

Summary
Autoimmune diabetes mellitus is a rare but significant side effect of treatment with immune checkpoint inhibitors. Immune checkpoint inhibitor‐induced diabetes mellitus (CPI‐DM) is characterized by acute onset of dramatic hyperglycemia with severe insulin deficiency and occurrence following exposure to programmed cell death‐1/programmed cell death ligand‐1 (PD‐1/PD‐L1) inhibitors rather than cytotoxic T lymphocyte‐associated antigen 4 (CTLA‐4) inhibitors. As a growing number of patients undergo immunotherapy, further understanding of the characteristics of CPI‐DM patients is needed for improved prognostic and diagnostic application in order to reduce overall morbidity for this already at‐risk population. Additionally, understanding of the features and mechanisms of CPI‐DM may contribute to understanding mechanisms of spontaneous type I diabetes mellitus (T1DM). Here, we summarize the clinical features of CPI‐DM and interrogate the genetic and cellular mechanisms that may contribute to the disease, as well as the clinical challenges for predicting and treating these patients as increasing cancer immunotherapies reach clinical utility.
Keywords: autoimmune diabetes mellitus, immune checkpoint inhibitor, immune‐related adverse event
Immune checkpoint inhibitor‐induced diabetes mellitus (CPI‐DM) is characterized by acute onset of dramatic hyperglycemia with severe insulin deficiency and occurrence following exposure to PD‐1/PD‐L1 inhibitors rather than CTLA‐4 inhibitors. Genetic predisposition is likely to play a role, as is a second trigger. Improved understanding of CPI‐DM is needed clinically to reduce overall morbidity and may also contribute to understanding mechanisms of spontaneous T1DM.
Introduction
The discovery of immune checkpoint inhibitors (CPIs), which include Food and Drug Administration (FDA)‐approved agents targeted against cytotoxic T lymphocyte‐associated protein‐4 (CTLA‐4), programmed cell death‐1 (PD‐1) and PD‐ligand 1 (PD‐L1) has changed the landscape of cancer therapeutics. At homeostasis, these immune checkpoints are important in the maintenance of peripheral tolerance but can be co‐opted or evaded by malignant cells to enable cancer growth (Fig. 1). CPIs were first approved for use in treatment of metastatic melanoma; now a growing number of cancer types and genetic anomalies receive survival benefit from these therapeutic modalities. Further clinical trials are under way to determine if CPIs in combination with other immunotherapies, conventional chemotherapies and radiation will provide greater benefit than CPIs on their own.
Figure 1.
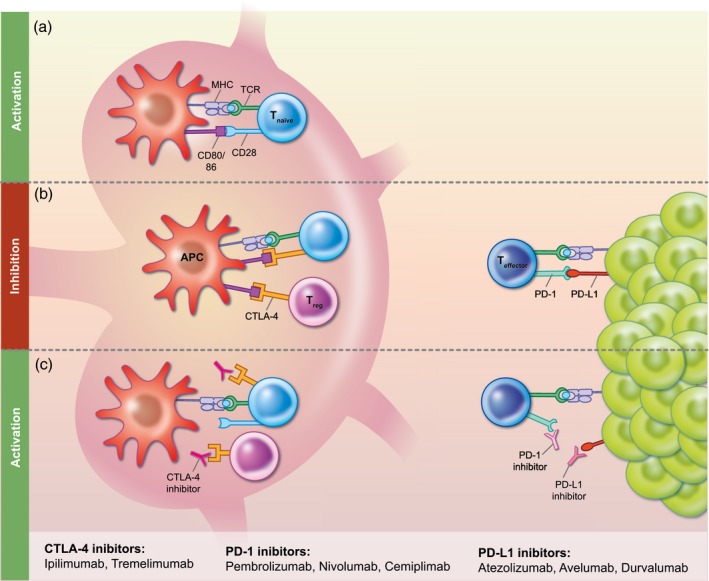
Immune Checkpoints and Immune Checkpoint Inhibitors (a) In the lymph node, naïve T cells are activated by a two signal system. The MHC complex on an antigen presenting cell (APC) presents antigen to the T cell receptor (TCR) on the naïve T cell. Costimulation via CD80 or CD86 on the APC binding to CD28 on the naïve T cell then leads to T cell activation. (b) In the lymph node, the presence of CTLA‐4 expression on either the naive T cell or regulatory T cells (Treg) prevents binding of CD80 or CD86 on the APC to CD28 on the T cell, thereby effectively inhibiting the immune response. In the periphery, the presence of PD‐L1 on the tumor leads to inhibition of effector T cells. (c) Immune checkpoint inhibitors prevent inhibition of the immune response. CTLA‐4 inhibitors act in the lymph tissues by blocking CTLA‐4, thereby allowing the second signal for activation of naïve T cells via CD28. At the tumor, PD‐1 inhibitors bind to PD‐1 expressed on the effector T cells and PD‐L1 inhibitors bind to PD‐L1 expressed by the tumor; this allows for activation of the immune response.
Despite their efficacy for tumor therapy, with the growing use of CPIs the frequency of autoimmune complications has become increasingly apparent. These autoimmune complications, called immune‐related adverse events (irAEs), often involve the endocrine tissues and include hypophysitis, thyroiditis and adrenalitis and autoimmune diabetes (CPI‐DM) 1, 2. Mechanisms of irAEs are proposed to be heterogeneous, including possible tissue destruction mediated by T cells (myocarditis, vitiligo), inflammatory cytokines (colitis), complement activation (hypophysitis) and autoantibodies (thyroiditis) 3. Endocrine irAEs are unique relative to other irAEs, as they are only rarely reversible and are not ameliorated by steroids. In CPI‐DM, as in type I diabetes mellitus (T1DM), steroids appear to both fail to reverse β cell dysfunction and worsen insulin resistance. Because of the high morbidity associated with diabetes, CPI‐DM is of particular clinical concern despite its rarity, with estimates between 0·2 and 1·4% 2, 4, 5, 6, 7 of CPI‐treated patients. As CPIs are increasingly used in lower stage cancers, including adjuvant settings and in complex therapeutic combinations, it is imperative to understand the risk of an adverse effect with such a high impact on quality of life. Furthermore, the emergence of autoimmune diabetes following targeted modulations of immune signaling may allow for insight into the factors contributing to development of conventional type 1 diabetes mellitus (T1DM).
In this review, we describe the key features of CPI‐DM and consider their utility in providing mechanistic understanding in the context of CPI‐DM, irAEs and general islet autoimmunity. To describe the key characteristics of CPI‐DM, we use a growing body of literature that includes a recent review by De Filette and colleagues 5 that summarizes 91 case reports, institution specific case–series that include an additional independent 53 cases [27 from University of California San Francisco (UCSF) and Yale (five of which were included in De Filette et al.’s case summary) 4, 21 from Mayo Clinic 7 and 10 from Melanoma Institute Australia 6)] and a recent analysis of the World Health Organization (WHO) pharmacovigilance database 8, which approaches CPI‐DM from a global perspective and has fewer patient‐specific details. Together, these papers encompass the current understanding of CPI‐DM from a clinical perspective.
Case definition and natural history
Various reports have used different case definitions for CPI‐DM, as there is no accepted version. Our preferred strict definition is a severe and persistent insulin deficiency as characterized by (1) presentation in diabetic ketoacidosis (DKA) or very low to absent insulin C‐peptide levels and (2) insulin dependence for at least weeks to months past the acute diagnostic period. CPI‐DM must be differentiated from exacerbated type II diabetes mellitus (T2DM). Key features of the natural history can help with this differentiation. CPI‐DM is often characterized by a severe and acute clinical presentation at the time of diagnosis. For example, DKA occurs in half to three‐quarters of cases at initial clinical presentation, the accompanying hyperglycemia can be more severe than in conventional T1DM and the onset is abrupt 4, 5, 8, 9. As overall awareness of CPI‐DM increases, presentation in DKA may be decreasing due to earlier recognition and intervention 6. CPI‐DM can occur despite pre‐existing T2DM, but it is exceptionally difficult to differentiate CPI‐DM from exacerbation of underlying T2DM unless the pre‐existing T2DM is well controlled without insulin 4, 6, 7. Despite marked hyperglycemia at diagnosis (at times more than 1000 mg/dL), hemoglobin A1c (HbA1c) levels are usually only mildly elevated at presentation (7·6–8·0% 4, 5, 6), which supports the acuity of this disease 9. Interestingly, it has also been observed that pancreatic lipase levels are elevated in a subset of patients at the time of presentation 4, 5, which may be accompanied by a decreased size of the pancreas 5. Taken together, this suggests that pancreatic exocrine dysfunction may play a role in disease onset in at least a subset of patients.
The length of time from CPI initiation to onset of CPI‐DM is highly variable. It occurs within days of the first treatment cycle to years following treatment initiation 4, 5, 7, 10, 11. Median time to diagnosis is between 7 and 17 weeks 4, 8, 10, 12. Onset has been reported to be faster in individuals with the following features at the time of CPI‐DM diagnosis: presentation in DKA 4, 5, 10, positive islet autoantibodies 4, 5, 10 and, possibly, in patients receiving combination PD‐1 and CTLA‐4 inhibitors rather than PD‐1 inhibitor monotherapy 5. These differences in timing of onset due to autoantibody presence and other clinical features suggest the possibility of mechanistic heterogeneity in CPI‐DM.
CPI‐DM is almost always permanent. Out of all published cases, only two cases report insulin therapy discontinuation. In the first reported case 13, a 58‐year‐old man was treated with pembrolizumab (a PD‐1 inhibitor) for metastatic melanoma when he developed severe fasting hyperglycemia [blood glucose (BG) over 400 mg/dL]. He was not in DKA and C‐peptide levels were not checked at that time, although detectable anti‐GAD65 autoantibodies were noted. Pembrolizumab was stopped and a multi‐dose insulin regimen started. Almost 3 weeks later, while on insulin, the patient had a mildly elevated fasting BG and a C‐peptide well within the normal range. Insulin therapy was then stopped, and a subsequent fasting BG was just barely in the diabetic range with a normal C‐peptide. While this case is consistent with acute hyperglycemia without clear etiology aside from CPI exposure, he did not meet our strict definition for CPI‐DM as he may have never been insulinopenic. In the second case 14, a 53‐year‐old man was initially treated with ipilimumab (a CTLA‐4 inhibitor) and nivolumab (a PD‐1 inhibitor) combination therapy for metastatic melanoma. After developing immune‐mediated hypoparathyroidism (a very rare endocrine irAE) and colitis, he stopped CPI therapy and was treated with oral corticosteroids and infliximab. Following resolution of his colitis, he restarted nivolumab monotherapy. Weeks later, he developed seronegative oligoarthritis, which was initially treated with corticosteroid injections. Days later, a random BG was 400 mg/dL with an inadequate C‐peptide of 2·2 ng/ml and detectable islet autoantibodies. A mixed meal tolerance test showed both impaired insulin secretion and reduced peripheral insulin sensitivity. He was started on insulin for his new diagnosis of diabetes and infliximab for the oligoarthritis. During this treatment window, he had reversal of his diabetes and was able to discontinue insulin. As in the first case, insulinopenia was never documented and, furthermore, there was indeed evidence of peripheral insulin resistance. As these cases did not meet our strict definition of CPI‐DM, it is unclear if these cases are examples of recovery from CPI‐DM despite autoantibody evidence of islet autoimmunity. They could instead be examples of partial β cell destruction (possible partial CPI‐DM) or recovery from impaired insulin sensitivity, such as is seen with steroid‐induced hyperglycemia.
Interestingly, CPI‐DM may portend an improved tumor response to CPI therapies 4, 6. Development of other irAEs has also been reported as a marker of improved tumor response 15, 16, 17, 18, but this is not a consistent finding among irAEs 19. These conflicting results may be from distinct effects of different irAEs in individual cancer types due to tissue and tumor‐specific mechanisms. For example, vitiligo in patients with melanoma 18 might be meaningful, given the potential for shared antigens that connect the tumor and autoimmune responses, while the mechanistic influence of CPI‐DM on a distinct cancer process such as melanoma remains obscure. Another possibility is that irAE treatments may diminish the anti‐tumor response to the same degree that autoimmunity had increased it. An attenuated anti‐tumor response has been reported in hypophysitis patients treated with high‐dose instead of low‐dose corticosteroids 15. Not all irAEs and tumors have shown such an association 19. Further complicating matters, the association between irAEs and cancer response is confounded by survival bias, in which patients must remain on treatment and survive long enough to develop an irAE. Nevertheless, it is important to evaluate this trend and consider what it may mean for the underlying mechanism of CPI‐DM.
Clinical attributes and epidemiology
The demographics of CPI‐DM cases probably reflect the demographics of patients receiving CPIs (Table 1). The average age is in the 60s 4, 5, 7, 8 and there is a slight male predominance 4, 5, 6, 7, 8. Non‐Hispanic Caucasians are the major race/ethnicity affected with CPI‐DM, but a meaningful minority of Asians are affected (15–25% 5, 8). Akin to T1DM, rates of personal history of thyroid disease, either prior to CPI initiation or as a thyroid irAE, are probably higher than the background population rates 4, 5, 8. Congruent with the longer FDA approval for CPI use, melanoma is the most common type of associated cancer, followed by lung cancer. However, it is important to note that it remains unknown whether this solely reflects the indications for use of CPIs rather than a connection between the site of cancer and irAE.
Table 1.
CPI‐DM clinical attributes reported in different cohorts
| Case report review [5] | UCSF and Yale cohort [4] | Melanoma Institute of Australia cohort [6] | Mayo Clinic cohort [7] | Pharmacovigilance cohort [8] | |
|---|---|---|---|---|---|
| Number of subjects (n) | 91 | 27 | 10 | 21 | 283 |
| Male (%) | 60 | 63 | 90 | 90 | 56 |
| Average age (years) | 61 | 64 | |||
| DKA at diagnosis (%) | 71 | 59 | 40 | 38 | 50 |
| Median time to diagnosis | |||||
| Median weeks (range) | 20 (1–228) | 25 (4–63) | 21 (3–103) | 17 (1–113) | |
| Median cycles (range) | 5 (1–17) | 6 (1–78) | 4 (1–17) | ||
| PD‐1/PD‐L1 exposed (%)a | 96 | 96 | 100 | 100 | 96 |
| Cancer type | |||||
| Melanoma (%) | 53 | 52 | 100 | 43 | 43 |
| Lung (%) | 15 | 18 | 0 | 24 | 32 |
| Other (%) | 32 | 30 | 0 | 33 | 24 |
| HLA type | |||||
| Subjects with full HLA typing (n) | 51 | 16 | 8 | – | – |
| Susceptible HLA (%)b | 61 | 69 | 25 | – | – |
| Protective and susceptible HLA (%)b | 4 | 0 | 13 | – | – |
| Protective HLA (%)b | 16 | 0 | 25 | – | – |
| HLA‐DR4 (%) | 49 | 76 | 38 | – | – |
| Islet autoantibodies | |||||
| Subjects with islet autoantibody testing (n) | 88 | 25 | 10 | 7 | – |
| Any islet autoantibody (%)c | 53 | 40 | 20 | 71 | – |
| Anti‐GAD65 autoantibody (%) | 49 | 36 | 20 | 57 | – |
PD‐1 inhibitor or PD‐L1 inhibitor either as monotherapy or combination therapy;
as defined by authors;
islet autoantibodies: GAD65 = zinc transport 8, IC 512/ IA2, insulin, ICA; – = data not available. PD‐1 = programmed cell death 1; PD‐L1 = programmed cell death ligand 1; HLA = human leukocyte antigen; DKA = diabetic ketoacidosis; CPI‐DM = immune checkpoint inhibitor induced diabetes mellitus; UCSF = University of California San Francisco.
Islet autoantibodies
Islet autoantibodies directed towards β cell antigens are a hallmark of conventional T1DM. Islet autoantibodies precede the development of T1DM by years 20 and are used to identify individuals at particularly high risk for T1DM prevention studies. By the time of diagnosis, more than 90% of those with T1DM will have at least one positive autoantibody if testing for all four known islet autoantigens is conducted [GAD65, zinc transporter 8, insulin, islet antigen 2 (islet cell antigen 512)] 21. In contrast, compiling autoantibody data from the reported cases and case–series, only 49% 4, 5, 6, 7 of CPI‐DM individuals are positive for any islet autoantibody; anti‐GAD65 is by far the most common at 45%.
Due to the rapidity of onset for CPI‐DM, it was hypothesized that autoantibodies may develop following irAE yet, in extended follow‐ups ranging from 1 to 32 months after diagnosis, islet autoantibodies remained negative 6. Within the UCSF cohort of our previously described case–series 4, a fraction have developed insulin autoantibodies since initial diagnosis while being treated with insulin (unpublished data). In the subset for whom autoantibodies were present at CPI‐DM diagnosis, it is unknown if they were present preceding treatment, potentially allowing for risk prediction, or whether they have developed during the course of treatment. Pretreatment serum was available for a limited number of cases, and these have had mixed results 4, 22, 23, 24. It is also unknown if the presence or absence of these autoantibodies represents different forms of CPI‐DM. Determining the frequency of autoantibodies in cancer patients prior to treatment and subsequent irAE development is of interest, and highlights the need for longitudinal collections. Notably, in thyroid irAEs, there is growing evidence that the presence of thyroid autoantibodies precedes the irAE 25. The presence of islet autoantibodies may identify a biomarker for risk of CPI‐DM that merits further investigation in the population.
Genetic predisposition
Genetic predisposition may contribute to development of CPI‐DM as it does to spontaneous T1DM. Human leukocyte antigen (HLA) genes contribute to encoding the major histocompatibility complex (MHC) I and II that enable antigen presentation to CD8+ and CD4+ T cells, respectively. They are significantly associated with a large number of autoimmune diseases and may also be associated with irAEs. There is evidence that specific individual HLA types were enriched in colitis 26, pruritis 26, rheumatoid arthritis 27 and hypophysitis 28. In conventional T1DM, the highest risk derives from the heterozygous combination of DR3‐DQ2 and DR4‐DQ8 with increased risk still conferred by each of these alleles on their own 29. In Asian populations, different HLA types confer high risk of T1DM and fulminant DM, most prominently DR4‐DQ4 and DR9‐DQ9 30, 31. There are also particular alleles that confer relative protection to T1DM in both Caucasian and Asian populations, exemplified by DR15‐DQ6 29, 31. HLA typing was performed in a subset of the published CPI‐DM cases and patterns in HLA susceptibility have emerged. We have previously reported that the prevalence of DR4 was significantly higher than expected in a Caucasian population and relative to a T1DM cohort 4. All but one case had either a DR4 or DR3 allele 4 and 69% had a high‐risk T1DM DR‐DQ haplotype, defined as DR4‐DQ8, DR3‐DQ2 or DR4‐DQ4 in an Asian individual. No one had a protective HLA type. De Filette and colleagues 5 defined susceptible HLA alleles as A2, DR3 or DR4, or DR9 in a Japanese population. They report similar results for susceptible haplotypes (65%) but a lower prevalence of HLA‐DR4 (49%). Interestingly, protective alleles may not be as robust in CPI‐DM as in the general population. Both Tsang and colleagues 6 and De Filette and colleagues 5 report CPI‐DM cases despite having protective HLA types, some but not all of whom also had susceptible HLA alleles 5, 6. At present, autoantibody presence has not been associated with HLA type 12, 32. With increasing case ascertainment, this association should be revisited to ensure that its absence is not merely due to a limited sample size.
Non‐HLA genes have also been associated with increased risk for T1DM. Some of these genes are essential to encode for immunomodulatory markers important for both anti‐tumor immunity and autoimmunity, such as CTLA‐4, interleukin (IL)‐2 and IL‐2 receptors 33. By combining a series of HLA and non‐HLA‐based single nucleotide polymorphisms (SNPs) in these high‐risk genes, a genetic risk score (GRS) has been developed that can differentiate between T1DM and T2DM 34. In a case study of a CPI‐DM patient, GRS was indeterminant 24. The GRS places the greatest weight on the HLA type for T1DM prediction; however, the high‐risk HLA types for CPI‐DM may be different and weights may need to be adjusted. Furthermore, non‐HLA SNPs may have differing importance; there may be new PD‐1/PD‐L1‐sensitive SNPs which, in combination with drug exposure, confer risk for release of immune constraints and breakdown of self‐tolerance in the pancreas. This highlights that while CPI‐DM may share some characteristics of T1DM, the genetic landscape of CPI‐induced autoimmune manifestations may not be reflective of the spontaneous conditions. Large‐scale studies are needed to elucidate the genetics of irAE and CPI response in the setting of cancer genetics.
PD‐1/PD‐L1 inhibitor exposure predominates
To date, the majority of CPI‐DM cases have been associated with the use of PD‐1 inhibitor therapy. Approximately three‐quarters of patients were on PD‐1 inhibitor monotherapy at the time of CPI‐DM diagnosis 4, 5, 6, 7, 8. A combination PD‐1 and CTLA‐4 inhibitors was the next most common (17% 4, 5, 6, 7, 8) followed by PD‐L1 inhibitors (6% of the combined cases 4, 5, 6, 7, 3% in the pharmacovigilance data 8). A small minority of the cases were on CTLA‐4 monotherapy (3% of the combined cases 4, 5, 6, 7, 4% in the pharmacovigilance data 8) at the time of diagnosis. Many, but not clearly all, the cases that occurred on CTLA‐4 inhibitors had prior exposure to alternate immunotherapies including nivolumab and IFN 4, 5. Of note, of the CPIs with the most FDA‐approved indications, pembrolizumab and nivolumab are both PD‐1 inhibitors. Combination and monotherapy with CTLA‐4 inhibitors has been FDA‐approved for only a limited number of cancers, which may account for the lower incidence of diagnoses in combination‐treated patients.
Potential mechanisms for PD‐1/PD‐L1 in the induction of CPI‐DM
The implication of PD‐1/PD‐L1 inhibitors in over 95% of the CPI‐DM cases highlights the importance of the PD‐1/PD‐L1 axis in mediating tolerance towards pancreatic islets (Fig. 2). This has been previously observed in mouse models using the non‐obese diabetic (NOD) mouse, in which PD‐1 inhibitors, but not CTLA‐4 inhibitors, rapidly induces diabetes in adult mice 35, 36. Similarly, in antigen‐specific tolerogenic models, PD‐1/PD‐L1 inhibition can disrupt long‐term anergy in the islets leading to autoimmune diabetes 37, 38. This suggests that the NOD model may provide utility to delineate mechanisms by which PD‐1/PD‐L1 inhibition induces CPI‐DM. In the tolerogenic setting, inhibition of PD‐1/PD‐L1 but not CTLA‐4 initiated antigen‐specific CD4+ T cells to swarm dendritic cells (DCs), leading to lower cellular velocity and prolonged T cell‐DC engagement within the pancreatic lymph node (LN) 38. In addition, using an autoreactive T cell transfer model, it was possible to examine the regulation and kinetics of BDC2·5+CD4+ T cells at physiological conditions in both autoimmune‐susceptible organs, such as the pancreatic islets, and the periphery 39. Notably, in response to PD‐L1 inhibition, antigen‐specific BDC2.5+CD4+ T cells, which are predominantly PD‐1+, displayed increased proliferation and a greater number of polyfunctional cells 39. These changes are similarly observed in PD‐1‐deficient BDC2·5+CD4+ T cells, but not those that are PD‐L1 deficient. PD‐1‐deficient NOD mice also accumulate insulin‐specific autoreactive CD4+ T cells in the pancreatic LN 40. Together, this data highlights that PD‐1 engagement in immune cells is critical for restraining antigen‐specific T cell responses.
Figure 2.
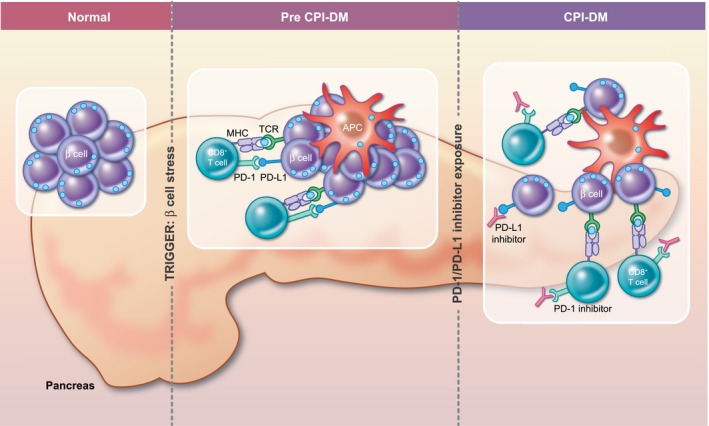
Hypothesized Mechanism for CPI‐DM. Genetic predisposition is likely to play a role in development of CPI‐DM. In individuals with a genetic predisposition, there are likely two components that are required. First, some unknown trigger that leads to b cell stress and priming of an immune response through PD‐L1 expression on the b cell along with recruitment of CD8+ T cells and APCs to the pancreas. Second, exposure to a PD‐1/ PD‐L1 inhibitor likely reinvigorates exhausted CD 8+ T cells. It is likely that these events may occur in either order. Once these two events have occurred, b cells fall victim to primed CD 8+ T cells.
In addition to the immune response to PD‐1/PD‐L1 inhibitor‐mediated autoimmune diabetes, it is important to consider changes to immunomodulatory markers in inflamed self‐tissue. In NOD mice, PD‐L1 expression in β cells correlated with increased age and immune infiltrate in the islets, with diabetic mice displaying the highest PD‐L1 levels 41, 42. An adaptive immune response within the islets appears critical for the up‐regulation of PD‐L1, as immunodeficient recombination activating gene 1 (RAG1)‐knock‐out mice did not express β cell‐derived PD‐L1; however, they were able to up‐regulate PD‐L1 in response to interferon (IFN)‐γ stimulation, and to a lesser extent IFN‐α, in vitro 41, 43. Notably, in the human pancreas, PD‐L1 expression in islets was present in autoantibody‐positive and T1DM patients at greater levels than T2DM patients and healthy controls 41. Therefore, PD‐L1 expression in the pancreas is likely to be indicative of an attempt to subdue an inflammatory response by engaging PD‐1‐expressing self‐reactive T cells.
Access to CPI‐DM pancreatic tissue is limited by clinical constraints, but one case report of pancreatic pathology in a CPI‐DM case is available 44. A 63‐year‐old man with a history of well‐controlled T2DM developed CPI‐DM while on nivolumab (a PD‐1 inhibitor) for metastatic renal cell carcinoma with tumor involvement of the pancreas leading to radical pancreatectomy. Immunohistochemical staining of non‐tumorous pancreas revealed increased T lymphocyte infiltration of islets and very few β cells. CD8+ T cells were found in and around the pancreatic islets more than CD4+ T cells with an absence of macrophages. Overall, most but not all islets were PD‐L1‐negative; however, none of the residual β cells expressed PD‐L1. These findings must be interpreted with caution. While this patient had a typical course for CPI‐DM (DKA at presentation, undetectable C‐peptide, high‐risk T1DM HLA type, negative islet autoantibodies), his pancreatic metastasis and history of T2DM make him relatively unique to other cases. Nonetheless, this is the only report, to our knowledge, of pathology of a CPI‐DM pancreas and suggests that CD8+ T cells may play an important role in mediating this irAE. The lack of PD‐L1 expression on the few surviving β cells raises questions as to whether injured β cells may have expressed PD‐L1 prior to and leading to their destruction. In other irAEs, the T cell immune infiltrate in the affected tissue has also been shown to play a critical role in mediating autoimmune disease. In two patients who developed myocarditis following combination CTLA‐4 and PD‐1 inhibitors, a T cell‐predominant infiltrate in the heart was noted, with both CD4+ and CD8+ T cells 45. T cell receptor sequencing in these individuals revealed an increase in a T cell clone shared in the tumor, cardiac muscle and skeletal muscle. Additionally, PD‐L1 was expressed on injured myocytes. CD8+ T cells have been found in colonic biopsy in PD‐1 inhibitor‐ but not CTLA‐4 inhibitor‐induced colitis 46, highlighting the differential patterns of irAE dictated by therapy. Further tissue samples from patients with CPI‐DM will be essential to understanding the immune phenotype of this irAE but will continue to be extremely rare unless uniform efforts to collect irAE affected tissue are employed.
Clinical challenges
Making the diagnosis
CPI‐DM can be challenging to diagnose. Often, patients have multiple random BG checked but may not have recent HbA1cs to help differentiate between T2DM exacerbation and new onset CPI‐DM. Autoantibody testing in CPI‐DM is not definitive for diagnosis, creating the potential for antibody‐negative individuals to be misclassified. C‐peptide levels are often not tested, and if tested too close to the initial event may be confounded by β cell dysfunction from glucotoxicity. In addition, patients often have generalized inflammation from metastatic cancer or exposure to corticosteroids, either for irAEs or as part of cancer therapy, both of which lead to increased insulin resistance. Furthermore, clinicians must be aware that not all new‐onset diabetes associated with CPIs is from autoimmunity directed towards the β cell—there are now case reports of diabetes due to CPI associated generalized lipodystrophy that is associated with severe insulin resistance rather than insulin deficiency 47, 48. Therefore, development of diagnostic assays would be extremely useful.
Future directions for immunotherapy
In addition to improving our understanding of islet autoimmunity, understanding the pathophysiology of CPI‐DM will allow for better clinical care of CPI‐treated patients. Knowledge of CPI‐DM risk factors and biomarker discovery will allow for an enhanced risk–benefit discussion with patients as they initiate cancer immunotherapies. Clinicians would be able to perform closer surveillance on patients at higher risk, which may include use of home glucometers or detailed anticipatory guidance that could prevent DKA and hospital admissions. As more cancer immunotherapies become available that target a greater number of immunomodulatory molecules, it may even contribute to oncologists’ treatment decisions. In the future, identification of very high‐risk patients or those with early‐onset disease may enable an intervention to change the CPI‐DM course. Recently, teplizumab, a humanized anti‐CD3 monoclonal antibody, was reported to delay the onset of T1DM in high‐risk individuals 49. Interestingly, teplizumab trials have identified an increased frequency of KLRG1+TIGIT+ CD8+ T cells 49, 50 in responders relative to non‐responders. It seems plausible that this particular exhausted T cell phenotype is what is reinvigorated by PD‐1/PD‐L1 inhibition. While teplizumab and many of the other agents tested in T1DM immunomodulation, such as abatacept (CTLA‐4‐Ig), would be unacceptable to use in the cancer population due to their interference with the T cell response that is critical for anti‐tumor immunity, other agents that have been reported to extend C‐peptide production, such as rituximab (anti‐CD20), imatinib (tyrosine kinase inhibitor) and tocilizumab (anti‐IL‐6) 51 may potentially be applicable for use in cancer patients. Biomarker discovery that would allow early detection of β cell destruction would be critical to identify patients prior to complete loss of insulin production. Possible biomarkers might include cytokines, novel autoantibodies and high‐risk genetics, among other factors. In the setting of T1DM, β cell‐specific DNA demethylation of insulin genes (INS1 and INS2) can identify β cell dysfunction prior to alterations in glycemic control in peripheral blood 52, 53, 54, 55; however, this would need to be validated in CPI‐DM. Use of these medications and biomarkers require further study in preclinical models prior to consideration in clinical trials.
As cancer immunotherapy progresses, novel combinations of agents are being tested in an attempt to find synergy between therapeutic regimens. It is essential that irAEs be considered within these combinations as they often limit the ability to continue treatment 1, 56, despite possibly being a sign of therapeutic response 4, 15, 16, 17, 18. Preclinical studies of these synergistic combinations should at least attempt to consider separating the effects of anti‐tumor immunity from autoimmunity, particularly as many immunomodulatory therapies that are moving towards clinical trials also appear critical for mediating autoimmunity 56. The non‐obese diabetic (NOD) mouse and our understanding of T1DM can play an important role in this. In both mice and humans, immune‐related loci have been implicated in the development of many forms of autoimmunity and are often associated with immune checkpoint genes 57, 58. For T1DM in NOD mice, these include insulin‐dependent diabetes (idd) susceptibility loci localized to relevant immune checkpoints, including IL‐2 and IL‐21 (idd3), CTLA‐4 and inducible co‐stimulatory molecule (ICOS) (idd5), and genes related to TNF signaling such as TNFRSF9 (CD137), TNFRSF8 (CD30) and TNFRSF1B (TNFR2) (idd9) that mediate T cell function in either regulatory T cells or CD8+ T cells 59, 60, 61. The ability to modify the onset of the autoimmune response, through sophisticated genetic models in which NOD mice are congenic for loci from autoimmune resistant C57BL/6, C57BL/10 strains, allows for interrogation of the contribution of these genes in mediating CPI‐DM. Notably, the idd3, idd5 and idd9 mice displayed reduced autoimmune diabetes onset following anti‐PD‐L1 treatment 62. This suggests that certain genetic loci control pathways critical for autoimmunity, and it remains to be seen whether abrogating the pathogenic autoimmune response impedes anti‐tumor immune responses. By evaluating pathways that may prevent autoimmune responses independently of anti‐tumor immune responses it may be possible to develop therapies that strategically inhibit irAEs, particularly as a greater arsenal of therapeutic modalities become available 56. However, as these new agents and novel combinations enter the clinic, clinicians must maintain a high level of vigilance to determine patients at high risk and the kinetics by which a new spectrum of irAEs may establish.
Conclusion
CPI‐DM is an emerging form of autoimmune diabetes that shares many but not all traits with forms of previously defined autoimmune diabetes. First, the age of onset is later than T1DM and even latent autoimmune diabetes in adults (LADA), due to the later age of exposure to CPIs. CPI‐DM occurs almost exclusively in the setting of exposure to PD‐1/PD‐L1 inhibitors, either alone or in combination with other immunotherapies, and occurs extremely rarely following CTLA‐4 inhibitor monotherapy. The time to onset of CPI‐DM can be many months after CPI initiation, suggesting that there may be an additional trigger required beyond exposure to PD‐1/PD‐L1 inhibition. It is not yet established what these triggers may be, so it is unclear if they are shared environmental exposures. The acuity of onset is faster than LADA, which is diagnosed at a later age than T1DM, similar to CPI‐DM, but has a slower progression to insulin dependence 63. It is also faster than conventional T1DM, where there is some C‐peptide produced even 2 years after diagnosis 64. The acute course may be most similar to the fulminant T1DM that has been described in Asian individuals 31. The presence of autoantibodies in only half the individuals with CPI‐DM is also quite different than T1DM and LADA in which autoantibodies are almost always present by the time of diagnosis 21, 63 but also different from the aforementioned fulminant T1DM in which autoantibodies are negative in almost all cases 31, suggesting the possibility of mechanistic heterogeneity. The known high‐risk T1DM HLA haplotypes can be used as a basis for analysis of CPI‐DM genetic risk. The majority of individuals who develop CPI‐DM have some genetic susceptibility to T1DM, most commonly harboring the HLA‐DR4 allele 4, 5. Given the similarities to fulminant T1DM seen in Asian populations, patients should also be assessed for HLA types that confer high risk to this subgroup of autoimmune diabetes. With these differences in mind, it is clear that CPI‐DM is a distinct form of autoimmune diabetes that should be studied to more clearly understand islet cell autoimmunity.
Disclosures
Z. Q. and A. Y. have no competing interests. Z. Q. is supported by American Diabetes Association grant no. 1‐19‐PDF‐131. A. Y. is supported by a NHMRC C. J. Martin Fellowship (1143981). M. A. is a stock holder in Medtronic and Merck.
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
Immune checkpoint inhibition: from molecules to clinical application. Clinical and Experimental Immunology 2020, 200: 105‐107.
TIGIT as an emerging immune checkpoint. Clinical and Experimental Immunology 2020, 200: 108‐119.
VISTA: Coming of age as a multi‐lineage immune checkpoint. Clinical and Experimental Immunology 2020, 200: 120‐130.
Mechanisms of checkpoint inhibition‐induced adverse events. Clinical and Experimental Immunology 2020, 200: 141‐154.
Role of inflammasome activation in tumor immunity triggered by immune checkpoint blockers. Clinical and Experimental Immunology 2020, 200: 155‐162.
References
- 1. June CH, Warshauer JT, Bluestone JA. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med 2017; 23:540–7. [DOI] [PubMed] [Google Scholar]
- 2. Barroso‐Sousa R, Barry WT, Garrido‐Castro AC et al Incidence of endocrine dysfunction following the use of different immune checkpoint inhibitor regimens a systematic review and meta‐analysis (Supplement). JAMA Oncol 2018; 4:173–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Postow MA, Sidlow R, Hellmann MD. Immune‐related adverse events associated with immune checkpoint inhibitors. New Engl J Med 2018; 378:158–68. [DOI] [PubMed] [Google Scholar]
- 4. Stamatouli AM, Quandt Z, Perdigoto AL et al Collateral damage: insulin‐dependent diabetes induced with checkpoint inhibitors. Diabetes 2018; 67:1471–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. De Filette J, Jansen Y, Schreuer M et al Incidence of thyroid‐related adverse events in melanoma patients treated with pembrolizumab. J Clin Endocrinol Metab 2016; 101:4431–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tsang VHM, McGrath RT, Clifton‐Bligh RJ et al Checkpoint inhibitor associated autoimmune diabetes (CIADM) is distinct from type 1 diabetes. J Clin Endocrinol Metab 2019; 104:5499–5506. [DOI] [PubMed] [Google Scholar]
- 7. Kotwal A, Haddox C, Block M, Kudva YC. Immune checkpoint inhibitors: an emerging cause of insulin‐dependent diabetes. BMJ Open Diabetes Res Care 2019; 7:e000591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wright JJ, Salem J, Johnson DB et al Increased reporting of immune checkpoint inhibitor–associated diabetes. Diabetes Care 2018; 41:e150–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Perdigoto AL, Quandt Z, Anderson M, Herold KC. Checkpoint inhibitor‐induced insulin‐dependent diabetes: an emerging syndrome. Lancet Diabetes Endocrinol 2019; 7:421–23. [DOI] [PubMed] [Google Scholar]
- 10. Akturk HK, Kahramangil D, Sarwal A, Hoffecker L, Murad MH, Michels AW. Immune checkpoint inhibitor‐induced Type 1 diabetes: a systematic review and meta‐analysis. Diabet Med 2019; 36:1075–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Galligan A, Xu W, Fourlanos S et al Diabetes associated with immune checkpoint inhibition: presentation and management challenges. Diabet Med 2018; 35:1283–90. [DOI] [PubMed] [Google Scholar]
- 12. Clotman K, Janssens K, Specenier P, Weets I, De Block CEM. Programmed cell death‐1 inhibitor‐induced type 1 diabetes mellitus. J Clin Endocrinol Metab 2018; 103:3144–54. [DOI] [PubMed] [Google Scholar]
- 13. Hansen E, Sahasrabudhe D, Sievert L. A case report of insulin‐dependent diabetes as immune‐related toxicity of pembrolizumab: presentation, management and outcome. Cancer Immunol Immunother 2016; 65:765–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Trinh B, Donath MY, Läubli H. Successful treatment of immune checkpoint inhibitor‐induced diabetes with infliximab. Diabetes Care 2019; 42:e153–4. [DOI] [PubMed] [Google Scholar]
- 15. Faje AT, Lawrence D, Flaherty K et al High‐dose glucocorticoids for the treatment of ipilimumab‐induced hypophysitis is associated with reduced survival in patients with melanoma. Cancer 2018; 124:3706–14. [DOI] [PubMed] [Google Scholar]
- 16. Haratani K, Hayashi H, Chiba Y et al Association of immune‐related adverse events with nivolumab efficacy in non‐small cell lung cancer. JAMA Oncol 2018; 4:374–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weber JS, Hodi FS, Wolchok JD et al Safety profile of nivolumab monotherapy: a pooled analysis of patients with advanced melanoma. J Clin Oncol 2017; 35:785–92. [DOI] [PubMed] [Google Scholar]
- 18. Teulings HE, Limpens J, Jansen SN et al Vitiligo‐like depigmentation in patients with stage III‐IV melanoma receiving immunotherapy and its association with survival: a systematic review and meta‐analysis. J Clin Oncol 2015; 33:773–81. [DOI] [PubMed] [Google Scholar]
- 19. Horvat TZ, Adel NG, Dang TO et al Immune‐related adverse events, need for systemic immunosuppression, and effects on survival and time to treatment failure in patients with melanoma treated with ipilimumab at Memorial Sloan Kettering Cancer Center. J Clin Oncol 2015; 33:3193–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bingley PJ, Bonifacio E, Williams AJK, Genovese S, Bottazzo GF, Gale EAM. Prediction of IDDM in the general population: strategies based on combinations of autoantibody markers. Diabetes 1997; 46:1701–10. [DOI] [PubMed] [Google Scholar]
- 21. Bingley PJ. Clinical applications of diabetes antibody testing. J Clin Endocrinol Metab 2010; 95:25–33. [DOI] [PubMed] [Google Scholar]
- 22. Gauci ML, Laly P, Vidal‐Trecan T et al Autoimmune diabetes induced by PD‐1 inhibitor—retrospective analysis and pathogenesis: a case report and literature review. Cancer Immunol Immunother 2017; 66:1399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Godwin JL, Jaggi S, Sirisena I et al Nivolumab‐induced autoimmune diabetes mellitus presenting as diabetic ketoacidosis in a patient with metastatic lung cancer. J Immunother Cancer 2017; 5:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lowe JR, Perry DJ, Salama AKS, Mathews CE, Moss LG, Hanks BA. Genetic risk analysis of a patient with fulminant autoimmune type 1 diabetes mellitus secondary to combination ipilimumab and nivolumab immunotherapy. J Immunother Cancer 2016; 4:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Osorio JC, Ni A, Chaft JE et al Antibody‐mediated thyroid dysfunction during T‐cell checkpoint blockade in patients with non‐small‐cell lung cancer. Ann Oncol 2017; 28:583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hasan Ali O, Berner F, Bomze D et al Human leukocyte antigen variation is associated with adverse events of checkpoint inhibitors. Eur J Cancer 2019; 107:8–14. [DOI] [PubMed] [Google Scholar]
- 27. Cappelli L, Dorak M, Bingham C III, Shah A. Association of HLA‐DRB1 shared epitope alleles with immune checkpoint inhibitor‐induced inflammatory arthritis [abstract]. Arthritis Rheumatol 2018; 70(Suppl 10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Heaney AP, Sumerel B, Rajalingam R, Bergsneider M, Yong WH, Liau LM. HLA markers DQ8 and DR53 are associated with lymphocytic hypophysitis and may aid in differential diagnosis. J Clin Endocrinol Metab 2015; 100:4092–7. [DOI] [PubMed] [Google Scholar]
- 29. Noble JA, Valdes AM. Genetics of the HLA region in the prediction of Type 1 diabetes. Curr Diab Rep 2011; 11:533–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tsutsumi C, Imagawa A, Ikegami H, Makino H, Kobayashi T, Hanafusa T. Class II HLA genotype in fulminant type 1 diabetes: a nationwide survey with reference to glutamic acid decarboxylase antibodies. J Diabetes Invest 2012; 3:62–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hanafusa T, Imagawa A. Fulminant type 1 diabetes: A novel clinical entity requiring special attention by all medical practitioners. Nat Clin Pract Endocrinol Metab 2007; 3:36–45. [DOI] [PubMed] [Google Scholar]
- 32. Stamatouli AM, Quandt Z, Perdigoto AL et al Collateral damage: insulin‐dependent diabetes induced with checkpoint inhibitors. Diabetes 2018; 67:1471–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Winkler C, Krumsiek J, Buettner F et al Feature ranking of type 1 diabetes susceptibility genes improves prediction of type 1 diabetes. Diabetologia 2014; 57:2521–9. [DOI] [PubMed] [Google Scholar]
- 34. Oram RA, Patel K, Hill A et al A type 1 diabetes genetic risk score can aid discrimination between type 1 and type 2 diabetes in young adults. Diabetes Care 2016; 39:337–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lühder F, Höglund P, Allison JP, Benoist C, Mathis D. Cytotoxic T lymphocyte‐associated antigen 4 (CTLA‐4) regulates the unfolding of autoimmune diabetes. J Exp Med 1998; 187:427–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ansari MJI, Salama AD, Chitnis T et al The programmed death‐1 (PD‐1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med 2003; 198:63–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fife BT, Guleria I, Gubbels Bupp M et al Insulin‐induced remission in new‐onset NOD mice is maintained by the PD‐1‐PD‐L1 pathway. J Exp Med 2006; 203:2737–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fife BT, Pauken KE, Eagar TN et al Interactions between PD‐1 and PD‐L1 promote tolerance by blocking the TCR‐induced stop signal. Nat Immunol 2009; 10:1185–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pauken KE, Jenkins MK, Azuma M, Fife BT. PD‐1, but not PD‐L1, expressed by islet‐reactive CD4+ T cells suppresses infiltration of the pancreas during type 1 diabetes. Diabetes 2013; 62:2859–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Martinov T, Spanier JA, Pauken KE, Fife BT. PD‐1 pathway‐mediated regulation of islet‐specific CD4+ T cell subsets in autoimmune diabetes. Immunoendocrinology 2016; 3:pii: e1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Osum KC, Burrack AL, Martinov T et al Interferon‐gamma drives programmed death‐ligand 1 expression on islet β cells to limit T cell function during autoimmune diabetes. Sci Rep 2018; 8:8295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rui J, Deng S, Arazi A, Perdigoto AL, Liu Z, Herold KC. β cells that resist immunological attack develop during progression of autoimmune diabetes in NOD mice. Cell Metab 2017; 25:727–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Colli ML, Hill JLE, Marroquí L et al PDL1 is expressed in the islets of people with type 1 diabetes and is up‐regulated by interferons‐α and‐γ via IRF1 induction. EBioMedicine 2018; 36:367–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yoneda S, Imagawa A, Hosokawa Y et al T‐lymphocyte infiltration to islets in the pancreas of a patient who developed type 1 diabetes after administration of immune checkpoint inhibitors. Diabetes Care 2019; 42:E116–E118. [DOI] [PubMed] [Google Scholar]
- 45. Johnson DB, Balko JM, Compton ML et al Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med 2016; 375:1749–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Coutzac C, Adam J, Soularue E et al Colon immune‐related adverse events: Anti‐CTLA‐4 and anti‐PD‐1 blockade induce distinct immunopathological entities. J Crohns Colitis 2017; 11:1238–46. [DOI] [PubMed] [Google Scholar]
- 47. Falcao CK, Cabral MCS, Mota JM et al Acquired lipodystrophy associated with nivolumab in a patient with advanced renal cell carcinoma. J Clin Endocrinol Metab 2019; 104:3245–8. [DOI] [PubMed] [Google Scholar]
- 48. Jehl A, Cugnet‐Anceau C, Vigouroux C et al Acquired generalized lipodystrophy: a new cause of anti‐PD‐1 immune‐related diabetes. Diabetes Care 2019; 42:2008–10. [DOI] [PubMed] [Google Scholar]
- 49. Herold KC, Bundy BN, Long SA et al An anti‐CD3 antibody, teplizumab, in relatives at risk for type 1 diabetes. N Engl J Med 2019; 381:603–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Long SA, Thorpe J, DeBerg HA et al Partial exhaustion of CD8 T cells and clinical response to teplizumab in new‐onset type 1 diabetes. Sci Immunol 2016; 1:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Matthews JB, Staeva TP, Bernstein PL, Peakman M, Von Herrath M. Developing combination immunotherapies for type 1 diabetes: recommendations from the ITN‐JDRF Type 1 Diabetes Combination Therapy Assessment Group. Clin Exp Immunol 2010; 160:176–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Akirav EM, Lebastchi J, Galvan EM et al Detection of β cell death in diabetes using differentially methylated circulating DNA. Proc Natl Acad Sci USA 2011; 108:19018–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Husseiny MI, Kaye A, Zebadua E, Kandeel F, Ferreri K. Tissue‐specific methylation of human insulin gene and PCR assay for monitoring beta cell death. PLOS One 2014; 9:e94591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Husseiny MI, Kuroda A, Kaye AN, Nair I, Kandeel F, Ferreri K. Development of a quantitative methylation‐specific polymerase chain reaction method for monitoring beta cell death in type 1 diabetes. PLOS One 2012; 7:e47942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rui J, Deng S, Lebastchi J, Clark PL, Usmani‐Brown S, Herold KC. Methylation of insulin DNA in response to proinflammatory cytokines during the progression of autoimmune diabetes in NOD mice. Diabetologia 2016; 59:1021–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Young A, Quandt Z, Bluestone JA. The Balancing act between cancer immunity and autoimmunity in response to immunotherapy. Cancer Immunol Res 2018; 6:1445–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Driver JP, Serreze DV, Chen Y‐G. Mouse models for the study of autoimmune type 1 diabetes: a NOD to similarities and differences to human disease. Semin Immunopathol 2011; 33:67–87. [DOI] [PubMed] [Google Scholar]
- 58. Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol 2005; 23:447–85. [DOI] [PubMed] [Google Scholar]
- 59. Todd JA, Wicker LS. Genetic protection from the inflammatory disease type 1 diabetes in humans and animal models. Immunity 2001; 15:387–95. [DOI] [PubMed] [Google Scholar]
- 60. Ivakine EA, Gulban OM, Mortin‐Toth SM et al Molecular genetic analysis of the Idd4 locus implicates the IFN response in type 1 diabetes susceptibility in nonobese diabetic mice. J Immunol 2006; 176:2976–90. [DOI] [PubMed] [Google Scholar]
- 61. Lyons PA, Hancock WW, Denny P et al The NOD Idd9 genetic interval influences the pathogenicity of insulitis and contains molecular variants of Cd30, Tnfr2, and Cd137. Immunity 2000; 13:107–15. [DOI] [PubMed] [Google Scholar]
- 62. Kochupurakkal NM, Kruger AJ, Tripathi S et al Blockade of the programmed death‐1 (PD1) pathway undermines potent genetic protection from type 1 diabetes. PLOS One 2014; 9:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Buzzetti R, Zampetti S, Maddaloni E. Adult‐onset autoimmune diabetes: current knowledge and implications for management. Nat Rev Endocrinol 2017; 13:674–86. [DOI] [PubMed] [Google Scholar]
- 64. Greenbaum CJ, Beam CA, Boulware D et al Fall in C‐peptide during first 2 years from diagnosis: Evidence of at least two distinct phases from composite type 1 diabetes trialnet data. Diabetes 2012; 61:2066–73. [DOI] [PMC free article] [PubMed] [Google Scholar]