

Abstract

Benzo[d]thiazole is widely used in synthetic and medicinal chemistry, and it is a component of many compounds and drugs that have several different bioactivities. Herein, we describe an elegant pathway for synthesis of methyl 4- and 5-hydroxy-2-amino-benzo[d]thiazole-6-carboxylates as building blocks that can be substituted at four different positions on the bicycle and thus offer the possibility to thoroughly explore the chemical space around the molecule studied as a ligand for the chosen target. A series of 12 new compounds was prepared using the described methods and Williamson ether synthesis.
1. Introduction
Heterocycles are a versatile set of scaffolds that are part of many natural products and are commonly used in synthetic and medicinal chemistry. They offer many ways of modification and substitution, and thus, there is a possibility to balance the physicochemical properties of the molecules. Benzo[d]thiazole is present in many bioactive compounds from a number of pharmacological areas. Compounds containing the benzo[d]thiazole heterocycle have been described as antibacterial,1 antifungal,2 and anticancer agents,3 and they have antidiabetic,4 antidepressant,5 anticonvulsant,6 and radioprotective activities,7 as well as neuroprotective properties useful for Alzheimer’s disease8 and Parkinson’s disease9 (Figure 1). Riluzole is an example of a marketed drug that contains benzo[d]thiazole10 (Figure 1); it has neuroprotective, anticonvulsant, and sedative properties11 and is used to treat amyotrophic lateral sclerosis.12 The benzo[d]thiazole moiety is also present in firefly luciferin, which is responsible for the bioluminescence of firefly species,13 which indicates that benzo[d]thiazole-based compounds can be used as fluorescent probes.14,15
Figure 1.

Representative examples of pharmacologically active benzo[d]thiazoles. (a) Fungal lanosterol 14α-demethylase (CYP51) inhibitor, with antifungal activity.2 (b) Dual inhibitor of phosphoinositide 3-kinase and mammalian target of rapamycin (mTOR), with anticancer activity.3 (c) Peroxisome proliferator-activated receptor (PPAR)α inhibitor, with antidiabetic activity.4 (d) Amyloid-binding alcohol dehydrogenase inhibitor, with anti-Alzheimer’s disease activity.8 (e) Riluzole, an inhibitor of glutamatergic neurotransmission in the central nervous system, with neuroprotective activity.11 (f) Firefly luciferin.13
Our research group has extensively studied the benzo[d]thiazole compounds in the context of the discovery of novel antibacterial compounds.16,17 Here, we present an efficient method for the preparation of benzo[d]thiazoles that can be conveniently substituted at four different positions: at the 2-amino group; at the 6-carboxy group; and at either the 4-hydroxy or 5-hydroxy groups. These can be used as building blocks toward the novel biologically active compounds that are urgently needed, especially as antibacterial, antifungal, and anticancer agents. In drug discovery, the availability of various building blocks is very important, as these enable efficient and rapid design and synthesis of bioactive analogues. Building blocks must be developable, and therefore, they must contain chemically addressable functional groups that can be further derivatized.18,19
There are several reported procedures to synthesize the benzo[d]thiazole bicycle via cyclization or condensation reactions using different catalysts (e.g., ammonium chloride, iodine, bromine, palladium acetate, and copper catalysts) and different reaction conditions (e.g., microwave irradiation, acidic or basic conditions, and resin- or polymer-supported condensation).20−26 In this paper, we describe a rapid and efficient preparation of a set of new benzo[d]thiazole-6-carboxylates via a cyclization reaction (see the proposed reaction mechanism below) that uses different methyl p-aminobenzoates, potassium thiocyanate, and bromine as reagents.27 Furthermore, we describe a convenient synthetic pathway to obtain first the benzo[d]thiazole bicycle with an unsubstituted hydroxyl group, which can later be easily derivatized with different alkyl substituents. This new approach enables more rapid generation of a higher number of benzo[d]thiazole derivatives than the cyclization of each methyl p-aminobenzoate individually. In this paper, we present 12 new compounds with six of them prepared using the new described approach.
2. Results and Discussion
We developed a convenient synthetic approach toward new methyl 2-aminobenzo[d]thiazole-6-carboxylates that can carry an −OR substituent or a free hydroxyl group on either position 4 or 5 and thus offer the possibility of O-substitution at these positions. The synthesis of methyl 2-aminobenzo[d]thiazole-6-carboxylate (Scheme 1, compound A) was selected as the model reaction. To synthesize compound A, 1 equiv of methyl 4-aminobenzoate and 4 equiv of KSCN were dissolved in glacial acetic acid, with the mixture stirred for 45 min at room temperature, and then cooled to 10 °C. Bromine (2 equiv) was dissolved in a small amount of acetic acid and added dropwise. The reaction mixture was then stirred at room temperature overnight. When the reaction was over, the reaction mixture was put on ice and basified to pH 8 using 25% NH3 solution, and the product was isolated using filtration (see Section 4.2 for detailed procedures).27
Scheme 1. Synthesis of Model Compound A.

Reagents and conditions: (a) KSCN (4 equiv), Br2 (2 equiv), CH3COOH, 10 °C, then rt, 15 h, 25% aq. NH3, yield: 55%.
This general procedure for cyclization was then applied to a series of 3- and 2-alkoxy-4-aminobenzoate compounds. The synthesis of 5a–f (Scheme 2) and 11a–b (Scheme 3) with different alkoxy substituents at positions 4 or 5 of the bicyclic structure proceeded as expected, with moderate to good yields (35–95%). To obtain 4-substituted compounds 5a–f, 3-hydroxy-4-nitrobenzoic acid (1) was first converted to a methyl ester 2 using H2SO4 in methanol. Compound 2 was then alkylated under conditions of the Williamson ether synthesis (3a–e) or the Mitsunobu reaction (3f). The nitro group of 3a–f was reduced to amino with catalytic hydrogenation (for 4a, 4c, 4d, and 4g) or using tin(II) chloride (for 4b and 4e). The described cyclization procedure was applied to 4a–f to obtain the desired products 5a–f (Scheme 2). For 5-substituted compounds, 7 was prepared with Fischer esterification of 4-aminosalicylic acid (6). The amino group of 7 was protected with the tert-butyloxycarbonyl-protecting group to obtain 8, which was alkylated with methyl iodide or benzyl bromide to obtain 9a–b. The tert-butyloxycarbonyl-protecting group was then removed by acidolysis, and finally, 10a–b that were obtained were then cyclized to 11a–b using the procedures described in Scheme 3.
Scheme 2. Synthesis of Compounds 5a–f.
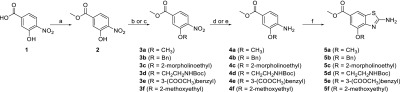
Reagents and conditions: (a) MeOH, H2SO4, 65 °C, 15 h, yield: 94%. (b) Corresponding alkyl halide, K2CO3, CH3CN or DMF, 60 °C, 15 h (for synthesis of 3a–e). (c) 2-Methoxyethanol, DIAD, PPh3, THF, rt, 15 h (for synthesis of 3f), yield: 22–97%. (d) H2, Pd/C, MeOH, rt, 2–5 h (for synthesis of 4a, 4c–d, 4f). (e) SnCl2, MeOH/EtOAc, 55 °C, 15 h (for synthesis of 4b, 4e), yield: 66–99%. (f) KSCN, Br2, CH3COOH, 10 °C, then rt, 15 h, 25% aq. NH3, yield: 35–95%.
Scheme 3. Synthesis of Compounds 11a–b.
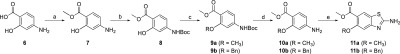
Reagents and conditions: (a) MeOH, H2SO4, 65 °C, 15 h, yield: 89%. (b) Boc2O, 70 °C, 48 h, yield: 43%. (c) Corresponding alkyl halide, K2CO3, CH3CN or DMF, 60–80 °C, 15 h, yield: 54–92%. (d) 4 M HCl in 1,4-Dioxane, 1,4-dioxane, rt, 3 d, yield: 51–84%. (e) KSCN, Br2, CH3COOH, 10 °C, then rt, 15 h, 25% aq. NH3, yield: 45–74%.
To define the reaction mechanism, we monitored formation of 5b with HPLC–MS analysis (Scheme 4; Supporting Information, Section S1). Before the addition of bromine, the reaction did not start (Supporting Information, Section S1, Figure S1), but immediately after the addition, the thiocyanate group was attached to the phenyl ring of the starting aniline (4b; Supporting Information, Section S1, Figure S2). Bromine was needed for the formation of the pseudohalogen thiocyanogen (Scheme 4), which was then involved in the thiocyanation of aniline.28 After 30 min, there was no more starting aniline in the reaction mixture (Supporting Information, Section S1, Figure S3), and the formation of intermediate I1 with the SCN group attached to the phenyl ring (Scheme 4) was confirmed using nuclear magnetic resonance (NMR) (Supporting Information, Section S1, Figure S4). After 60 min, the formation of benzo[d]thiazole 5b was detected. Although intermediate I1 and product 5b have exactly the same mass (Supporting Information, Section S1, Figure S5), they have different retention times in the HPLC chromatograms, as well as significantly different chemical shifts for NH2 protons in the NMR spectra (6.40 ppm for I1; 7.91 ppm for 5b; Supporting Information, Section S1, Figure S6). After 15 h, the reaction was complete (Supporting Information, Section S1, Figure S7), and 5b could be isolated.
Scheme 4. Proposed Mechanism for Formation of the Benzo[d]thiazole Bicycle.

As the cyclization of each individual methyl p-aminobenzoate to obtain different substituents at positions 4 and 5 was time consuming, and therefore not so efficient, we developed a synthetic route for preparation of 4- and 5-hydroxy benzo[d]thiazoles, which could be synthesized on a large scale and derivatized at later synthetic stages. This would provide a rapid route for preparation of a central benzo[d]thiazole core as a building block around which the chemical space could be explored by the introduction of different chemical groups. To achieve this, we first looked for a suitable hydroxyl-protecting group that would not be cleaved under the acidic conditions that are used for cyclization and that would be easy to remove afterward. Benzyl- and acetyl-protecting groups were first used, but neither was optimal because of the unsuccessful removal after cyclization (in the case of benzyl protection), the migration of the acetyl group between OH and NH2 (in the case of acetyl protection) or the non-regioselective cyclization (in the case of acetyl protection). Detailed description and results of the exploration of these protecting groups can be found in Section S2.1 in the Supporting Information.
In a novel approach, the tert-butyldimethylsilyl group was used for protection of the hydroxyl group (Scheme 5; Supporting Information, Section S2.2, Scheme S3), which was easily introduced into the methyl 3-hydroxy-4-nitrobenzoate (2; Scheme 5) using tert-butyldimethylsilyl chloride as the reagent. The nitro group of 12 (Scheme 5) was reduced to the amino group using catalytic hydrogenation (13; Scheme 5). The details of the exploration of the cyclization reaction using the tert-butyldimethylsilyl-protecting group can be found in Supporting Information, Section S2.2. To successfully prepare the desired products, the cyclization reaction was set up following the general procedure, although halved amounts of KSCN and bromine (from initial 4 equiv of KSCN and 2 equiv of Br2) were used. Using ammonia solution for the neutralization during the isolation process resulted in successfully prepared tert-butyldimethylsilyl-protected product 14 (Scheme 5) that can be used as a convenient building block. On the other hand, using saturated aq. NaHCO3 as the base instead of ammonia, we successfully obtained the desired product with an unsubstituted OH group at position 4 (15; Scheme 5).
Scheme 5. Synthesis of Methyl 2-Amino-4-hydroxybenzo[d]thiazole-6-carboxylate (15) and 2-Amino-5-hydroxybenzo[d]thiazole-6-carboxylate (18) Using the tert-Butyldimethylsilyl-Protecting Group.

Reagents and conditions: (a) TBDMSCl, pyridine, rt, 15 h. (b) H2, Pd/C, MeOH, rt, 5 h, yield: 56%. (c) KSCN (2 equiv), Br2 (1 equiv), CH3COOH, 10 °C, then rt, 15 h, 25% aq. NH3 (for synthesis of 14) or sat. aq. NaHCO3 (for synthesis of 15, 17, and 18), yield: 13–60%. (d) TBDMSCl, 4-methylimidazole, DCM, rt, 96 h, yield: 75%.
The same reaction conditions were later applied for the preparation of benzo[d]thiazoles with an unsubstituted OH group at position 5 (Scheme 5). First, the OH group of 7 was selectively protected with the tert-butyldimethylsilyl-protecting group in the presence of NH2, to obtain 16, thus avoiding an additional step of Boc-protection/deprotection. After cyclization, a mixture of tert-butyldimethylsilyl-protected (17) and -deprotected (18) products substituted at position 5 was obtained (Scheme 5). Apparently, the bulky silyl protection group sterically hindered cyclization to position 7, which was observed in the case of the acetyl-protecting group (Supporting Information, Section S2.1, Scheme S2), and this thus allowed the cyclization to proceed only in the desired direction. The mixture of products 17 and 18 obtained after the reaction was then easily separated. Detailed exploration and procedures for the separation can be found in Supporting Information, Sections S2.3 and S4. Overall, we developed a convenient new method for preparation of both benzo[d]thiazole with an unsubstituted 5-OH group and benzo[d]thiazole with a tert-butyldimethylsilyl-protected 5-OH group, which were used in the further reactions, where the hydroxy group had to be either protected or not.
A successful synthesis of the desired hydroxybenzo[d]thiazoles 15 and 18 enabled the preparation of a series of new compounds that were alkylated at the 4-OH or 5-OH groups (products 5b, 19a–d, and 20; Scheme 6). The etherification of the OH group could be performed selectively in the presence of an unprotected 2-amino group, with the Williamson ether synthesis method using different alkylating agents and K2CO3 in acetonitrile or dimethylformamide as the solvent (Scheme 6).
Scheme 6. Synthesis of Compounds 5b, 19a–d, and 20.
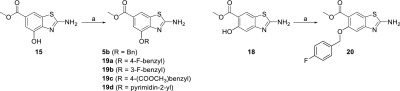
Reagents and conditions: (a) corresponding alkyl halide, K2CO3, CH3CN or DMF, 60–80 °C, 15 h, yield: 28–45%.
3. Conclusions
In summary, we explored a common procedure for cyclization of benzothiazoles and then modified this to develop a simple method for synthesis of methyl 2-aminobenzo[d]thiazole-6-carboxylates with an OH group on either position 4 or 5 of the bicycle. The benzo[d]thiazole ring formation proceeded from 4-aminobenzoates with KSCN and bromine in acetic acid. Different protecting groups for the hydroxyl group during cyclization were explored. The best protection/deprotection was achieved with a tert-butyldimethylsilyl group, which can be removed easily during the isolation process. The obtained methyl 2-amino-4-hydroxybenzo[d]thiazole-6-carboxylate (15) and methyl 2-amino-5-hydroxybenzo[d]thiazole-6-carboxylate (18) were then selectively derivatized on the hydroxy groups in the presence of an unprotected 2-amino group. In the case of the 4-substituted product, we also developed two selective isolation procedures to either remove the tert-butyldimethylsilyl-protecting group or to keep the product protected.
In conclusion, as benzo[d]thiazole is an important scaffold in many bioactive compounds, the convenient preparation of building blocks described in this study offers the elegant possibility of rapidly exploring the chemical space at various positions of the bicycle. Thus, the new products described herein can serve as useful starting points in the synthesis of novel bioactive compounds.
4. Experimental Section
4.1. General Experimental Details
Chemicals were obtained from Acros Organics (Geel, Belgium), Sigma-Aldrich (St. Louis, MO, USA), and Apollo Scientific (Stockport, UK) and used without further purification. Analytical TLC was performed on silica gel Merck 60 F254 plates (0.25 mm), using visualization with UV light and spray reagents. Column chromatography was carried out on silica gel 60 (particle size 240–400 mesh). 1H and 13C NMR spectra were recorded at 400 and 100 MHz, respectively, on a Bruker AVANCE III 400 spectrometer (Bruker Corporation, Billerica, MA, USA) in DMSO-d6 or CDCl3 solutions, with TMS as the internal standard. HPLC–MS analyses were performed on an Agilent Technologies 1260 Infinity II LC System (Agilent Technologies, Inc., Santa Clara, CA, USA) coupled to an ADVION expression CMSL mass spectrometer (Advion Inc., Ithaca, USA). The column used was Waters XBridge C18 column (3.5 μm, 4.6 mm × 150 mm), a flow rate of 1.5 mL/min, and sample injection volume of 10 μL. The mobile phase consisted of acetonitrile (as solvent A) and 0.1% formic acid and 1% acetonitrile in ultrapure water (as solvent B). The gradient (for solvent A) was 0–1 min, 25%; 1–6 min, 25–98%; 6–6.5 min, 98%; 6.5–7.5 min, 98–25%; 7.5–10.5 min, 25%. Mass spectra were obtained using the Exactive Plus Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, Massachusetts, ZDA) or Advion expression CMSL mass spectrometer (Advion Inc., Ithaca, USA).
4.2. General Procedures
4.2.1. General Procedure A. Synthesis of Example Compound A
To a solution of methyl 4-aminobenzoate (500 mg, 3.31 mmol) in acetic acid (12 mL), KSCN (1.28 g, 13.2 mmol) was added and the solution was stirred at rt for 45 min. The reaction mixture was cooled to 10 °C, and bromine (0.339 mL, 6.62 mmol) in acetic acid was added dropwise upon which the solution turned to a yellow suspension. The reaction mixture was then stirred at room temperature overnight. The reaction mixture was neutralized with 25% aqueous NH3 solution (50 mL) to pH = 8. The precipitate was filtered off, excessively washed with water, and dried. The solid was suspended in methanol, heated, filtered off, and dried.
4.2.1.1. Methyl 2-Aminobenzo[d]thiazole-6-carboxylate (A)29
Yield: 369 mg (55%); yellow solid. 1H NMR (400 MHz, DMSO-d6): δ 3.83 (s, 3H), 7.38 (d, J = 8.0 Hz, 1H), 7.83 (dd, J = 1.6, 8.0 Hz, 1H), 7.92 (s, 2H), 8.30 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 52.3, 117.6, 122.2, 123.0, 127.5, 131.6, 157.4, 166.6, 170.2.
4.2.2. General Procedure B. Synthesis of Example Compound 2
To a solution of 3-hydroxy-4-nitrobenzoic acid (1, 10.0 g, 54.6 mmol) in methanol (200 mL), conc. H2SO4 (6 mL, 112.6 mmol) was added and the mixture was stirred at 65 °C overnight. The solvent was evaporated under reduced pressure. The residue was neutralized with saturated aqueous NaHCO3 solution and extracted with ethyl acetate (200 mL). The organic phase was washed with brine (2 × 50 mL), dried over Na2SO4, and filtered; the solvent was removed in vacuo.
4.2.2.1. Methyl 3-Hydroxy-4-nitrobenzoate (2)30
Yield: 10.1 g (94%); yellow crystals. 1H NMR (400 MHz, CDCl3): δ 3.97 (s, 3H), 7.62 (dd, J = 8.8, 1.8 Hz, 1H), 7.84 (d, J = 1.7 Hz, 1H), 8.18 (d, J = 8.8 Hz, 1H), 10.51 (s, 1H).
4.2.3. General Procedure C. Synthesis of Example Compound 3a
To a suspension of methyl 3-hydroxy-4-nitrobenzoate (2, 3.00 g, 15.2 mmol) and K2CO3 (3.16 g, 22.8 mmol) in DMF (20 mL), methyl iodide (1.9 mL, 30.5 mmol) was added dropwise and the reaction mixture was stirred at 60 °C overnight. The solvent was removed in vacuo, and the residue was dissolved in ethyl acetate (30 mL) and washed with water (2 × 20 mL) and brine (20 mL). The organic phase was dried over Na2SO4, filtered, and the solvent was evaporated under reduced pressure.
4.2.3.1. Methyl 3-Methoxy-4-nitrobenzoate (3a)31
Yield: 3.00 g (93%); off-white crystals. 1H NMR (400 MHz, DMSO-d6): δ 3.92 (s, 3H), 4.00 (s, 3H), 7.68 (dd, J = 8.3, 1.6 Hz, 1H), 7.78 (d, J = 1.6 Hz, 1H), 8.01 (d, J = 8.6 Hz, 1H).
4.2.3.2. Methyl 3-(2-Methoxyethoxy)-4-nitrobenzoate (3f)
To a stirred solution of compound 2 (0.70 g, 3.55 mmol) and triphenylphosphine (1.86 g, 7.10 mmol) in anhydrous tetrahydrofuran (20 mL), 2-methoxyethan-1-ol (0.310 mL, 3.91 mmol) was added and the mixture was stirred at rt for 10 min. Diisopropyl azodicarboxylate (DIAD, 1.40 mL, 7.10 mmol) was added dropwise, and the mixture was stirred at rt for 15 h under the argon atmosphere. The solvent was evaporated in vacuo, and the residue was purified with flash column chromatography using hexane/ethyl acetate (2:1) as the eluent. The crude product was used in the next step without further purification.
4.2.4. General Procedure D. Synthesis of Example Compound 4a
Methyl 3-methoxy-4-nitrobenzoate (3a 2.98 g, 14.1 mmol) was dissolved in methanol/tetrahydrofuran (7:3, 100 mL) under the argon atmosphere; Pd/C (500 mg) was added, and the reaction mixture was stirred at room temperature under hydrogen atmosphere for 5 h. The catalyst was filtered off, and the solvent was removed in vacuo.
4.2.4.1. Methyl 4-Amino-3-methoxybenzoate (4a)31
Yield: 2.39 g (93%); light gray crystals. 1H NMR (400 MHz, DMSO-d6): δ 3.76 (s, 3H), 3.81 (s, 3H), 5.61 (br s, 2H), 6.65 (d, J = 8.2 Hz, 1H), 7.29 (d, J = 1.8 Hz, 1H), 7.39 (dd, J = 8.2, 1.8 Hz, 1H).
4.2.5. General Procedure E. Synthesis of Example Compound 4b
To a solution of methyl 3-(benzyloxy)-4-nitrobenzoate (3b, 1.48 g, 5.17 mmol) in ethyl acetate/methanol (1.5:1, 25 mL), SnCl2 (4.90 g, 25.8 mmol) was added and the reaction mixture was stirred at 55 °C overnight. The solvent was removed in vacuo, and to the residue, NaHCO3 (220 mL) was added dropwise on an ice bath. The obtained white suspension was sonicated for 30 min. Ethyl acetate was added, and the precipitate was filtered off. The phases in the mother liquid were separated, and the water phase was extracted with additional ethyl acetate. The precipitate was also resuspended in ethyl acetate and filtered again. The combined organic phases were washed with brine, dried over Na2SO4, and filtered and the solvent was removed in vacuo.
4.2.5.1. Methyl 4-Amino-3-(benzyloxy)benzoate (4b)32
Yield: 1.23 g (93%); dark yellow solid. 1H NMR (400 MHz, DMSO-d6): δ 3.75 (s, 3H), 5.17 (s, 2H), 5.68 (br s, 2H), 6.68 (d, 1H), 7.29–7.45 (m, 5H), 7.49–7.56 (m, 2H).
4.2.5.2. Methyl 4-((tert-Butoxycarbonyl)amino)-2-hydroxybenzoate (8)33
To a solution of methyl 4-amino-2-hydroxybenzoate (9.57 g, 57.3 mmol), di-tert-butyl dicarbonate (13.8 g, 63.0 mmol) was added and the mixture was stirred at 70 °C for 48 h. The solvent was removed under reduced pressure; to the residue, ethyl acetate (100 mL) and water were added, and the phases were separated. The organic phase was washed with 1 M HCl (3 × 40 mL) and brine (3 × 40 mL), dried over Na2SO4, filtered, and the solvent removed in vacuo. The crude product was purified with flash column chromatography using ethyl acetate/hexane (1:7) as an eluent. Yield: 6.52 g (43%); white crystals. 1H NMR (400 MHz, CDCl3): δ 1.54 (s, 9H), 3.94 (s, 3H), 6.62 (s, 1H), 6.95 (dd, J = 8.7, 2.2 Hz, 1H), 7.01 (d, J = 2.1 Hz, 1H), 7.76 (d, J = 8.7 Hz, 1H), 10.86 (s, 1H).
4.2.5.3. Methyl 4-Amino-2-methoxybenzoate (10a)34
To a solution of compound 9a (0.867 g, 3.08 mmol) in dichloromethane (15 mL), trifluoroacetic acid (5 mL) was added and the reaction mixture was stirred at rt for 4 h. To the reaction mixture, dichloromethane (40 mL) was added and neutralized with saturated aqueous NaHCO3 solution (60 mL). The phases were separated, and the organic phase was washed with saturated aqueous NaHCO3 solution (2 × 35 mL) and brine (3 × 30 mL), dried over Na2SO4, filtered, and the solvent was removed in vacuo. Yield: 471 mg (84%); white crystals. mp 128–132 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.66 (s, 3H), 3.71 (s, 3H), 5.94 (s, 2H), 6.14 (dd, J = 8.5, 2.0 Hz, 1H), 6.20 (d, J = 2.0 Hz, 1H), 7.50 (d, J = 8.5 Hz, 1H).
4.2.5.4. Methyl 4-Amino-2-(benzyloxy)benzoate (10b)35
Compound 9b (0.605 g, 1.69 mmol) was dissolved in 2 M HCl in diethyl ether (5 mL) and stirred at rt for 15 h. The precipitate in the reaction mixture was filtered off and suspended in ethyl acetate (50 mL) which was washed with saturated aqueous NaHCO3 solution (30 mL) and brine (30 mL), dried over Na2SO4, filtered, and the solvent was removed in vacuo. Yield: 223 mg (51%); light brown solid. mp 111–112 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.71 (s, 3H), 5.10 (s, 2H), 5.62 (br s, 2H), 6.30 (dd, J = 8.5, 2.0 Hz, 1H), 6.44 (d, J = 2.0 Hz, 1H), 7.29–7.36 (m, 1H), 7.38–7.45 (m, 2H), 7.49–7.57 (m, 2H), 7.59 (d, 1H).
4.2.5.5. Methyl 3-((tert-Butyldimethylsilyl)oxy)-4-nitrobenzoate (12)
To a solution of methyl 3-hydroxy-4-nitrobenzoate (2, 2.00 g, 10.4 mmol) in pyridine (25 mL), tert-butyldimethylsilyl chloride (4.59 g, 30.4 mmol) was added. The reaction mixture was stirred at room temperature overnight. Then, ethyl acetate (85 mL) was added, and the solution was washed with 1 M HCl (4 × 50 mL). The organic phase was dried over Na2SO4 and filtered, and the solvent was removed in vacuo. A crude oily product was used in the next step without further purification.
4.2.5.6. Methyl 4-Amino-2-((tert-butyldimethylsilyl)oxy)benzoate (16)
A solution of compound 7 (9.79 g, 58.6 mmol) in dichloromethane (150 mL), tert-butylchlorodimethylsilane (17.7 g, 117 mmol), and 4-methylimidazole was stirred at rt 96 h. The reaction mixture was washed with water (2 × 20 mL), and organic phases were dried over Na2SO4 and filtered, and the solvent was evaporated under reduced pressure. The residue was purified with flash column chromatography using ethyl acetate/hexane (1/4) as an eluent to give compound 17 (12.3 g) as pink-brown crystals. Yield 12.3 g (75%); mp 64–66 °C; 1H NMR (400 MHz, DMSO-d6): δ 0.17 (s, 6H), 0.97 (s, 9H), 3.66 (s, 3H), 5.86 (s, 2H), 6.10 (d, J = 2.1 Hz, 1H), 6.18 (dd, J = 8.6, 2.1 Hz, 1H), 7.48 (d, J = 8.6 Hz, 1H). MS (ESI+) m/z: 282.2 ([M + H]+).
Acknowledgments
The work was supported by the Slovenian Research Agency (grant no. P1-0208). The authors wish to thank Christopher Berrie for scientific editing of the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c00768.
LC–MS analysis, experimental details, characterization data for all of the products, and 1H and 13C NMR spectra for representative compounds (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Gjorgjieva M.; Tomašič T.; Kikelj D.; Mašič L. P. Benzothiazole-Based Compounds in Antibacterial Drug Discovery. Curr. Med. Chem. 2019, 25, 5218–5236. 10.2174/0929867324666171009103327. [DOI] [PubMed] [Google Scholar]
- Zhao S.; Zhao L.; Zhang X.; Liu C.; Hao C.; Xie H.; Sun B.; Zhao D.; Cheng M. Design, Synthesis, and Structure-Activity Relationship Studies of Benzothiazole Derivatives as Antifungal Agents. Eur. J. Med. Chem. 2016, 123, 514–522. 10.1016/j.ejmech.2016.07.067. [DOI] [PubMed] [Google Scholar]
- D’Angelo N.-D.; Kim T.-S.; Andrews K.; Booker S.-K.; Caenepeel S.; Chen K.; D’Amico D.; Freeman D.; Jiang J.; Liu L.; McCarter J.-D.; San Miguel T.; Mullady E.-L.; Schrag M.; Subramanian R.; Tang J.; Wahl R.-C.; Wang L.; Whittington D.-A.; Wu T.; Xi N.; Xu Y.; Yakowec P.; Yang K.; Zalameda L.-P.; Zhang N.; Hughes P.; Norman M.-H. Discovery and Optimization of a Series of Benzothiazole Phosphoinositide 3-Kinase (PI3K)/Mammalian Target of Rapamycin (MTOR) Dual Inhibitors. J. Med. Chem. 2011, 54, 1789–1811. 10.1021/jm1014605. [DOI] [PubMed] [Google Scholar]
- Ammazzalorso A.; Giancristofaro A.; D’Angelo A.; Filippis B. D.; Fantacuzzi M.; Giampietro L.; Maccallini C.; Amoroso R. Benzothiazole-Based N-(Phenylsulfonyl)Amides as a Novel Family of PPARα Antagonists. Bioorg. Med. Chem. Lett. 2011, 21, 4869–4872. 10.1016/j.bmcl.2011.06.028. [DOI] [PubMed] [Google Scholar]
- Demir Özkay Ü.; Kaya C.; Acar Çevik U.; Can Ö. Synthesis and Antidepressant Activity Profile of Some Novel Benzothiazole Derivatives. Molecules 2017, 22, 1490. 10.3390/molecules22091490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.-C.; Zhang H.-J.; Jin C.-M.; Quan Z.-S. Synthesis and Biological Evaluation of Novel Benzothiazole Derivatives as Potential Anticonvulsant Agents. Molecules 2016, 21, 164. 10.3390/molecules21020164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prouillac C.; Vicendo P.; Garrigues J.-C.; Poteau R.; Rima G. Evaluation of New Thiadiazoles and Benzothiazoles as Potential Radioprotectors: Free Radical Scavenging Activity in Vitro and Theoretical Studies (QSAR, DFT). Free Radical Biol. Med. 2009, 46, 1139–1148. 10.1016/j.freeradbiomed.2009.01.016. [DOI] [PubMed] [Google Scholar]
- Hroch L.; Benek O.; Guest P.; Aitken L.; Soukup O.; Janockova J.; Musil K.; Dohnal V.; Dolezal R.; Kuca K.; Smith T. K.; Gunn-Moore F.; Musilek K. Design, Synthesis and in Vitro Evaluation of Benzothiazole-Based Ureas as Potential ABAD/17β-HSD10 Modulators for Alzheimer’s Disease Treatment. Bioorg. Med. Chem. Lett. 2016, 26, 3675–3678. 10.1016/j.bmcl.2016.05.087. [DOI] [PubMed] [Google Scholar]
- Ilgın S.; Osmaniye D.; Levent S.; Sağlık B.; Acar Çevik U.; Çavuşoğlu B.; Özkay Y.; Kaplancıklı Z. Design and Synthesis of New Benzothiazole Compounds as Selective HMAO-B Inhibitors. Molecules 2017, 22, 2187. 10.3390/molecules22122187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyanarayana B.; Saravanan M.; Siva Kumari K.; Lokamaheshwari D.-P.; Sridhar C.; Ravishankar R.; Nageswari A.; Pratap Reddy P. Synthesis and Spectral Characterization of Related Compounds of Riluzole, an Amyotrophic Lateral Sclerosis Drug Substance. Arkivoc 2008, 2008, 109. 10.3998/ark.5550190.0009.e12. [DOI] [Google Scholar]
- Doble A. The Pharmacology and Mechanism of Action of Riluzole. Neurology 1996, 47, 233S–241S. 10.1212/wnl.47.6_suppl_4.233s. [DOI] [PubMed] [Google Scholar]
- Phukan J.; Pender N. P.; Hardiman O. Cognitive Impairment in Amyotrophic Lateral Sclerosis. Lancet Neurol. 2007, 6, 994–1003. 10.1016/s1474-4422(07)70265-x. [DOI] [PubMed] [Google Scholar]
- Kanie S.; Nishikawa T.; Ojika M.; Oba Y. One-Pot Non-Enzymatic Formation of Firefly Luciferin in a Neutral Buffer from p-Benzoquinone and Cysteine. Sci. Rep. 2016, 6, 24794. 10.1038/srep24794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrobáriková V.; Hrobárik P.; Gajdoš P.; Fitilis I.; Fakis M.; Persephonis P.; Zahradník P. Benzothiazole-Based Fluorophores of Donor−π-Acceptor−π-Donor Type Displaying High Two-Photon Absorption. J. Org. Chem. 2010, 75, 3053–3068. 10.1021/jo100359q. [DOI] [PubMed] [Google Scholar]
- Ren Y.; Fan D.; Ying H.; Li X. Rational design of the benzothiazole-based fluorescent scaffold for tunable emission. Tetrahedron Lett. 2019, 60, 1060–1065. 10.1016/j.tetlet.2019.03.029. [DOI] [Google Scholar]
- Tomašič T.; Barančoková M.; Zidar N.; Ilaš J.; Tammela P.; Kikelj D. Design, Synthesis, and Biological Evaluation of 1-Ethyl-3-(Thiazol-2-Yl)Urea Derivatives as Escherichia Coli DNA Gyrase Inhibitors. Arch. Pharm. 2018, 351, 1700333. 10.1002/ardp.201700333. [DOI] [PubMed] [Google Scholar]
- Gjorgjieva M.; Tomašič T.; Barančokova M.; Katsamakas S.; Ilaš J.; Tammela P.; Peterlin Mašič L.; Kikelj D. Discovery of Benzothiazole Scaffold-Based DNA Gyrase B Inhibitors. J. Med. Chem. 2016, 59, 8941–8954. 10.1021/acs.jmedchem.6b00864. [DOI] [PubMed] [Google Scholar]
- Wang J.; Hou T. Drug and Drug Candidate Building Block Analysis. J. Chem. Inf. Model. 2010, 50, 55–67. 10.1021/ci900398f. [DOI] [PubMed] [Google Scholar]
- Boström J.; Brown D. G.; Young R. J.; Keserü G. M. Expanding the Medicinal Chemistry Synthetic Toolbox. Nat. Rev. Drug Discovery 2018, 17, 709–727. 10.1038/nrd.2018.116. [DOI] [PubMed] [Google Scholar]
- Prajapati N. P.; Vekariya R. H.; Borad M. A.; Patel H. D. Recent Advances in the Synthesis of 2-Substituted Benzothiazoles: A Review. RSC Adv. 2014, 4, 60176–60208. 10.1039/c4ra07437h. [DOI] [Google Scholar]
- Fortenberry C.; Nammalwar B.; Bunce R. A. Ammonium Chloride-catalyzed Synthesis of Benzo-fused Heterocycles from o-Substituted Anilines and Orthoesters. Org. Prep. Proced. Int. 2013, 45, 57–65. 10.1080/00304948.2013.743751. [DOI] [Google Scholar]
- Liu Y.; Yuan X.; Guo X.; Zhang X.; Chen B. Efficient 2-aryl benzothiazole formation from acetophenones, anilines, and elemental sulfur by iodine-catalyzed oxidative C(CO)-C(alkyl) bond cleavage. Tetrahedron 2018, 74, 6057–6062. 10.1016/j.tet.2018.08.047. [DOI] [Google Scholar]
- Shaikh A.; Ravi O.; Pushpa Ragini S.; Sadhana N.; Reddy Bathula S. Benzimidazoles and benzothiazoles from styrenes and N-vinylimidazole via palladium catalysed oxidative CC and CN bond cleavage. Tetrahedron Lett. 2020, 61, 151356. 10.1016/j.tetlet.2019.151356. [DOI] [Google Scholar]
- Satish G.; Reddy K. H. V.; Ramesh K.; Karnakar K.; Nageswar Y. V. D. Synthesis of 2-N-substituted benzothiazoles via domino condensation-hetero cyclization process, mediated by copper oxide nanoparticles under ligand-free conditions. Tetrahedron Lett. 2012, 53, 2518–2521. 10.1016/j.tetlet.2012.03.012. [DOI] [Google Scholar]
- Chakraborti A. K.; Selvam C.; Kaur G.; Bhagat S. An Efficient Synthesis of Benzothiazoles by Direct Condensation of Carboxylic Acids with 2-Aminothiophenol under Microwave Irradiation. Synlett 2004, 5, 0851–0855. 10.1055/s-2004-820012. [DOI] [Google Scholar]
- Yu X.; Yin Q.; Zhang Z.; Huang T.; Pu Z.; Bao M. Synthesis of 2-substituted benzothiazoles via the Brønsted acid catalyzed cyclization of 2-amino thiophenols with nitriles. Tetrahedron Lett. 2019, 60, 1964–1966. 10.1016/j.tetlet.2019.06.039. [DOI] [Google Scholar]
- Saeed A.; Rafique H.; Hameed A.; Rasheed S. Synthesis and Antibacterial Activity of Some New 1-Aroyl-3-(Substituted-2-Benzothiazolyl)Thioureas. Pharm. Chem. J. 2008, 42, 191–195. 10.1007/s11094-008-0094-x. [DOI] [Google Scholar]
- Kaufmann H. P.; Oehring W.; Clauberg A. Wissenschaftlicher Teil.: Die Bildung von Thiazol-Derivaten bei der Rhodanierung von Aminen. Arch. Pharm. 1928, 266, 197–218. 10.1002/ardp.192800069. [DOI] [Google Scholar]
- Venter J.; Perez C.; van Otterlo W. A. L.; Martínez A.; Blackie M. A. L. 1-Aryl-3-(4-methoxybenzyl)ureas as potentially irreversible glycogen synthase kinase 3 inhibitors: Synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2019, 29, 1597–1600. 10.1016/j.bmcl.2019.04.049. [DOI] [PubMed] [Google Scholar]
- Walker D. P.; Wishka D. G.; Piotrowski D. W.; Jia S.; Reitz S. C.; Yates K. M.; Myers J. K.; Vetman T. N.; Margolis B. J.; Jacobsen E. J.; Acker B. A.; Groppi V. E.; Wolfe M. L.; Thornburgh B. A.; Tinholt P. M.; Cortes-Burgos L. A.; Walters R. R.; Hester M. R.; Seest E. P.; Dolak L. A.; Han F.; Olson B. A.; Fitzgerald L.; Staton B. A.; Raub T. J.; Hajos M.; Hoffmann W. E.; Li K. S.; Higdon N. R.; Wall T. M.; Hurst R. S.; Wong E. H. F.; Rogers B. N. Design, Synthesis, Structure–Activity Relationship, and in Vivo Activity of Azabicyclic Aryl Amides as Α7 Nicotinic Acetylcholine Receptor Agonists. Bioorg. Med. Chem. 2006, 14, 8219–8248. 10.1016/j.bmc.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Ishikawa M.; Tsushima M.; Kubota D.; Yanagisawa Y.; Hiraiwa Y.; Kojima Y.; Ajito K.; Anzai N. A Scalable Synthesis of MN-447, an Antagonist for Integrins αv β3 and αIIbβ3. Org. Process Res. Dev. 2008, 12, 596–602. 10.1021/op800073z. [DOI] [Google Scholar]
- Demont E.; Charrier N.; Dunsdon R.; Maile G.; Naylor A.; O’Brien A.; Redshaw S.; Theobald P.; Vesey D.; Walter D. Synthesis of Indoles: Efficient Functionalisation of the 7-Position. Synthesis 2006, 3467–3477. 10.1055/s-2006-950223. [DOI] [Google Scholar]
- Chen J.; Zhao M.; Jiang X.; Sizovs A.; Wang M. C.; Provost C. R.; Huang J.; Wang J. Genetically anchored fluorescent probes for subcellular specific imaging of hydrogen sulfide. Analyst 2016, 141, 1209–1213. 10.1039/c5an02497h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke L.; Zhu G.; Qian H.; Xiang G.; Chen Q.; Chen Z. Catalytic Selective Oxidative Coupling of Secondary N-Alkylanilines: An Approach to Azoxyarene. Org. Lett. 2019, 21, 4008–4013. 10.1021/acs.orglett.9b01200. [DOI] [PubMed] [Google Scholar]
- Chen J.; Lu W.; Chen H.; Bian X.; Yang G. A New Series of Salicylic Acid Derivatives as Non-saccharide α-Glucosidase Inhibitors and Antioxidants. Biol. Pharm. Bull. 2019, 42, 231–246. 10.1248/bpb.b18-00661. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.