

Abstract
Aims
Myeloid differentiation protein 1 (MD1) is expressed in the mammalian heart and exerts an anti‐arrhythmic effect. Atrial fibrillation (AF) is closely related to heart failure with preserved ejection fraction (HFpEF). The potential impact of MD1 on AF vulnerability in an HFpEF model is not clear.
Methods and results
MD1 knock‐out and wild‐type (WT) mice were subjected to uninephrectomy and continuous saline or d‐aldosterone infusion and given 1% sodium chloride drinking water for 4 weeks. Echocardiographic and haemodynamic measurements, electrophysiological studies, Masson staining, and molecular analysis were performed. Aldosterone‐infused WT mice develop HFpEF with left ventricular hypertrophy, moderate hypertension, pulmonary congestion, and diastolic dysfunction. Aldosterone infusion increased the vulnerability of WT mice to AF, as shown by a prolonged interatrial conduction time, shortened effective refractory period, and higher incidence of AF. In addition, aldosterone infusion increased myocardial fibrosis and inflammation, decreased sarcoplasmic reticulum Ca2+‐ATPase 2a protein expression and the phosphorylation of phospholamban at Thr17, and increased sodium/calcium exchanger 1 protein expression and the phosphorylation of ryanodine receptor 2 in WT mice. All of the above adverse effects of aldosterone infusion were further exacerbated in MD1 knock‐out mice compare with WT mice. Mechanistically, MD1 deletion increased the activation of the toll‐like receptor 4/calmodulin‐dependent protein kinase II signalling pathway in in vivo and in vitro experiments.
Conclusions
MD1 deficiency increases the vulnerability of HFpEF mice to AF. This is mainly caused by aggravated maladaptive left atrial fibrosis and inflammation and worsened dysregulation of calcium handling, which is induced by the enhanced activation of the toll‐like receptor 4/calmodulin‐dependent protein kinase II signalling pathway.
Keywords: Myeloid differentiation protein 1, Fibrosis, Inflammation, Calcium handling proteins, Atrial fibrillation, Heart failure with preserved ejection fraction
Introduction
Heart failure (HF) with presented ejection fraction (HFpEF) has a high prevalence and poor prognosis.1, 2 Clinical trials and registries have shown that atrial fibrillation (AF) is highly prevalent in HFpEF, and patients with HFpEF are at increased risk of developing AF.3, 4, 5 Independent of several common risk factors, such as diabetes, obstructive sleep apnoea, smoking, hypertension, and obesity, there appear to be several mechanisms that drive HFpEF patients to develop AF.6 It is vital to note that much of the current understanding of the mechanisms behind AF in patients with HFpEF has been derived from experiment models of HF with reduced EF (HFrEF). Unlike HFrEF, in which multiple therapeutic agents have been shown to improve survival, to date, all drugs and devices that have been tested rigorously have failed in patients with HFpEF.7, 8 Therefore, there is currently a need for experimental animal models of HFpEF population and identify valid therapeutic targets for improving the prognosis of HFpEF and preventing the occurrence of AF.
Previous experimental models of HF have also demonstrated that calcium handling disorder plays a critical role in arrhythmogenic mechanisms for the initiation of AF. The dysregulation of calcium handling in the failing heart can increase the ectopic triggered activity of atrial myocytes.9, 10 Calmodulin‐dependent protein kinase II (CaMKII), a pro‐arrhythmic signalling molecule, is critically involved in calcium handling disorder, in which altered sarcoplasmic reticulum (SR) Ca2+ handling proteins contribute to enhanced SR Ca2+ leak and AF development.11, 12, 13 In addition, a recent study found that the toll‐like receptor (TLR4)/CaMKII signalling pathway can affect calcium homoeostasis in atrial myocytes and increase the susceptibility of high fat‐diet (HFD)‐fed mice to AF.14
Myeloid differentiation protein 1 (MD1) is a type of secreted glycoprotein that can form a complex with radioprotective protein 105 (RP105) called MD1‐RP105.15 The MD1‐RP105 complex can directly interact with the MD2‐TLR4 complex by lateral binding, acting as a negative physiological regulator of the TLR4 signalling pathway.16 Our previous study showed that MD1 is widely expressed in the heart, and MD1 deletion leads to a more pronounced activation of the TLR4/CaMKII signalling pathway, which may further significantly influence the expression levels of Ca2+ handling proteins, increasing the vulnerability of HF mice to ventricular arrhythmia.17 Therefore, we hypothesized that MD1 deletion can alter calcium homoeostasis in atrial myocytes and increase susceptibility to AF in HFpEF by enhancing the activation of TLR4/CaMKII signalling.
To further investigate this possibility, we used loss‐of‐function approaches and observed that MD1 deletion can aggravate maladaptive left atrial (LA) fibrosis and inflammation, deteriorate dysregulation of calcium handling, and increase the vulnerability of HFpEF mice to AF. Mechanistically, MD1 deletion markedly enhances the activation of the TLR4/CaMKII signalling pathway.
Methods
An expanded Methods section describing all procedures and protocols can be found in the Supporting Information.
Myeloid differentiation protein 1 knock‐out mice
Myeloid differentiation protein 1 knock‐out (MD1‐KO) mice were generated as described in previous studies.17, 18 In brief, MD1‐KO mice were purchased from the Japan RIKEN BioResource Centre Mouse (BRC) (B6.129P2‐MD‐1 < tm1Kmiy>). The deletion of MD1 was confirmed in the obtained MD1‐KO mice by western blot analysis of LA tissues.
Experimental Model
The use of all experimental mice were approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University and was consistent with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (the eighth edition, National Resource Center 2011). The HFpEF mice underwent uninephrectomy and aldosterone infusion as previously described.19, 20 Briefly, 8‐week‐old MD1‐KO mice and wild‐type (WT) littermates were anaesthetized with pentobarbital sodium (50 mg/kg, intraperitoneally). They were subjected to uninephrectomy and intraperitoneal implantation of osmotic mini‐pumps (Durect Corp, Cupertino, CA) that delivered a continuous infusion of either saline or 0.15‐μg/h aldosterone (IA0700, Solarbio Co., China) for 4 weeks, accompanied by 1% sodium chloride (NaCl) intake. The four groups studied were as follows: (i) the WT‐Sal group (n = 20), WT mice infused with saline; (ii) the KO‐Sal group (n = 21), MD1‐KO mice infused with saline; (iii) the WT‐Aldo group (n = 20), WT mice infused with aldosterone; and (iv) the KO‐Aldo group (n = 21), MD1‐KO mice infused with aldosterone.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5.0 (GraphPad, San Diego, CA). Continuous variables are shown as the means ± standard error of the mean. Statistical comparisons among multiple groups were performed with one‐way analysis of variance followed by Tukey's post hoc test and AF incidence across groups was compared using Fisher exact test. A value of P < 0.05 was considered statistically significant.
Results
The characteristics of the heart failure with preserved ejection fraction mouse model
None of the mice died during the 4 weeks of experiment. The characteristics of WT and KO mice 4 weeks after saline or aldosterone infusion are summarized in Tables 1 and 2. First, aldosterone infusion significantly increased the heart weight/body weight (BW) of WT‐Aldo and KO‐Aldo mice. There were no significant differences in cardiac hypertrophy between WT‐Sham and KO‐Sham mice. However, cardiac hypertrophy in KO‐Aldo mice was significantly higher than that in WT‐Aldo mice. There was no difference in LA chamber size or left ventricular ejection fraction (LVEF) between WT and KO mice regardless of saline or aldosterone infusion. Second, haemodynamics revealed that the end‐systolic pressure and end‐diastolic pressure (EDP) were significantly increased after 4 weeks of aldosterone infusion in WT‐Aldo (125.08 ± 1.56 and 7.91 ± 0.4; Table 2) and KO‐Aldo mice (127.1 ± 2.32 and 10.4 ± 0.56; Table 2) compared with their respective saline‐infused controls. Third, aldosterone infusion significantly increased the lung weight (LW)/BW and LW/tibial length (TL), the indicator of pulmonary congestion, of WT‐Aldo mice vs. WT‐Sham mice (P < 0.05) and KO‐Aldo mice vs. KO‐Sham mice (P < 0.05; Table 1). There were no differences in the LW/BW and LW/TL between saline‐infused WT and KO mice. However, the LW/TL of KO‐Aldo mice were markedly higher than that of WT‐Aldo mice, indicating more pulmonary congestion (P < 0.05; Table 1). In addition, haemodynamics revealed diastolic dysfunction with increased LVEDP (dp/dt min) and LV pressure isovolumetric relaxation constant (tau) in Aldo mice vs. sham mice at 4 weeks (Table 2). Finally, serum aldosterone levels in WT‐Aldo and KO‐Aldo mice were markedly elevated compared with those in the saline‐infused mice. There was no difference in serum aldosterone levels between WT and KO mice regardless of saline or aldosterone infusion.
Table 1.
Characteristics of wild‐type or myeloid differentiation protein 1 knock‐out mice 4 Weeks after saline/aldosterone infusion
| Groups | WT‐Saline (n = 8) | KO‐Saline (n = 8) | WT‐Aldo (n = 8) | KO‐Aldo (n = 8) |
|---|---|---|---|---|
| BW (g) | 21.56 ± 0.35 | 22.28 ± 0.31 | 22.81 ± 0.34 | 23.59 ± 0.44* |
| HW/BW (mg/g) | 4.56 ± 0.06 | 4.48 ± 0.07 | 5.82 ± 0.09* | 6.82 ± 0.16*,** |
| LW/BW (mg/g) | 4.98 ± 0.06 | 5.08 ± 0.1 | 6.96 ± 0.17* | 7.77 ± 0.21*,** |
| LW/TL (mg/g) | 5.85 ± 0.07 | 6.09 ± 0.1 | 8.5 ± 0.12* | 9.94 ± 0.15*,** |
| LVEDd (mm) | 3.9 ± 0.08 | 3.83 ± 0.09 | 3.85 ± 0.1 | 3.96 ± 0.07 |
| LVESd (mm) | 2.18 ± 0.06 | 2.11 ± 0.07 | 2.15 ± 0.07 | 2.35 ± 0.13 |
| LVEF (%) | 81.63 ± 0.8 | 82.12 ± 0.95 | 80.13 ± 1.91 | 77.13 ± 2.95 |
| LVFS (%) | 44.38 ± 0.01 | 44.63 ± 0.01 | 43.88 ± 0.02 | 40.63 ± 0.03 |
| LAD (mm) | 1.55 ± 0.04 | 1.59 ± 0.04 | 1.56 ± 0.1 | 1.59 ± 0.07 |
| Serum aldosterone levels (pg/mL) | 235.3 ± 19.09 | 238.79 ± 15.13 | 438.47 ± 12.85* | 458.48 ± 10.8* |
Aldo, Aldosterone; BW, body weight; HW, heart weight; KO, knock‐out; LAD, left atrial diameter; LVEDd, left ventricle end‐diastolic dimension; LVEF, left ventricular ejection fraction; LVFS, left ventricular fractional shortening; LVESd, left ventricular end‐systolic diameter; LW, lung weight; TL, tibial length.
Data are expressed as Mean ± standard error of the mean.
P < 0.05 vs. WT‐Sal group.
P < 0.05 vs. WT‐Aldo group.
Table 2.
Invasive haemodynamic measurements
| Parameters | WT‐Sham (n = 6) | KO‐Sham (n = 6) | WT‐Aldo (n = 6) | KO‐Aldo (n = 6) |
|---|---|---|---|---|
| Pressure volume measurements | ||||
| HR (bpm) | 479.91 ± 16.79 | 487.18 ± 22.53 | 468 ± 19.17 | 451.15 ± 11.3 |
| LVESP (mmHg) | 114.38 ± 2.28 | 113.18 ± 2.09 | 125.08 ± 1.56* | 127.1 ± 2.32* |
| LVEDP (mmHg) | 6.16 ± 0.48 | 6.12 ± 0.69 | 7.91 ± 0.4* | 10.4 ± 0.56*,** |
| LVESV (uL) | 28.13 ± 0.62 | 27.82 ± 0.72 | 25.26 ± 0.83* | 25.22 ± 0.76* |
| LVEDV (uL) | 45.02 ± 0.74 | 45.22 ± 1.16 | 40.8 ± 1.04* | 40.45 ± 0.66* |
| SV (uL) | 20.29 ± 0.78 | 19.74 ± 0.54 | 18.32 ± 0.72* | 17.57 ± 0.84*,** |
| CO (mL/min) | 10 378 ± 255.71 | 9964.25 ± 306.88 | 8277.63 ± 286.63* | 7349.63 ± 300.32*,** |
| Ea (mmHg/uL) | 5.82 ± 0.24 | 5.93 ± 0.27 | 7.06 ± 0.27* | 8.05 ± 0.18*,** |
| Systolic function | ||||
| EF (%) | 43.15 ± 0.98 | 42.74 ± 1.59 | 43.8 ± 1.32 | 40.98 ± 1.38 |
| SW (mmHg*uL) | 1881 ± 96.38 | 1737.88 ± 76.67 | 1766.5 ± 75.19 | 1666.13 ± 52.61 |
| dp/dt max (mmHg/s) | 14 182.75 ± 678.67 | 14 441.25 ± 703.76 | 13 833.75 ± 702.75 | 12 286.25 ± 356.47* |
| Diastolic function | ||||
| TAU (ms) | 6 ± 0.34 | 6.27 ± 0.33 | 8.59 ± 0.22* | 10.51 ± 0.49*,** |
| dp/dt min (mmHg/s) | −10625.75 ± 305.95 | −11497.75 ± 697.78 | −9057.25 ± 200.11* | −7546 ± 321.78*,** |
CO, cardiac output; dp/dt max, maximum rate of increase in left ventricular pressure; dp/dt min, maximum rate of decrease in left ventricular pressure; Ea, arterial elastance (measure of ventricular afterload); EF, ejection fraction; HR, heart rate; LVEDP, left ventricular end‐diastolic pressure; LVEDV, left ventricular end‐diastolic volume; LVESP, left ventricular end‐systolic pressure; LVESV, left ventricular end‐systolic volume; SV, stroke volume; SW, stroke work; TAU, left ventricular isovolumetric relaxation constant (relaxation time constant calculated by the Weiss method).
Data express as Mean ± SEM.
P < 0.05 vs. WT‐Sal group.
P < 0.05 vs. WT‐Aldo group.
Therefore, all of the above data showed that mice subjected to uninephrectomy and aldosterone infusion for 4 weeks developed HFpEF with LV hypertrophy, moderate hypertension, pulmonary congestion, and diastolic dysfunction while maintaining a normal/preserved LVEF, and the above adverse effects were further exacerbated by MD1 deletion.
Myeloid differentiation protein 1 deletion increased the vulnerability of heart failure with preserved ejection fraction mice to atrial fibrillation
To clarify the underlying role of MD1 in susceptibility to HFpEF‐related AF, we first monitored electrical signals by surface electrocardiogram. There was no difference in RR intervals, P waves, PR intervals, or QRS durations between the WT and KO mice regardless of saline or aldosterone infusion (Figure 1 A, B). However, aldosterone infusion significantly increased QTc intervals in the WT‐Aldo and KO‐Aldo mice. There was no significant difference in QTc intervals between the WT‐Sal and KO‐Sal mice (Figures 1 A and 1 B).
Figure 1.
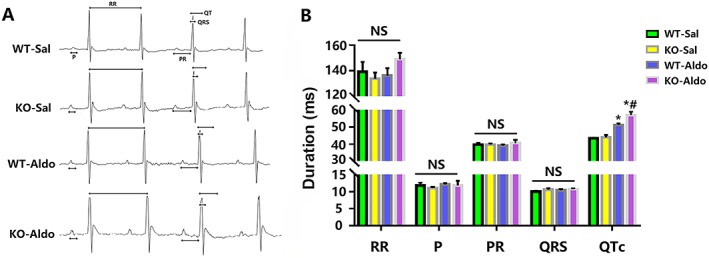
Analysis of surface electrocardiograph in wide‐type (WT) and myeloid differentiation protein 1 knock‐out mice 4 weeks after saline or aldosterone infusion. (A) A representative trace of the surface electrocardiography. RR interval, P wave duration, PR interval, QRS duration, and QTc interval were indicated as an arrow. (B) Surface electrocardiography parameters (n = 8). Data are expressed as mean ± standard error of the mean. * P < 0.05 vs. WT‐Sal group and # P < 0.05 vs. WT‐Aldo group.
In addition, we further characterized changes in electrophysiological parameters [the atrial effective refractory period (ERP), intra‐atrial conduction time (IACT), and incidence of AF] in Langendorf‐perfused hearts. Aldosterone infusion significantly prolonged the IACT of 40, 60, 80, and 100‐ms cycles in WT‐Aldo mice vs. WT‐Sal mice (P < 0.05) and KO‐Aldo mice vs. KO‐Sal mice (P < 0.05). The increase in the IACT in KO‐Aldo mice was significantly attenuated in WT‐Aldo mice (Figures 2 A–2 D). Moreover, aldosterone infusion significantly decreased the LA ERP of 40, 60, 80, 100‐ms basic cycles tested in WT‐Aldo mice vs. WT‐Sal mice (P < 0.05) and KO‐Aldo mice vs. KO‐Sal mice (P < 0.05). The shortening of the IACT in KO‐Aldo mice was significantly attenuated in WT‐Aldo mice (Figures 2 A–D). Moreover, aldosterone infusion significantly increased the AF induction rate in WT‐Aldo mice vs. WT‐Sal mice (P < 0.05) and KO‐Aldo mice vs. KO‐Sal mice (P < 0.05). The increase in the AF induction rate in WT‐Aldo mice (7/13, 53.8%) was significantly decreased compared with KO‐Aldo mice (11/12, 91.7%) (Figure 2 F). These results indicated that loss of MD1 increased the vulnerability of HFpEF mice to AF.
Figure 2.
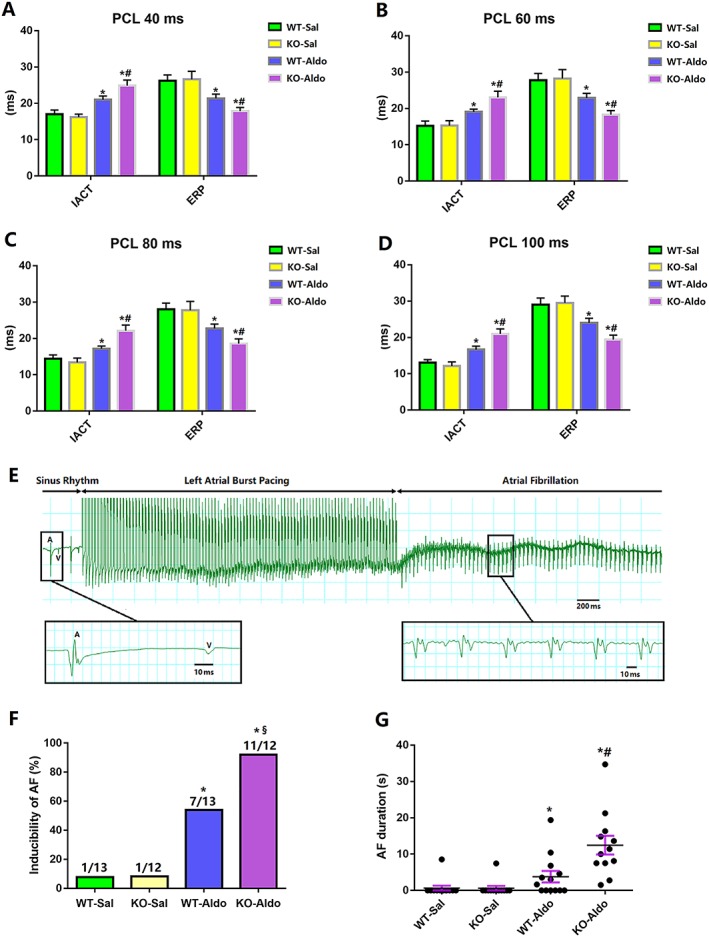
The electrophysiological properties of the atrial in wide‐type (WT) and myeloid differentiation protein 1 knock‐out mice 4 weeks after saline or aldosterone infusion. (A–D) Intra‐atrial conduction time (IACT) and effective refractory period (ERP) of the left atrium evaluated in the study using isolated perfused heart mice (n = 8). (E) Representative electrograms of atrial fibrillation (AF) induction after atrial burst pacing in the isolated perfused heart using Langendorff apparatus in a knock‐out (KO)‐Aldo mouse. (F,G) Inducibility and duration of AF in the study using isolated perfused heart (n = 12–13). Data are expressed as mean ± standard error of the mean. * P < 0.05 vs. wild‐type (WT)‐Sal group, # P < 0.05 vs. WT‐Aldo group, and § P < 0.1 vs. WT‐Aldo group.
Myeloid differentiation protein 1 knock‐out aggravated left atrial fibrosis in heart failure with preserved ejection fraction mice
Atrial fibrosis is a common process by which HFpEF promotes AF, which ultimately creates heterogeneity of conduction within the atria and thus acts as a substrate for re‐entry.21, 22 Therefore, we also detected the state of fibrosis in four groups. Western blot analysis of LA tissue samples indicated successful knock‐out of MD1 in WT‐KO mice (Figure 3 A). Aldosterone infusion significantly increased the area of myocardial fibrosis in WT‐Aldo mice vs. WT‐Sal mice (P < 0.05) and KO‐Aldo mice vs. KO‐Sal mice (P < 0.05; Figures 3 B and 3 C). Myocardial fibrosis in KO‐Aldo mice was significantly increased compared with that in WT‐Aldo mice (P < 0.05; Figures 3 B and 3 C). Consistent with these findings, the mRNA expression of molecular markers (collagen I, collagen III, and transforming growth factor‐β1) of cardiomyocyte fibrosis was increased in the LA of WT‐Aldo mice vs. WT‐Sal mice (P < 0.05) and KO‐Aldo mice vs. KO‐Sal mice (P < 0.05; Figures 3 C and 3 D).
Figure 3.
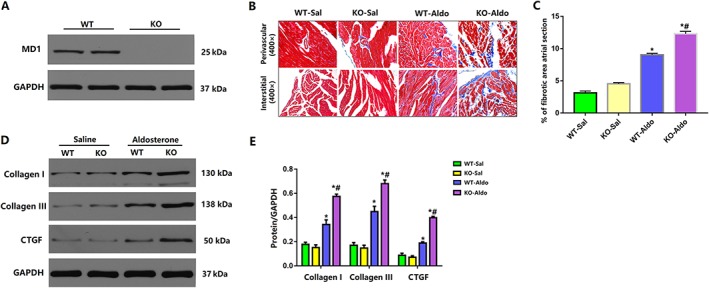
Myocardial fibrosis in wild‐type (WT) and Myeloid differentiation protein 1 (MD1)‐knock‐out mice 4 weeks after saline or aldosterone infusion. (A) Representative western blots of MD1 expression in left atrial tissues from WT and MD1‐KO mice (n = 6). (B) Representative Masson trichrome staining. Original magnification ×400; (C) myocardial fibrosis area (n = 4); and (D,E) representative western blots and statistical analysis of the fibrosis‐related proteins levels (n = 4). Data are expressed as mean ± standard error of the mean. * P < 0.05 vs. WT‐Sal group and # P < 0.05 vs. WT‐Aldo group.
Myeloid differentiation protein 1 knock‐out aggravated left atrial inflammation in heart failure with preserved ejection fraction mice
Inflammation is the most crucial pathological response to cardiac damage and repair.23 Moreover, there is extensive evidence that inflammation contributes to the pathophysiology of AF.24 Therefore, we also explored the expression levels of cytokines secreted by inflammatory cells. The mRNA levels of the proinflammatory cytokines interleukin (IL)‐1β, IL‐6, and tumour necrosis factor (TNF)‐α were increased in the LA of WT‐Aldo mice vs. WT‐Sal mice (P < 0.05) and KO‐Aldo mice vs. KO‐Sal mice (P < 0.05). The increase in proinflammatory cytokines in KO‐Aldo mice was markedly attenuated in WT‐Aldo mice (P < 0.05) (Figure 4 A). Our previous study reported that the nuclear factor kappa B (NF‐κB) signalling pathway play a vital role in regulating the inflammatory response and that MD1 has an effect on NF‐κB suppression in the setting of obesity.18 Therefore, the NF‐κB signalling pathway was investigated to confirm MD1 function after aldosterone infusion. As shown in Figure 4 B, the levels of phosphorylated p65 and inhibitor of kappa B (IκBα) were substantially increased in KO‐Aldo mice compared with WT‐Aldo mice, indicating that the NF‐κB signalling pathway was strongly activated by MD1 deletion. These data suggested that MD1 modulates atrial inflammation by interfering with the NF‐κB signalling pathway to some extent.
Figure 4.
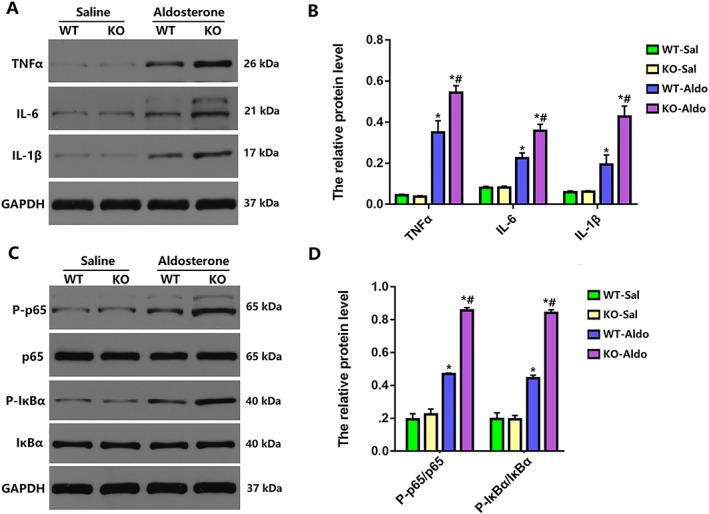
Myocardial inflammation in wild‐type (WT) and myeloid differentiation protein 1 knock‐out mice 4 weeks after saline or aldosterone infusion. (A,B) Representative western blots and statistical analysis of the inflammation‐related protein levels (n = 4). (C,D) Representative western blots and statistical analysis of p65, P‐p65, inhibitor of kappa B (IκBα), and P‐IκBα (n = 4). Data are expressed as mean ± standard error of the mean. * P < 0.05 vs. WT‐Sal group and # P < 0.05 vs. WT‐Aldo group.
Loss of myeloid differentiation protein 1 knock‐out worsened the dysregulation of calcium handling in heart failure with preserved ejection fraction mice
Extensive studies have suggested that alterations in calcium‐handling proteins, including ryanodine receptor 2 (RyR2), SR Ca2+‐ATPase 2a (SERCA2a), phospholamban (PLB), and sodium/calcium exchanger 1 (NCX1), contribute to changes in intracellular calcium transients and diastolic SR Ca2+ release that, in turn, lead to Ca2+‐triggered arrhythmogenesis in HF animal models.9, 25, 26, 27 Therefore, we next detected the protein expression of RyR2, SERCA2a, PLB, and NCX1 in each group.
First, the phosphorylation of RyR2 at Ser 2808 and Ser 2814 was markedly increased by aldosterone infusion in both WT and MD1‐KO hearts, and the adverse effect of phosphorylation at Ser 2808 and Ser 2814 was further aggravated in KO‐Aldo mice compared with WT‐Aldo mice (Figures 5 A and 5 B). However, there was no difference in RyR2 protein expression between WT and KO mice regardless of saline or aldosterone infusion.
Figure 5.
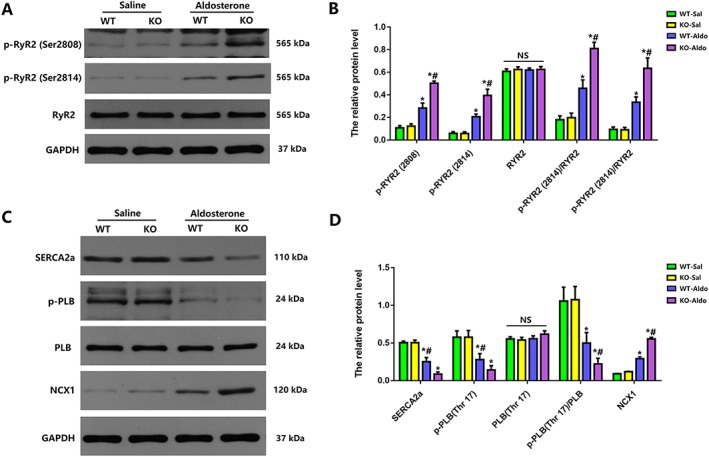
Expression of calcium handling regulatory proteins in wild‐type (WT) and and myeloid differentiation protein 1 knock‐out mice 4 weeks after saline or aldosterone infusion. (A,B) Representative western blots and statistical analysis of ryanodine receptor 2 (RyR2) phosphorylation at Ser2814 and RyR2 phosphorylation at Ser2808, RyR2, and GAPDH (n = 4). (C,D) Representative western blots and statistical analysis of sarcoplasmic reticulum Ca2+‐ATPase 2a (SERCA2a) and phospholamban (PLB) phosphorylation at Thr17, PLB, sodium/calcium exchanger (NCX), and GAPDH (n = 4). Data are expressed as mean ± standard error of the mean. * P < 0.05 vs. WT‐Sal group and # P < 0.05 vs. WT‐Aldo group.
In addition, aldosterone infusion significantly decreased the protein expression of SERCA2a and phosphorylated PLB (Thr17) in WT‐Aldo mice vs. WT‐Sal mice (P < 0.05) and KO‐Aldo mice vs. KO‐Sal mice (P < 0.05). The decrease in the protein expression of SERCA2a and phosphorylated PLB (Thr17) in KO‐Aldo mice was significantly attenuated in WT‐Aldo mice. However, there was no difference in PLB protein expression between WT and MD1‐KO mice regardless of saline or aldosterone infusion (Figures 5 C and D).
Finally, aldosterone infusion significantly increased NCX protein expression in WT‐Aldo mice vs. WT‐Sal mice (P < 0.05) and KO‐Aldo mice vs. KO‐Sal mice (P < 0.05). The increase in NCX1 protein expression in KO‐Aldo mice was markedly attenuated in WT‐Aldo mice (Figures 5 C and 5 D). Therefore, our data showed that the loss of MD1 worsened the dysregulation of SERCA2, the phosphorylation of RyR2 at Ser 2808 and Ser 2814, and the phosphorylation of PLB at Thr17 in HFpEF mice.
Myeloid differentiation protein 1 knock‐out regulated the activation of the toll‐like receptor 4/calmodulin‐dependent protein kinase II signalling pathway in vivo and in vitro
The results above indicate that MD1‐KO HFpEF mice may have increased vulnerability to AF through facilitated fibrosis and inflammation and the dysregulation of calcium handling. However, the underlying mechanism by which MD1‐KO exerts its pro‐arrhythmia response is unclear. The TLR4/CaMKII signalling pathway has been reported to play an important role in atrial arrhythmia through regulating atrial fibrosis, inflammation, and the function of calcium‐handling proteins.14, 28 Besides, according to our previous studies, MD1 has a suppressive effect on TLR4/CaMKII signalling pathway.17, 29 Thus the TLR4/CaMKII signalling pathway was investigated to confirm the function of MD1. As shown in Figures 6 A and 6 B, the levels of TLR4 and phosphorylated CaMKII were substantially increased in KO‐Aldo mice, indicating that the TLR4/CaMKII signalling pathway was strongly activated by MD1 deletion. These data suggested that MD1 modulated atrial fibrosis and calcium‐handling proteins by affecting the TLR4/CaMKII signalling pathway.
Figure 6.
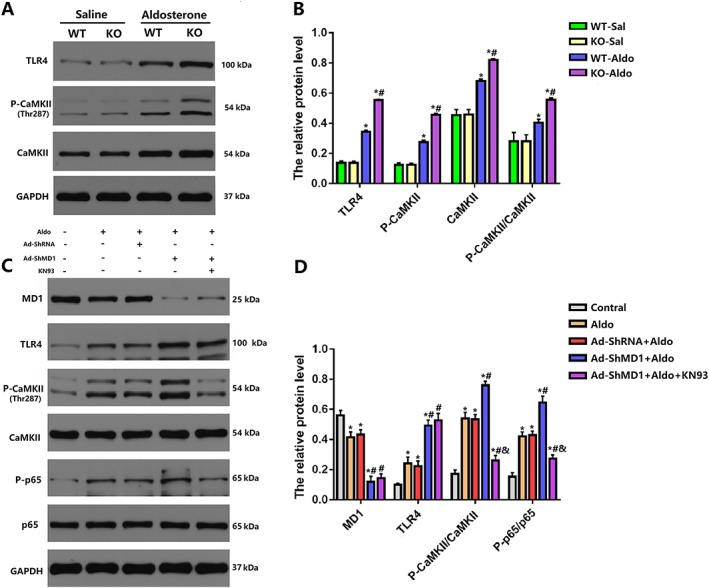
Myeloid differentiation protein 1 (MD1) regulates toll‐like receptor 4 (TLR4)/calmodulin‐dependent protein kinase II (CaMKII) signalling in in vivo and in vitro experiments. (A,B) Representative western blots and statistical analysis of TLR4, CaMKII, and P‐CaMKII in wild‐type (WT) and MD1‐knock‐out (KO) mice 4 weeks after saline or aldosterone infusion (n = 4). Data are expressed as mean ± standard error of the mean, * P < 0.05 vs. WT‐Sal group and # P < 0.05 vs. WT‐Aldo group. (C, D) Representative western blots and statistical analysis of MD1, TLR4, P‐CaMKII, CaMKII, P‐p65, and p65 in aldosterone‐induced H9C2 cell (n = 4). Data are expressed as mean ± standard error of the mean. * P < 0.05 vs. Control, # P < 0.05 vs. Aldo, and & P < 0.05 vs. Ad‐ShMD1.
To confirm the modulation of the TLR4/CaMKII signalling by MD1, the CaMKII inhibitor KN93 was used to pre‐treat H9c2 cells before aldosterone stimulation. H9C2 cells were infected with Ad‐shMD1 or Ad‐shRNA and then treated with 1‐μM aldosterone for 18 h.30, 31 Consistent with the in vivo results, the aldosterone‐induced activation of CaMKII and P65 was enhanced in MD1 knockdown cells. In addition, we exposed cultured H9C2 cells that had been previously infected with Ad‐shMD1 to a CaMKII inhibitor, KN93, for 30 min and then treated with aldosterone for 18 h. The protein phosphorylation levels of CaMKII and P65 were increased following aldosterone stimulation, and this was significantly suppressed by KN93 compared with the no treatment (Figures 6 C and 6 D). Therefore, the in vitro experiment further verified that the inactivation of the TLR4/CaMKII signalling pathway rescued the adverse effect of MD1 deficiency in aldosterone‐induced myocyte remodelling. In brief, all of the above data demonstrated that MD1 modulated the TLR4/CaMKII signalling pathway in aldosterone‐induced pathological myocyte remodelling.
Discussion
In the present study, aldosterone‐infused WT mice developed HFpEF with LV hypertrophy, moderate hypertension, pulmonary congestion, and diastolic dysfunction while maintaining a preserved LVEF, and the above adverse effects were further exacerbated by MD1 deletion. Moreover, the loss of MD1 increased the vulnerability of aldosterone‐induced HFpEF mice to AF, as shown by the prolonged IACT, shortened EDP, and higher incidence of AF. In addition, aldosterone infusion markedly increased myocardial fibrosis and inflammation, decreased SERCA2a protein expression and the phosphorylation of PLB at Thr17, and increased NCX1 protein expression and the phosphorylation of RyR2 in MD1‐KO mice. Finally, MD1 deletion markedly enhanced the activation of the TLR4/CaMKII signalling pathway in vivo and in vitro. These findings suggest that MD1 may be a potential therapeutic target for reducing the incidence of AF in HFpEF patients.
An ideal animal model should meet various requirements for mimicking human disease, including cardiac, haemodynamic, neurohormonal, and peripheral aberrations commonly seen in HFpEF patients. A recent review32 comprehensively summarized current models of HFpEF and found that a mouse model of uninephrectomy, aldosterone infusion, and 1% NaCl administration recapitulates clinical HFpEF phenotypes and features molecular changes that have been described clinically. Several studies have consistently shown that the combination of uninephrectomy, aldosterone infusion, and 1% NaCl administration induces an experimental model that mimics HFpEF and can be used to study the pathogenesis of HFpEF in vivo.31, 33, 34 These studies showed that HFpEF mice exhibit moderate hypertension, LV hypertrophy, pulmonary congestion, and diastolic dysfunction while maintaining a preserved LVEF. Consistent with our observations, aldosterone‐infused WT and MD1‐KO mice showed cardiac hypertrophy, a higher end‐systolic pressure and EDP, increased LW/TL, lower dp/dt min, and preserved LVEF. The above adverse effects in WT‐Aldo mice were significantly decreased in KO‐Aldo mice. Therefore, the HFpEF mouse model was successfully established, and MD1‐KO mice exhibited worsened cardiac diastolic function than that of WT‐Aldo mice.
Clinical studies have shown that over 30% of patients with incident HFpEF have prevalent AF, and the presence of AF is more strongly linked to incident HFpEF than to HFrEF.35, 36 Consistent with these data, our study showed that the AF induction rate was significantly increased in aldosterone‐induced HFpEF mice. In addition, the IACT and LA ERP were markedly increased and decreased, respectively, in aldosterone‐induced HFpEF mice. All of the above changes were worse in KO‐Aldo mice. Several studies have shown that, before the onset of AF, shorter atrial ERPs and a longer IACT are associated with higher inducibility of AF in HF patients and animals.37, 38, 39 Moreover, earlier work from our group showed that MD1 deletion can increase the AF induction rate in the setting of HFD.18 Therefore, a deficiency in MD1 can increase vulnerability to AF caused by aldosterone‐induced HFpEF.
The importance of atrial fibrosis in the initiation and re‐entrance of AF, especially in the matter of conduction disturbances, is widely understood.40 Atrial interstitial fibrosis has been shown to increase AF vulnerability in animal models of HF.22, 41, 42 According to previous research, ERP shortening may be explained by an increase in the number of fibroblasts, which shortens the duration of the action potential.43 In the present study, aldosterone infusion significantly shortened the ERP and increased LA fibrosis in WT‐Aldo mice compared with the WT‐Sal mice. The shortened ERP and increased atrial fibrosis in KO‐Aldo mice were substantially worse than those in WT‐Aldo mice. According to the findings of this study, loss of MD1 exacerbated atrial fibrosis and increased the vulnerability of aldosterone‐induced HFpEF mice to AF.
Human and animal studies have demonstrated that inflammation plays an essential role in the pathophysiology of HFpEF.44, 45, 46 Many studies have shown that inflammation plays a vital role in triggering AF. Li et al. observed that TNF‐α blood levels were higher in patients with AF compared with those in sinus rhythm and in persistent and permanent AF compared with paroxysmal AF.47 Moreover, a recent study also suggested that higher IL‐6 blood levels are associated with greater AF risk in the general population.48 Consistent with this possibility, our present correlative evidence suggested that chronic aldosterone infusion increased the levels of proinflammatory cytokines IL‐1β, IL‐6, and TNF‐α and vulnerability to AF. Besides, in our study, chronic aldosterone infusion substantially increased the levels of phosphorylated p65 and IκBα in KO‐Aldo mice compared with WT‐Aldo mice, indicating that the NF‐κB pathway was markedly activated by MD1 deletion. Supporting these data, our previous study has shown that MD1 deficiency increases the vulnerability HFD‐fed mice to AF, mainly due to an increase in the proinflammatory IL‐1β, IL‐6, and TNF‐α, a decrease in the levels of the anti‐inflammatory cytokine IL‐10, and the facilitation of atrial fibrosis. This mainly occurs through an increase in the levels of phosphorylated p65 and IκBα because of the enhanced activation of the TLR4 signalling pathway.18 These observations are consistent with a recent review by Queisser and Schupp showing that aldosterone can activate the NF‐κB pathway in humans and different experimental models.49 Some studies have reported that NF‐κB can regulate the expression of sodium channel subunits that contribute to AF‐associated electrical remodelling.50 Although ion channels were not investigated in this study, we are confident that the loss of MD1 increases AF susceptibility by regulating cardiac inflammation.
Extensive studies have suggested that alterations in Ca2+ handling proteins contribute to changes in intracellular Ca2+ transients and diastolic SR Ca2+ release that, in turn, lead to Ca2+‐triggered atrial arrhythmogenesis.11, 12, 25, 51 Recent studies have indicated that alterations in CaMKII‐dependent RyR2 phosphorylation are also exhibited in the atrium of chronic AF patients.11, 13 Abnormal diastolic RyR2 Ca2+ release may be the primary cause of abnormal Ca2+ handling in HF and chronic AF.9, 26, 27 In addition, increased diastolic SR Ca2+ leakage along with impaired function of Ca2+ uptake because of lower expression of the SERCA inhibitory protein PLB in HF and increased Na influx via NCX for Ca2+ removal can abnormally trigger activity and initiate atrial arrhythmias.52, 53 In our study, chronic aldosterone infusion decreased SERCA2a protein expression and the phosphorylation of PLB at Thr17 and increased NCX1 protein expression and the phosphorylation of RyR2 but did not alter RyR2 or PLB protein expression. Phosphorylation of PLB at Thr17 site is a specific target of CaMKII, and activated CaMKII is associated with increased phosphorylation of PLB at Thr17 site.54 However, in our study, the level of phosphorylation of PLB at Thr17 is markedly decreased in the aldosterone‐induced heart, where activated CaMKII was observed, although no apparent change in total PLB abundance. Interestingly, this decrease occurred despite an increase in CaMKII activity characteristic of HF. The observed differences in p‐PLB could be because of the balance in protein phosphatase and/or protein phosphatase inhibitor. It has been proposed that in human failing myocardium, phosphorylation of Thr17 decreased because of increased activity of protein phosphatase 2B.55 All of the above changes lead to Ca2+‐triggered atrial arrhythmogenesis. In addition, alterations in Ca2+ handling proteins in WT‐Aldo mice were significantly worsened in KO‐Aldo mice. A recent study indicated that MD1‐KO can interfere with the expression of Ca2+ handling proteins in pressure‐induced HF mice.17 Therefore, there is strong and growing evidence that MD1 deletion worsens the dysregulation of calcium handling and increases the vulnerability of aldosterone‐induced HFpEF mice to AF.
The underlying mechanisms by which MD1 regulates the vulnerability of aldosterone‐induced HFpEF mice to AF may be associated with its downstream TLR4/CaMKII signalling pathway. Several studies have consistently shown that activated CaMKII can increase the phosphorylation of RyR2 and induce an imbalance between NCX1 and SERCA2, which lead to a disturbance in intracellular Ca2+ homoeostasis‐triggered activity and arrhythmia initiation in the setting of HF.27, 56, 57, 58 Furthermore, a recent study found that inhibiting CaMKII can attenuate atrial fibrosis, which creates heterogeneity of conduction within the atria and thus acts as a substrate for re‐entry.21, 22, 59 Additionally, the activation of CaMKII in cardiomyocytes triggers the activation of NF‐κB and the expression of inflammatory chemokines and cytokines and ultimately increased AF vulnerability.60, 61 Several studies have demonstrated that CaMKII can regulate NF‐κB activation under pathological conditions, such as myocardial infarction, ischaemia/reperfusion injury, and obesity, which suggests that CaMKII serves to trigger and sustain subsequent changes in inflammatory gene expression that contribute to cardiac stress.28, 62, 63 Our previous studies showed that MD1 deletion leads to a more pronounced activation of the TLR4/CaMKII signalling pathway, which may further significantly influence the expression levels of Ca2+ handling proteins, increasing the vulnerability of HF mice to ventricular arrhythmia.14 Consistent with our current research, the protein expression levels of P‐CaMKII/CaMKII were markedly increased in LA tissues from KO‐Aldo mice compared with WT‐Aldo mice, indicating that the TLR4/CaMKII signalling pathway was strongly activated by MD1 deletion. In addition, the in vitro experiment further verified that the inactivation of the TLR4/CaMKII signalling pathway rescued the adverse effect of MD1 deficiency by decreasing the phosphorylation levels of CaMKII and p65 in aldosterone‐induced myocyte remodelling. Therefore, we believe that the loss of MD1 enhanced the activation of the TLR4/CaMKII signalling pathway to facilitate cardiomyocyte arrhythmogenesis following aldosterone‐induced HFpEF.
Novelty and limitations
We propose for the first time that the loss of MD1 can increase the vulnerability of a mouse model of HFpEF to AF and that MD1 may represent a novel therapeutic target for the treatment of HFpEF‐related remodelling of the atrium. However, the current study has some limitations. First, clinical and experimental evidences have been used to postulate the underlying complex pathophysiological mechanisms of AF, including electrical remodelling, structural remodelling, autonomic nervous system changes, and Ca2+ handling abnormalities.40, 64, 65, 66 In the current study, we mainly discussed the regulation of calcium homoeostasis by MD1 in HFpEF mice, but the relationship between MD1 and ion channels or the autonomic nervous system has not been studied. In addition, our recent study found that MD1 can regulate cardiac ion channels in pressure overload‐induced HF mice and obese mice.17, 29 Therefore, we cannot exclude the possibility that MD1 alters AF susceptibility by affecting the expression of ion channels. Second, although we used the H9C2 cell line to verify the relevant mechanism, H9C2 cells are ventricular myocytes. Given some differences between atrial myocytes and ventricular myocytes, the use of HL‐1 mouse atrial myocytes or MD1‐overexpressing mice or MD1 specific agonists may be more beneficial for elucidating the mechanism. Third, because the heart rate of normal mice is approximately 600 beats per minute, our results have shown that the heart rate was 450–500 beats per minute, which indicated that the anaesthesia maybe too stronger. Therefore, the measurement of haemodynamic and electrophysiological parameter may not be close to the values of the physiological conditions. Finally, because of the species difference between humans and mice, additional studies are needed to determine whether the regulation of vulnerability to AF by MD1 observed in a mouse model of HFpEF is likely to be beneficial in humans with HFpEF.
Conclusions
In brief, our study demonstrated that MD1 deficiency increases the vulnerability of HFpEF mice to AF. This is mainly caused by exacerbated maladaptive LA fibrosis and inflammation of the TLR4/CaMKII signalling pathway to some extent. These findings further suggest that MD1 may be a potential therapeutic target for reducing the susceptibility of patients with HFpEF to AF.
Conflict of interest
None declared.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81570306) and National Key R&D Program of China (2017YFC1700504).
Supporting information
Data S1. Supporting Information.
Shuai, W. , Kong, B. , Yang, H. , Fu, H. , and Huang, H. (2020) Loss of myeloid differentiation protein 1 promotes atrial fibrillation in heart failure with preserved ejection fraction. ESC Heart Failure, 7: 626–638. 10.1002/ehf2.12620.
References
- 1. McCullough PA, Philbin EF, Spertus JA, Kaatz S, Sandberg KR, Weaver WD, Resource utilization among congestive heart failures . Confirmation of a heart failure epidemic: findings from the Resource Utilization Among Congestive Heart Failure (REACH) study. J Am Coll Cardiol 2002; 39: 60–69. [DOI] [PubMed] [Google Scholar]
- 2. Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. New Engl J Med 2006; 355: 251–259. [DOI] [PubMed] [Google Scholar]
- 3. Shah KS, Xu HL, Matsouaka RA, Bhatt DL, Heidenreich PA, Hernandez AF, Devore AD, Yancy CW, Fonarow GC. Heart failure with preserved, borderline, and reduced ejection fraction 5‐year outcomes. J Am Coll Cardiol 2017; 70: 2476–2486. [DOI] [PubMed] [Google Scholar]
- 4. Lewis GA, Schelbert EB, Williams SG, Cunnington C, Ahmed F, McDonagh TA, Miller CA. Biological phenotypes of heart failure with preserved ejection fraction. J Am Coll Cardiol 2017; 70: 2186–2200. [DOI] [PubMed] [Google Scholar]
- 5. Massie BM, Carson PE, McMurray JJ, Komajda M, McKelvie R, Zile MR, Anderson S, Donovan M, Iverson E, Staiger C, Ptaszynska A, Investigators I‐P. Irbesartan in patients with heart failure and preserved ejection fraction. New Engl J Med 2008; 359: 2456–2467. [DOI] [PubMed] [Google Scholar]
- 6. Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B, Clausell N, Desai AS, Diaz R, Fleg JL, Gordeev I, Harty B, Heitner JF, Kenwood CT, Lewis EF, O'Meara E, Probstfield JL, Shaburishvili T, Shah SJ, Solomon SD, Sweitzer NK, Yang S, McKinlay SM, Investigators T. Spironolactone for heart failure with preserved ejection fraction. New Engl J Med 2014; 370: 1383–1392. [DOI] [PubMed] [Google Scholar]
- 7. Vaduganathan M, Michel A, Hall K, Mulligan C, Nodari S, Shah SJ, Senni M, Triggiani M, Butler J, Gheorghiade M. Spectrum of epidemiological and clinical findings in patients with heart failure with preserved ejection fraction stratified by study design: a systematic review. Eur J Heart Fail 2016; 18: 54–65. [DOI] [PubMed] [Google Scholar]
- 8. Ling LH, Kistler PM, Kalman JM, Schilling RJ, Hunter RJ. Comorbidity of atrial fibrillation and heart failure. Nat Rev Cardiol 2016; 13: 131–147. [DOI] [PubMed] [Google Scholar]
- 9. Yeh YH, Wakili R, Qi XY, Chartier D, Boknik P, Kaab S, Ravens U, Coutu P, Dobrev D, Nattel S. Calcium‐handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ Arrhythm Electrophysiol 2008; 1: 93–102. [DOI] [PubMed] [Google Scholar]
- 10. Stambler BS, Fenelon G, Shepard RK, Clemo HF, Guiraudon CM. Characterization of sustained atrial tachycardia in dogs with rapid ventricular pacing‐induced heart failure. J Cardiovasc Electrophysiol 2003; 14: 499–507. [DOI] [PubMed] [Google Scholar]
- 11. Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Muller FU, Schmitz W, Schotten U, Anderson ME, Valderrabano M, Dobrev D, Wehrens XH. Calmodulin kinase II‐mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest 2009; 119: 1940–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chiang DY, Kongchan N, Beavers DL, Alsina KM, Voigt N, Neilson JR, Jakob H, Martin JF, Dobrev D, Wehrens XHT, Li N. Loss of microRNA‐106b‐25 cluster promotes atrial fibrillation by enhancing ryanodine receptor type‐2 expression and calcium release. Circ Arrhythm Electrophysiol 2014; 7: 1214–U391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Neef S, Dybkova N, Sossalla S, Ort KR, Fluschnik N, Neumann K, Seipelt R, Schondube FA, Hasenfuss G, Maier LS. CaMKII‐dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ Res 2010; 106: 1134–1144. [DOI] [PubMed] [Google Scholar]
- 14. Zhong P, Quan D, Huang Y, Huang H. CaMKII activation promotes cardiac electrical remodeling and increases the susceptibility to arrhythmia induction in high‐fat diet‐fed mice with hyperlipidemia conditions. J Cardiovasc Pharmacol 2017; 70: 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Miyake K, Shimazu R, Kondo J, Niki T, Akashi S, Ogata H, Yamashita Y, Miura Y, Kimoto M. Mouse MD‐1, a molecule that is physically associated with RP105 and positively regulates its expression. J Immunol 1998; 161: 1348–1353. [PubMed] [Google Scholar]
- 16. Yoon SI, Hong M, Wilson IA. An unusual dimeric structure and assembly for TLR4 regulator RP105‐MD‐1. Nat Struct Mol Biol 2011; 18: 1028–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Peng JY, Liu Y, Xiong XJ, Huang CX, Mei Y, Wang ZQ, Tang YH, Ye J, Kong B, Liu WL, Wang T, Huang H. Loss of MD1 exacerbates pressure overload‐induced left ventricular structural and electrical remodelling. Sci Rep 2017; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shuai W, Kong B, Fu H, Shen CJ, Jiang XB, Huang H. MD1 Deficiency promotes inflammatory atrial remodelling induced by high‐fat diets. Can J Cardiol 2019; 35: 208–216. [DOI] [PubMed] [Google Scholar]
- 19. Wilson RM, De Silva DS, Sato K, Izumiya Y, Sam F. Effects of fixed‐dose isosorbide dinitrate/hydralazine on diastolic function and exercise capacity in hypertension‐induced diastolic heart failure. Hypertension 2009; 54: 583–590. [DOI] [PubMed] [Google Scholar]
- 20. Sam F, Duhaney TA, Sato K, Wilson RM, Ohashi K, Sono‐Romanelli S, Higuchi A, De Silva DS, Qin F, Walsh K, Ouchi N. Adiponectin deficiency, diastolic dysfunction, and diastolic heart failure. Endocrinology 2010; 151: 322–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hanna N, Cardin S, Leung TK, Nattel S. Differences in atrial versus ventricular remodeling in dogs with ventricular tachypacing‐induced congestive heart failure. Cardiovasc Res 2004; 63: 236–244. [DOI] [PubMed] [Google Scholar]
- 22. Li D, Fareh S, Leung TK, Nattel S. Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sort. Circulation 1999; 100: 87–95. [DOI] [PubMed] [Google Scholar]
- 23. Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 2013; 62: 263–271. [DOI] [PubMed] [Google Scholar]
- 24. Harada M, Van Wagoner DR, Nattel S. Role of inflammation in atrial fibrillation pathophysiology and management. Circ J 2015; 79: 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schulman H, Hanson PI, Meyer T. Decoding calcium signals by multifunctional CaM kinase. Cell Calcium 1992; 13: 401–411. [DOI] [PubMed] [Google Scholar]
- 26. Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin‐dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res 2005; 97: 1314–1322. [DOI] [PubMed] [Google Scholar]
- 27. Respress JL, van Oort RJ, Li N, Rolim N, Dixit SS, deAlmeida A, Voigt N, Lawrence WS, Skapura DG, Skardal K, Wisloff U, Wieland T, Ai X, Pogwizd SM, Dobrev D, Wehrens XH. Role of RyR2 phosphorylation at S2814 during heart failure progression. Circ Res 2012; 110: 1474–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhong P, Quan D, Peng J, Xiong X, Liu Y, Kong B, Huang H. Role of CaMKII in free fatty acid/hyperlipidemia‐induced cardiac remodeling both in vitro and in vivo. J Mol Cell Cardiol 2017; 109: 1–16. [DOI] [PubMed] [Google Scholar]
- 29. Shuai W, Kong B, Fu H, Shen C, Huang H. Loss of MD1 increases vulnerability to ventricular arrhythmia in diet‐induced obesity mice via enhanced activation of the TLR4/MyD88/CaMKII signalling pathway. Nutr Metab Cardiovasc Dis 2019; 29: 991–998. [DOI] [PubMed] [Google Scholar]
- 30. Garcia AG, Wilson RM, Heo J, Murthy NR, Baid S, Ouchi N, Sam F. Interferon‐gamma ablation exacerbates myocardial hypertrophy in diastolic heart failure. Am J Physiol Heart Circ Physiol 2012; 303: H587–H596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tanaka K, Valero‐Munoz M, Wilson RM, Essick EE, Fowler CT, Nakamura K, van den Hoff M, Ouchi N, Sam F. Follistatin like 1 regulates hypertrophy in heart failure with preserved ejection fraction. JACC Basic Transl Sci 2016; 1: 207–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Valero‐Munoz M, Backman W, Sam F. Murine models of heart failure with preserved ejection fraction: a “fishing expedition”. JACC Basic Transl Sci 2017; 2: 770–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Valero‐Munoz M, Li S, Wilson RM, Boldbaatar B, Iglarz M, Sam F. Dual endothelin‐a/endothelin‐b receptor blockade and cardiac remodeling in heart failure with preserved ejection fraction. Circ Heart Fail 2016; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Valero‐Munoz M, Li SP, Wilson RM, Hulsmans M, Aprahamian T, Fuster JJ, Nahrendorf M, Scherer PE, Sam F. Heart failure with preserved ejection fraction induces beiging in adipose tissue. Circ Heart Fail 2016; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Campbell RT, McMurray JJ. Comorbidities and differential diagnosis in heart failure with preserved ejection fraction. Heart Fail Clin 2014; 10: 481–501. [DOI] [PubMed] [Google Scholar]
- 36. Santhanakrishnan R, Wang N, Larson MG, Magnani JW, McManus DD, Lubitz SA, Ellinor PT, Cheng S, Vasan RS, Lee DS, Wang TJ, Levy D, Benjamin EJ, Ho JE. Atrial fibrillation begets heart failure and vice versa: temporal associations and differences in preserved versus reduced ejection fraction. Circulation 2016; 133: 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rensma PL, Allessie MA, Lammers WJ, Bonke FI, Schalij MJ. Length of excitation wave and susceptibility to reentrant atrial arrhythmias in normal conscious dogs. Circ Res 1988; 62: 395–410. [DOI] [PubMed] [Google Scholar]
- 38. Huang JL, Tai CT, Chen JT, Ting CT, Chen YT, Chang MS, Chen SA. Effect of atrial dilatation on electrophysiologic properties and inducibility of atrial fibrillation. Basic Res Cardiol 2003; 98: 16–24. [DOI] [PubMed] [Google Scholar]
- 39. Sanders P, Morton JB, Davidson NC, Spence SJ, Vohra JK, Sparks PB, Kalman JM. Electrical remodeling of the atria in congestive heart failure: electrophysiological and electroanatomic mapping in humans. Circulation 2003; 108: 1461–1468. [DOI] [PubMed] [Google Scholar]
- 40. Nattel S, Burstein B, Dobrev D. Atrial remodeling and atrial fibrillation: mechanisms and implications. Circ Arrhythm Electrophysiol 2008; 1: 62–73. [DOI] [PubMed] [Google Scholar]
- 41. Guerra JM, Everett TH, Lee KW, Wilson E, Olgin JE. Effects of the gap junction modifier rotigaptide (ZP123) on atrial conduction and vulnerability to atrial fibrillation. Circulation 2006; 114: 110–118. [DOI] [PubMed] [Google Scholar]
- 42. Lee KW, Everett TH, Rahmutula D, Guerra JM, Wilson E, Ding C, Olgin JE. Pirfenidone prevents the development of a vulnerable substrate for atrial fibrillation in a canine model of heart failure. Circulation 2006; 114: 1703–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ashihara T, Haraguchi R, Nakazawa K, Namba T, Ikeda T, Nakazawa Y, Ozawa T, Ito M, Horie M, Trayanova NA. The role of fibroblasts in complex fractionated electrograms during persistent/permanent atrial fibrillation: implications for electrogram‐based catheter ablation. Circ Res 2012; 110: 275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nunes RB, Alves JP, Kessler LP, Dal LP. Aerobic exercise improves the inflammatory profile correlated with cardiac remodeling and function in chronic heart failure rats. Clinics 2013; 68: 876–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tamaki S, Mano T, Sakata Y, Ohtani T, Takeda Y, Kamimura D, Omori Y, Tsukamoto Y, Ikeya Y, Kawai M, Kumanogoh A, Hagihara K, Ishii R, Higashimori M, Kaneko M, Hasuwa H, Miwa T, Yamamoto K, Komuro I. Interleukin‐16 promotes cardiac fibrosis and myocardial stiffening in heart failure with preserved ejection fraction. PLoS ONE 2013; 8: e68893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Glezeva N, Baugh JA. Role of inflammation in the pathogenesis of heart failure with preserved ejection fraction and its potential as a therapeutic target. Heart Fail Rev 2014; 19: 681–694. [DOI] [PubMed] [Google Scholar]
- 47. Li J, Solus J, Chen Q, Rho YH, Milne G, Stein CM, Darbar D. Role of inflammation and oxidative stress in atrial fibrillation. Heart Rhythm 2010; 7: 438–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Conway DS, Buggins P, Hughes E, Lip GY. Prognostic significance of raised plasma levels of interleukin‐6 and C‐reactive protein in atrial fibrillation. Am Heart J 2004; 148: 462–466. [DOI] [PubMed] [Google Scholar]
- 49. Queisser N, Schupp N. Aldosterone, oxidative stress, and NF‐kappaB activation in hypertension‐related cardiovascular and renal diseases. Free Radic Biol Med 2012; 53: 314–327. [DOI] [PubMed] [Google Scholar]
- 50. Shang LL, Sanyal S, Pfahnl AE, Jiao Z, Allen J, Liu H, Dudley SC Jr. NF‐kappaB‐dependent transcriptional regulation of the cardiac SCN5a sodium channel by angiotensin II. Am J Physiol Cell Physiol 2008; 294: C372–C379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. DeSantiago J, Maier LS, Bers DM. Frequency‐dependent acceleration of relaxation in the heart depends on CaMKII, but not phospholamban. J Mol Cell Cardiol 2002; 34: 975–984. [DOI] [PubMed] [Google Scholar]
- 52. Bers DM. Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res 2000; 87: 275–281. [DOI] [PubMed] [Google Scholar]
- 53. Bers DM. Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu Rev Physiol 2014; 76: 107–127. [DOI] [PubMed] [Google Scholar]
- 54. Tzimas C, Terrovitis J, Lehnart SE, Kranias EG, Sanoudou D. Calcium/calmodulin‐dependent protein kinase II (CaMKII) inhibition ameliorates arrhythmias elicited by junctin ablation under stress conditions. Heart Rhythm 2015; 12: 1599–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Munch G, Bolck B, Karczewski P, Schwinger RH. Evidence for calcineurin‐mediated regulation of SERCA 2a activity in human myocardium. J Mol Cell Cardiol 2002; 34: 321–334. [DOI] [PubMed] [Google Scholar]
- 56. Greiser M, Neuberger HR, Harks E, El‐Armouche A, Boknik P, de Haan S, Verheyen F, Verheule S, Schmitz W, Ravens U, Nattel S, Allessie MA, Dobrev D, Schotten U. Distinct contractile and molecular differences between two goat models of atrial dysfunction: AV block‐induced atrial dilatation and atrial fibrillation. J Mol Cell Cardiol 2009; 46: 385–394. [DOI] [PubMed] [Google Scholar]
- 57. Sossalla S, Fluschnik N, Schotola H, Ort KR, Neef S, Schulte T, Wittkopper K, Renner A, Schmitto JD, Gummert J, El‐Armouche A, Hasenfuss G, Maier LS. Inhibition of elevated Ca2+/calmodulin‐dependent protein kinase II improves contractility in human failing myocardium. Circ Res 2010; 107: 1150–1161. [DOI] [PubMed] [Google Scholar]
- 58. Lu YM, Huang JY, Shioda N, Fukunaga K, Shirasaki Y, Li XM, Han F. CaMKII delta B mediates aberrant NCX1 expression and the imbalance of NCX1/SERCA in transverse aortic constriction‐induced failing heart. PLoS ONE 2011; 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu Z, Finet JE, Wolfram JA, Anderson ME, Ai X, Donahue JK. Calcium/calmodulin‐dependent protein kinase II causes atrial structural remodeling associated with atrial fibrillation and heart failure. Heart Rhythm 2019; 16: 1080–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ikeda S, Matsushima S, Okabe K, Ikeda M, Ishikita A, Tadokoro T, Enzan N, Yamamoto T, Sada M, Deguchi H, Morimoto S, Ide T, Tsutsui H. Blockade of L‐type Ca(2+) channel attenuates doxorubicin‐induced cardiomyopathy via suppression of CaMKII‐NF‐kappaB pathway. Sci Rep 2019; 9: 9850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Aschar‐Sobbi R, Izaddoustdar F, Korogyi AS, Wang Q, Farman GP, Yang F, Yang W, Dorian D, Simpson JA, Tuomi JM, Jones DL, Nanthakumar K, Cox B, Wehrens XH, Dorian P, Backx PH. Increased atrial arrhythmia susceptibility induced by intense endurance exercise in mice requires TNFalpha. Nat Commun 2015; 6: 6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Singh MV, Kapoun A, Higgins L, Kutschke W, Thurman JM, Zhang R, Singh M, Yang J, Guan X, Lowe JS, Weiss RM, Zimmermann K, Yull FE, Blackwell TS, Mohler PJ, Anderson ME. Ca2+/calmodulin‐dependent kinase II triggers cell membrane injury by inducing complement factor B gene expression in the mouse heart. J Clin Invest 2009; 119: 986–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ling H, Gray CB, Zambon AC, Grimm M, Gu Y, Dalton N, Purcell NH, Peterson K, Brown JH. Ca2+/Calmodulin‐dependent protein kinase II delta mediates myocardial ischemia/reperfusion injury through nuclear factor‐kappaB. Circ Res 2013; 112: 935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Heeringa J, van der Kuip DA, Hofman A, Kors JA, van Herpen G, Stricker BH, Stijnen T, Lip GY, Witteman JC. Prevalence, incidence and lifetime risk of atrial fibrillation: the Rotterdam study. Eur Heart J 2006; 27: 949–953. [DOI] [PubMed] [Google Scholar]
- 65. Wakili R, Voigt N, Kaab S, Dobrev D, Nattel S. Recent advances in the molecular pathophysiology of atrial fibrillation. J Clin Invest 2011; 121: 2955–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nattel S, Dobrev D. The multidimensional role of calcium in atrial fibrillation pathophysiology: mechanistic insights and therapeutic opportunities. Eur Heart J 2012; 33: 1870–1877. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information.