

Summary
Annexins are well‐known Ca2+ phospholipid‐binding proteins, which have a wide variety of cellular functions. The role of annexin A1 (AnxA1) in the innate immune system has focused mainly on the anti‐inflammatory and proresolving properties through its binding to the formyl‐peptide receptor 2 (FPR2)/ALX receptor. However, studies suggesting an intracellular role of AnxA1 are emerging. In this study, we aimed to understand the role of AnxA1 for interleukin (IL)‐1β release in response to activators of the nucleotide‐binding domain leucine‐rich repeat (NLR) and pyrin domain containing receptor 3 (NLRP3) inflammasome. Using AnxA1 knockout mice, we observed that AnxA1 is required for IL‐1β release in vivo and in vitro. These effects were due to reduction of transcriptional levels of IL‐1β, NLRP3 and caspase‐1, a step called NLRP3 priming. Moreover, we demonstrate that AnxA1 co‐localize and directly bind to NLRP3, suggesting the role of AnxA1 in inflammasome activation is independent of its anti‐inflammatory role via FPR2. Therefore, AnxA1 regulates NLRP3 inflammasome priming and activation in a FPR2‐independent manner.
Keywords: annexin A1, IL‐1β, inflammasome, NLRP3
Annexin A1 directly binds to NLRP3 and is required for inflammasome priming and IL‐1β release independent of its anti‐inflammatory role via FPR2.

Abbreviations
- AnxA1
annexin A1
- ASC
apoptosis‐associates speck‐like protein containing CARD
- ATP
adenosine triphosphate
- BMDM
bone marrow‐derived macrophages
- FPR2
formyl‐peptide receptor 2
- IL‐1β
interleukin‐1β
- LPS
lipopolysaccharides
- LTA
lipoteichoic acid
- MAMP
microbe‐associated molecular patterns
- MSU
monosodium urate crystals
- NLRP3
nucleotide‐binding domain leucine‐rich repeat (NLR) and pyrin domain containing receptor 3
- PRR
pattern recognition receptors
- TLR
Toll‐like receptor
Introduction
The annexin family is a well‐known group of Ca2+ phospholipid‐binding proteins, which have a wide variety of cellular functions. Annexin A1 (AnxA1) functions in cells are related with proliferation, apoptosis, vesicle trafficking, differentiation and inflammatory responses. 1 This molecule is regulated by glucocorticoids, and its first described anti‐inflammatory action was inhibition of cytosolic phospholipase A2 (PLA2), subsequently blocking the release of arachidonic acid and, in turn, preventing synthesis of eicosanoids. 2
Accumulating years of studies on the actions of AnxA1 have shown the undoubtedly anti‐inflammatory/proresolving activities of this protein. 3 , 4 AnxA1 modulates inflammation mainly through binding to a receptor named formyl‐peptide receptor 2 (FPR2)/ALXR. Activation of FPR2 by AnxA1 regulates neutrophil recruitment to inflammatory sites, induces neutrophil apoptosis, induces monocyte recruitment and increases efferocytosis. 5 However, recent studies have shown that AnxA1 may contribute to the pathogenesis of certain conditions via activation of FPR2. 6 Some of these effects are associated with proliferation of fibroblasts, 7 activation and differentiation of T‐cells, 8 leucocyte transendothelial migration through increased ICAM expression, 9 phosphorylation of PLA2, and increased eicosanoids production on mast cells. 10 The molecular mechanisms responsible for the different roles of AnxA1 in different scenarios, including innate immunity, are not fully understood. Some studies have suggested a receptor‐independent and intracellular function of AnxA1. 11 , 12 However, the intracellular effects and roles of AnxA1 remain to be fully investigated.
The innate immune system is characterized by a family of pattern recognition receptors (PRRs) that are involved in sensing damage signals or microbe‐associated molecular patterns (MAMPS). Among the sensors, there is a group of cytosolic sensors named nucleotide‐binding domain leucine‐rich repeat containing proteins (NLR), of which NLRP3 (pyrin domain containing 3) is the most studied. After activation, NLRP3 oligomerizes and recruits the adaptor ASC (apoptosis‐associates speck‐like protein containing CARD), which promotes caspase 1 (CASP1) activation forming a complex called inflammasome. 13 , 14 Once activated, the inflammasome complex induces the cleavage and secretion of the pro‐inflammatory cytokines interleukin (IL)‐1β and IL‐18. 19 It is well known that the production of these cytokines relies on two steps: the first one is provided by Toll‐like receptor (TLR) ligands or cytokines that induce transcriptional regulation of the genes for the cytokines and inflammasome components – a process called priming. 15 The second signal is induced by a variety of molecules that can then activate the inflammasome and lead to the cleavage for the pro‐cytokine proteins. For the NLRP3 inflammasome, activators include adenosine triphosphate (ATP), reactive oxygen species and crystals, such as uric acid crystals. 13 Moreover, MAMPS have also been shown to trigger inflammasome assembly. 16 , 17 , 18
Interleukin‐1 family members, which involve ligands and receptors, have the dual role of defending the host against microbes or injury, through innate immune response inducing or reducing inflammation. 20 IL‐1β exerts its pro‐inflammatory properties by inducing expression of adhesion molecules and chemokines, and promoting infiltration of leucocytes from blood to the tissues. It is well known that IL‐1β is involved in a variety of inflammatory conditions, such as arthritis, 21 diabetes mellitus, 22 gout 23 and severe asthma. 24 Therefore, understanding how inflammasome activation is regulated is critical for the development of novel treatment strategies for inflammatory diseases. It has been previously shown that the direct interaction of certain proteins with the NLRP3 is required for inflammasome assembly and/or signalling. 25 , 26 Here, we describe the role of AnxA1 in regulating IL‐1β production in the specific context of NLRP3 activation.
Materials and methods
Animals
Male BALB/c and male C57Bl/6 (8–12 weeks) mice were obtained from the Center of bioterism of Universidade Federal de Minas Gerais (UFMG) Brazil, and were housed under standard conditions, and had free access to commercial food and water. Male AnxA1‐deficient mice (AnxA1−/−‐Balb/C background) 27 and FPR2/3‐deficient mice (FPR2/3−/−‐C57Bl/6 background) 28 were maintained in the animal facilities at Universidade Federal de Minas Gerais (UFMG), Brazil. This study was carried out in accordance with the recommendations of the law no. 11.794 from National Council for Control of Animals Experimentation – CONCEA, Brazil. The protocol was approved by the Animal Ethics Council – CEUA – at Universidade Federal de Minas Gerais (Protocol Number 2/2015), Brazil.
In vivo models
Gout model
Monosodium urate crystals (MSU) were prepared from uric acid (Sigma‐Aldrich, St Louis, MO), and the model was performed as previously described. 29 Mice were anaesthetized i.p. with a solution containing 80 mg/kg ketamine : 15 mg/kg xylazine, and received an intra‐articular injection of 100 μg/10 μl into knee cavity. The peri‐articular tissue was collected 12 hr after the injection (the peak of MSU‐induced inflammation) and processed for IL‐1β quantification by ELISA.
Silicosis model
Mice were anaesthetized i.p. with a solution containing 80 mg/kg ketamine : 15 mg/kg xylazine, and were instilled intra‐nasally with 10 mg of silica (Sigma‐Aldrich) in 40 μl of phosphate buffer as previously described. 30 Mice were killed at 24 hr post‐challenge (the peak of silica‐induced inflammation) and lungs were collected for IL‐1β quantification by ELISA.
Cell culture
Bone marrow‐derived macrophages (BMDMs) were obtained as previously described. 31 Briefly, bone marrow cells were collected from femurs of wild‐type (WT), AnxA1−/− and FPR2/3−/− mice and differentiated for 7 days in RPMI 1640 supplemented with 20% of fetal bovine serum (FBS) and 30% L929 conditioned medium. Differentiated BMDMs were plated on day 7 for treatment the following day. Cells were maintained at 37° in a humidified atmosphere of 5% CO2. Cells were detached with cold phosphate‐buffered saline (PBS), resuspended in RPMI 1640 supplemented with 10% FBS and seeded in a tissue culture plate overnight to ensure adherence. On the next day, cells were stimulated as described below.
Inflammasome activation
Bone marrow‐derived macrophages (1 × 106 cells/well) were seeded in 24‐well plates for cytokine. The following day BMDMs were stimulated with the TLR4 agonist lipopolysaccharide (LPS; from Escherichia coli serotype O:111:B4; 1 μg/ml; Sigma‐Aldrich) for 1 hr as previously described, 23 or stimulated with the TLR2 agonist lipoteichoic acid (LTA; 10 μg/ml; Sigma‐Aldrich). 32 The AnxA1 peptidomimetic Ac2‐26 (32 μm; GeneScript) 33 and a selective FPR2 antagonist, WRW4 (10 μm; Calbiochem) 34 were added into the cells prior to inflammasome activation. Then BMDMs were stimulated with inflammasome activators in RPMI 1640 medium without FBS: MSU crystals (300 μg/ml for 6 hr); 29 Silica (250 μg/ml for 6 hr); 35 adenosine 5′‐triphosphate disodium salt (ATP 5 mm for 30 min); 36 Poly(dA:dT) (Sigma‐Aldrich; 500 ng/ml for 2 hr); 37 Legionella pneumophila bacteria (strain JR32 – MOI of 10 for 4 hr). 37 Supernatants were removed and analysed using ELISA kits according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN, USA).
Quantitative reverse transcriptase‐quantitative polymerase chain reaction
Total RNA was extracted from 1 × 106 cells BMDMs and synovial tissue using TRizol reagent (Ambion, Life Technologies, Themo Fisher Scientific, Grand Island, NY, USA). Quantitative polymerase chain reactions (qPCRs) were performed on a 7500 Fast Real‐Time PCR system using Power SYBR Green PCR Master Mix (Applied Biosystems, Thermo Fisher Scientific) after reverse transcription reactions of 0·5 μg RNA using SuperScript III reverse transcriptase (Invitrogen, Life Technologies, Carlsbad, CA, USA). The relative level of gene expression was determined by the comparative threshold cycle method, as described by the manufacturer, whereby data for each sample were normalized to a GAPDH constitutive gene and expressed as a fold change (2^−Δct). The following primer pairs were used: for Gapdh, 5′‐ACG GCC GCATCT TCT TGT GCA‐3′ (forward) and 5′‐CGG CCA AAT CCG TTC ACA CCGA‐3′ (reverse); for Il‐1β, 5′‐CTA CAG GCT CCG AGA TGA ACA AC‐3′ (forward) and 5′‐TCC ATT GAG GTG GAG AGC TTT C‐3′ (reverse); for Caspase 1, 5′ ‐ CAT GCG CAC ACA GCA ATT GTG GT‐3′ (forward) and 5′‐TGC CCT CAG GAT CTT GTC AGC C‐3′ (reverse); for Asc, 5′‐CAG AGT ACA GCC AGA ACA GGA CAC‐3′ (forward) and 5′‐GTG GTC TCT GCA CGA ACT GCC TG‐3′ (reverse); for Nlrp3, 5′‐GCT GCT GTC TCA CAT CCG CGT‐3′ (forward) and 5′‐GGC CGG AAT TCA CCA ACC CCA‐3′ (reverse).
Western Blot analysis
Cells lysate
Cells were lysed in lysis buffer (100 mm Tris/HCl, 200 mm NaCl, 10% glycerol 1% Triton X‐100 and 5 mm EDTA) containing anti‐proteases, as described. 38 , 39 Protein amounts were quantified with the Bradford assay reagent from Bio‐Rad (Hercules, CA). Extracts were separated by electrophoresis on a denaturing, 8−12% polyacrylamide−sodium dodecyl sulphate (SDS) gel and electrotransferred to nitrocellulose membranes. Membranes were incubated with specific primary antibodies [anti‐ASC, anti‐IL‐1β purchased from Santa Cruz; anti‐NLRP3 (Cryo2) purchased from Adipogen; anti‐caspase 1 p‐20 (Genentech; kindly donated by Dario Zamboni, University of São Paulo)] and then incubated with appropriated horseradish peroxidase (HRP)‐conjugated secondary antibody. Immunoreactive bands were visualized by using an ECL detection system, as described by the manufacturer (GE Healthcare, Piscataway, NJ). For loading control, membranes were re‐probed with anti‐β‐actin (Sigma). For densitometric analysis, membranes were scanned, quantified using Fiji imaging software, and normalized by β‐actin. Values are expressed as arbitrary units.
Supernatant
Cell culture supernatants were precipitated by the addition of an equal volume of methanol and 0·25 volumes of chloroform, as described previously. 40 These mixtures were vortexed and then centrifuged for 10 min at 8960 g. The upper phase was discarded and 500 ml methanol was added to the interphase. This mixture was centrifuged for 10 min at 8960 g and the protein pellet was dried at 55°, resuspended in Laemmli buffer and boiled for 5 min at 99°. Samples were separated by 12% SDS–polyacrylamide gel electrophoresis (PAGE) and were transferred onto nitrocellulose membranes. Blots were probed with rat monoclonal‐anti‐mouse caspase‐1 p20 antibody.
Co‐immunoprecipitation
The co‐immunoprecipitation was evaluated as previously described. 41 BMDMs (5 × 107 cells) were lysed in lysis solution (0·5 m HEPES, 2·5 m NaCl, 0·5 m MgCl2, 0·5 m EDTA, 1% Triton X‐100, pH 7·4) containing anti‐proteases (Sigma Fast protease inhibitor tablets, Sigma‐Aldrich). Protein amounts were quantified using the Bradford assay reagent from Bio‐Rad (Hercules, CA). One milligram of protein was incubated with anti‐AnxA1 (Santa Cruz) for 30 min at 4° and then incubated with 80 μl of protein G Sepharose 4 fast flow beads (GE Healthcare) for 2 hr at 4°. Beads were washed, 50 μl of Laemmli sample buffer was added to the beads, which were then incubated for 10 min at 50°. Proteins were subjected to SDS−PAGE, followed by electroblotting onto nitrocellulose membranes and, finally, analysed by Western blot. Anti‐P‐MYPT was used as immunoprecipitation control (CT‐).
Confocal analysis
After stimulation, BMDMs were recovered from the plate and 5 × 105 cells were cytocentrifuged using Shandon CytoSpin III (Thermo Shandon) in a coverslip. Cells were fixed with 4% paraformaldehyde for 20 min at room temperature, permeabilized with Triton X‐100 0·1% for 10 min and intracellular stained with rabbit anti‐AnxA1 (Santa Cruz) and mouse anti‐NLRP3 (Adipogen) overnight. Then, secondary anti‐rabbit IgG Alexa Fluor 647 and anti‐mouse IgG FITC were incubated for 1 hr, and DAPI (1 : 1000; BD) was used to stain the nucleus. Finally, coverslips were prepared with fluoromount (Sigma‐Aldrich) for confocal microscopy analysis. Images were obtained using Nikon Eclipse Ti with a A1 confocal head equipped with four different lasers (excitation at four wavelengths: 405, 488, 546 and 647 nm) and emission bandpass filters at 450/50, 515/30, 584/50 and 663/738 nm. Objective Plan Apo 60 ×; scale bar: 100 μm. Z‐sections were also performed. Image analysis was performed using Fiji imaging software and Volocity software 6.3 (Perkin‐Elmer, Waltham, MA).
Statistical analysis
Results are expressed as means ± SEM. Data were analysed by one‐way anova followed by Holm‐Sidak’s multiple comparison post‐test, using Prism 7.0 software (GraphPad, San Diego, CA), with significance accepted at values of P < 0·05.
Results
AnxA1 is required for the release of IL‐1β in vivo
To assess the effects of AnxA1 on the release of IL‐1β, we used two in vivo models, in which IL‐1β is shown to be implicated in the pathogenesis: Gout and Silicosis. WT and AnxA1−/− mice were injected with an intra‐articular injection of uric acid crystals (MSU 100 μg/cavity) into the tibiofemoral knee joint, and peri‐articular tissues were collected to measure IL‐1β. To induce silicosis, mice were instilled with silica (10 mg/40 μl) and lungs were collected to measure IL‐1β. In both models, WT mice showed increased IL‐1β levels after challenge; however, IL‐1β levels in both models were significantly lower in AnxA1−/− mice (Fig. 1a,b). These data suggest that AnxA1 may contribute to the release of IL‐1β in vivo.
Figure 1.
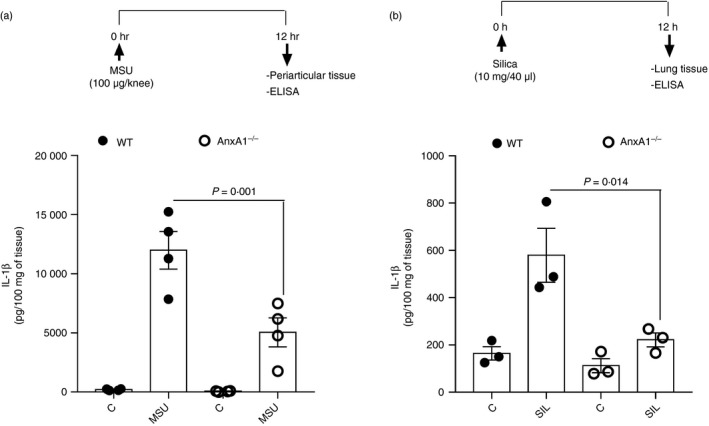
Annexin A1 (AnxA1) is necessary for the production of interleukin (IL)‐1β in vivo. Wild‐type (WT) and AnxA1−/− mice were injected with 100 μg/knee of uric acid crystals (MSU) in the tibiofemoral joint and, 12 hr after injection, peri‐articular tissue was collected to measure IL‐1β levels (a). WT and AnxA1−/− mice were instilled with 10 mg/40 μl of silica and, 24 hr after instillation, lungs were collected to measure IL‐1β levels (b). Bars show the means ± SEM of 5 mice/group and are from one experiment representative of two independent experiments. Significance was calculated using anova followed by Holm‐Sidak’s multiple comparison test.
AnxA1 is required for NLRP3 inflammasome‐dependent IL‐1β release
Next, we looked at the in vitro release of IL‐1β by BMDMs that were isolated from both WT and AnxA1−/− mice. Cells were treated with the TLR4 agonist LPS alone or in combination with the NLRP3 inflammasome activators (MSU, ATP or Silica), or a NLRC4 inflammasome activator (Legionella), or an AIM2 inflammasome activator [deoxyadenylic‐deoxythymidylic Poly(dA:dT)]. IL‐1β was secreted in large amounts by BMDMs obtained from WT mice in response to LPS plus all stimuli tested (Fig. 2a–d). However, secretion of IL‐1β was significantly lower in macrophages from AnxA1−/− when these were activated with NLRP3 inflammasome activators, but not NLRC4 (Legionella) or AIM2 activators [Poly(dA:dT)] (Fig. 2a,b). Interestingly, secretion of TNF in response to LPS was unaffected in AnxA1−/− BMDMs (Fig. S1). These data suggest that the effect of AnxA1 on IL‐1β release seems to be specific for the NLRP3 inflammasome.
Figure 2.
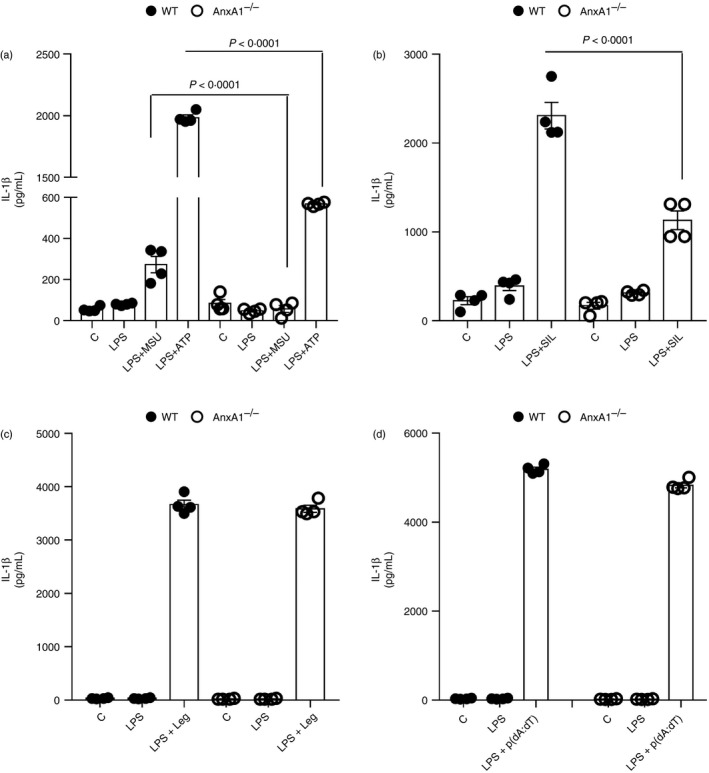
Effects of annexin A1 (AnxA1) on interleukin (IL)‐1β release seem to be specific for activators of the NLRP3 inflammasome. Bone marrow‐derived macrophages (BMDMs) from wild‐type (WT) and AnxA1−/− mice were stimulated with lipopolysaccharide (LPS; 1 μg/ml) for 1 hr followed by the indicated stimulus: 6 hr of uric acid crystals (MSU – 300 μg/ml) (a), 30 min of adenosine 5′‐triphosphate disodium salt (ATP – 5 mm) (A), 6 hr of Silica (SIL – 250 μg/ml) (b); 4 hr of Legionella pneumophila bacteria (Leg ‐ strain JR32 – MOI of 10) (c) and 2 hr of Poly(dA:dT) (p(dA:dT) – 5 μg/ml) (d). Supernatants were collected and IL‐1β levels were measured. Bars show the means ± SEM of 4 replicates/group and are from one experiment representative of three independent experiments. Significance was calculated using anova followed by Holm‐Sidak’s multiple comparison test.
AnxA1 does not require FPR2
To investigate whether the effects of AnxA1 on the NLRP3 inflammasome were dependent on AnxA1 receptor FPR2, WT and FPR2/3−/− mice were injected with an intra‐articular injection of uric acid crystals (MSU 100 μg/cavity) into the tibiofemoral knee joint to induce gout, and peri‐articular tissue was collected to measure IL‐1β. In contrast to the phenotype found in AnxA1−/− mice, FPR2/3−/− mice had increased levels of IL‐1β, as compared with WT mice (Fig. 3a). Moreover, we investigated the IL‐1β release in BMDMs from WT and FPR2/3−/− mice after stimulation with LPS alone or in combination with the NLRP3 inflammasome activators (MSU and ATP). There was a significant increase of IL‐1β release by BMDMs derived from FPR2/3−/− mice when compared with BMDMs derived from WT mice (Fig. 3b). Additionally, BMDMs obtained from WT mice treated with WRW4, a selective inhibitor of FPR2 receptor, showed the same pattern of results: significant increase of IL‐1β release in WRW4‐treated group when compared with untreated macrophages (Fig. S2a).
Figure 3.
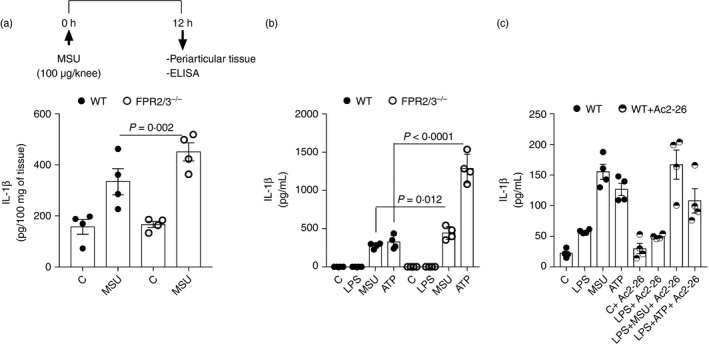
Effect of formyl‐peptide receptor 2 (FPR2) and Ac2‐26 on interleukin (IL)‐1β release. Wild‐type (WT) and FPR2/3−/− mice were injected with 100 μg/knee of uric acid crystals (MSU) in the tibiofemoral joint and, 12 hr after injection, peri‐articular tissue was collected to measure IL‐1β levels (a). Bone marrow‐derived macrophages (BMDMs) from WT and FPR2/3−/− mice were stimulated with lipopolysaccharide (LPS; 1 μg/ml) for 1 hr followed by the indicated stimulus: 6 hr of uric acid crystals (MSU – 300 μg/ml), 30 min of adenosine 5′‐triphosphate disodium salt (ATP – 5 mm). Supernatants were collected and IL‐1β levels were measured (b). BMDMs from WT mice were treated with the AnxA1 peptidomimetic Ac2‐26 (32 μm) and were stimulated with LPS (1 μg/ml) for 1 hr followed by stimulation with uric acid crystals (MSU – 300 μg/ml for 6 hr) or adenosine 5′‐triphosphate disodium salt (ATP − 5 mm for 30 min). Supernatants were collected to measure IL‐1β level (c). Bars show the means ± SEM of 5 mice/group (in vivo), and 4 replicates/group (in vitro) and are from one experiment representative of three independent experiments. Significance was calculated using anova followed by Holm‐Sidak’s multiple comparison test.
To determine whether exogenous AnxA1 could reconstitute NLRP3‐dependent IL‐1β release, we treated BMDMs from WT mice with the AnxA1 peptidomimetic Ac2‐26, and then stimulated the cells with LPS and MSU or ATP. The treatment with Ac2‐26 had no effect on IL‐1β release (Fig. 3c). Moreover, BMDMs obtained from AnxA1−/− mice treated with Ac2‐26 showed the same results: no effect on IL‐1β release in response to LPS and MSU or ATP (Fig. S2b). Together, these data suggest that AnxA1 seems to be acting independently of FPR2 and in an intracellular manner, once exogenously given Ac2‐26 did not reverse NLRP3 activation.
AnxA1 contributes to NLRP3 inflammasome priming
The absence of AnxA1 specifically reduced NLRP3‐dependent IL‐1β release, suggesting that AnxA1 could be involved in NLRP3 inflammasome activation/or assembly or influencing IL‐1β production. To investigate this, mRNA expression of Il‐1β, Nlrp3, Asc and Caspase‐1 was assessed in BMDMs from WT and AnxA1−/− stimulated with LPS. Although there was no significant effect of each genotype on gene expression (Fig. 4a–d), there was a trend for decreased expression of Il‐1β and Nlrp3 in BMDMs from AnxA1−/− mice (Fig. 4a,b). Furthermore, the same trend was also observed for Il‐1β mRNA expression in synovial tissue of AnxA1−/− mice injected with MSU compared with WT mice (Fig. S3a). In order to investigate if the priming process was TLR4 dependent, we evaluated the release of IL‐1β after priming BMDMs from WT and AnxA1−/− with LTA, a TLR2 agonist. We observed that IL‐1β was secreted in both WT and AnxA1−/− BMDMs, in response to LTA plus the NLRP3 stimuli tested (MSU or ATP). However, secretion of IL‐1β was significantly lower in AnxA1−/− macrophages after priming with LTA (Fig. S3b), as it was observed after LPS priming (Fig. 2a).
Figure 4.
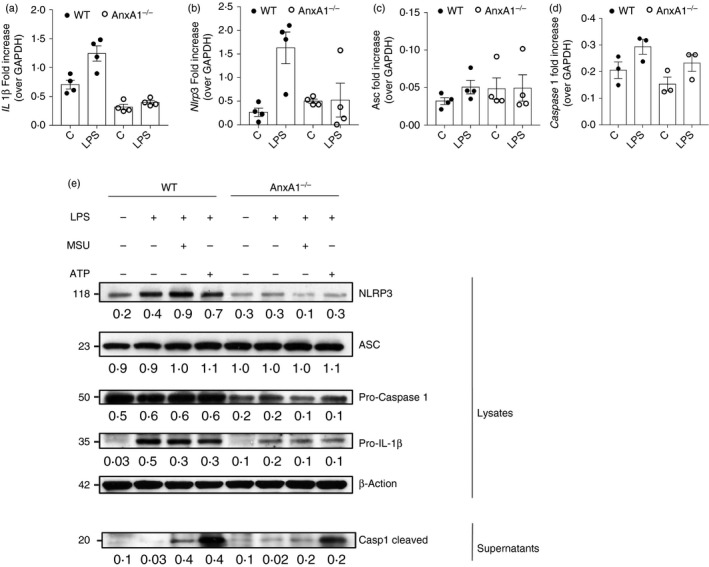
Annexin A1 (AnxA1) is involved in NLRP3 priming. Bone marrow‐derived macrophages (BMDMs) from wild‐type (WT) and AnxA1−/− mice were stimulated with lipopolysaccharide (LPS; 1 μg/ml) for 1 hr, and cells were collected to RNA extraction and quantitative polymerase chain reaction (qPCR) analysis for Il‐1β (a), Nlrp3 (b), Asc (c) and Caspase 1 (d) normalized by GAPDH. BMDMs from WT and AnxA1−/− mice were stimulated with LPS (1 μg/ml) for 1 hr and then stimulated with uric acid crystals (MSU – 300 μg/ml – 6 hr) and adenosine 5′‐triphosphate disodium salt (ATP – 5 mm – 30 min). Cells were collected for protein extraction and Western blot analysis for pro‐IL‐1β, NLRP3, ASC and pro‐caspase 1 (e). Supernatant were collected to analyse cleaved‐caspase 1 expression by Western blot (e). For loading control, membranes were re‐probed with anti‐β‐actin. Densitometry was performed, normalized by β‐actin and values (arbitrary units) are represented below respective lines. Bars show the means ± SEM of 4 replicates/group and are from one experiment representative of three independent experiments. Significance was calculated using anova followed by Holm‐Sidak’s multiple comparison test.
Next, we tested if the absence of AnxA1 affected translation and processing of proteins, which are pivotal for NLRP3 inflammasome activation. Intracellular levels of pro‐IL‐1β, NLRP3 and pro‐caspase‐1 protein were reduced in BMDMs from AnxA1−/− as compared with WT mice (Fig. 4e). In agreement with the data shown in Fig. 2, no difference was observed on ASC protein. In addition, to evaluate activation of inflammasome, levels of cleaved‐caspase‐1 in the supernatant of BMDMs stimulated with LPS, LPS + MSU or LPS + ATP were measured. There was a decrease in the release of cleaved‐caspase 1 in the supernatant from AnxA1−/− BMDMs (Fig. 4e). Together, these data suggest that AnxA1 contributes to NLRP3 inflammasome priming through NLRP3‐dependent processing of pro‐IL‐1β and pro‐caspase‐1 associated with an effect on protein expression.
AnxA1 interacts with NLRP3
The data gathered so far revealed a critical role of AnxA1 in NLRP3 priming and assembly. Next, we evaluated whether AnxA1 interacted with NLRP3. Confocal analysis of AnxA1 and NLRP3 staining in BMDMs from WT and AnxA1−/− mice (Fig. 5a,b) showed co‐localization of both proteins after LPS treatment followed by MSU only in BMDMs from WT mice (Fig. 5a). In order to demonstrate the interaction between AnxA1 and NLRP3, z‐sections were performed and a 3D video showed the co‐localization (Movie S1). To confirm whether AnxA1 interacted with NLRP3, we performed co‐immunoprecipitation (Co‐IP) experiments using AnxA1 antibody in BMDMs lysates from WT mice, after LPS priming plus MSU. Co‐IP revealed a direct interaction between AnxA1 and NLRP3 (Figs 6 and S4). These data suggest, for the first time, that AnxA1 interacts with NLRP3 and may be important for NLRP3 inflammasome priming and assembly.
Figure 5.
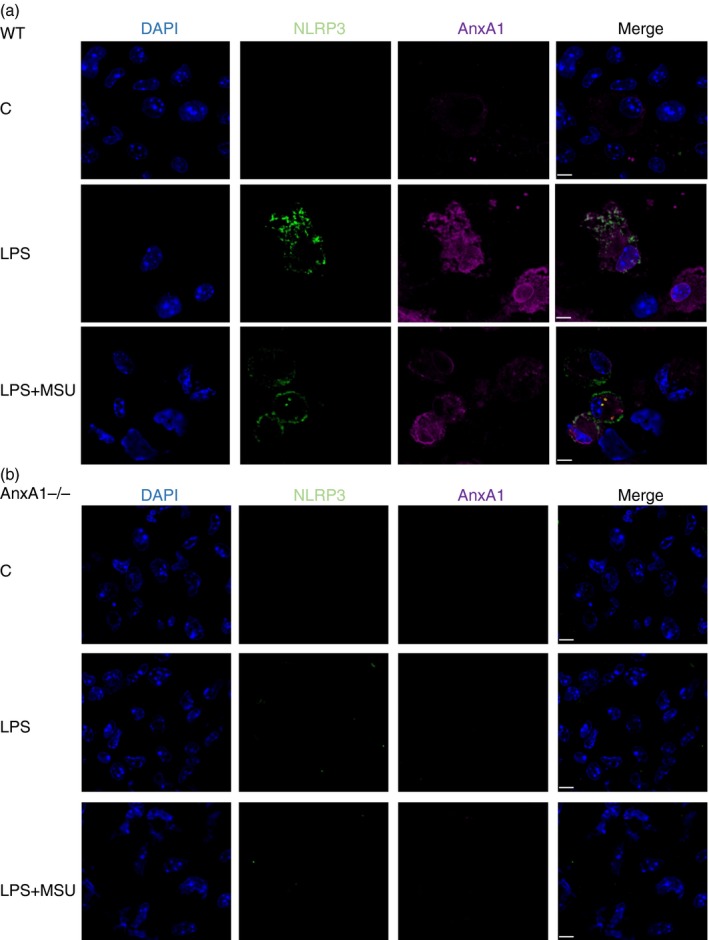
Annexin A1 (AnxA1) co‐localizes with NLRP3. Bone marrow‐derived macrophages (BMDMs) from wild‐type (WT) and AnxA1−/− mice were stimulated with lipopolysaccharide (LPS; 1 μg/ml) for 1 hr and then stimulated with uric acid crystals (MSU – 300 μg/ml ‐ 4 hr). Cells were collected and cytocentrifuged in a coverslip. After fixation and permeabilization, cells were stained with rabbit anti‐AnxA1 (Santa Cruz) and mouse anti‐NLRP3 (Adipogen) overnight. Then, secondary anti‐rabbit IgG Alexa Fluor 647, and anti‐mouse IgG FITC were incubated for 1 hr, and DAPI (1 : 1000 – BD) was used to stain nucleus. Co‐localization of AnxA1 (purple) and NLRP3 (green) were visualized using confocal microscopy. Scale bar: 100 μm. Figure is representative of at least 500 cells/glass slides analysed.
Figure 6.
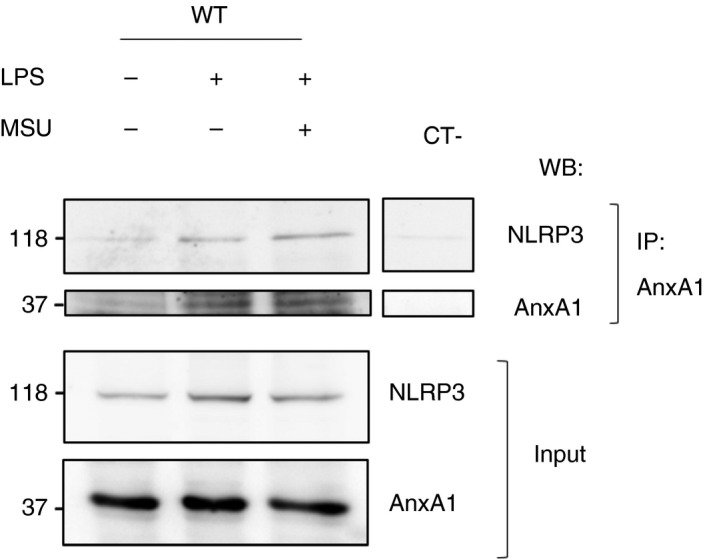
Annexin A1 (AnxA1) interacts directly with NLRP3. Bone marrow‐derived macrophages (BMDMs) from wild‐type (WT) mice were stimulated with lipopolysaccharide (LPS; 1 μg/ml) for 1 hr and then stimulated with uric acid crystals (MSU – 300 μg/ml ‐ 4 hr). Cells were collected to protein extraction. 1 mg of protein was incubated with anti‐AnxA1 (Santa Cruz) and then incubated with protein G Sepharose 4 fast flow beads (GE Healthcare), and Western blot analyses were performed. Representative immunoblot shows AnxA1 co‐immunoprecipitates NLRP3, and also shows AnxA1 and NLRP3 expression in the cell lysate (input). Immunoprecipitation control (p‐MYPT) is represented as CT‐. The immunoblots are representative of two independent experiments.
Discussion
Annexin A1 is a 37‐kDa glucocorticoid‐regulated protein member of the annexin superfamily. It has been characterized as an anti‐inflammatory/proresolving protein due to its actions in the modulation of the immune response. In particular, AnxA1 limits neutrophil recruitment, induces neutrophil apoptosis, enhances clearance of apoptotic cells and modulates cytokine synthesis. 5 The majority of the pharmacological properties described for AnxA1 have been shown to be mediated through binding to FPR2. 42 , 43 The role of intracellular AnxA1 is poorly understood. In this study, we investigated the production of IL‐1β in response to NLRP3 inflammasome activators in the presence or absence of AnxA1. Our major findings can be summarized as follows: (i) AnxA1 is required for IL‐1β release in response to NLRP3 activators; (ii) the need for AnxA1 for IL‐1β release in response to NLRP3 activators appears not to be dependent on its FPR2 receptor; (iii) mechanistically, AnxA1 contributes to NLRP3 inflammasome priming and directly interacts with NLRP3. Therefore, we have shown here that AnxA1 might be involved in NLRP3 priming and activation, regulating IL‐1β release and modulating the immune response.
Hannon et al. (2003) demonstrated that AnxA1‐deficient mice generated increased levels of IL‐1β 2 hr after zymosan injection in the peritoneum. However, this difference disappeared by 4 hr, 27 suggesting that IL‐1β levels did not follow the kinetics of inflammation in AnxA1‐deficient mice. In addition, peritoneal macrophages from AnxA1−/− mice presented delayed IL‐1β release after LPS stimulation, suggesting a dysregulated response of macrophages. 44 Using NLRP3‐dependent stimulate, we have observed in two different models and compartments, joint and lung, that AnxA1‐deficient mice had impaired IL‐1β release at the peak of inflammation, suggesting that AnxA1 was required, at least partially, for the increase of IL‐1β concentrations in a tissue.
Secretion of IL‐1β is tightly regulated. It is produced as a precursor, pro‐IL‐1β, that needs to be proteolytically cleaved into a mature and biologically active form, IL‐1β. To convert this cytokine, the activation of a cytosolic protein complex called inflammasome is necessary. The most studied inflammasome complex is NLRP3. 45 However, depending on the insult, other PRRs can be activated to cleave pro‐IL‐1β. 23 , 40 , 46 Here we observed that AnxA1 is required for IL‐1β release only after NLRP3 stimulation, suggesting that AnxA1 may specifically regulate IL‐1β release by NLRP3. Indeed, other molecules have been described for NLRP3‐dependent IL‐1β release, such as macrophage inhibitory factor (MIF), 25 microtubule‐affinity regulating kinase 4 (MARK4), 47 NEK7 48 and Vimentin, 26 which suggest that NLRP3 needs other regulatory pathways in order to avoid deleterious effects.
Annexin A1 might be required for optimal activation of macrophages, once AnxA1 was identified as a regulator of TLR‐induced IFN‐β and CXCL10 secretion. 49 Bits et al. demonstrated that AnxA1 associated with TBK1, a downstream signalling‐associated molecule of the TRIF signalling pathway, which was independent on FPR2. Similar to these findings, our results demonstrated that the impairment on IL‐1β release was independent on the AnxA1 receptor. Indeed, our results showed that FPR2/3 knockout mice and pharmacological inhibition of FPR2 by using WRW4 led not to inhibition but an increase of the levels of IL‐1β. In fact, exogenous AnxA1 acting on FPR2 may downmodulate the Myd88/NFκB pathway and downregulate IL‐1β secretion, 50 although we found here that pre‐treatment of BMDMs with Ac2‐26 had no effect on IL‐1β release.
For NLRP3 inflammasome activation, two distinct pro‐inflammatory stimuli are required. In the first step, TLR signalling activates NF‐κB and upregulates NLRP3 and pro‐IL‐1β. In resting conditions, macrophages have low levels of NLRP3 and pro‐IL‐1β. 51 Here, we described that AnxA1 is required for this first step of priming, once AnxA1 knockout mice presented low proteins levels of NLRP3, pro‐caspase‐1 and pro‐IL‐1β. Priming does not change ASC expression, 15 as we confirmed in our result. The priming step is not only dependent on TLR4 stimulation, as TLR2 agonists can also work as a priming signal activating NFκB‐inducing transcription of pro‐IL‐1β and NLRP3. 15 The priming activation step is dependent on NFκB activation. A previous study has demonstrated that AnxA1 associated physically with NFκB. AnxA1 interacted with p65 and recruited it to the nucleus to trigger IL‐1β transcription and caspase‐1 cleavage to affect the maturation of IL‐1β. 52 Similar to these findings, our results showed that AnxA1 knockout mice had an impaired post‐transcriptional step.
It has been shown that AnxA1 regulates TNF mRNA stability, 53 suggesting that AnxA1 might participate in the regulation of mRNA half‐life from other cytokines and could explain the mRNA levels observed here. However, mRNA levels were not significantly different when comparing WT and AnxA1 knockout mice. The reason why the mRNA levels did not follow the protein levels was not clear, and previous studies have demonstrated similar results to ours. 54 Nevertheless, transcription‐independent roles of the priming signal have been recently shown. 55 LPS can prime the NLRP3 inflammasome activation independently of NLRP3 induction, once a short‐term pre‐treatment with LPS did not change NLRP3 expression. 56 Together, the priming step seems to be more involved in licensing NLRP3 inflammasome activation beyond the transcriptional level, 57 and AnxA1 seems to participate in this license step.
Annexin A1 is not only capable of binding phospholipid‐containing membranes, but also to mediate membrane vesicle aggregation and interact with cellular protein ligands. 58 Analysis of AnxA1 proteins interactions has identified 436 proteins associated with AnxA1 being S100A9 and Vimentin two key AnxA1‐interacted proteins confirmed by Co‐IP. 59 Here, we have shown that AnxA1 directly interacts with NLRP3 by Co‐IP. This interaction seems to occur in the cytoplasm, as observed by the co‐localization of NLRP3 and AnxA1 in the confocal analysis. It is important to highlight that vimentin also binds to NLRP3, and might serve as a scaffold for the inflammasome by binding to one or more proteins in the inflammasome complex. 26 However, whether AnxA1 needs vimentin to bind to NLRP3 remains to be investigated and is beyond the scope of this work.
Annexin A1 can bind to membrane phospholipids in a Ca2+‐dependent manner. 58 Moreover, constitutive AnxA1 defines Ca2+ homeostasis, and any change in AnxA1 levels can cause alteration of intracellular Ca2+ release. 60 Interestingly, the increased cytoplasmic Ca2+ promotes the assembly of inflammasome. 61 Our results suggest that AnxA1 has a role not only on the priming step, but also in the assembling step, which could be explained by AnxA1 inducing changes in Ca2+ homeostasis. However, the direct association of AnxA1, Ca2+ and NLRP3 assembly should be better investigated.
In conclusion, the present study showed that AnxA1 plays a role in the production, processing and release of IL‐1β. The absence of AnxA1 resulted in reduced production and release of IL‐1β in response of NLRP3 stimulus. Moreover, AnxA1 directly interacts with NLRP3, suggesting that AnxA1 is involved in NLRP3 inflammasome priming and assembly.
Author contributions
I.G., F.M.R., G.B.M.., D.S.Z., L.P.S., M.M.T. designed the research and wrote the paper. I.G., R.N.H.C., J.P.V., A.L.N.S., T.G.C., M.M.A. performed the experiments and analysed the data.
Disclosure
The authors declare the submitted work was not carried out in the presence of any personal, professional and financial relationships that could potentially be construed as a conflict of interest.
Supporting information
Figure S1. LPS‐induced TNF release in BMDMs from AnxA1−/− and WT mice.
{kind=link}
Figure S2. Effects of the treatment with a FPR2 antagonist (WRW4) or an AnxA1 peptidomimetic (Ac2‐26) on IL‐1β release from WT and AnxA1−/− BMDMs.
{kind=link}
Figure S3. NLRP3 priming process in AnxA1−/− mice.
{kind=link}
Figure S4. Original Western blot from NLRP3 Co‐IP.
{kind=link}
Movie S1. Co‐localization between AnxA1 and NLRP3.
Acknowledgements
The authors would like to thank Frankcinéia Assis and Ilma Marçal for technical assistance. This work was supported by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil), Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG, Brazil), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brazil).
References
- 1. Mussunoor S, Murray GI. The role of annexins in tumour development and progression. J Pathol 2008; 216:131–40. [DOI] [PubMed] [Google Scholar]
- 2. Flower RJ, Rothwell NJ. Lipocortin‐1: cellular mechanisms and clinical relevance. Trends Pharmacol Sci 1994; 15:71–6. [DOI] [PubMed] [Google Scholar]
- 3. Sheikh MH, Solito E. Annexin A1: uncovering the many talents of an old protein. Int J Mol Sci 2018;19:e1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. D'Acquisto F, Perretti M, Flower RJ. Annexin‐A1: a pivotal regulator of the innate and adaptive immune systems. Br J Pharmacol. 2008; 155:152–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sugimoto MA, Vago JP, Teixeira MM, Sousa LP. Annexin A1 and the resolution of inflammation: modulation of neutrophil recruitment, apoptosis, and clearance. J Immunol Res 2016; 2016:8239258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. D'Acquisto F, Piras G, Rattazzi L. Pro‐inflammatory and pathogenic properties of Annexin‐A1: the whole is greater than the sum of its parts. Biochem Pharmacol 2013; 85:1213–8. [DOI] [PubMed] [Google Scholar]
- 7. Tagoe CE, Marjanovic N, Park JY, Chan ES, Abeles AM, Attur M et al Annexin‐1 mediates TNF‐alpha‐stimulated matrix metalloproteinase secretion from rheumatoid arthritis synovial fibroblasts. J Immunol 2008; 181:2813–20. [DOI] [PubMed] [Google Scholar]
- 8. D'Acquisto F, Merghani A, Lecona E, Rosignoli G, Raza K, Buckley CD et al Annexin‐1 modulates T‐cell activation and differentiation. Blood 2007; 109(3):1095–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Williams SL, Milne IR, Bagley CJ, Gamble JR, Vadas MA, Pitson SM et al A proinflammatory role for proteolytically cleaved annexin A1 in neutrophil transendothelial migration. J Immunol 2010; 185:3057–63. [DOI] [PubMed] [Google Scholar]
- 10. Kwon JH, Lee JH, Kim KS, Chung YW, Kim IY. Regulation of cytosolic phospholipase A2 phosphorylation by proteolytic cleavage of annexin A1 in activated mast cells. J Immunol 2012; 188:5665–73. [DOI] [PubMed] [Google Scholar]
- 11. Bist P, Leow SC, Phua QH, Shu S, Zhuang Q, Loh WT et al Annexin‐1 interacts with NEMO and RIP1 to constitutively activate IKK complex and NF‐kappaB: implication in breast cancer metastasis. Oncogene 2011; 30:3174–85. [DOI] [PubMed] [Google Scholar]
- 12. Zhang Z, Huang L, Zhao W, Rigas B. Annexin 1 induced by anti‐inflammatory drugs binds to NF‐kappaB and inhibits its activation: anticancer effects in vitro and in vivo. Cancer Res 2010; 70:2379–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature 2006; 442:39–44. [DOI] [PubMed] [Google Scholar]
- 14. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol 2009; 27:229–65. [DOI] [PubMed] [Google Scholar]
- 15. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D et al Cutting edge: NF‐kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 2009; 183:787–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de Carvalho RVH, Andrade WA, Lima‐Junior DS, Dilucca M, de Oliveira CV, Wang K et al Leishmania lipophosphoglycan triggers caspase‐11 and the non‐canonical activation of the NLRP3 inflammasome. Cell Rep 2019; 26:429–37. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J et al Non‐canonical inflammasome activation targets caspase‐11. Nature 2011; 479:117–21. [DOI] [PubMed] [Google Scholar]
- 18. de Castro‐Jorge LA, de Carvalho RVH, Klein TM, Hiroki CH, Lopes AH, Guimaraes RM et al The NLRP3 inflammasome is involved with the pathogenesis of Mayaro virus. PLoS Pathog 2019; 15:e1007934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Man SM, Kanneganti TD. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat Rev Immunol 2016; 16:7–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dinarello CA. Immunological and inflammatory functions of the interleukin‐1 family. Annu Rev Immunol 2009; 27:519–50. [DOI] [PubMed] [Google Scholar]
- 21. van den Berg WB. Arguments for interleukin 1 as a target in chronic arthritis. Ann Rheum Dis 2000; 59(Suppl 1):i81–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ruscitti P, Cipriani P, Di Benedetto P, Liakouli V, Berardicurti O, Carubbi F et al Monocytes from patients with rheumatoid arthritis and type 2 diabetes mellitus display an increased production of interleukin (IL)‐1beta via the nucleotide‐binding domain and leucine‐rich repeat containing family pyrin 3(NLRP3)‐inflammasome activation: a possible implication for therapeutic decision in these patients. Clin Exp Immunol 2015; 182:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout‐associated uric acid crystals activate the NALP3 inflammasome. Nature 2006; 440:237–41. [DOI] [PubMed] [Google Scholar]
- 24. Kim RY, Pinkerton JW, Essilfie AT, Robertson AAB, Baines KJ, Brown AC et al Role for NLRP3 inflammasome‐mediated, IL‐1beta‐dependent responses in severe, steroid‐resistant asthma. Am J Respir Crit Care Med 2017; 196:283–97. [DOI] [PubMed] [Google Scholar]
- 25. Lang T, Lee JPW, Elgass K, Pinar AA, Tate MD, Aitken EH et al Macrophage migration inhibitory factor is required for NLRP3 inflammasome activation. Nat Commun 2018; 9:2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. dos Santos G, Rogel MR, Baker MA, Troken JR, Urich D, Morales‐Nebreda L et al Vimentin regulates activation of the NLRP3 inflammasome. Nat Commun 2015; 6:6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hannon R, Croxtall JD, Getting SJ, Roviezzo F, Yona S, Paul‐Clark MJ et al Aberrant inflammation and resistance to glucocorticoids in annexin 1‐/‐ mouse. FASEB J 2003; 17:253–5. [DOI] [PubMed] [Google Scholar]
- 28. Dufton N, Hannon R, Brancaleone V, Dalli J, Patel HB, Gray M et al Anti‐inflammatory role of the murine formyl‐peptide receptor 2: ligand‐specific effects on leukocyte responses and experimental inflammation. J Immunol 2010; 184:2611–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Amaral FA, Costa VV, Tavares LD, Sachs D, Coelho FM, Fagundes CT et al NLRP3 inflammasome‐mediated neutrophil recruitment and hypernociception depend on leukotriene B(4) in a murine model of gout. Arthritis Rheum 2012; 64:474–84. [DOI] [PubMed] [Google Scholar]
- 30. Trentin PG, Ferreira TP, Arantes AC, Ciambarella BT, Cordeiro RS, Flower RJ et al Annexin A1 mimetic peptide controls the inflammatory and fibrotic effects of silica particles in mice. Br J Pharmacol 2015; 172:3058–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marim FM, Silveira TN, Lima DS Jr, Zamboni DS. A method for generation of bone marrow‐derived macrophages from cryopreserved mouse bone marrow cells. PLoS One 2010; 5:e15263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Islam MA, Proll M, Holker M, Tholen E, Tesfaye D, Looft C et al Alveolar macrophage phagocytic activity is enhanced with LPS priming, and combined stimulation of LPS and lipoteichoic acid synergistically induce pro‐inflammatory cytokines in pigs. Innate Immun 2013; 19:631–43. [DOI] [PubMed] [Google Scholar]
- 33. Machado MG, Tavares LP, Souza GVS, Queiroz‐Junior CM, Ascencao FR, Lopes ME et al The Annexin A1/FPR2 pathway controls the inflammatory response and bacterial dissemination in experimental pneumococcal pneumonia. FASEB J 2019; 34:2749–64. [DOI] [PubMed] [Google Scholar]
- 34. Lima KM, Vago JP, Caux TR, Negreiros‐Lima GL, Sugimoto MA, Tavares LP et al The resolution of acute inflammation induced by cyclic AMP is dependent on annexin A1. J Biol Chem 2017; 292:13 758–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL et al Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 2008; 9:847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sadatomi D, Nakashioya K, Mamiya S, Honda S, Kameyama Y, Yamamura Y et al Mitochondrial function is required for extracellular ATP‐induced NLRP3 inflammasome activation. J Biochem 2017; 161:503–12. [DOI] [PubMed] [Google Scholar]
- 37. Goncalves AV, Margolis SR, Quirino GFS, Mascarenhas DPA, Rauch I, Nichols RD et al Gasdermin‐D and Caspase‐7 are the key Caspase‐1/8 substrates downstream of the NAIP5/NLRC4 inflammasome required for restriction of Legionella pneumophila. PLoS Pathog 2019; 15:e1007886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vago JP, Nogueira CR, Tavares LP, Soriani FM, Lopes F, Russo RC et al Annexin A1 modulates natural and glucocorticoid‐induced resolution of inflammation by enhancing neutrophil apoptosis. J Leukoc Biol 2012; 92:249–58. [DOI] [PubMed] [Google Scholar]
- 39. Sousa LP, Silva BM, Brasil BS, Nogueira SV, Ferreira PC, Kroon EG et al Plasminogen/plasmin regulates alpha‐enolase expression through the MEK/ERK pathway. Biochem Biophys Res Commun 2005; 337:1065–71. [DOI] [PubMed] [Google Scholar]
- 40. Fernandes‐Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009; 458:509–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Raka F, Di Sebastiano AR, Kulhawy SC, Ribeiro FM, Godin CM, Caetano FA et al Ca(2+)/calmodulin‐dependent protein kinase II interacts with group I metabotropic glutamate and facilitates receptor endocytosis and ERK1/2 signaling: role of beta‐amyloid. Mol Brain 2015; 8:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gavins FN, Hughes EL, Buss NA, Holloway PM, Getting SJ, Buckingham JC. Leukocyte recruitment in the brain in sepsis: involvement of the annexin 1‐FPR2/ALX anti‐inflammatory system. FASEB J 2012; 26:4977–89. [DOI] [PubMed] [Google Scholar]
- 43. Yazid S, Norling LV, Flower RJ. Anti‐inflammatory drugs, eicosanoids and the annexin A1/FPR2 anti‐inflammatory system. Prostaglandins Other Lipid Mediat 2012; 98:94–100. [DOI] [PubMed] [Google Scholar]
- 44. Damazo AS, Yona S, D'Acquisto F, Flower RJ, Oliani SM, Perretti M. Critical protective role for annexin 1 gene expression in the endotoxemic murine microcirculation. Am J Pathol 2005; 166:1607–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci 2016; 41:1012–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pereira MS, Morgantetti GF, Massis LM, Horta CV, Hori JI, Zamboni DS. Activation of NLRC4 by flagellated bacteria triggers caspase‐1‐dependent and ‐independent responses to restrict Legionella pneumophila replication in macrophages and in vivo. J Immunol 2011; 187:6447–55. [DOI] [PubMed] [Google Scholar]
- 47. Li X, Thome S, Ma X, Amrute‐Nayak M, Finigan A, Kitt L et al MARK4 regulates NLRP3 positioning and inflammasome activation through a microtubule‐dependent mechanism. Nat Commun. 2017; 8:15 986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schmid‐Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E et al A genome‐wide CRISPR (clustered regularly interspaced short palindromic repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J Biol Chem 2016; 291:103–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bist P, Shu S, Lee H, Arora S, Nair S, Lim JY et al Annexin‐A1 regulates TLR‐mediated IFN‐beta production through an interaction with TANK‐binding kinase 1. J Immunol 2013; 191:4375–82. [DOI] [PubMed] [Google Scholar]
- 50. Pantaleao L, Rocha GHO, Reutelingsperger C, Tiago M, Maria‐Engler SS, Solito E et al Connections of annexin A1 and translocator protein‐18kDa on toll like receptor stimulated BV‐2 cells. Exp Cell Res 2018; 367(2):282–90. [DOI] [PubMed] [Google Scholar]
- 51. Bauernfeind F, Bartok E, Rieger A, Franchi L, Nunez G, Hornung V. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol 2011; 187:613–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhao Y, Li X, Gong J, Li L, Chen L, Zheng L et al Annexin A1 nuclear translocation induces retinal ganglion cell apoptosis after ischemia‐reperfusion injury through the p65/IL‐1beta pathway. Biochim Biophys Acta Mol Basis Dis 2017; 1863:1350–8. [DOI] [PubMed] [Google Scholar]
- 53. Nair S, Arora S, Lim JY, Lee LH, Lim LH. The regulation of TNFalpha production after heat and endotoxin stimulation is dependent on Annexin‐A1 and HSP70. Cell Stress Chaperones 2015; 20:583–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schindler R, Clark BD, Dinarello CA. Dissociation between interleukin‐1 beta mRNA and protein synthesis in human peripheral blood mononuclear cells. J Biol Chem 1990; 265:10 232–7. [PubMed] [Google Scholar]
- 55. de Carvalho RVH, Silva ALN, Santos LL, Andrade WA, de Sa KSG, Zamboni DS. Macrophage priming is dispensable for NLRP3 inflammasome activation and restriction of Leishmania amazonensis replication. J Leukoc Biol 2019; 106:631–40. [DOI] [PubMed] [Google Scholar]
- 56. Schroder K, Sagulenko V, Zamoshnikova A, Richards AA, Cridland JA, Irvine KM et al Acute lipopolysaccharide priming boosts inflammasome activation independently of inflammasome sensor induction. Immunobiology 2012; 217:1325–9. [DOI] [PubMed] [Google Scholar]
- 57. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci 2019; 20:3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gerke V, Moss SE. Annexins: from structure to function. Physiol Rev 2002; 82:331–71. [DOI] [PubMed] [Google Scholar]
- 59. Xiao Y, Ouyang C, Huang W, Tang Y, Fu W, Cheng A. Annexin A1 can inhibit the in vitro invasive ability of nasopharyngeal carcinoma cells possibly through Annexin A1/S100A9/Vimentin interaction. PLoS One 2017; 12:e0174383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Frey BM, Reber BF, Vishwanath BS, Escher G, Frey FJ. Annexin I modulates cell functions by controlling intracellular calcium release. FASEB J 1999; 13:2235–45. [DOI] [PubMed] [Google Scholar]
- 61. Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach‐Mansky R, Sacks DB et al The calcium‐sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012; 492:123–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. LPS‐induced TNF release in BMDMs from AnxA1−/− and WT mice.
Figure S2. Effects of the treatment with a FPR2 antagonist (WRW4) or an AnxA1 peptidomimetic (Ac2‐26) on IL‐1β release from WT and AnxA1−/− BMDMs.
Figure S3. NLRP3 priming process in AnxA1−/− mice.
Figure S4. Original Western blot from NLRP3 Co‐IP.
Movie S1. Co‐localization between AnxA1 and NLRP3.