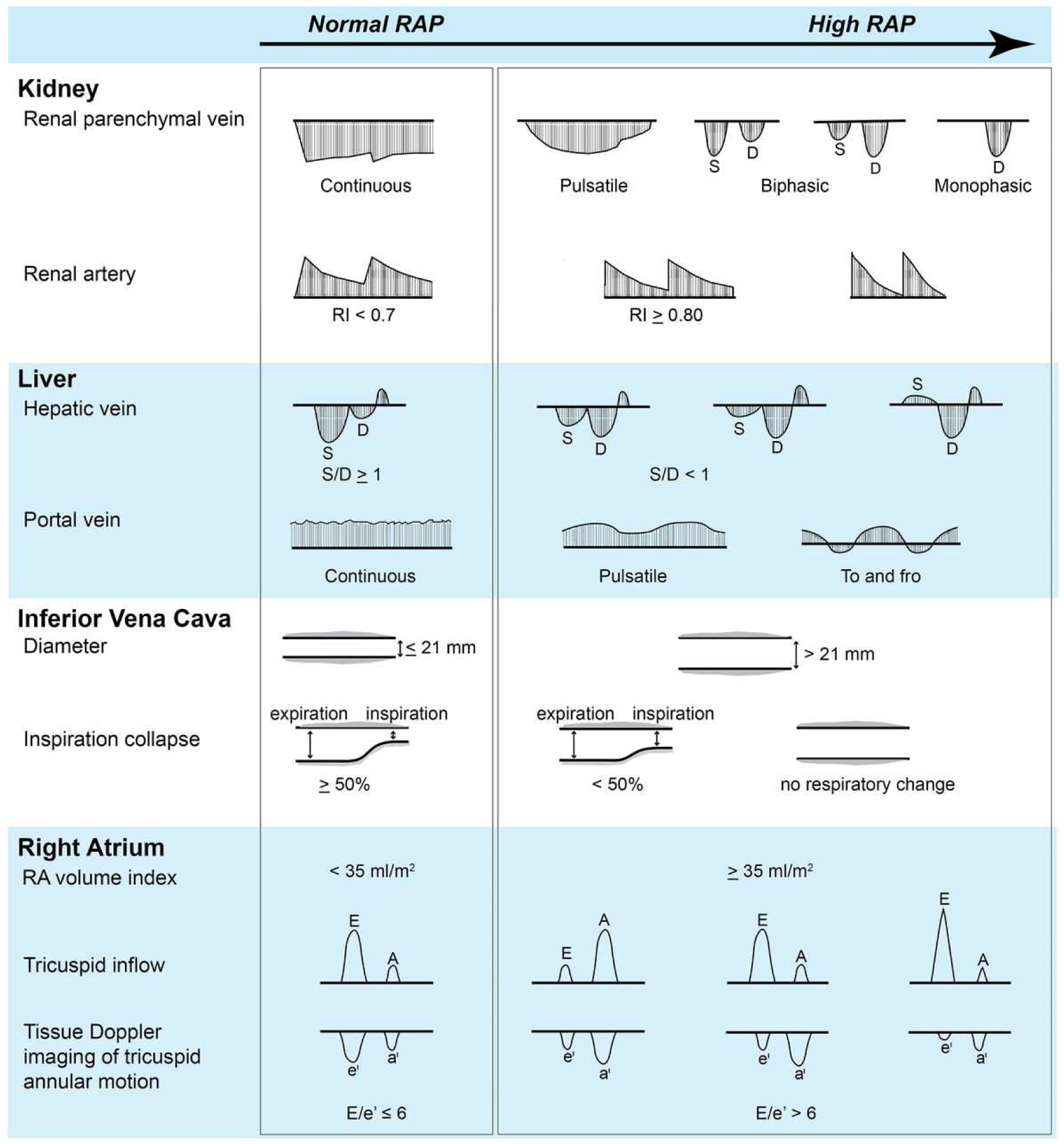
Abbreviation: RAP, right atrial pressure. (From Tang WH, Kitai T. Intrarenal Venous Flow: A Window Into the Congestive Kidney Failure Phenotype of Heart Failure? JACC Heart Fail. 2016;4(8):683–6; with permission.)
Intra-abdominal Pressure via Indwelling Urine Catheter
Splanchnic congestion secondary to backward pressure in RHF can be evaluated by measure of IAP as mentioned before. Measurements of IAP could be obtained by measuring intra-bladder pressure using the intra-bladder catheter connected to a transducer. Increased IAP is defined as elevated pressure greater than normal range of 5–7 mmHg (81).
Medical Treatment Options for Right Heart Failure and Cardio-Renal Syndrome
Decongestion is the cornerstone in CRS treatment to reduce systemic venous congestion and return balance in hemodynamic, neurohormonal and biological activation. Hence, improvement of renal perfusion pressure by reducing CVP, renal venous pressure and RV volume overload thereby improve RV and LV performance(138). Decongestion strategies include diuretics, ultrafiltration and dialysis. Oral diuretic is the first line strategy and has been used for years with natriuretic effect and volume reduction leading to immediate relief of HF symptoms(139). Although, the most challenging of decongestion in CRS is diuretic resistance due to several mechanisms, such as impaired renal function, reduced cardiac output leading to low delivery of diuretic to the site of action (kidney), inadequate dose of diuretic or inadequate substrate (sodium and chloride) at the renal tubules(138, 140).
Loop diuretics
Loop diuretics inhibit Na+-K+−2Cl− co-transporter at the thick ascending limb of the loop of Henle and are the most common diuretics used in clinical practice since they have a short peak of action (10–30 minutes and 1–1.5 hours for intravenous and oral administration, respectively)(141). Loop diuretics cause natriuresis, thereby causing net negative water and salt balance and reduced volume overload. The most commonly used loop diuretics are furosemide, torsemide and bumetanide. Torsemide has greater and more consistent oral bioavailability (90%) than furosemide (10–90%)(141–143). Although, the oral bioavailability of furosemide can be improved when taken before a meal, since furosemide has been shown to have a 30% reduction in oral bioavailability with food(144). Intravenous or novel subcutaneous furosemide ensure 100% bioavailability (145, 146). However, torsemide, with a longer half-life, leads to less frequent doses compared to furosemide (142). Hence, torsemide is suggested as a more effective and well tolerated diuretic in CHF compared to furosemide in several studies including a meta-analysis (147–149). Moreover, loop diuretics are protein-bound anions; >90% bound to plasma proteins and are secreted in the proximal tubules to reach their site of action(150). The ability of the diuretic to be protein-bound can have competition from exogenous anions such as non-steroidal anti-inflammatory drugs (NSAIDs) and endogenous anions such as bile acid and uremic toxin (116, 138) resulting in reduced diuretic efficacy. In addition, hypoalbuminemia, which is common in advanced HF, could contribute to decreased loop diuretic transportation to the site of action(138).
Indeed, dose-response to diuretic curve in HF shifts downward and to the right; therefore, a higher dose of diuretic is needed to achieve the same therapeutic effect in HF(151). A single dose of furosemide elicits transient natriuresis(152). Hence, increased frequency or continuous use of a loop diuretic could be considered. However, continuous dosing of loop diuretics showed no difference in symptom relief or change in renal function in The Diuretic Optimization Strategies Evaluation in Acute HF (DOSE-AHF) trial compared with a bolus strategy, while high doses were associated with greater diuresis, weight loss, and transient WRF(153). A bolus dose of loop diuretic also showed no difference in mortality compared with continuous infusion. However, continuous infusion is associated with more hyponatremia, need for vasopressors, rehospitalization and death at 6 months (154). Starting with an intravenous diuretic dose 2.5 times the daily oral equivalent diuretic dose is reasonable as advised in the DOSE-AHF trial(138). Upon transition to oral therapy, the dosing frequency should depend on medication half-life which is every 4–6 hour for furosemide and bumetanide and every 8–12 hours for torsemide(155).
A stepwise pharmacologic strategy has been proposed and studied in post hoc analysis of 3 randomized controlled trials in acute HF with CRS, including DOSE-AHF(153), the Cardiorenal Rescue Study in Acute Decompensated Heart Failure (CARRESS-HF)(156) and Renal Optimization Strategies Evaluation in AHF (ROSE-AHF) trial(157). These studies showed superiority to standard decongestive therapy (including non-adjusted diuretic dose) without WRF and superiority to ultrafiltration in preservation of renal function at 96 hours (158). The stepwise diuretic regimen is shown in Table 3. In the setting of significant venous congestion as a result of RHF leading to progressive impedance of venous return from the kidneys, effective decongestion may improve renal perfusion and thereby increase diuresis and natriuresis. On the other hand, excessive diuresis without adequate right heart reserve can reduce preload and impair cardiac output, leading to relative intravascular hypovolemia and decreased diuresis and natriuresis. Regardless, the rise in serum creatinine may or may not be indicative of intrinsic injury or damage to the kidneys; and the ultimate delicate balance can be impacted by the status of the right heart.
Table 3.
Stepped diuretic strategy: Treatment algorithm from CARRESS-HF
| Daily UO assessment | ||
| UO > 5 L/day : reduce current diuretic regimen if desired | ||
| UO3–5 L/day : continue current diuretic regimen | ||
| UO < 3 L/ day : see diuretic table | ||
| At 24 hours assessment | ||
| If persistent volume overload | ||
| Assessed daily UO as above | ||
| Advance to the next step on diuretic table if UO < 3 L/ day | ||
| At 48 hours assessment | ||
| If persistent volume overload | ||
| Assessed daily UO as above | ||
| Advance to the next step on diuretic table if UO < 3 L/ day and consider | ||
| : Dobutamine or dopamine at 2 μg/kg/min if SBP < 100 mmHg and LVEF < 40% or | ||
| RV systolic dysfunction | ||
| : Nitroglycerin or nesiritide if SBP > 120 mmHg and severe symptoms | ||
| At 72–96 hours assessment | ||
| If persistent volume overload | ||
| Assessed daily UO as above | ||
| Advance to the next step on diuretic table if UO < 3L/ day and consider | ||
| : Dobutamine or dopamine at 2 μg/kg/min if SBP < 100 mmHg and LVEF < 40% or | ||
| RV systolic dysfunction | ||
| : Nitroglycerin or nesiritide if SBP > 120 mmHg and severe symptoms | ||
| - Hemodynamic guided IV-therapy | ||
| - LVAD | ||
| - UF or dialysis | ||
| Diuretic table | ||
| Current loop diuretic | Suggested dose | |
| dose | Loop diuretic dose | Thiazide dose |
| ± thiazide | ||
| A. ≤80 mg | 40 mg IV bolus + 5 mg/hr | 0 |
| B. 81–160 mg | 80 mg Iv bolus + 10 mg/hr | 5 mg metolazone QD |
| C. 161–240 mg | 80 mg Iv bolus + 20 mg/hr | 5 mg metolazone BID |
| D. ≥240 mg | 80 mg Iv bolus + 30 mg/hr | 5 mg metolazone BID |
CARRESS-HF: Cardiorenal Rescue Study in Acute Decompensated Heart Failure, UO: Urine output, LVEF: Left ventricular ejection fraction, RV: Right ventricle, SBP: Systolic blood pressure, LVAD: Left ventricular assist device, UF: Ultrafiltration (From https://biolincc.nhlbi.nih.gov/media/studies/carress/Protocol.pdf?link_time=2020-01-15_23:30:43.304569.)
Diuretic resistance
To date, the definition of diuretic resistance is not clear and no standard definition is available. However, it has been defined as diminished or loss of diuretic response before the therapeutic goal of relief from edema has been achieved(159). There are various metrics to measure either diuretic efficacy or resistance, including weight loss, net fluid loss, urine output per 40 mg of intravenous furosemide-equivalent doses and natriuresis(155). These measures account for loop diuretics, the most commonly used diuretics in volume overload, but do not account for other diuretics(155). Moreover, there is no definite cut-off for those metrics for either diuretic resistance or efficacy. However, diuretic efficacy in HF has been shown as a strong predictor for mortality and morbidity including all-cause death, HF readmission and renal related readmission after correction with baseline eGFR(140, 160–165). This was attributed to GFR and diuretic efficacy represented in a different part of kidney function; while GFR is the glomerular function, diuretic efficacy is the tubular function(155).
Braking phenomena, diminished diuretic-induced natriuresis by hemodynamic and neurohormonal responses, have all been implied as contributors to diuretic resistance(138). Hemodynamic braking develops when diuretic reduces extracellular fluid volume thereby causing SNS and RAAS activation which increases sodium reabsorption at proximal tubules (166). Neurohormonal braking develops when diuretic increases urine sodium and activates TGF causing renin production thereby causing afferent arteriolar vasoconstriction which indirectly reduces sodium filtration(167). Nephron remodeling (distal tubular hypertrophy and hyperplasia) as a consequence of prolonged use of loop diuretic is also considered a determinant of diuretic efficacy (168, 169). In addition, furosemide-treatment in HF has demonstrated enhanced distal sodium transport more than proximal which could attenuate the loop diuretic response (170). Hence, addition of non-loop diuretics (i.e. thiazide or potassium sparing diuretic), which is termed “segmental nephron blockage,” may be reasonable and also might overcome the braking phenomenon and nephron remodeling thereby augmenting natriuresis(171) without compromising GFR(156). However, the evidence of this approach in CRS is still lacking, let alone in the setting of RHF. Moreover, renal dysfunction in the context of CRS reduces excretion of the diuretic into the tubular lumen and natriuresis in CKD is reduced by decreased sodium filtration (172, 173).
Other medical therapies
Vasoactive and inotropic drugs have been used extensively in clinical practice for significant RHF, although clinical trial evidence has been lacking (especially regarding preference of milrinone over other vasoactive drugs). Much of the literature is based upon cardio-centric optimization of hemodynamic parameters in advanced HF patients. In the acute setting where pulmonary hypertension is a major contributor, selective pulmonary vasodilators have been used with success (e.g., inhaled nitric oxide, prostacyclin and iloprost), although there is limited data to support the role of phosphodiesterase type 5 inhibitors for this indication.
Summary/Conclusion
The heart and kidney have complex bidirectional interlinks termed CRS type 1 and 2 that represent the renal dysfunction secondary to acute and chronic heart problems, respectively. Contemporary data have shown more correlation of venous congestion and renal dysfunction in HF which represents the significant influence from the right heart. Prevention of CRS should be the most important goal. This requires understanding of the pathophysiology of CRS which involves several interfaces and is not just limited to heart and kidney. Splanchnic organs play an important role in this such as pressure effect of venous congestion, inflammatory responses and neurohormonal activation. Decongestion is still the mainstay strategy in HF with CRS and is clinically challenging. These could be caused by reduced diuretic efficiency and diuretic resistance. However, dedicated diuretic strategy in CRS or diuretic resistance remain unclear and further randomized trials are needed. In addition, clinical assessment of extracellular fluid status remains important to keep the balance between hypervolemia and dehydration.
Synopsis.
Cardio-renal syndrome is a complex interplay of dysregulated heart and kidney interaction that leads to multiorgan system dysfunction, which is not an uncommon occurrence in the setting of right heart failure. The traditional concept of impaired perfusion and forward flow has recently been modified to include the recognition of systemic venous congestion as a contributor, with direct and indirect mechanisms including elevated renal venous pressure, reduced renal perfusion pressure, increased renal interstitial pressure, tubular dysfunction, splanchnic congestion and neurohormonal and inflammatory activation. Treatment options beyond diuretics and vasoactive drugs remain limited and lack supportive evidence.
Key Points.
The physiologic definition of cardio-renal syndrome refers to cardio-renal dysregulation as therapy for congestive symptoms and is limited by decline in renal function.
Right heart failure contributes to the traditional concept of “forward failure” by providing inadequate preload to maintain cardiac output, thereby creating arterial under filling and impaired renal perfusion.
Systemic venous congestion as a result of “backward failure” has gained better recognition as an important contributor to increased renal venous pressure, renal interstitial pressure, and increased intra-abdominal pressure as part of splanchnic congestion.
Current diagnostic strategies, besides bedside assessment, include novel serum or urine biomarkers, renal ultrasonography, and intra-abdominal pressure measurements.
Effective relief of congestion and maintenance of circulatory organ perfusion remains the primary treatment goal along with the lack of targeted specific therapy improvement in right ventricular reserve.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ronco C, Haapio M, House AA, Anavekar N, Bellomo R. Cardiorenal syndrome. J Am Coll Cardiol. 2008;52(19):1527–39. [DOI] [PubMed] [Google Scholar]
- 2.Ronco C, McCullough P, Anker SD, Anand I, Aspromonte N, Bagshaw SM, et al. Cardio-renal syndromes: report from the consensus conference of the acute dialysis quality initiative. Eur Heart J. 2010;31(6):703–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Damman K, Valente MA, Voors AA, O’Connor CM, van Veldhuisen DJ, Hillege HL. Renal impairment, worsening renal function, and outcome in patients with heart failure: an updated meta-analysis. Eur Heart J. 2014;35(7):455–69. [DOI] [PubMed] [Google Scholar]
- 4.Nohria A, Hasselblad V, Stebbins A, Pauly DF, Fonarow GC, Shah M, et al. Cardiorenal interactions: insights from the ESCAPE trial. J Am Coll Cardiol. 2008;51(13):1268–74. [DOI] [PubMed] [Google Scholar]
- 5.Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Secular trends in renal dysfunction and outcomes in hospitalized heart failure patients. J Card Fail. 2006;12(4):257–62. [DOI] [PubMed] [Google Scholar]
- 6.Forman DE, Butler J, Wang Y, Abraham WT, O’Connor CM, Gottlieb SS, et al. Incidence, predictors at admission, and impact of worsening renal function among patients hospitalized with heart failure. J Am Coll Cardiol. 2004;43(1):61–7. [DOI] [PubMed] [Google Scholar]
- 7.Gottlieb SS, Abraham W, Butler J, Forman DE, Loh E, Massie BM, et al. The prognostic importance of different definitions of worsening renal function in congestive heart failure. J Card Fail. 2002;8(3):136–41. [DOI] [PubMed] [Google Scholar]
- 8.Krumholz HM, Chen YT, Vaccarino V, Wang Y, Radford MJ, Bradford WD, et al. Correlates and impact on outcomes of worsening renal function in patients > or =65 years of age with heart failure. Am J Cardiol. 2000;85(9):1110–3. [DOI] [PubMed] [Google Scholar]
- 9.Weinfeld MS, Chertow GM, Stevenson LW. Aggravated renal dysfunction during intensive therapy for advanced chronic heart failure. Am Heart J. 1999;138(2 Pt 1):285–90. [DOI] [PubMed] [Google Scholar]
- 10.National Heart L, and Blood institute Cardio-Renal Connections in Heart Failure and Cardiovascular Disease. 2004. [updated August 20, 2004. Available from: https://www.nhlbi.nih.gov/events/2004/cardio-renal-connections-heart-failure-and-cardiovascular-disease.
- 11.Schrier RW, Abraham WT. Hormones and hemodynamics in heart failure. N Engl J Med. 1999;341(8):577–85. [DOI] [PubMed] [Google Scholar]
- 12.Ljungman S, Laragh JH, Cody RJ. Role of the kidney in congestive heart failure. Relationship of cardiac index to kidney function. Drugs. 1990;39 Suppl 4:10–21; discussion 2–4. [DOI] [PubMed] [Google Scholar]
- 13.Hanberg JS, Sury K, Wilson FP, Brisco MA, Ahmad T, Ter Maaten JM, et al. Reduced Cardiac Index Is Not the Dominant Driver of Renal Dysfunction in Heart Failure. J Am Coll Cardiol. 2016;67(19):2199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mullens W, Abrahams Z, Francis GS, Sokos G, Taylor DO, Starling RC, et al. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol. 2009;53(7):589–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Damman K, van Deursen VM, Navis G, Voors AA, van Veldhuisen DJ, Hillege HL. Increased central venous pressure is associated with impaired renal function and mortality in a broad spectrum of patients with cardiovascular disease. J Am Coll Cardiol. 2009;53(7):582–8. [DOI] [PubMed] [Google Scholar]
- 16.Guglin M, Rivero A, Matar F, Garcia M. Renal dysfunction in heart failure is due to congestion but not low output. Clin Cardiol. 2011;34(2):113–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adams KF Jr., Fonarow GC, Emerman CL, LeJemtel TH, Costanzo MR, Abraham WT, et al. Characteristics and outcomes of patients hospitalized for heart failure in the United States: rationale, design, and preliminary observations from the first 100,000 cases in the Acute Decompensated Heart Failure National Registry (ADHERE). Am Heart J. 2005;149(2):209–16. [DOI] [PubMed] [Google Scholar]
- 18.Sweitzer NK, Lopatin M, Yancy CW, Mills RM, Stevenson LW. Comparison of clinical features and outcomes of patients hospitalized with heart failure and normal ejection fraction (> or =55%) versus those with mildly reduced (40% to 55%) and moderately to severely reduced (<40%) fractions. Am J Cardiol. 2008;101(8):1151–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Damman K, Voors AA, Hillege HL, Navis G, Lechat P, van Veldhuisen DJ, et al. Congestion in chronic systolic heart failure is related to renal dysfunction and increased mortality. Eur J Heart Fail. 2010;12(9):974–82. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka M, Yoshida H, Furuhashi M, Togashi N, Koyama M, Yamamoto S, et al. Deterioration of renal function by chronic heart failure is associated with congestion and oxidative stress in the tubulointerstitium. Intern Med. 2011;50(23):2877–87. [DOI] [PubMed] [Google Scholar]
- 21.Damman K, Navis G, Smilde TD, Voors AA, van der Bij W, van Veldhuisen DJ, et al. Decreased cardiac output, venous congestion and the association with renal impairment in patients with cardiac dysfunction. Eur J Heart Fail. 2007;9(9):872–8. [DOI] [PubMed] [Google Scholar]
- 22.Uthoff H, Thalhammer C, Potocki M, Reichlin T, Noveanu M, Aschwanden M, et al. Central venous pressure at emergency room presentation predicts cardiac rehospitalization in patients with decompensated heart failure. Eur J Heart Fail. 2010;12(5):469–76. [DOI] [PubMed] [Google Scholar]
- 23.Testani JM, Khera AV, St John Sutton MG, Keane MG, Wiegers SE, Shannon RP, et al. Effect of right ventricular function and venous congestion on cardiorenal interactions during the treatment of decompensated heart failure. Am J Cardiol. 2010;105(4):511–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winton FR. The influence of venous pressure on the isolated mammalian kidney. J Physiol. 1931;72(1):49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schrier RW, De Wardener HE. Tubular reabsorption of sodium ion: influence of factors other than aldosterone and glomerular filtration rate. 2. N Engl J Med. 1971;285(23):1292–303. [DOI] [PubMed] [Google Scholar]
- 26.Kastner PR, Hall JE, Guyton AC. Renal hemodynamic responses to increased renal venous pressure: role of angiotensin II. Am J Physiol. 1982;243(3):F260–4. [DOI] [PubMed] [Google Scholar]
- 27.Schrier RW. Blood urea nitrogen and serum creatinine: not married in heart failure. Circ Heart Fail. 2008;1(1):2–5. [DOI] [PubMed] [Google Scholar]
- 28.Komuro K, Seo Y, Yamamoto M, Sai S, Ishizu T, Shimazu K, et al. Assessment of renal perfusion impairment in a rat model of acute renal congestion using contrast-enhanced ultrasonography. Heart Vessels. 2018;33(4):434–40. [DOI] [PubMed] [Google Scholar]
- 29.Ruggenenti P, Remuzzi G. Worsening kidney function in decompensated heart failure: treat the heart, don’t mind the kidney. Eur Heart J. 2011;32(20):2476–8. [DOI] [PubMed] [Google Scholar]
- 30.Ross EA. Congestive renal failure: the pathophysiology and treatment of renal venous hypertension. J Card Fail. 2012;18(12):930–8. [DOI] [PubMed] [Google Scholar]
- 31.Ganda A, Onat D, Demmer RT, Wan E, Vittorio TJ, Sabbah HN, et al. Venous congestion and endothelial cell activation in acute decompensated heart failure. Curr Heart Fail Rep. 2010;7(2):66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verbrugge FH, Dupont M, Steels P, Grieten L, Malbrain M, Tang WH, et al. Abdominal contributions to cardiorenal dysfunction in congestive heart failure. J Am Coll Cardiol. 2013;62(6):485–95. [DOI] [PubMed] [Google Scholar]
- 33.Maxwell MH, Breed ES, Schwartz IL. Renal Venous Pressure in Chronic Congestive Heart Failure. J Clin Invest. 1950;29(3):342–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Afsar B, Ortiz A, Covic A, Solak Y, Goldsmith D, Kanbay M. Focus on renal congestion in heart failure. Clin Kidney J. 2016;9(1):39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gottschalk CW, Mylle M. Micropuncture study of pressures in proximal tubules and peritubular capillaries of the rat kidney and their relation to ureteral and renal venous pressures. Am J Physiol. 1956;185(2):430–9. [DOI] [PubMed] [Google Scholar]
- 36.Fiksen-Olsen MJ, Strick DM, Hawley H, Romero JC. Renal effects of angiotensin II inhibition during increases in renal venous pressure. Hypertension. 1992;19(2 Suppl):II137–41. [DOI] [PubMed] [Google Scholar]
- 37.Braam B, Cupples WA, Joles JA, Gaillard C. Systemic arterial and venous determinants of renal hemodynamics in congestive heart failure. Heart Fail Rev. 2012;17(2):161–75. [DOI] [PubMed] [Google Scholar]
- 38.Schirmer HK, Marshall RE, Jackson MP. The effect of altered renal venous pressure on urine flow and cortical metabolism. J Urol. 1968;100(3):205–8. [DOI] [PubMed] [Google Scholar]
- 39.Joles JA, Bongartz LG, Gaillard CA, Braam B. Renal venous congestion and renal function in congestive heart failure. J Am Coll Cardiol. 2009;54(17):1632; author reply −3. [DOI] [PubMed] [Google Scholar]
- 40.Clausen G, Oien AH, Aukland K. Myogenic vasoconstriction in the rat kidney elicited by reducing perirenal pressure. Acta Physiol Scand. 1992;144(3):277–90. [DOI] [PubMed] [Google Scholar]
- 41.Bradley SE, Bradley GP. The Effect of Increased Intra-Abdominal Pressure on Renal Function in Man. J Clin Invest. 1947;26(5):1010–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Firth JD, Raine AE, Ledingham JG. Raised venous pressure: a direct cause of renal sodium retention in oedema? Lancet. 1988;1(8593):1033–5. [DOI] [PubMed] [Google Scholar]
- 43.Boberg U, Persson AE. Tubuloglomerular feedback during elevated renal venous pressure. Am J Physiol. 1985;249(4 Pt 2):F524–31. [DOI] [PubMed] [Google Scholar]
- 44.Burnett JC, Jr., Knox FG. Renal interstitial pressure and sodium excretion during renal vein constriction. Am J Physiol. 1980;238(4):F279–82. [DOI] [PubMed] [Google Scholar]
- 45.Blake WD, Wegria R, et al. Effect of increased renal venous pressure on renal function. Am J Physiol. 1949;157(1):1–13. [DOI] [PubMed] [Google Scholar]
- 46.Doty JM, Saggi BH, Sugerman HJ, Blocher CR, Pin R, Fakhry I, et al. Effect of increased renal venous pressure on renal function. J Trauma. 1999;47(6):1000–3. [DOI] [PubMed] [Google Scholar]
- 47.Rowntree LG MDFR MD; Geraghty JT,MD. The effects of experimental chronic passive congestion on renal function. Arch Intern Med (Chic). 1913;11(2):121–47. [Google Scholar]
- 48.Carmines PK, Inscho EW, Gensure RC. Arterial pressure effects on preglomerular microvasculature of juxtamedullary nephrons. Am J Physiol. 1990;258(1 Pt 2):F94–102. [DOI] [PubMed] [Google Scholar]
- 49.Abildgaard U, Henriksen O, Amtorp O. Sympathetic reflex-induced vasoconstriction during renal venous stasis elicited from the capsule in the dog kidney. Acta Physiol Scand. 1985;123(1):1–8. [DOI] [PubMed] [Google Scholar]
- 50.Abildgaard U, Amtorp O, Agerskov K, Sjontoft E, Christensen NJ, Henriksen O. Renal vascular adjustments to partial renal venous obstruction in dog kidney. Circ Res. 1987;61(2):194–202. [DOI] [PubMed] [Google Scholar]
- 51.Dilley JR, Corradi A, Arendshorst WJ. Glomerular ultrafiltration dynamics during increased renal venous pressure. Am J Physiol. 1983;244(6):F650–8. [DOI] [PubMed] [Google Scholar]
- 52.Haddy FJ. Effect of elevation of intraluminal pressure on renal vascular resistance. Circ Res. 1956;4(6):659–63. [DOI] [PubMed] [Google Scholar]
- 53.Kishimoto T, Maekawa M, Abe Y, Yamamoto K. Intrarenal distribution of blood flow and renin release during renal venous pressure elevation. Kidney Int. 1973;4(4):259–66. [DOI] [PubMed] [Google Scholar]
- 54.Gudmundsson FF, Gislason HG, Myking OL, Viste A, Grong K, Svanes K. Hormonal changes related to reduced renal blood flow and low urine output under prolonged increased intra-abdominal pressure in pigs. Eur J Surg. 2002;168(3):178–86. [DOI] [PubMed] [Google Scholar]
- 55.Bloomfield GL, Blocher CR, Fakhry IF, Sica DA, Sugerman HJ. Elevated intra-abdominal pressure increases plasma renin activity and aldosterone levels. J Trauma. 1997;42(6):997–1004; discussion −5. [DOI] [PubMed] [Google Scholar]
- 56.Guazzi M, Gatto P, Giusti G, Pizzamiglio F, Previtali I, Vignati C, et al. Pathophysiology of cardiorenal syndrome in decompensated heart failure: role of lung-right heart-kidney interaction. Int J Cardiol. 2013;169(6):379–84. [DOI] [PubMed] [Google Scholar]
- 57.Fudim M, Hernandez AF, Felker GM. Role of Volume Redistribution in the Congestion of Heart Failure. J Am Heart Assoc. 2017;6(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Greenway CV, Lister GE. Capacitance effects and blood reservoir function in the splanchnic vascular bed during non-hypotensive haemorrhage and blood volume expansion in anaesthetized cats. J Physiol. 1974;237(2):279–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Greenway CV. Role of splanchnic venous system in overall cardiovascular homeostasis. Fed Proc. 1983;42(6):1678–84. [PubMed] [Google Scholar]
- 60.Rapaport E, Weisbart MH, Levine M. The splanchnic blood volume in congestive heart failure. Circulation. 1958;18(4 Part 1):581–7. [DOI] [PubMed] [Google Scholar]
- 61.Fallick C, Sobotka PA, Dunlap ME. Sympathetically mediated changes in capacitance: redistribution of the venous reservoir as a cause of decompensation. Circ Heart Fail. 2011;4(5):669–75. [DOI] [PubMed] [Google Scholar]
- 62.Greenway CV, Oshiro G. Comparison of the effects of hepatic nerve stimulation on arterial flow, distribution of arterial and portal flows and blood content in the livers of anaesthetized cats and dogs. J Physiol. 1972;227(2):487–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rzouq F, Alahdab F, Olyaee M. New insight into volume overload and hepatorenal syndrome in cirrhosis, “the hepatorenal reflex hypothesis“. Am J Med Sci. 2014;348(3):244–8. [DOI] [PubMed] [Google Scholar]
- 64.Kostreva DR, Castaner A, Kampine JP. Reflex effects of hepatic baroreceptors on renal and cardiac sympathetic nerve activity. Am J Physiol. 1980;238(5):R390–4. [DOI] [PubMed] [Google Scholar]
- 65.Koyama S, Nishida K, Terada N, Shiojima Y, Takeuchi T. Reflex renal vasoconstriction on portal vein distension. Jpn J Physiol. 1986;36(3):441–50. [DOI] [PubMed] [Google Scholar]
- 66.Ming Z, Smyth DD, Lautt WW. Decreases in portal flow trigger a hepatorenal reflex to inhibit renal sodium and water excretion in rats: role of adenosine. Hepatology. 2002;35(1):167–75. [DOI] [PubMed] [Google Scholar]
- 67.Vallon V, Muhlbauer B, Osswald H. Adenosine and kidney function. Physiol Rev. 2006;86(3):901–40. [DOI] [PubMed] [Google Scholar]
- 68.Hamza SM, Kaufman S. Splenorenal reflex modulates renal blood flow in the rat. J Physiol. 2004;558(Pt 1):277–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hamza SM, Kaufman S. Role of spleen in integrated control of splanchnic vascular tone: physiology and pathophysiology. Can J Physiol Pharmacol. 2009;87(1):1–7. [DOI] [PubMed] [Google Scholar]
- 70.de Bold AJ, Borenstein HB, Veress AT, Sonnenberg H. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Reprinted from Life Sci. 28:89–94, 1981. J Am Soc Nephrol. 2001;12(2):403–9; discussion −8, 8–9. [DOI] [PubMed] [Google Scholar]
- 71.Sultanian R, Deng Y, Kaufman S. Atrial natriuretic factor increases splenic microvascular pressure and fluid extravasation in the rat. J Physiol. 2001;533(Pt 1):273–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Takala J Determinants of splanchnic blood flow. Br J Anaesth. 1996;77(1):50–8. [DOI] [PubMed] [Google Scholar]
- 73.Sandek A, Rauchhaus M, Anker SD, von Haehling S. The emerging role of the gut in chronic heart failure. Curr Opin Clin Nutr Metab Care. 2008;11(5):632–9. [DOI] [PubMed] [Google Scholar]
- 74.Sandek A, Bauditz J, Swidsinski A, Buhner S, Weber-Eibel J, von Haehling S, et al. Altered intestinal function in patients with chronic heart failure. J Am Coll Cardiol. 2007;50(16):1561–9. [DOI] [PubMed] [Google Scholar]
- 75.Ding J, Magnotti LJ, Huang Q, Xu DZ, Condon MR, Deitch EA. Hypoxia combined with Escherichia coli produces irreversible gut mucosal injury characterized by increased intestinal cytokine production and DNA degradation. Shock. 2001;16(3):189–95. [DOI] [PubMed] [Google Scholar]
- 76.Lekawanvijit S Role of Gut-Derived Protein-Bound Uremic Toxins in Cardiorenal Syndrome and Potential Treatment Modalities. Circ J. 2015;79(10):2088–97. [DOI] [PubMed] [Google Scholar]
- 77.Evenepoel P, Meijers BK, Bammens BR, Verbeke K. Uremic toxins originating from colonic microbial metabolism. Kidney Int Suppl. 2009(114):S12–9. [DOI] [PubMed] [Google Scholar]
- 78.Vanholder R, De Smet R, Glorieux G, Argiles A, Baurmeister U, Brunet P, et al. Review on uremic toxins: classification, concentration, and interindividual variability. Kidney Int. 2003;63(5):1934–43. [DOI] [PubMed] [Google Scholar]
- 79.Hietbrink F, Besselink MG, Renooij W, de Smet MB, Draisma A, van der Hoeven H, et al. Systemic inflammation increases intestinal permeability during experimental human endotoxemia. Shock. 2009;32(4):374–8. [DOI] [PubMed] [Google Scholar]
- 80.Niebauer J, Volk HD, Kemp M, Dominguez M, Schumann RR, Rauchhaus M, et al. Endotoxin and immune activation in chronic heart failure: a prospective cohort study. Lancet. 1999;353(9167):1838–42. [DOI] [PubMed] [Google Scholar]
- 81.Mullens W, Abrahams Z, Skouri HN, Francis GS, Taylor DO, Starling RC, et al. Elevated intra-abdominal pressure in acute decompensated heart failure: a potential contributor to worsening renal function? J Am Coll Cardiol. 2008;51(3):300–6. [DOI] [PubMed] [Google Scholar]
- 82.Mohmand H, Goldfarb S. Renal dysfunction associated with intra-abdominal hypertension and the abdominal compartment syndrome. J Am Soc Nephrol. 2011;22(4):615–21. [DOI] [PubMed] [Google Scholar]
- 83.De Waele JJ, De Laet I, Kirkpatrick AW, Hoste E. Intra-abdominal Hypertension and Abdominal Compartment Syndrome. Am J Kidney Dis. 2011;57(1):159–69. [DOI] [PubMed] [Google Scholar]
- 84.Mullens W, Abrahams Z, Francis GS, Taylor DO, Starling RC, Tang WH. Prompt reduction in intra-abdominal pressure following large-volume mechanical fluid removal improves renal insufficiency in refractory decompensated heart failure. J Card Fail. 2008;14(6):508–14. [DOI] [PubMed] [Google Scholar]
- 85.Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323(4):236–41. [DOI] [PubMed] [Google Scholar]
- 86.Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res. 2015;116(7):1254–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Carlstedt F, Lind L, Lindahl B. Proinflammatory cytokines, measured in a mixed population on arrival in the emergency department, are related to mortality and severity of disease. J Intern Med. 1997;242(5):361–5. [DOI] [PubMed] [Google Scholar]
- 88.Ferrari R, Bachetti T, Confortini R, Opasich C, Febo O, Corti A, et al. Tumor necrosis factor soluble receptors in patients with various degrees of congestive heart failure. Circulation. 1995;92(6):1479–86. [DOI] [PubMed] [Google Scholar]
- 89.Testa M, Yeh M, Lee P, Fanelli R, Loperfido F, Berman JW, et al. Circulating levels of cytokines and their endogenous modulators in patients with mild to severe congestive heart failure due to coronary artery disease or hypertension. J Am Coll Cardiol. 1996;28(4):964–71. [DOI] [PubMed] [Google Scholar]
- 90.Colombo PC, Ganda A, Lin J, Onat D, Harxhi A, Iyasere JE, et al. Inflammatory activation: cardiac, renal, and cardio-renal interactions in patients with the cardiorenal syndrome. Heart Fail Rev. 2012;17(2):177–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hedayat M, Mahmoudi MJ, Rose NR, Rezaei N. Proinflammatory cytokines in heart failure: double-edged swords. Heart Fail Rev. 2010;15(6):543–62. [DOI] [PubMed] [Google Scholar]
- 92.Dhingra S, Sharma AK, Arora RC, Slezak J, Singal PK. IL-10 attenuates TNF-alpha-induced NF kappaB pathway activation and cardiomyocyte apoptosis. Cardiovasc Res. 2009;82(1):59–66. [DOI] [PubMed] [Google Scholar]
- 93.Engel D, Peshock R, Armstong RC, Sivasubramanian N, Mann DL. Cardiac myocyte apoptosis provokes adverse cardiac remodeling in transgenic mice with targeted TNF overexpression. Am J Physiol Heart Circ Physiol. 2004;287(3):H1303–11. [DOI] [PubMed] [Google Scholar]
- 94.Haudek SB, Taffet GE, Schneider MD, Mann DL. TNF provokes cardiomyocyte apoptosis and cardiac remodeling through activation of multiple cell death pathways. J Clin Invest. 2007;117(9):2692–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kaur K, Sharma AK, Dhingra S, Singal PK. Interplay of TNF-alpha and IL-10 in regulating oxidative stress in isolated adult cardiac myocytes. J Mol Cell Cardiol. 2006;41(6):1023–30. [DOI] [PubMed] [Google Scholar]
- 96.Radeke HH, Meier B, Topley N, Floge J, Habermehl GG, Resch K. Interleukin 1-alpha and tumor necrosis factor-alpha induce oxygen radical production in mesangial cells. Kidney Int. 1990;37(2):767–75. [DOI] [PubMed] [Google Scholar]
- 97.Kim YS, Morgan MJ, Choksi S, Liu ZG. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol Cell. 2007;26(5):675–87. [DOI] [PubMed] [Google Scholar]
- 98.Kalra D, Sivasubramanian N, Mann DL. Angiotensin II induces tumor necrosis factor biosynthesis in the adult mammalian heart through a protein kinase C-dependent pathway. Circulation. 2002;105(18):2198–205. [DOI] [PubMed] [Google Scholar]
- 99.Ruiz-Ortega M, Ruperez M, Lorenzo O, Esteban V, Blanco J, Mezzano S, et al. Angiotensin II regulates the synthesis of proinflammatory cytokines and chemokines in the kidney. Kidney Int Suppl. 2002(82):S12–22. [DOI] [PubMed] [Google Scholar]
- 100.Moriyama T, Fujibayashi M, Fujiwara Y, Kaneko T, Xia C, Imai E, et al. Angiotensin II stimulates interleukin-6 release from cultured mouse mesangial cells. J Am Soc Nephrol. 1995;6(1):95–101. [DOI] [PubMed] [Google Scholar]
- 101.Tsutamoto T, Wada A, Maeda K, Mabuchi N, Hayashi M, Tsutsui T, et al. Angiotensin II type 1 receptor antagonist decreases plasma levels of tumor necrosis factor alpha, interleukin-6 and soluble adhesion molecules in patients with chronic heart failure. J Am Coll Cardiol. 2000;35(3):714–21. [DOI] [PubMed] [Google Scholar]
- 102.Prabhu SD, Chandrasekar B, Murray DR, Freeman GL. beta-adrenergic blockade in developing heart failure: effects on myocardial inflammatory cytokines, nitric oxide, and remodeling. Circulation. 2000;101(17):2103–9. [DOI] [PubMed] [Google Scholar]
- 103.Anker SD, Egerer KR, Volk HD, Kox WJ, Poole-Wilson PA, Coats AJ. Elevated soluble CD14 receptors and altered cytokines in chronic heart failure. Am J Cardiol. 1997;79(10):1426–30. [DOI] [PubMed] [Google Scholar]
- 104.Charalambous BM, Stephens RC, Feavers IM, Montgomery HE. Role of bacterial endotoxin in chronic heart failure: the gut of the matter. Shock. 2007;28(1):15–23. [DOI] [PubMed] [Google Scholar]
- 105.Rossol M, Heine H, Meusch U, Quandt D, Klein C, Sweet MJ, et al. LPS-induced cytokine production in human monocytes and macrophages. Crit Rev Immunol. 2011;31(5):379–446. [DOI] [PubMed] [Google Scholar]
- 106.Peschel T, Schonauer M, Thiele H, Anker SD, Schuler G, Niebauer J. Invasive assessment of bacterial endotoxin and inflammatory cytokines in patients with acute heart failure. Eur J Heart Fail. 2003;5(5):609–14. [DOI] [PubMed] [Google Scholar]
- 107.Colombo PC, Onat D, Sabbah HN. Acute heart failure as “acute endothelitis”--Interaction of fluid overload and endothelial dysfunction. Eur J Heart Fail. 2008;10(2):170–5. [DOI] [PubMed] [Google Scholar]
- 108.Cheng JJ, Wung BS, Chao YJ, Wang DL. Cyclic strain enhances adhesion of monocytes to endothelial cells by increasing intercellular adhesion molecule-1 expression. Hypertension. 1996;28(3):386–91. [DOI] [PubMed] [Google Scholar]
- 109.Kawai M, Naruse K, Komatsu S, Kobayashi S, Nagino M, Nimura Y, et al. Mechanical stress-dependent secretion of interleukin 6 by endothelial cells after portal vein embolization: clinical and experimental studies. J Hepatol. 2002;37(2):240–6. [DOI] [PubMed] [Google Scholar]
- 110.Wang BW, Chang H, Lin S, Kuan P, Shyu KG. Induction of matrix metalloproteinases-14 and −2 by cyclical mechanical stretch is mediated by tumor necrosis factor-alpha in cultured human umbilical vein endothelial cells. Cardiovasc Res. 2003;59(2):460–9. [DOI] [PubMed] [Google Scholar]
- 111.Colombo PC, Rastogi S, Onat D, Zaca V, Gupta RC, Jorde UP, et al. Activation of endothelial cells in conduit veins of dogs with heart failure and veins of normal dogs after vascular stretch by acute volume loading. J Card Fail. 2009;15(5):457–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Colombo PC, Onat D, Harxhi A, Demmer RT, Hayashi Y, Jelic S, et al. Peripheral venous congestion causes inflammation, neurohormonal, and endothelial cell activation. Eur Heart J. 2014;35(7):448–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Konstam MA, Kiernan MS, Bernstein D, Bozkurt B, Jacob M, Kapur NK, et al. Evaluation and Management of Right-Sided Heart Failure: A Scientific Statement From the American Heart Association. Circulation. 2018;137(20):e578–e622. [DOI] [PubMed] [Google Scholar]
- 114.Dini FL, Demmer RT, Simioniuc A, Morrone D, Donati F, Guarini G, et al. Right ventricular dysfunction is associated with chronic kidney disease and predicts survival in patients with chronic systolic heart failure. Eur J Heart Fail. 2012;14(3):287–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Husain-Syed F, McCullough PA, Birk HW, Renker M, Brocca A, Seeger W, et al. Cardio-Pulmonary-Renal Interactions: A Multidisciplinary Approach. J Am Coll Cardiol. 2015;65(22):2433–48. [DOI] [PubMed] [Google Scholar]
- 116.Rangaswami J, Bhalla V, Blair JEA, Chang TI, Costa S, Lentine KL, et al. Cardiorenal Syndrome: Classification, Pathophysiology, Diagnosis, and Treatment Strategies: A Scientific Statement From the American Heart Association. Circulation. 2019;139(16):e840–e78. [DOI] [PubMed] [Google Scholar]
- 117.Brisco MA, Testani JM. Novel renal biomarkers to assess cardiorenal syndrome. Curr Heart Fail Rep. 2014;11(4):485–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.McCullough PA, Duc P, Omland T, McCord J, Nowak RM, Hollander JE, et al. B-type natriuretic peptide and renal function in the diagnosis of heart failure: an analysis from the Breathing Not Properly Multinational Study. Am J Kidney Dis. 2003;41(3):571–9. [DOI] [PubMed] [Google Scholar]
- 119.Jiang K, Shah K, Daniels L, Maisel AS. Review on natriuretic peptides: where we are, where we are going. Expert Opin Med Diagn. 2008;2(10):1137–53. [DOI] [PubMed] [Google Scholar]
- 120.Palazzuoli A, Ruocco G, Pellegrini M, Martini S, Del Castillo G, Beltrami M, et al. Patients with cardiorenal syndrome revealed increased neurohormonal activity, tubular and myocardial damage compared to heart failure patients with preserved renal function. Cardiorenal Med. 2014;4(3–4):257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Colbert G, Jain N, de Lemos JA, Hedayati SS. Utility of traditional circulating and imaging-based cardiac biomarkers in patients with predialysis CKD. Clin J Am Soc Nephrol. 2015;10(3):515–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tang WH, Shrestha K, Shao Z, Borowski AG, Troughton RW, Thomas JD, et al. Usefulness of plasma galectin-3 levels in systolic heart failure to predict renal insufficiency and survival. Am J Cardiol. 2011;108(3):385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Waring WS, Moonie A. Earlier recognition of nephrotoxicity using novel biomarkers of acute kidney injury. Clin Toxicol (Phila). 2011;49(8):720–8. [DOI] [PubMed] [Google Scholar]
- 124.Haase M, Bellomo R, Devarajan P, Schlattmann P, Haase-Fielitz A. Accuracy of neutrophil gelatinase-associated lipocalin (NGAL) in diagnosis and prognosis in acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis. 2009;54(6):1012–24. [DOI] [PubMed] [Google Scholar]
- 125.Mortara A, Bonadies M, Mazzetti S, Fracchioni I, Delfino P, Chioffi M, et al. Neutrophil gelatinase-associated lipocalin predicts worsening of renal function in acute heart failure: methodological and clinical issues. J Cardiovasc Med (Hagerstown). 2013;14(9):629–34. [DOI] [PubMed] [Google Scholar]
- 126.Knight EL, Verhave JC, Spiegelman D, Hillege HL, de Zeeuw D, Curhan GC, et al. Factors influencing serum cystatin C levels other than renal function and the impact on renal function measurement. Kidney Int. 2004;65(4):1416–21. [DOI] [PubMed] [Google Scholar]
- 127.Shlipak MG, Matsushita K, Arnlov J, Inker LA, Katz R, Polkinghorne KR, et al. Cystatin C versus creatinine in determining risk based on kidney function. N Engl J Med. 2013;369(10):932–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Manzano-Fernandez S, Boronat-Garcia M, Albaladejo-Oton MD, Pastor P, Garrido IP, Pastor-Perez FJ, et al. Complementary prognostic value of cystatin C, N-terminal pro-B-type natriuretic Peptide and cardiac troponin T in patients with acute heart failure. Am J Cardiol. 2009;103(12):1753–9. [DOI] [PubMed] [Google Scholar]
- 129.Masson S, Latini R, Milani V, Moretti L, Rossi MG, Carbonieri E, et al. Prevalence and prognostic value of elevated urinary albumin excretion in patients with chronic heart failure: data from the GISSI-Heart Failure trial. Circ Heart Fail. 2010;3(1):65–72. [DOI] [PubMed] [Google Scholar]
- 130.van de Wal RM, Asselbergs FW, Plokker HW, Smilde TD, Lok D, van Veldhuisen DJ, et al. High prevalence of microalbuminuria in chronic heart failure patients. J Card Fail. 2005;11(8):602–6. [DOI] [PubMed] [Google Scholar]
- 131.Jackson CE, Solomon SD, Gerstein HC, Zetterstrand S, Olofsson B, Michelson EL, et al. Albuminuria in chronic heart failure: prevalence and prognostic importance. Lancet. 2009;374(9689):543–50. [DOI] [PubMed] [Google Scholar]
- 132.Tang WH, Kitai T. Intrarenal Venous Flow: A Window Into the Congestive Kidney Failure Phenotype of Heart Failure? JACC Heart Fail. 2016;4(8):683–6. [DOI] [PubMed] [Google Scholar]
- 133.Bateman GA, Cuganesan R. Renal vein Doppler sonography of obstructive uropathy. AJR Am J Roentgenol. 2002;178(4):921–5. [DOI] [PubMed] [Google Scholar]
- 134.Oktar SO, Yucel C, Ozdemir H, Karaosmanoglu D. Doppler sonography of renal obstruction: value of venous impedance index measurements. J Ultrasound Med. 2004;23(7):929–36. [DOI] [PubMed] [Google Scholar]
- 135.Jeong SH, Jung DC, Kim SH. Renal venous doppler ultrasonography in normal subjects and patients with diabetic nephropathy: value of venous impedance index measurements. J Clin Ultrasound. 2011;39(9):512–8. [DOI] [PubMed] [Google Scholar]
- 136.Iida N, Seo Y, Sai S, Machino-Ohtsuka T, Yamamoto M, Ishizu T, et al. Clinical Implications of Intrarenal Hemodynamic Evaluation by Doppler Ultrasonography in Heart Failure. JACC Heart Fail. 2016;4(8):674–82. [DOI] [PubMed] [Google Scholar]
- 137.Beaubien-Souligny W, Benkreira A, Robillard P, Bouabdallaoui N, Chasse M, Desjardins G, et al. Alterations in Portal Vein Flow and Intrarenal Venous Flow Are Associated With Acute Kidney Injury After Cardiac Surgery: A Prospective Observational Cohort Study. J Am Heart Assoc. 2018;7(19):e009961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chitturi C, Novak JE. Diuretics in the Management of Cardiorenal Syndrome. Adv Chronic Kidney Dis. 2018;25(5):425–33. [DOI] [PubMed] [Google Scholar]
- 139.Faris RF, Flather M, Purcell H, Poole-Wilson PA, Coats AJ. Diuretics for heart failure. Cochrane Database Syst Rev. 2012(2):CD003838. [DOI] [PubMed] [Google Scholar]
- 140.Valente MA, Voors AA, Damman K, Van Veldhuisen DJ, Massie BM, O’Connor CM, et al. Diuretic response in acute heart failure: clinical characteristics and prognostic significance. Eur Heart J. 2014;35(19):1284–93. [DOI] [PubMed] [Google Scholar]
- 141.Oh SW, Han SY. Loop Diuretics in Clinical Practice. Electrolyte Blood Press. 2015;13(1):17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Vargo DL, Kramer WG, Black PK, Smith WB, Serpas T, Brater DC. Bioavailability, pharmacokinetics, and pharmacodynamics of torsemide and furosemide in patients with congestive heart failure. Clin Pharmacol Ther. 1995;57(6):601–9. [DOI] [PubMed] [Google Scholar]
- 143.Sica DA. Pharmacotherapy in congestive heart failure: drug absorption in the management of congestive heart failure: loop diuretics. Congest Heart Fail. 2003;9(5):287–92. [DOI] [PubMed] [Google Scholar]
- 144.McCrindle JL, Li Kam Wa TC, Barron W, Prescott LF. Effect of food on the absorption of frusemide and bumetanide in man. Br J Clin Pharmacol. 1996;42(6):743–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gilotra NA, Princewill O, Marino B, Okwuosa IS, Chasler J, Almansa J, et al. Efficacy of Intravenous Furosemide Versus a Novel, pH-Neutral Furosemide Formulation Administered Subcutaneously in Outpatients With Worsening Heart Failure. JACC Heart Fail. 2018;6(1):65–70. [DOI] [PubMed] [Google Scholar]
- 146.Rangaswami J, McCullough PA. Efficacy of Subcutaneous Versus Intravenous Administration of Furosemide in Patients With Worsening Heart Failure: The Devil Is in the Details. JACC Heart Fail. 2018;6(3):266–7. [DOI] [PubMed] [Google Scholar]
- 147.Cosin J, Diez J. Torasemide in chronic heart failure: results of the TORIC study. Eur J Heart Fail. 2002;4(4):507–13. [DOI] [PubMed] [Google Scholar]
- 148.DiNicolantonio JJ. Should torsemide be the loop diuretic of choice in systolic heart failure? Future Cardiol. 2012;8(5):707–28. [DOI] [PubMed] [Google Scholar]
- 149.Wargo KA, Banta WM. A comprehensive review of the loop diuretics: should furosemide be first line? Ann Pharmacother. 2009;43(11):1836–47. [DOI] [PubMed] [Google Scholar]
- 150.Brater DC. Diuretic therapy. N Engl J Med. 1998;339(6):387–95. [DOI] [PubMed] [Google Scholar]
- 151.Michael Felker G Diuretic management in heart failure. Congest Heart Fail. 2010;16 Suppl 1:S68–72. [DOI] [PubMed] [Google Scholar]
- 152.Francis GS, Siegel RM, Goldsmith SR, Olivari MT, Levine TB, Cohn JN. Acute vasoconstrictor response to intravenous furosemide in patients with chronic congestive heart failure. Activation of the neurohumoral axis. Ann Intern Med. 1985;103(1):1–6. [DOI] [PubMed] [Google Scholar]
- 153.Felker GM, Lee KL, Bull DA, Redfield MM, Stevenson LW, Goldsmith SR, et al. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med. 2011;364(9):797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Palazzuoli A, Pellegrini M, Ruocco G, Martini G, Franci B, Campagna MS, et al. Continuous versus bolus intermittent loop diuretic infusion in acutely decompensated heart failure: a prospective randomized trial. Crit Care. 2014;18(3):R134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Verbrugge FH, Mullens W, Tang WH. Management of Cardio-Renal Syndrome and Diuretic Resistance. Curr Treat Options Cardiovasc Med. 2016;18(2):11. [DOI] [PubMed] [Google Scholar]
- 156.Bart BA, Goldsmith SR, Lee KL, Givertz MM, O’Connor CM, Bull DA, et al. Ultrafiltration in decompensated heart failure with cardiorenal syndrome. N Engl J Med. 2012;367(24):2296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chen HH, Anstrom KJ, Givertz MM, Stevenson LW, Semigran MJ, Goldsmith SR, et al. Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: the ROSE acute heart failure randomized trial. JAMA. 2013;310(23):2533–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Grodin JL, Stevens SR, de Las Fuentes L, Kiernan M, Birati EY, Gupta D, et al. Intensification of Medication Therapy for Cardiorenal Syndrome in Acute Decompensated Heart Failure. J Card Fail. 2016;22(1):26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kramer BK, Schweda F, Riegger GA. Diuretic treatment and diuretic resistance in heart failure. Am J Med. 1999;106(1):90–6. [DOI] [PubMed] [Google Scholar]
- 160.Testani JM, Brisco MA, Turner JM, Spatz ES, Bellumkonda L, Parikh CR, et al. Loop diuretic efficiency: a metric of diuretic responsiveness with prognostic importance in acute decompensated heart failure. Circ Heart Fail. 2014;7(2):261–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Singh D, Shrestha K, Testani JM, Verbrugge FH, Dupont M, Mullens W, et al. Insufficient natriuretic response to continuous intravenous furosemide is associated with poor long-term outcomes in acute decompensated heart failure. J Card Fail. 2014;20(6):392–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Aronson D, Burger AJ. Diuretic Response: Clinical and Hemodynamic Predictors and Relation to Clinical Outcome. J Card Fail. 2016;22(3):193–200. [DOI] [PubMed] [Google Scholar]
- 163.ter Maaten JM, Dunning AM, Valente MA, Damman K, Ezekowitz JA, Califf RM, et al. Diuretic response in acute heart failure-an analysis from ASCEND-HF. Am Heart J. 2015;170(2):313–21. [DOI] [PubMed] [Google Scholar]
- 164.Verbrugge FH, Dupont M, Bertrand PB, Nijst P, Penders J, Dens J, et al. Determinants and impact of the natriuretic response to diuretic therapy in heart failure with reduced ejection fraction and volume overload. Acta Cardiol. 2015;70(3):265–73. [DOI] [PubMed] [Google Scholar]
- 165.Kumar D, Bagarhatta R. Fractional excretion of sodium and its association with prognosis of decompensated heart failure patients. J Clin Diagn Res. 2015;9(4):OC01–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ellison DH. The physiologic basis of diuretic synergism: its role in treating diuretic resistance. Ann Intern Med. 1991;114(10):886–94. [DOI] [PubMed] [Google Scholar]
- 167.Komlosi P, Fintha A, Bell PD. Current mechanisms of macula densa cell signalling. Acta Physiol Scand. 2004;181(4):463–9. [DOI] [PubMed] [Google Scholar]
- 168.Kaissling B, Bachmann S, Kriz W. Structural adaptation of the distal convoluted tubule to prolonged furosemide treatment. Am J Physiol. 1985;248(3 Pt 2):F374–81. [DOI] [PubMed] [Google Scholar]
- 169.Ellison DH, Velazquez H, Wright FS. Adaptation of the distal convoluted tubule of the rat. Structural and functional effects of dietary salt intake and chronic diuretic infusion. J Clin Invest. 1989;83(1):113–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Rao VS, Planavsky N, Hanberg JS, Ahmad T, Brisco-Bacik MA, Wilson FP, et al. Compensatory Distal Reabsorption Drives Diuretic Resistance in Human Heart Failure. J Am Soc Nephrol. 2017;28(11):3414–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Knauf H, Mutschler E. Functional state of the nephron and diuretic dose-response--rationale for low-dose combination therapy. Cardiology. 1994;84 Suppl 2:18–26. [DOI] [PubMed] [Google Scholar]
- 172.Rudy DW, Gehr TW, Matzke GR, Kramer WG, Sica DA, Brater DC. The pharmacodynamics of intravenous and oral torsemide in patients with chronic renal insufficiency. Clin Pharmacol Ther. 1994;56(1):39–47. [DOI] [PubMed] [Google Scholar]
- 173.Gehr TW, Rudy DW, Matzke GR, Kramer WG, Sica DA, Brater DC. The pharmacokinetics of intravenous and oral torsemide in patients with chronic renal insufficiency. Clin Pharmacol Ther. 1994;56(1):31–8. [DOI] [PubMed] [Google Scholar]