

Abstract
Objective
To describe changes in Japanese clinical trial regulations after the implementation of the Clinical Trials Act in April 2018.
Methods
First, how to apply multiple regulations after the enforcement of Clinical Trials Act was described. Second, the changes in the number of clinical trials in the National Cancer Center Hospital under each regulation were compared before and after the implementation of Clinical Trials Act. Third, new requirements imposed by Clinical Trials Act and their influences were discussed.
Results
In April 2018, Clinical Trials Act was enacted and academic clinical trials were classified into the following three categories: (i) investigator-initiated registration-directed trial under the Pharmaceuticals and Medical Devices Act; (ii) clinical trial under Clinical Trials Act; and (iii) clinical trial under the Ethical Guidelines. While 90% (205/227) of interventional studies were conducted under the Ethical Guidelines before the implementation of Clinical Trials Act in 2018, 46% (94/204) were subject to Clinical Trials Act in 2019 at the National Cancer Center Hospital. Under the Clinical Trials Act, investigators receive a scientific/ethical review by a certified review board (CRB). The identification of investigators in charge is mandated and they are required to submit the conflict of interest management plan to CRB. After the CRB review, the principal investigator must submit the trial plan to the government, and the content is uploaded to the newly established clinical trial registry site, the Japan Registry of Clinical Trials.
Conclusions
The enforcement of the new Clinical Trials Act was supposed to improve the reliability of academic clinical trials in Japan; however, the financial and administrative burden may reduce clinical trial activity in the years to come.
Keywords: Japanese regulation, Clinical Trials Act, Ethical Guidelines, Advanced Medical Care, Specified clinical trial
After the implementation of Clinical Trials Act, 46% of the examined interventional studies were subject to the Act. Its financial and administrative burden may reduce clinical trial activity in Japan.
Background
Japan is the second largest drug market in the world (1) and one of the three countries/economic blocs that originally make up the International Committee on Harmonization. While registration-directed clinical trials are regulated by the Pharmaceuticals and Medical Devices Act (PMD Act), Japanese Good Clinical Practice (GCP) and other relevant ordinances, non-registration clinical trials were previously regulated using less stringent guidelines: the Ethical Guidelines for Medical and Health Research Involving Human Subjects (‘Ethical Guidelines’, hereafter) (2).
However, due to several instances of scientific misconduct, such as the Valsartan case (3,4), more strict regulation was demanded. Therefore, the new Clinical Trials Act (CTA) was adopted by the National Diet in 2017 and enacted in April 2018 (5). The essential issues in the Valsartan case were the lack of a mechanism to ensure the study quality and the unclear relationship between academia and the funding industry. Following the enforcement of the new CTA, the landscape of Japanese regulations surrounding academic clinical trials has become more complicated, which may have a negative influence on international academic clinical trials that include Japanese institutions. However, as far as we are aware, there has been no report comprehensively describing this complicated regulatory situation.
In this manuscript, details of updated Japanese regulations for academic clinical trials are described and their influence on Japanese clinical trial activity is discussed.
Applicable regulations for academic clinical trials in Japan
Before the enforcement of CTA, academic clinical trials were simply classified into two categories from a regulatory perspective: (i) investigator-initiated registration-directed trial (IIRDT) under the PMD Act and (ii) clinical trial under the Ethical Guidelines (Fig. 1A). Clinical trials using unapproved or off-label regenerative medical products are regulated by another law, the Act to Ensure the Safety of Regenerative Medicine; however, these details are omitted in this manuscript because the number of the trials is relatively law. Although most trials were classified as being under the Ethical Guidelines, these were not laws adopted by the National Diet, but simply an administrative guidance jointly issued by the Ministry of Education, Culture, Sports, Science and Technology and the Ministry of Health, Labour and Welfare (MHLW); thus, the Ethical Guidelines did not have legal binding force, and this led the establishment of CTA.
Figure 1.
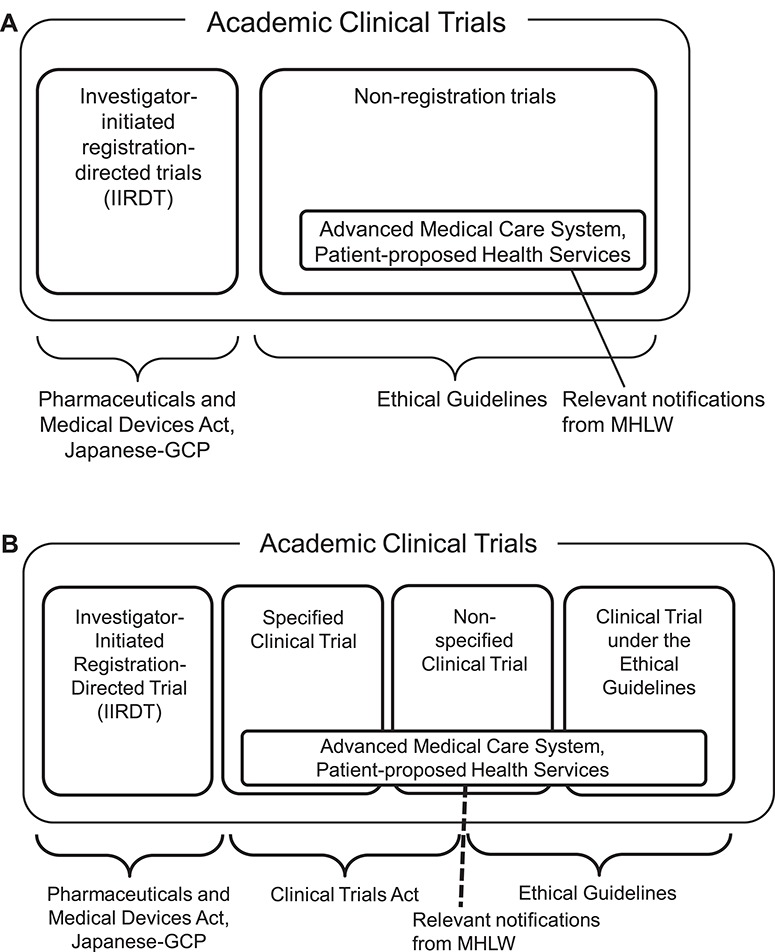
(A) Past classification and applicable regulations of Japanese academic clinical trials applied before April 2018. (B) New classification and applicable regulations of Japanese academic clinical trials applied after April 2018.
In April 2018, CTA was enacted and academic clinical trials came to be classified into the following four categories: (i) IIRDT under the PMD Act; (ii) clinical trial obliged to comply with CTA, which is called a specified clinical trial; (iii) clinical trial required to make effort to follow CTA; and iv) clinical trial under the Ethical Guidelines (Fig. 1B).
A categorization flowchart is shown in Fig. 2. First, when the investigator intends to establish clinical trial data for obtaining a new pharmaceutical application or expanding indication, it is classified into IIRDT and must follow the PMD Act. Next, when investigators intend to clarify the efficacy or safety of pharmaceuticals, medical devices or regenerative products (‘pharmaceuticals’, hereafter) in humans, the clinical trial is subject to CTA. If they use unapproved/off-label pharmaceuticals or they receive research funding by a manufacturer with marketing approval for pharmaceuticals, the clinical trial is called a specified clinical trial and investigators must comply with CTA. If the clinical trial does not fulfill the above two conditions, it is called a non-specific clinical trial and investigators are required to ‘make effort’ to conduct the trial in compliance with CTA. In other words, the applicability of the act is limited if investigators have an appropriate reason for not complying (e.g. lack of research funding). While investigators must file a clinical trial application to the MHLW in the specified clinical trials, it is not required for non-specified clinical trials. Due to the soft application of the act, some non-specified clinical trials were conducted under the Ethical Guidelines. Finally, when investigators do not intend to clarify the efficacy or safety of pharmaceuticals (for example, surgical trials or simple radiotherapy trials), the trials are conducted under the Ethical Guidelines. Non-interventional studies are also subjected to the Ethical Guidelines.
Figure 2.
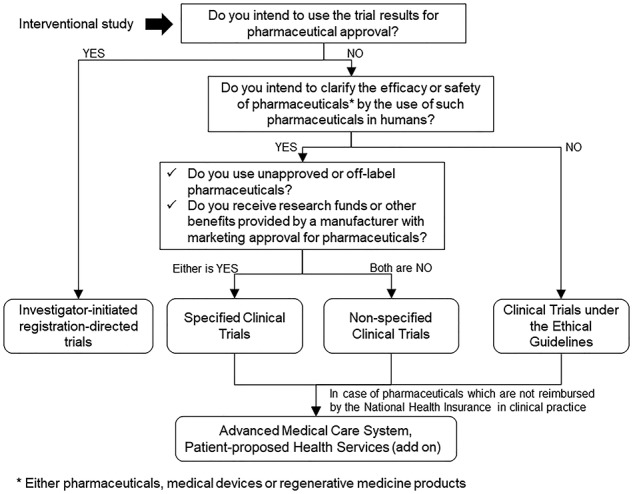
Flowchart of the regulatory categorization of Japanese academic clinical trials.
Number of clinical trials by regulations
Figure 3 shows the recent trend in the number of academic clinical trials (interventional studies) in the National Cancer Center Hospital. Although the investigator has discretion in deciding whether to comply with CTA or the Ethical Guidelines in non-specified clinical trials, most trials are performed in accordance with the Ethical Guidelines due to budget limitations. While 90% (205/227) of interventional studies were conducted under the Ethical Guidelines before the implementation of CTA in 2018, 46% (94/204) were subject to CTA in 2019 at the National Cancer Center Hospital.
Figure 3.
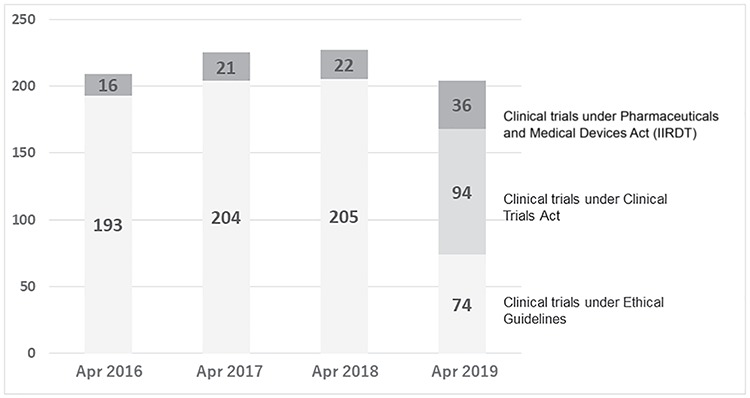
Trend in the number of academic interventional studies in the National Cancer Center Hospital, Japan (2016–2019).
Investigator-initiated registration-directed trials
When the purpose of the clinical trial is a new drug application for the Pharmaceuticals and Medical Devices Agency (PMDA), clinical trials should be conducted under the PMD Act and also comply with Japanese GCP and other relevant ordinances. These registration-directed trials are also known as ‘Chiken’. Previously, Chiken was allowed only to pharmaceutical companies; however, Chiken has become available even to investigators following the amendment of the Pharmaceutical Affairs Law in 2003, and the Japanese GCP has been modified specifically for IIRDT. The Japanese GCP is almost compatible with ICH-GCP, yet there are some differences in the IIRDT. One of the major differences is the concept of a study sponsor. In the Japanese GCP, principal investigators in all participating institutions serve as the study sponsor for IIRDT and the hospital directors take some responsibility for study conduct and the related contract. These differences can present an obstacle for international clinical trials.
Ethical Guidelines for Medical and Health Research Involving Human Subjects
The original Ethical Guidelines for clinical researches were established in 2003. Most parts of the original guidelines focused on the protection of research subjects, such as institutional ethical reviews, informed consent and protection of personal information; however, the guidelines did not mention study quality. In response to the Valsartan case, the Ethical Guidelines were amended in 2014, which introduced requirements to ensure study quality such as mandating monitoring for interventional studies, and also reinforced the conflict of interest (COI) management. Due to this amendment, the Ethical Guidelines came much closer to ICH-GCP, but they were not fully compatible with ICH-GCP. In addition, there was no documented decision criteria by PMDA concerning whether the data collected with ICH-GCP could be used for a new drug application. Thus, clinical trial data under the Ethical Guidelines could generally not be used for a new drug application.
Changes in the CTA
Although CTA covers most essential elements required under ICH-GCP and these are also covered in the Ethical Guidelines, some unique requirements have been newly imposed under CTA.
Single review by a certified review board
One of the largest changes in the act is that a single scientific/ethical review has become necessary. Even before the implementation of the act, a central review had been acceptable under the Ethical Guidelines. However, each hospital traditionally had its own institutional review board (IRB) with the hospital director as part of its consulting body and, in most cases, the in-house IRB performed the review even in multi-institutional clinical trials. The involvement of too many IRBs and the inconsistent quality of the ethical reviews were two of the major issues in the Valsartan case. As a result, a certification system for the review board has been introduced by the MHLW and single scientific/ethical review is required by these certified review boards (CRB). As of 2019, more than 90 CRB have been certified by the MHLW (6). Although most IRBs under the Ethical Guidelines are free of charge, all CRBs impose review fees, which range from 800 USD to more than 50 000 USD (6). This financial burden may reduce the activity of academic clinical trials.
Strict COI management
The unclear relationship between investigators and the industry was criticized in the Valsartan case. Under the Ethical Guidelines, there was no need to clearly identify the investigators who were in charge of the clinical trial. The identification of the principal investigator and sub-investigators is mandated under the CTA, and all participating sites are required to submit a COI management plan to CRB. Each participating site is obliged to confirm the accuracy of each investigator’s COI declaration, which has increased the burden on each site.
Submission of a trial plan and entry to new clinical trial registry
Under CTA, the representative principal investigator must submit the trial plan to the MHLW before the start of the clinical trial. This trial plan includes not only the contents specified by the WHO Registry Network but also the information of participating sites, such as the contact information of the principal investigator, the site contact details for inquiries concerning the trial, the name of the hospital director and a status of approval from the hospital director about the trial’s implementation, CRB approval date and facility details. If any of the information in the trial plan is changed, it must be reviewed by the CRB and submitted to the MHLW. Since such information changes frequently, the representative principal investigator is forced to manage the frequent revision of the trial plan. Moreover, the potentially related industries who will sell the pharmaceuticals used in the trial should be listed in the trial plan, even if there is no financial support. Such burden is one of the factors which is decreasing the number of academic clinical trials.
In addition, the contents of the trial plan must be uploaded to the newly established clinical trial registry website, the Japan Registry of Clinical Trials (jRCT). Previously, there were three official clinical trial registries in Japan (UMIN-CTR, JapicCTI, JMACCT-CTR), and jRCT has become the fourth one. Because all clinical trials conducted under CTA are obliged to register with the jRCT, unification of other three clinical trial registries into jRCT is expected.
Advanced Medical Care system and Patient-Proposed Health Services
Advanced Medical Care (AMC) and Patient-Proposed Health Services (PHS) are the systems that enable investigators to conduct clinical trials with unapproved/off-label treatments, partially covered by the National Health Insurance (Fig. 2). In Japan, the National Health Insurance is basically applied to all citizens, yet medical care including unapproved/off-label treatment is not covered by the insurance, except for a few exceptions such as IIRDT, AMC and PHS. The National Health Insurance, similar to the Medicare Clinical Trial Policy system in the United States, will cover the routine cost when a clinical trial notification IIRDT is accepted by PMDA or an AMC/PHS application to MHLW is approved by the relevant committees in MHLW. The difference between AMC and PHS is that AMC is planned by an investigator to examine the usefulness of the treatment whereas PHS is proposed by a patient to receive an unapproved/off-label drugs. Because the applicable regulation for the AMC is the AMC notification issued by MHLW in addition to CTA or the Ethical Guidelines, which require less stringent quality control and data traceability than the PMD Act, AMC is utilized whenever the budget is not enough to conduct IIRDT. On the other hand, the possibility of AMC trial results being utilized for drug approval/label change is not always high. Only when the effectiveness of the test treatment in the AMC trial is publicly endorsed, for example it appeared in public treatment guidelines, the test treatment is likely to be approved by MHLW as ‘Kouchi-Shinsei’ (7), i.e. a supplemental new drug application is publicly endorsed. Since there are few examples of AMC that have been approved through ‘Kouchi-Shinsei’, IIRDT is still the most effective way to achieve drug approval/label change, assuming the investigators have sufficient budget.
Discussion
The enforcement of the new CTA is expected to improve the reliability of academic clinical trials in Japan; however, the regulatory classification has become more complicated. While the Ethical Guidelines were just an administrative guidance by MHLW, the new CTA is a law with legal binding force. Therefore, investigators must classify their clinical trials accurately and the flowchart in this manuscript will help them to do so.
Even in an international academic clinical trial which falls under CTA, Japanese investigators should be compliant with CTA. Of course ICH-GCP is the regulatory cornerstone, but, for a specified clinical trial under CTA, such Japan-specific consideration would be needed as the CRB review fee, COI management plan, trial plan notification to MHLW, and registry to jRCT. In an international academic trial, a country-specific appendix is often needed to document regulatory differences. This will be utilized more in Japan, because about half of the academic clinical trials are expected to be classified under CTA.
Unfortunately, academic clinical trial activity is expected to decrease in Japan in the years to come because CTA increases the financial burden for CRB review, local site burden for the new COI management system and the central management burden for the frequent revision of the trial plan. The cost of academic clinical trials has enormously increased due to the financial burden, and investigators sometimes hesitate to plan new clinical trials due to the management burden. Nevertheless, public funding to boost such academic trials is limited, so academic investigators have come to rely on funding from pharmaceutical companies more, delegating the trial management to a contract research organization. As pharmaceutical companies rarely provide funding for the trials that focus on academic or patients’ needs but do not make any profit for them, such investigator-initiated trials would decrease and industry-funded trials would dominate in Japan in the near future. As a consequence, it will give a detrimental influence on evidence-based clinical practice.
Similar crisis on academic clinical trials were observed in European countries when the EU directive was introduced in 2004. At that moment, the number of clinical trials in the UK decreased by 50%, the clinical trial cost was doubled, the clinical trial management staff increased by 85% and the time until the start of patient accrual was prolonged from 5 to 10 months (8). Since a similar situation is expected to occur in Japanese academic clinical trials from now on, a collaborative effort between academia, the government and the regulatory authorities to lower the burden of CTA is urgently needed.
Conclusions
The enforcement of the new CTA was supposed to improve the reliability of academic clinical trials in Japan; however, the financial and administrative burden may reduce Japanese clinical trial activity in the years to come.
Acknowledgment
We thank all the members of the JCOG Data Center, JCOG Operations and Clinical Research Support Office in the National Cancer Center Hospital for their useful suggestions.
Funding
This work was supported by the National Cancer Center Research and Development Fund (29-A-3, 29-A-15).
Conflict of interest statement
The authors declare no conflict of interest.
References
- 1. Patented Medicine Prices Review Board Annual Report, 2017.
- 2. Ministry of Education, Culture, Sports, Science and Technology, Ministry of Health, Labour and Welfare Ethical Guidelines for Medical and Health Research Involving Human Subjects. 2015. https://www.mhlw.go.jp/file/06-Seisakujouhou-10600000-Daijinkanboukouseikagakuka/0000080278.pdf(7 December 2019, date last accessed).
- 3. Tanimoto T, Kami M, Shibuya K. Research misconduct and scientific integrity: a call for a global forum. Lancet 2013;382:14–20. [DOI] [PubMed] [Google Scholar]
- 4. MuCurry J. Former Novartis employee arrested over Valsartan data. Lancet 2014;383:2111. [DOI] [PubMed] [Google Scholar]
- 5. Ministry of Health, Labour and Welfare Clinical Trials Act. 2017. https://www.mhlw.go.jp/file/06-Seisakujouhou-10800000-Iseikyoku/0000213334.pdf(7 December 2019, date last accessed).
- 6. Certified Review Board Information Service (in Japanese) https://jcrb.niph.go.jp/(29 December 2019, date last accessed).
- 7. Shimozawa R, Ikeda M. Japanese regulatory system for approval of off-label drug use: evaluation of safety and effectiveness in literature-based applications. Pharmacotherapy 2012;34:2104–16. [DOI] [PubMed] [Google Scholar]
- 8. European Commission Impact assessment on the revision of the “Clinical Trials Directive” 2001/20/EC. https://ec.europa.eu/health/sites/health/files/files/clinicaltrials/2012_07/impact_assessment_part1_en.pdf(7 December 2019, date last accessed).