

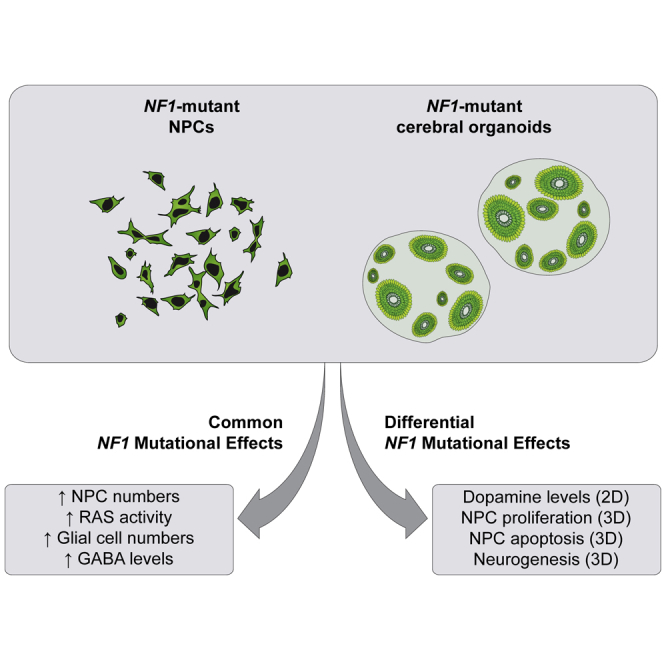
Summary
Neurofibromatosis type 1 (NF1) is a common neurodevelopmental disorder caused by a spectrum of distinct germline NF1 gene mutations, traditionally viewed as equivalent loss-of-function alleles. To specifically address the issue of mutational equivalency in a disease with considerable clinical heterogeneity, we engineered seven isogenic human induced pluripotent stem cell lines, each with a different NF1 patient NF1 mutation, to identify potential differential effects of NF1 mutations on human central nervous system cells and tissues. Although all mutations increased proliferation and RAS activity in 2D neural progenitor cells (NPCs) and astrocytes, we observed striking differences between NF1 mutations on 2D NPC dopamine levels, and 3D NPC proliferation, apoptosis, and neuronal differentiation in developing cerebral organoids. Together, these findings demonstrate differential effects of NF1 gene mutations at the cellular and tissue levels, suggesting that the germline NF1 gene mutation is one factor that underlies clinical variability.
Keywords: neurofibromatosis type 1, disease modeling, human iPSCs, neurodevelopment
Graphical Abstract
Highlights
-
•
All NF1 patient mutations increase RAS activity and cell proliferation
-
•
NF1-mutant neurons have both shared and differential abnormalities in 2D cultures
-
•
NF1 mutations differentially affect proliferation/apoptosis in 3D organoid-NPCs
-
•
NF1 mutations differentially affect neuronal differentiation in 3D cerebral organoids
To critically evaluate NF1 mutational equivalency in human brain cells, Anastasaki and colleagues generated an isogenic series of seven human induced pluripotent stem cell lines harboring different NF1 patient NF1 mutations. Although all mutations increased 2D NPC and astrocyte RAS activity and cell proliferation, distinct NF1 mutations had differential effects on NPC proliferation, apoptosis and neuronal differentiation within 3D cerebral organoids.
Introduction
Neurofibromatosis 1 (NF1; OMIM 162200) is a neurogenetic condition characterized by remarkable phenotypic variability, where affected children develop a wide variety of central nervous system (CNS) pathologies, ranging from brain tumors and motor delays to learning difficulties, attention deficits, and autism (Fisher et al., 2018, Hyman et al., 2006, Jett and Friedman, 2010, Korf, 2013, Morris and Gutmann, 2018). One of many potential factors underlying this clinical variability could be the specific NF1 germline mutation, a notion suggested by population-based studies (Anastasaki et al., 2017, Bolcekova et al., 2013, Kehrer-Sawatzki et al., 2017, Koczkowska et al., 2018, Pinna et al., 2015, Rojnueangnit et al., 2015, Sharif et al., 2011, Trevisson et al., 2019, Upadhyaya et al., 2007). For example, patients harboring the c.2970–2971_delAAT, c.5425C > T, and c.3112A > G NF1 germline mutations lack dermal and plexiform neurofibromas, the signature peripheral nervous system tumors in NF1 (Pinna et al., 2015, Trevisson et al., 2019, Upadhyaya et al., 2007).
Although these studies raise the possibility that not all NF1 gene mutations are functionally equivalent, they do not establish differential effects of NF1 patient germline mutations at the cellular or tissue levels, a critical step in interpreting the significance of reported genotype-phenotype associations. To specifically evaluate differential NF1 mutation effects on human CNS cells and tissues, while controlling for important confounding factors (e.g., sex, genomic differences), we generated an isogenic series of human induced pluripotent stem cells (hiPSCs) harboring seven representative NF1-patient NF1 mutations.
Results and Discussion
Generation of Isogenic NF1-Mutant hiPSCs
The seven NF1 pathogenic mutations, derived from patients in our clinic population at Washington University/St. Louis Children's Hospital, represent the spectrum of mutations typically seen in individuals with NF1. In this regard, the selected mutations were interspersed throughout the NF1 protein (neurofibromin) coding sequence, were both proximal and distal to the well-characterized RAS GTPase-activating protein (RAS-GAP) domain (GRD), and included four nonsense (c.1149C > A, c.2041C > T, c.6513T > A, c.6619C > T), one splice site (c.1185+1G > A), one missense (c.5425C > T), and one frameshift (c.3431-32_dupGT) mutation (Figures 1A, S1, and S2). All of the engineered isogenic hiPSCs harbored only a single NF1 mutation (“NF1-mutant”), retained expression of the remaining wild-type NF1 allele as confirmed by DNA and RNA sequencing (Figure S1), and expressed similar levels of NF1 mRNA (Figure 1B). For all hiPSC lines with two clones, identical results were obtained using numerous independently generated biological replicates, as well as with three NF1 patient-derived hiPSC lines generated from somatic cells (fibroblasts; Figure S3, Table S1).
Figure 1.

Isogenic NF1-Mutant hiPSC-Derived NPCs Exhibit Increased RAS Activity and Cell Proliferation
(A) Schematic diagram illustrating the position of the engineered NF1 patient mutations within the NF1 gene. The location of the RAS-GAP domain is highlighted in black.
(B) Relative NF1 mRNA expression in isogenic NF1-mutant NPCs is similar to the controls.
(C–E) (C) Quantitation demonstrating increased RAS activity (RAS-GTP) in isogenic NF1-mutant NPCs relative to controls (CTL) before and after the addition of (D) 10 μg/mL FGF or (E) BDNF. A minimum of three independent replicates was performed for each treatment condition.
(F) BrdU incorporation is increased by 2.6- to 3.2-fold in NF1-mutant NPCs relative to control NPCs.
(G) 1.9- to 2-fold increases in total cell numbers were observed in NF1-mutant NPCs compared with controls.
(H) Representative bright-field images of embryoid bodies and cerebral organoids at 16 and 56 DIV.
(I and J) (I) Quantitation demonstrating increased RAS activity (2.8- to 3.2-fold) and (J) increased numbers of SOX2+ NPCs per ventricular zone (1.6- to 2.2-fold) in 16 DIV NF1-mutant cerebral organoids relative to control organoids.
Each dot represents an independently generated data point derived from separate experiments and the two different clones for each line are denoted as black versus gray dots. All data are represented as means ± SEM. (B, C, F, G, I, and J) One-way ANOVA with Tukey post-test. (D and E) Two-way ANOVA with Bonferroni post-test. n.s., not significant. Scale bar, 1 mm.
Isogenic NF1-Mutant hiPSC-Derived NPCs and Astroglia Have Increased RAS Activity and Proliferation
To determine the consequence of the different NF1 gene mutations on neurofibromin signal transduction and function in human CNS cells, NF1-mutant and control hiPSCs were first differentiated into neural progenitor cells (NPCs) capable of generating both neurons (TUJ1+ cells) and glia (S100β+ cells) (Figure S2B). Because neurofibromin primarily functions as an RAS-GAP to control cell proliferation, we initially assessed RAS activity. Consistent with this negative RAS regulatory property, all NF1-mutant NPCs exhibited a comparable 1.8- to 2.2-fold increase in RAS-GTP relative to the isogenic control (Figure 1C). Importantly, the addition of growth factors (fibroblast growth factor [FGF] or brain-derived neurotrophic factor [BDNF]) did not further increase RAS activity in the NF1-mutant lines, but resulted in greater RAS-GTP levels in the control lines, equivalent to the levels observed in the unstimulated NF1-mutant lines (Figures 1D and 1E). These findings demonstrate that a heterozygous NF1 mutation phenocopies the effect of exogenous growth factor stimulation on RAS activation. In addition, all NF1-mutant NPCs exhibited increased cell division, as demonstrated by increased bromodeoxyuridine (BrdU) incorporation (2.6- to 3.2-fold increase; Figure 1F) and total cell number (1.9- to 2-fold increase; Figure 1G). To evaluate the effects of distinct NF1 gene mutations on the production of NPCs in a 3D model of human brain development, we generated cerebral organoids from the control and NF1-mutant hiPSC lines. Despite repeated efforts, we were unable to derive organoids from two of the seven NF1-mutant hiPSC lines (c.2041C > T and c.6513T > A), but successfully generated organoids from the control and five of the seven NF1-mutant hiPSC lines (c.1149C > A, c.1185+1G > A, c.3431-32_dupGT, c.5425C > T, c.6619C > T; Figure 1H). The organoids formed radially organized ventricle-like structures populated by SOX2+ NPCs by 16 days in vitro (DIV). Similar to the 2D cultures, all NF1-mutant organoids exhibited a 2.8- to 3.2-fold increase in RAS activity (Figure 1I), as well as a 1.6- to 2.2-fold increase in total NPCs per ventricular zone at 16 DIV (Figure 1J).
Next, we sought to determine whether these heterozygous NF1 mutational effects were observed in another proliferating CNS cell type by differentiating the NPCs into astrocytes (Figure 2A). Similar to the NPCs, NF1-mutant astrocytes exhibited 2- to 2.3-fold increased RAS activity (Figure 2B), 2.3- to 2.7-fold increased cell division (Figure 2C), and 2.1- to 2.5-fold greater total cell number (Figure 2D) relative to the control line. Consistent with the 2D astrocytes, 56 DIV NF1-mutant organoids had more EAAT1- and GFAP-expressing cells (astrocytes) (Figure 2E) compared with control organoids. Importantly, isogenic NF1-mutant NPCs and organoids were similar to those of their respective patient-derived NPCs (c.1185+1G > A; c.5425C > T; c.6513T > A) and organoids (c.1185+1G > A; c.5425C > T) (Figures S3 and S4; Table S1) in RAS activity and NPC proliferation, as well as to whole-brain lysates from genetically engineered mice harboring the analogous germline Nf1 gene mutations (c.1149C > A, c.2041C > T, c.3431-32_dupGT, and c.5425C > T; Figure 3A). Taken together, these data illustrate that all heterozygous NF1 mutations increase RAS activity and RAS-regulated cell proliferation in both human and murine CNS cells.
Figure 2.

hiPSC-Derived NF1-Mutant Astroglia Exhibit Increased RAS Activity and Cell Proliferation
(A) NF1-mutant and control NPCs were differentiated into GFAP+, S100+, EAAT1+, and EAAT2+ astrocytes in 2D cultures. Scale bar, 100 μm.
(B) RAS-GTP was increased by 2- to 2.3-fold in NF1-mutant astrocytes relative to controls (CTL).
(C) Proliferation of NF1-mutant astrocytes was 2.3- to 2.7-fold higher relative to controls.
(D) Direct cell counting demonstrated a 2.1- to 2.5-fold increase in NF1-mutant astrocytes compared with controls.
(E) NF1-mutant cerebral organoids grown for 56 DIV had increased cells with GFAP+ fibers and increased numbers of EAAT1+ glial cells compared with isogenic controls. Scale bars, 50 μm.
Each dot represents an independently generated data point derived from separate experiments and the two different clones for each line are denoted as black versus gray dots. All data are represented as means ± SEM. One-way ANOVA with Tukey post-test.
Figure 3.

hiPSC-Derived NF1-Mutant Neurons, NPCs, and Nf1-Mutant Mice Display Molecular Similarities and Differences
(A) Nf1-mutant (1149C > A, 2041C > T, 3431-32_dupGT, 5425C > T) genetically engineered mouse brain lysates exhibit increased RAS activity compared with wild-type littermate controls.
(B) GABA levels are increased in all NF1-mutant NPC-derived neurons relative to controls.
(C and D) Dopamine levels are differentially reduced in (C) NF1-mutant NPCs relative to controls and (D) Nf1-mutant genetically engineered mouse brain lysates compared with WT littermate controls.
Each dot represents an independently generated data point derived from separate experiments and the two different clones for each line are denoted as black versus gray dots. All data are represented as means ± SEM. One-way ANOVA with Tukey post-test.
hiPSC-Derived NF1-Mutant Neurons Exhibit Both Shared and Differential Deficits in 2D Cultures
As some children with NF1 exhibit cognitive deficits and neurodevelopmental delays (Hyman et al., 2005, Hyman et al., 2006, Jett and Friedman, 2010, Morris and Gutmann, 2018), we sought to determine the effects of distinct NF1 germline mutations on human CNS neuronal function and differentiation. Based on the observation that Nf1-mutant (Nf1+/−) mice exhibit increased GABAergic tone that contributes to the observed deficits in learning and spatial memory (Costa et al., 2002, Cui et al., 2008), we assayed GABA levels in NPC-derived GABAergic neurons (Figures 3B and S3E). In all NF1-mutant neurons (2D cultures), GABA levels were increased (6.5- to 7.8-fold) relative to isogenic control neurons, revealing a shared abnormality in all NF1-mutant GABAergic neurons.
In contrast, NF1-mutant NPCs in 2D cultures displayed striking differences in dopamine (DA) (Figure 3C) levels. DA levels were reduced by >70% in the c.1149C > A, c.2041C > T, and c.6619C > T NF1 mutants, but by <40% in the c.1185+1G > A, c.3431-32_dupGT, c.5425C > T, and c.6513T > A NF1 mutants relative to the control line. These differential effects mirror findings using patient-derived NPCs (Figures S3E and S3F; Table S1) (Anastasaki et al., 2015), as well as mice engineered with NF1 patient-specific Nf1 germline mutations (Figure 3D) (Toonen et al., 2016). Taken together, these findings demonstrate the existence of differential effects of NF1 germline mutations on neuronal differentiation in vitro.
Differential Effects of NF1 Mutations on Cerebral Organoid NPC Proliferation, Apoptosis and Differentiation
To further explore the differential effects of NF1 mutations in the developing human brain, we used the more contextually relevant cerebral organoid platform. Examination of NPC proliferation, apoptosis and neuronal differentiation in 16 DIV cerebral organoids revealed two distinct groups of NF1 mutants (Figure 4): group 1 (c.1185+1G > A; c.5425C > T; c.6619C > T) NF1 mutants exhibited increased NPC proliferation (1.3- to 1.4-fold) and apoptosis (2- to 3-fold), but had similar numbers of early (NeuroD1+, TUJ1+) and late (MAP2+) immature neurons relative to controls. In this manner, group 1 NF1 mutations increased both proliferation and apoptosis during NPC differentiation, allowing neurogenesis to proceed normally. In contrast, group 2 (c.1149C > A; c.3431-32_dupGT) NF1-mutant organoids had normal NPC proliferation, but reduced NPC apoptosis (70%–92% reduction) and very few immature neurons relative to the isogenic controls (73%–84% reduction). In this latter group, the reduction in NPC death was coupled with a delay in neurogenesis, suggesting that inappropriate survival of NPC subpopulations creates a barrier to initiating timely neuronal differentiation. Importantly, these observations persist in patient-derived cerebral organoids harboring the same mutations (Figure S4; Table S1).
Figure 4.

Differential Effects of NF1 Mutations on Cerebral Organoid Progenitor Cell Dynamics and Neurogenesis
(A and B) SOX2+ NPCs in the ventricular zones of group 1 NF1-mutant cerebral organoids exhibit (A) 1.3- to 1.4-fold increased proliferation (Ki67+ cells; white arrowheads) and (B) 2- to 3-fold increased cell apoptosis (cleaved caspase-3; white arrowheads) compared with control and group 2 cerebral organoids at 16 DIV.
(C–E) Decreased numbers of (C and D) early immature neurons (NeuroD1; TUJ1 white arrowheads) and (E) late immature neurons (MAP2; white arrowheads) migrating into the periventricular zone of group 2 compared with group 1 and control cerebral organoids at 16 DIV.
(F) Quantifications of %Ki67+ NPCs, %cleaved caspase-3+ NPCs and NeuroD1+ immature neurons in NF1-mutant cerebral organoids compared with controls at 16 DIV.
Each dot represents an independently generated data point derived from separate experiments and the two different clones for each line are denoted as black versus gray dots. All data are represented as means ± SEM. One-way ANOVA with Dunnett post-test. n.s., not significant. Scale bars, 10 μm (C) and 50 μm (A, B, D, and E).
Conclusions
The findings described in this report, in combination with compelling population-based genotype-phenotype associations, suggest that the germline NF1 gene mutation is one of the factors that underlies clinical heterogeneity in patients with NF1. Using an isogenic series of NF1-mutant hiPSC lines, we identified differential NF1 mutational effects on human CNS cells and tissues. Importantly, unlike previous studies, the use of an isogenic series of hiPSCs eliminates other contributing factors, such as sex and genomic variation (potential modifier genes), and permits a direct examination of the effects of different NF1 gene mutations. Moreover, this study raises several important points relevant to NF1 pathobiology.
First, we established that all heterozygous NF1 gene mutations similarly increase CNS NPC and astroglial cell proliferation and RAS activity, which is consistent with numerous reports demonstrating that neurofibromin controls cell proliferation largely by regulating RAS activity in mouse, swine, and Drosophila cells and tissues. Moreover, the regulation of RAS-mediated cell proliferation by neurofibromin is further supported using paired hiPSC-derived NPCs, heterozygous and homozygous for the same NF1 gene mutation, where a clear gene dose dependency was revealed (C. Anastasaki, Personal Communication). Therefore, the vast majority of human clinical trials for NF1-null tumors have appropriately used molecularly targeted therapies that inhibit RAS and RAS downstream effectors (e.g., MEK) (Dombi et al., 2016).
Second, we demonstrated differential effects of NF1 germline mutations on neuronal differentiation. These differential effects could reflect the fact that neurofibromin functions as a high-affinity dimer, where different mutations could change the overall architecture of the dimer interface (Sherekar et al., 2020). Because neurons from individuals with NF1 harbor only a single NF1 germline mutation, different NF1 mutations likely cause unique neuron-related pathologies. Therefore, the use of isogenic hiPSCs revealed differential effects of distinct NF1 gene mutations on NPC proliferation, apoptosis, and neuronal differentiation not previously reported in the developing Nf1-knockout (Nf1wt/neo) mouse brain. Given the high degree of mutational specificity for autism symptomatology in children with NF1 (Morris and Gutmann, 2018), these findings suggest that investigations using Nf1 mice with different patient germline Nf1 mutations might uncover unique behavioral abnormalities not appreciated using conventional Nf1 knockout mice (Costa et al., 2002, Omrani et al., 2015) and identify causative underlying molecular mechanisms.
Third, the fact that the observed differences in neuronal differentiation in cerebral organoids and NPC DA levels do not correlate with RAS activation supports the existence of non-RAS-mediated neurofibromin functions. In this regard, neurofibromin also directly binds to several proteins important for neuronal differentiation, spinogenesis, and serotonin receptor activity, including collapsin response mediator protein-2 (Patrakitkomjorn et al., 2008), syndecan (Hsueh et al., 2001), and the 5-hydroxyltryptamine-6 receptor (Deraredj Nadim et al., 2016), through domains distinct from the GRD. Moreover, the notion that non-RAS-mediated neurofibromin functions exist in neurons is reinforced by the presence of a neurofibromin isoform containing an additional amino terminal exon (11alt12), whose expression is restricted to postnatal brain neurons (Gutmann et al., 1999). Future investigations aimed at discovering novel neuron-specific neurofibromin binding partners will be critical to understanding how NF1 mutations differentially affect cognition and behavior in children with NF1.
Finally, although population and murine studies provided the first evidence for NF1 genotype-phenotype correlations, there had been no direct demonstration of the primary effect of the NF1 mutation at the cellular and tissue levels in humans. The use of this experimental human iPSC platform revealed NF1 mutational abnormalities in human NPCs and neurons. Collectively, these studies establish a foundational basis for future studies aimed at unraveling mechanistic etiologies responsible for NF1-specific CNS phenotypes, discovering new therapeutic targets, and assessing treatments relevant to precision medicine.
Experimental Procedures
Human iPSCs
Seven distinct NF1-patient germline NF1 gene mutations (Transcript ID NM_000267; c.1149C > A, c.1185+1G > A, c.2041C > T, c.3431-32_dupGT, c.5425C > T, c.6513T > A, c.6619C > T) were individually engineered using CRISPR/Cas9 technology into a single commercially available male control human iPSC line (BJFF.6) by the Washington University Genome Engineering and iPSC Core (GEiC) facility. Heterozygous mutations were confirmed by NGS sequencing (Bell et al., 2014), and two different clones were expanded for each of the six NF1-mutant and the control lines (Figures S1A–S1C). Only a single clone heterozygous for the c.6619C > T NF1 mutation could be generated without any additional genomic insertions or deletions. Retention of heterozygosity in the hiPSCs was confirmed by sequencing after five passages, as well as in all of the derivative cell lines by RAS activity assays. Similar results were obtained after each passage. In addition, iPSCs reprogrammed from the fibroblasts of three NF1 patients (c.1185+1G > A; c.5425C > T; c.6513T > A) and one control subject (Anastasaki et al., 2015) were used for subsequent analyses. For NPC differentiation, hiPSCs were passaged onto PLO/Laminin (Millipore Sigma)-coated plates using ReLeSR (STEMCELL Technologies), and seeded at 200,000 cells/cm2 in NPC induction medium (50% DMEM F12 [Gibco], 50% Neurobasal medium [Gibco], supplemented with N2, B27 [Fisher], 2 mM GlutaMAX [Gibco], 10 ng/mL hLIF, 4 μM CHIR99021, 3 μM SB431541, and 0.1 μM Compound E [all from STEMCELL Technologies]). Cells were maintained in this medium supplemented with 2 μM dorsomorphin for 3 days and without dorsomorphin (STEMCELL Technologies) for an additional 5 days. NPCs were subsequently incubated in NPC maturation medium (50% DMEM/F12, 50% Neurobasal medium supplemented with N2, B27, 2 mM GlutaMAX, 10 ng/mL hLIF, 3 μM CHIR99021 and 2 μM SB431541), and were passaged weekly following Accutase (STEMCELL Technologies) dissociation according to the manufacturer's instructions. NPCs were treated for 24 h with 10 μg/μL of FGF or BDNF (both STEMCELL Technologies) to assess growth factor-induced cell proliferation. GABAergic neurons were differentiated as described previously (Liu et al., 2013). For astrocytic differentiation, NPCs were plated on Primaria-coated plates in Astrocyte Growth Media (ScienCell) for a minimum of 2 weeks and a maximum of ten passages (Tcw et al., 2017). Cerebral organoids were generated as described previously (Lancaster and Knoblich, 2014) with minor modifications: embryoid bodies were cultured in NIM (STEMCELL Technologies) supplemented with 20 μM Rock inhibitor Y27632 (Millipore) and 4 ng/mL basic FGF (Peprotech) for the first 5 days, followed by NIM alone for an additional 4 days, before direct transfer to cerebral organoid media without Matrigel embedding. Cerebral organoids were maintained for up to 56 DIV. All experiments used at least three biological replicates from two independently generated hiPSC clones.
RNA Extraction, cDNA Production, qPCR, and Targeted Allele Expression Analysis
Total RNA was extracted from snap-frozen cell pellets of three independent passages of two clones per hiPSC line, using QIAGEN RNeasy Mini Kit. Total RNA was reverse-transcribed into cDNA using an Applied Biosystems High-Capacity cDNA Reverse Transcription Kit as per the manufacturer's instructions. Real-time quantitative PCR (qPCR) was performed using TaqMan Gene Expression assays NF1 (Hs01035108_m1) and GAPDH (Hs02786624_g1; internal control), and relative NF1 expression was calculated using the ΔΔCT analysis method following the manufacturer's instructions (Thermo Fisher Scientific). For allele-specific expression analyses, primer pairs including Illumina adapter sequences concatenated to their 5′ ends were used for all NF1-mutant hiPSCs to initially amplify the mutation-surrounding region, and later to add the P5 sites, P7 sites, and sample-specific index (Supplemental Experimental Procedures). The samples were pooled and the amplicons were deep-sequenced on a MiSeq machine. Illumina adapters, 5' and 3′ bases with quality scores <25, as well as sequences <25 bases long were trimmed using Trimmomatic v.0.331 software. Trimmed reads were aligned to the human reference genome hg19 using STARv2.52 software and allele reads were calculated using Integrative Genomics Viewer v.2.3.293.
Immunohistochemistry and ELISA Assays
Immunocytochemistry on NPCs, astrocytes, and neurons was performed following established protocols (Anastasaki et al., 2015) using the antibodies described (Table S2). RAS activity (Thermo Fisher Scientific), GABA, dopamine (both Rocky Mountain Diagnostics) detection (Anastasaki et al., 2015), BrdU proliferation assays (Roche), and direct cell counting were performed as described previously (Toonen et al., 2016). Immunohistochemistry on cryosections of cerebral organoids was performed as described previously (Sloan et al., 2018). A minimum of three independent samples representing different passages of two separate clones were used for each line.
Mice
All animals were maintained on an inbred C57BL/6 background using a 12-h light/dark cycle with ad libitum access to food and water. Heterozygous Nf1 mice were generated to harbor point mutations corresponding to the human c.1149C > A, c.2041C > T, c.3431_32dupGT, and c.5425C > T mutations. The c.1149C > A, c.2041C > T, and c.3431_32dupGT mice were generated using C57BL/6 embryonic stem cells backcrossed a minimum of 10 times to wild-type C57BL/6 mice, while the c.5425C > T mice were generated by CRISPR/Cas9 engineering on a C57BL/6 genetic background and heterozygous mutation was confirmed by direct sequencing. All mice were used in accordance with an approved Animal Studies Protocol at the Washington University School of Medicine.
Statistics
All statistical tests were performed using GraphPad Prism 5 software. We performed t tests, one- or two-way analysis of variance (ANOVA) with Dunnett or Bonferroni post-test correction using GraphPad Prism 5 software. Statistical significance was set at p < 0.05.
Accession Numbers
The accession number for the deep sequencing data reported in this paper is GEO: GSE144601.
Author Contributions
C.A., M.L.W., and D.H.G. designed and analyzed the experiments. C.A., M.L.W., K.H., J.B.P., N.D.K., J.C., O.C., and J.D.D. conducted and/or interpreted the experiments. The article was assembled by C.A., M.L.W., and D.H.G. D.H.G. was responsible for the final production of the manuscript.
Acknowledgments
This work was supported by an R50 Research Specialist Award from the National Cancer Institute, USA (1-R50-CA233164-01 to C.A.), a Young Investigator's Award grant from the Children's Tumor Foundation, USA (2018-01-003 to M.L.W.), and a Research Program Award grant from the National Insitute of Neurological Disorders and Stroke, USA (1-R35-NS07211-01 to D.H.G.). The GEiC Center at WUSM engineered the hiPSCs and is subsidized by National Cancer Institute, USA Cancer Center Support grant (P30-CA091842). Dr. Gutmann has a licensing agreement for the GFAP-Cre mouse strain with the Tuberous Sclerosis Alliance.
Published: April 2, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2020.03.007.
Supplemental Information
References
- Anastasaki C., Morris S.M., Gao F., Gutmann D.H. Children with 5'-end NF1 gene mutations are more likely to have glioma. Neurol. Genet. 2017;3:e192. doi: 10.1212/NXG.0000000000000192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasaki C., Woo A.S., Messiaen L.M., Gutmann D.H. Elucidating the impact of neurofibromatosis-1 germline mutations on neurofibromin function and dopamine-based learning. Hum. Mol. Genet. 2015;24:3518–3528. doi: 10.1093/hmg/ddv103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell C.C., Magor G.W., Gillinder K.R., Perkins A.C. A high-throughput screening strategy for detecting CRISPR-Cas9 induced mutations using next-generation sequencing. BMC Genomics. 2014;15:1002. doi: 10.1186/1471-2164-15-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolcekova A., Nemethova M., Zatkova A., Hlinkova K., Pozgayova S., Hlavata A., Kadasi L., Durovcikova D., Gerinec A., Husakova K. Clustering of mutations in the 5' tertile of the NF1 gene in Slovakia patients with optic pathway glioma. Neoplasma. 2013;60:655–665. doi: 10.4149/neo_2013_084. [DOI] [PubMed] [Google Scholar]
- Costa R.M., Federov N.B., Kogan J.H., Murphy G.G., Stern J., Ohno M., Kucherlapati R., Jacks T., Silva A.J. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature. 2002;415:526–530. doi: 10.1038/nature711. [DOI] [PubMed] [Google Scholar]
- Cui Y., Costa R.M., Murphy G.G., Elgersma Y., Zhu Y., Gutmann D.H., Parada L.F., Mody I., Silva A.J. Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell. 2008;135:549–560. doi: 10.1016/j.cell.2008.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deraredj Nadim W., Chaumont-Dubel S., Madouri F., Cobret L., De Tauzia M.L., Zajdel P., Benedetti H., Marin P., Morisset-Lopez S. Physical interaction between neurofibromin and serotonin 5-HT6 receptor promotes receptor constitutive activity. Proc. Natl. Acad. Sci. U S A. 2016;113:12310–12315. doi: 10.1073/pnas.1600914113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombi E., Baldwin A., Marcus L.J., Fisher M.J., Weiss B., Kim A., Whitcomb P., Martin S., Aschbacher-Smith L.E., Rizvi T.A. Activity of selumetinib in neurofibromatosis type 1-related plexiform neurofibromas. N. Engl. J. Med. 2016;375:2550–2560. doi: 10.1056/NEJMoa1605943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher M.J., Belzberg A.J., de Blank P., De Raedt T., Elefteriou F., Ferner R.E., Giovannini M., Harris G.J., Kalamarides M., Karajannis M.A. 2016 Children's Tumor Foundation conference on neurofibromatosis type 1, neurofibromatosis type 2, and schwannomatosis. Am. J. Med. Genet. A. 2018;176:1258–1269. doi: 10.1002/ajmg.a.38675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutmann D.H., Zhang Y., Hirbe A. Developmental regulation of a neuron-specific neurofibromatosis 1 isoform. Ann. Neurol. 1999;46:777–782. doi: 10.1002/1531-8249(199911)46:5<777::aid-ana15>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Hsueh Y.P., Roberts A.M., Volta M., Sheng M., Roberts R.G. Bipartite interaction between neurofibromatosis type I protein (neurofibromin) and syndecan transmembrane heparan sulfate proteoglycans. J. Neurosci. 2001;21:3764–3770. doi: 10.1523/JNEUROSCI.21-11-03764.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman S.L., Shores A., North K.N. The nature and frequency of cognitive deficits in children with neurofibromatosis type 1. Neurology. 2005;65:1037–1044. doi: 10.1212/01.wnl.0000179303.72345.ce. [DOI] [PubMed] [Google Scholar]
- Hyman S.L., Arthur Shores E., North K.N. Learning disabilities in children with neurofibromatosis type 1: subtypes, cognitive profile, and attention-deficit-hyperactivity disorder. Dev. Med. Child Neurol. 2006;48:973–977. doi: 10.1017/S0012162206002131. [DOI] [PubMed] [Google Scholar]
- Jett K., Friedman J.M. Clinical and genetic aspects of neurofibromatosis 1. Genet. Med. 2010;12:1–11. doi: 10.1097/GIM.0b013e3181bf15e3. [DOI] [PubMed] [Google Scholar]
- Kehrer-Sawatzki H., Mautner V.F., Cooper D.N. Emerging genotype-phenotype relationships in patients with large NF1 deletions. Hum. Genet. 2017;136:349–376. doi: 10.1007/s00439-017-1766-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koczkowska M., Callens T., Gomes A., Sharp A., Chen Y., Hicks A.D., Aylsworth A.S., Azizi A.A., Basel D.G., Bellus G. Expanding the clinical phenotype of individuals with a 3-bp in-frame deletion of the NF1 gene (c.2970_2972del): an update of genotype-phenotype correlation. Genet. Med. 2018;21:867–876. doi: 10.1038/s41436-018-0269-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf B.R. Neurofibromatosis. Handb. Clin. Neurol. 2013;111:333–340. doi: 10.1016/B978-0-444-52891-9.00039-7. [DOI] [PubMed] [Google Scholar]
- Lancaster M.A., Knoblich J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014;9:2329–2340. doi: 10.1038/nprot.2014.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Liu H., Sauvey C., Yao L., Zarnowska E.D., Zhang S.C. Directed differentiation of forebrain GABA interneurons from human pluripotent stem cells. Nat. Protoc. 2013;8:1670–1679. doi: 10.1038/nprot.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris S.M., Gutmann D.H. A genotype-phenotype correlation for quantitative autistic trait burden in neurofibromatosis 1. Neurology. 2018;90:377–379. doi: 10.1212/WNL.0000000000005000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omrani A., van der Vaart T., Mientjes E., van Woerden G.M., Hojjati M.R., Li K.W., Gutmann D.H., Levelt C.N., Smit A.B., Silva A.J. HCN channels are a novel therapeutic target for cognitive dysfunction in neurofibromatosis type 1. Mol. Psychiatry. 2015;20:1311–1321. doi: 10.1038/mp.2015.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrakitkomjorn S., Kobayashi D., Morikawa T., Wilson M.M., Tsubota N., Irie A., Ozawa T., Aoki M., Arimura N., Kaibuchi K. Neurofibromatosis type 1 (NF1) tumor suppressor, neurofibromin, regulates the neuronal differentiation of PC12 cells via its associating protein, CRMP-2. J. Biol. Chem. 2008;283:9399–9413. doi: 10.1074/jbc.M708206200. [DOI] [PubMed] [Google Scholar]
- Pinna V., Lanari V., Daniele P., Consoli F., Agolini E., Margiotti K., Bottillo I., Torrente I., Bruselles A., Fusilli C. p.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur. J. Hum. Genet. 2015;23:1068–1071. doi: 10.1038/ejhg.2014.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojnueangnit K., Xie J., Gomes A., Sharp A., Callens T., Chen Y., Liu Y., Cochran M., Abbott M.A., Atkin J. High incidence of Noonan syndrome features including short stature and pulmonic stenosis in patients carrying NF1 missense mutations affecting p.Arg1809: genotype-phenotype correlation. Hum. Mutat. 2015;36:1052–1063. doi: 10.1002/humu.22832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif S., Upadhyaya M., Ferner R., Majounie E., Shenton A., Baser M., Thakker N., Evans D.G. A molecular analysis of individuals with neurofibromatosis type 1 (NF1) and optic pathway gliomas (OPGs), and an assessment of genotype-phenotype correlations. J. Med. Genet. 2011;48:256–260. doi: 10.1136/jmg.2010.081760. [DOI] [PubMed] [Google Scholar]
- Sherekar M., Han S.W., Ghirlando R., Messing S., Drew M., Rabara D., Waybright T., Juneja P., O’Neill H., Stanley C.B. Biochemical and structural analyses reveal that the tumor suppressor neurofibromin (NF1) forms a high-affinity dimer. J. Biol. Chem. 2020;295:1105–1119. doi: 10.1074/jbc.RA119.010934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan S.A., Andersen J., Pasca A.M., Birey F., Pasca S.P. Generation and assembly of human brain region-specific three-dimensional cultures. Nat. Protoc. 2018;13:2062–2085. doi: 10.1038/s41596-018-0032-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tcw J., Wang M., Pimenova A.A., Bowles K.R., Hartley B.J., Lacin E., Machlovi S.I., Abdelaal R., Karch C.M., Phatnani H. An efficient platform for astrocyte differentiation from human induced pluripotent stem cells. Stem Cell Reports. 2017;9:600–614. doi: 10.1016/j.stemcr.2017.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toonen J.A., Anastasaki C., Smithson L.J., Gianino S.M., Li K., Kesterson R.A., Gutmann D.H. NF1 germline mutation differentially dictates optic glioma formation and growth in neurofibromatosis-1. Hum. Mol. Genet. 2016;25:1703–1713. doi: 10.1093/hmg/ddw039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevisson E., Morbidoni V., Forzan M., Daolio C., Fumini V., Parrozzani R., Cassina M., Midena E., Salviati L., Clementi M. The Arg1038Gly missense variant in the NF1 gene causes a mild phenotype without neurofibromas. Mol. Genet. Genomic Med. 2019;7:e616. doi: 10.1002/mgg3.616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyaya M., Huson S.M., Davies M., Thomas N., Chuzhanova N., Giovannini S., Evans D.G., Howard E., Kerr B., Griffiths S. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation. Am. J. Hum. Genet. 2007;80:140–151. doi: 10.1086/510781. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.