

Summary
Immune checkpoint blockers improve the overall survival of a limited number of patients among different cancers. Identifying pathways that influence the immunological and clinical response to treatment is critical to improve the therapeutic efficacy and predict clinical responses. Recently, a key role has been assigned to innate immune mechanisms in checkpoint blockade‐driven anti‐tumor responses. However, inflammatory pathways can both improve and impair anti‐tumor immunity. In this review, we discuss how different inflammatory pathways, particularly inflammasome activation, can influence the clinical outcome of immune checkpoint blockers. Inflammasome activation may reinforce anti‐tumor immunity by boosting CD8+ T cell priming as well as by enhancing T helper type 17 (Th17) responses. In particular, we focus on the modulation of the cation channel transmembrane protein 176B (TMEM176B) and the ectonucleotidase CD39 as potential targets to unleash inflammasome activation leading to reinforced anti‐tumor immunity and improved efficacy of immune checkpoint blockers. Future studies should be aimed at investigating the mechanisms and cell subsets involved in inflammasome‐driven anti‐tumor responses.
Keywords: Cancer, checkpoint blockade, immunotherapy, inflammasome
Innate immunity controls anti‐tumoral responses triggered by immune checkpoint blockers (ICB). Here, we discuss recent findings suggesting a role for inflammasome activation in ICB‐based cancer immunotherapy.
Inflammation and immune checkpoint blockers
Immunotherapy based on monoclonal antibodies has revolutionized oncology therapy by unleashing the breaks on T cells through the blockade of cytotoxic T lymphocyte antigen‐4 (CTLA‐4) and programmed cell death‐1/PD‐ligand‐1 (PD‐1/PD‐L1) pathways [1, 2]. However, a minority of patients experience clinical benefit when receiving these treatments. Thus, understanding primary and secondary resistance mechanisms is urgently needed to improve the clinical efficacy of immune checkpoint blockers (ICB) [3]. However, despite considerable research efforts, the precise molecular and cellular pathways that mediate tumor immunity elicited by ICBs remain incompletely understood [4]. Moreover, an in‐depth investigation of the fundamental mechanisms triggered by ICB will be critical for developing predictive biomarkers of response to therapy. In this context, inflammation is certainly a key player in shaping ICB‐triggered anti‐tumoral immunity [5]. Nevertheless, the role of inflammation in cancer is well known to be ambiguous [6, 7]. Inflammatory mediators impact upon cancer hallmarks such as increased survival, proliferation, angiogenesis, invasion and even immune escape. Conversely, the innate immune system also plays a critical role in developing anti‐tumor adaptive immune responses.
Most human solid tumors show one of three distinct immunological phenotypes: immune inflamed, immune excluded or immune desert [8]. There is now growing evidence that inflamed tumors are associated with enhanced clinical responses to ICBs [8, 9, 10]. Thus, there is an increasing interest in modulating inflammatory pathways to enhance tumor immunity, particularly in the context of ICB. For instance, blocking transforming growth factor (TGF)‐β in colon and urothelial cancer overcomes immune exclusion [11, 12]. Furthermore, Toll‐like receptor (TLR) ligands can trigger innate immune pathways which potentiate anti‐PD‐1 therapy. In fact, intratumoral and peritumoral injection of the TLR‐9 ligand cytosine–phosphate–guanosine (CpG) increased the survival of anti‐PD‐1‐treated tumor‐bearing mice [13, 14, 15]. A Phase Ib multi‐center study showed that intratumor injection of a synthetic CpG combined with anti‐PD‐1 blockade was well tolerated, and had a high response rate in a small number of patients who were naive to PD‐1 blockade at baseline in advanced melanoma [16]. Combined treatment resulted in enhanced tumor infiltration by CD8+ T cells. Alternative intratumoral injection of TLR‐9 or TLR‐7 agonists, combined with anti‐PD‐1 therapy, suppressed growth of experimental head and neck squamous cell carcinoma (HNSCC) at the primary tumor and metastatic sites [17]. Nanoparticles (NP) loaded with the TLR‐7 and TLR‐8 agonist R848 polarized tumor‐associated macrophages (TAMs) to an M1 phenotype and sensitized mice to the anti‐tumoral effects of PD‐1 blockade [18]. Also, PD‐1‐targeted delivery of NPs loaded with R848 improved the survival of MC38 colon cancer‐bearing mice treated with anti‐PD‐1 antibody [19]. Furthermore, the TLR‐3‐specific RNA agonist, ARNAX, triggers cross‐priming of antigen‐specific CD8+ T cells by dendritic cells (DCs) in an Ifnar‐dependent manner [20]. Systemic co‐injection of ARNAX+ tumor antigens followed by therapy with anti‐PD‐L1 antibodies effectively controlled tumor growth. This anti‐tumoral effect was associated with enhanced tumor infiltration by total and tumor‐specific CD8+ T cells [20]. Thus, combination therapies using ICB and TLR agonists are expected to generate more efficient and durable anti‐tumor responses.
With regard to other proinflammatory mediators, it has been appreciated that proinflammatory cytokines enhance the anti‐tumor efficacy of ICBs [19]. Accordingly, lowering the cytotoxicity threshold of TNF‐α by TNF receptor‐associated factor 2 (TRAF2) inactivation in melanoma cells enhanced tumor necrosis factor (TNF)‐α‐dependent anti‐tumor efficacy of anti‐PD‐1 therapy [21]. Furthermore, high TNF‐α in tumor biopsies was associated with clinical responses to PD‐1 blockade in melanoma patients [21]. However, the role of TNF‐α in anti‐tumor immunity seems to be complex, as contradictory observations have been reported. Thus, concomitant blockade of TNF‐α and PD‐1 has been proposed to reinforce the anti‐tumor efficacy of anti‐PD‐1 antibodies [22, 23].
Type I IFNs constitute another relevant inflammatory pathway. Type I IFNs play a critical role in tumor immunity triggered by ICBs. In human melanoma, a type I IFN signature was associated with clinical responses to anti‐CTLA‐4 [24]. The combination of anti‐CTLA‐4 + anti‐PD‐L1 therapy in mouse melanoma significantly lost efficacy in animals deficient in TMEM173 (stimulator of interferon genes; STING) [25]. Moreover, activation of STING by intratumoral injection of cyclic dinucleotide‐adenosine monophosphate (cGAMP) before anti‐CTLA‐4/anti‐PD‐1 therapy significantly enhanced anti‐tumor immunity triggered by ICBs [26]. Accordingly, a STING‐activating nanovaccine showed great synergy with PD‐1 blockade in controlling murine lung cancer growth [27]. Interestingly, the cGAS–STING axis controlled the nucleotide‐binding oligomerization domain and leucine‐rich repeat receptors (NLR)P3 inflammasome activation in human myeloid cells [28]. Thus, inflammasome activation might enhance tumor immunity in the context of ICBs.
Is there a role for inflammasome activation in cancer immunotherapy?
Inflammasomes are cytosolic multi‐protein complexes that sense cellular stress [29]. Sensor proteins can be divided into nucleotide‐binding oligomerization domain and leucine‐rich repeat receptors (NLRs), absent in melanoma 2 (AIM2)‐like receptors as well as pyrin. Once activated, they cleave caspase‐1, which then processes pro‐IL‐interleukin (IL)‐1β and pro‐IL‐18 to give the active and secreted forms of these proinflammatory cytokines [29]. Furthermore, active caspase‐1 cleaves gasdermin D (GSDMD), which forms pores in the cell membrane leading to cell death [30]. Inflammasomes are involved in different hallmarks of cancer development, performing tumor‐promoting as well as tumor‐suppressive functions [31, 32, 33]. The inflammasome‐related cytokines IL‐1β and IL‐18 promote proliferation and survival of malignant cells. IL‐18 has been shown to control caspase 8‐mediated apoptosis in gastric cancer cells [34]. In breast cancer, IL‐1β induces malignant cell proliferation by inducing nuclear translocation of β‐catenin [35]. Additionally, IL‐1β has been shown to regulate angiogenesis in several settings [36, 37] as well as immune suppression mechanisms, by inducing myeloid‐derived suppressor cells [38]. The Canakinumab Anti‐Inflammatory Thrombosis Outcome Study (CANTOS) has shown a significant decrease in lung cancer incidence and lung cancer mortality when IL‐1β was blocked compared to placebo in patients with atherosclerosis [39]. In contrast, NLRP3‐dependent secretion of IL‐18 triggers tumoricidal activity of NK cells against metastatic colon cancer cells in the liver in murine models [40]. In the context of tumor therapies, inflammasomes play an anti‐tumoral role in immunogenic chemotherapy [14] and probably in response to BRAF inhibitors [15]. Furthermore, pharmacological activation of inflammasome components has been proposed as a potential strategy to enhance the anti‐tumor efficacy of ICBs [41].
The NLRP3 inflammasome activation is tightly regulated by cytosolic levels of ions such as K+, Ca++ and Cl− [42, 43, 44, 45, 46, 47]. We have recently shown that unleashing inflammasome activation by targeting transmembrane protein 176B (TMEM176B), a cation channel expressed on antigen‐presenting cells, reinforces tumor immunity triggered by CTLA‐4 and PD‐1 blockade [48]. TMEM176B inhibits adenosine triphosphate (ATP) and nigericin‐induced NLRP3 inflammasome activation through ionic mechanisms in human and mouse DCs and macrophages. Furthermore, anti‐PD‐1 and anti‐CTLA‐4 therapies lost anti‐tumor efficacy in Nlrp3−/− and Casp1/11−/−. Accordingly, an inflammasome signature in tumor biopsies was associated with a response to PD‐1 blockade in two cohorts of advanced melanoma patients [48].
The NLRP3 inflammasome is activated, among other stimuli, by extracellular ATP (eATP) [42]. eATP levels increase by 100–1000‐fold as a consequence of tissue stress such as inflammation, hypoxia or ischemia in the tumor microenvironment [49, 50, 51]. ATP release can occur not only upon membrane damage, but also independently of cell death through ATP‐binding cassette (ABC) transporters, vesicular release or pannexins and connexins [51]. eATP is recognized through its interaction with the ionotropic P2X and metabotropic purinergic P2Y receptors [52]. Dying tumor cells release ATP, which binds to P2X7R on DCs and reinforces CD8+ T cell responses in an inflammasome‐dependent manner [14]. Interestingly, breast cancer patients with a loss‐of‐function substitution (E496A) in P2X7R showed shorter metastatic disease‐free survival [14]. The levels of eATP are also determined by CD39, the rate‐limiting enzyme in the hydrolysis of this nucleoside. CD39 plays a critical immunoregulatory role by modulating effector and regulatory T cells, macrophages, natural killer (NK) and myeloid‐derived suppressor cells (MDSCs), among other mechanisms [49, 50]. Thus, it would be expected that CD39 blockade with specific antibodies may lead to increased eATP levels within the tumor microenvironment, enhanced inflammasome activation and probably reinforced tumor immunity triggered by ICBs. In agreement with this hypothesis, anti‐CD39 therapy showed synergistic effects with PD‐1 and CTLA‐4 blockade in controlling lung metastasis of B16F10 murine melanoma [53]. More recently, Li et al. have shown in MC38 tumors that anti‐CD39 is less effective in Casp1/11−/−, Nlrp3−/−, Pycard−/− and P2x7r−/− mice [54]. Using syngeneic and humanized tumor models, the authors also showed that CD39 and PD‐1 blockade have synergistic anti‐tumoral effects [54].
In contrast to those studies, IL‐1β blockade was shown to improve the anti‐tumoral efficacy of anti‐PD‐1 therapy in a mouse model of breast cancer [55]. In this model, IL‐1β was neutralized before the commencement of anti‐PD‐1 therapy. Thus, although the inflammasome/IL‐1β pathway may play different roles in different tumor types [48, 55], unknown mechanisms may explain these apparent contradictory observations.
How does inflammasome activation reinforce tumor immunity triggered by ICBs?
In spite of considerable progress showing that CD8+ T cells mediate anti‐tumor immune responses when inflammasome is unleashed [48, 54], the mechanisms by which inflammasomes promote anti‐tumor immunity in the context of ICBs remain unclear. Inflammasomes may regulate priming of cytotoxic T lymphocytes. The response of some PD‐L1 negative tumors, which also lack tumor‐infiltrating T cells to anti PD‐1 therapy, suggest that PD‐1 blockade can trigger de‐novo anti‐tumor responses. Mice devoid of Sec22b, which lack the ability to cross‐present antigens to CD8+ T cells, showed a compromised response to anti‐PD‐1 therapy [56]. This observation suggests that anti‐PD‐1 may facilitate the priming of CD8+ T cells, although antigen cross‐presentation may be necessary before PD‐1 blockade. In non‐small‐cell lung cancer (NSCLC), neoadjuvant anti‐PD‐1 induced the expansion of mutation‐associated, neoantigen‐specific T cell clones in peripheral blood [57]. Accordingly, PD‐1 blockade has been shown to enhance early stages of T cell activation in lymph nodes [58]. However, anti‐PD‐1 therapy in transplantable mouse tumor models such as MC38 colon cancer does not depend upon lymph node priming [59, 60]. Nevertheless, recent single‐cell RNA and T cell receptor sequencing data from basal or squamous cell carcinoma tumors showed that the expansion of T cell clones did not derive from pre‐existing tumor‐infiltrating T lymphocytes [61]. Novel clones may, therefore, derive from tumor‐extrinsic sources including lymph nodes. Similarly, inflammasome activation may enhance anti‐PD‐1 therapy by reinforcing CD8+ T cell de‐novo priming. In fact, the expression of IL‐1R1 enhances CD8+ T cell responses in viral infections [62, 63], Mycobacterium tuberculosis [64] and tumors [14]. Accordingly, IL‐1 reinforces expansion, effector function, tissue localization and memory response of CD8+ T cells in response to antigen stimulation [65]. Interestingly, activated CD8+ T cells can promote inflammasome activation in antigen‐presenting cells (APCs) through perforin‐dependent mechanisms, suggesting a positive feedback leading to tumor rejection [66].
Conversely, anti‐tumoral CD8+ T cell responses can be reinforced by T helper type 17 (Th17) cells [67]. Different lines of evidence suggest that Th17 cells may play an important role as downstream effectors of unleashed inflammasome activation. Accordingly, genetic deletion or pharmacological blockade of TMEM176B are associated with increased numbers of CD4+ retinoid‐related orphan receptor γt (RORγt+) T cells in tumor‐draining lymph nodes [48]. Moreover, blockade of IL‐17A undermined the capacity of TMEM176B−/− mice to control tumor growth [48]. However, the role played by Th17 cells in cancer is controversial [68]. Intratumoral Th17 cells have been associated not only with a good, but also with a bad prognosis [68, 69]. In fact, Th17 cells are known to be a heterogeneous population behaving as either regulatory or effector cells, depending on the dominant cytokine microenvironment. Importantly, IL‐1β is a key factor in determining effector properties of Th17 cells [70, 71]. Interestingly, CD39 and CD73 are markers of regulatory Th17 cells which promote tumor growth, suggesting that the ATP/inflammasome pathway promotes effector Th17 responses [70, 72, 73]. Adoptive transfer of intratumoral CD39+CD4+RORγt+ intratumoral T cells promoted tumor growth in murine models, whereas this effect was lost in cells from CD39−/− animals [73]. Furthermore, eATP inhibits the suppressive potential of regulatory T cells (Tregs) through P2X7R expressed on these cells [74]. eATP also induces the differentiation of Tregs into Th17 cells [74]. Conversely, CD39 expression by Tregs prevented conversion into Th17 cells [75, 76]. Interestingly, CD39+ Tregs were proposed to specifically suppress pathogenic Th17 cells [77, 78]. Thus, TMEM176B and CD39 may serve as two different physiological strategies to impair ATP‐induced inflammation mediated by inflammasomes and Th17 cells. Pharmacological blockade of TMEM176B and CD39 led to inflammasome‐dependent tumor immunity and enhanced anti‐tumor efficacy of ICBs [48, 54]. Thus, concomitant blockade of both TMEM176B and CD39 may trigger Th17‐dependent responses and enhance the efficacy of ICBs. However, it remains to be determined whether inflammasome activation in vivo skews Th17 differentiation into effector cells as well as whether bona fide effector Th17 responses promote tumor immunity in the context of ICBs. In melanoma as well as in prostate cancer patients, clinical responses to PD‐1 blockade have been associated with increased peripheral CD4+IL‐17+ T cells [79, 80]. Although these observations suggest a role for Th17 cells in anti‐PD‐1 therapy, the effector or regulatory nature of these cells has not been assessed. In this regard, it has been recently reported that IL‐17A blockade abrogates immune‐related adverse events in a 50‐year‐old man with metastatic colon cancer treated with anti‐PD‐1 antibodies. However, IL‐17A blockade was also associated with an increase in the tumor marker carcinoembryonic antigen (CEA) in plasma, suggesting that IL‐17A was, in fact, mediating an anti‐tumoral immune response [81]. Recently, Sharma’s team showed that CD4+ T cells polarize to Th17 rather than Th1 in a TGF‐β‐dependent manner in bone metastases of prostate cancer patients, leading to the failure of ICB [82]. It remains to be determined whether unleashing inflammasome activation by blocking TMEM176B and/or CD39 can commit Th17 cells into an effector phenotype that may reinforce tumor immunity in the context of ICBs (Fig. 1).
Fig. 1.
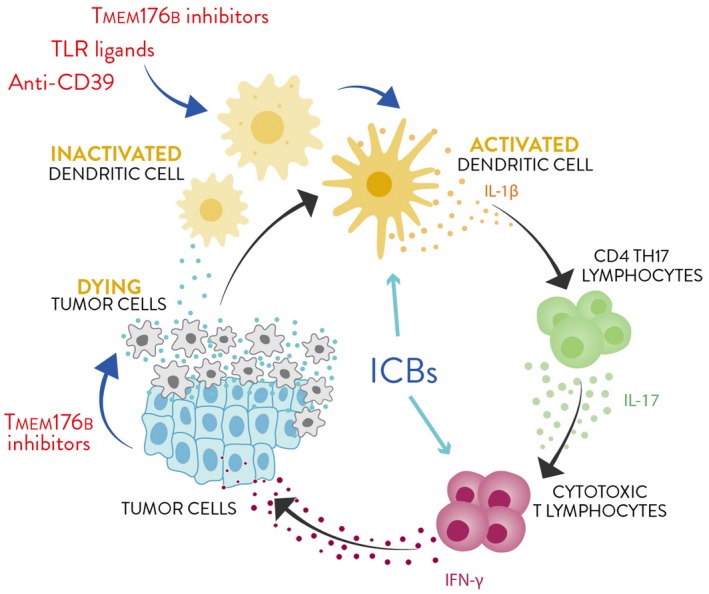
Manipulating innate players to improve the efficacy of immune checkpoint blockers (ICBs). Anti‐CD39 antibodies, Toll‐like receptor (TLR) ligands and transmembrane protein 176B (TMEM176B) inhibitors trigger dendritic cell (DC) activation and tumor immunity through the cancer immunity cycle. Interleukin (IL)‐1β secreted by activated DCs may reinforce T helper type 17 (Th17) cells leading to improved cytotoxic T lymphocyte (CTL) responses. Alternatively, IL‐1β might directly impact upon CD8+ T cells priming to promote the differentiation of CTLs. Moreover, TMEM176B inhibitors might induce malignant cell death fueling the whole process.
TMEM176B as a potential target in cancer therapy
A growing body of studies are being conducted to develop and characterize inflammasome inhibitors to control immune‐mediated inflammatory diseases. There are, however, far fewer examples of pharmacological strategies aiming at inducing inflammasome activation to trigger tumor immunity. We will focus on an emerging potential target, TMEM176B, the therapeutic blockade of which leads to enhancement of anti‐tumor responses.
TMEM176B and TMEM176A are members of the CD20‐like MS4A family [83, 84]. These are ubiquitous proteins highly expressed by macrophages and dendritic cells [84, 85], although they can also be expressed by NK and NK T cells [86] and by RORγt+ [type 3 innate lymphoid cells (ILC3), Th17 and γδT] cells [87]. TMEM176B is associated with allograft tolerance [84], and its expression is strongly down‐regulated during DC maturation [84, 85]. TMEM176B controls the immunoregulatory properties of tolerogenic DCs [88] and is localized in the endophagosomal membrane [88, 89] as well as in the trans‐Golgi network [53]. In DCs, TMEM176B controls phagosomal pH and promotes antigen cross‐presentation to activate CD8+ Treg cells [88]. TMEM176A and TMEM176B interact physically to form cation channels [87, 90]. Their expression is dysregulated in different cancers when compared to normal tissues [90]. In human gastric cancer, low mRNA expression of TMEM176A and TMEM176B are correlated with better overall survival [91]. Accordingly, low TMEM176B protein levels in the stroma were associated with improved overall survival in colorectal carcinoma [48]. In glioblastoma, TMEM176A was shown to inhibit Bcl2 expression and to induce apoptosis [92]. As well, TMEM176B was up‐regulated in tumor vessels of human renal cell carcinoma specimens, suggesting a role as a potential target for anti‐angiogenic intervention in these tumors [93]. In contrast, epigenetic silencing of TMEM176A has been associated with tumor progression in colorectal [94], esophageal [95] and hepatocellular [96] cancer.
Thus, targeting TMEM176B has the potential to reinforce anti‐tumoral CD8+ T cell responses in an inflammasome‐dependent manner while directly killing malignant cells, at least in some tumor types. Although not directly demonstrated, pharmacological inhibition of TMEM176B might also impact upon tumor angiogenesis [93]; therefore, TMEM176B seems so far to be a safe target. Our studies have not demonstrated acute toxicity upon pharmacological inhibition of TMEM176B or spontaneous autoimmunity in our TMEM176B−/− mice [48].
Concluding remarks
Recent evidence suggests that inflammasome activation leading to IL‐1β and IL‐18 secretion can modulate the immunological outcome of ICB in cancer immunotherapy settings. However, further work is needed to understand the mechanisms underlying these effects. Defining the timing of this process and determining which DC subsets and helper T cells are critical for inflammasome activation is of paramount importance to improve the clinical efficacy of immunotherapy. Moreover, characterization of molecular targets critical to trigger inflammasome activation is needed to design more selective and potent inhibitors. Combinatorial approaches aimed at blocking TMEM176B and CD39 may help unleash inflammasome activation, leading to enhancement of anti‐tumor immunity. It remains to be determined, however, whether Th17 cells are downstream in‐vivo effectors, and whether and how these cells may promote anti‐tumoral responses upon blockade of immune checkpoints.
Disclosures
M. H. is founder and CSO of ARDAN Pharma.
Acknowledgements
Work in M. H.’s laboratory is supported by Uruguay INNOVA‐2, FMV from ANII, CABBIO, PEDECIBA, ECOS‐SUD AUF/FAPESP and FOCEM (MERCOSUR Structural Convergence Fund) COF 03/11 grants to M. H., CSIC UDELAR and FCE from ANII to M. S. and FCE from ANII to S. R. Work in G. A. R.’s laboratory is supported by grants from Agencia Nacional de Promoción Científica y Tecnológica (2017‐0494 to G. A. R) as well as Bunge & Born, Sales and Richard Lounsbery Foundations.
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
Immune checkpoint inhibition: from molecules to clinical application. Clinical and Experimental Immunology 2020, 200: 105‐107.
TIGIT as an emerging immune checkpoint. Clinical and Experimental Immunology 2020, 200: 108‐119.
VISTA: Coming of age as a multi‐lineage immune checkpoint. Clinical and Experimental Immunology 2020, 200: 120‐130.
Immune checkpoint inhibitor diabetes mellitus: a novel form of autoimmune diabetes. Clinical and Experimental Immunology 2020, 200: 131‐140.
Mechanisms of checkpoint inhibition‐induced adverse events. Clinical and Experimental Immunology 2020, 200: 141‐154.
VISTA: Coming of age as a multi‐lineage immune checkpoint. Clinical and Experimental Immunology 2020, 200: 120–130.
References
- 1. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA‐4 blockade. Science 1996; 271:1734–6. [DOI] [PubMed] [Google Scholar]
- 2. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD‐L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD‐L1 blockade. Proc Natl Acad Sci USA 2002; 99:12293–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Syn NL, Teng MWL, Mok TSK, Soo RA. De‐novo and acquired resistance to immune checkpoint targeting. Lancet Oncol 2017; 18:e731–41. [DOI] [PubMed] [Google Scholar]
- 4. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov 2018; 8:1069–86. [DOI] [PubMed] [Google Scholar]
- 5. Demaria O, Cornen S, Daëron M, Morel Y, Medzhitov R, Vivier E. Harnessing innate immunity in cancer therapy. Nature 2019; 574:45–56. [DOI] [PubMed] [Google Scholar]
- 6. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010; 140:883–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Berraondo P, Minute L, Ajona D, Corrales L, Melero I, Pio R. Innate immune mediators in cancer: between defense and resistance. Immunol Rev 2016; 274:290–306. [DOI] [PubMed] [Google Scholar]
- 8. Hegde PS, Karanikas V, Evers S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin Cancer Res 2016; 22:1865–74. [DOI] [PubMed] [Google Scholar]
- 9. Tumeh PC, Harview CL, Yearley JH et al PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014; 515:568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol 2017; 17:559–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tauriello DVF, Palomo‐Ponce S, Stork D et al TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018; 554:538–43. [DOI] [PubMed] [Google Scholar]
- 12. Mariathasan S, Turley SJ, Nickles D et al TGFβ attenuates tumour response to PD‐L1 blockade by contributing to exclusion of T cells. Nature 2018; 554:544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mangsbo SM, Sandin LC, Anger K, Korman AJ, Loskog A, Tötterman TH. Enhanced tumor eradication by combining CTLA‐4 or PD‐1 blockade with CpG therapy. J Immunother 2010; 33:225–35. [DOI] [PubMed] [Google Scholar]
- 14. Ghiringhelli F, Apetoh L, Tesniere A et al Activation of the NLRP3 inflammasome in dendritic cells induces IL‐1beta‐dependent adaptive immunity against tumors. Nat Med 2009; 15:1170–8. [DOI] [PubMed] [Google Scholar]
- 15. Hajek E, Krebs F, Bent R et al BRAF inhibitors stimulate inflammasome activation and interleukin 1 beta production in dendritic cells. Oncotarget. 2018; 9:28294–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ribas A, Medina T, Kummar S et al SD‐101 in combination with pembrolizumab in advanced melanoma: results of a phase Ib, multicenter study. Cancer Discov 2018; 8:1250–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sato‐Kaneko F, Yao S, Ahmadi A et al Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight 2017; 2:e93397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rodell CB, Arlauckas SP, Cuccarese MF et al TLR7/8‐agonist‐loaded nanoparticles promote the polarization of tumour‐associated macrophages to enhance cancer immunotherapy. Nat Biomed Eng 2018; 2:578–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schmid D, Park CG, Hartl CA et al T cell‐targeting nanoparticles focus delivery of immunotherapy to improve antitumor immunity. Nat Commun 2017; 8:1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Takeda Y, Kataoka K, Yamagishi J, Ogawa S, Seya T, Matsumoto M. A TLR3‐specific adjuvant relieves innate resistance to PD‐L1 blockade without cytokine toxicity in tumor vaccine immunotherapy. Cell Rep 2017; 19:1874–87. [DOI] [PubMed] [Google Scholar]
- 21. Vredevoogd DW, Kuilman T, Ligtenberg MA et al Augmenting immunotherapy impact by lowering tumor TNF cytotoxicity threshold. Cell 2019; 178:585–99.e15. [DOI] [PubMed] [Google Scholar]
- 22. Perez‐Ruiz E, Minute L, Otano I et al Prophylactic TNF blockade uncouples efficacy and toxicity in dual CTLA‐4 and PD‐1 immunotherapy. Nature 2019; 569:428–32. [DOI] [PubMed] [Google Scholar]
- 23. Bertrand F, Montfort A, Marcheteau E et al TNFα blockade overcomes resistance to anti‐PD‐1 in experimental melanoma. Nat Commun 2017; 8:2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chiappinelli KB, Strissel PL, Desrichard A et al Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 2015; 162:974–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Woo S‐R, Fuertes MB, Corrales L et al STING‐dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014; 41:830–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Demaria O, De Gassart A, Coso S et al STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci USA 2015; 112:15408–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luo M, Wang H, Wang Z et al A STING‐activating nanovaccine for cancer immunotherapy. Nat Nanotechnol 2017; 12:648–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gaidt MM, Ebert TS, Chauhan D et al The DNA inflammasome in human myeloid cells is initiated by a STING‐cell death program upstream of NLRP3. Cell 2017; 171:1110–24.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rathinam VAK, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell 2016; 165:792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kesavardhana S, Malireddi RKS, Kanneganti T‐D. Caspases in cell death, inflammation, and gasdermin‐induced pyroptosis. Annu Rev Immunol 2020;38 10.1146/annurev-immunol-073119-095439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Karki R, Man SM, Kanneganti T‐D. Inflammasomes and cancer. Cancer Immunol Res 2017; 5:94–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Karki R, Kanneganti T‐D. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat Rev Cancer. 2019; 19:197–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Christgen S, Kanneganti T‐D. Inflammasomes and the fine line between defense and disease. Curr Opin Immunol 2020; 62:39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Deswaerte V, Nguyen P, West A et al Inflammasome adaptor ASC suppresses apoptosis of gastric cancer cells by an IL18‐mediated inflammation‐independent mechanism. Cancer Res 2018; 78:1293–307. [DOI] [PubMed] [Google Scholar]
- 35. Perez‐Yepez EA, Ayala‐Sumuano J‐T, Lezama R, Meza I. A novel β‐catenin signaling pathway activated by IL‐1β leads to the onset of epithelial–mesenchymal transition in breast cancer cells. Cancer Lett 2014; 354:164–71. [DOI] [PubMed] [Google Scholar]
- 36. Saijo Y, Tanaka M, Miki M et al Proinflammatory cytokine IL‐1β promotes tumor growth of Lewis lung carcinoma by induction of angiogenic factors. In vivo analysis of tumor–stromal interaction. J Immunol 2002; 169:469–75. [DOI] [PubMed] [Google Scholar]
- 37. Jung Y‐J, Isaacs JS, Lee S, Trepel J, Neckers L. IL‐1β mediated up‐regulation of HIF‐lα via an NFkB/COX‐2 pathway identifies HIF‐1 as a critical link between inflammation and oncogenesis. FASEB J 2003; 17:1–22. [DOI] [PubMed] [Google Scholar]
- 38. van Deventer HW, Burgents JE, Wu QP et al The inflammasome component Nlrp3 impairs antitumor vaccine by enhancing the accumulation of tumor‐associated myeloid‐derived suppressor cells. Cancer Res 2010; 70:10161–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ridker PM, MacFadyen JG, Thuren T et al Effect of interleukin‐1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double‐blind, placebo‐controlled trial. Lancet 2017; 390:1833–42. [DOI] [PubMed] [Google Scholar]
- 40. Dupaul‐Chicoine J, Arabzadeh A, Dagenais M et al The Nlrp3 inflammasome suppresses colorectal cancer metastatic growth in the liver by promoting natural killer cell tumoricidal activity. Immunity 2015; 43:751–63. [DOI] [PubMed] [Google Scholar]
- 41. Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov 2018; 17:588–606. [DOI] [PubMed] [Google Scholar]
- 42. Mariathasan S, Weiss DS, Newton K et al Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006; 440:228–32. [DOI] [PubMed] [Google Scholar]
- 43. Muñoz‐Planillo R, Kuffa P, Martínez‐Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013; 38:1142–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Murakami T, Ockinger J, Yu J et al Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci USA 2012; 109:11282–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tang T, Lang X, Xu C et al CLICs‐dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat Commun 2017; 8:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gong T, Yang Y, Jin T, Jiang W, Zhou R. Orchestration of NLRP3 inflammasome activation by Ion Fluxes. Trends Immunol 2018; 39:393–406. [DOI] [PubMed] [Google Scholar]
- 47. Eugenia Schroeder M, Russo S, Costa C et al Pro‐inflammatory Ca++‐activated K+ channels are inhibited by hydroxychloroquine. Sci Rep. 2017; 7:1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Segovia M, Russo S, Jeldres M et al Targeting TMEM176B enhances antitumor immunity and augments the efficacy of immune checkpoint blockers by unleashing inflammasome activation. Cancer Cell 2019; 35:767–781.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Allard D, Allard B, Stagg J. On the mechanism of anti‐CD39 immune checkpoint therapy. J Immunother Cancer 2020; 8:e000186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev 2017; 276:121–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol 2013; 31:51–72. [DOI] [PubMed] [Google Scholar]
- 52. Burnstock G, Knight GE. Cellular distribution and functions of P2 receptor subtypes in different systems. Int Rev Cytol 2004; 240:31–304. [DOI] [PubMed] [Google Scholar]
- 53. Zhang H, Vijayan D, Li X‐Y et al The role of NK cells and CD39 in the immunological control of tumor metastases. OncoImmunology 2019; 8:e1593809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li X‐Y, Moesta AK, Xiao C et al Targeting CD39 in cancer reveals an extracellular ATP‐ and inflammasome‐driven tumor immunity. Cancer Discov 2019; 9:1754–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kaplanov I, Carmi Y, Kornetsky R et al Blocking IL‐1β reverses the immunosuppression in mouse breast cancer and synergizes with anti–PD‐1 for tumor abrogation. Proc Natl Acad Sci USA 2019; 116:1361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Alloatti A, Rookhuizen DC, Joannas L et al Critical role for Sec22b‐dependent antigen cross‐presentation in antitumor immunity. J Exp Med 2017; 214:2231–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Forde PM, Chaft JE, Pardoll DM. Neoadjuvant PD‐1 blockade in resectable lung cancer. N Engl J Med 2018; 379:e14. [DOI] [PubMed] [Google Scholar]
- 58. Goldberg MV, Maris CH, Hipkiss EL et al Role of PD‐1 and its ligand, B7–H1, in early fate decisions of CD8 T cells. Blood 2007; 110:186–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Garris CS, Arlauckas SP, Kohler RH et al Successful anti‐PD‐1 cancer immunotherapy requires T cell‐dendritic cell crosstalk involving the cytokines IFN‐γ and IL‐12. Immunity 2018; 49:1148–61.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chow MT, Ozga AJ, Servis RL et al Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti‐PD‐1 therapy. Immunity 2019; 50:1498–512.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yost KE, Satpathy AT, Wells DK et al Clonal replacement of tumor‐specific T cells following PD‐1 blockade. Nat Med 2019; 25:1251–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med 2009; 206:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Joeckel LT, Wallich R, Metkar SS, Froelich CJ, Simon MM, Borner C. Interleukin‐1R signaling is essential for induction of proapoptotic CD8 T cells, viral clearance, and pathology during lymphocytic choriomeningitis virus infection in mice. J Virol 2012; 86:8713–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fremond CM, Togbe D, Doz E et al IL‐1 receptor‐mediated signal is an essential component of MyD88‐dependent innate response to Mycobacterium tuberculosis infection. J Immunol 2007; 179:1178–89. [DOI] [PubMed] [Google Scholar]
- 65. Ben‐Sasson SZ, Hogg A, Hu‐Li J et al IL‐1 enhances expansion, effector function, tissue localization, and memory response of antigen‐specific CD8 T cells. J Exp Med 2013; 210:491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yao Y, Chen S, Cao M et al Antigen‐specific CD8+ T cell feedback activates NLRP3 inflammasome in antigen‐presenting cells through perforin. Nat Commun 2017; 8:15402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Martin‐Orozco N, Muranski P, Chung Y et al T Helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity 2009; 31:787–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Martin F, Apetoh L, Ghiringhelli F. Controversies on the role of Th17 in cancer: a TGF‐β‐dependent immunosuppressive activity? Trends Mol Med 2012; 18:742–9. [DOI] [PubMed] [Google Scholar]
- 69. Bailey SR, Nelson MH, Himes RA, Li Z, Mehrotra S, Paulos CM. Th17 cells in cancer: the ultimate identity crisis. Front Immunol 2014; 5:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chatterjee S, Thyagarajan K, Kesarwani P et al Reducing CD73 expression by IL1 ‐programmed Th17 cells improves immunotherapeutic control of tumors. Cancer Res 2014; 74:6048–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ghoreschi K, Laurence A, Yang X‐P et al Generation of pathogenic TH17 cells in the absence of TGF‐β signalling. Nature 2010; 467:967–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Longhi MS, Moss A, Bai A et al Characterization of human CD39+ Th17 cells with suppressor activity and modulation in inflammatory bowel disease. PLOS ONE 2014; 9:e87956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chalmin F, Mignot G, Bruchard M et al Stat3 and Gfi‐1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity 2012; 36:362–73. [DOI] [PubMed] [Google Scholar]
- 74. Schenk U, Frascoli M, Proietti M et al Inhibits the generation and function of regulatory T cells through the activation of purinergic P2X receptors. Sci Signal 2011; 4:ra12. [DOI] [PubMed] [Google Scholar]
- 75. Zhou Q, Yan J, Putheti P et al Isolated CD39 expression on CD4+ T cells denotes both regulatory and memory populations. Am J Transplant 2009; 9:2303–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dwyer KM, Hanidziar D, Putheti P et al Expression of CD39 by human peripheral blood CD4+CD25+ T cells denotes a regulatory memory phenotype: CD39 expression on regulatory memory T cells. Am J Transplant 2010; 10:2410–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Grant CR, Liberal R, Holder BS et al Dysfunctional CD39 POS regulatory T cells and aberrant control of T‐helper type 17 cells in autoimmune hepatitis: Grant et al . Hepatology 2014; 59:1007–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fletcher JM, Lonergan R, Costelloe L et al CD39+ Foxp3+ regulatory T cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J Immunol 2009; 183:7602–10. [DOI] [PubMed] [Google Scholar]
- 79. Krieg C, Nowicka M, Guglietta S et al High‐dimensional single‐cell analysis predicts response to anti‐PD‐1 immunotherapy. Nat Med 2018; 24:144–53. [DOI] [PubMed] [Google Scholar]
- 80. Dulos J, Carven GJ, van Boxtel SJ et al PD‐1 blockade augments Th1 and Th17 and suppresses Th2 responses in peripheral blood from patients with prostate and advanced melanoma cancer. J Immunother 2012; 35:169–78. [DOI] [PubMed] [Google Scholar]
- 81. Esfahani K, Miller WH. Reversal of autoimmune toxicity and loss of tumor response by interleukin‐17 blockade. N Engl J Med 2017; 376:1989–91. [DOI] [PubMed] [Google Scholar]
- 82. Jiao S, Subudhi SK, Aparicio A et al Differences in tumor microenvironment dictate T helper lineage polarization and response to immune checkpoint therapy. Cell 2019; 179:1177–1190.e13. [DOI] [PubMed] [Google Scholar]
- 83. Eon Kuek L, Leffler M, Mackay GA, Hulett MD. The MS4A family: counting past 1, 2 and 3. Immunol Cell Biol 2016; 94:11–23. [DOI] [PubMed] [Google Scholar]
- 84. Louvet C, Chiffoleau E, Heslan M et al Identification of a new member of the CD20/FcepsilonRIbeta family overexpressed in tolerated allografts. Am J Transplant 2005; 5:2143–53. [DOI] [PubMed] [Google Scholar]
- 85. Condamine T, Le Texier L, Howie D et al Tmem176B and Tmem176A are associated with the immature state of dendritic cells. J Leukoc Biol 2010; 88:507–15. [DOI] [PubMed] [Google Scholar]
- 86. The Immunological Genome Project Consortium , Bezman NA, Kim CC, Sun JC et al Molecular definition of the identity and activation of natural killer cells. Nat Immunol 2012; 13:1000–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Drujont L, Lemoine A, Moreau A et al RORγt+ cells selectively express redundant cation channels linked to the Golgi apparatus. Sci Rep 2016; 6:23682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Segovia M, Louvet C, Charnet P et al Autologous dendritic cells prolong allograft survival through Tmem176b‐dependent antigen cross‐presentation: immunoregulatory mechanisms of autologous DCs. Am J Transplant 2014; 14:1021–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Shui W, Sheu L, Liu J et al Membrane proteomics of phagosomes suggests a connection to autophagy. Proc Natl Acad Sci USA 2008; 105:16952–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Cuajungco MP, Podevin W, Valluri VK, Bui Q, Nguyen VH, Taylor K. Abnormal accumulation of human transmembrane (TMEM)‐176A and 176B proteins is associated with cancer pathology. Acta Histochem 2012; 114:705–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sun L, Zhang Y, Zhang C. Distinct expression and prognostic value of MS4A in gastric cancer. Open Med 2018; 13:178–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Liu Z, An H, Song P et al Potential targets of TMEM176A in the growth of glioblastoma cells. OncoTargets Ther 2018; 11:7763–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Otsubo T, Hida Y, Ohga N et al Identification of novel targets for antiangiogenic therapy by comparing the gene expressions of tumor and normal endothelial cells. Cancer Sci 2014; 105:560–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Gao D, Han Y, Yang Y et al Methylation of TMEM176A is an independent prognostic marker and is involved in human colorectal cancer development. Epigenetics 2017; 12:575–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Wang Y, Zhang Y, Herman JG, Linghu E, Guo M. Epigenetic silencing of TMEM176A promotes esophageal squamous cell cancer development. Oncotarget 2017; 8:70035–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Li H, Zhang M, Linghu E et al Epigenetic silencing of TMEM176A activates ERK signaling in human hepatocellular carcinoma. Clin Epigenet 2018; 10:137. [DOI] [PMC free article] [PubMed] [Google Scholar]