

Abstract
Aims
Although distinct DNA methylation patterns have been reported, its localization and roles remain to be defined in heart failure. We investigated the cellular and subcellular localization of DNA methylation and its pathophysiological significance in human failing hearts.
Methods and results
Using left ventricular (LV) endomyocardial biopsy specimens from 75 patients with dilated cardiomyopathy (DCM; age: 58 ± 14 years old, %female: 32%) and 20 patients without heart failure (controls; age: 56 ± 17 years old, %female: 45%), we performed immunohistochemistry and immunoelectron microscopy for methylated DNA, 5‐methylcytosine (5‐mC). We next investigated possible relations of the incidence of 5‐mC‐positive (%5‐mC+) cardiomyocytes with clinicopathological parameters. Immunopositivity for 5‐mC was detected in the cardiomyocytes and other cell types. The %5‐mC+ cardiomyocytes was significantly greater in DCM hearts than in controls (57 ± 13% in DCM vs. 25 ± 12% in controls, P < 0.0001). The localization of 5‐mC immunopositivity in cardiomyocyte nuclei coincided well with that of heterochromatin, as confirmed by immunoelectron microscopy. Substantial DNA methylation was also observed in interstitial non‐cardiomyocytes, but the incidences did not differ between control and DCM hearts (39 ± 7.9% in DCM vs. 41 ± 10% in controls, P = 0.4099). In DCM patients, the %5‐mC+ cardiomyocytes showed a significant inverse correlation with LV functional parameters such as heart rate (r = 0.2391, P = 0.0388), end‐diastolic pressure (r = 0.2397, P = 0.0397), and ejection fraction (r = −0.2917, P = 0.0111) and a positive correlation with LV dilatation (volume index at diastole; r = 0.2442, P = 0.0347; and volume index at systole; r = 0.3136, P = 0.0062) and LV hypertrophy (mass index; r = 0.2287, P = 0.0484)—that is, LV remodelling parameters. No significant correlations between DNA methylation and the histological parameters of the biopsies, including cardiomyocyte hypertrophy, fibrosis, and inflammatory cell infiltration, were noted.
Conclusions
The present study revealed increased nuclear DNA methylation in cardiomyocytes, but not other cell types, from DCM hearts, with predominant localization in the heterochromatin. Its significant relations with LV functional and remodelling parameters imply a pathophysiological significance of DNA methylation in heart failure.
Keywords: Chromatin remodelling; Dilated cardiomyopathy; DNA methylation, Epigenetics
1. Introduction
Clinical and experimental studies suggest epigenetic mechanisms play a pivotal role in the regulation of cardiac gene expression patterns and in the progression of cardiac hypertrophy and heart failure.1, 2, 3, 4, 5, 6, 7 DNA methylation and histone modification are the most common mechanisms underlying epigenetic modulation of gene expression. It has been suggested that histone modification may contribute to the reprogramming of cardiac gene expression in heart failure,1 but the role of DNA methylation is far less defined. Recent studies have shown that distinct epigenomic DNA methylation patterns are present at important DNA elements within the cardiac genome in human end‐stage cardiomyopathy2 and that differential DNA methylation correlates with differential expression of angiogenic factors in human heart failure.3, 4 Subsequent studies also using human cardiac tissue revealed differences in methylation of genes involved in pathways related to heart disease, which may provide an opportunity for differential diagnosis of heart failure.5, 6, 7, 8 Gilsbach et al.9 provided information on how DNA methylation patterns change during developmental and pathological remodelling; interestingly, in a pressure overload model of heart failure, the DNMT3A‐catalyzed patterns of DNA methylation detected in isolated cardiomyocytes were similar to those seen during development.
However, basic information such as cellular and subcellular localization of methylated DNA remains elusive. Because genome‐wide studies using failing hearts analysed DNA in myocardial cells without distinguishing among cell types, it is impossible to know in which cell type(s), cardiomyocytes, or other interstitial cells, including fibroblasts, vascular cells, and inflammatory cells, undergo alterations in DNA methylation. It should be noted that non‐cardiomyocytes, including interstitial and vascular cells account for 65–75% of the cells within the normal heart.10, 11, 12 It will therefore be important to investigate the state of the DNA methylation in these cell types. In addition, nuclear structure is markedly altered in cardiomyocytes of failing hearts, characterized as being hypertrophied with severe crenation of nuclear membrane and widespread clumping of the chromatin, two hallmarks of nuclear hypertrophy.13, 14, 15, 16 We hypothesized that nuclear hypertrophy is associated with altered DNA methylation in cardiomyocyte nuclei in human failing hearts and may also be related to cardiac anatomical and functional alterations. To test that idea, we compared the DNA methylation levels and intranuclear localization of DNA methylation in endomyocardial biopsy specimens from human hearts with dilated cardiomyopathy (DCM) with non‐failing hearts. And then, we investigated the possible association of DNA methylation with the haemodynamics and pathology of failing hearts with DCM.
2. Methods
The protocol of the present study was approved by the ethics committee of Gifu University Graduate School of Medicine (Approval No. 30‐059). The investigation conformed to the principles outlined in the Declaration of Helsinki (BMJ 1964; ii: 177).
2.1. Patient profile
After obtaining approval for this study from our local ethics committees, patients with DCM were selected from among those who underwent left ventricular biopsy at Gifu University Hospital during the period from 2004 to 2013. All patients were evaluated clinically using both non‐invasive and invasive methods. Diagnoses of DCM were made according to the definition and classification proposed by the World Health Organization‐International Society and Federation of Cardiology task force.17 A total of 75 patients were enrolled in the study, including 51 men and 24 women with a mean age of 58 ± 14 years (range: 17–78 years). Patients with severe coronary artery stenosis (>75% luminal narrowing) and those with a history of apparent hypertension were excluded from this study. All patients were given medications, including various combinations of a digitalis glycoside, diuretic, angiotensin converting enzyme inhibitor, angiotensin II type 1 receptor blocker, β‐blocker, and L‐type calcium channel blocker. However, no drugs were given on the day of biopsy collection. The control group without heart failure included 20 patients who had been clinically suspected of some cardiac disease because of chest pain, minimal electrocardiographic change, or arrhythmia, but for whom both non‐invasive and invasive examinations of coronary angiography and biopsy findings were not diagnostic. The specimens were processed in the same way as those from patients with DCM.
2.2. Echocardiographic, haemodynamic, and angiographic evaluation
With all patients, two‐dimensional echocardiographic examinations were performed no more than 3 days before invasive examinations using SSD‐3500 (ALOKA, Tokyo, Japan) until March 2010 and iE33 (PHILIPS, Amsterdam, Netherlands) afterwards. The ventricular septal thickness and left ventricular (LV) posterior wall thickness were recorded during the diastolic and systolic phases. All patients underwent both right‐heart and left‐heart catheterization, biplane left ventriculography, and selective coronary angiography using standard techniques. The heart rate and arterial pressures from the right and left heart were recorded, and the cardiac index was estimated using the thermodilution method. Left ventricular end‐diastolic and end‐systolic volume indexes (LVEDVI and LVESVI) and ejection fraction (LVEF) were calculated from the LV cineangiogram obtained in the right anterior oblique projection.
2.3. Endomyocardial biopsy procedure and histologic evaluation
From each patient, one to four biopsy specimens were collected from the left ventricular free wall during the cardiac catheterization. One or two specimens were immediately fixed in a 10% buffered‐formalin solution, dehydrated, embedded in paraffin for light microscopy (Olympus BX53, Tokyo, Japan). In 4 μm thick paraffin sections stained with haematoxylin and eosin or Masson's trichrome, cardiomyocyte size (mean diameter of the transversely sectioned cells, 30 to 50 cardiomyocytes in random fields of view per section) and degree of fibrosis (from 0 to 3) were evaluated.18 In addition, the mean numbers of inflammatory cells (total polymorphonuclear leukocytes, lymphocytes, and plasma cells) per high power field (×400) were calculated.
2.4. Immunohistochemistry
After deparaffinization, the 4 μm thick serial sections neighbouring the haematoxylin and eosin‐stained ones were soaked in citrate buffer and heated for 10 min in a microwave oven for antigen retrieval and incubated with a primary antibody against 5‐methylcytosine (5‐mC; dilution 1/500, clone 33D3, Epigentek Group, Farmingdale, NY, USA). Acid denaturation was not performed to the tissue slides. A Vectastain Elite ABC system (Vector Laboratories, Burlingame, CA, USA) was then used to immunostain the sections; diaminobenzidine served as the chromogen, and the nuclei were counterstained with haematoxylin. The presence of immunoreactive 5‐mC was assessed by light microscopic examination. Using all of the endomyocardial biopsy specimens, 5‐mC in the nucleus was graded as positive or negative, and the percentage of 5‐mC‐immunopositive nuclei (%5‐mC+ nuclei) to a total of 30–50 cardiomyocyte nuclei in each specimen was calculated. Similarly, the %5‐mC+ nuclei were calculated in non‐cardiomyocyte interstitial cell nuclei.
In order to specify cell types contained in the endomyocardial biopsy specimens, we performed immunohistochemical stain for dystrophin, a specific marker for skeletal muscle cells including cardiomyocytes, which makes it easy to distinguish cardiomyocytes from non‐cardiomyocytes. After deparaffinization, the 4 μm thick sections were incubated with a primary antibody against dystrophin (dilution 1/100, ab15277, Abcam, Cambridge, UK). Antigen retrieval was not performed. A Vectastain Elite ABC system was then used to immunostain the sections; diaminobenzidine served as the chromogen, and the nuclei were counterstained with haematoxylin. Searching high power fields, we determined percentage of the cardiomyocyte population in specimens from six pairs of age‐matched and gender‐matched patients from the control and DCM groups.
2.5. Electron microscopy
Endomyocardial biopsy specimens other than those used for light microscopy were immediately fixed for 4 h in 2.5% glutaraldehyde in 0.1 mol/L phosphate buffer. The specimens were then post‐fixed in 1% osmium tetroxide for 1 h, dehydrated through graded ethanol and propylene oxide series, and embedded in Epon. Thereafter, the specimens were thin‐sectioned (70 nm) using an ultramicrotome, mounted on plain nickel grids, stained with uranyl acetate and lead citrate, and examined with an electron microscope (HT7700, Hitachi, Tokyo, Japan).
In the endomyocardial specimens, cell types were identified by their specific ultrastructure and/or position: cardiomyocytes by the presence of sarcomeres; endothelial cells by the innermost layer position of the vessels; fibroblasts by the spindle shaped cells containing rough endoplasmic reticulum, scattering in the interstitium; vascular smooth muscle cells by the wrapping position around the vascular endothelial cells of small arteries, containing dense actin filaments in the cutoplasm; pericytes by the wrapping position around the vessels; macropahges by the amorphous shape having pseudopods and containing foreign bodies, scattering in the interstitium; and polymorphonuclear cells by the segmented nucleus and cytoplasmic granules.
2.6. Immunoelectron microscopy
Thin sections (70 nm) were mounted on bare 200‐mesh nickel grids. The methylated DNA was labelled using a mouse anti‐5‐mC antibody at a dilution of 1:100 in 2% horse serum‐containing PB for 1 h at room temperature. The grids were then incubated with colloidal gold (particle size: 15 nm)–conjugated goat anti‐mouse IgG (GAF‐011‐15, EY Laboratories, San Mateo, CA) at a dilution of 1:5 in 2% horse serum‐containing phosphate buffer for 1 h at room temperature. After rinsing the grids with distilled water, they were counterstained with lead citrate and examined using an electron microscope. The grids were washed with phosphate buffer between each step. We checked the validity of this method by omitting the primary antibody (negative control).
Localization of immunogold‐labelled 5‐mC within cardiomyocyte nuclei was determined in 30 nuclei per specimen using specimens from six pairs of age‐matched and gender‐matched patients from the control and DCM groups: the density of the immunogold particles (per μm2) was calculated manually per micrograph, as were the fractions of immunogold particles localized in the heterochromatin and euchromatin within the nuclei. We similarly determined the density of nuclear immunogold particles labelling 5‐mC in vascular endothelial cells and myocardial fibroblasts from those patients; 20 cells each were counted per specimen.
2.7. Statistical analysis
All statistical analyses were performed using Graph Pad PRISM Version 6.0 statistical software (GraphPad Software, CA). Data are expressed as means ± SD. Group comparisons were made using Student's t‐tests. Spearman's analysis was used to determine correlations between parameters. Values of P < 0.05 were considered significant.
3. Results
3.1. Clinical and histological findings
Table 1 summarizes the patients' clinical, echocardiographic, haemodynamic, and angiographic findings. The hearts of all 75 study participants had dilated LV cavities characterized by enlarged left ventricles (i.e. increased LVEDVI and LVESVI) or hypertrophied left ventricles (i.e. increased LV mass index). In addition, LV end‐diastolic pressure was elevated, and LV systolic function was impaired, as indicated by the small LVEF (38 ± 10% in DCM vs. 64 ± 6% in controls) and increased heart rate. The biopsy specimens from patients with DCM showed enlarged cardiomyocytes and increased fibrosis, varying degrees of inflammatory cell infiltration (Table 1). Cardiomyocyte population was 33 ± 5.3% in the control hearts and 23 ± 3.6% in DCM hearts (n = 6 each, P = 0.0056) (Figures 1A and 1B), being compatible with earlier reports.10‐12
Table 1.
Clinicopathological data
| Non‐failing hearts (n = 20) | DCM (n = 75) | |||||
|---|---|---|---|---|---|---|
| Mean ± SD or n | Limits | Mean ± SD or n | Limits | Correlation with %5‐mC+ cardiomyocyte | ||
| r value | P value | |||||
| Clinical data | ||||||
| Age (year old) | 56 ± 17 | 14–77 | 58 ± 14 | 17‐81 | −0.0661 | 0.5730 |
| Gender (M/F) | 11/9 | — | 51/24 | — | — | — |
| LVSP (mm Hg) | 137 ± 29 | 100–200 | 135 ± 25 | 75–200 | −0.0314 | 0.7892 |
| LVEDP (mm Hg) | 13 ± 5.9 | 4–25 | 16 ± 8.2 | 4–40 | 0.2397 | 0.0397 |
| Heart rate (bpm) | 68 ± 13 | 47–94 | 77 ± 16* | 53–132 | 0.2391 | 0.0388 |
| LVEF (%) | 64 ± 6 | 55–76 | 38 ± 10* | 10–54 | −0.2917 | 0.0111 |
| LVEDVI (ml/m2) | 90 ± 19 | 57–135 | 124 ± 39* | 30–222 | 0.2442 | 0.0347 |
| LVESVI (ml/m2) | 32 ± 10 | 12–57 | 78 ± 33* | 15–169 | 0.3136 | 0.0062 |
| LVMI (g/m2) | 101 ± 42 | 57–206 | 137 ± 41* | 65–248 | 0.2287 | 0.0484 |
| Histological data | ||||||
| Cardiomyocyte size (μm) | 17 ± 2.6 | 12.8–21.8 | 26 ± 4.7* | 13.9–40.3 | 0.0248 | 0.8325 |
| Degree of fibrosis (0–3) | 1.0 ± 0.7 | 0–2 | 1.7 ± 0.7* | 0.5–3 | 0.0296 | 0.8009 |
| Inflammatory cells (per HPF) | 0.4 ± 0.5 | 0–1 | 0.9 ± 0.6* | 0–2 | 0.0513 | 0.6621 |
| Cardiomyocyte population (%)a | 33 ± 5.3 | 24–38 | 23 ± 3.6* | 18–28 | — | — |
LVSP and LVEDP, left ventricular peak systolic and end‐diastolic pressure; LVEDVI and LVESVI, left ventricular end‐diastolic and end‐systolic volume index; LVEF, left ventricular ejection fraction; LVMI, left ventricular mass index; HPF, high power field.
P < 0.05 vs. non‐failing hearts.
n = 6 in each group.
3.2. DNA methylation in cardiac cells
All specimens positively immunostained for 5‐mC, which was exclusively confined to the nuclei in both cardiomyocytes and non‐cardiomyocytes (Figures 1 C to 1 D′). In cardiomyocytes, the incidence of 5‐mC‐immunopositive (5‐mC+) nuclei was significantly greater in DCM patients (57 ± 13%, range: 24–95%) than in controls (25 ± 12%, range: 1.9–44%, P < 0.0001) (Figure 1 E). In non‐cardiomyocytes, on the other hand, the incidences were not different between the two groups (39 ± 7.9% in controls vs. 41 ± 10% in DCM, P = 0.4099) (Figure 1F).
Figure 1.
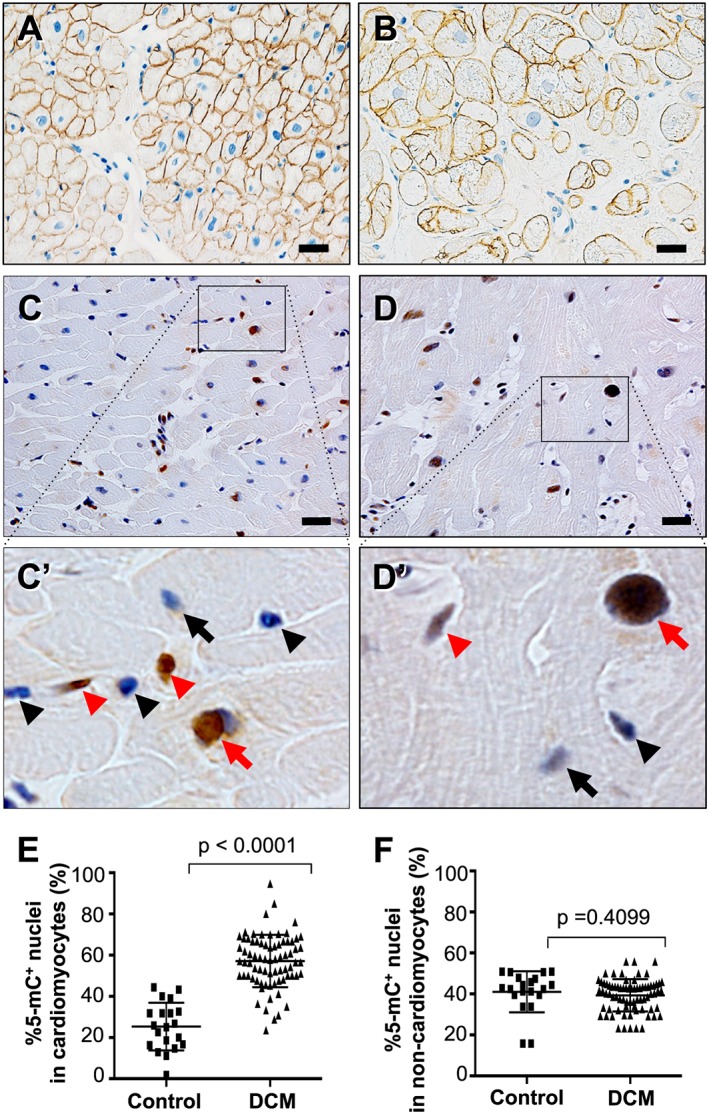
Cell type specification and immunohistochemical detection of 5‐mC in endomyocardial biopsy specimens at the light microscopic level. (A and B) Immunohistochemistry for dystrophin in a control specimen in panel A and a DCM specimen in panel B. Plasma membranes and T‐tubules are stained specifically in cardiomyocytes. Scale bars, 20 μm. (C and D) 5‐mC‐positive nuclei are stained brown while 5‐mC‐negative nuclei are stained blue in both a control specimen in panel C and a DCM specimen in panel D. Panels A′ and B′ are highly magnified images of the boxed areas in panels A and B, respectively. Red arrows, 5‐mC‐positive cardiomyocyte nuclei; black arrows, 5‐mC‐negative cardiomyocyte nuclei; red arrowheads, 5‐mC‐positive non‐cardiomyocyte nuclei; black arrowheads, 5‐mC‐negative non‐cardiomyocyte nuclei. Scale bars, 20 μm. (E) Graph showing comparison of the %5‐mC‐positive nuclei in cardiomyocytes between controls (n = 20) and DCM patients (n = 75). (F) Graph showing comparison of the %5‐mC‐positive nuclei in non‐cardiomyocyte interstitial cells between controls (n = 20) and DCM patients (n = 75).
3.3. Nuclear localization of methylated DNA and its relation to chromatin remodelling
Cardiomyocyte nuclei in specimens from DCM patients generally exhibited degenerative changes whereby they were hypertrophied or bizarrely shaped and contained abundant heterochromatin.13, 14, 15, 16 To evaluate the spatial relationship between the heterochromatin and methylated DNA, we performed an immunoelectron microscopic analysis of the 5‐mC. Immunogold particles bound to 5‐mC concentrated predominantly on electron dense heterochromatin; electron lucent euchromatin was rarely stained (Figure 2 ). Quantitative analysis further confirmed that immunogold particles bound to 5‐mC were more densely concentrated within cardiomyocyte nuclei affected by DCM (106 ± 27 particles/μm2) than within control nuclei (33 ± 4 particles/μm2, P < 0.0001). Moreover, the particles localized almost entirely in the heterochromatin; approximately 94% of the immunogold particles were localized on heterochromatin in both controls (94 ± 2.0%) and DCM (94 ± 2.1%).
Figure 2.
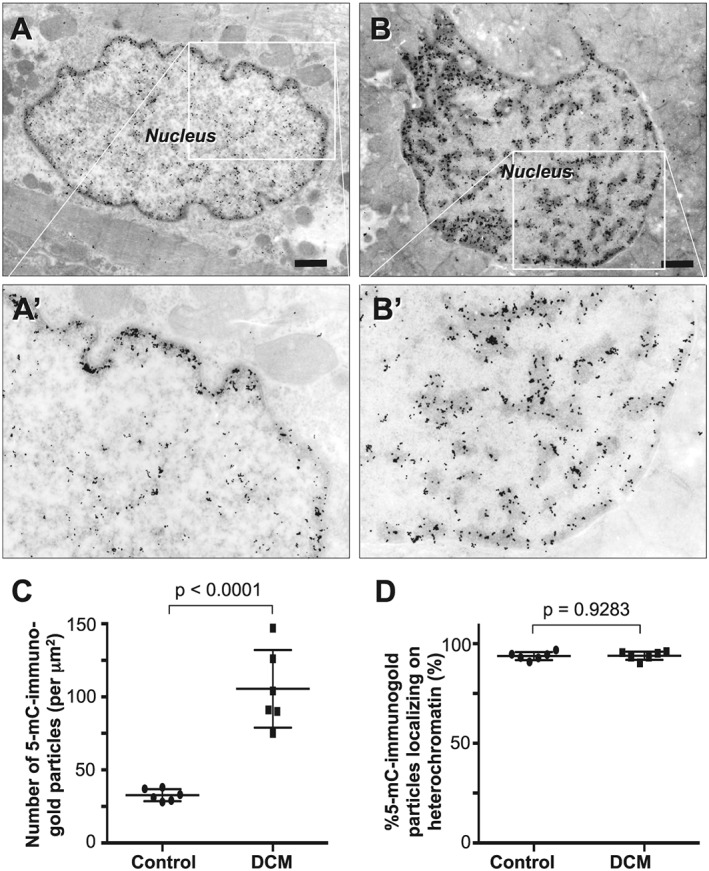
Immunocytochemical detection of 5‐mC in cardiomyocytes in endomyocardial biopsy specimens at the electron microscopic level. Immunogold particles bound to 5‐mC localize predominantly in heterochromatin in both normal (A) and bizarrely shaped (B) nuclei. The bizarrely shaped nucleus is rich in heterochromatin where immunogold particles are abundant. Panels A′ and B′ are highly magnified and lightly printed images of the boxed areas in panels A and B, respectively. They highlight the relationship between heterochromatin and immunogold particles. Scale bars, 1 μm. (C) Graph showing comparison of the number of 5‐mC bound immunogold particles between controls (n = 6) and DCM patients (n = 6). (D) Graph showing fraction of the 5‐mC bound immunogold particles localized on heterochromatin in controls (n = 6) and DCM patients (n = 6).
Immunoelecron microscopy confirmed the accumulation of immunogold particles bound to 5‐mC in nuclear heterochromatin in all types of non‐cardiomyocytes, including vascular endothelial cells, vascular pericytes, vascular smooth muscle cells, fibroblasts, macrophages, and circulating leukocytes (Figure 3 ). However, no apparent differences in the nuclear density of immunogold particles were noted among the different cell types between DCM and controls. Quantitative analysis revealed no difference between specimens from control and DCM hearts with respect to the number of immunogold particles within the nuclei of vascular endothelial cells (controls, 114 ± 32 particles/μm2 vs. DCM, 113 ± 23 particles/μm2, P = 0.9356) or myocardial fibroblasts (controls, 111 ± 23 particles/μm2 vs. DCM, 113 ± 20 particles/μm2, P = 0.8937).
Figure 3.
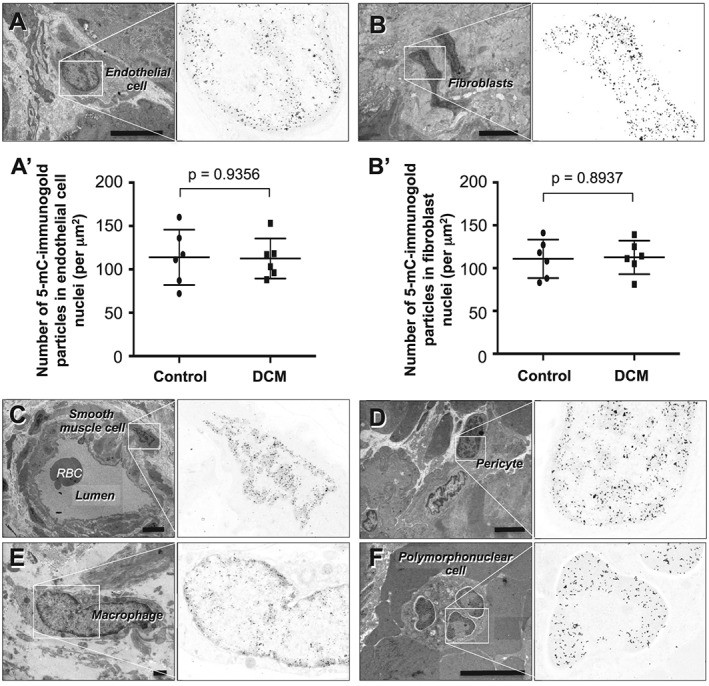
Immunocytochemical detection of 5‐mC in non‐cardiomyocyte interstitial cells in endomyocardial biopsy specimens from patients with DCM. (A) Capillary endothelial cell. (B) Fibroblast. (A′ and B′) Graphs showing comparison of the number of 5‐mC bound immunogold particles in capillary endothelial cell nuclei (A′) and in fibroblast nuclei (B′) between controls (n = 6) and DCM patients (n = 6). Group comparisons were made using Student's t‐tests. (C) Smooth muscle cell in a small artery. (D) Pericyte in an arteriole. (E) Macrophage. (F) Polymorphonuclear cell within a vascular lumen. Scale bars, 1 μm.
Figure 4.
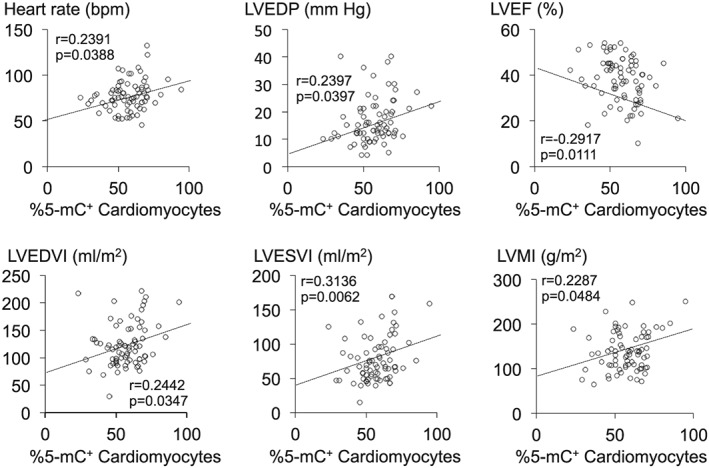
Plots showing the correlations between the percent 5‐mC‐positive (%5‐mC+) cardiomyocytes and the indicated haemodynamic parameters in patients with DCM (n = 75 in each graph). LVEDP, LV end‐diastolic pressure; LVEF, LV ejection fraction; LVEDVI, LV end‐diastolic volume index; LVESVI, LV end‐systolic volume index; LVMI, LV mass index.
3.4. Relations between methylated DNA and clinicopathological parameters
We investigated the relations between DNA methylation in cardiomyocyte nuclei and several clinicopathological parameters (age, gender, cardiac structure and function, and cardiac histology) in the DCM patients. There was no difference between male and female in the degree of DNA methylation in DCM patients (male: 57 ± 1.8% vs. female: 57 ± 2.5%, P = 0.7785). On the other hand, we found that there was a significant positive correlation between the percent 5‐mC‐positive cardiomyocytes and heart rate (Table 1 and Figure 4). We also found a significant positive correlation between the percent 5‐mC‐positive cardiomyocytes and LV remodelling parameters, including LV dilatation (LVEDVI and LVESVI) and LV hypertrophy (LV mass index). There was an inverse correlation with LV systolic function (LVEF). No significant correlations between DNA methylation and the histological parameters of the biopsies, including cardiomyocyte hypertrophy, fibrosis, and inflammatory cell infiltration, were noted (Table 1). In the control hearts, no significant correlation was noted between the degree of DNA methylation and any clinicopathological parameters (P > 0.05 in all).
4. Discussion
The major findings of the present study of human endomyocardial biopsy specimens are the following: (i) nuclear DNA methylation detected through immunohistochemical labelling of 5‐mC was increased in the failing hearts of patients with DCM, but the increase in nuclear DNA methylation was noted only in cardiomyocytes but not in other cell types; (ii) visualized at the subcellular level, 5‐mC localized to heterochromatin; and (iii) DNA methylation was related to haemodynamic parameters associated with LV remodelling and LV dysfunction.
4.1. Increased DNA methylation in the cardiomyocytes from failing hearts
DNA methylation and histone modifications are the most common mechanisms of epigenetic modulation of gene expression. It has been suggested that histone modification may contribute to the reprogramming of cardiac gene expression in heart failure,9, 19 but the role of DNA methylation is far less well defined. Several studies have investigated genome‐wide DNA methylation in failing hearts. Movassagh et al. reported that DNA methylation is altered in human failing hearts, and this was confirmed in subsequent studies.4, 5, 6, 7, 8 However, all of those studies assessed DNA methylation biochemically using immunoprecipitation chips, where DNA from not only cardiomyocytes but also other cell types within the myocardium was analysed without distinguishing among them. In the present study, we assessed DNA methylation through immunohistochemical staining for 5‐mC, which enabled us to selectively evaluate DNA methylation at each cell type within the heart. Our findings indicate that cardiomyocyte‐specific alteration (enhancement) of nuclear DNA methylation occurs in the hearts of DCM patients. No change in DNA methylation was noted in non‐cardiomyocyte interstitial cells. It is somewhat unexpected that DNA methylation status was unchanged in cardiac fibroblasts from DCM patients, as it was previously suggested that fibroblasts are activated through modification of DNA methylation in response to transforming growth factor‐β1 or hypoxia, both of which play roles within failing hearts.20, 21
4.2. DNA methylation and chromatin remodelling
Cardiac myocytes display dramatic alterations of their phenotype in human hypertrophic and failing hearts. For example, the cytoplasm shows an increased subcellular organelle fraction accompanied by a decreased myofilament fraction, mitochondriosis, deposition of glycogen granules, and intracytoplasmic vacuoles.13 Nuclear structure is also markedly altered and is characterized as being ‘hypertrophied’, with severe crenation of the nuclear membrane (bizarre shape) and widespread clumping of the chromatin, two hallmarks of ‘nuclear hypertrophy’.14, 15, 16 The molecular biological alterations within the hypertrophied nuclei in human failing hearts are largely unknown, though the nuclear structural changes are reminiscent of chromatin remodelling. The widespread chromatin clumping cytologically resembles heterochromatin, which appears as small, darkly staining, irregular particles scattered throughout the nucleus or accumulated adjacent to the nuclear envelope.22 Because chromatin remodelling is one phenotype of epigenetic modification, epigenetic mechanisms are supposed to be significantly modified in hypertrophied nuclei within cardiomyocytes.
We examined the distribution of DNA methylation within nuclei through immunoelectron microscopic detection of 5‐mC, which revealed that 5‐mC‐immunopositive particles gathered specifically in nuclear heterochromatin in both cardiomyocytes and interstitial non‐cardiomyocytes. Heterochromatin is also associated with the di‐methylation and tri‐methylation of https://en.wikipedia.org/wiki/Histone_code in certain portions of the genome.23 In addition, a special coincidence between DNA methylation and chromatin condensed as a result of its remodelling has been suggested.24 Lamin A/C, a nuclear membrane protein, interacts with genome through lamin‐associated domains and regulates gene expression, which is known to influence spatial reorganization of the chromatin.25, 26 A recent study revealed rearrangement of lamin A/C‐chromatin interaction and association of lamin‐associated domain with increased DNA methylation in DCM.27 Our findings seem to be consistent with those ideas.
4.3. Pathophysiological significance of DNA methylation in heart failure
Abnormal regulation of genes and/or proteins involved in sarcomere organization, expression of cardiac hormones and proto‐oncogenes, and extracellular matrix remodelling that directly influences cardiac function and structure has been described in numerous studies of DCM and heart failure.24, 28, 29, 30 Moreover, it was recently suggested that epigenetic mechanisms play a pivotal role in the regulation of cardiac gene expression patterns during the progression of cardiac hypertrophy and failure.1, 2, 3, 4, 5, 6, 7, 8 In the present study, nuclear DNA methylation in cardiomyocytes was found to correlate significantly with parameters of LV remodelling (LV dilatation and LV hypertrophy) and LV dysfunction. There may thus be a significant association between nuclear DNA methylation in cardiomyocytes and the pathophysiology of heart failure.
However, recent studies have demonstrated the therapeutic potential of inhibiting DNA methylation in models of cardiac hypertrophy and reduced cardiac contractility. For example, treatment with the DNA methyltransferase inhibitor 5‐aza‐2′‐deoxycytidine blunted hypertrophic growth in a model of norepinephrine‐induced hypertrophy;31 while the inhibitor 5‐azacytidine exerted antifibrotic and antihypertrophic effects in a spontaneously hypertensive rat model.32 In addition, treatment with 5‐aza‐2′‐deoxycytidine ameliorated cellular autophagy failure in Danon disease with progressive fatal cardiomyopathy by reactivating the silent LAMP2 gene allele.33 Thus far, however, the link between DNA methylation and heart failure remains correlative. Additional studies will be needed to determine whether altered DNA methylation is causative and/or consequential, which will be essential for their development as therapeutic targets in the treatment of heart failure.2
4.4. Study limitations
It is generally not possible to determine a cause and effect relationship between the two in human studies. Owing to the limitations of human biopsy study, it was also difficult to determine in this study which genes were dysregulated by the augmented DNA methylation in failing hearts.
5. Conclusions
The present study revealed increased nuclear DNA methylation in cardiomyocytes, but not in non‐cardiomyocytes, in biopsy specimens from the failing hearts of patients with DCM. The increased DNA methylation was predominantly localized in the heterochromatin and was significantly related to LV functional and structural parameters, although the link remains correlative. These findings are suggestive of the potential pathophysiological significance of DNA methylation in heart failure.
Conflict of interest
None declared.
Funding
This study was supported in part by Research Grant from Asahi University and Grant‐in‐Aid for Scientific Research (16K09509) from the Ministry of Educational, Cultural, Sports, Science and Technology of Japan.
Acknowledgements
We thank Chika Ogawa, Ayaka Kanbara, Kazuho Niwa, and Akiho Kimura at Gifu University and Akiko Niwa, Rieko Hori, Norie Soga, and Yasuaki Hotta at Asahi University for their assistance.
Watanabe, T. , Okada, H. , Kanamori, H. , Miyazaki, N. , Tsujimoto, A. , Takada, C. , Suzuki, K. , Naruse, G. , Yoshida, A. , Nawa, T. , Tanaka, T. , Kawasaki, M. , Ito, H. , Ogura, S. , Okura, H. , Fujiwara, T. , Fujiwara, H. , and Takemura, G. (2020) In situ nuclear DNA methylation in dilated cardiomyopathy: an endomyocardial biopsy study. ESC Heart Failure, 7: 493–502. 10.1002/ehf2.12593.
References
- 1. Marín‐García J, Akhmedov AT. Epigenetics of the failing heart. Heart Fail Rev 2015; 20: 435–459. [DOI] [PubMed] [Google Scholar]
- 2. Kim SY, Morales CR, Gillette TG, Hill JA. Epigenetic regulation in heart failure. Curr Opin Cardiol 2016; 31: 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rosa‐Garrido M, Chapski DJ, Vondriska TM. Epigenomes in cardiovascular disease. Circ Res 2018; 122: 1586–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Movassagh M, Vujic A, Foo R. Genome‐wide DNA methylation in human heart failure. Epigenomics 2011; 3: 103–109. [DOI] [PubMed] [Google Scholar]
- 5. Haas J, Frese KS, Park YJ, Keller A, Vogel B, Lindroth AM, Weichenhan D, Franke J, Fischer S, Bauer A, Marquart S, Sedaghat‐Hamedani F, Kayvanpour E, Köhler D, Wolf NM, Hassel S, Nietsch R, Wieland T, Ehlermann P, Schultz JH, Dösch A, Mereles D, Hardt S, Backs J, Hoheisel JD, Plass C, Katus HA, Meder B. Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO Mol Med 2013; 5: 413–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Koczor CA, Lee EK, Torres RA, Boyd A, Vega JD, Uppal K, Yuan F, Fields EJ, Samarel AM, Lewis W. Detection of differentially methylated gene promoters in failing and nonfailing human left ventricle myocardium using computation analysis. Physiol Genomics 2013; 45: 597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Meder B, Haas J, Sedaghat‐Hamedani F, Kayvanpour E, Frese K, Lai A, Nietsch R, Scheiner C, Mester S, Bordalo DM, Amr A, Dietrich C, Pils D, Siede D, Hund H, Bauer A, Holzer DB, Ruhparwar A, Mueller‐Hennessen M, Weichenhan D, Plass C, Weis T, Backs J, Wuerstle M, Keller A, Katus HA, Posch AE. Epigenome‐wide association study identifies cardiac gene patterning and a novel class of biomarkers for heart failure. Circulation 2017; 136: 1528–1544. [DOI] [PubMed] [Google Scholar]
- 8. Glezeva N, Moran B, Collier P, Moravec CS, Phelan D, Donnellan E, Russell‐Hallinan A, O'Connor DP, Gallagher WM, Gallagher J, McDonald K, Ledwidge M, Baugh J, Das S, Watson CJ. Targeted DNA Methylation profiling of human cardiac tissue reveals novel epigenetic traits and gene deregulation across different heart failure patient subtypes. Circ Heart Fail 2019; 12: e005765. [DOI] [PubMed] [Google Scholar]
- 9. Gilsbach R, Schwaderer M, Preissl S, Grüning BA, Kranzhöfer D, Schneider P, Nührenberg TG, Mulero‐Navarro S, Weichenhan D, Braun C, Dreßen M, Jacobs AR, Lahm H, Doenst T, Backofen R, Krane M, Gelb BD, Hein L. Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat Commun 2018; 9: 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nag AC. Study of non‐muscle cells of the adult mammalian heart: a fine structural analysis and distribution. Cytobios 1980; 28: 41–61. [PubMed] [Google Scholar]
- 11. Weber KT, Anversa P, Armstrong PW, Brilla CG, Burnett JC Jr, Cruickshank JM, Devereux RB, Giles TD, Korsgaard N, Leier CV, Mendelsohn FAO, Motz WH, Mulvany MJ, Strauer BE. Remodeling and reparation of the cardiovascular system. J Am Coll Cardiol 1992; 20: 3–16. [DOI] [PubMed] [Google Scholar]
- 12. Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol 2007; 293: H1883–H1891. [DOI] [PubMed] [Google Scholar]
- 13. Olsen EGJ. Structural and ultrasound basis of myocardial diseases. Proc R Soc Med 1976; 69: 195–197. [PMC free article] [PubMed] [Google Scholar]
- 14. Kuhn H, Breithardt G, Knieriem HJ, Loongen F, Both A, Schmidt WA, Stroobandt R, Gleichmann U. Die Bedeutung der endomyokardialen Kathetherbiopsie fur die Diagnostik und die Beurteilung der Prognose der kongestiven Kardiopathie. Deutsch Med Wochenschr 1975; 100: 717–723. [DOI] [PubMed] [Google Scholar]
- 15. Baandrup U, Florio RA, Roters F, Olsen EG. Electron microscopic investigation of endomyocardial biopsy samples in hypertrophy and cardiomyopathy: a semiquantitative study in 48 patients. Circulation 1981; 63: 1289–1298. [DOI] [PubMed] [Google Scholar]
- 16. Koda M, Takemura G, Okada H, Kanoh M, Maruyama R, Esaki M, Li Y, Miyata S, Kanamori H, Li L, Ogino A, Kondo T, Minatoguchi S, Fujiwara T, Fujiwara H. Nuclear hypertrophy reflects increased biosynthetic activities in myocytes of human hypertrophic hearts. Circ J 2006; 70: 710–718. [DOI] [PubMed] [Google Scholar]
- 17. Report of the WHO/ISFC task force on the definition and classification of cardiomyopathies. Br Heart J 1980; 44: 672–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Takemura G, Fujiwara H, Horike K, Mukoyama M, Saito Y, Nakao K, Matsuda M, Kawamura A, Ishida M, Kida M, Uegaito T, Tanaka M, Matsumori A, Fujiwara Y, Fujiwara T, Imura H, Kawai C. Ventricular expression of atrial natriuretic polypeptide and its relations with hemodynamics and histology in dilated human hearts. Immunohistochemical study of the endomyocardial biopsy specimens. Circulation 1989; 80: 1137–1147. [DOI] [PubMed] [Google Scholar]
- 19. Ooi JY, Tuano NK, Rafehi H, Gao XM, Ziemann M, Du XJ, El‐Osta A. HDAC inhibition attenuates cardiac hypertrophy by acetylation and deacetylation of target genes. Epigenetics 2015; 10: 418–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pan X, Chen Z, Huang R, Yao Y, Ma G. Transforming growth factor β1 induces the expression of collagen type I by DNA methylation in cardiac fibroblasts. PLoS ONE 2013; 8: e60335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Watson CJ, Collier P, Tea I, Neary R, Watson JA, Robinson C, Phelan D, Ledwidge MT, McDonald KM, McCann A, Sharaf O, Baugh JA. Hypoxia‐induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast‐like phenotype. Hum Mol Genet 2014; 23: 2176–2188. [DOI] [PubMed] [Google Scholar]
- 22. Bian Q, Belmont AS. Revisiting higher‐order and large‐scale chromatin organization. Curr Opin Cell Biol 2012; 24: 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mathiyalagan P, Keating ST, Du XJ, El‐Osta A. Chromatin modifications remodel cardiac gene expression. Cardiovasc Res 2014; 103: 7–16. [DOI] [PubMed] [Google Scholar]
- 24. Chen H, Orozco LD, Wang J, Rau CD, Rubbi L, Ren S, Wang Y, Pellegrini M, Lusis AJ, Vondriska TM. DNA methylation indicates susceptibility toisoproterenol‐induced cardiac pathology and is associated with chromatin states. Circ Res 2016; 118: 786–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gruenbaum Y, Foisner R. Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu Rev Biochem 2015; 84: 131–164. [DOI] [PubMed] [Google Scholar]
- 26. van Steensel B, Belmont AS. Lamina‐associated domains: Links with chromosome architecture, heterochromatin, and gene repression. Cell 2017; 169: 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cheedipudi SM, Matkovich SJ, Coarfa C, Hu X, Robertson MJ, Sweet M, Taylor M, Mestroni L, Cleveland J, Willerson JT, Gurha P, Marian AJ. genomic reorganization of lamin‐associated domains in cardiac myocytes is associated with differential gene expression and dna methylation in human dilated cardiomyopathy. Circ Res 2019; 124: 1198–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kittleson MM, Minhas KM, Irizarry RA, Ye SQ, Edness G, Breton E, Conte JV, Tomaselli G, Garcia JG, Hare JM. Gene expression analysis of ischemic and nonischemic cardiomyopathy: shared and distinct genes in the development of heart failure. Physiol Genomics 2005; 21: 299–307. [DOI] [PubMed] [Google Scholar]
- 29. Dirkx E, da Costa Martins PA, De Windt LJ. Regulation of fetal gene expression in heart failure. Biochim Biophys Acta 1832; 2013: 2414–2424. [DOI] [PubMed] [Google Scholar]
- 30. Schirone L, Forte M, Palmerio S, Yee D, Nocella C, Angelini F, Pagano F, Schiavon S, Bordin A, Carrizzo A, Vecchione C, Valenti V, Chimenti I, De Falco E, Sciarretta S, Frati G. A review of the molecular mechanisms underlying the development and progression of cardiac remodeling. Oxid Med Cell Longev 2017; 2017: 3920195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xiao D, Dasgupta C, Chen M, Zhang K, Buchholz J, Xu Z, Zhang L. Inhibition of DNA methylation reverses norepinephrine‐induced cardiac hypertrophy in rats. Cardiovasc Res 2014; 101: 373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Watson CJ, Horgan S, Neary R, Glezeva N, Tea I, Corrigan N, McDonald K, Ledwidge M, Baugh J. Epigenetic therapy for the treatment of hypertension‐induced cardiac hypertrophy and fibrosis. J Cardiovasc Pharmacol Ther 2016; 21: 127–137. [DOI] [PubMed] [Google Scholar]
- 33. Ng KM, Mok PY, Butler AW, Ho JC, Choi SW, Lee YK, Lai WH, Au KW, Lau YM, Wong LY, Esteban MA, Siu CW, Sham PC, Colman A, Tse HF. Amerioration of X‐linked related autophagy failure in Danon disease with DNA methylation inhibitor. Circulation 2016; 134: 1373–1389. [DOI] [PubMed] [Google Scholar]