

Summary
The absence of adenosine deaminase (ADA) causes severe combined immune deficiency (SCID), which has been treated with PEGylated bovine‐extracted ADA (ADAGEN). ADAGEN was recently replaced by a PEGylated recombinant bovine ADA, expressed in Escherichia coli (elapegademase, ELA–ADA). Limited information on ELA–ADA is available. ADA enzymatic activity of ELA–ADA and ADAGEN was assessed in vitro at diverse dilutions. ADA activity and immune reconstitution in an ADA–SCID patient treated with ELA–ADA were compared with age‐matched patients previously treated with ADAGEN. ADA activity and thymus reconstitution were evaluated in ADA‐deficient mice following ELA–ADA or ADAGEN administered from 7 days postpartum. In vitro, ADA activity of ELA–ADA and ADAGEN were similar at all dilutions. In an ADA–SCID patient, ELA–ADA treatment led to a marked increase in trough plasma ADA activity, which was 20% higher than in a patient previously treated with ADAGEN. A marked increase in T cell numbers and generation of naive T cells was evident following 3 months of ELA–ADA treatment, while T cell numbers increased following 4 months in 3 patients previously treated with ADAGEN. T cell proliferations stimulation normalized and thymus shadow became evident following ELA–ADA treatment. ADA activity was significantly increased in the blood of ADA‐deficient mice following ELA–ADA compared to ADAGEN, while both treatments improved the mice weights, the weight, number of cells in their thymus and thymocyte subpopulations. ELA–ADA has similar in‐ vitro and possibly better in‐vivo activity than ADAGEN. Future studies will determine whether ELA–ADA results in improved long‐term immune reconstitution.
Keywords: adenosine deaminase deficiency, elapegademase, enzyme replacement therapy, pegademase, severe combined immunodeficiency
1. Elapegademase, a PEGylated recombinant form of bovine adenosine deaminase, has similar in vitro and possibly better in vivo activity than PEGylated bovine adenosine deaminase, ADAGEN.2. Elapegademase treatment led to a marked increase of T cell numbers and generation of naïve T cells within 3 months in an ADA‐deficient patient with severe combined immune deficiency.

Introduction
Adenosine deaminase (ADA) is an important enzyme in the purine degradation and salvage pathway. Complete or near‐complete absence of ADA activity cause accumulation of the enzyme’s substrates adenosine and 2′‐deoxyadenosine, as well as the phosphorylated derivatives, often considered together as total deoxyadenosine nucleotides (dAXP) 1. The perturbed purine metabolism leads to abnormal DNA synthesis and repair, enhanced apoptosis of cells and impaired intracellular signaling 2. Inherited defects in the ADA gene cause severe combined immune deficiency (ADA–SCID) with progressive decline in the number of T, B and natural killer (NK) lymphocytes leading to increased susceptibility to infections 2, 3. In addition, ADA–SCID patients might develop skeletal dysplasia, manifesting primarily in scapular spurring and costo‐chondral cupping 4, developmental delay 5, sensory‐neural hearing defects 6 respiratory distress 7, hepatic failure 8 and neutropenia 9. Allogeneic hematopoietic stem cell transplants (HSCT) from ADA‐proficient donors 10 or hematopoietic stem cell gene therapy (HSC‐GT) using the patients’ own cells that were transduced with a normal ADA gene 11 can correct the immune and systemic abnormalities associated with ADA–SCID.
Appreciation that ADA deficiency is a systemic metabolic disease and that purine nucleosides can cross biological membranes in accordance with concentration gradients led to the development of ADA enzyme replacement therapy (ERT) 12. ADA ERT has been employed to improve patients’ clinical status prior to HSCT and HSC‐GT or as a long‐term therapeutic option for ADA–SCID patients who failed or are not candidates for definitive treatments 13. Since the 1980s, ADA ERT has relied primarily on ADA extracted from calf intestines that is covalently conjugated to 11–17 strands of monomethoxy‐polyethylene glycol (PEG) in a process called PEGylation 14. PEG itself is considered non‐toxic and non‐immunogenic, although recent studies suggest that some individuals might develop antibodies to PEG 15. PEGylation via a linker succinimidyl succinate to the lysine residues in bovine ADA conferred several beneficial properties 16. Animal studies showed that the circulatory half‐life of pegylated ADA (pegademase, ADAGEN®) was prolonged from minutes to 28 h, as clearance from the circulation was inhibited 14. In ADA‐deficient patients, ADAGEN half‐life varied from 3 to more than 6 days 12. ADAGEN was found to have reduced immunogenicity, which further helped to extend its circulating life 17. ADAGEN was shown to rapidly correct the metabolic, immune and systemic abnormalities in ADA‐deficient patients 13, 18, 19, and recent guidelines recommend administering ADAGEN to all newly diagnosed ADA–SCID 20. In mice lacking ADA [ADA‐knock‐out (KO) mice], which recapitulate many of the features observed in the human condition including profound T and B cell deficiency, ADAGEN can correct the metabolic, immune and non‐immune abnormalities when treatment is initiated at an early age 21, 22.
To overcome the reliance on bovine products and the associated risk of infections, such as bovine spongiform encephalopathy, to ensure consistent quality and supply and to possibly extend the shelf life of the enzyme, a recombinant form of bovine ADA (rADA) was developed. The protein was modified (Cys74Ser), expressed in Escherichia coli and conjugated to PEG using a different linker, succinimidyl carbonate 23. The new PEGylated rADA, labeled initially as EZN‐2279 and subsequently as elapegademase (Revcovi™, abbreviated ELA–ADA) was recently approved for use in the United States and Japan for ADA‐deficient patients. In parallel, ADAGEN was removed from the North American markets. In the application to the US Food and Drug Administration (FDA), brief descriptions of ELA–ADA treatment in 10 ADA‐deficient patients was provided, including a comparison in adult patients of circulating ADA activity levels achieved with a comparable dose of ADAGEN 24. However, only two patients were ADA–SCID infants. ELA–ADA treatment was initiated in the two ADA–SCID patients at 4·4 months and 3·4 months of age, and continued for 148 and 107 days, respectively, without significant improvement in T cell numbers. One of the patients succumbed to respiratory failure, possibly related to pre‐existing cytomegalovirus infection, while clinical and laboratory data on the other patient were not provided 24. No other reports on the use of ELA–ADA could be found. Thus, only scarce information is available on the effects of ELA–ADA in ADA–SCID. Moreover, there is no direct comparison of the two PEGylated ADA products, thereby limiting clinicians and scientists from using the extensive experience gained with ADAGEN.
Here we compare ELA–ADA and ADAGEN activity in‐vitro and the effects of ELA–ADA in a newly diagnosed ADA–SCID infant with that of ADAGEN used in three previous patients who received ERT from an early age. In addition, we assess the enzyme activity of both products in ADA‐KO mice, and their effects on thymus reconstitution in the mice.
Methods and results
Activity of 4 µl ELA–ADA and ADAGEN, serially diluted in phosphate‐buffered saline (PBS), was determined by the percentage of 1 μmol [8‐14C]adenosine conversion to inosine for 30 min in three separate non‐expired vials of ELA–ADA and ADAGEN that were stored and handled similarly. ADA assays (50 microl) contained 0·2 M potassium phosphate (pH 7.0) and 10 µM [8‐14C]adenosine (Moravek Biochemicals, Brea, CA, USA). No significant differences (P < 0·05) were noted between ELA–ADA and ADAGEN (Fig. 1).
Figure 1.
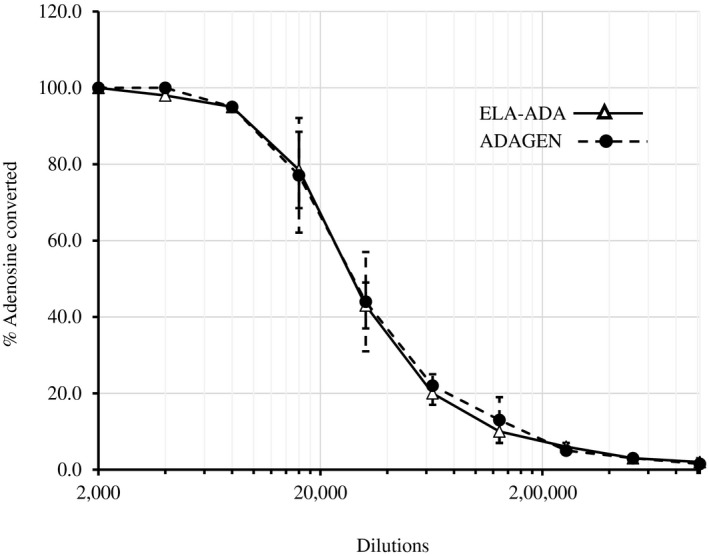
Elapegademase and pegademase enzyme activity in vitro. Activity of 4 µl elapegademase (ELA–ADA) and pegademase (ADAGEN) serially diluted in phosphate‐buffered saline (PBS) was determined by the percentage of 1 µmol [8‐14C]adenosine converted during 30 min. Results are the mean ± standard deviation of three independent experiments using three vials of each product.
A female patient, who was born at term, with a birth weight of 3260 g, to non‐consanguineous parents of Somalian descent, presented at 4 months of age with pyomyositis of the left gastrocnemius muscle, failure to thrive (weight and length both below the 3% for age), Pneumocystis jirovecii pneumonia and developmental delay. The patient was born in a region that had not yet implemented newborn screening for SCID. Liver enzymes and neutrophil numbers were normal (data not shown). Chest X‐ray revealed absence of a thymus, without clear costo‐chondral or scapular abnormalities. Immunoglobulin (Ig)G level was 2·37 g/l (normal range for age = 2·00–7·00 g/l), possibly still representing transplacental passage of maternal IgG, while IgA, IgM and IgE were all below the level of detection. There was marked lymphopenia (0·2 × 109/l), absent CD19+ B cells and only 0·07 × 109/L CD3–16+56+ NK cells, 0·02 × 109/l CD3+CD4+ and 0·01 × 109/L CD3+CD8+ T cells. ADA activity in the erythrocytes of the patient, who had not received blood transfusions, was below the level of detection at < 0·2 nmol/min/mg protein (normal range = 0·48–1·32 nmol/min/mg protein). Sequencing of the ADA gene revealed a homozygous Q3X mutation, previously reported as damaging 25, confirming the diagnosis of ADA–SCID. The patient, who did not have any human leukocyte antigen (HLA)‐matched siblings, was treated with trimethoprim–sulfamethoxazole and intravenous immunoglobulin prophylaxis. While definitive treatment options were being explored, the patient received ELA–ADA 0·2 mg/kg twice a week from 5 months of age (for a total weekly dose of 0·4 mg/kg) in accordance with the manufacturer’s guidelines 26. The patient’s clinical status improved rapidly, her weight percentile increased and she was catching up on her development. Throughout the treatment period, there was no evidence for autoimmune hemolytic anemia or thrombocytopenia, previously reported as potential adverse effects of ADA ERT 20. There were no other hematological, hepatic, skeletal or pulmonary complications that have been associated with ADA deficiency 20. After 6 months of ELA–ADA treatment, the patient’s plasma ADA activity was 143–146 mmol/h/l, and erythrocyte dAXP were undetectable (normal range = < 0·002 mmol/l). These parameters were measured in samples sent to the laboratory of MS Hershfield at Duke University, as previously described 12). The ADA activity following ELA–ADA was 15–20% higher than that in another previously reported ADA–SCID 27, also carrying the Q3X mutation, who had received 30 units/kg body weight ADAGEN, where ADA activity was only 121–126 mmol/h/l, albeit sufficient to normalize erythrocyte dAXP. Naive B and T cells appeared rapidly following ELA–ADA (Table 1). Recent thymic emigrants, characterized by expression of CD3+CD4+CD45Ra+CD31+ 28, which were practically undetectable upon diagnosis, increased to 0·8 × 109/l after 3 months of ELA–ADA (normal range for age = 0·8–6·2 × 109/l) and to 1·0 × 109/l after 6 months of treatment. Chest X‐ray, performed 6 months after initiating ERT demonstrated appearance of a thymus silhouette (Fig. 2). ELA–ADA led to a rapid increase in the number of CD19+ cells (Fig. 3a) and CD3–CD16+CD56+ cells (Fig. 3b) followed by increase in CD3+CD4+ and CD3+CD8+ T cells (Fig. 3c and d, respectively). The immune reconstitution with ELA–ADA was similar, and the recovery of T cells possibly faster and to higher values, in comparison to three other age‐matched ADA–SCID patients who previously received 30 units/kg body weight ADAGEN twice a week (Fig. 3). Patients’ data were compiled prospectively and retrospectively in accordance to institutional Research Ethics Boards approved protocols.
Table 1.
Naive T and B lymphocytes in an ADA–SCID patient treated with elapegademase
| Time after elapegademase initiation | 0 month | 1 months | 2 months | 3 months | 6 months |
|---|---|---|---|---|---|
| CD3+CD4+CD45+CD27+ | 0·002 | 0·011 | 0·078 | 0·686 | 0·996 |
| CD3+CD8+CD45+CD27+ | 0·002 | 0 | 0·009 | 0·259 | 0·396 |
| CD19+IgD+CD27− | 0 | 1·999 | 0·74 | 0·52 | 0·604 |
Absolute counts (×109/l). ADA–SCID = adenosine deaminase–severe combined immune deficiency; Ig = immunoglobulin. Bold type indicates results that are abnormal for age.
Figure 2.
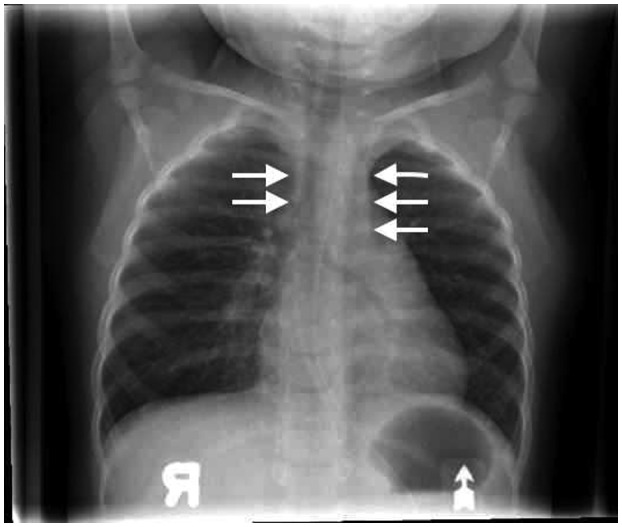
Thymus recovery in an adenosine deaminase–severe combined immune deficiency (ADA–SCID) patient following elapegademase treatment. Thymus silhouette (arrows) in an ADA–SCID patient following 6 months of twice‐weekly elapegademase enzyme replacement therapy (ERT).
Figure 3.
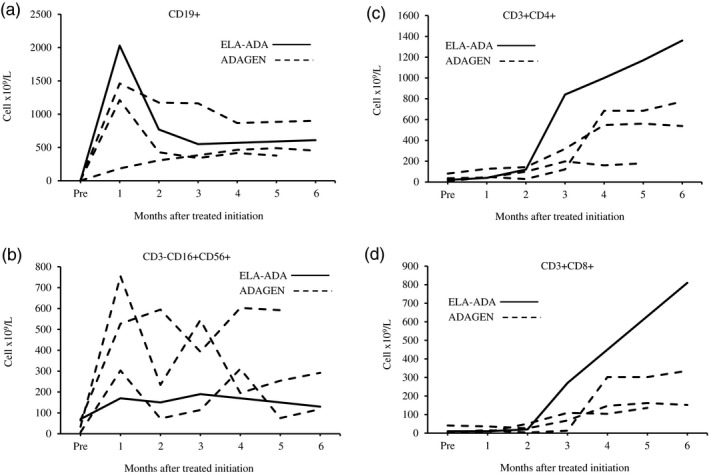
Elapegademase and pegademase effects on immune reconstitution in adenosine deaminase–severe combined immune deficiency (ADA–SCID) patients. The number of lymphocyte subpopulations, assessed by flow cytometry at the indicated time [months after initiation of enzyme replacement therapy (ERT)] in a patient receiving elapegademase (ELA–ADA, solid line) and three patients receiving pegademase (ADAGEN, dashed lines). (a) CD19+ B cells; (b) CD3−16+56+ natural killer (NK) cells; (c) CD3+CD4+ T cells; (d) CD3+CD8+ T cells.
ADA‐KO mice [FB, 129‐Adatm1Mw‐TgN (PLADA)], which lack ADA activity in their blood, were treated with 0·25 ml/kg body weight ADAGEN (62·5 units/kg) or ELA–ADA (0·4 mg/kg), both from Leadiant Bioscience Inc. (Gaithersburg, MD, USA). ADAGEN and ELA–ADA were administered by intraperitoneal injections at 7, 10 and 13 days post‐partum (pp). Activity of the products was determined prior to and after completion of the experiments to ensure the enzyme potency has not changed. ADA activity in the blood of the treated mice was measured at the same time during the day prior to 1, 2 and 3 days after the first injection, as well as prior to the third injection and daily thereafter for 5 days (13–18 days pp). ADA activity was also measured in ADA+/– and ADA+/+ mice to establish normal ranges for the mouse strain (16–32 mmol/h/l). All animal procedures were approved by the Institute’s Animal Care Committee and performed in accordance with the Canadian Council for Animal Care guidelines. ADA activity following ELA–ADA and ADAGEN (Fig. 4a) was similar during the first and second days after the injections; however, by 3 days, activity was significantly (P = 0·02) higher in mice treated with ELA–ADA (15·3 ± 6·1 mmol/h/l) than in mice treated with ADAGEN (6·6 ± 4·2 mmol/h/l). Trough ADA activity was significantly higher (P < 0·01) at 13 days pp in ADA‐KO mice treated with ELA–ADA than with ADAGEN, which became more pronounced (P < 0·001) at 2, 3 and 4 days after the third injection (Fig. 4b). Consequently, ADA activity in ADA‐KO mice treated with ELA–ADA remained above or within normal range for more than 4 days after injection, in contrast to fewer than 3 days after ADAGEN treatment. Body and thymus weights, the number of thymocytes and flow cytometry analysis of thymocyte subpopulation were assessed at 17–19 days pp in treated and non‐treated ADA‐KO and normal littermate mice, as previously reported 22. Both ELA–ADA and ADAGEN treatments significantly increased thymus weights and the number of thymocytes (Fig. 5a and b, respectively) and the percentage of CD4+CD8+ (Fig. 5c), as well as the body weights (Fig. 5d). Similarly, higher blood ADA activity and markedly improved thymus values were observed with higher doses of ELA–ADA (1·6 and 3·2 mg/kg) and ADAGEN (250 and 500 units/kg) administered at 7, 10 and 13 days pp (data not shown).
Figure 4.
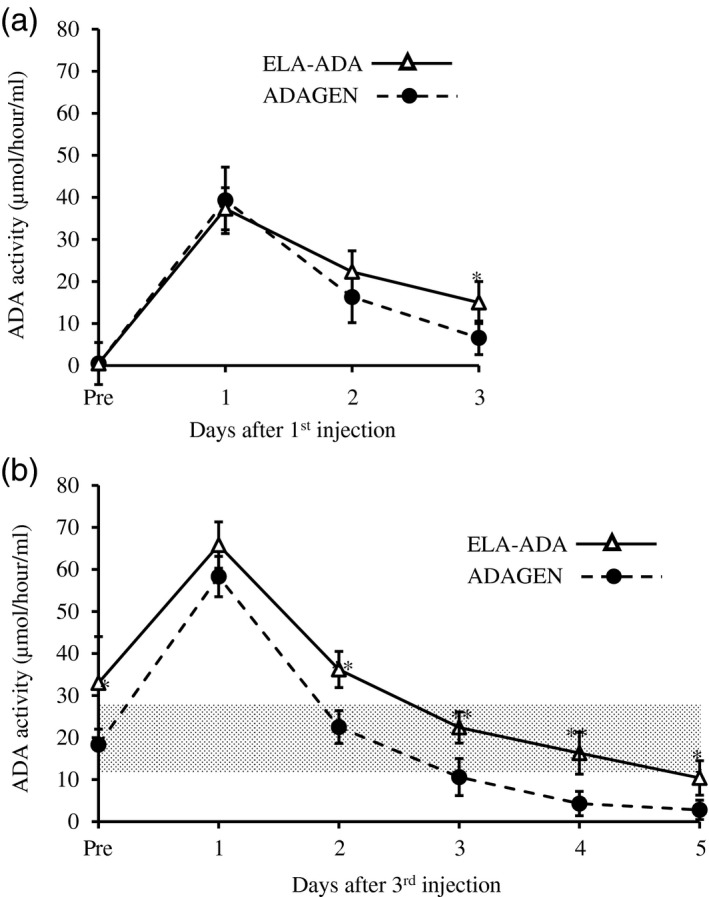
Elapegademase and pegademase enzyme activity in adenosine deaminase (ADA)‐deficient mice. (a) ADA‐deficient [knock‐out (KO)] mice received 0·25 ml//kg body weight pegademase (ADAGEN, 62·5 units/kg) or elapegademase (ELA–ADA, 0·4 mg/kg) at 7 days postpartum. ADA activity (conversion of [8‐14C]adenosine to hypoxanthine) in the blood was determined at the indicated hours after treatment; n = 6 mice in each group from two independent experiments. Results are the mean ± standard deviation. *P = 0·02. (b) ADA‐deficient (KO) mice received 0·25 ml//kg body weight pegademase (ADAGEN, 62·5 units/kg) or elapegademase (ELA–ADA, 0·4 mg/kg) at 7, 10 and 13 days postpartum. ADA activity (conversion of [8‐14C]adenosine to hypoxanthine) in the blood was measured at the indicated hours after the third injection. Hatched area represents ADA activity in ADA+/− and ADA+/+ mice littermates, n = 6–9 mice in each group from two independent experiments. Results are the mean ± standard deviation. *P < 0·01, **P < 0·001 between ADA‐KO mice treated with ELA–ADA or ADAGEN.
Figure 5.

Elapegademase and pegademase effects on the weight and thymus of adenosine deaminase (ADA)‐deficient mice. ADA‐deficient [knock‐out (KO)] mice received 0·25 ml//kg body weight pegademase (ADAGEN, 62·5 units/kg) or elapegademase (ELA–ADA, 0·4 mg/kg) at 7, 10 and 13 days postpartum. Treated and non‐treated ADA‐KO as well as normal littermate mice were assessed at 17–19 days postpartum. Results are the mean ± standard deviation with n = 3–6 in all groups. (a) Thymus weights; (b) number of thymocytes; (c) thymocyte subpopulation CD4−CD8−, CD4+CD8+, CD4+CD8− and CD4−CD8+ assessed by flow cytometry; (d) body weights. *P < 0·01, ** P < 0·001 compared to untreated ADA‐KO mice.
Discussion
Extensive experience has been gained with the use of ADAGEN, with more than 200–250 ADA‐deficient patients estimated to have been treated with ADAGEN in recent decades 20. Hence, the limited data on ELA–ADA, particularly regarding its use in the medically fragile ADA–SCID infants, has raised concerns by patients, families, physicians and regulatory authorities. Reassuringly, we find that replacing the native cysteine present in ADAGEN with serine at position 74 of the bovine ADA to create ELA–ADA has no significant effect on the enzyme’s ability to convert adenosine in vitro at a wide dilution range. This finding is expected, as it is likely that extensive screening of various amino acid substitutions throughout the bovine ADA was performed prior to embarking on the commercial production of ELA–ADA.
The patient detailed here presented with classical features of SCID, which will become less common with the expansion of newborn screening programs for SCID and earlier identification of affected infants. The T–B–NK and the patient’s Somalian descent, a population with particularly frequent mutations in the ADA gene 29, led to the rapid enzymatic and genetic diagnosis of ADA–SCID. As recommended in recent consensus guidelines 20, ADA ERT was initiated while definitive treatment with allogeneic HSCT or autologous GT were explored.
The dose of ELA–ADA, 0·2 mg/kg twice a week, was based on the manufacturer’s recommendation for patients who had not received prior ERT 24. The rapid clinical improvement observed in our patient, and the absence of hepatic, pulmonary, skeletal, neurological and hematological complications during treatment that typically occur in inadequately detoxified ADA–SCID, suggests that ELA–ADA dosing was sufficient. Indeed, the regimen used in our patient led to a trough ADA plasma level of more than 140 mmol/h/l, markedly higher than the 30 mmol/h/l, recommended when treating ADA‐deficient patients 24. Whether such doses are required to achieve maximal clinical improvement is not clear, as the ADA activity in our patient was 10–20 times higher than the 6–11 mmol/h/l ADA activity in the plasma of healthy individuals. Moreover, the effective dose of ELA–ADA needed for ADA–SCID patients diagnosed through the newborn screening programs, prior to massive accumulation of toxic purine metabolites and before the development of organ damage occurs, might be even lower. Other important factors to consider are concomitant infections, kidney function, weight changes and compliance, which also contribute to the individual variability seen in ADA activity and purine metabolites in ADA–SCID patients. Hence, it is important to monitor ADA activity and dAXP frequently during ELA–ADA treatment. Interestingly, ADA activity following ELA–ADA was higher than those previously measured by us in another ADA–SCID with the same mutation who received equivalent doses of ADAGEN, suggesting a longer in‐vivo half‐life of ELA–ADA in ADA–SCID. An extended biological half‐life was previously reported in six older (age = 16–37 years) ADA‐deficient patients mentioned in the manufacturer’s application for ELA–ADA approval 24. In these patients, half‐life of ELA–ADA was 6–21 days, whereas that of ADAGEN was 3 to > 6 days. Whether such differences also exist in ADA–SCID will need to be verified by prospectively studying larger number of patients.
Lymphoid reconstitution following ELA–ADA in our patient was rapid, and followed the pattern we and other groups 18 have observed in ADA–SCID who received ADAGEN. B cells increased significantly in the initial month of therapy followed by decline in the second month, while increases in CD4 and CD8 T cells were observed after 2–3 months of ELA–ADA. Harbingers of the B and T cell reconstitution were the emergence of naive cells of the relevant lineages and normalization of the number of recent thymic emigrants. The thymus shadow also appeared, although infections and other stress factors might have contributed to the initial inability to detect the thymus by chest X‐ray, which is not the best modality to visualize the presence and size of the thymus. Measurement of T cell receptor excision circles could have assisted in diagnosing and following T cell deficiency and recovery; however, this assay was not available for the patient reported here. Remarkably, the number of CD4 and CD8 lymphocytes continued to increase until the last measurement at 6 months after ELA–ADA was initiated, reaching absolute numbers that were higher than in our three previous patients treated with ADAGEN. The number of CD3+ T cells in our patient (2·17 × 109/l) was also higher than the number of such cells in many other ADA–SCID patients reported previously 30, 31, 32, 33, 34. Whether the differences in T cell recovery reflect individual variation or are caused by better immune reconstitution with ELA–ADA will need to be determined through larger prospective studies.
To further explore the relationship between ELA–ADA and ADAGEN, we took advantage of the ADA‐KO mice. The mouse model was generated a decade after the approval of ADAGEN 35, and has since been instrumental in evaluating the different treatment options for ADA deficiency, including HSCT, HSC‐GT and ERT 36, 37, 38. In contrast to previous studies that explored ‘low’ and ‘high’ as well as ‘early’ and ‘late’ ERT 21, 22, we examined regimens reminiscent of that used in ADA–SCID patients, initiating treatment at 7 days pp. Importantly, we found that ELA–ADA circulating half‐life, estimated at 24–28 h, was consistently longer than the 16–20 h estimated for ADAGEN. The difference in circulating half‐life, although not evident in the initial day after injection, eventually led to higher trough ADA levels in mice treated with ELA–ADA, similar to our observation in the ADA–SCID patient. Moreover, in accordance with data provided by the manufacturer in the application for approval 39, following a single injection in rats the circulating half‐life of ELA–ADA was 4–15 h longer than that of ADAGEN. Hence, it appears that the substitution of cysteine by serine and replacing the linker between ADA and PEG from succinimidyl succinate to succinimidyl carbonate, which reduced spontaneous de‐PEGylation, resulted in a markedly enhanced circulatory half‐life. Such dramatic effects have been reported previously for other PEGylated molecules such as uricase, asparginase and others 16.
Despite the extended circulatory life of ELA–ADA and the longer periods that ADA activity was maintained above normal range in comparison to ADAGEN, both treatments had similar effects on the T cell immunity and general status of ADA‐KO mice. Within 10 days of starting ELA–ADA and ADAGEN ERT, the thymus weights, thymocyte numbers and the percentages of CD4+CD8+ thymocytes improved, reflecting the marked sensitivity of rapidly proliferating thymocytes to toxic purine metabolites, as shown previously 40. The body weight of ADA‐KO mice normalized, probably indicating reversal of the lung‐related hypoxia associated with ADA deficiency. Interestingly, the data submitted with the application for approval of ELA–ADA 39 ADA‐KO mice who received a single injection of the lowest dosage of ELA–ADA showed a better life span than mice treated with ADAGEN. The differences between the results might be attributed to the late (18 days) and lower dose (estimated at less than 0·05 unit/kg) used by the manufacturer, compared to the earlier (7 days) and higher (0·4 mg/kg) dosing that we used. Most ADA–SCID are expected to be identified and treated early in life, therefore we postulated that the regimen used here could reflect clinical scenarios more clearly.
A major concern with ADAGEN treatment has been the decline in immunity with prolonged use and the lack of improvement observed in up to 20% of patients who received ADAGEN 30, 41. This phenomenon has been attributed in part to the development of antibodies to the bovine ADA 17, which were not assessed in the current study. The enhanced PEGylation and potential epitope changes of the rADA have been proposed to also reduce the immune response to ELA–ADA, as has been observed with other modified proteins such as purine nucleoside phosphorylase 42. To investigate these putative added advantages of ELA–ADA, prospective studies with a larger number of patients followed over much longer periods of time are needed.
In conclusion, we demonstrate here that ELA–ADA has similar in‐vitro and possibly better in‐vivo activity compared to ADAGEN, supporting the use of ELA–ADA in patients with ADA–SCID until definitive treatment.
Disclosures
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author contributions
L. M. F. and E. G. conceptualized this study and designed the experiments. L. M. F. and E. G. managed the patients. All authors contributed to writing and reviewing the manuscript and data visualization. W. M., R. L. and M. L. P. conducted the experiments.
Acknowledgements
This work was supported in part by The Campbell Chair for Immunology Research (E. G.). The authors thank the patients and their families as well as the medical teams for their kind cooperation. The authors also thank Dr Michael S. Hershfield for his useful comments and contributions to the development of this manuscript.
References
- 1. Grunebaum E, Cohen A, Roifman CM. Recent advances in understanding and managing adenosine deaminase and purine nucleoside phosphorylase deficiencies. Curr Opin Allergy Clin Immunol 2013; 13:630–8. [DOI] [PubMed] [Google Scholar]
- 2. Hershfield MS. New insights into adenosine‐receptor‐mediated immunosuppression and the role of adenosine in causing the immunodeficiency associated with adenosine deaminase deficiency. Eur J Immunol 2005; 35:25–30. [DOI] [PubMed] [Google Scholar]
- 3. Giblett ER, Anderson JE, Cohen F, Pollara B, Meuwissen HJ. Adenosine‐deaminase deficiency in two patients with severely impaired cellular immunity. Lancet 1972; 2:1067–9. [DOI] [PubMed] [Google Scholar]
- 4. Manson D, Diamond L, Oudjhane K, Hussain FB, Roifman C, Grunebaum E. Characteristic scapular and rib changes on chest radiographs of children with ADA‐deficiency SCIDS in the first year of life. Pediatr Radiol 2013; 43:589–92. [DOI] [PubMed] [Google Scholar]
- 5. Whitmore KV, Gaspar HB. Adenosine deaminase deficiency – more than just an immunodeficiency. Front Immunol 2016; 16:314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Albuquerque W, Gaspar HB. Bilateral sensorineural deafness in adenosine deaminase‐deficient severe combined immunodeficiency. J Pediatr 2004; 144:278–80. [DOI] [PubMed] [Google Scholar]
- 7. Grunebaum E, Cutz E, Roifman CM. Pulmonary alveolar proteinosis in patients with adenosine deaminase deficiency. J Allergy Clin Immunol 2012; 129:1588–93. [DOI] [PubMed] [Google Scholar]
- 8. Bollinger ME, Arredondo‐Vega FX, Santisteban I, Schwarz K, Hershfield MS, Lederman HM. Brief report: hepatic dysfunction as a complication of adenosine deaminase deficiency. N Engl J Med 1996; 334:1367–71. [DOI] [PubMed] [Google Scholar]
- 9. Kim VH, Pham‐Huy A, Grunebaum E. Neutropenia among patients with adenosine deaminase deficiency. J Allergy Clin Immunol 2019; 143:403–5. [DOI] [PubMed] [Google Scholar]
- 10. Hassan A, Booth C, Brightwell A et al Outcome of hematopoietic stem cell transplantation for adenosine deaminase‐deficient severe combined immunodeficiency. Blood 2012; 120:3615–24. [DOI] [PubMed] [Google Scholar]
- 11. Kohn DB, Gaspar HB. How we manage adenosine deaminase‐deficient severe combined immune deficiency (ADA SCID). J Clin Immunol 2017; 37:351–6. [DOI] [PubMed] [Google Scholar]
- 12. Hershfield MS, Buckley RH, Greenberg ML et al Treatment of adenosine deaminase deficiency with polyethylene glycol‐modified adenosine deaminase. N Engl J Med 1987; 316:589–96. [DOI] [PubMed] [Google Scholar]
- 13. Gaspar HB, Aiuti A, Porta F, Candotti F, Hershfield MS, Notarangelo LD. How I treat ADA deficiency. Blood 2009; 114:3524–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Davis S, Abuchowski A, Park YK, Davis FF. Alteration of the circulating life and antigenic properties of bovine adenosine deaminase in mice by attachment of polyethylene glycol. Clin Exp Immunol 1981; 46:649–52. [PMC free article] [PubMed] [Google Scholar]
- 15. Ganson NJ, Povsic TJ, Sullenger BA et al Pre‐existing anti‐polyethylene glycol antibody linked to first‐exposure allergic reactions to pegnivacogin, a PEGylated RNA aptamer. J Allergy Clin Immunol 2016; 137:1610–1613.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Turecek PL, Bossard MJ, Schoetens F, Ivens IA. PEGylation of biopharmaceuticals: a review of chemistry and nonclinical safety information of approved drugs. J Pharm Sci 2016; 105:460–75. [DOI] [PubMed] [Google Scholar]
- 17. Chaffee S, Mary A, Stiehm ER, Girault D, Fischer A, Hershfield MS. IgG antibody response to polyethylene glycol‐modified adenosine deaminase in patients with adenosine deaminase deficiency. J Clin Invest 1992; 89:1643–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weinberg K, Hershfield MS, Bastian J, Kohn D, Sender L, Parkman R, Lenarsky C. T lymphocyte ontogeny in adenosine deaminase‐deficient severe combined immune deficiency after treatment with polyethylene glycol‐modified adenosine deaminase. J Clin Invest 1993; 92:596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Scott O, Kim VH, Reid B et al Long‐term outcome of adenosine deaminase‐deficient patients – a single‐center experience. J Clin Immunol 2017; 37:582–91. [DOI] [PubMed] [Google Scholar]
- 20. Kohn DB, Hershfield MS, Puck JM et al Consensus approach for the management of severe combined immune deficiency caused by adenosine deaminase deficiency. J Allergy Clin Immunol 2019; 143:852–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Blackburn MR, Aldrich M, Volmer JB et al. The use of enzyme therapy to regulate the metabolic and phenotypic consequences of adenosine deaminase deficiency in mice. Differential impact on pulmonary and immunologic abnormalities. J Biol Chem 2000; 275:32114–21. [DOI] [PubMed] [Google Scholar]
- 22. Xu X, Negandhi J, Min W et al Early enzyme replacement therapy improves hearing and immune defects in adenosine deaminase deficient‐mice. Front Immunol 2019; 13:416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Food and Drug Administration . Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761092Orig1s000ChemR.pdf (accessed 15 September 2019).
- 24. Food and Drug Administration . Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761092s000lbl.pdf (accessed 15 September 2019).
- 25. Santisteban I, Arredondo‐Vega FX, Kelly S et al Three new adenosine deaminase mutations that define a splicing enhancer and cause severe and partial phenotypes: implications for evolution of a CpG hotspot and expression of a transduced ADA cDNA. Hum Mol Genet 1995; 4:2081–7. [DOI] [PubMed] [Google Scholar]
- 26. Revcovi™ Fact Sheet . Available at: https://revcovi.com/wp-content/uploads/2019/02/Revcovi-Fact-Sheet-Jan-31-2019.pdf (accessed 15 September 2019).
- 27. Husain M, Grunebaum E, Naqvi A et al Burkitt’s lymphoma in a patient with adenosine deaminase deficiency‐severe combined immunodeficiency treated with polyethylene glycol‐adenosine deaminase. J Pediatr 2007; 151:93–5. [DOI] [PubMed] [Google Scholar]
- 28. Ravkov E, Slev P, Heikal N. Thymic output: assessment of CD4(+) recent thymic emigrants and T‐Cell receptor excision circles in infants. Cytometry B Clin Cytom 2017; 92:249–57. [DOI] [PubMed] [Google Scholar]
- 29. Sanchez JJ, Monaghan G, Børsting C, Norbury G, Morling N, Gaspar HB. Carrier frequency of a nonsense mutation in the adenosine deaminase (ADA) gene implies a high incidence of ADA‐deficient severe combined immunodeficiency (SCID) in Somalia and a single, common haplotype indicates common ancestry. Ann Hum Genet 2007; 71:336–47. [DOI] [PubMed] [Google Scholar]
- 30. Chan B, Wara D, Bastian J et al Long‐term efficacy of enzyme replacement therapy for adenosine deaminase (ADA)‐deficient severe combined immunodeficiency (SCID). Clin Immunol 2005; 117:133–43. [DOI] [PubMed] [Google Scholar]
- 31. Kaufman DA, Hershfield MS, Bocchini JA, Moissidis IJ, Jeroudi M, Bahna SL. Cerebral lymphoma in an adenosine deaminase‐deficient patient with severe combined immunodeficiency receiving polyethylene glycol‐conjugated adenosine deaminase. Pediatrics 2005; 116:e876–e879. [DOI] [PubMed] [Google Scholar]
- 32. Serana F, Sottini A, Chiarini M et al The different extent of B and T cell immune reconstitution after hematopoietic stem cell transplantation and enzyme replacement therapies in SCID patients with adenosine deaminase deficiency. J Immunol 2010; 185:7713–22. [DOI] [PubMed] [Google Scholar]
- 33. Nakazawa Y, Kawai T, Uchiyama T et al Effects of enzyme replacement therapy on immune function in ADA deficiency patient. Clin Immunol 2015; 161:391–3. [DOI] [PubMed] [Google Scholar]
- 34. Tartibi HM, Hershfield MS, Bahna SL. A 24‐year enzyme replacement therapy in an adenosine‐deaminase‐deficient patient. Pediatrics 2016; 137:e20152169. [DOI] [PubMed] [Google Scholar]
- 35. Blackburn MR, Datta SK, Kellems RE. Adenosine deaminase‐deficient mice generated using a two‐stage genetic engineering strategy exhibit a combined immunodeficiency. J Biol Chem 1998; 273:5093–100. [DOI] [PubMed] [Google Scholar]
- 36. Carbonaro DA, Jin X, Cotoi D et al Neonatal bone marrow transplantation of ADA‐deficient SCID mice results in immunologic reconstitution despite low levels of engraftment and an absence of selective donor T lymphoid expansion. Blood 2008; 111:5745–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carbonaro DA, Jin X, Wang X et al Gene therapy/bone marrow transplantation in ADA‐deficient mice: roles of enzyme‐replacement therapy and cytoreduction. Blood 2012; 120:3677–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Carbonaro DA, Zhang L, Jin X et al Preclinical demonstration of lentiviral vector‐mediated correction of immunological and metabolic abnormalities in models of adenosine deaminase deficiency. Mol Ther 2014; 22:607–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Food and Drug Administration . Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761092Orig1s000MicroR.pdf.
- 40. Apasov SG, Blackburn MR, Kellems RE, Smith PT, Sitkovsky MV. Adenosine deaminase deficiency increases thymic apoptosis and causes defective T cell receptor signaling. J Clin Invest 2001; 108:131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Booth C, Gaspar HB. Pegademase bovine (PEG‐ADA) for the treatment of infants and children with severe combined immunodeficiency (SCID). Biologics 2009; 3:349–58. [PMC free article] [PubMed] [Google Scholar]
- 42. Hershfield MS, Chaffee S, Koro‐Johnson L, Mary A, Smith AA, Short SA. Use of site‐directed mutagenesis to enhance the epitope‐shielding effect of covalent modification of proteins with polyethylene glycol. Proc Natl Acad Sci USA 1991; 88:7185–9. [DOI] [PMC free article] [PubMed] [Google Scholar]