

Summary
Carbon-carbon bond formation through polarity reversal ketyl radical anion coupling of carbonyls has inspired new reaction modes to this cornerstone carbonyl group and played significant roles in organic chemistry. The introduction of this resplendent polarity reversal ketyl strategy into polymer chemistry will inspire new polymerization mode with unpredicted discoveries. Here we show the successful introduction of polarity reversal ketyl approach to polymer chemistry to realize self-condensing ketyl polymerization with polymerization-induced emission. In this polarity reversal approach, it exhibits intriguing reversed polymerizability, where traditional excellent leaving groups are not suitable for polymerization but challenging polymerizations involving the cleavage of challenging C-F and C-CF3 bonds are realized under mild Barbier conditions. This polarity reversal approach enables the polymer chemistry with polarity reversal ketyl mode, opens up a new avenue toward the polymerization of challenging C-X bonds under mild conditions, and sparks design inspiration of new reaction, polymerization, and functional polymer.
Subject Areas: Organic Chemistry, Optical Property, Polymers
Graphical Abstract
Highlights
-
•
Self-condensing ketyl polymerization
-
•
Polymerization-induced emission
-
•
C-F/C-CF3 activation polymerization
-
•
Polarity reversal and reversed polymerizability
Organic Chemistry; Optical Property; Polymers
Introduction
Carbon-carbon (C-C) bond formation plays a central role in modern organic synthesis. In comparison with C-C bond formation through classic polar coupling mechanism, C-C bond formation through polarity reversal ketyl radical anion (ketyl) coupling of carbonyls enables access to a polarity-reversed platform with reactivity umpolung and has inspired new reaction modes to this cornerstone carbonyl group in organic chemistry (Hart, 1984, Wang et al., 2017, Wang et al., 2018). The development of new polymerization methodology based on this resplendent polarity reversal ketyl strategy will inspire new polymerization mode with unpredicted discoveries, expand the structure and functionality libraries of monomer and polymer, and open up a new avenue for the design and application of polymer materials.
Classical C-C bond formation reactions, including atom transfer radical addition reaction (Pintauer and Matyjaszewski, 2008, Wang and Matyjaszewski, 1995), radical addition-fragmentation reaction (Chiefari et al., 1998, Moad et al., 2008), olefin metathesis reaction (Bielawski and Grubbs, 2000, Bielawski and Grubbs, 2007, Vougioukalakis and Grubbs, 2010), Suzuki coupling reaction (Baggett et al., 2015, Kotha et al., 2002, Littke et al., 2000, Miyaura et al., 1981, Schluter, 2001, Yokoyama et al., 2007), Michael addition reaction (Hong et al., 2007, Liu et al., 2003, Wang et al., 2005), Stille coupling reaction (Bao et al., 1995, Guo et al., 2014, Littke and Fu, 1999, Yin et al., 2016), click chemistry reactions (He et al., 2016, He et al., 2017), multiple components reactions (Deng et al., 2012, Deng et al., 2016, Kreye et al., 2011, Wei et al., 2017, Wu et al., 2017, Xue et al., 2016), the Barbier reaction (Jing et al., 2019, Sun et al., 2017), and radical cascade reaction (Zhu et al., 2020), have been introduced to polymer chemistry to develop polymerization methodologies (Hawker and Wooley, 2005, Huang et al., 2019, Iha et al., 2009, Jiang et al., 2018, Song et al., 2018, Tsao and Wooley, 2017). Generally, C-C bond formation is initiated by activation-cleavage of C-X bonds, such as C-I, C-Br, C-Cl, C-S, C-O, and C-B, and these C-X bond activation-cleavage polymerizations have been well established in polymer chemistry with X as traditional leaving group. In comparison, both organic reaction and polymerization involving C-F or C-CF3 bond activation-cleavage have not been well realized yet and are significantly challenging, taking Barbier reaction as an example (Figure 1A), (Berger et al., 2018, Blomberg, 1993, Blomberg and Hartog, 1977, Li and Zhang, 1998, Sergeev et al., 1982, Zhang and Li, 1999, Zhou and Li, 2014) stemming from the extremely high bond dissociation energy (Ahrens et al., 2015, Amii and Uneyama, 2009, Cui et al., 2018, Meissner et al., 2017, Tian et al., 2015).
Figure 1.

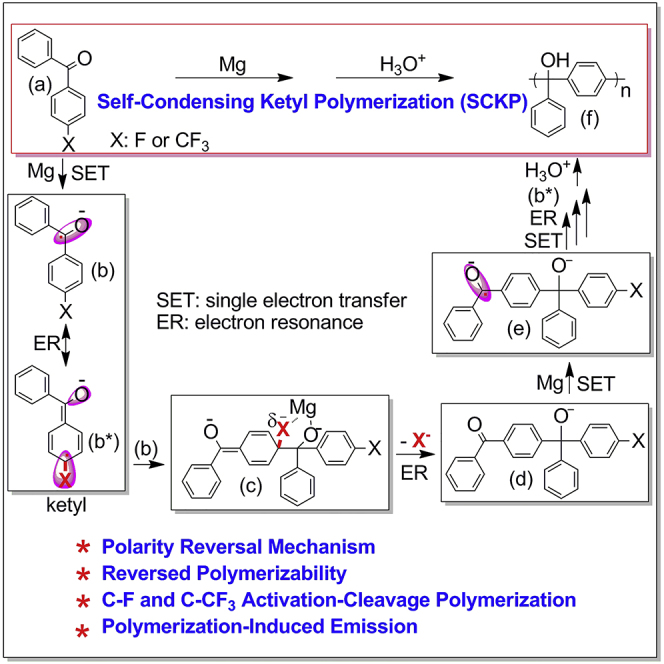
The Introduction of Polarity Reversal Ketyl Strategy into Polymer Chemistry
The introduction of polarity reversal ketyl strategy into polymer chemistry to realize self-condensing ketyl polymerization triggered by electron resonance, enabling reversed polymerizability toward C-F and C-CF3 activation-cleavage polymerization.
(A) Previous work of the Barbier reaction involving challenging C-F or C-CF3.
(B) Polarity reversal strategy via self-condensing ketyl polymerization in this work.
Owing to the high bond dissociation energy of the C-F and C-CF3 bonds, activation-cleavage of these bonds through direct insertion of Mg into the C-F or C-CF3 bond is difficult. Taking C-F activation-cleavage in Barbier reaction as an example, we hypothesize that, instead, Mg will react with the carbonyl group in the formation of ketyl through a reductive polarity reversal mechanism via single electron transfer (SET) and Mg will interact with C-F bond through van der Waals forces as well, resulting in nucleophilic addition of a ketyl to the C-F bond and formation of a five-membered ring intermediate (Figure S1). Mg-mediated C-F activation-cleavage and addition to a carbonyl group can therefore be realized in a one-pot Barbier reaction through polarity reversal ketyl mechanism. Furthermore, in the case of 4-fluorobenzophenone ([a] in Figure 1B), a SET between Mg and 4-fluorobenzophenone (a) will produce a ketyl (b) through reductive polarity reversal mechanism, where (b∗) is another electron resonance (ER) structure of ketyl of (b). Self-condensing coupling between (b) and (b∗) will initiate the self-condensing dimerization of ketyl, which involves formation of (c), cleavage of the C-F bond and ER in the formation of a carbonyl end group (d), and a SET between (d) and Mg forming a ketyl dimer (e). Continuous chain propagation with (b∗) as monomer followed by ER and SET processes, Mg-mediated C-F activation-cleavage polymerization can be realized by polarity reversal strategy through self-condensing ketyl polymerization (SCKP) of (a), similar to the self-condensing concept of self-condensing vinyl polymerization proposed by Fréchet (Fréchet et al., 1995, Hawker et al., 1995, Liu et al., 1999).
Herein, we demonstrate the successful introduction of polarity reversal ketyl strategy to polymer chemistry to realize SCKP. Through this polarity reversal SCKP, the polymerizability of monomers gets reversed, where traditional excellent leaving groups are not suitable for polymerization but challenging polymerizations involving the cleavage of challenging C-F and C-CF3 bonds are realized under mild Barbier conditions. This new polymerization mode also exhibits intriguing tunable polymerization-induced emission properties by simply adjusting monomer structure and polymerization time. This work therefore enables the polymer chemistry with polarity reversal ketyl mode with reversed polymerizability and opens up a reversed and feasible strategy for the polymerization of challenging monomers, which might inspire new reaction, polymerization, and luminescent polymer design.
Results and Discussion
To demonstrate the above hypothesis of reductive polarity reversal mechanism, the reaction between fluorobenzene and carbonyl compounds was carried out under Barbier conditions, where the reaction exhibits reversed reactivity. The reactivity order of aromatic halides in the polarity reversal ketyl mechanism is C-F > C-Cl (Figure S1 and Tables S1–S3), whereas the conventional reactivity order of aromatic halides is C-Cl >> C-F. Such reaction involving Mg and fluorobenzene is thought of as sluggish, and this result therefore confirms the significance of polarity reversal ketyl mechanism and is therefore of significant importance in organic chemistry. To further confirm the hypothesis of SCKP by introducing polarity reversal ketyl strategy into polymer chemistry, the polymerization of 4-fluorobenzophenone as monomer was carried out in the presence of Mg (1.2 eq.) and 1, 2-dibromoethane (0.2 eq.) at 45°C in THF for 24 h (Figure 4 entry 1). As shown in Figure 2A, the C-F bond is a dormant bond before polarity reversal of reactive C=O bond. After reductive polarity reversal of C=O bond in the formation of ketyl, followed by ER, the dormant C-F bond becomes reactive bond. The activation-cleavage of challenging C-F bond is therefore realized in the polarity reversal strategy, which further enables the SCKP of challenging monomer containing C-F bond.
Figure 4.

Results of Self-Condensing Ketyl Polymerization
Reaction conditions: monomer (1.0 g, 1.0 eq.), Mg (1.2 eq.), 1, 2-dibromoethane (0.2 eq.), THF (10 mL), 45°C, 24 h; [a]measured by GPC; [b]polymerization-induced emission.
(A) Substrate range of self-condensing ketyl polymerization.
(B) Emission digital photos of polymerization process of 2-fluorobenzophenone at different polymerization times (under irradiation with UV lamp at 365 nm).
(C) Emission digital photos of polymerization process of 4-fluorobenzophenone at different polymerization times (under irradiation with UV lamp at 365 nm).
(D) Emission digital photos of polymerization process of 4-trifluoromethylbenzophenone at different polymerization times (under irradiation with UV lamp at 365 nm).
Figure 2.

Process of Self-Condensing Ketyl Polymerization and Characterizations of Fluoro-poly(triphenylmethanol) (fluoro-PTPM) Synthesized with 4-fluorobenzophenone as Monomer
(A) Proposed mechanism of self-condensing ketyl polymerization.
(B) 1H NMR spectrum of 4-fluorobenzophenone in CDCl3.
(C) 1H NMR spectrum of fluoro-PTPM in CDCl3.
(D) Complete MALDI-TOF spectrum of fluoro-PTPM.
(E) Comparison between observed and calculated MALDI-TOF mass spectra of pentamer [M].
From the characterizations of the products shown in Figures 2 and S3, the successful formation of polymer can be verified obviously from the comparison of NMR spectra, where sharp 1H NMR signals of monomer disappear and broader aromatic and hydroxide signals of polymer appear at 7.60–6.75 and 3.83–3.35 ppm, respectively. From comparison of the 13C NMR spectra of the monomer and the polymer, the carbonyl signal at 195.33 ppm can be seen to disappear and the fluorobenzene signals at 166.67 and 164.15 ppm decrease, which confirms that the polymer contains fluorobenzene as an end group. The existence of this fluorobenzene end group is confirmed by 19F NMR signal at 115.46 ppm. The successful formation of the polymer is further verified by gel permeation chromatography (GPC) characterization with a Mn of 3,400 g/mol and Ð of 1.40. The chemical structure of the polymer is further verified by the Fourier transform infrared (FTIR) spectrum, which contains groups like C-OH, phenyl, and fluorobenzene. The characteristic glass transition temperature (Tg) of fluoro-poly(triphenylmethanol) (fluoro-PTPM) was measured by differential scanning calorimetry (DSC) with a Tg of ~104.7°C (Figure S3).
To confirm the polymerization mechanism, matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry was utilized to characterize the polymer sample obtained after 10 min of polymerization, as shown in Figures 2D–2F and S4. From the complete spectrum, an m/z difference of 182.196 can be clearly observed as the molecular weight of the repeating unit, which is consistent with the molecular weight of diphenylmethanol group (182.073 Da). From the enlarged spectrum of pentamer, every peak can be identified and assigned as [M]-3OH, [M]-2OH, [M]-OH-nH, [M], and [M]+Na+. The chemical structure of fluoro-PTPM is further supported by comparison of the similarity between the observed and calculated isotope peaks of [M], [M]-OH-nH, and [M]-2OH, signals that show a very similar isotope pattern. All these results confirm the hypothesized polarity reversal SCKP mechanism shown in Figures 1B and 2A, which relies on the polarity reversal of reactive ketone group in the formation of ketyl intermediates and ER triggered C-F bond activation-cleavage, resulting in polymerization through further SCKP.
To further confirm the above polymerization mechanism, a deuterium labeling experiment was carried out in THF-d8 with 3 eq. of Mg and 1 eq. of 1, 2-dibromoethane at 45°C. The greatly increased amounts of Mg and 1, 2-dibromoethane will influence the reductive polarity reversal process via SET and interrupt the polymerization process, so that the intermediate should be captured. Through the deuterium labeling experiment, para-deuterated diphenylethylmethanol with 6% isolated yield and 35% deuterated ratio was successfully captured (Figure 3 and Figures S28–S32). According to the analysis shown in Figure 3, the only possible pathway to the formation of para-deuterated diphenylethylmethanol requires the reductive polarity reversal of carbonyl via a SET in the formation of a ketyl radical anion intermediate, the C-F activation via ER of the ketyl radical anion intermediate, the hydrogen/deuterium abstraction reaction between the ER ketyl radical anion intermediate and THF-d8 as found in the literature (Clerici et al., 2005, Pryor et al., 1983), the C-F cleavage via ER in the formation of carbonyl compound, and the addition of the Grignard reagent. To further clarify the polymerization process of SCKP through polarity reversal ketyl mechanism, 1H NMR and FTIR techniques were carried out with different substituents as leaving groups (Figures S8–S12 and 4A). The failure of polymerization of AB-type dimer containing both C-F and C=O moieties indicates the importance of C-F activation-cleavage triggered by ER in SCKP (Figure 4A entry 2). This also indicates that the polymerization mechanism of SCKP is not a traditional AB-type polycondensation, which is further confirmed by controlled experiments of polymerization of AB-type bifunctional 4-fluorobenzophenone in the presence of 1 eq. of monofunctional fluorobenzene or benzophenone. In this nontraditional AB-type polycondensation, monofunctional fluorobenzene did not inhibit the polymerization but monofunctional benzophenone did (Figure 4A entries 3 and 4). The evidences of obvious polymer signal observed at 15 min with conversion as low as 28.8%, successful isolation of 3.2% yield of dimer (d) after 24 h of polymerization, and still more than 10% of monomer observed after 24 h of polymerization also confirm this SCKP is not a traditional AB-type polycondensation (Figures S5 and S6). FTIR tracing experiments verify that this SCKP process contains carbonyl addition process and C-F activation-cleavage process with decreased carbonyl signal, increased C-O signal, and decreased fluorophenyl signal (Figure S6).
Figure 3.

Validation of the Self-Condensing Ketyl Polymerization Mechanism via a Deuterium Labeling Experiment
These above results therefore confirm the proposed polymerization mechanism shown in Figure 1B. In the reductive polarity reversal mechanism, carbonyl addition happens first via a SET between carbonyl and Mg in the formation of ketyl. C-F activation happens second via ER of ketyl and, third, is followed by a coupling reaction between (b) and (b∗) to initiate the polymerization of (b∗). The fourth step is the C-F cleavage via ER in the formation of the carbonyl end group, which will further react with Mg via SET to complete chain propagation with one monomer inserted. Attributed to the continuous relay of reactions in the order of carbonyl addition via SET in the formation of ketyl, C-F activation via ER, and ketyl coupling and C-F cleavage via ER, the SCKP is realized through polarity reversal ketyl mechanism. This polarity reversal ketyl mechanism exhibits intriguing reversed polymerizability as well. With traditional leaving groups like acetoxy (OAc), tosylate (OTs), and triflate (OTf), which could react with Mg directly, their failures in polymerization indicate that the direct activation-cleavage of leaving groups by Mg would interrupt SCKP (Figure 4A entry 7–9). However, with even more challenging trifluoromethyl (CF3) substituent as leaving group, its SCKP is realized successfully (Figure 4A entry 6). These results indicate that polarity reversal strategy of SCKP enables the polymerization of challenging monomers through activation-cleavage of challenging bonds under mild conditions. It is worth mentioning that this discovery might open up a reversed and feasible strategy for the polymerization of challenging monomers, which might further inspire new reaction and polymerization.
Photophysical properties are intriguing phenomena with potential optical, electronic, and sensory applications (Lv et al., 2017, Lv et al., 2019, Wan et al., 2017, Wan et al., 2018). To illustrate the advantage of this SCKP, photophysical properties of the obtained PTPMs and the polymerization process were investigated (Figures 4B–4D and S13–S16). From the results, these PTPMs exhibit obviously site-specific luminescence that can be simply adjusted by monomer structures. When the phenyl rings of obtained para-PTPMs are rotation free, they are aggregation-induced emission (AIE)-type luminescence, similar to Tang's reports (Hong et al., 2011, Liu et al., 2020, Luo et al., 2001, Mei et al., 2015, Zhang et al., 2017). When the rotation about the phenyl rings is hindered, the ortho-PTPM exhibits traditional luminescence with aggregation caused quenching (ACQ) effect. These results confirm that PTPM is a special type of luminescent polymer deriving from the intramolecular and intermolecular through-space conjugation effect under polymer chain constraint and intramolecular charge-transfer effect (Jing et al., 2019, Sun et al., 2017). Interestingly, the terminal groups of PTPMs are significant to their luminescence. When the terminal group is a fluoro group, para-PTPM is AIE with cyan color, whereas that of trifluoromethyl group is AIE with yellow color. Time-dependent density functional theory calculations indicate terminal groups have significant influences on the band gaps and dihedral angles, which further cause the differences on luminescence (Figures S17 and S18). The band gaps of both F-PTPM and CF3-PTPM decrease with the increase of chain length, which indicates the through-space conjugation under polymer chain constraint plays important roles in the luminescent properties. Besides, obviously different HOMO, LUMO, and band gaps are observed between F-PTPMs and CF3-PTPMs with different chain lengths, causing the differences in luminescent properties of PTPMs with different terminal groups. SCKP also exhibits polymerization-induced emission property, which can be tuned from ACQ type to AIE type by simply adjusting monomer structures (Figures 4B–4D). The luminescence color of AIE-type polymerization-induced emission can be tuned as well from cyan to yellow by simply adjusting monomer structures and polymerization time. This tunable polymerization-induced emission is due to the formation of luminescent non-conjugated PTPM during polymerization. In comparison, luminescent polymers are generally designed by polymerization of luminescent monomers, where polymerization has a limited effect on the design of luminescent polymers. This result therefore opens up a new avenue to design luminescent polymers through polymerization of nonemissive monomers in situ.
Conclusion
In conclusion, the successful introduction of polarity reversal ketyl strategy into polymer chemistry has been realized through SCKP. In this polarity reversal approach, it exhibits intriguing reversed polymerizability, where traditional excellent leaving groups are not suitable for polymerization but challenging polymerizations involving the cleavage of C-F and C-CF3 bonds are realized under mild Barbier conditions. The SCKP involves continuous relay of reactions in the order of carbonyl addition via SET in the formation of ketyl intermediate, C-F activation via ER, and ketyl coupling and C-F cleavage via ER in one polymerization cycle. This SCKP also exhibits intriguing polymerization-induced emission capability to construct luminescent polymers from nonemissive monomers and the emissive properties vary from traditional luminescence to AIE-type luminescence with emission color varying from blue to cyan and yellow, which can be simply adjusted by adjusting the monomer structure and polymerization time. This work therefore provides polymer chemistry with polarity reversal ketyl strategy and opens up a new avenue toward the polymerization of challenging C-X bonds, which might inspire new reaction, polymerization, and luminescent polymer design.
Limitations of the Study
This study focuses on the demonstration of the introduction of polarity reversal strategy into polymer chemistry to demonstrate the advantage of resplendent polarity reversal mechanism with reversed polymerizability. The substrates used in this study limit on carbonyl compounds, and further investigations on other groups will expand the utilization of this polymerization methodology. Fortunately, carbonyl compounds account for a high portion in organic chemicals and have played significant cornerstone roles in organic chemistry, which might expand the monomer library of polymer materials and inspire the design of novel polymer from carbonyl chemicals.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We acknowledge funding support from NSFC (21971236, 21922112, 21672213 and 21871258), the National Key R&D Program of China (Grant No. 2017YFA0700103), and the Haixi Institute of CAS (Grant No. CXZX-2017-P01). This work is dedicated to Professor Cai-Yuan Pan, University of Science and Technology of China, for his 80th birthday in May 2020.
Author Contributions
S.-S.L. performed the polymerization experiments and analyzed the data. N.Z. performed the organic chemistry experiments and analyzed the data. Y.-N.J. assisted on polymerization and performed calculations. W.-M.W. conceived the concept. Y.L., H.B., and W.-M.W. designed the study and wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: April 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101031.
Supplemental Information
References
- Ahrens T., Kohlmann J., Ahrens M., Braun T. Functionalization of fluorinated molecules by transition-metal-mediated C-F bond activation to access fluorinated building blocks. Chem. Rev. 2015;115:931–972. doi: 10.1021/cr500257c. [DOI] [PubMed] [Google Scholar]
- Amii H., Uneyama K. C-F bond activation in organic synthesis. Chem. Rev. 2009;109:2119–2183. doi: 10.1021/cr800388c. [DOI] [PubMed] [Google Scholar]
- Baggett A.W., Guo F., Li B., Liu S.Y., Jäkle F. Regioregular synthesis of Azaborine oligomers and a polymer with a syn Conformation stabilized by NH⋅⋅⋅π interactions. Angew. Chem. Int. Ed. 2015;54:11191–11195. doi: 10.1002/anie.201504822. [DOI] [PubMed] [Google Scholar]
- Bao Z., Chan W.K., Yu L. Exploration of the Stille coupling reaction for the synthesis of functional polymers. J. Am. Chem. Soc. 1995;117:12426–12435. [Google Scholar]
- Berger A.L., Donabauer K., Konig B. Photocatalytic Barbier reaction - visible-light induced allylation and benzylation of aldehydes and ketones. Chem. Sci. 2018;9:7230–7235. doi: 10.1039/c8sc02038h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielawski C.W., Grubbs R.H. Highly efficient ring-opening metathesis polymerization (ROMP) using new ruthenium catalysts containing N-heterocyclic carbene ligands. Angew. Chem. Int. Ed. 2000;39:2903–2906. doi: 10.1002/1521-3773(20000818)39:16<2903::aid-anie2903>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Bielawski C.W., Grubbs R.H. Living ring-opening metathesis polymerization. Prog. Polym. Sci. 2007;32:1–29. [Google Scholar]
- Blomberg C. Springer-Verlag; 1993. The Barbier Reaction and Related One-step Processes. [Google Scholar]
- Blomberg C., Hartog F.A. Barbier reaction - one-step alternative for syntheses via organomagnesium compounds. Synthesis (Stuttg.) 1977:18–30. [Google Scholar]
- Chiefari J., Chong Y.K., Ercole F., Krstina J., Jeffery J., Le T.P.T., Mayadunne R.T.A., Meijs G.F., Moad C.L., Moad G. Living free-radical polymerization by reversible addition−fragmentation chain Transfer: the RAFT process. Macromolecules. 1998;31:5559–5562. [Google Scholar]
- Clerici A., Cannella R., Panzeri W., Pastori N., Regolini E., Porta O. TiCl3/PhN2+-mediated radical addition of ethers to aldimines generated in situ under aqueous conditions. Tetrahedron Lett. 2005;46:8351–8354. [Google Scholar]
- Cui B.Q., Jia S.C., Tokunaga E., Shibata N. Defluorosilylation of fluoroarenes and fluoroalkanes. Nat. Commun. 2018;9:4393. doi: 10.1038/s41467-018-06830-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X.X., Li L., Li Z.L., Lv A., Du F.S., Li Z.C. Sequence regulated poly(ester-amide)s based on Passerini reaction. ACS Macro Lett. 2012;1:1300–1303. doi: 10.1021/mz300456p. [DOI] [PubMed] [Google Scholar]
- Deng H.Q., Han T., Zhao E.G., Kwok R.T.K., Lam J.W.Y., Tang B.Z. Multicomponent click polymerization: a facile strategy toward fused heterocyclic polymers. Macromolecules. 2016;49:5475–5483. [Google Scholar]
- Fréchet J.M.J., Henmi M., Gitsov I., Aoshima S., Leduc M.R., Grubbs R.B. Self-condensing vinyl polymerization - an approach to dendritic materials. Science. 1995;269:1080–1083. doi: 10.1126/science.269.5227.1080. [DOI] [PubMed] [Google Scholar]
- Guo F., Yin X., Pammer F., Cheng F., Fernandez D., Lalancette R.A., Jäkle F. Regioregular organoborane-functionalized poly(3-alkynylthiophene)s. Macromolecules. 2014;47:7831–7841. [Google Scholar]
- Hart D.J. Free-radical carbon-carbon bond formation in organic synthesis. Science. 1984;223:883–887. doi: 10.1126/science.223.4639.883. [DOI] [PubMed] [Google Scholar]
- Hawker C.J., Frechet J.M.J., Grubbs R.B., Dao J. Preparation of hyperbranched and star polymers by a "living", self-condensing free radical polymerization. J. Am. Chem. Soc. 1995;117:10763–10764. [Google Scholar]
- Hawker C.J., Wooley K.L. The convergence of synthetic organic and polymer chemistries. Science. 2005;309:1200–1205. doi: 10.1126/science.1109778. [DOI] [PubMed] [Google Scholar]
- He B.Z., Zhen S.J., Wu Y.W., Hu R.R., Zhao Z.J., Qin A.J., Tang B.Z. Cu(I)-Catalyzed amino-yne click polymerization. Polym. Chem. 2016;7:7375–7382. [Google Scholar]
- He B.Z., Su H.F., Bai T.W., Wu Y.W., Li S.W., Gao M., Hu R.R., Zhao Z.J., Qin A.J., Ling J. Spontaneous amino-yne click polymerization: a powerful tool toward regio- and stereospecific poly(beta-aminoacrylate)s. J. Am. Chem. Soc. 2017;139:5437–5443. doi: 10.1021/jacs.7b00929. [DOI] [PubMed] [Google Scholar]
- Hong C.Y., You Y.Z., Wu D.C., Liu Y., Pan C.Y. Thermal control over the topology of cleavable polymers: from linear to hyperbranched structures. J. Am. Chem. Soc. 2007;129:5354–5355. doi: 10.1021/ja070871+. [DOI] [PubMed] [Google Scholar]
- Hong Y., Lam J.W.Y., Tang B.Z. Aggregation-induced emission. Chem. Soc. Rev. 2011;40:5361–5388. doi: 10.1039/c1cs15113d. [DOI] [PubMed] [Google Scholar]
- Huang H., Wang W., Zhou Z., Sun B., An M., Haeffner F., Niu J. Radical ring-closing/ring-opening cascade polymerization. J. Am. Chem. Soc. 2019;141:12493–12497. doi: 10.1021/jacs.9b05568. [DOI] [PubMed] [Google Scholar]
- Iha R.K., Wooley K.L., Nystrom A.M., Burke D.J., Kade M.J., Hawker C.J. Applications of orthogonal "click" chemistries in the synthesis of functional soft materials. Chem. Rev. 2009;109:5620–5686. doi: 10.1021/cr900138t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang K.M., Zhang L., Zhao Y.C., Lin J., Chen M. Palladium-Catalyzed cross-coupling polymerization: a new access to cross-conjugated polymers with modifiable structure and tunable optical/conductive properties. Macromolecules. 2018;51:9662–9668. [Google Scholar]
- Jing Y.N., Li S.S., Su M., Bao H., Wan W.M. Barbier hyperbranching polymerization-induced emission toward facile fabrication of white LED and light harvesting film. J. Am. Chem. Soc. 2019;141:16839–16848. doi: 10.1021/jacs.9b08065. [DOI] [PubMed] [Google Scholar]
- Kotha S., Lahiri K., Kashinath D. Recent applications of the Suzuki–Miyaura cross-coupling reaction in organic synthesis. Tetrahedron. 2002;58:9633–9695. [Google Scholar]
- Kreye O., Toth T., Meier M.A.R. Introducing multicomponent reactions to polymer science: Passerini reactions of renewable monomers. J. Am. Chem. Soc. 2011;133:1790–1792. doi: 10.1021/ja1113003. [DOI] [PubMed] [Google Scholar]
- Li C.J., Zhang W.C. Unexpected Barbier−Grignard allylation of aldehydes with magnesium in water. J. Am. Chem. Soc. 1998;120:9102–9103. [Google Scholar]
- Littke A.F., Dai C., Fu G.C. Versatile catalysts for the Suzuki cross-coupling of arylboronic acids with aryl and vinyl halides and triflates under mild conditions. J. Am. Chem. Soc. 2000;122:4020–4028. [Google Scholar]
- Littke A.F., Fu G.C. The first general method for Stille cross-couplings of aryl chlorides. Angew. Chem. Int. Ed. 1999;38:2411–2413. doi: 10.1002/(sici)1521-3773(19990816)38:16<2411::aid-anie2411>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Liu B., Zhang H., Liu S., Sun J., Zhang X., Tang B.Z. Polymerization-induced emission. Mater. Horiz. 2020 doi: 10.1039/c1039mh01909j. [DOI] [Google Scholar]
- Liu M., Vladimirov N., Fréchet J.M.J. A new approach to hyperbranched polymers by ring-opening polymerization of an AB monomer: 4-(2-Hydroxyethyl)-ϵ-caprolactone. Macromolecules. 1999;32:6881–6884. [Google Scholar]
- Liu Y., Wu D.C., Ma Y.X., Tang G.P., Wang S., He C.B., Chung T.S., Goh S. Novel poly(amino ester)s obtained from Michael addition polymerizations of trifunctional amine monomers with diacrylates: safe and efficient DNA carriers. Chem. Commun. 2003:2630–2631. doi: 10.1039/b309487a. [DOI] [PubMed] [Google Scholar]
- Luo J.D., Xie Z.L., Lam J.W.Y., Cheng L., Chen H.Y., Qiu C.F., Kwok H.S., Zhan X.W., Liu Y.Q., Zhu D.B. Aggregation-induced emission of 1-methyl-1,2,3,4,5-pentaphenylsilole. Chem. Commun. 2001:1740–1741. doi: 10.1039/b105159h. [DOI] [PubMed] [Google Scholar]
- Lv X.H., Li S.S., Tian C.Y., Yang M.M., Li C., Zhou Y., Sun X.L., Zhang J., Wan W.M. Borinic acid polymer: simplified synthesis and enzymatic biofuel cell application. Macromolecular Rapid Commun. 2017;38:1600687. doi: 10.1002/marc.201600687. [DOI] [PubMed] [Google Scholar]
- Lv X.H., Wang X.Y., Zhou Y., Xu H., Wan W.M. Promoting water dissociation performance by borinic acid for the strong-acid/base-free hydrogen evolution reaction. Chem. Commun. 2019;55:9821–9824. doi: 10.1039/c9cc04569d. [DOI] [PubMed] [Google Scholar]
- Mei J., Leung N.L.C., Kwok R.T.K., Lam J.W.Y., Tang B.Z. Aggregation-induced emission: together we shine, united we soar! Chem. Rev. 2015;115:11718–11940. doi: 10.1021/acs.chemrev.5b00263. [DOI] [PubMed] [Google Scholar]
- Meissner G., Kretschmar K., Braun T., Kemnitz E. Consecutive transformations of tetrafluoropropenes: hydrogermylation and catalytic C-F activation steps at a Lewis acidic aluminum fluoride. Angew. Chem. Int. Ed. 2017;56:16338–16341. doi: 10.1002/anie.201707759. [DOI] [PubMed] [Google Scholar]
- Miyaura N., Yanagi T., Suzuki A. The palladium-catalyzed cross-coupling reaction of phenylboronic acid with haloarenes in the presence of bases. Synth. Commun. 1981;11:513–519. [Google Scholar]
- Moad G., Rizzardo E., Thang S.H. Radical addition–fragmentation chemistry in polymer synthesis. Polymer. 2008;49:1079–1131. [Google Scholar]
- Pintauer T., Matyjaszewski K. Atom transfer radical addition and polymerization reactions catalyzed by ppm amounts of copper complexes. Chem. Soc. Rev. 2008;37:1087–1097. doi: 10.1039/b714578k. [DOI] [PubMed] [Google Scholar]
- Pryor W.A., Ohto N., Church D.F. Reaction of cumene with ozone to form cumyl hydrotrioxide and the kinetics of decomposition of cumyl hydrotrioxide. J. Am. Chem. Soc. 1983;105:3614–3622. [Google Scholar]
- Schluter A.D. The tenth anniversary of Suzuki polycondensation (SPC) J. Polym. Sci. A Polym. Chem. 2001;39:1533–1556. [Google Scholar]
- Sergeev G.B., Smirnov V.V., Badaev F.Z. Low-temperature reaction of magnesium with fluorobenzene. J. Organomet. Chem. 1982;224:C29–C30. [Google Scholar]
- Song Y., Ji X., Dong M., Li R., Lin Y.N., Wang H., Wooley K.L. Advancing the development of highly-functionalizable glucose-based polycarbonates by tuning of the glass transition temperature. J. Am. Chem. Soc. 2018;140:16053–16057. doi: 10.1021/jacs.8b10675. [DOI] [PubMed] [Google Scholar]
- Sun X.L., Liu D.M., Tian D., Zhang X.Y., Wu W., Wan W.M. The introduction of the Barbier reaction into polymer chemistry. Nat. Commun. 2017;8:1210. doi: 10.1038/s41467-017-01472-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian P.P., Feng C., Loh T.P. Rhodium-catalysed C(sp(2))-C(sp(2)) bond formation via C-H/C-F activation. Nat. Commun. 2015;6:7472. doi: 10.1038/ncomms8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao Y.T., Wooley K.L. Synthetic, functional thymidine-derived polydeoxyribonucleotide analogues from a six-membered cyclic phosphoester. J. Am. Chem. Soc. 2017;139:5467–5473. doi: 10.1021/jacs.7b01116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vougioukalakis G.C., Grubbs R.H. Ruthenium-based heterocyclic carbene-coordinated olefin metathesis catalysts. Chem. Rev. 2010;110:1746–1787. doi: 10.1021/cr9002424. [DOI] [PubMed] [Google Scholar]
- Wan W.M., Li S.S., Liu D.M., Lv X.H., Sun X.L. Synthesis of electron-deficient borinic acid polymers with multiresponsive properties and their application in the fluorescence detection of alizarin red S and electron-rich 8-hydroxyquinoline and fluoride ion: substituent effects. Macromolecules. 2017;50:6872–6879. [Google Scholar]
- Wan W.M., Tian D., Jing Y.N., Zhang X.Y., Wu W., Ren H., Bao H.L. NBN-doped conjugated polycyclic aromatic hydrocarbons as an AIEgen class for extremely sensitive detection of explosives. Angew. Chem. Int. Ed. 2018;57:15510–15516. doi: 10.1002/anie.201809844. [DOI] [PubMed] [Google Scholar]
- Wang J.S., Matyjaszewski K. Controlled/"living" radical polymerization. atom transfer radical polymerization in the presence of transition-metal complexes. J. Am. Chem. Soc. 1995;117:5614–5615. [Google Scholar]
- Wang D., Liu Y., Hu Z.C., Hong C.Y., Pan C.Y. Michael addition polymerizations of trifunctional amines with diacrylamides. Polymer. 2005;46:3507–3514. [Google Scholar]
- Wang H.N., Dai X.J., Li C.J. Aldehydes as alkyl carbanion equivalents for additions to carbonyl compounds. Nat. Chem. 2017;9:374–378. doi: 10.1038/nchem.2677. [DOI] [PubMed] [Google Scholar]
- Wang L., Lear J.M., Rafferty S.M., Fosu S.C., Nagib D.A. Ketyl radical reactivity via atom transfer catalysis. Science. 2018;362:225–229. doi: 10.1126/science.aau1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei B., Li W.Z., Zhao Z.J., Qin A.J., Hu R.R., Tang B.Z. Metal-free multicomponent tandem polymerizations of alkynes, amines, and formaldehyde toward structure- and sequence-controlled luminescent polyheterocycles. J. Am. Chem. Soc. 2017;139:5075–5084. doi: 10.1021/jacs.6b12767. [DOI] [PubMed] [Google Scholar]
- Wu H.B., Wang Z.M., Tao L. The Hantzsch reaction in polymer chemistry: synthesis and tentative application. Polym. Chem. 2017;8:7290–7296. [Google Scholar]
- Xue H.D., Zhao Y., Wu H.B., Wang Z.L., Yang B., Wei Y., Wang Z.M., Tao L. Multicomponent combinatorial polymerization via the Biginelli reaction. J. Am. Chem. Soc. 2016;138:8690–8693. doi: 10.1021/jacs.6b04425. [DOI] [PubMed] [Google Scholar]
- Yin X., Guo F., Lalancette R.A., Jäkle F. Luminescent main-chain organoborane polymers: highly robust, electron-deficient poly(oligothiophene borane)s via Stille coupling polymerization. Macromolecules. 2016;49:537–546. [Google Scholar]
- Yokoyama A., Suzuki H., Kubota Y., Ohuchi K., Higashimura H., Yokozawa T. Chain-growth polymerization for the synthesis of polyfluorene via Suzuki-Miyaura coupling reaction from an externally added initiator unit. J. Am. Chem. Soc. 2007;129:7236–7237. doi: 10.1021/ja070313v. [DOI] [PubMed] [Google Scholar]
- Zhang H.K., Zheng X.Y., Xie N., He Z.K., Tiu J.K., Leung N.L.C., Niu Y.L., Huang X.H., Wong K.S., Kwok R.T.K. Why do simple molecules with "isolated" phenyl rings emit visible light? J. Am. Chem. Soc. 2017;139:16264–16272. doi: 10.1021/jacs.7b08592. [DOI] [PubMed] [Google Scholar]
- Zhang W.C., Li C.J. Magnesium-mediated carbon−carbon bond formation in aqueous media: Barbier−Grignard allylation and pinacol coupling of aldehydes. J. Org. Chem. 1999;64:3230–3236. doi: 10.1021/jo982497p. [DOI] [PubMed] [Google Scholar]
- Zhou F., Li C.J. The Barbier-Grignard-type arylation of aldehydes using unactivated aryl iodides in water. Nat. Commun. 2014;5:4254. doi: 10.1038/ncomms5254. [DOI] [PubMed] [Google Scholar]
- Zhu N., Chiou M.-F., Xiong H., Su M., Su M., Li Y., Wan W.-M., Bao H. The introduction of the radical cascade reaction into polymer chemistry: a one-step strategy for synchronized polymerization and modification. iScience. 2020;23:100902. doi: 10.1016/j.isci.2020.100902. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.