

Abstract
OBJECTIVE: Intestinal microbiota plays a vital role in the pathogenesis of colorectal cancer (CRC), which is crucial for assessing the risk and prognosis of CRC. Most studies regarding human gut microbiota mainly based on the feces, but the exact composition of microbiota vary significantly due to fecal composition is easily affected by many factors. We aim to evaluate whether intestinal lavage fluid (IVF) is a better substitution mirroring the gut microbiota. METHODS: We performed 16S rRNA gene analysis on fecal and IVF samples from 30 CRC patients and 25 healthy individuals, comparison in luminal (feces) / mucosal (IVF) adherent bacterial community profiles were analyzed. RESULTS: The difference between feces and IVF were observed, including the diversity and abundance of pathogenic bacteria (either in single strain or in co-occurrence pattern). IVF group shared 605 OTUs with the fecal group, but there was 94 OTUs only observed in fecal samples, while 247 OTUs were mainly existing in the IVF group. Among them, 27 vital bacterial species detected in IVF, while 10 critical species detected in fecal samples. The co-occurrence bacteria Fusobacteria Cluster and Proteobacteria Cluster 2 significantly increased in IVF than in control (P < .01), while Firmicutes Cluster 1, Firmicutes Cluster 2 and Proteobacteria Cluster 1 were markedly lower in IVF than in control (P < .001). In CRC feces, Fusobacteria Cluster was higher than in control (P < .05), but Firmicutes Cluster 1 was of substantially less abundance than in control (P < .001). Proteobacteria Cluster 2 was increased dramatically in IVF than in feces (P < .05), Firmicutes Cluster 1 were of substantially less abundance than in feces (P < .05). CONCLUSION: Pathogenic microbiota is more abundant in IVF than in feces. Microbiota of IVF may closely be related to the mucosal-associated microbial communities, which benefit from elucidating the relationship of the intestinal microbiota and CRC carcinogenesis.
Background
Colorectal cancer (CRC) is the second most common cancer in females and the third in males worldwide [1]. One of the crucial factors associated with CRC is the intestinal microbiota [2]. The human gut is the host to roughly a thousand different bacterial species, which contains beneficial commensal bacteria and potentially pathogenic bacteria. The intestinal dysbiosis may be associated with the overgrowth of opportunistic pathogens that are typically inhibited by commensal bacteria [3]. CRC carcinogenesis can be a result of dysbiosis in the colonic microbiota with an increased proportion of bacteria whose metabolism elaborates cytotoxic or genotoxic compounds that cause DNA damage either through the production of free radicals or through abnormal activation of resident immune cells [4]. Additionally, specific intestinal bacterial agents may be significant factors that contribute to the accumulated mutations that often manifest during cancer cell differentiation and development in the gut [5]. Accumulating evidence suggests that the gut microbiota or its metabolites may be the proximate environmental modifiers of risk for CRC [[6], [7], [8]].
The role of pathogen in colorectal carcinogenesis has been proved. Fusobacterium nucleate (Fn) has been pointed out as initial triggers in the development of CRC [9], which elicit a pro-inflammatory microenvironment around the tumor, driving tumor formation and progression [10,11]. Besides Fn, Escherichia coli, Enterococcus faecalis, Streptococcus gallolyticus, and enterotoxigenic Bacteroides fragilis are candidate microorganisms closely associated with CRC carcinogenesis [9]. Compare to a single pathogen, and intestinal microbiota may have more strength to influences the intestinal microenvironment, which may play a crucial role in CRC carcinogenesis. The intimate crosstalk between gut microbial community and the host's epithelium layer is a critical factor for cell proliferation and differentiation, gene expression in host epithelial cells, as well as the regulation of inflammation [12,13]. Nowadays, the understanding of intestinal microbiota complexity and dynamics is still in its infancy [14], and the inconsistency results observed in the gut microbiome.
The intestinal microbiota is continually changing by various factors, including diet, antibiotic use, sampling, and detection methods. Various microorganisms have been reported in association with human CRC, but the findings have not unified in the reports nor conclusive. Some of the variances may occur due to the methodology: sampling differences (feces vs. mucosal tissue) or due to different stages or differences between the right and left colon [8]. Given the above factors, the exact composition of intestinal microbiota and its function in CRC carcinogenesis remain unclear. Gut microbiota can be divided into two groups depending on their anatomical localization: luminal and mucosa-associated microbiota [15]. The luminal compartment consists mostly of transients, while the mucosa-adherent chamber consists of entrenched residents [16]. Compared with the mucosa-associated microbiome, the fecal-luminal microbiota is more affected by diet [9] and sampling time. Mucosa-associated microbiota may closely be related to CRC by directly interacting with the mucosal epithelial cells, principally acting through co-metabolism or metabolic exchange [15,17,18]. Theoretically, the mucosa-associated microbiome is more accurate in studying the role of the intestinal microbiome in CRC. However, the detection of mucosa-associated microbiota need obtains mucosa sample that required bowel preparation and colonoscopy biopsy. The procedure of taking sample is invasive and difficultly accepted by the subjects, both of which hindered its extensive application in clinical practice. Fecal-luminal microbiota can be acquired relatively easily by collecting feces. Therefore, most large-scale studies regarding human gut microbiota including some key studies, such as MeTaHIT cohort and Human Microbiome Project, analyzed fecal-luminal microbiota [19,20]. Even though, there is study suggested that the common use of fecal samples to study microbial communities may not reflect the tumor microenvironment [21]. Thus, study focusing on fecal samples, was unable to detect differences in microbial community diversity or richness between normal and cancer-associated microbiomes [22]. The discrepancy of two types of samples is worth further study.
The intestinal lavage fluid (IVF) is obtained from patients preparing the laparoscopic colorectal resection. Some species of mucosa-associated microbiota (either in surface or cavities) maybe get after a few times intestinal rinse. Thus, microbiota in IVF may represent mucosa-associated microbiota to some extent. In this study, the difference of microbiota composition between IVF and feces were analyzed, try to reveal whether IVF is a better substitution mirroring the gut microbiota.
Methods
Patients
The study was approved by the Shantou University Medical College Institutional Review Board that including all procedures (participant recruitment and all experimental protocols.). Participants came from the Second Affiliated Hospital of Shantou University, who provided written informed consent. Thirty patients with CRC were confirmed by pathological diagnosis. Patients with a history of polyps, adenomas, or non-primary colorectal cancer were excluded. The normal control derived from 25 healthy individuals. All participants are Chinese living in Guangdong Province, China. No antibiotics or prebiotics used in these patients before the sample collection (Table 1).
Table 1.
Clinical characteristics of participants
| Indexes | Control feces (n = 25) | CRC IVF (n = 20) | CRC Feces (n = 10) |
|---|---|---|---|
| Age (year) | 57.44 ± 2.56 | 58.40 ± 2.36 | 56.70 ± 6.77 |
| Gender | |||
| Female | 11 (44%) | 8 (40%) | 6 (60%) |
| Male | 14 (56%) | 12 (60%) | 4 (40%) |
| BMI | 22.36 ± 0.62 | 23.86 ± 1.00 | 22.94 ± 1.26 |
| TNM | |||
| 0-II | - | 9 (45%) | 7 (70%) |
| III-IV | - | 11 (55%) | 3 (30%) |
***P < .001; **P < .01; *P < .05.
Sample Collection
IVF: The patient fasted for 1 day before the sample collected. Then, all patients were given 500 ml enema, and all liquid discharged from the intestine was collected. Samples were centrifuged (4 ° C, 10000 G, 10 min) within 2 hours. The sediments were collected and stored at −80 ° C. About 2 g of the fresh fecal specimens of healthy and CRC people were obtained and stored at −80 ° C. Feces: about 5 g feces were obtained from patients and the controls.
DNA Extraction From Feces and IVF and 16S rRNA Amplicon Sequencing
Bacterial genetic DNA was isolated from fecal / IVF samples using the AllPrep DNA/RNA kit (Qiagen, German). DNA quality test had been done firstly, and then the qualified DNA was used to construct the libraries. For PCR products, the jagged ends of DNA fragment would be converted into blunt ends by using a T4 DNA polymerase, Klenow fragment, and T4 Polynucleotide Kinase. Ampure beads removed too short Fragments. For genetic DNA, we use fusion primer with dual index and adapters for PCR. Only the qualified library can be used for sequencing. Build a library with qualified samples: Paired-end sequencing reads were obtained as demultiplexed libraries per sample. Briefly, 16S amplicon PCR forward primer (V3 region 341F): ACTCCTACGGGAGGCAGCAG; 16S amplicon PCR reverse primer (V4 region 806R): GGACTACHVGGGTWTCTAAT; (V4 region 515F): GTGCCAGCMGCCGCGGTA A; (V4 region 806R): GGACTACHVGGGTWTCTAAT.
Bioinformatics Analysis Workflow
For subsequent bioinformatics analysis, the raw data will be pre-processed to get clean data by the in-house procedure. And tags were clustered to OTU at 97% sequence similarity. Taxonomic ranks were assigned to the OTU representative sequence using the Ribosomal Database Project (RDP) Native Bayesian Classifier v.2.2. The indices calculated by Mothur (v1.31.2, and the corresponding rarefaction curve was drawn by software R (v3.1.1). Alpha diversity, beta diversity, and the different species screening were analyzed based on OTU and taxonomic ranks. For the pooling library with barcode samples mixed, the clean reads were assigned to corresponding samples by allowing 0 base mismatch to barcode sequences with in-house scripts.
Statistical Analyses
Categorical and continuous variables analyzed by the chi-square test and the two-sample t-test. In multivariate analysis, significant variables from univariate analysis were selected and manually entered the model step by step. Data were analyzed using R(v3.1.1) software, QIIME (v1.80), Unifrac software, SPSS 20.0 software, and P-values represent two-sided statistical tests. All graphics made with GraphPad Prism 7.04 and R (v3.1.1).
Results
Microbiota Diversity in CRC Patients and Healthy Individuals
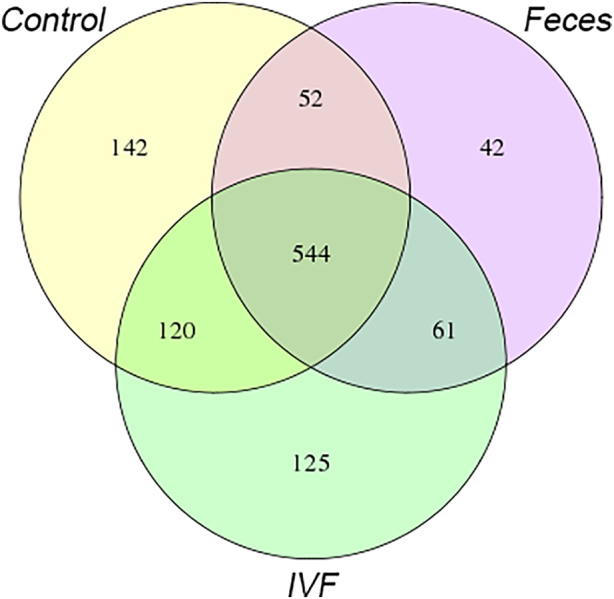
A total of 3433151*2 clean reads, with an average of 624218*2 clean reads per sample, were obtained. 3,420,613 tags were obtained by sequencing 55 samples, with an average of 61,643 ± 475 Tags per control fecal sample, 63,243 ± 556 Tags per CRC fecal sample, and 62,981 ± 431 tags per IVF sample. By using UPARSE to cluster at 97% similarity, the representative sequence of OTU was obtained. The high-quality sequence was clustered in 1086 bacterial OTUs. A relative abundance of OTU in each sample will be calculated. To further analyzed the features of OTUs between various groups, the Venn diagram analysis performed using R language software. Five hundred forty-four common OTUs found in the three groups; 142 OTUs found in the healthy fecal group, 125 OTUs in IVF, and 42 OTUs in CRC fecal group. The CRC fecal group shared 596 OTUs with the control fecal groups, while the CRC IVF group shared 664 OTUs with the control fecal groups. In the CRC group, the IVF group shared 605 OTUs with the fecal group. There were 94 OTUs only observed in fecal samples, while 245 OTUs mainly existed in the IVF group (Figure 1).
Figure 1.
For comparing the diversity of microbiota between CRC patients (IVF and fecal samples) and healthy individuals, ɑ and β diversity were analyzed. ɑ diversity includes Observed species, Chao, Ace, Shannon, and Simpson indices. Species diversity index in the CRC fecal group was similar to the control group, while the diversity index in the IVF group was lower than the other two fecal groups, though the difference did not reach the statistical significance. Of which the Shannon index was close to 0.05. Simpson's index was the opposite meaning with the previous index; Simpson's index was the highest in the IVF group (P > .05). In the three groups, the Shannon index in the IVF group was significantly lower compared to the fecal group, demonstrating that the reduction of microbiome diversity was significant in IVF than that in fecal samples. Boxplot is used to visually display the differences of the ɑ diversity among three groups (Figure 2).
Figure 2.

Boxplots represent the indices of observed species. Chao, Ace, Shannon, Simpson, Good's coverage in the fecal group and IVF group. Control: feces of the healthy group, IVF: IVF of CRC patients, Feces: feces of CRC patients.
β diversity was analyzed by software QIIME (v1.80) to reveal the differences of samples in species complexity. By using unweighted Unifrac Beta diversity analyses, our results showed that β diversity in the control group significantly different from the CRC group, either in fecal (P < .01) or in the IVF group (P < .01) (Figure 3).
Figure 3.

Unweighted Unifrac Beta diversity analysis demonstrated that differences in β diversity existed in the IVF and fecal groups. Control: feces of the healthy group, IVF: IVF of CRC patients, Feces: feces of CRC patients. **P < .01.
PCA and PcoA Analyses
Unifrac software, principal component analysis (PCA), principal coordinate analysis (PCoA), and UPGMA clustering tree analysis were used for further study of the bacterial community complexity. PCA reflects the flora diversity between samples by the distance between the points. The more similar the composition of the two sample groups, the closer they are in the PCA. PCoA was used to show the differences between the samples according to the matrix of beta diversity distance. UPGMA clustering tree analysis revealed most of the bacterial species in regular group huddles together, which separated with CRC samples (either in IVF or in feces).
CRC patients and healthy controls were divided into different branches in the cluster map (without considering OTU abundance), demonstrating the difference of flora was obvious between CRC patients and healthy controls. PCA and PoCA results showed that bacteria in healthy individuals significantly separated by those in CRC patients(either in the feces or IVF). In CRC patients, microbiota various between feces and IVF, the distributed area was divided into each other but had some extent overlap area (Figure 4). To conclude, our results showed that gut microbiota was the difference in CRC and healthy individuals, and the diversity in IVF samples had more significance than the fecal samples.
Figure 4.

Beta diversity among the three groups. (A) UPGMA clustering tree analysis. Bacterial species in normal control huddle together, which separated with CRC samples (either in IVF or feces). (B) PCA analyses. The abscissa represents the contribution rate of the first principal component to the sample difference (17.68%); the ordinate represents the second principal component, and the percentage represents the contribution rate of the second principal component to the sample (11.37%). (C) PcoA analyses. The abscissa is a principal component, the ordinate is another principal component, and the percentage on the coordinate axis indicates the contribution rate of the two principal components to the sample difference.
Comparison of Single Bacterium Between the Feces and the IVF in CRC Patients
The single bacterium was compared between IVF and the fecal samples. The OTUs in different samples were summarized in a profiling table or histogram with the software R (v3.1.1). The taxonomics composition distribution histograms of each sample in three groups were shown at Phylum, Order, Class, Family, Genus, and Species level. The species of which abundance was less than 0.5% in all samples were classified into ‘others’ in other ranks. False discovery rate (FDR) was adopted to assess the significance of differences between microbial communities. Above the level of phylum, Firmicutes was significantly decreased in CRC (IVF and Feces) group compared with the control group, while Fusobacteria significantly increased. In Order level, Actinomycetales, Aeromonadales, Burkholderiales, Campylobacterales, Enterobacteriales, Erysipelotrichales, Flavobacteriales, Fusobacteriales, Lactobacillales, Neisseriales, Pasteurellales, and Pseudomonadales in IVF was significantly higher than those in feces (abundance of bacteria was more than at least two folds), while Bacteroidales, Clostridiales, Coriobacteriales and Desulfovibrionales in IVF was significantly lower than those of feces. In the genus level, Roseburia, Lachnospira, Faecalibacterium, Coprococcus, and Lachnobacterium were reduced considerably in the CRC IVF group and fecal group, while Blautia, Collinsella, Cloacibacillus, Holdemania, and Bifidobacterium reduced only in IVF group. Fusobacterium, Neisseria, Campylobacter, and Enterococcus increased significantly in the IVF group, and the fecal group, Proteobacteria, and Escherichia only risen substantially in the IVF group, and Parabacteroides only increased dramatically in CRC fecal group. In CRC, Proteobacteria, Roseburia, Lachnospira, and Bilophila showed significant differences between the IVF group and Feces. Our results demonstrated that the bacterial strain was significantly different between IVF and fecal samples at the genus level. Of note, higher levels of Campylobacter Hemophilus, Prevotella, Veillonella, and Streptococcus were only observed in the IVF samples, indicating that these bacteria may locate near mucosal tissue instead in luminal(Figure 5).
Figure 5.

IVF: intestinal lavage fluid. *P < .05.
Key Species of Intestinal Flora in IVF and Feces of CRC Patients
To determine which bacterial taxa were primary drivers of the microbiome between CRC and controls, LDA was used to calculate the effect size of different bacterial taxa. In the phylogenetic tree, the circle radiating from the inside to the outside represents the classification level from the phylum to the genus. Each node represents a species, and the diameter of each small circle is proportional to the relative abundance of the taxon. The yellow node indicated that the species had no significant difference in the three groups. The red node demonstrated the flora that plays an important role in the normal human fecal specimen, and the blue node indicated that the species that plays an important role in the specimen of CRC, while the green node represents a microbial group that plays an important role in the IVF of CRC. Our results revealed that more key species existed in the IVF group than that in the fecal group, indicating that IVF contained more bacterial species. LDA score histogram and evolutionary branch diagram were shown in Figure 6, Figure 7. That LDA value greater than 2 indicates significantly different existing in species, and the larger LDA means, the greater the difference. Accordingly, 27 key species (LDA >2) detected in IVF, two of them with LDA >4, including Proteobacteria and Fusobacteria at the phylum level. Four classes detected in class level, including Erysipelotrichi, Fusobacteriia (LDA >4), Epsilonproteobacteria, and Gammaproteobacteria (LDA >4). In order levels, there were Erysipelotrichales, Fusobacteriales (LDA >4), Neisseriales, Campylobacterales, Enterobacteriales, Pseudomonadales. In family levels, there were Enterococcaceae, Neisseriaceae, Erysipelotrichaceae, Moraxellaceae, Helicobacteraceae, Camobacteriaceae, Peptostreptococcaceae, Tissierellaceae, Fusobacteriaceae (LDA >4), Leptotrichiaceae, Comamonadaceae, Campylobacteraceae, Moraxellaceae, and Leptotrichiaceae. In the genus level, there were Neisseria, Acinetobacter, Campylobacter, Granulicatella, Fusobacterium, Schlegelella, Tepidimonas, and Leptotrichia. On the other side, only 10 key species detected in fecal samples. At the family level, there were Porphyromonadaceae (LDA >4), Enterococcaceae, Peptostreptococcaceae, Tissierellaceae, and Peptostreptococcaceae. Compared with the fecal samples, nine more bacteria families found in IVF, including some pathogenic species, i.e., Neisseriaceae, Helicobacteraceae, Fusobacteriaceae, and Campylobacteraceae. Contrary to samples of CRC, feces from normal individuals had 18 key species, there were Firmicutes (LDA >4) in phylum level, Clostridia (LDA >4) and Deltaproteobacteria in class level, Bifidobacteriales, Clostridiales (LDA >4), and Desulfovibrionales in order level, Bifidobacteriaceae, Lachnospiraceae (LDA >4), Ruminococcaceae (LDA >4), Veillonellaceae (LDA >4) and Desulfovibrionaceae in family level, and Faecalibacterium (LDA >4), Roseburia (LDA >4), Coprococcus, Bifidobacterium, Lachnobacterium, Anaerostipes in genus level.
Figure 6.

System clustering tree. The red color indicates the healthy control group (Control), the green color represents the CRC group(IVF), and the blue color indicates the CRC fecal group (Feces). The red nodes represent the key species in the control group; the green nodes represent the key species in the IVF group, while the blue nodes represent the key species in a fecal group. The yellow nodes represent normal species. The name of the species was on the right side of the figure.
Figure 7.

The key species in the three groups were analyzed by LDA analysis. The LDA score was represented.
Co-Occurrence Bacterial Group in CRC and Healthy Controls
Recent studies have suggested the feasibility of using a combination of bacteria (or microbiota signature) in the fecal microbiota of patients with CRC as a marker for detecting the disease [23,24]. According to previous study and statistical analyses, six co-occurrence bacterial clusters in the OTUs data set were obtained and named them according to the most notable characteristic (Figure 8).
Figure 8.

Hierarchical Ward-linkage clustering based on the Pearson correlation coefficients of the relative abundance of operational taxonomic units in fecal and IVF microbiota of 30 individuals with CRC and feces of 25 healthy controls. Bacterial clusters were defined based on the clusters in the vertical tree and named after their most notable characteristic. Column color coding is according to the legend below. The right showed the most abundant bacterial genera, as well as the most strongly connected genera in each Bacterial cluster (i.e., genera with the highest numbers of significant positive correlations with other members of each respective group), were listed.
The Fusobacteria Cluster and Proteobacteria Cluster 1, 2 were significantly increased abundance in individuals with IVF (P < .001, P < 0 .001and P < .01, respectively). The Fusobacteria Cluster and Proteobacteria Cluster 2 were also significantly increased in CRC fecal group (P < .001and P < .01, respectively). On the contrary, the significantly less abundance of Firmicutes Cluster 1 and Firmicutes Cluster 2 were observed in the CRC group (either in IVF or fecal), but both were more abundant in healthy individuals (P < .01; Figure 9).
Figure 9.

Boxplots of relative abundances of the six bacterial clusters. The abundance of Fusobacteria Cluster (P < .001), Proteobacteria Cluster 1 (P < .001), and Proteobacteria Cluster 2 (P < .01)) were significantly increased in the IVF group. Two clusters (Fusobacteria Cluster, Proteobacteria Cluster 2) were significantly increased in individuals with CRC fecal group. Two clusters (Firmicutes Cluster 1 and Firmicutes Cluster 2) were significantly decreased in CRC IVF and fecal groups but more abundant in healthy individuals. ***P < .001; **P < .01; *P < .05.
ROC Curve Analysis of the Bacterial Clusters
ROC curve was used to evaluate the diagnostic significance of the bacterial clusters. When the area under the ROC curve (AUC) is more significant than 0.85, the markers have higher accuracy for diagnosis. Compared with the fecal group, two bacterial clusters (Firmicutes cluster1 and Fusobacteria cluster) in the IVF group have higher diagnostic accuracy, AUC was 93%, and 87.5%, respectively. For the fecal group, the Fusobacteria cluster has the highest AUC (94.4%) in CRC diagnosis. (Table 2).
Table 2.
Performance of bacterial cluster in the diagnosis of CRC
| AUC | Bacteroidetes | Firmicutes1 | Firmicutes 2 | Fusobacteria | Proteobacteria 1 | Proteobacteria 2 |
|---|---|---|---|---|---|---|
| F/IVF (Control vs CRC) |
55.4% | 93% | 75.6% | 87.5% | 79.1% | 65.4% |
| F/F (Control vs CRC) |
60.4% | 83.6% | 68.8% | 94.4% | 69.2% | 69.2% |
IVF: intestinal lavage fluid, F: feces.
Discussion
A novel aspect of CRC carcinogenesis is the association of biofilm-forming bacterial communities and their capacity to modulate cancer metabolism [25,26]. However, it is difficult to describe the intestinal microbiota due to its composition accurately is highly dynamic during life [27]. The stability of intestinal microbiota achieved in part through the ability of these microbes to attach to the mucosa [28]. The biofilm-like architecture of the mucosal microbiota, in close contact with the underlying gut epithelium, facilitates beneficial functions, including nutrient exchange and induction of host innate immunity [16]. Gut microbiota can form a specific niche, which may be a response to an altered colonic mucosal microenvironment [29]. Therefore, the mucosa-associated microbes are believed to be relatively stable in individuals throughout a lifetime [30], which is likely to contribute essential influences on host physiology in health and the development of disease [31].
Proper sampling of mucosal-associated microbiota, in their natural state, is essential for a better understanding of the host-microbial relationships in health and disease [28]. A mucosal biopsy may be the most appropriate method, but it is an invasive test that was significantly reducing its clinical applicability. Moreover, the biopsy specimen only represents a site rather than the entire intestine. Bowel preparation before biopsy may change the microbiota composition. There was a decrease in richness and microbial structure similarity after extensive colonic lavage [28]. The study suggests that colonic lavage reflected the microbial composition of a mucosal biopsy adequately [32]. Our results showed that 247 OTUs mainly exist in IVF group, while 94 OTUs only observed in fecal samples, much more OTUs in IVF indicating that the fecal microbiota partially reflects mucosal microbiota in CRC [8,33], both of which may fulfill different roles within the gut ecosystem [14]. The previous study revealed that mucosal microbiota of patients with CRC differs significantly from that of controls throughout the colon, while in CRC, mucosal microbiota differs in rectal, distal and proximal area. Therefore, samples taken by mucosal biopsy may not mirror entire microenvironment of the gut. Moreover, the altered microbiota composition in the mucosa of CRC patients is not restricted to cancerous tissue [8]. The study reveals that intestinal mucosa ecosystem may keep consistent within the same individual, in which microbiota may fulfill different roles [14]. IVF enrich the mucosal microbes from the entire colonic environment by repeating rinse the whole intestine, which could enlarge the detecting area that tissue biopsy could not reach.
The diversity of the intestinal microbiota is a crucial factor in CRC carcinogenesis. Intestinal microbiome represented by ɑ- and β-diversity. ɑ diversity is the diversity within an individual's microbiota, while β- diversity reflects the microbial composition between individuals. Presently, few data demonstrated that the degree of the fecal microbiota differs from mucosal microbiota [14]. Inconsistent reports on the flora diversity, either in healthy or CRC individuals, were reported. Decreased the ɑ-diversity was observed in the CRC patients' feces compared with the controls' [33,34]. On the other side, a study demonstrated that there was a significant increase in diversity within carcinomas as compared with adenomas [29]. When tissue samples from patients with colon adenomas compared with patient-matched healthy tissues, the ɑ-diversity at the site of the lesion was increased [35]. The methodology of sampling may be one of the reasons inducing different results [8]. In our study, the ɑ diversity had not significantly different between fecal samples of CRC and healthy individuals, but the decreased ɑ-diversity was observed in IVF of CRC. A wide variety might reflect a feature of a healthy gut enabling a species-rich ecosystem that deals with environmental challenges that promote disease processes [36]. Inter-individual variations in the tumor-associated mucosal microbiome have posed a long-standing problem for deciphering microbial signatures implicated in colorectal tumorigenesis [29]. When comparing the β diversity between healthy control and CRC, with either fecal or IVF samples, a definite separation of gut microbiota was found in the study. Gut microbiota of CRC patients was separated from that of the healthy group, while in the CRC group, microbiota in fecal samples was separated from in IVF, though some overlap existed. Our results demonstrated that different results might arouse by sampling. Nowadays, many gut microbiome studies were still based on fecal samples that may reflect the disease state but possibly not the tumor microenvironment [29].
It is now emerging that specific bacteria implicated in the risk of CRC [11,37]. Tumor microbiota data sets from the American/ European/ Vietnamese cohort 21 displayed very high abundances of Fusobacterium spp. [38]. In this study, the plenty of Fusobacteria was also higher in CRC than those in control, both in feces and in IVF, indicating that similar abundance of Fusobacteria in surface-adherent and luminal. A higher proportion of Proteobacteria exists in mucosa-associated microbiota than in fecal-luminal microbiota [39]. In this study, the abundance of fecal Proteobacteria were similar in CRC and healthy controls, but the abundance of Proteobacteria was higher in IVF than those in feces, indicating IVF contained a higher abundance of Proteobacteria that plays a critical role in CRC carcinogenesis. An increase in the abundance of Proteobacteria bacteria drastically enhanced the permeability of the ordinarily sterile mucus inner layer to more penetrable regions, resulting in bacterial infiltration into the inner layer close to epithelium [39]. Enterobacteriaceae, a branch of Proteobacteria, containing genera of E. coli, Klebsiella spp., and Proteus spp. E. coli and Klebsiella spp., have the potential for overgrowth and intestinal domination during dysbiosis [40]. E. coli, promoting invasive carcinoma in mice, and occurring at increased frequency in patients with CRC and inflammatory bowel disease, suggests the active involvement of enterobacterial toxins in tumorigenesis [41]. Given the mucosa-associated E. coli is significantly prevalent in CRC tissue and correlates with tumor stage and prognosis [42], a higher abundance of E. coli observed in IVF may have a significant clinical significance. Besides E.coli, high plenty of Pasteurellaceae, Streptococcaceae, and Hemophilus(belongs to the phylum Proteobacteria) were also observed in IVF, suggesting that the bacterial species in IVF may be related to the occurrence and development of CRC [43]. Bacteroides fragilis (B. fragilis) is a minority member of the healthy colonic microbiota, but it has been suggested to act as a “keystone pathogen” in the development of CRC [36,44]. In this study, B. fragilis was higher in fecal samples than in IVF, indicating that B. fragilis was distributed differently at different locations in the gut. Our results suggest that the position of bacteria in the intestine may be crucial in CRC carcinogenesis.
The bacteria population in the gut can be classified into predominant, subdominant, and transit bacteria [45]. In CRC, the relationship between the microbiota and disease may be complicated; combinations or co-abundance groups (CAGs) of organisms may be operative [14,46]. Firmicutes and Bacteroidetes are the dominant phyla of the gut [20,47], followed by Actinobacteria and Verrucomicrobia [48]. Certain bacteria, including Fusobacteria, Alistipes, Porphyromonadaceae, Coriobacteridae, Staphylococcaceae, Akkermansia spp. and Methanobacteriales) were consistently augmented, while some other (such as Bifidobacterium, Lactobacillus, Ruminococcus, Faecalibacterium spp., Roseburia, and Treponema) were underrepresented in CRC [49]. F. prausnitzii exhibited a progressively stronger positive association with members of the Ruminococcaceae toward carcinogenesis. Conversely, the co-occurrence of Blautia and Bacteroides was remarkably more robust in normal mucosae but weakened with tumor development [29]. Presently, identification and characterization of the significant patterns related to human gut microbiota configurations remain challenging [50]. Comparing the microbial co-occurrence populations between fecal and IVF samples, we found that Firmicutes Cluster 1 and 2 were significantly lower in the IVF group, while Proteobacteria Cluster 1 and 2 were considerably higher in IVF group when compared with the fecal group. Both E. coli (Enterobacteriaceae) and Streptococcus gallolyticus (Streptococcaceae) as protagonists of tumor development due to the correlation of their presence and increased risks of CRC [46]. Our results revealed that the enrichment of Proteobacteria and depletion of Firmicutes in CRC was significant in the IVF group. Given a lack of understanding of how microbiome profiles change during the transition from normal mucosae, adenomatous to malignant lesions, assigning individual members, or a consortium of the gut microbes with potential causative roles in CRC remains a grand challenge [29]. Analysis of co-occurrence flora in the IVF may help build the model reflecting predominate bacteria composition in CRC. Establish a niche through the formation of biofilms and the creation of selection pressures that prevent the expansion of other microbial communities may play a significant role in CRC carcinogenesis [28].
Accurate analysis of the intestinal microbiota will facilitate the establishment of an evaluating system for assessing CRC risk and prognosis. Microbial markers may represent an essential strategy for CRC detection in the future, screening patients with a “high-risk” microbial pattern to other further diagnostic procedures such as colonoscopy [51]. Samples taken from the tumor microenvironment are preferable, at least at the initial phase [33]. To testify the possibility of co-occurrence flora in IVF mirror the composition and overall structure of mucosa microbiota in CRC patients, studies of large sample sizes in both IVF and biopsy specimens are needed.
This study is preliminary, and the main limitations are the lack of healthy control of intestinal lavage specimens and the small sample size. It's better to validate the sequencing results by using the qPCR method in the large independent clinical samples in both IVF and feces. Moreover, analyze the ratio of single (or cluster) pathogenic bacterium and probiotics, for example, Fn/Bifidobacterium, Fn/E.coli, or Fusobacteria Cluster and Proteobacteria Cluster 1, 2. Combined clinical data, including tumor stage, parameters of prognosis with intestinal flora, will benefit from setting up the risk evaluating the system for predicting CRC development, providing more valuable information for the treatment of CRC.
Conclusion
The detection of IVF reveals more pathogenic factors of CRC patients, which has important significance in the exploration of the etiology of CRC.
Declarations
Ethical Approval and Consent to Participate
The Ethics Committee of Shantou University Medical College approved the study. The human investigations were performed after approved by an institutional review board. Informed written consent obtained from each subject or each subject's guardian.
Consent for Publication
Not Applicable.
Availability of Data and Materials
All data generated or analyzed during this study are included in this published article.
Competing Interests
The authors declare that they have no competing interests.
Funding
This work supported by the Shantou Science and Technology Project (grant numbers. 180709174010328) and the Shantou Science and Technology Project (grant numbers. 201718239) .
Authors' Contributions
Weitao Shen and Jiayu Sun contributed equally to this paper.
WTS and JYS: designed research; WTS and XYJ: conducted research; JYS and FY analyzed data; XYJ, KHL, YMY, YXC, ZYL, HH, and JZ: collect samples and conducted experiment; and all authors reviewed the draft manuscript and read and approved the final manuscript.
Contributor Information
Weitao Shen, Email: 646957018@qq.com.
Jiayu Sun, Email: 18jysun1@stu.edu.cn.
Fen Yao, Email: fyao@stu.edu.cn.
Kaihuang Lin, Email: linkaihuang@163.com.
Yumeng Yuan, Email: 671421696@qq.com.
Yexi Chen, Email: chenyexi@vip.sina.com.
Hui Han, Email: hanhui0571@126.com.
Zhiyang Li, Email: medyzy@163.com.
Juan Zou, Email: 15271689450@163.com.
Xiaoyang Jiao, Email: xyjiao@stu.edu.cn.
References
- 1.Jemal A. Global cancer statistics. CA Cancer J. Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Azcarate-Peril M.A., Sikes M., Bruno-Barcena J.M. The intestinal microbiota, gastrointestinal environment and colorectal cancer: a putative role for probiotics in prevention of colorectal cancer? Am. J. Physiol. Gastrointest. Liver Physiol. 2011;301(3):G401–G424. doi: 10.1152/ajpgi.00110.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barman M. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect. Immun. 2008;76(3):907–915. doi: 10.1128/IAI.01432-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akin H., Tozun N. Diet, microbiota, and colorectal cancer. J. Clin. Gastroenterol. 2014;48(Suppl. 1):S67–S69. doi: 10.1097/MCG.0000000000000252. [DOI] [PubMed] [Google Scholar]
- 5.Geng J. Co-occurrence of driver and passenger bacteria in human colorectal cancer. Gut Pathog. 2014;6:26. doi: 10.1186/1757-4749-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burns M.B. Virulence genes are a signature of the microbiome in the colorectal tumor microenvironment. Genome Med. 2015;7(1):55. doi: 10.1186/s13073-015-0177-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Candela M. Human intestinal microbiota: cross-talk with the host and its potential role in colorectal cancer. Crit. Rev. Microbiol. 2011;37(1):1–14. doi: 10.3109/1040841X.2010.501760. [DOI] [PubMed] [Google Scholar]
- 8.Flemer B. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut. 2017;66(4):633–643. doi: 10.1136/gutjnl-2015-309595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoon H. Comparisons of gut microbiota among healthy control, patients with conventional adenoma, sessile serrated adenoma, and colorectal cancer. J Cancer Prev. 2017;22(2):108–114. doi: 10.15430/JCP.2017.22.2.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castellarin M. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22(2):299–306. doi: 10.1101/gr.126516.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Irrazabal T. The multifaceted role of the intestinal microbiota in colon cancer. Mol. Cell. 2014;54(2):309–320. doi: 10.1016/j.molcel.2014.03.039. [DOI] [PubMed] [Google Scholar]
- 12.Hasegawa M. A role of lipophilic peptidoglycan-related molecules in induction of Nod1-mediated immune responses. J. Biol. Chem. 2007;282(16):11757–11764. doi: 10.1074/jbc.M700846200. [DOI] [PubMed] [Google Scholar]
- 13.Arthur J.C. Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nat. Commun. 2014;5:4724. doi: 10.1038/ncomms5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eckburg P.B. Diversity of the human intestinal microbial flora. Science. 2005;308(5728):1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fung T.C., Artis D., Sonnenberg G.F. Anatomical localization of commensal bacteria in immune cell homeostasis and disease. Immunol. Rev. 2014;260(1):35–49. doi: 10.1111/imr.12186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sonnenburg J.L., Angenent L.T., Gordon J.I. Getting a grip on things: how do communities of bacterial symbionts become established in our intestine? Nat. Immunol. 2004;5(6):569–573. doi: 10.1038/ni1079. [DOI] [PubMed] [Google Scholar]
- 17.van der Waaij L.A. Bacterial population analysis of human colon and terminal ileum biopsies with 16S rRNA-based fluorescent probes: commensal bacteria live in suspension and have no direct contact with epithelial cells. Inflamm. Bowel Dis. 2005;11(10):865–871. doi: 10.1097/01.mib.0000179212.80778.d3. [DOI] [PubMed] [Google Scholar]
- 18.Chen W. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS One. 2012;7(6) doi: 10.1371/journal.pone.0039743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qin J. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Human Microbiome Project, C., Structure, function and diversity of the healthy human microbiome. Nature, 2012. 486(7402): p. 207–14. [DOI] [PMC free article] [PubMed]
- 21.Thomas A.M. Tissue-associated bacterial alterations in rectal carcinoma patients revealed by 16S rRNA community profiling. Front. Cell. Infect. Microbiol. 2016;6:179. doi: 10.3389/fcimb.2016.00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zeller G. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 2014;10:766. doi: 10.15252/msb.20145645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zackular J.P. The human gut microbiome as a screening tool for colorectal cancer. Cancer Prev. Res. (Phila.) 2014;7(11):1112–1121. doi: 10.1158/1940-6207.CAPR-14-0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu J. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut. 2017;66(1):70–78. doi: 10.1136/gutjnl-2015-309800. [DOI] [PubMed] [Google Scholar]
- 25.Johnson C.H. Metabolism links bacterial biofilms and colon carcinogenesis. Cell Metab. 2015;21(6):891–897. doi: 10.1016/j.cmet.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu J. Metabolic co-dependence gives rise to collective oscillations within biofilms. Nature. 2015;523(7562):550–554. doi: 10.1038/nature14660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lozupone C.A. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489(7415):220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harrell L. Standard colonic lavage alters the natural state of mucosal-associated microbiota in the human colon. PLoS One. 2012;7(2) doi: 10.1371/journal.pone.0032545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakatsu G. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat. Commun. 2015;6:8727. doi: 10.1038/ncomms9727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zoetendal E.G., Akkermans A.D., De Vos W.M. Temperature gradient gel electrophoresis analysis of 16S rRNA from human fecal samples reveals stable and host-specific communities of active bacteria. Appl. Environ. Microbiol. 1998;64(10):3854–3859. doi: 10.1128/aem.64.10.3854-3859.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Backhed F. Host-bacterial mutualism in the human intestine. Science. 2005;307(5717):1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 32.Watt E. Extending colonic mucosal microbiome analysis-assessment of colonic lavage as a proxy for endoscopic colonic biopsies. Microbiome. 2016;4(1):61. doi: 10.1186/s40168-016-0207-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mira-Pascual L. Microbial mucosal colonic shifts associated with the development of colorectal cancer reveal the presence of different bacterial and archaeal biomarkers. J. Gastroenterol. 2015;50(2):167–179. doi: 10.1007/s00535-014-0963-x. [DOI] [PubMed] [Google Scholar]
- 34.Ahn J. Human gut microbiome and risk for colorectal cancer. J. Natl. Cancer Inst. 2013;105(24):1907–1911. doi: 10.1093/jnci/djt300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen X.J. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut Microbes. 2010;1(3):138–147. doi: 10.4161/gmic.1.3.12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tilg H. The intestinal microbiota in colorectal cancer. Cancer Cell. 2018;33(6):954–964. doi: 10.1016/j.ccell.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 37.Marinelli L., Tenore G.C., Novellino E. Probiotic species in the modulation of the anticancer immune response. Semin. Cancer Biol. 2017;46:182–190. doi: 10.1016/j.semcancer.2017.08.007. [DOI] [PubMed] [Google Scholar]
- 38.Kostic A.D. Genomic analysis identifies association of fusobacterium with colorectal carcinoma. Genome Res. 2012;22(2):292–298. doi: 10.1101/gr.126573.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ringel Y. High throughput sequencing reveals distinct microbial populations within the mucosal and luminal niches in healthy individuals. Gut Microbes. 2015;6(3):173–181. doi: 10.1080/19490976.2015.1044711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taur Y., Pamer E.G. The intestinal microbiota and susceptibility to infection in immunocompromised patients. Curr. Opin. Infect. Dis. 2013;26(4):332–337. doi: 10.1097/QCO.0b013e3283630dd3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwabe R.F., Wang T.C. Cancer. Bacteria deliver a genotoxic hit. Science. 2012;338(6103):52–53. doi: 10.1126/science.1229905. [DOI] [PubMed] [Google Scholar]
- 42.Bonnet M. Colonization of the human gut by E. coli and colorectal cancer risk. Clin. Cancer Res. 2014;20(4):859–867. doi: 10.1158/1078-0432.CCR-13-1343. [DOI] [PubMed] [Google Scholar]
- 43.Rowland I.R. The role of the gastrointestinal microbiota in colorectal cancer. Curr. Pharm. Des. 2009;15(13):1524–1527. doi: 10.2174/138161209788168191. [DOI] [PubMed] [Google Scholar]
- 44.Hajishengallis G., Darveau R.P., Curtis M.A. The keystone-pathogen hypothesis. Nat Rev Microbiol. 2012;10(10):717–725. doi: 10.1038/nrmicro2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bourlioux P. The intestine and its microflora are partners for the protection of the host: report on the Danone Symposium "The Intelligent Intestine," held in Paris, June 14, 2002. Am. J. Clin. Nutr. 2003;78(4):675–683. doi: 10.1093/ajcn/78.4.675. [DOI] [PubMed] [Google Scholar]
- 46.Sears C.L., Garrett W.S. Microbes, microbiota, and colon cancer. Cell Host Microbe. 2014;15(3):317–328. doi: 10.1016/j.chom.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faith J.J. The long-term stability of the human gut microbiota. Science. 2013;341(6141):1237439. doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tlaskalova-Hogenova H. Commensal bacteria (normal microflora), mucosal immunity and chronic inflammatory and autoimmune diseases. Immunol. Lett. 2004;93(2–3):97–108. doi: 10.1016/j.imlet.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 49.Vipperla K., O'Keefe S.J. Diet, microbiota, and dysbiosis: a 'recipe' for colorectal cancer. Food Funct. 2016;7(4):1731–1740. doi: 10.1039/c5fo01276g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Costea P.I. Enterotypes in the landscape of gut microbial community composition. Nat. Microbiol. 2018;3(1):8–16. doi: 10.1038/s41564-017-0072-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eklof V. Cancer-associated fecal microbial markers in colorectal cancer detection. Int. J. Cancer. 2017;141(12):2528–2536. doi: 10.1002/ijc.31011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.