

Abstract
Though molecules exhibiting thermally activated delayed fluorescence (TADF) have seen extensive development in organic light-emitting diodes, their incorporation into polymer nanomaterials and thin films has led to a range of applications in sensing and imaging probes. Triplet quenching can be used to probe oxygen concentration, and the reverse intersystem crossing mechanism which gives rise to TADF can also be used to measure temperature. Moreover, the long emission lifetimes of TADF materials allows for noise reduction in time-gated microscopy, making these compounds ideal for time-resolved fluorescence imaging (TRFI). A polymer matrix enables control over energy transfer between molecules, and can be used to modulate TADF behavior, solubility, biocompatibility, or desirable mechanical properties. Additionally, a polymer's oxygen permeability can be tuned to suit imaging applications in a range of media. Here we review the applications of polymer nanoparticles and films exhibiting TADF in sensing and imaging, demonstrating that this class of materials has great potential beyond electroluminescent devices still waiting to be explored.
Keywords: TADF, imaging, oxygen sensing, polymer films, polymer nanostructure, temperature sensing
Introduction
Since the demonstration of their utility in organic light-emitting diodes (OLEDs) in 2011 (Endo et al., 2011), molecules exhibiting thermally activated delayed fluorescence (TADF) have generated tremendous research interest (Wu et al., 2009; Goushi et al., 2012; Zhang et al., 2012; Cho et al., 2015; Li et al., 2017; Steinegger et al., 2017; Wong and Zysman-Colman, 2017; Yang et al., 2017; Chen et al., 2018). When used as emitters in OLEDs, internal quantum efficiencies up to 100% can be achieved due to their ability to harvest both singlet and triplet excitons (Jankus et al., 2014; Wu et al., 2016). Concurrently, the incorporation of TADF materials into polymer nanostructures led to a range of emerging applications as sensors and imaging probes (Kochmann et al., 2013; Xiong et al., 2014; Gan et al., 2017; Li et al., 2017; Steinegger et al., 2017; Zhu et al., 2018; Tsuchiya et al., 2019; Tonge et al., 2020). TADF emitters can be incorporated into polymer nanostructures either as small molecules dispersed in a polymeric matrix or covalently incorporated into the polymer itself. By incorporating TADF materials into polymers, the solubility, biocompatibility, and oxygen permeability of the material can be readily tuned, and rates of energy transfer within the material can be controlled as well. In this way, the desirable properties of TADF materials can be applied to a wider range of media and combined with other chemical functionality for cellular uptake, targeting, or drug delivery.
In TADF materials, forward and reverse intersystem crossing processes (ISC/RISC) interconvert excitons between singlet and triplet excited states. While transitions between states of unlike spin are generally forbidden, a suitably small singlet-triplet energy gap (ΔEST) allows for state mixing such that these transitions become observable. To design molecules with a small ΔEST, donor (D) and acceptor (A) moieties are arranged so as to limit the overlap between the HOMO and LUMO, minimizing the exchange energy which causes the singlet-triplet energy gap (Endo et al., 2011). TADF is realized when ΔEST is small enough such that thermal energy alone is sufficient to promote RISC from a triplet state to a higher-energy singlet (Penfold et al., 2018). In applications where a high triplet concentration is detrimental to the material's function, a short emission lifetime and, therefore, high RISC rate (kRISC) is desirable. El-Sayed's rule for intersystem crossing (ISC) states that transitions between a pure triplet charge transfer state (3CT) and pure singlet charge transfer state (1CT) are prohibited, and thus even with a small ΔEST, the 3CT 1CT RISC process is generally slow (Lim et al., 1981). Recent research, however, has found that kRISC can be increased by the presence of energetically proximate locally excited triplet (3LE) states, such that a small 3LE-3CT energy gap (|ΔE3LE−3CT|) can facilitate conversion between 3CT and 1CT (Chen et al., 2015; Dias et al., 2016; Gibson et al., 2016; Marian, 2016; Gibson and Penfold, 2017; Hosokai et al., 2017). The converse of this observation is that molecules can also be intentionally designed to have low kRISC values, and thus long emissive lifetimes, increasing sensitivity in numerous imaging applications.
The fluorescent mechanism of TADF dyes has significant potential for applications beyond electroluminescent devices, including in sensing and fluorescence imaging. TADF emission lifetimes are typically biexponential, with a short component (<10 ns) due to fluorescence directly from the first excited singlet state (S1) as well as a second, delayed component (μs to ms) arising from the RISC process (Dias, 2015; Palmeira and Berberan-Santos, 2018). The involvement of triplet excited states in TADF makes these emitters particularly susceptible to fluorescence quenching from triplet oxygen. This response can be exploited to develop emitters that are sensitive to the O2 concentration in a given medium by measuring the change in emission intensity and lifetime of delayed fluorescence (Baleizão et al., 2008; DeRosa et al., 2015). Similarly, kRISC depends directly on the temperature of the environment in which the emitter is located, meaning the temperature of a sample can be determined directly from a fluorescence lifetime (Palmeira and Berberan-Santos, 2018). In applications such as these, longer emission lifetimes result in probes with higher sensitivity.
The presence of delayed emission lifetimes without the costly or potentially toxic metallic elements typically found in phosphorescent dyes also makes TADF materials well-suited to time-resolved fluorescence imaging (TRFI) (Zhang et al., 2019). In typical biological systems, autofluorescence from background processes and structures within the cell can significantly reduce the resolution of fluorescent cell imaging (He et al., 2018). These background processes, however, are generally complete in <100 ns, while TADF emission may be detected over a significantly longer timescale. This makes photostable TADF dyes with high brightness highly desirable for TRFI, as background fluorescence can be filtered out by resolving the integrated signal over time or gating the detected luminescence.
In sensing or imaging applications, seclusion of the TADF emitter to control aggregation, limit the influence of environmental factors, and introduce solubility in aqueous systems is the primary goal in host material selection and can be efficiently achieved through incorporation in a polymer host. Characteristics such as charge mobility have minimal effect on sensors unlike small molecule hosts typically used in TADF OLEDs (Chatterjee and Wong, 2019). Such polymer encapsulation can be performed by covalent or non-covalent interactions, with minimal effect on the absorption spectrum of the TADF material. The emission spectrum, however, can be tuned depending on the ability of the polymer matrix to stabilize the emitter's CT excited state. Quantum yield (ΦF) values will also vary depending on the polarity of the host polymer and the impact of aggregation on the dye (Gan et al., 2017; Li et al., 2017; Marghad et al., 2019). Indeed, encapsulation within a polymer host results in a reduction of rotational and vibrational degrees of freedom, often resulting in enhanced ΦF and elongated fluorescence lifetimes.
Herein, we discuss recent examples of TADF in polymer nanostructures and thin films. The application of these systems in oxygen and thermal sensing is discussed, as well as methods by which polymer structure can be used to modulate TADF behavior. Progress and current areas of research focus are highlighted, alongside potential avenues for further advancement.
TADF in Sensors
TADF Sensors for Oxygen
Luminescent sensors for oxygen can make use of polymeric hosts to control the interaction of O2 with a fluorophore, helping to define the sensitivity and dynamic range of the sensor (Kochmann et al., 2013; Steinegger et al., 2017). TADF was first exploited in oxygen sensing by Wolfbeis and coworkers in 2013, by doping 13C70 into a series of polymer hosts (Kochmann et al., 2013). 13C70 has a ΦF of 9%, the highest of any fullerene known, due to increased TADF efficiency from the increased nuclear mass of 13C, as well as a nuclear magnetic effect that increases RISC rates (Baleizão and Berberan-Santos, 2011). Moreover, 13C70 exhibits a high triplet quantum yield of 0.994 and a 170-fold enhancement in ΦF under inert atmosphere (Baleizão and Berberan-Santos, 2011). In this study, 13C70 was doped into a series of hosts, specifically polystyrene (PS), ethyl cellulose (EC), and organically modified silica (Ormosil), to investigate a range of oxygen permeabilities (PO2) from moderate to high. A thermostated flow cell (298 or 333 K) was used to prevent interference from temperature variations, and decay lifetimes in the wavelength range 670–700 nm were monitored as O2 concentrations were varied (Figure 1A). In these experiments, Stern-Volmer plots of vs. O2 concentration could be fit using a two-site model which accounted for two distinct probe environments (Figures 1B–D). The linearity of the fit was found to decrease in all films with increasing temperature, due to increased efficiency of RISC and an increased oxygen quenching rate. The lowest limit of detection (LOD) for all films was measured at 0.25 ppm by volume (ppmv) in EC, with upper detection limits of 4,900 ppmv giving the sensors a four order of magnitude dynamic range. These limits of detection were competitive with state-of-the-art sensors using phosphorescent palladium(II) porphyrin complexes in fluorinated polymer hosts, without the use of transition metals.
Figure 1.
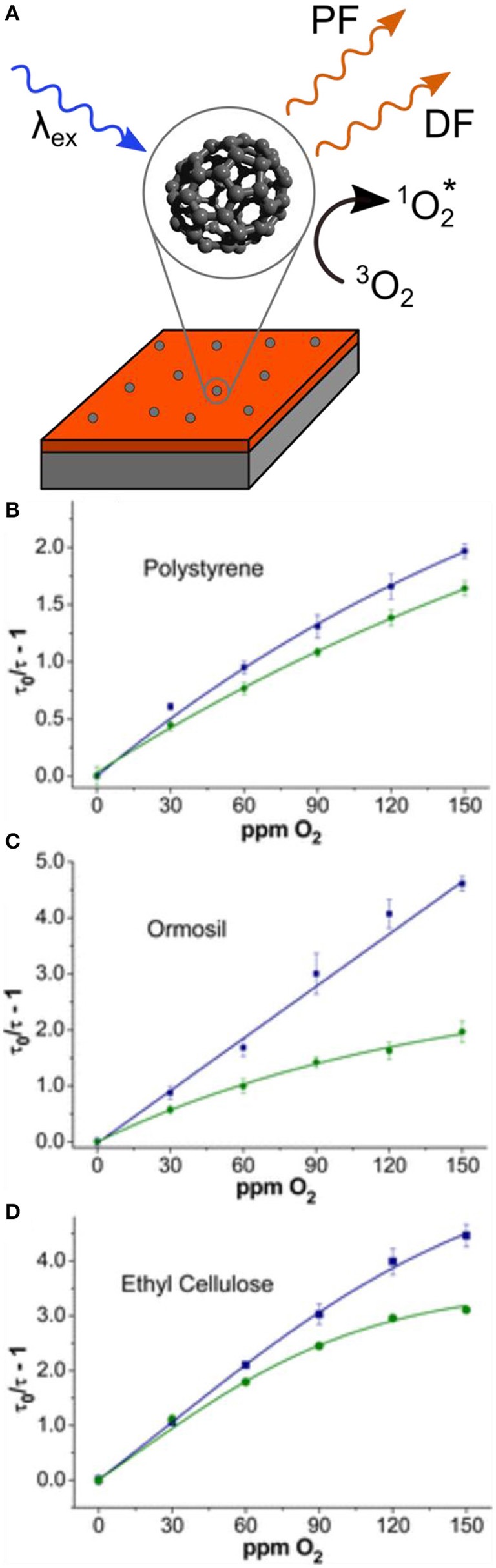
(A) Schematic representation of 13C70 films with excited state quenching by triplet oxygen; (B–D) lifetime-based Stern-Volmer plots at 298 K (blue squares) and 333 K (green circles) for 13C70 in (B) PS, (C) Ormosil, and (D) EC. Adapted from reference (Kochmann et al., 2013) with permission from the American Chemical Society.
Despite the success of the 13C70-based films, their low ΦF presents a barrier to more widespread use. An improved sensor design was demonstrated by Borisov and coworkers, who used TADF compounds based on dicyanobenzene (DCB) and anthraquinone (AQ) electron acceptors with arylamine-based electron donors (Figure 2) doped into PS films (Steinegger et al., 2017). The combination of these donors and acceptors affords compounds with absorption in the visible region, high ΦF, and good photostability (Zhang et al., 2015). Compounds 1–14 dispersed in PS films have π- π* absorption maxima ranging from 329 to 375 nm and charge transfer bands ranging from 440 to 490 nm. Compared to toluene solution the PS host results in blue shifted emission, with DCB dyes 11–14 showing emission maxima from 493 to 531 nm and AQ dyes 1–10 ranging from 577 to 614 nm in PS films. A significant enhancement in ΦF and excited state lifetimes of 1–10 is also observed upon polymer encapsulation due to a restriction in rotational degrees of freedom, decreasing the rate of non-radiative decay. Multicomponent fluorescence lifetimes for films containing 1–14 were observed, including a delayed fluorescence component in the μs-ms range. Photodegradation studies revealed that DCB dyes 11–14 are highly resistant to photobleaching, while AQ dyes 1–10 degrade 2–5 times faster.
Figure 2.

Structures of TADF compounds used by Borisov and coworkers to prepare thin films for oxygen and temperature sensing (Steinegger et al., 2017).
To investigate 1–14 as potential oxygen sensors, the PS films were tested using a flow cell thermostated at 298 K to eliminate cross-sensitivity to temperature. 11–14 have relatively short delayed lifetimes (9–40 μs) and formed devices with low oxygen sensitivity, while 1–5 (2,830–5,560 μs lifetimes) gave sensors with sensitivities up to 1.15 hPa−1. Similarly to 13C70-based sensors, a two-site model was used to fit the Stern-Volmer quenching behavior of 1–14 in thin films. The Stern-Volmer constant (KSV) ranged from 0.00169 to 1.15 hPa−1 and results in devices with similar sensitivities to common metal-based phosphorescent oxygen sensors in PS such as platinum(II) pentafluorophenylphophyrin (KSV = 1.81 Pa−1) (Borisov and Klimant, 2007), platinum(II) tetraphenyltetrabenzoporphyrins (KSV = 1.65–2.18 Pa−1) (Borisov et al., 2008), and ruthenium(II) tris(4,7-diphenyl)-1,10-phenanthroline (KSV = 0. 215 Pa−1) (Borisov and Klimant, 2007), however, sensitivities were significantly lower than palladium(II) tetraphenyltetrabenzoporphyrins (KSV = 9.1–10.4 Pa−1) (Borisov et al., 2008). Temperature was found to have significant cross-sensitivity with oxygen in these materials however, hindering their application in situations where temperature is uncontrolled.
Polymers exhibiting TADF were recently developed in our laboratory that act as ratiometric sensors for oxygen, based on the dual-emissive nature of a TADF chromophore. A series of TADF dopants were prepared using an oxadiazole acceptor paired with either phenoxazine (POZ, 15), phenothiazine (PTZ, 16), dimethylacridan (DMAC, 17), or N-methylphenazine (MPAZ, 18) donors to achieve emission ranging from blue to orange (Figures 3A,B) (Tonge et al., 2020). These materials were doped into a carbazole-based host using copper(0) reversible deactivation radical polymerization (Cu(0)-RDRP), giving molecular weights of ~20 kDa with dispersities from 1.10 to 1.45 (Anastasaki et al., 2016; Sauv et al., 2018; Tonge et al., 2019). Using this method, polymers were synthesized containing 5–15 wt.% TADF emitter, with ΦF values as high as 96%.
Figure 3.
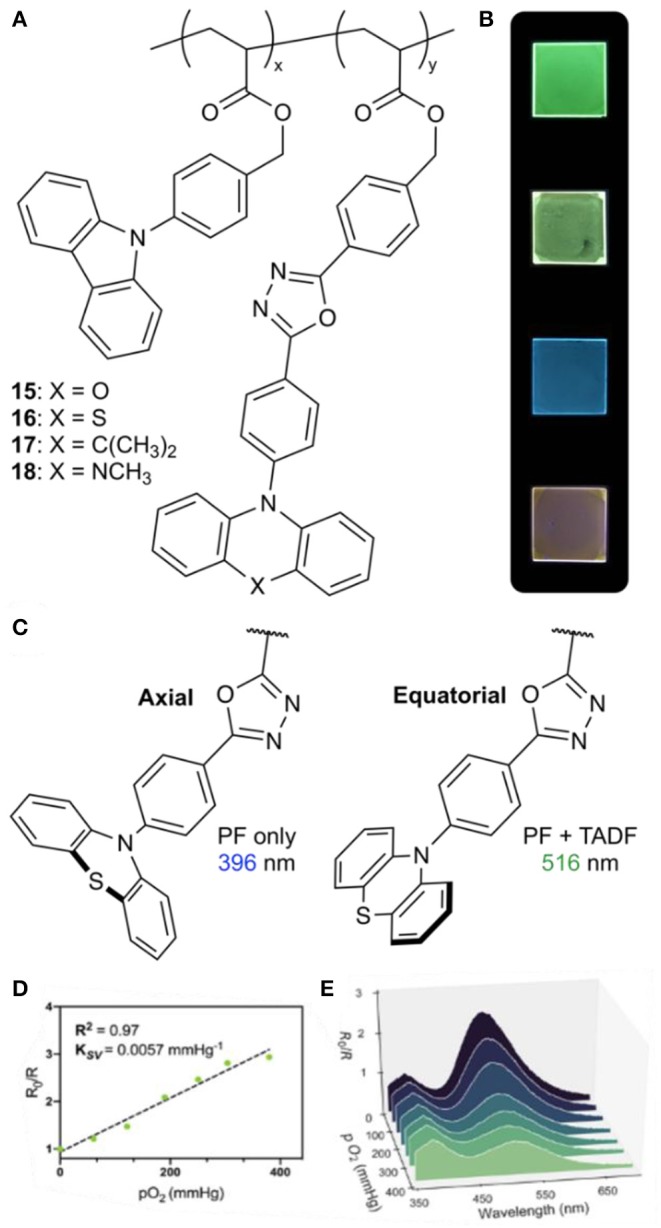
(A) Donor-acceptor TADF polymers; (B) films of 15–18 (from top to bottom); (C) illustration of pseudoaxial and pseudoequatorial conformers of phenothiazine and their respective emission wavelengths; (D) Stern-Volmer plot comparing the ratio of emission intensity at 516 nm to emission intensity at 396 nm (I516/I396) at various partial pressures of O2 for a film of 16; (E) fluorescence emission response of 16 to O2 concentration.
The PTZ-containing polymer 16 was found to exhibit dual emission in both solution and the solid state, with emission maxima at 396 and 516 nm. The higher-energy violet emission was found to remain constant regardless of oxygen concentration, while the green emission exhibited oxygen sensitivity characteristic of TADF. There have been several literature reports of PTZ-based donor-acceptor emitters exhibiting dual emission (Daub et al., 2001; Stockmann et al., 2002; Acar et al., 2003; Tanaka et al., 2014; Okazaki et al., 2017; Marghad et al., 2019), arising from two stable conformers of the phenothiazine moiety (Malrieu and Pullman, 1964; Bodea and Silberg, 1968). The pseudoaxial conformer gives a locally excited state with higher-energy emission, while the pseudoequatorial conformer participates in charge transfer, leading to lower-energy emission and TADF (Figure 3C). By taking advantage of the stable emission of the pseudoaxial conformer, oxygen concentration can be determined using the relative intensity of the locally excited and charge transfer emission bands. Stern-Volmer behavior (KSV = 0.0057 mmHg−1) was observed using neat thin films of 16 with a linear relationship observed between the emission intensity ratio and O2 concentration up to 50% oxygen by volume at 1 atm (Figures 3D,E).
While oxygen-sensitive thin films have potential applications as standalone oxygen sensors, oxygen sensing in aqueous solution would enable applications in biological imaging as well. To demonstrate O2-responsiveness in aqueous environments, polymer nanoparticles were prepared by coprecipitating 16 with an amphiphilic polymer, polystyrene-co-poly(maleic anhydride) (PS-co-PMA). This gave water-soluble “polymer dots” (Pdots) with diameters of 82 ± 38 nm, whose fluorescent properties were similar to those of 16 in thin films. The dual emission of the Pdots showed a responsive color change through the full range of O2 solubility in water (up to 44 ppm at 25°C). In contrast to the thin films, this response was non-linear due the distribution of TADF chromophores in the interior of the Pdots and at the surface, giving environments with variable accessibility to O2.
TADF Temperature Probes
In order to employ TADF materials as temperature-sensing probes, the access of oxygen to the dyes must be limited. Borisov and coworkers developed TADF-based temperature probes by encapsulating compounds 1–14 in the low-oxygen permeability polymer poly(vinylidene chloride-co-acrylonitrile (P(VDC-co-AN) (Steinegger et al., 2017). The use of P(VDC-co-AN) gives similar absorption maxima compared with PS films, however, a slight red shift in emission is observed due to the higher polarity of the P(VDC-co-AN) host. ΦF and lifetimes are reduced compared to PS with the decrease most pronounced in 6–9 having ΦF values dropping to ~0.05. Lifetimes as a function of temperature could be fit by an Arrhenius model (Equation 1):
| (1) |
where k0 is the temperature-independent decay rate, k1is a pre-exponential factor, kB is the Boltzman constant, and T the temperature in kelvin. The relative sensitivity (% change in τ per K) of the sensors was found to be −1.4 to −4.2 % K−1, exceeding the sensitivity of similar europium(III) (Khalil et al., 2004), ruthenium(II) (Liebsch et al., 1999), or chromium(III)-doped (Borisov et al., 2010) yttrium aluminum borate-based thermal sensors (−0.6 to −2.3% K−1). 12 and 14 were then incorporated into cationic Eudragit RL100 (ERL) nanoparticles, which are cell-penetrating and provide a route to intracellular fluorescence measurements. ERL is moderately oxygen permeable, and dyes 12 and 14 were chosen to minimize oxygen cross-sensitivity. 12 and 14 ERL dots retained a high sensitivity to temperature, with sensitivities of −2.2 and −2.8 % K−1 at 298 K, respectively (Figure 4). A small oxygen cross-sensitivity error of ± 4 K was found for 12 with 14 having a reduced error of ± 1.5 K.
Figure 4.
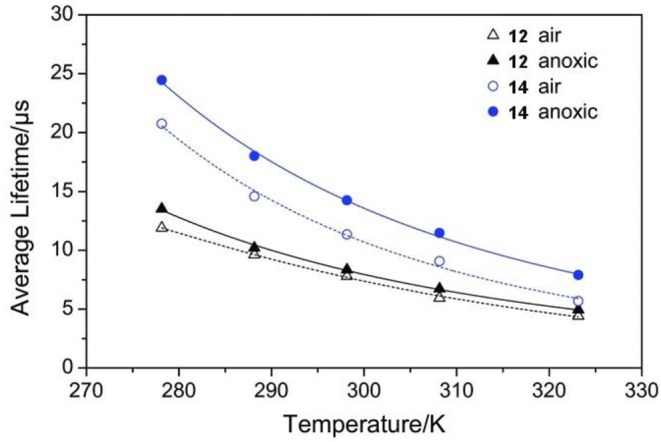
Temperature dependence of the average luminescence decay times of 12 and 14 ERL NPs. Adapted from reference (Steinegger et al., 2017) with permission from Wiley.
Fluorescence Lifetime-based Imaging Agents
Delayed emission from compounds free from phosphorescent heavy-metal dopants are of significant interest in biological imaging. The absence of metals decreases the toxicity and reduces the cost of emitters used as imaging probes. In typical biological systems, autofluorescence and other background fluorescence processes cause significant noise that is detrimental to high resolution imaging. These fluorescent processes have lifetimes in the ns range, however, allowing the use of time gating to remove background fluorescence and allowing only emission from species with long excited-state lifetimes to be detected.
In 2014, Peng and coworkers reported the use of a purely organic fluorophore for TRFI, using fluorescein derivative 19 (Figure 5A) (Xiong et al., 2014). 19 has two absorption features at 485 and 550 nm which density functional theory (DFT) calculations suggest result from transitions between the xanthene and pyran moieties (S0 → S1) and within the xanthene moiety itself (S0 → S4). Steady-state emission in aerated acetonitrile gives emission peaks at 525 and 649 nm and a ΦF of 0.28 is observed in ethanol (Xiong et al., 2013). In deoxygenated solution the peak at 649 nm enhances in intensity 24-fold, and with the application of a 100 μs gating time, the 525 nm peak disappears entirely. A multicomponent decay curve is observed with a two-component portion in the μs time scale, with an average lifetime of 22.11 μs. Unsurprisingly 19 shows no DF in aerated 10 mM phosphate-buffered saline (PBS), however, upon the addition of BSA DF is observed. 19 has a suitable size and polarity to enter the hydrophobic cavity of BSA, which secludes the fluorophore from molecular oxygen. Additionally, it has been found that BSA has cell-penetrating qualities that enhance cellular uptake (Xiong et al., 2014). TRFI experiments with nanoparticles (NPs) of 19 encapsulated in BSA using MCF-7 cancer cells demonstrated a large reduction of background fluorescence (Figures 5B–E). Moreover, confocal fluorescence imaging showed extensive permeation of the cell membrane by the TADF NPs, with fluorescence signals localized in the lysosomes. Colocalization experiments using a commercial lysosome tracking dye (LysoSensor Green DND-189) also showed good signal overlap from the two emitters. Finally, biological toxicity of free 19 using the MCF-7 cell line showed minimal cell death observed after 10 h, making this compound promising for applications in TADF-based TRFI.
Figure 5.

(A) Fluorescein derivative 19; (B) bright-field image and (C) steady-state luminescence (510–560 nm excitation) of MCF-7 cells immunostained with 19 (20 μM) and BSA (40 μL, 10 mM) at 37°C; (D) bright-field image and (E) time-resolved luminescence (510–560 nm excitation) of MCF-7 cells stained with 19 (20 μM) and BSA (40 μL, 10 mM) at 37° C. Adapted from reference (Xiong et al., 2014) with permission from the American Chemical Society.
While the use of BSA as a biopolymer encapsulation matrix enables imaging in aqueous environments, a low ΦF is often observed from the resulting NPs. Tang and coworkers argued that aggregation-caused quenching (ACQ) resulting from high concentrations of fluorophore inside the BSA NPs was responsible, and proposed that this problem could be resolved using TADF molecules that exhibit aggregation-induced emission (AIE) (Gan et al., 2017). Compounds with a benzophenone acceptor and POZ or PTZ donors were investigated, giving materials 20–23 with highly twisted conformations which inhibit π stacking (Figures 6A,B). Solid-state emission maxima for these compounds ranged from 544 to 558 nm, with ΦF values from 0.14 to 0.24 and delayed lifetimes between 0.66 and 1.36 μs. Encapsulation in BSA gave TADF NPs in water with diameters ranging from 109 to 150 nm. Confocal laser scanning microscopy (CLSM) showed that the TADF NPs effectively stain HeLa cells and show green to yellow emission within the cytoplasm (Figures 6C–H). Moreover, differences in intracellular viscosities result in the measurement of two distinctive lifetime regions (~1,000 and ~2,000 ps, respectively) by TRFI.
Figure 6.

(A) Fabrication of BSA NPs; (B) AIE TADF compounds 20–23; confocal fluorescence images of HeLa cells after incubation with (C–E) BSA NPs of 21 (10 μM of 21) and (F–H) BSA NPs of 22 (10 μM of 22) at 37°C for 0.5 h; (C,F) bright-field images; (D,G) fluorescence images; (E,H) merged bright field and fluorescence images; scale bar = 10 μm. Adapted from reference (Gan et al., 2017) with permission from The Royal Society of Chemistry.
While the use of AIE materials facilitates imaging at high brightness within BSA nanoparticles, the cross-sensitivity with intracellular viscosity suggests that the TADF dyes are not well-isolated from the cytoplasm. Alternatively, the use of Pdots formed from amphiphilic polymers offers the ability to encapsulate organic molecules within a hydrophobic core, surrounded by a hydrophilic, biocompatible corona (Kuo et al., 2015; Massey et al., 2015; Yu et al., 2017). The amphiphilic polymer acts as a barrier against oxygen and polar molecules, allowing for time-gated imaging under biological conditions. Moreover, the exclusion of TADF dyes from the cytoplasm prevents cell damage from singlet oxygen produced through triplet quenching or innate toxicity from the fluorophores themselves.
Huang and coworkers investigated the use of TADF Pdots for biological imaging by employing the amphiphilic polymer 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG, Figure 7) and the TADF material 2,3,5,6-tetracarbazole-4-cyano-pyridine (24) (Li et al., 2017). 24 has been shown to have increased emission in the solid state (ΦF = 54.9%) compared with chloroform solution (ΦF = 24.7%) and, therefore, would not be negatively affected by aggregation within the Pdot. 24 displays green emission, absorbs in the visible range, and exhibits a DF lifetime of 8.3 μs in deoxygenated toluene. DSPE-PEG and 24 were precipitated to form Pdots which had a spherical morphology with an average diameter of 17 nm by TEM and 23 ± 5 by DLS. Pdots of 24 show a CT absorption band at ~450 nm in water which is red-shifted compared to free 24 in THF, and emission at 535 nm with ΦF = 0.38. In aerated water a biexponential decay is observed with a long lifetime component of 9.4 μs. Photobleaching studies revealed Pdots of 24 retained 90% of their original fluorescence intensity after 45 min of laser irradiation (55.6 mW cm−2 at 405 nm).
Figure 7.

The amphiphilic polymer DSPE-PEG and TADF 24 used by Huang and coworkers (Li et al., 2017).
Pdots of 24 show good ΦF, long lifetimes, and resistance to photobleaching, however, their stability and biocompatibility had to be investigated. To test the chemical stability of the Pdots fluorescence measurements were made in Milli-Q water, PBS, and tris-acetate buffer. Dulbeco's modified Eagle medium (DMEM) was also used to simulate physiological conditions. Compared to the Milli-Q water solution, the fluorescence intensity was minimally affected by PBS and tris-acetate buffer and only decreased by 20% in DMEM. Moreover, in all cases the fluorescence intensity remained stable after 48 h in solution at 37°C. Toxicity studies were also conducted by incubating HeLa cells with variable concentrations of Pdots for 24 h. A maximum concentration of 9 μM could be used while retaining >80% cell viability and concentrations of 4.5 μM or lower resulted in minimal cell death.
The effectiveness of 24 as an imaging probe for in vitro studies was determined using HeLa cells as an example system. The cells were incubated with Pdots of 24 (0.5 μM) in PBS for 2 h. Short-lived background fluorescence is observed from the cytoplasm while emission from 24 is detected from the cell membrane with an average lifetime of 165 ns. Co-staining with 24 Pdots and a cell membrane marker, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiIC18), gives overlapping emission confirming the localization of the Pdots. It was also proposed that the lipophilicity of DSPE-PEG causes Pdots near the cell to interact with the cell membrane. During this process, dye 24 can transfer from the Pdot into the cell membrane, where it is retained. This mechanism is consistent with the observation that excited state lifetimes shortened significantly during imaging compared to free Pdots of 24 measured in water. Finally, the membrane-labeling property of Pdots of 24 was explored by confocal imaging and TRFI of a living zebrafish, giving a clear picture of the vascular network of the animal with minimal background fluorescence (Figure 8).
Figure 8.

Confocal fluorescence images of zebrafish: (A–C) zebrafish injected with 24 Pdots; (D–F) zebrafish without any Pdots injected; (A,D) bright field images; (B,E) confocal fluorescence images recorded with 480–580 nm bandpass filters for 24 Pdots upon excitation at 405 nm; (C,F) fluorescence lifetime images. Reproduced from reference (Li et al., 2017) under the Creative Commons CC-BY license.
While polymer encapsulation often reduces ΦF values and red-shifts the emission of TADF dyes, Adachi and coworkers recently applied concepts from OLED design to address these issues and improve the performance of TADF Pdots (Tsuchiya et al., 2019). Organic semiconductor host materials are commonly used in OLEDs to isolate emitters and prevent aggregation quenching effects, and it was hypothesized that these same ideas could be used to increase ΦF, reduce aggregation, and enhancing the stability of the TADF dyes in Pdots. By incorporating TADF dyes into the glassy semiconductor host 25 within DSPE-PEG, TADF dye 12 (Figure 9A) could be effectively protected from the surrounding medium and aggregation could be prevented. The combination of 12+25 had previously been shown to be an effective dye/matrix combination in the emissive layer of OLEDs (Uoyama et al., 2012; Nakanotani et al., 2013), suggesting that energy transfer from 25 to 12 should be highly favorable. Aqueous mixtures of 12, 25, and DSPE-PEG were heated to 180°C under pressure and then rapidly cooled to melt and subsequently solidify 12 and 25 within the Pdot. This produced spherical particles (Figure 9B) with an average diameter of 371 nm by DLS. The use of host 26 in place of 25 was also attempted, but it was found that the high melting point of 26 (268.7°C) and lower crystallization temperature (131.3°C) resulted in microcrystal formation during processing. Aqueous glassy Pdots of 12+25 gave slightly red-shifted emission and higher ΦF (λem = 516 nm and ΦF = 0.94) compared with 12 alone in toluene solution (λem = 498 nm and ΦF = 0.83), suggesting that the strategy was promising. Importantly, the fabrication of glassy Pdots in argon-saturated water was found to be essential as the use of aerated water during fabrication resulted in a drastic decrease in ΦF of 32%. A delayed lifetime of 3.1 μs was observed from the glassy Pdots which was nearly insensitive to the presence of oxygen. High photo-stability was observed with intensity dropping by only 25% after 115 min of irradiation. Minimal cytotoxicity was observed with HEK293 cells, consistent with the work of Huang and coworkers (Li et al., 2017) and the known biocompatibility of DSPE-PEG. The Pdots showed strong fluorescence in HEK293 cells (Figure 9C) with cells retaining their emission after 7 days of culturing. Long-term traceability experiments were also conducted with HEK293 cells to utilize the high stability and low cytotoxicity of the glassy Pdots. The glassy Pdots were observable for 21 days with cell viability matching that of the control cells. Interestingly, cell membrane labeling was not observed in this study, despite the use of the same amphiphilic polymer employed in the earlier study by Huang and coworkers (Li et al., 2017).
Figure 9.

(A) TADF emitter and host materials; (B) SEM image of 6 wt.% glassy Pdots (2 μm scale bar); (C) combined phase contrast image of glassy Pdots in HEK293 cells after 12 h of incubation (100 μm scale bar). Adapted from reference (Tsuchiya et al., 2019) with permission from The Royal Society of Chemistry.
Despite the utility of non-glassy and glassy Pdots in TRFI imaging, both require several hours of incubation time to effectively stain cells due to their low membrane permeability. Zhao and coworkers used a cell-penetrating peptide (CPP) as an alternative encapsulating amphiphile to address this issue (Zhu et al., 2018). The CPP (F6G6(rR)3R2) consisted of a hydrophilic octamer [(rR)3R2] formed from L-arginine (R) and D-arginine (r), a hexamer of glycine (G), and a hydrophobic hexamer of phenylalanine (F) (Figure 10) (Zhu et al., 2018). (rR)3R2 was used as the hydrophilic moiety of the CPP amphiphile due to previous research showing the high biocompatibility of the fragment (Ma et al., 2012). The TADF dyes 12, 27, and 28 were used as they have been successfully incorporated into high performance OLEDs and give a range of emission wavelengths. Aqueous CPP Pdot solutions were formed using a reprecipitation method giving particles 92 to 177 nm in diameter by DLS. 12, 27, and 28 Pdots showed emission at 555, 607, and 657 nm with ΦF values of 0.12, 0.025, and 0.008, respectively. Emission is red-shifted and ΦF values were drastically reduced compared to their values in toluene solution due to the effects of aggregation and the polar amide backbone of F6G6(rR)3R2. All fluorescence decays were multi-exponential with delayed lifetimes ranging from 1.8 to 93.7 μs. No significant change in lifetime was observed between aerated and deoxygenated solution.
Figure 10.

Schematic of CPP Pdots fabricated from F6G6(rR)3R2 and TADF emitters 12, 27, and 28. CLSM images of HeLa cells after incubation with CPP encapsulated (a,d) 12, (b,e) 27, (c,f), and 28 Pdots for (a–c) 5 min and (d–f) 15 min. [12] = [27] = [28] = 2 μg mL−1. Adapted from reference (Zhu et al., 2018) with permission from the American Chemical Society.
HeLa and 3T3 cells were used to test the cell-penetrating TADF PDots as imaging agents. As expected, low cytotoxicity was observed from all three TADF Pdots using both cell lines with 24 h of incubation at 37°C. Cell viabilities remained above 80% with up to 10 μg/mL of TADF dye. After only 5 min of incubation with these Pdots (2 μg/mL of TADF dye) HeLa cells showed fluorescence by CLSM, with a 44% or higher fluorescence-positive rate by flow cytometry (Figures 10A–C). With an incubation time of 15 min the fluorescence-positive-rate rose to a minimum of 54%, cellular accumulation of the TADF Pdots increased dramatically (Figures 10D–F). Cellular uptake was also conducted with 3T3 cells with similar results. In this study, TRFI could be used with a delay time of only 50 ns to remove background fluorescence, yet delays as long as 500 ns could also be used with observable emission.
Controlling TADF With Polymer Morphology
Given the applications of TADF materials in polymers for imaging and sensing, our group has recently explored methods for modulating TADF behavior in polymers by controlling polymer morphology (Tonge and Hudson, 2019). With typical TADF emitters it can be challenging to achieve turn-on/off delayed fluorescence emission using a small molecule emitter in which the donor and acceptor are bound covalently. In 2017, Wang and coworkers reported that TADF behavior could be observed in non-conjugated copolymers of discrete donor and acceptor monomers using a through-space charge transfer (TSCT) mechanism (Shao et al., 2017). Wang and coworkers hypothesized that minimization of donor-acceptor overlap and a small ΔEST could be achieved simply by copolymerizing electron-rich and electron-poor organic semiconductors, using a polymer backbone to achieve close D-A contact. This strategy was used successfully to fabricate high-efficiency blue OLEDs, as well as achieving color-tunable emission by modifying the acceptor electronic properties (Hu et al., 2019). Excited by this new paradigm in TADF polymer research, we sought to determine if alternative polymer morphologies could be used to control TSCT emission, using bottlebrush copolymers (BBCPs). Consisting of polymeric side chains attached to a linear backbone, the steric interactions of the densely packed side chains induce an extended backbone conformation, allowing for discrete nanoscale domains to be prepared along the length of the polymer.
Using this approach, we designed a series of BBCPs composed of acridine-based donors and triazine-based acceptors, varying the degree of donor-acceptor blending along the BBCP to control TSCT (Figure 11) (Tonge and Hudson, 2019). Grafting density along the bottlebrush backbone was maximized by employing a grafting-through polymerization strategy, in which short linear polymers functionalized with reactive norbornene end groups (termed “macromonomers”) are prepared first, then polymerized using ring-opening metathesis polymerization to give the BBCPs. Three bottlebrush copolymers containing 50% acridine-based donor and 50% triazine-based acceptor were prepared: one bottlebrush in which the macromonomers themselves were random D-A copolymers, another mixed arm (or “miktoarm”) bottlebrush consisting of a random copolymer of D or A macromonomers, and a third in which the D and A macromonomers were polymerized to give a diblock BBCP.
Figure 11.
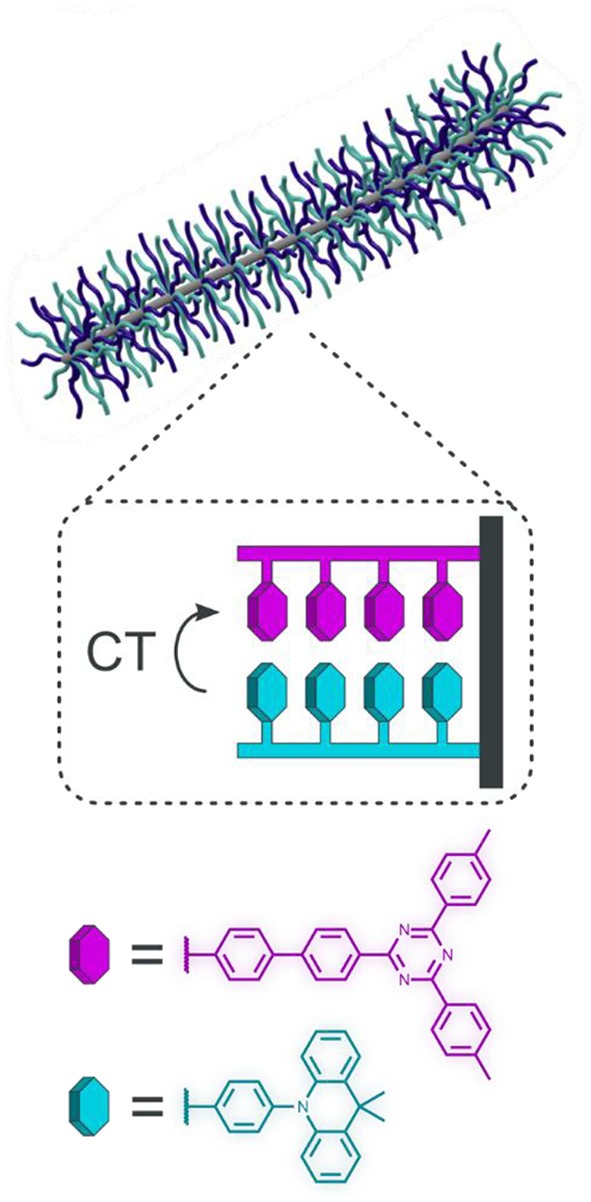
Schematic of TSCT within BBCPs.
These blended BBCPs were found to exhibit unique photophysical properties as a direct result of their morphology. The diblock copolymer displayed only prompt fluorescence in both the solution and solid state, while the miktoarm and random copolymers showed some degree of TSCT emission. The fully blended BBCP exhibited only TSCT emission in both the solid state and in solution, as near-complete mixing was ensured by the microstructure of the bottlebrush. Interestingly, by segregating the donor and acceptor functionalities in the diblock polymer, minimal interpenetration of these blocks was observed in the solid state or in solution, resulting in emission from only the monomers themselves. This result is noteworthy as a simple spin-cast film of a blend of linear donor and acceptor polymers gives both monomer and TSCT emission. This implies that using a bottlebrush backbone, the degree of interaction between these monomers can be controlled simply by modulating polymer morphology.
The TSCT in these bottlebrushes was also found to exhibit a substantial AIE effect, particularly in the case of the miktoarm copolymer (Figure 12A). When dissolved in THF, the miktoarm polymer demonstrates an approximately equal mix of prompt emission and TSCT emission, as adjacent chains are spread out due to favorable interactions with the solvent. Upon aggregation in the presence of water, however, the solvent is excluded, leading to a significant increase in TSCT emission and a corresponding decrease in prompt fluorescence (Figures 12B–D). This results in a 15.8-fold increase in brightness for the delayed fluorescence peak, which is not observed in the case of the diblock bottlebrush where TSCT is prevented by the polymer morphology. The fluorescence lifetime was also found to increase substantially upon aggregation of the random and miktoarm BBCPs, a feature that can potentially be taken advantage of for fluorescence lifetime imaging and other sensing applications.
Figure 12.

(A) Schematic of conformational changes in the aggregated state; (B) lifetime increase from increasing water fraction (f w = 0 to 90%) at a concentration of 0.02 mg mL−1; (C) aggregation induced emission of miktoarm bottlebrush copolymer (0.02 mg mL−1), normalized such that the maximum intensity at f w = 0% is set to 1 and all other spectra are scaled accordingly; (D) image of the fluorescent emission of the corresponding aggregated samples from f w = 0–90%. λex = 313 nm.
Conclusions and Outlook
Alongside their applications in OLEDs, TADF materials are becoming increasingly attractive as emitters for sensors and imaging probes. Embedding TADF materials in polymer films or nanostructures has unlocked many such applications, allowing these materials to be compatible for applications such as biological imaging, and presenting new avenues for controlling the photophysics of the TADF dopant. Taking advantage of triplet quenching processes, polymer thin films have been demonstrated as effective sensors for molecular oxygen with tunable sensitivity depending on the polymer host chosen. Using a TADF material with dual emission, it has been demonstrated that oxygen sensing is also possible without internal calibrants, enabling color-contrast imaging with water-soluble nanoparticles. Using polymers with low oxygen permeability, effective temperature sensors with high sensitivity have also been developed with dynamic ranges tunable simply by the TADF dopant used. The effect of TADF dopant concentration on oxygen permeability and TADF is understudied and would bring benefit to the community.
Using BSA to encapsulate TADF emitters, water-soluble, biocompatible nanoparticles can be formed which permit the use of TADF compounds in TRFI. By using TADF compounds exhibiting AIE behavior or by incorporating glassy semiconductor hosts into the nanoparticle, the detrimental effects of aggregation can be alleviated and imaging using TADF nanoparticles can be improved. TADF Pdots can be formed from amphiphilic polymers to retain the high brightness of the dyes in aqueous media, and cell-penetrating peptides can be used as effective encapsulating agents to increase cellular uptake. Finally, polymer morphology alone has recently been found to be an effective method for controlling TSCT TADF, either by aggregation of TADF-based bottlebrush fibers, or by controlling the extent of D-A mixing along a bottlebrush backbone to give the level of TADF desired.
Stimuli-responsive polymers exploiting TADF have now opened up a wide range of future avenues for research in sensors and biological imaging. Time-gated detection is now possible in aqueous environments at high brightness without the use of phosphorescent metal complexes, and in some cases without the need for colocalization dyes for calibration. This is sure to create opportunities for TADF materials in the growing field of theranostics, wherein chemistries for cell targeting, imaging, and therapy are combined onto a single nanoparticle carrier. In imaging applications, the potential for TADF dopant leakage has yet to be addressed, with current implementations relying on the poor solubility of TADF dyes in water to prevent emitter loss from the polymer particle. Covalent incorporation of TADF dyes into the polymer structure would prevent this entirely, and presents a promising strategy deserving of further research. Moreover, imaging probes with high stability to photobleaching, high ΦF, and enhanced cellular uptake have yet to be achieved and are sure to be actively sought by the community. Overall, we believe that stimuli-responsive polymers exhibiting TADF have only begun to be explored, and are sure to have a bright future in biological, analytical, and photophysical research.
Author Contributions
This review was written by NP, CT, and ZH.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the Natural Sciences and Engineering Council of Canada (NSERC) for financial support. NP thanks NSERC for a postgraduate scholarship, and ZH is grateful for support from the Canada Research Chairs Program.
References
- Acar N., Kurzawa J., Fritz N., Stockmann A., Roman C., Schneider S., et al. (2003). Phenothiazine—pyrene dyads: photoinduced charge separation and structural relaxation in the CT state. J. Phys. Chem. A 107, 9530–9541. 10.1021/jp036250u [DOI] [Google Scholar]
- Anastasaki A., Nikolaou V., Nurumbetov G., Wilson P., Kempe K., Quinn J. F., et al. (2016). Cu(0)-mediated living radical polymerization: a versatile tool for materials synthesis. Chem. Rev. 116, 835–877. 10.1021/acs.chemrev.5b00191 [DOI] [PubMed] [Google Scholar]
- Baleizão C., Berberan-Santos M. N. (2011). The brightest fullerene: a new isotope effect in molecular fluorescence and phosphorescence. ChemPhysChem 12, 1247–1250. 10.1002/cphc.201100156 [DOI] [PubMed] [Google Scholar]
- Baleizão C., Nagl S., Schäferling M., Berberan-Santos M. N., Wolfbeis O. S. (2008). Dual fluorescence sensor for trace oxygen and temperature with unmatched range and sensitivity. Anal. Chem. 80, 6449–6457. 10.1021/ac801034p [DOI] [PubMed] [Google Scholar]
- Bodea C., Silberg I. (1968). Recent advances in the chemistry of phenothiazines. Adv. Heterocycl. Chem. 9, 321–460. 10.1016/S0065-2725(08)60375-X [DOI] [PubMed] [Google Scholar]
- Borisov S. M., Gatterer K., Bitschnau B., Klimant I. (2010). Preparation and characterization of chromium(III)-activated yttrium aluminum borate: a new thermographic phosphor for optical sensing and imaging at ambient temperatures. J. Phys. Chem. C 114, 9118–9124. 10.1021/jp1016467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borisov S. M., Klimant I. (2007). Ultrabright oxygen optodes based on cyclometalated iridium(III) coumarin complexes. Anal. Chem. 79, 7501–7509. 10.1021/ac0710836 [DOI] [PubMed] [Google Scholar]
- Borisov S. M., Nuss G., Klimant I. (2008). Red light-excitable oxygen sensing materials based on platinum(II) and palladium(II) benzoporphyrins. Anal. Chem. 80, 9435–9442. 10.1021/ac801521v [DOI] [PubMed] [Google Scholar]
- Chatterjee T., Wong K. T. (2019). Perspective on host materials for thermally activated delayed fluorescence organic light emitting diodes. Adv. Opt. Mater. 4:1800565 10.1002/adom.201800565 [DOI] [Google Scholar]
- Chen X.-K., Kim D., Brédas J.-L. (2018). Thermally Activated Delayed Fluorescence (TADF) path toward efficient electroluminescence in purely organic materials: molecular level insight. Acc. Chem. Res. 51, 2215–2224. 10.1021/acs.accounts.8b00174 [DOI] [PubMed] [Google Scholar]
- Chen X. K., Zhang S. F., Fan J. X., Ren A. M. (2015). Nature of highly efficient thermally activated delayed fluorescence in organic light-emitting diode emitters: nonadiabatic effect between excited states. J. Phys. Chem. C 119, 9728–9733. 10.1021/acs.jpcc.5b00276 [DOI] [Google Scholar]
- Cho Y. J., Jeon S. K., Chin B. D., Yu E., Lee J. Y. (2015). The design of dual emitting cores for green thermally activated delayed fluorescent materials. Angew. Chem. Int. Ed. 54, 5201–5204. 10.1002/anie.201412107 [DOI] [PubMed] [Google Scholar]
- Daub J., Engl R., Kurzawa J., Miller S. E., Schneider S., Stockmann A., et al. (2001). Competition between conformational relaxation and intramolecular electron transfer within phenothiazine-pyrene dyads. J. Phys. Chem. A 105, 5655–5665. 10.1021/jp0037293 [DOI] [Google Scholar]
- DeRosa C. A., Samonina-Kosicka J., Fan Z., Hendargo H. C., Weitzel D. H., Palmer G. M., et al. (2015). Oxygen sensing difluoroboron dinaphthoylmethane polylactide. Macromolecules 48, 2967–2977. 10.1021/acs.macromol.5b00394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias F. B. (2015). Kinetics of thermal-assisted delayed fluorescence in blue organic emitters with large singlet–triplet energy gap. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 373:20140447. 10.1098/rsta.2014.0447 [DOI] [PubMed] [Google Scholar]
- Dias F. B., Santos J., Graves D. R., Data P., Nobuyasu R. S., Fox M. A., et al. (2016). The role of local triplet excited states and D-A relative orientation in thermally activated delayed fluorescence: photophysics and devices. Adv. Sci. 3:1600080. 10.1002/advs.201600080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo A., Sato K., Yoshimura K., Kai T., Kawada A., Miyazaki H., et al. (2011). Efficient up-conversion of triplet excitons into a singlet state and its application for organic light emitting diodes. Appl. Phys. Lett. 98:083302 10.1063/1.3558906 [DOI] [Google Scholar]
- Gan S., Zhou J., Smith T. A., Su H., Luo W., Hong Y., et al. (2017). New AIEgens with delayed fluorescence for fluorescence imaging and fluorescence lifetime imaging of living cells. Mater. Chem. Front. 1, 2554–2558. 10.1039/C7QM00286F [DOI] [Google Scholar]
- Gibson J., Monkman A. P., Penfold T. J. (2016). The importance of vibronic coupling for efficient reverse intersystem crossing in thermally activated delayed fluorescence molecules. ChemPhysChem 17, 2956–2961. 10.1002/cphc.201600662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson J., Penfold T. J. (2017). Nonadiabatic coupling reduces the activation energy in thermally activated delayed fluorescence. Phys. Chem. Chem. Phys. 19, 8428–8434. 10.1039/C7CP00719A [DOI] [PubMed] [Google Scholar]
- Goushi K., Yoshida K., Sato K., Adachi C. (2012). Organic light-emitting diodes employing efficient reverse intersystem crossing for triplet-to-singlet state conversion. Nat. Photonics 6, 253–258. 10.1038/nphoton.2012.31 [DOI] [Google Scholar]
- He T., Ren C., Li Z., Xiao S., Li J., Lin X., et al. (2018). Thermally activated delayed fluorescence organic dots for two-photon fluorescence lifetime imaging. Appl. Phys. Lett. 112:211102 10.1063/1.5034375 [DOI] [Google Scholar]
- Hosokai T., Matsuzaki H., Nakanotani H., Tokumaru K., Tsutsui T., Furube A., et al. (2017). Evidence and mechanism of efficient thermally activated delayed fluorescence promoted by delocalized excited states. Sci. Adv. 3:e1603282. 10.1126/sciadv.1603282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J., Li Q., Wang X., Shao S., Wang L., Jing X., et al. (2019). Developing through-space charge transfer polymers as a general approach to realize full-color and white emission with thermally activated delayed fluorescence. Angew. Chem. Int. Ed. 58, 8405–8409. 10.1002/anie.201902264 [DOI] [PubMed] [Google Scholar]
- Jankus V., Data P., Graves D., McGuinness C., Santos J., Bryce M. R., et al. (2014). Highly efficient TADF OLEDs: how the emitter-host interaction controls both the excited state species and electrical properties of the devices to achieve near 100% triplet harvesting and high efficiency. Adv. Funct. Mater. 24, 6178–6186. 10.1002/adfm.201400948 [DOI] [Google Scholar]
- Khalil G. E., Lau K., Phelan G. D., Carlson B., Gouterman M., Callis J. B., et al. (2004). Europium beta-diketonate temperature sensors: effects of ligands, matrix, and concentration. Rev. Sci. Instrum. 75, 192–206. 10.1063/1.1632997 [DOI] [Google Scholar]
- Kochmann S., Baleizão C., Berberan-Santos M. N., Wolfbeis O. S. (2013). Sensing and imaging of oxygen with parts per billion limits of detection and based on the quenching of the delayed fluorescence of 13C 70 fullerene in polymer hosts. Anal. Chem. 85, 1300–1304. 10.1021/ac303486f [DOI] [PubMed] [Google Scholar]
- Kuo S.-Y., Li H.-H., Wu P.-J., Chen C.-P., Huang Y.-C., Chan Y.-H. (2015). Dual colorimetric and fluorescent sensor based on semiconducting polymer dots for ratiometric detection of lead ions in living cells. Anal. Chem. 87, 4765–4771. 10.1021/ac504845t [DOI] [PubMed] [Google Scholar]
- Li T., Yang D., Zhai L., Wang S., Zhao B., Fu N., et al. (2017). Thermally activated delayed fluorescence organic dots (TADF Odots) for time-resolved and confocal fluorescence imaging in living cells and in vivo. Adv. Sci. 4:1600166. 10.1002/advs.201600166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebsch G., Klimant I., Wolfbeis O. S. (1999). Luminescence lifetime temperature sensing based on sol-gels and poly(acrylonitrile)s dyed with ruthenium metal-ligand complexes. Adv. Mater. 11, 1296–1299. [DOI] [Google Scholar]
- Lim B. T., Okajima S., Chandra A. K., Lim E. C. (1981). Radiationless transitions in electron donor-acceptor complexes: selection rules for S1 → T intersystem crossing and efficiency of S1 → S0 internal conversion. Chem. Phys. Lett. 79, 22–27. 10.1016/0009-2614(81)85280-3 [DOI] [Google Scholar]
- Ma Y., Gong C., Ma Y., Fan F., Luo M., Yang F., et al. (2012). Direct cytosolic delivery of cargoes in vivo by a chimera consisting of D- and L-arginine residues. J. Control. Release 162, 286–294. 10.1016/j.jconrel.2012.07.022 [DOI] [PubMed] [Google Scholar]
- Malrieu J. P., Pullman B. (1964). Sur les propriétés électroniques de la phénothiazine et de son radical. Theor. Chim. Acta 2, 293–301. 10.1007/BF00527056 [DOI] [Google Scholar]
- Marghad I., Bencheikh F., Wang C., Manolikakes S., Rérat A., Gosmini C., et al. (2019). Control of the dual emission from a thermally activated delayed fluorescence emitter containing phenothiazine units in organic light-emitting diodes. RSC Adv. 9, 4336–4343. 10.1039/C8RA10393C [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marian C. M. (2016). Mechanism of the triplet-to-singlet upconversion in the assistant dopant ACRXTN. J. Phys. Chem. C 120, 3715–3721. 10.1021/acs.jpcc.6b00060 [DOI] [Google Scholar]
- Massey M., Wu M., Conroy E. M., Algar W. R. (2015). Mind your P's and Q's: the coming of age of semiconducting polymer dots and semiconductor quantum dots in biological applications. Curr. Opin. Biotechnol. 34, 30–40. 10.1016/j.copbio.2014.11.006 [DOI] [PubMed] [Google Scholar]
- Nakanotani H., Masui K., Nishide J., Shibata T., Adachi C. (2013). Promising operational stability of high-efficiency organic light-emitting diodes based on thermally activated delayed fluorescence. Sci. Rep. 3:2127. 10.1038/srep02127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki M., Takeda Y., Data P., Pander P., Higginbotham H., Monkman A. P., et al. (2017). Thermally activated delayed fluorescent phenothiazine–dibenzo[a,j]phenazine–phenothiazine triads exhibiting tricolor-changing mechanochromic luminescence. Chem. Sci. 8, 2677–2686. 10.1039/C6SC04863C [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmeira T., Berberan-Santos M. N. (2018). “TADF kinetics and data analysis in photoluminescence and in electroluminescence,” in Highly Efficient OLEDs (Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA; ), 229–255. 10.1002/9783527691722.ch7 [DOI] [Google Scholar]
- Penfold T. J., Dias F. B., Monkman A. P. (2018). The theory of thermally activated delayed fluorescence for organic light emitting diodes. Chem. Commun. 54, 3926–3935. 10.1039/C7CC09612G [DOI] [PubMed] [Google Scholar]
- Sauv,é E. R., Tonge C. M., Paisley N. R., Cheng S., Hudson Z. M. (2018). Cu(0)-RDRP of acrylates based on p-type organic semiconductors. Polym. Chem. 9, 1397–1403. 10.1039/C8PY00295A [DOI] [Google Scholar]
- Shao S., Hu J., Wang X., Wang L., Jing X., Wang F. (2017). Blue thermally activated delayed fluorescence polymers with nonconjugated backbone and through-space charge transfer effect. J. Am. Chem. Soc. 139, 17739–17742. 10.1021/jacs.7b10257 [DOI] [PubMed] [Google Scholar]
- Steinegger A., Klimant I., Borisov S. M. (2017). Purely organic dyes with thermally activated delayed fluorescence-a versatile class of indicators for optical temperature sensing. Adv. Opt. Mater. 5:1700372 10.1002/adom.201700372 [DOI] [Google Scholar]
- Stockmann A., Kurzawa J., Fritz N., Acar N., Schneider S., Daub J., et al. (2002). Conformational control of photoinduced charge separation within phenothiazine-pyrene dyads. J. Phys. Chem. A 106, 7958–7970. 10.1021/jp0142987 [DOI] [Google Scholar]
- Tanaka H., Shizu K., Nakanotani H., Adachi C. (2014). Dual intramolecular charge-transfer fluorescence derived from a phenothiazine-triphenyltriazine derivative. J. Phys. Chem. C 118, 15985–15994. 10.1021/jp501017f [DOI] [Google Scholar]
- Tonge C. M., Hudson Z. M. (2019). Interface-dependent aggregation-induced delayed fluorescence in bottlebrush polymer nanofibers. J. Am. Chem. Soc. 141, 13970–13976. 10.1021/jacs.9b07156 [DOI] [PubMed] [Google Scholar]
- Tonge C. M., Paisley N. R., Polgar A. M., Lix K., Algar W. R., Hudson Z. M. (2020). Color-tunable thermally activated delayed fluorescence in oxadiazole-based acrylic copolymers: photophysical properties and applications in ratiometric oxygen sensing. ACS Appl. Mater. Interfaces 12, 6525–6535. 10.1021/acsami.9b22464 [DOI] [PubMed] [Google Scholar]
- Tonge C. M., Yuan F., Lu Z.-H., Hudson Z. M. (2019). Cu(0)-RDRP as an efficient and low-cost synthetic route to blue-emissive polymers for OLEDs. Polym. Chem. 10, 3288–3297. 10.1039/C9PY00294D [DOI] [Google Scholar]
- Tsuchiya Y., Ikesue K., Nakanotani H., Adachi C. (2019). Photostable and highly emissive glassy organic dots exhibiting thermally activated delayed fluorescence. Chem. Commun. 55, 5215–5218. 10.1039/C9CC01420A [DOI] [PubMed] [Google Scholar]
- Uoyama H., Goushi K., Shizu K., Nomura H., Adachi C. (2012). Highly efficient organic light-emitting diodes from delayed fluorescence. Nature 492, 234–238. 10.1038/nature11687 [DOI] [PubMed] [Google Scholar]
- Wong M. Y., Zysman-Colman E. (2017). Purely organic thermally activated delayed fluorescence materials for organic light-emitting diodes. Adv. Mater. 29:1605444. 10.1002/adma.201605444 [DOI] [PubMed] [Google Scholar]
- Wu H., Ying L., Yang W., Cao Y. (2009). Progress and perspective of polymer white light-emitting devices and materials. Chem. Soc. Rev. 38, 3391–3400. 10.1039/b816352a [DOI] [PubMed] [Google Scholar]
- Wu Z., Yu L., Zhou X., Guo Q., Luo J., Qiao X., et al. (2016). Management of singlet and triplet excitons: a universal approach to high-efficiency all fluorescent WOLEDs with reduced efficiency roll-off using a conventional fluorescent emitter. Adv. Opt. Mater. 4, 1067–1074. 10.1002/adom.201600117 [DOI] [Google Scholar]
- Xiong X., Song F., Sun S., Fan J., Peng X. (2013). Red-emissive fluorescein derivatives and detection of bovine serum albumin. Asian J. Org. Chem. 2, 145–149. 10.1002/ajoc.201200109 [DOI] [Google Scholar]
- Xiong X., Song F., Wang J., Zhang Y., Xue Y., Sun L., et al. (2014). Thermally activated delayed fluorescence of fluorescein derivative for time-resolved and confocal fluorescence imaging. J. Am. Chem. Soc. 136, 9590–9597. 10.1021/ja502292p [DOI] [PubMed] [Google Scholar]
- Yang Z., Mao Z., Xie Z., Zhang Y., Liu S., Zhao J., et al. (2017). Recent advances in organic thermally activated delayed fluorescence materials. Chem. Soc. Rev. 46, 915–1016. 10.1039/C6CS00368K [DOI] [PubMed] [Google Scholar]
- Yu J., Rong Y., Kuo C.-T., Zhou X.-H., Chiu D. T. (2017). Recent advances in the development of highly luminescent semiconducting polymer dots and nanoparticles for biological imaging and medicine. Anal. Chem. 89, 42–56. 10.1021/acs.analchem.6b04672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Chen R., Zhu Z., Adachi C., Zhang X., Lee C.-S. (2015). Highly stable near-infrared fluorescent organic nanoparticles with a large stokes shift for noninvasive long-term cellular imaging. ACS Appl. Mater. Interfaces 7, 26266–26274. 10.1021/acsami.5b08539 [DOI] [PubMed] [Google Scholar]
- Zhang Q., Li J., Shizu K., Huang S., Hirata S., Miyazaki H., et al. (2012). Design of efficient thermally activated delayed fluorescence materials for pure blue organic light emitting diodes. J. Am. Chem. Soc. 134, 14706–14709. 10.1021/ja306538w [DOI] [PubMed] [Google Scholar]
- Zhang Q., Xu S., Li M., Wang Y., Zhang N., Guan Y., et al. (2019). Rationally designed organelle-specific thermally activated delayed fluorescence small molecule organic probes for time-resolved biological applications. Chem. Commun. 55, 5639–5642. 10.1039/C9CC00898E [DOI] [PubMed] [Google Scholar]
- Zhu Z., Tian D., Gao P., Wang K., Li Y., Shu X., et al. (2018). Cell-penetrating peptides transport noncovalently linked thermally activated delayed fluorescence nanoparticles for time-resolved luminescence imaging. J. Am. Chem. Soc. 140, 17484–17491. 10.1021/jacs.8b08438 [DOI] [PubMed] [Google Scholar]