

Abstract
Background
Somatic alterations in circulating tumor DNA (ctDNA) may be associated with treatment response or prognosis in prostate cancer (PCa). The goal was to characterize androgen receptor gene (AR) amplifications and mutations detected in ctDNA from patients with PCa and to further understand the somatic genetic heterogeneity of advanced prostate cancer.
Patients and Methods
This study included a heterogeneous group of 892 patients with advanced PCa (predominantly castrate‐resistant prostate cancer) with AR alterations detected in ctDNA that underwent next‐generation sequencing of 54 to 73 genes via Guardant360 testing (Guardant Health, Inc., Redwood City, CA). Distribution and summary of AR alterations detected, the association of AR alterations with other genes, and a pathway analysis are reported.
Results
The median absolute plasma copy number of AR amplifications was 3.3 (range, 1.2–165.2). Many patients had multiple AR mutations; a total of 112 unique mutations were identified in AR, including L702H (25%), T878A (14%), H875Y (11%), W742C (8%), W742L (4%), F877L (2%), and T878S (2%). Other ctDNA gene alterations in the Guardant assays included TP53 (50%), MYC (34%), BRAF (32%), PIK3CA (29%), MET (25%), CDK6 (26%), EGFR (24%), FGFR1 (21%), and APC (12%). Many of these non‐AR alterations are not tissue verified in other studies. AR amplification cosegregated with alterations in MYC (p < .001), BRAF (p < .001), PIK3CA (p < .001), MET (p < .001), CDK6 (p < .001), EGFR (p < .001), FGFR1 (p = .391), and more. Alterations in APC were significantly associated with mutations in AR (p < .001).
Conclusion
Several AR alterations and concomitant non‐AR alterations that associate with drug resistance were detected. These findings provide additional insights into the heterogeneity of advanced prostate cancer.
Implications for Practice
The goal was to characterize androgen receptor gene (AR) amplifications and mutations detected in circulating tumor DNA (ctDNA) from patients with prostate cancer in relation to non‐AR gene alterations detected in the ctDNA landscape. The study included 892 patients with prostate cancer with AR alterations in ctDNA. AR alterations were significantly associated with other gene alterations detected in ctDNA. The common AR mutations found are linked to resistance to abiraterone, enzalutamide, or bicalutamide. Characterization of the circulating AR landscape and gene alterations provides potential additional insight into the somatic genetic heterogeneity of advanced prostate cancer.
Keywords: Prostate cancer, Androgen receptor gene, Circulating tumor DNA, Abiraterone, Enzalutamide, Drug resistance
Short abstract
This article characterizes AR amplifications and mutations detected in circulating tumor DNA from patients with prostate cancer, providing insight into the heterogeneity of advanced prostate cancer.
Introduction
The androgen receptor (AR) has long been understood to be central to prostate cancer pathophysiology. Since 1941, androgen deprivation therapy has played a key role in treatment of prostate cancer 1. Over the last 76 years, manipulations of the androgen axis have continued to improve the management of patients with prostate cancer, with agents such as enzalutamide, apalutamide, and darolutamide now being approved for nonmetastatic castrate‐resistant disease 2 and abiraterone acetate (abiraterone) plus prednisone for castration‐sensitive metastatic disease 3. The novel antiandrogens work as classic androgen receptor antagonists with high potency, and abiraterone is a CYP17 inhibitor that decreases androgen synthesis. After being approved for treatment of castrate‐resistant prostate cancer (CRPC), the development of novel androgen axis inhibitors (e.g., enzalutamide, apalutamide, darolutamide) has moved to metastatic castration‐sensitive prostate cancer settings 1, 4.
Despite these advances, drug resistance to novel androgen axis inhibitors invariably happens. Patients resistant to the novel androgen axis inhibitors represent a particular challenge, as sequential treatment with similar androgen axis inhibitors is unlikely to result in meaningful clinical benefit 5, 6, 7. A variety of adaptive responses in the androgen axis have been well annotated 8, 9, 10, 11, and resistance can involve a variety of mechanisms. These include upregulation of androgen synthesis, upregulation and/or amplification of the androgen receptor gene (AR), development of splice variants leading to ligand independent activation of the AR, and a variety of alterations in coactivators or repressors that can dramatically alter the AR‐induced transcriptional program. Overcoming these resistance mechanisms is an area of intense interest.
Other mechanisms of resistance clearly exist and involve alterations in genes outside of the androgen axis pathway. This represents an area of active investigation and has clear clinical implications on the development of novel drug regimens for these patients. Improved understanding of alteration in the AR pathway and the concomitant alteration in non‐AR pathway genes will have the potential to guide development of novel monotherapy and combinatorial regimens targeting these alterations. This is especially important in the context of metastatic CRPC, for which no new drug or regimen has been approved since 2013. This is a large patient population with a very high unmet need. Herein we report the spectrum of alterations in AR and concomitant alterations in non‐AR pathways in men with advanced prostate cancer, predominantly CRPC, as revealed through analysis of circulating tumor DNA (ctDNA).
Materials and Methods
De‐identified ctDNA data were obtained from a heterogeneous group of 892 unique patients with advanced prostate cancer who underwent a targeted next‐generation sequencing assay performed by Guardant360 (Guardant Health, Inc., Redwood City, CA) between July 2, 2014, and August 15, 2017, a total of 37% of the total samples received had AR abnormalites. These samples were derived from a “real‐world” setting and not from an established protocol. Treatment histories were not available, but discussions with clinicians involved with this research indicated that the vast majority of patients had advanced cancer and CRPC (exact percentages were not ascertainable). Guardant Health is a Clinical Laboratory Improvement Amendments (CLIA)–licensed, College of American Pathologists–accredited, New York State Department of Health–approved clinical laboratory. Testing was performed using the Guardant Health standard collection protocol, in which peripheral venous blood, collected in two 10‐cc Streck tubes, was used to obtain 5–30 ng of ctDNA from isolated plasma and analyzed as previously described 12, 13. Guardant360 uses digital sequencing to detect single nucleotide variants (SNVs), insertions/deletions (indels), copy number amplifications (CNAs), and fusions in select exons and genes from ctDNA. Regarding CNAs, plasma copy number is dependent on both the copy number in tissue and the amount of tumor‐derived DNA shed into blood; this tumor copy number in plasma is diluted by circulating germline DNA from leukocytes with an expected normal copy number of 2.0 for genes that are not X‐linked, or 1.0 for X‐linked genes in males.
Throughout the course of the study period, four versions of the assay (54‐, 68‐, 70‐, and 73‐gene panels) were used with expanding coverage of genes and alterations. The composition of the panel has changed over time, with the current panel assessing SNVs in 73 genes, indels in 23 genes, amplifications in 18 genes, and fusions in 6 genes. Of note, all exons of the AR gene were assessed for SNVs on all four panel versions; CNA of the AR gene was not assessed on the earliest 54‐gene panel but was on all following panel versions. All mutational calls and reports are part of the commercial process used in the Guardant ctDNA assays.
The distribution of AR alterations throughout the gene was assessed with MutationMapper (version 1.0.1; cBioPortal). The cosegregation of other genetic alterations within the AR positive population was evaluated with OncoPrinter (version 1.0.1; cBioPortal) 14, 15. Chi‐square tests and Fisher's exact test were used to evaluate the association(s) between genetic alterations and AR alterations including amplifications and/or SNVs. A value of p < .05 was considered significant.
The patient population consisted of those men with prostate cancer tested with the Guardant360 assay clinically, and this data set includes only those individuals with AR mutations or amplifications as reported by Guardant. Details on their stage and treatment histories were not available, but the vast majority were patients with advanced CRPC.
In order to discover genetic alterations correlated with patients with AR mutations only, AR amplifications only, and patients with both AR mutations and amplifications, chi‐squared statistics were calculated on a gene‐by‐gene basis. Standardized residuals were calculated in order to control for false discovery rate (FDR) less than 0.05. Genetic alterations with standardized residuals deviating more than two SDs were selected for gene ontology enrichment in order to identify statistically overrepresented biological processes 16. Statistical significance of biological processes was calculated using FDR‐adjusted p values by Fisher's exact test.
Results
The sample assessed in this database were derived exclusively from samples with AR alterations; thus AR amplifications and/or mutations were identified in all 892 patients. Access to data from patients with non‐AR mutations was not provided by Guardant, and these data are thus not reported herein. Median age of patients was 70 years (range, 41–93) at testing. Patients had a median of 6 (range, 1–63) ctDNA alterations with a median mutant allele frequency of 0.64% (range, 0.01%–97.66%). Amplifications among all amplified genes had a median absolute plasma copy number of 3.33 (range, 1.19–165.18).
Of the 892 patients studied with AR alterations, there were 436 cases (49%) with only AR amplifications and a median of 5 additional alterations (95% confidence interval [CI], 6.0–7.0; p = .1985). A total of 283 patients (32%) had nonsynonymous mutations (SNV or indel) only, with a median of 5 alterations (95% CI, 5.9–7.3; p = .1985). In 165 patients (18%), both amplifications and nonsynonymous mutations were identified, with a median number of 9 non‐AR alterations (95% CI, 9.4–11.6; p < .0001). Synonymous AR alterations only were present in <1% (8/892) of patients. Patients with only synonymous AR alterations were excluded from further analyses. A higher frequency of non‐AR genetic alterations was significantly associated with patients that had both AR mutation and amplification (p < .0001; Table 1).
Table 1.
Frequency of genetic alterations per patient and AR status in circulating tumor DNA from patients with advanced prostate cancer
| Patient subgroup | Number of cases | Alterations, median ± SE (95% CI) | p value (Mann‐Whitney) |
|---|---|---|---|
| AR amplification only | 436 | 5 ± 0.25 (6.0–7.0) | .1985 |
| AR amplification and mutation | 165 | 9 ± 0.55 (9.4–11.6) | <.0001 |
| AR mutation only | 283 | 5 ± 0.36 (5.9–7.3) | .1985 |

Abbreviations: AR, androgen receptor gene; CI, confidence interval.
A total of 112 unique mutations were identified in the AR gene (supplemental online Table 1). Of the 112 AR mutations detected, most were missense mutations (90.20%, n = 101). Truncating alterations (8.90%, n = 10) and splice site mutations (1.80%, n = 2) were also annotated. The most prevalent AR alterations are shown in Table 2; these included L702H (24.55%, n = 219), T878A (14.46%, n = 129), H875Y (11.43%, n = 102), W742C (8.41%, n = 75), W742L (3.81%, n = 34), F877L (2.13%, n = 19), T878S (1.57%, n = 14), V716M (1.23%, n = 11), D891H (0.78%, n = 7), M750V (0.67%, n = 6), M750T (0.45%, n = 4), and S889G (0.45%, n = 4). Mutations of high frequency were clustered in the ligand binding domain of the AR protein (supplemental online Fig. 1).
Table 2.
Most common AR mutations and allelic fractions in circulating tumor DNA
| AR alteration | Patients, n (%) | Average mutant allele fraction, % ± SD | Median mutant allele fraction, % (range) |
|---|---|---|---|
| L702H | 219 (24.55) | 8.98 ± 14.58 | 2.35 (0.05–79.66) |
| T878A | 129 (14.46) | 10.75 ± 19.91 | 2.29 (0.16–97.66) |
| H875Y | 102 (11.43) | 12.03 ± 19.82 | 3.07 (0.11–92.86) |
| W742C | 75 (8.41) | 6.22 ± 14.3 | 1.44 (0.03–96.35) |
| W742L | 34 (3.81) | 2.69 ± 3.94 | 1.26 (0.12–19.31) |
| F877L | 19 (2.13) | 4.78 ± 10.02 | 1.17 (0.11–43.7) |
| T878S | 14 (1.57) | 2.52 ± 3.94 | 1.09 (0.14–15.43) |
| V716M | 11 (1.23) | 3.92 ± 4.37 | 2.3 (0.22–13.83) |
| D891H | 7 (0.78) | 0.9 ± 1 | 0.43 (0.18–2.79) |
| M750V | 6 (0.67) | 7.15 ± 10.3 | 3.07 (0.25–26.83) |
| M750T | 4 (0.45) | 4.5 ± 4.65 | 3.92 (0.48–9.7) |
| S889G | 4 (0.45) | 0.66 ± 0.86 | 0.3 (0.22–2.2) |

Abbreviation: AR, androgen receptor gene.
Repetitive AR alterations with the highest average mutant allele fractions (MAFs; supplemental online Tables 1 and 2) were H875Y (MAF, 12.03% ± 19.82), T878A (MAF, 10.75% ± 19.91), D891V (MAF, 9.31% ± 8.05), L702H (MAF, 8.98% ± 14.88), M750V (MAF, 7.14% ± 10.30), W742C (MAF, 6.22% ± 14.30), M750T (MAF, 4.50% ± 4.65), and V716M (MAF, 3.92% ± 4.37). Mutations at codon 750 (M750V and M750T) and codon 716 were uncommonly described but, when present in this data set, had a high allelic fraction.
A number of patients had multiple AR alterations detected. These alterations are shown in Table 3 and include the double mutants L702H + H875H, T878S + T875Y, T878S + W742C, W742C + W742L, and L702H + T878A (supplemental online Fig. 2). One triple mutant population was repeatedly detected (L702H + T878A + H875Y). AR amplifications (AMPs) co‐occurring with AR mutations include AMP + L702H, AMP + W742C, AMP + T878A, and AMP + H875Y. AR amplifications also co‐occurred with double mutants, including AMP + L702H + T878A, AMP + W742C + W742L, and AMP + L702H + H875Y. The assay results do not determine if the doublet and triplet AR alterations were present in one clone or several clones.
Table 3.
Co‐occurrence of androgen receptor gene amplifications and mutations in circulating tumor DNA
| Alteration | n | Percent of total patients | Percent of patients with co‐mutation |
|---|---|---|---|
| Amplification only | 165 | 18.67 | |
| Isolated mutation | 368 | 41.63 | |
| Co‐mutation | 351 | 39.71 | |
| AMP + L702H | 83 | 9.39 | 23.65 |
| L702H + H875Y | 32 | 3.62 | 9.12 |
| AMP + L702H + T878A | 21 | 2.38 | 5.98 |
| AMP + W742C | 21 | 2.38 | 5.98 |
| L702H + T878A + H875Y | 17 | 1.92 | 4.84 |
| AMP + T878A | 15 | 1.70 | 4.27 |
| T878S + H875Y | 14 | 1.58 | 3.99 |
| AMP + W742C + W742L | 13 | 1.47 | 3.70 |
| T878S + W742C | 13 | 1.47 | 3.70 |
| AMP + H875Y | 11 | 1.24 | 3.13 |
| W742C + W742L | 9 | 1.02 | 2.56 |
| L702H + T878A | 8 | 0.90 | 2.28 |
| AMP + L702H + H875Y | 7 | 0.79 | 1.99 |

Abbreviation: AMP, amplification.
Among patients with AR alterations, additional amplifications and alterations were identified in 74 other genes, including TP53 (50%, n = 444), MYC (34%, n = 300), BRAF (32%, n = 286), PIK3CA (29%, n = 258), MET (25%, n = 223), CDK6 (26%, n = 226), EGFR (24%, n = 208), FGFR1 (21%, n = 187), and APC (12%, n = 111; Table 4). DNA repair gene alterations were detected in combination with AR alterations in a substantial portion of these patients, including BRCA2 (8%, n = 68), BRCA1 (5%, n = 42), and ATM (3%, n = 28; supplemental online Table 4 and supplemental online Fig. 3). The distribution of missense mutations or amplifications identified in other genes was associated with either AR mutation or AR amplification, respectively (Table 4).
Table 4.
Frequency of additional gene alterations in circulating tumor DNA segregated by AR status in individual patients
| Gene | AR amplification only (n = 436), n (%) | AR mutation only (n = 283), n (%) | Both (n = 165), n (%) | p value |
|---|---|---|---|---|
| TP53 | 227 (52.06) | 132 (46.64) | 85 (51.52) | .3413 |
| MYC | 176 (40.37) | 53 (18.73) | 71 (43.03) | <1 × 10 −6 |
| BRAF | 152 (34.86) | 53 (18.73) | 81 (49.09) | <1 × 10 −6 |
| PIK3CA | 136 (31.19) | 53 (18.73) | 69 (41.82) | 1 × 10 −6 |
| MET | 109 (25.00) | 54 (19.08) | 60 (36.36) | 2.57 × 10 −4 |
| CDK6 | 131 (30.05) | 40 (14.13) | 55 (33.33) | <1 × 10 −6 |
| EGFR | 114 (26.15) | 43 (15.19) | 51 (30.91) | 1.52 × 10 −4 |
| FGFR1 | 106 (24.31) | 41 (14.49) | 40 (24.24) | 3.91 × 10 −3 |
| APC | 33 (7.57) | 53 (18.73) | 25 (15.15) | 3.20 × 10 −5 |
| PDGFRA | 66 (15.14) | 26 (9.19) | 42 (25.45) | 2.20 × 10 −5 |
| CCNE1 | 75 (17.20) | 15 (5.30) | 27 (16.36) | 1.10 × 10 −5 |
| RAF1 | 62 (14.22) | 25 (8.83) | 34 (20.61) | 4.27 × 10 −3 |
| KIT | 55 (12.61) | 25 (8.83) | 28 (16.97) | 3.76 × 10 −2 |
| NF1 | 42 (9.63) | 31 (10.95) | 23 (13.94) | .3166 |
| ERBB2 | 39 (8.94) | 18 (6.36) | 20 (12.12) | .1103 |
| BRCA2 | 24 (5.50) | 29 (10.25) | 15 (9.09) | .0498 |
| CCND1 | 53 (12.16) | 9 (3.18) | 26 (15.76) | 1 × 10 −5 |
| CTNNB1 | 28 (6.42) | 26 (9.19) | 17 (10.30) | .2029 |
| ARID1A | 29 (6.65) | 24 (8.48) | 13 (7.88) | .6440 |

Bold p values are those that are statistically significant.
Abbreviation: AR, androgen receptor gene.
For many genes, the frequency of alteration was significantly associated with either AR amplification or mutation (Table 4). AR amplification cosegregated with alterations in MYC (p < 1 × 10−6), BRAF (p < 1 × 10−6), PIK3CA (p < 1 × 10−6), MET (p = 2.57 × 10−4), CDK6 (p < 1 × 10−6), EGFR (p = 1.52 × 10−4), FGFR1 (p = 3.91 × 10−3), and more. Alterations in APC were significantly associated with mutations in AR (p = 3.20 × 10−5). TP53, despite being the most frequently altered gene, was detected regardless of AR alteration type (p = .4313). Chi‐squared analysis was performed according to frequency of a genetic alteration and sorted by AR gene status (supplemental online Fig. 4). Gene ontology term enrichment of statistically significant genes from chi‐squared analysis and associated with AR status is shown in Table 5. Results indicate that genes involved in signal transduction were altered in patients with AR mutation, whereas genes involved in cell cycle progression were associated with AR amplification. CNA was the most frequent category of alteration identified in significantly altered genes involved in cell cycle and associated with AR amplification. In contrast, none of the significantly altered genes involved in signal transduction and associated with AR mutation was copy number amplified (supplemental online Table 2 and supplemental online Figs. 5–6).
Table 5.
Gene ontology term enrichment of statistically significant genes altered in patients with prostate cancer with either AR mutation or AR amplification
| PANTHER GO‐Slim biological process | Homo sapiens reference list (21042) | Input (4) | Fold enrichment | Raw p value | FDR‐adjusted p value |
|---|---|---|---|---|---|
| AR mutation | |||||
| Intracellular signal transduction (GO:0035556) | 1,071 | 3 | 14.74 | 5.10 × 10−4 | 4.15 × 10−2 |
| Cell surface receptor signaling pathway (GO:0007166) | 1,206 | 3 | 13.09 | 7.24 × 10−4 | 4.42 × 10−2 |
| Signal transduction (GO:0007165) | 2,318 | 4 | 9.08 | 1.48 × 10−4 | 3.61 × 10−2 |
| Cell communication (GO:0007154) | 2,686 | 4 | 7.83 | 2.66 × 10−4 | 3.25 × 10−2 |
| AR amplification | |||||
| Cell proliferation (GO:0008283) | 67 | 2 | >100 | 1.05 × 10−4 | 1.28 × 10−2 |
| Regulation of cell cycle (GO:0051726) | 186 | 2 | 45.25 | 7.80 × 10−4 | 4.76 × 10−2 |
| Cell cycle (GO:0007049) | 723 | 4 | 23.28 | 6.87 × 10−6 | 1.68 × 10−3 |
| Phosphate‐containing compound metabolic process (GO:0006796) | 1,595 | 4 | 10.55 | 1.56 × 10−4 | 1.27 × 10−2 |
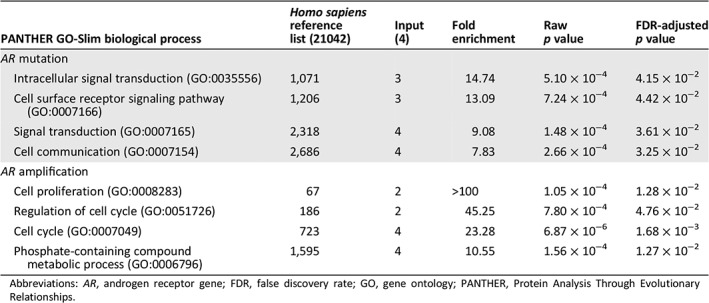
Abbreviations: AR, androgen receptor gene; FDR, false discovery rate; GO, gene ontology; PANTHER, Protein Analysis Through Evolutionary Relationships.
Discussion
Some of the AR alterations identified by ctDNA analysis in this population of patients with advanced prostate cancer (predominantly CRPC) have direct pharmacologic relevance and thus may be relevant to the development of drug resistance and patient selection for ongoing trials. Newer drugs such enzalutamide and apalutamide can serve as agonists for AR with the F877L mutation. Hence, detection of this F877L mutation may warrant precluding the use of enzalutamide and apalutamide but may suggest inclusion of these men in an ongoing trial (NCT02987829) with a drug TRC253, a potent inhibitor of F877L mutant AR 17, 18. Bicalutamide can confer agonist activities to W742C/L, T878S, S889G, and D891H mutations in AR 19, 20. Responses to withdrawal of enzalutamide, bicalutamide, nilutamide, and flutamide are well described 19, 21, 22, 23, although most studies did not annotate the underlying molecular defects. We note that the mutations described herein are typically associated with AR gain of function, not loss of function, when considering ligand‐mediated transcriptional activity.
Certain mutations seen in our data set allow promiscuous activation of AR, including the H875Y, T878S, D891H, and S889G mutations, all of which can be activated by progesterone and estrogens as well as by various androgens 11, 20, 24, 25, 26, 27. Progesterone is elevated in abiraterone‐treated patients and may account for some cases of abiraterone resistance 24. Withdrawal of progestational agents can result in prostate‐specific antigen declines 28, 29. Withdrawal of estrogens can be associated with prostate‐specific antigen declines in selected patients with AR mutations 30.
Glucocorticoids are frequently used with multiple U.S. Food and Drug Administration (FDA)–approved therapeutic regimens (e.g., in combination with abiraterone, docetaxel, mitoxantrone, and cabazitaxel). Studies of glucocorticoids are relatively limited in AR mutants, but data suggest that L702H mutations may interact with a variety of glucocorticoids, including hydrocortisone and methylprednisolone 20, 31, 32, 33.
Mutations at the 716 (V716M) and 750 positions (both M750T and M750V), though relatively rare, have a significant allelic fraction in these studies. Prior functional analyses of V716M are relatively limited, and studies of M750T and M750V mutants are absent. Potentially, additional functional studies of these mutants may give insights into AR function and/or pharmacology. Further studies of rare mutations are beyond the scope of this manuscript.
Of particular interest in this data set, we have characterized the distribution of gene alterations associated with AR alteration status (amplification, mutation, or both) in considerable detail in a large cohort of patients and note extensively the association between various AR mutations and amplifications in a detailed analysis. We must note with caution, however, that tissue corroboration of these changes is not present and that a number of genetic alterations detected in Guardant are not detected in tissue‐based assays. The subcohort harboring both AR amplification and mutation has a higher frequency of genetic alterations (p < .0001) compared with the other two cohorts. In contrast, copy number amplification of cell cycle oncogenes is significantly associated with the AR amplification–only subcohort. The two most frequently altered cell cycle oncogenes in the AR amplification subcohort were MYC and CDK6. MYC has been difficult to target but is being explored experimentally 34, whereas CDK4/6 inhibitors are now FDA approved in breast cancer 35. The AR mutated–only subset of patients was enriched for APC alterations, and, of note, WNT pathway inhibitors are now in development 36. Limitations are apparent in these data. Although the clinical assay used in this cohort is indicated in men with advanced prostate cancer harboring detectable ctDNA, further details regarding the specific stage of disease and prior treatments and the exact proportion of patients with CRPC, factors that segregate with frequency of AR mutations and alterations in prior studies, are not available in this study. Furthermore, we were able to analyze only those patients that had AR alterations in this data set, and thus patients without AR mutations were a distinct subset that were not studied herein. We also note that inclusion in this patient cohort was also only predicated on ctDNA being detected by this clinical assay; several factors, including low volume disease or ctDNA suppression from current treatment at the time of blood draw, can limit the detection of AR splice variants, such as AR‐V7 37, 38, 39, which are of potential interest in this patient population. Matched tissue genotyping results, clinical signs of drug resistance, and clinical outcomes, which may vary as a function of AR status 40, 41, 42, 43, were not available for this cohort. Furthermore, we note that that the Guardant assays’ report of aberrations in certain genes such as BRAF and EGFR are not consistent with tissue‐based reports; thus we are uncertain if these alterations represent true genetic events. Functional studies of the various mutants were not performed herein. Certain important genes such as PTEN and RB are known to be deleted in prostate cancer 44, 45; however, ctDNA assays are limited in their ability to detect gene deletions, and thus genetic losses are likely underrepresented in this report. We also note that ctDNA analyses are restricted to 73 genes, some of which include all exons, and many other genes were not assessed in these assays.
Consistency of results between ctDNA assays are not always apparent 46 and may differ based on biological factors, assay technological specifications, and laboratory reporting practices. Lack of comparison between the assays used herein are a limitation that cannot be assessed with the currently available data; however, we note that the Guardant assay has recently been compared with the Wyatt assay (which has been tissue correlated), and the findings for AR are highly concordant 47.
Conclusion
Despite the noted limitations, there are many strengths. The size of the database is large, and the detection of these alterations in ctDNA provides an insight on prevailing AR alterations, potentially related to therapy resistance, in a real‐world population. The selected common mutations reported in this data set, such as L702H, W742C and W742L, H875Y, T878S, and F877L, are all in the ligand‐binding domain of AR, are all characterized (to varying degrees), and all bind to relevant physiological or pharmacologic ligands encountered in the course of patient care 20. Ultimately, characterization of the circulating AR landscape may provide an accessible and clinically relevant insight into prostate cancer progression, have implications on therapy selection in the real‐world setting, and provide guidance on further drug development strategies in this population with high unmet need.
Author Contributions
Conception/design: Elisa M. Ledet, Michael B. Lilly, Guru Sonpavde, Neeraj Agarwal, Oliver Sartor
Provision of study material or patients: Elisa M. Ledet, Michael B. Lilly, Guru Sonpavde, Rebecca J. Nagy, Lesli Kiedrowski, Neeraj Agarwal, Oliver Sartor
Collection and/or assembly of data: Elisa M. Ledet, Michael B. Lilly, Guru Sonpavde, Rebecca J. Nagy, Lesli Kiedrowski, Neeraj Agarwal, Oliver Sartor
Data analysis and interpretation: Elisa M. Ledet, Michael B. Lilly, Guru Sonpavde, Edwin Lin, Roberto H. Nussenzveig, Pedro C. Barata, Mark Yandell, Rebecca J. Nagy, Lesli Kiedrowski, Neeraj Agarwal, Oliver Sartor
Manuscript writing: Elisa M. Ledet, Michael B. Lilly, Guru Sonpavde, Edwin Lin, Roberto H. Nussenzveig, Pedro C. Barata, Mark Yandell, Rebecca J. Nagy, Lesli Kiedrowski, Neeraj Agarwal, Oliver Sartor
Final approval of manuscript: Elisa M. Ledet, Michael B. Lilly, Guru Sonpavde, Edwin Lin, Roberto H. Nussenzveig, Pedro C. Barata, Mark Yandell, Rebecca J. Nagy, Lesli Kiedrowski, Neeraj Agarwal, Oliver Sartor
Disclosures
Guru Sonpavde: Merck, Bristol‐Myers Squibb, Sanofi, Bayer, Genentech, Novartis, Pfizer, Astellas/Agensys, Janssen, Amgen, AstraZeneca, Argos, Eisai, Exelixis, EMD Serono (SAB), Onyx/Amgen, Sanofi, Bayer, Boehringer Ingelheim, Celgene, Merck, Pfizer (RF—institution), AstraZeneca, Bristol‐Myers Squibb, Bavarian‐Nordic, Astellas (other—steering committee); Rebecca J. Nagy: Guardant Health, Inc. (E, OI); Lesli Kiedrowski: Guardant Health, Inc. (E, OI); Neeraj Agarwal: Astellas, AstraZeneca, Argos, Bristol‐Myers Squibb, Bayer, Clovis, Eisai, Exelixis, EMD Serono, Eli Lilly & Co., Foundation One, Genentech, Merck, Medivation, Novartis, Nektar, Pfizer, Pharmacyclics (C/A), Active Biotech, AstraZeneca, Bavarian‐Nordic, Bristol‐Myers Squibb, Calithera, Celldex, Eisai, Exelixis, Genentech, GlaxoSmithKline, Immunomedics, Janssen, Medivation, Merck, New Link Genetics, Novartis, Pfizer, Prometheus, Rexahn, Sanofi, Takeda, Tracon (RF—institution); Oliver Sartor: AstraZeneca, Bayer, Bellicum, Bristol‐Myers Squibb, Celgene, Dendreon, EMD Serono, Johnson & Johnson, Oncogenex, Pfizer, Sanofi‐Aventis, Constellation, Endocyte, Advanced Accelerator Applications, Bavarian‐Nordic (C/A), Bayer, Endocyte, Innocrin, Johnson & Johnson, Invitae, Sanofi‐Aventis, Merck (RF—institution). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Supplemental Figures
Supplemental Tables
Disclosures of potential conflicts of interest may be found at the end of this article.
Contributor Information
Neeraj Agarwal, Email: neeraj.agarwal@hci.utah.edu.
Oliver Sartor, Email: osartor@tulane.edu.
References
- 1. Huggins C, Hodges CV. Studies on prostatic cancer: Effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res 1941;1:293–297. [Google Scholar]
- 2. Higano C. Enzalutamide, apalutamide, or darolutamide: Are apples or bananas best for patients? Nat Rev Urol 2019;16:335–336. [DOI] [PubMed] [Google Scholar]
- 3. Fizazi K, Tran N, Fein L et al. Abiraterone plus prednisone in metastatic, castration‐sensitive prostate cancer. N Engl J Med 2017;377:352–360. [DOI] [PubMed] [Google Scholar]
- 4. James ND, de Bono JS, Spears MR et al. Abiraterone for prostate cancer not previously treated with hormone therapy. N Engl J Med 2017;377:338–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bianchini D, Lorente D, Rodriguez‐Vida A et al. Antitumour activity of enzalutamide (MDV3100) in patients with metastatic castration‐resistant prostate cancer (CRPC) pre‐treated with docetaxel and abiraterone. Eur J Cancer 2014;50:78–84. [DOI] [PubMed] [Google Scholar]
- 6. Cheng HH, Gulati R, Azad A et al. Activity of enzalutamide in men with metastatic castration‐resistant prostate cancer is affected by prior treatment with abiraterone and/or docetaxel. Prostate Cancer Prostatic Dis 2015;18:122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Noonan KL, North S, Bitting RL et al. Clinical activity of abiraterone acetate in patients with metastatic castration‐resistant prostate cancer progressing after enzalutamide. Ann Oncol 2013;24:1802–1807. [DOI] [PubMed] [Google Scholar]
- 8. Egan A, Dong Y, Zhang H et al. Castration‐resistant prostate cancer: Adaptive responses in the androgen axis. Cancer Treat Rev 2014;40:426–433. [DOI] [PubMed] [Google Scholar]
- 9. Karantanos T, Evans CP, Tombal B et al. Understanding the mechanisms of androgen deprivation resistance in prostate cancer at the molecular level. Eur Urol 2015;67:470–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Robinson D, Van Allen EM, Wu YM et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161:1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Taplin ME, Bubley GJ, Ko YJ et al. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res 1999;59:2511–2515. [PubMed] [Google Scholar]
- 12. Lanman RB, Mortimer SA, Zill OA et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell‐free circulating tumor DNA. PLoS One 2015;10:e0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Odegaard JI, Vincent JJ, Mortimer S et al. Validation of a plasma‐based comprehensive cancer genotyping assay utilizing orthogonal tissue‐ and plasma‐based methodologies. Clin Cancer Res 2018;24:3539–3549. [DOI] [PubMed] [Google Scholar]
- 14. Gao J, Aksoy BA, Dogrusoz U et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cerami E, Gao J, Dogrusoz U et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mi H, Huang X, Muruganujan A et al. PANTHER version 11: Expanded annotation data from gene ontology and reactome pathways, and data analysis tool enhancements. Nucleic Acids Res 2017;45:D183–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Joseph JD, Lu N, Qian J et al. A clinically relevant androgen receptor mutation confers resistance to second‐generation antiandrogens enzalutamide and ARN‐509. Cancer Discov 2013;3:1020–1029. [DOI] [PubMed] [Google Scholar]
- 18. Korpal M, Korn JM, Gao X et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov 2013;3:1030–1043. [DOI] [PubMed] [Google Scholar]
- 19. Hara T, Miyazaki J, Araki H et al. Novel mutations of androgen receptor: A possible mechanism of bicalutamide withdrawal syndrome. Cancer Res 2003;63:149–153. [PubMed] [Google Scholar]
- 20. Lallous N, Volik SV, Awrey S et al. Functional analysis of androgen receptor mutations that confer anti‐androgen resistance identified in circulating cell‐free DNA from prostate cancer patients. Genome Biol 2016;17:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rodriguez‐Vida A, Bianchini D, Van Hemelrijck M et al. Is there an antiandrogen withdrawal syndrome with enzalutamide? BJU Int 2015;115:373–380. [DOI] [PubMed] [Google Scholar]
- 22. Sartor AO, Tangen CM, Hussain MH et al. Antiandrogen withdrawal in castrate‐refractory prostate cancer: A Southwest Oncology Group trial (SWOG 9426). Cancer 2008;112:2393–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schellhammer PF, Venner P, Haas GP et al. Prostate specific antigen decreases after withdrawal of antiandrogen therapy with bicalutamide or flutamide in patients receiving combined androgen blockade. J Urol 1997;157:1731–1735. [PubMed] [Google Scholar]
- 24. Chen EJ, Sowalsky AG, Gao S et al. Abiraterone treatment in castration‐resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin Cancer Res 2015;21:1273–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fenton MA, Shuster TD, Fertig AM et al. Functional characterization of mutant androgen receptors from androgen‐independent prostate cancer. Clin Cancer Res 1997;3:1383–1388. [PubMed] [Google Scholar]
- 26. Steketee K, Timmerman L, Ziel‐van der Made AC et al. Broadened ligand responsiveness of androgen receptor mutants obtained by random amino acid substitution of H874 and mutation hot spot T877 in prostate cancer. Int J Cancer 2002;100:309–317. [DOI] [PubMed] [Google Scholar]
- 27. Veldscholte J, Voorhorst‐Ogink MM, Bolt‐de Vries J et al. Unusual specificity of the androgen receptor in the human prostate tumor cell line LNCaP: High affinity for progestagenic and estrogenic steroids. Biochim Biophys Acta 1990;1052:187–194. [DOI] [PubMed] [Google Scholar]
- 28. Dawson NA, McLeod DG. Dramatic prostate specific antigen decrease in response to discontinuation of megestrol acetate in advanced prostate cancer: Expansion of the antiandrogen withdrawal syndrome. J Urol 1995;153:1946–1947. [PubMed] [Google Scholar]
- 29. Sartor O, Eastham JA. Progressive prostate cancer associated with use of megestrol acetate administered for control of hot flashes. South Med J 1999;92:415–416. [DOI] [PubMed] [Google Scholar]
- 30. Vasudevamurthy AK, Ledet E, Garvey C et al. Estrogen‐mediated activation of H875Y androgen receptor mutation in a prostate cancer patient. Clin Genitourin Cancer 2017;15:e111–e113. [DOI] [PubMed] [Google Scholar]
- 31. Carreira S, Romanel A, Goodall J et al. Tumor clone dynamics in lethal prostate cancer. Sci Transl Med 2014;6:254ra125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Culig Z, Hobisch A, Cronauer MV et al. Mutant androgen receptor detected in an advanced‐stage prostatic carcinoma is activated by adrenal androgens and progesterone. Mol Endocrinol 1993;7:1541–1550. [DOI] [PubMed] [Google Scholar]
- 33. Zhao XY, Malloy PJ, Krishnan AV et al. Glucocorticoids can promote androgen‐independent growth of prostate cancer cells through a mutated androgen receptor. Nat Med 2000;6:703–706. [DOI] [PubMed] [Google Scholar]
- 34. Chen H, Liu H, Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther 2018;3:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Goel S, DeCristo MJ, McAllister SS et al. CDK4/6 inhibition in cancer: Beyond cell cycle arrest. Trends Cell Biol 2018;28:911–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krishnamurthy N, Kurzrock R. Targeting the Wnt/beta‐catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat Rev 2018;62:50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Antonarakis ES, Lu C, Luber B et al. Androgen receptor splice variant 7 and efficacy of taxane chemotherapy in patients with metastatic castration‐resistant prostate cancer. JAMA Oncol 2015;1:582–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ho Y, Dehm SM. Androgen receptor rearrangement and splicing variants in resistance to endocrine therapies in prostate cancer. Endocrinology 2017;158:1533–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Scher HI, Lu D, Schreiber NA et al. Association of AR‐V7 on circulating tumor cells as a treatment‐specific biomarker with outcomes and survival in castration‐resistant prostate cancer. JAMA Oncol 2016;2:1441–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Azad AA, Volik SV, Wyatt AW et al. Androgen receptor gene aberrations in circulating cell‐free DNA: Biomarkers of therapeutic resistance in castration‐resistant prostate cancer. Clin Cancer Res 2015;21:2315–2324. [DOI] [PubMed] [Google Scholar]
- 41. Romanel A, Gasi Tandefelt D, Conteduca V et al. Plasma AR and abiraterone‐resistant prostate cancer. Sci Transl Med 2015;7:312re310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wyatt AW, Azad AA, Volik SV et al. Genomic alterations in cell‐free DNA and enzalutamide resistance in castration‐resistant prostate cancer. JAMA Oncol 2016;2:1598–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Steinkamp MP, O'Mahony OA, Brogley M et al. Treatment‐dependent androgen receptor mutations in prostate cancer exploit multiple mechanisms to evade therapy. Cancer Res 2009;69:4434–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Aparicio A, Den RB, Knudsen KE. Time to stratify? The retinoblastoma protein in castrate‐resistant prostate cancer. Nat Rev Urol 2011;8:562–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Whang YE, Wu X, Suzuki H et al. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc Natl Acad Sci USA 1998;95:5246–5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Torga G, Pienta KJ. Patient‐paired sample congruence between 2 commercial liquid biopsy tests. JAMA Oncol 2018;4:868–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Taavitsainen S, Annala M, Ledet E et al. Evaluation of commercial circulating tumor DNA test in metastatic prostate cancer. JCO Precis Oncol 2019. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Supplemental Figures
Supplemental Tables