

Abstract
Glioblastoma (GBM) is the most aggressive adult brain tumor. While GBM typically occurs sporadically, familial GBM can be associated with certain hereditary disorders and isolated familial GBMs in the absence of syndrome are rare. Relevant hereditary factors have remained elusive in these cases. Understanding specific genetic abnormality may potentially lead to better treatment strategies in these patients. Here, we analyzed GBM tissue from our patient and 2 afflicted family members, with next generation sequencing to better understand the genetic alterations associated with this disease development. DNA was extracted and sequenced and the data were then analyzed. Results revealed 2 common mutations in afflicted family members; PDGFRA and HRAS. In addition, both siblings showed a mutation of the SMARCB1 gene. The sister of our patient exhibited a homozygous mutation, while our patient had heterozygous mutation of this gene in the tumor tissue. This result suggests that mutation of SMARCB1, either alone or in the presence of PDGFRA and HRAS mutations, is associated with earlier onset GBM.
Keywords: Familial GBM, NGS, SMARCB1
INTRODUCTION
Glioblastoma (GBM) is one of the most aggressive brain tumors in humans. Approximately 12%–15% of all intracranial neoplasms, as well as 50%–60% of all astrocytic tumors are GBM (1). The incidence rate of GBM is 2–3 cases per 100 000 in Europe and North America (2). Despite multi-modality treatment with surgical resection, radiation and chemotherapy, the median survival of patients with GBM is only ∼15 months. In spite of an increase in basic and clinical research over the last few decades, little improvement is noted with regards to the poor prognosis of patients with GBM.
It is estimated that ∼5%–10% of gliomas are familial (3). Specifically, familial gliomas can be found in association with certain hereditary disorders, such as Turcot syndrome, tuberous sclerosis, neurofibromatosis, and Li-Fraumeni syndrome (4). Isolated familial GBMs, in the absence of a syndrome, do occur but are exceedingly rare. Understanding the genetic alterations of familial GBM may uncover some of the unknown mechanistic pathways and potentially lead to better understanding of familial GBM pathogenesis.
The current report presents the case of a 19-year-old, Hispanic male, who initially presented with recurrent bouts of severe headaches and a significant family history of GBM. His initial computed tomography and magnetic resonance imaging scans revealed a left frontal lobe mass, which was then resected and histopathologically confirmed as GBM, WHO grade IV, with unmethylated MGMT status. He was initially treated with the standard care of Stupp protocol (radiation co-administered with temozolomide followed by temozolomide maintenance for 6 months), but his tumor recurred after 2 years. He was then treated with Lomustine and Novo TTF (tumor treating fields) magnetic field scalp electrodes. Unfortunately, his tumor progressed rapidly and he died one month after recurrent disease treatment was initiated. Interestingly, both his father (38 years old) and his sister (6 years old) were diagnosed with GBM 7 years prior to his diagnosis and passed away within a year from their diagnosis (Fig. 1). His mother, however, is in good health without any significant medical conditions. Given his significant family history of GBM, we performed next generation sequencing of extracted GBM tumor tissues from our patient, his father and sister. We proposed that we could identify a familial gene that may link diagnosis among these family members with GBM.
FIGURE 1.
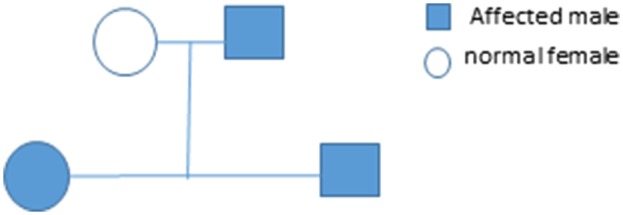
The family pedigree is shown. The patient (filled square bottom row), his sister (filled circle bottom row), and his father (filled square, top row) had GBM. The patient’s mother (unfilled circle, top row) was not affected by GBM.
MATERIALS AND METHODS
Resected GBM tissue was formalin-fixed and embedded in paraffin for hematoxylin and eosin (H&E) and MIB-1 staining. This staining allowed for identification of neoplastic from nonneoplastic tissue by our institutional neuropathologist. The neoplastic tissue was then carefully separated and isolated from the nonneoplastic tissue to avoid contamination. Tissue was digested and cells were isolated. Cell samples from neoplastic tissue and nonneoplastic tissue were submitted for next generation sequencing to determine if common hotspot mutations were present. Nonneoplastic tissue from the patient’s sister was not available for analysis.
To perform next generation sequencing, DNA was extracted from neoplastic and nonneoplastic formalin-fixed paraffin-embedded brain biopsy specimens, using Ambion Recover All Total Nucleic Acid Isolation Kit (Life Technologies, Carlsbad, CA). Extractions were sequenced using the Ampliseq Library Prep kit with the Comprehensive Cancer Hotspot Panel v2.0 (Life Technologies). Library preparation for each specimen was equalized to 100 pM using the Ion Library Equalizer kit then amplified and sequenced using the Ion Chef and Ion PGM systems, respectively (Life Technologies). Data were analyzed using Variant Caller v5.0.2.1 (Life Technologies).
RESULTS
The histopathologic slide of our patient is shown in Figure 2. We noted neoplastic cells of astrocytic phenotype with an atypical mitosis shown in circle (Fig. 2A) as well as prominent pleomorphism (Fig. 2B). Other characteristic features of GBM, such as microvascular hyperplasia (Fig. 2C) and geographic necrosis (Fig. 2D), were also noted in the sections of his tumor tissue. Both family members (father and sister) showed similar histopathology including a pleomorphic astrocytic phenotype, necrosis and microvascular hyperplasia. The common hotspot mutations identified in the tumors of these 3 patients are shown in Table 1. The tissue from the father’s brain tumor demonstrated hotspot mutations in 3 genes: Heterozygous mutations in PDGFRA (platelet derived growth factor receptor A) and TP53 (tumor protein p53) on chromosomes 4 and 17, respectively, and a homozygous mutation in HRAS (Harvey rat sarcoma viral oncogene homolog) on chromosome 11. The sibling’s tumor had homozygous mutations in PDGFRA, HRAS and SMARCB1 (SW1/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1) on chromosomes 4, 11, and 22, respectively. Interestingly, the 19-year-old patient’s tumor showed heterozygous mutations in PDGFRA, HRAS, and SMARCB1 on chromosomes 4, 11, and 22, respectively.
FIGURE 2.

Representative tumor sections from the patient are shown. Hematoxylin and eosin (H&E)-stained section demonstrates neoplastic cells of astrocytic phenotype with an atypical mitosis as shown in the circle (A). Neoplastic cells show prominent pleomorphism (B). There are areas of florid microvascular hyperplasia (C) and geographic necrosis (D), which confirm the diagnosis of glioblastoma.
TABLE 1.
Next Generation Sequencing Data on Neoplastic Tissue (Hotspot Mutations)
| Chromosome | Position | Reference | Variant | Allele Call | Type | Allele Source | Gene ID |
|---|---|---|---|---|---|---|---|
| Father | |||||||
| Chr4 | 55152040 | C | T | Heterozygous | SNP | Hotspot | PDGFRA |
| Chr 11 | 534242 | A | G | Homozygous | SNP | Hotspot | HRAS |
| Chr 17 | 7578211 | C | T | Heterozygous | SNP | Hotspot | TP53 |
| Sister | |||||||
| Chr4 | 55152040 | C | T | Homozygous | SNP | Hotspot | PDGFRA |
| Chr 11 | 534242 | A | G | Homozygous | SNP | Hotspot | HRAS |
| Chr 22 | 24176287 | G | A | Homozygous | SNP | Hotspot | SMARCB1 |
| Patient | |||||||
| Chr4 | 55152040 | C | T | Heterozygous | SNP | Hotspot | PDGFRA |
| Chr 11 | 534242 | A | G | Heterozygous | SNP | Hotspot | HRAS |
| Chr 22 | 24176287 | G | A | Heterozygous | SNP | Hotspot | SMARCB1 |
Common hotspot mutations identified in neoplastic brain tissue shared by 2 or more family members with glioblastoma are shown. All family members shared mutations in PDGFRA and HRAS and 2 children share mutations in SMARCB1.
Nonneoplastic brain tissues from the patient and his father were available and sequenced. The mutations that the 2 individuals shared in common are shown in Table 2. The patient and his father shared homozygous mutations in FGFR3 (fibroblast growth factor receptor 3) and RET (rearranged during transfection) on chromosomes 4 and 10, respectively, as well as a heterozygous mutation in HRAS on chromosome 11. The father had homozygous mutations in EGFR (epidermal growth factor receptor) and FLT3 (FMS-related tyrosine kinase 3) on chromosomes 7 and 13, respectively, while the son had heterozygous mutations in these genes. In addition, the father had homozygous multiple nucleotide pleomorphisms in CSF1R (colony factor 1 receptor) on chromosome 5 while the son had a heterozygous single nucleotide polymorphism mutation in the same gene. In comparing mutations observed both in neoplastic and nonneoplastic tissues from the patient and his father, HRAS was the only mutation identified in both neoplastic as well as nonneoplastic tissues in these individuals.
TABLE 2.
Next Generation Sequencing Data on Nonneoplastic Brain Tissue
| Chromosome | Position | Reference | Variant | Allele Call | Type | Allele Source | Gene ID |
|---|---|---|---|---|---|---|---|
| Father | |||||||
| Chr 11 | 534242 | A | G | Heterozygous | SNP | Novel | HRAS |
| Chr4 | 1807894 | G | A | Homozygous | SNP | Novel | FGFR3 |
| Chr5 | 1494533049 | G | A | Heterozygous | SNP | Novel | CSFIR |
| Chr7 | 55249063 | G | A | Homozygous | SNP | Novel | EGFR |
| Chr10 | 43613843 | G | T | Homozygous | SNP | Novel | RET |
| Chr13 | 28610183 | A | G | Homozygous | SNP | Novel | FLT3 |
| Patient | |||||||
| Chr 11 | 534242 | A | G | Heterozygous | SNP | Novel | HRAS |
| Chr4 | 1807894 | G | A | Homozygous | SNP | Novel | FGFR3 |
| Chr5 | 149433596 | TG | GA | Homozygous | MNP | Novel | CSFIR |
| Chr7 | 55249063 | G | A | Heterozygous | SNP | Novel | EGFR |
| Chr10 | 43613843 | G | T | Homozygous | SNP | Novel | RET |
| Chr13 | 28610183 | A | G | Heterozygous | SNP | Novel | FLT3 |
Common novel mutations identified in nonneoplastic brain tissue shared between the patient and his father are shown. Nonneoplastic tissue from the sister was not available. Of note, HRAS is the only mutation identified in both neoplastic and nonneoplastic tissues for the patient and his father.
DISCUSSION
In summary, all the family members demonstrated 2 common hotspot mutations of PDGFRA and HRAS genes within neoplastic tissues. Additionally, our patient and his sister had a SMARCB1 mutation within their tumor tissue. Compared with his father (GBM diagnosed at 38 years), who had no mutation on the SMARCB1 gene, the heterozygous mutation of SMARCB1 gene in our patient can be associated with an earlier GBM diagnosis at the age of 19. Interestingly, the homozygous mutation of SMARCB1 in his sister’s tumor was associated with the earliest diagnosis of GBM at the age of 6. Thus, mutation of SMARCB1 in the presence of PDGFRA and HRAS, resulted in an earlier appearance of GBM in our patient and his sister when compared with his father. The result suggests that SMARCB1 may play a critical role in earlier development/presentation of GBM.
PDGFRA is located on chromosome 4q12 and has an established association with a subset of somatic gastrointestinal stromal tumors, chronic eosinophilic leukemia, and idiopathic hypereosinophilic syndrome (5). In addition, overexpression of PDGFRA is noted in GBM with proneural subtype (6). Imatinib is a FDA approved PDGFRA inhibitor and has been reported to exhibit therapeutic potential to treat solid tumors with PDGFR mutations (7).
HRAS mutations have been noted previously in gliomas (8). HRAS is located on chromosome 11p15.5, and belongs to RAS family of oncogenes. In response to stimulation from growth factors, such as EGFR, HRAS promotes cell division through ERK-MAPK and PI3 kinase pathways. RAS acts as a molecular switch that stays either in active state (when bound to GTP) or inactive state (when bound to GDP). Guanine nucleotide exchange factor (GEF) exchanges GDP with GTP, whereas GTPase-activating protein (GAP) hydrolyzes the GTP to GDP. Mutational activation of HRAS can lead to uncontrolled cell division and tumor genesis. HRAS has association with somatic bladder cancer, somatic follicular thyroid carcinoma, Schimmelpenning-Feuerstein-Mims syndrome (linear nevus sebaceous syndrome), Costello syndrome and congenital myopathy with excess of muscle spindles (9). Potential drugs targeting HRAS associated with certain cancers are under investigations and development (10).
SMCARCB1, also known as INI1, BAF47, and hSNF5, is located on chromosome 22q11.2. SMARCB1 is one of the evolutionarily conserved core subunits of switch/sucrose non-fermentable (SWI/SNF), which is a nucleosome remodeling complex in eukaryotes (11). The function of SMARCB1 within the SWI/SNF is not fully understood; however, the current consensus in the scientific community is that SMARCB1 acts as tumor suppressor epigenetically by regulating gene transcription (11). It was shown that SMARCB1 suppresses the action of Cyclin D1 transcription and inhibits the action of CDK 4/6 by binding and recruiting HDAC activity in G1 of cell cycle; all resorting in cell cycle progression (12). SMCARCB1 is frequently deleted in malignant rhabdoid tumors including atypical teratoid rhabdoid tumor (ATRT) (13). It is also associated with rhabdoid predisposition syndrome 1 and schwannomatosis 1 (13). Renal medullary carcinoma, a highly malignant cancer, has been reported to be associated with complete loss of SMARCB1/INI1 protein expression (14). While SMARCB1 missense mutation has been reported in GBM, its occurrence is rare (15). A study showed that SMARCB1 inhibits sonic hedgehog (SHH) pathway by preventing transcription of glioma-associated oncogene homolog (GLI), and antagonizes polycomb complexes (16). In this report, the heterozygous mutation of SMARCB1 gene led to the early onset of GBM in our patient, which may suggest a gain-of-function mutation of this gene. The patient’s sister, who had homozygous mutation of SMARCB1 gene, had brain tumor morphologically classic for GBM; however, the small possibility that she actually had ATRT cannot be ruled out. Unfortunately, we were unable to perform an immunostaining of SMARCB1 protein expression due to the lack of adequate tumor tissue.
The result of our clinical report suggests that SMARCB1 mutation, either alone or in conjunction with the mutations of PDGFRA and HRAS genes, may lead to the earlier development of the GBM. This is a novel finding that has not been previously reported. The result of this report may aid proper counseling and work-up of familial GBM cases. These findings may assist in evaluating glioma risk and may lead to the development of better treatment strategies for SMARCB1 mutated GBM patients; akin to those being developed for SMARCB1 mutated rhabdoid tumors (17).
This study was funded by Baylor Scott & White Healthcare.
The authors have no duality or conflicts of interest to declare.
REFERENCES
- 1. Bondy ML, Scheurer ME, Malmer B, et al. Brain tumor epidemiology: From the Brain Tumor Epidemiology Consortium. Cancer 2008;113:1953–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barnard RO, Geddes JF.. The incidence of multifocal cerebral gliomas. A histologic study of large hemisphere sections. Cancer 1987;60:1519–31 [DOI] [PubMed] [Google Scholar]
- 3. Malmer B, Henriksson R, Gronberg H.. Familial brain tumours—Genetics or environment? A nationwide cohort study of cancer risk in spouses and first-degree relatives of brain tumour patients. Int J Cancer 2003;106:260–3 [DOI] [PubMed] [Google Scholar]
- 4. Burger PC, Green SB.. Patient age, histologic features, and length of survival in patients with glioblastoma multiforme. Cancer 1987;59:1617–25 [DOI] [PubMed] [Google Scholar]
- 5. Velghe AI, Van Cauwenberghe S, Polyansky AA, et al. PDGFRA alteration in cancer: Characterization of a gain of function V326E transmembrane mutant as well as loss of function and passenger mutations. Oncogene 2014;33:2568–76 [DOI] [PubMed] [Google Scholar]
- 6. Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes if glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR and NFI. Cancer Cell 2010;17:98–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Malavaki CJ, Roussidis AE, Gialeli C, et al. Imatinib as a key inhibitor of the platelet-derived growth factor receptor mediated expression of cell surface haparan sulfate proteoglycans and functional properties of breast cancer cells. FEBS J 2013;280:2477–89 [DOI] [PubMed] [Google Scholar]
- 8. Lymbouridou R, Soufla G, Chatzinikola AM, et al. Down-regulation of K-ras and H-ras in human brain gliomas. Eur J Cancer 2009;45:1294–303 [DOI] [PubMed] [Google Scholar]
- 9.190020 HRAS PROTOONCOGENE, GTPase; HRAS. Available at: https://www.omim.org/entry/190020.
- 10. Suqita S, Enokida H, Yoshino H, et al. HRAS as potential therapeutic target of salirasib RAS inhibitor in bladder cancer. Int J Oncol 2018;53:725–36 [DOI] [PubMed] [Google Scholar]
- 11. Kim KH, Roberts CW.. Mechanism by which SMARCB1 loss drives rhabdoid tumor growth. Cancer Genet 2014;207:365–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang Z, Davies KP, Allen J, et al. Cell cycle arrest and repression of cyclin D1 transcription by INI1/hSNF5. Mol Cell Biol 2002;22:5975–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.601607 SWI/SNF-RELATED, MATRIX-ASSOCIATED, ACTIN-DEPENDENT REGULATOR OF CHROMATIN, SUBFAMILY B, MEMBER 1; SMARCB1. Available at: https://www.omim.org/entry/601607. [DOI] [PubMed]
- 14. Cheng JX, Tretiakova M, Gong C, et al. Renal medullary carcinoma: Rhabdoid features and the absence of INH expression as markers of aggressive behavior. Mod Pathol 2008;21:647–52 [DOI] [PubMed] [Google Scholar]
- 15. Williams EA, Miller JJ, Tummala SS, et al. TERT promoter wild-type glioblastomas show distinct clinical features and frequent PI3K pathway mutations. Acta Neuropathol Commun 2018;6:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jagani Z, Mora-Blanco EL, Sansam CG, et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat Med 2010;16:1429–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Geller JI, Roth JJ, Biegel JA.. Biology and treatment of rhabdoid tumor. Crit Rev Oncog 2015;201:199–216 [DOI] [PMC free article] [PubMed] [Google Scholar]