

Abstract
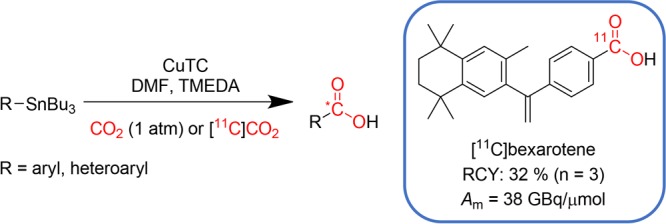
A novel copper-mediated carboxylation strategy of aryl- and heteroaryl-stannanes is described. The method serves as a mild (i.e., 1 atm) carboxylation method using stable carbon dioxide and is transferable as a radiosynthetic approach for carbon-11-labeled aromatic and heteroaromatic carboxylic acids using sub-stoichiometric quantities of [11C]CO2. The methodology was applied to the radiosynthesis of the retinoid X receptor agonist, [11C]bexarotene, with a decay-corrected radiochemical yield of 32 ± 5% and molar activity of 38 ± 23 GBq/μmol (n = 3).
Introduction
Novel transition-metal-catalyzed carboxylation reactions with CO2 in organic synthesis have been extensively applied for large-scale industrial applications1,2 as well as for accessing isotopically labeled carboxylic acids from [13C]CO2 or [14C]CO2.3,4 [11C]CO2 is a cyclotron-generated building block produced by the proton bombardment of nitrogen gas (14N(p,α)11C; t1/2 = 20.3 min) in the presence of trace oxygen. Recent years have seen a renaissance in [11C]CO2 fixation strategies for the synthesis of 11C=O-labeled compounds and radiopharmaceuticals and have been applied to in vivo medical imaging with positron emission tomography (PET).5−8 Organic bases capable of efficiently trapping [11C]CO2, such as 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and 2-tert-butylimino-2-diethylamino-1,3-dimethyl-perhydro-1,3,2-diazaphosphorine (BEMP), enable mild (room temperature) one-pot radiosyntheses of [11C]carbamates, [11C]ureas, and [11C]oxazolidones.6,7,9−11 The 11C-carbamate [11C]CURB12,13 and 11C-oxazolidone [11C]SL25.118814,15 were developed at our laboratories for imaging fatty acid amide hydrolase and monoamine oxidase B, respectively, and have been translated for human PET studies. In contrast, the synthesis of 11C-carboxylic acids via [11C]CO2 fixation has proven to be more challenging. Traditional methods to produce carboxylic acids from [11C]CO2 using air- and moisture-sensitive Grignard, organolithium, or silanamine reagents are limited by poor functional group tolerance and the need to continuously exclude atmospheric CO2.6 Metal-catalyzed direct carboxylation of aryl halides and pseudo-halides employing CO2 in drug discovery has recently been reviewed in the context of identifying opportunities for the radiolabeling of [11C]carboxylic acid based drugs via [11C]CO2 fixation.2 Only a handful of aromatic 11C-carboxylic acids have been radiolabeled and translated for preclinical PET imaging,16−21 and new 11C-carboxylation approaches are avidly sought. For example, retinoid X receptor (RXR) agonists and partial agonists are a class of drugs for oncology and neurodegenerative diseases that generally contain an aromatic carboxylic acid moiety.22 In particular, the RXR agonist bexarotene (Targretin) is an FDA-approved drug for the treatment of cutaneous T-cell lymphoma (CTCL)23 and has more recently been investigated for the treatment of Alzheimer’s disease.24Figure 1 illustrates three examples of 11C-carboxyl-based imaging agents that were synthesized via [11C]CO2 fixation methods for neuroimaging of RXRs: [11C]bexarotene (A; [11C]13b),16,17 [11C]6-[ethyl-(4-isobutoxy-3-isopropylphenyl)amino]pyridine-3-carboxylic acid (B),18 and [11C]1-[(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)-amino]benzotriazole-5-carboxylic acid (C).20
Figure 1.
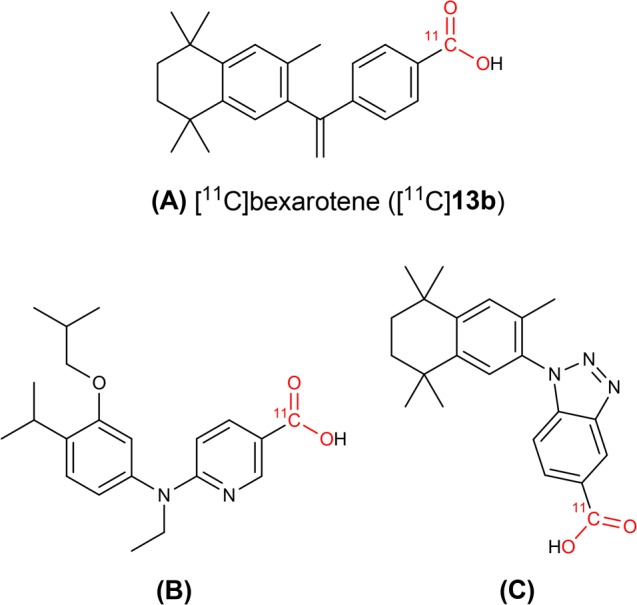
Examples of RXR agonists and partial agonists radiolabeled using [11C]CO2 fixation: [11C]bexarotene ([11C]13b; A), [11C]6-[ethyl-(4-isobutoxy-3-isopropylphenyl)amino]pyridine-3-carboxylic acid (B), and [11C]1-[(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)-amino]benzotriazole-5-carboxylic acid (C).
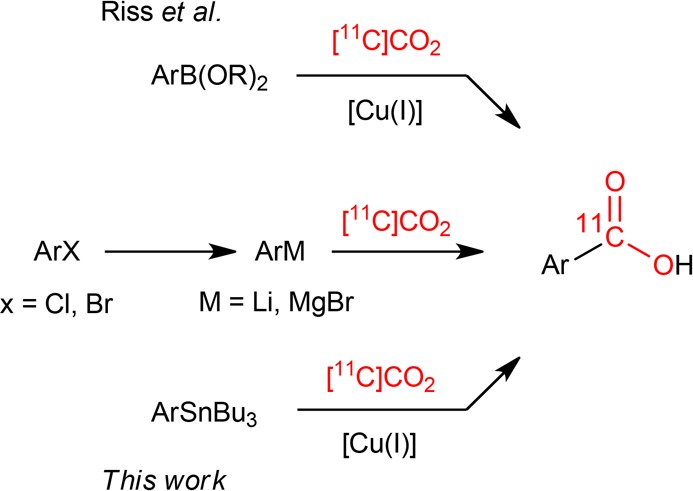
Riss et al. reported a mild one-pot procedure for the conversion of boronic esters to [11C]carboxylic acids mediated by a copper(I) source and [11C]CO2 as the radiochemical feedstock (Scheme 1).25 The strategy has advantages for the preparation of [11C]carboxylic acids, including a broad functional group tolerance and the ability to generate aryl, vinyl, and alkynyl 11C-carboxylic acids in high RCYs (>70%). This strategy also offers a novel route to 11C-labeled esters and amides.25 The procedure was adapted by our group to synthesize the prototypical RXR agonist, [11C]bexarotene (Figure 1A), and was translated for preclinical PET imaging in nonhuman primates.16,17 Despite these initial advances, Kakuta et al. found that aryl boronic esters afforded low yields (<1%) for the 11C-radiolabeling of the common aromatic carboxylic moiety in next-generation RXR agonists and partial agonists (Figure 1B,C), and the authors reverted to traditional [11C]CO2 fixation reactions via organolithiation procedures to prepare these labeled compounds in low yields.18,20 More recently, Rafique et al. also reported that [11C]CO2 fixation via Grignard reagents in a three-step route to 11C-amides was preferred over the use of boronic esters.26 Thus, we were motivated to explore alternative 11C-carboxylation strategies that are transferrable for both CO2 at 1 atm pressure as well as sub-stoichiometric quantities of [11C]CO2. We hypothesized that organostannanes could also be suitable precursors to carry out copper(I)-catalyzed 11C-carboxylation reactions. Organostannanes offer several advantages including their general stability to air and moisture, established synthetic methods, and commercial availability with widespread use as precursors in PET radiochemistry. Organotin compounds have also found utility alongside boronic esters27 in Pd(0)-mediated cross-coupling reactions with [11C]CH3I in the Stille and Suzuki reactions, respectively,28 as well as in Cu(II)-mediated 11C-cyanation reactions.29,30 The activation of organostannanes (RSnBu3; R = aryl, vinyl) using Cu(I) has also been utilized in Stille-type cross-coupling reactions with vinyl and aryl iodides,31−33 where formation of an organocuprate (RCu(I)) is considered as a key step in these reactions.34,35 The knowledge that organocuprates readily undergo CO2 insertion, also a key step in the Cu-catalyzed carboxylation of boronic acids36 and esters,37,38 supports our hypothesis that organostannanes would be suitable for this transformation.
Scheme 1. Radiosynthesis of (Hetero)aromatic Carboxylic Acids from [11C]CO2.
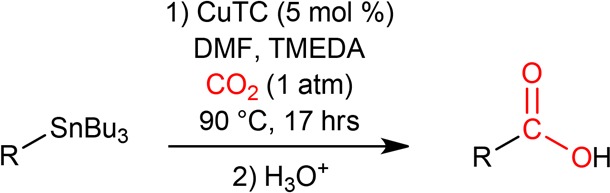
In the present study, we report the unprecedented Cu(I)-mediated carboxylation of (hetero)arylstannanes (Scheme 1). After first evaluating the reaction using CO2 at atmospheric pressure, the radiochemical yield for [11C]benzoic acid ([11C]1b) formation from tributylphenylstannane (1a), using [11C]CO2,was next optimized. This was followed by an assessment of the substrate scope with the aim of evaluating the sensitivity of the reaction to electron-withdrawing and -donating substituents as well as oxygen-, sulfur-, and nitrogen-containing heteroarenes. Finally, the methodology was applied to the labeling of [11C]bexarotene.
Results and Discussion
The feasibility of the Cu(I)-mediated carboxylation of (hetero)arylstannanes was first investigated using stable CO2, and the reaction conditions described in Table 1. Our general procedure was adapted from the work of Takaya et al.37 for the carboxylation of aryl boronic esters: a DMF solution (2.5 mL) containing organostannane (0.08 M, 0.2 mmol, 1.0 equiv.), N,N,N′,N′-tetramethylethylenediamine (TMEDA; 0.67 M, 1.7 mmol, 8.4 equiv.), and copper(I) thiophene-2-carboxylate (CuTC; 3.8 mM, 9.4 μmol, 0.05 equiv.) was sealed under an atmosphere of CO2 and heated at 90 °C for 17 h. Isolated yields of 58 and 33% were obtained for the formation of carboxylic acids 1b and 4b, respectively. Both carboxylic acids were isolated with reasonably high purity without the need for chromatographic separation. Analytical HPLC analyses of the reaction mixtures revealed lower conversions for 11b (8%) and 13b (6%). The relatively high yields of 1b and 4b suggest the active copper(I) species (present in ca. 5 mol % relative to organostannane) to be catalytic in these reactions but stoichiometric in the latter two instances. Ohishi et al.38 and Nayal et al.36 have both reported that the addition of an exogenous alkoxide is required for the carboxylation of boronic acids and esters to be catalytic in Cu(I) species. We did not pursue the effect of alkoxide addition as Riss et al. demonstrated that [11C]CO2 was rendered unreactive in the copper-mediated carboxylation of boronic esters upon addition of potassium tert-butoxide.25
Table 1. Carboxylation of Arylstannanes Using 1 atm CO2a.

Reactions were carried out using the following conditions: DMF (2.5 mL); [TMEDA] = 0.67 M; [ArSnBu3] = 0.08 M; [CuTC] = 3.8 mM; 90 °C; 17 h.
Yields determined by HPLC analyses of the crude reaction mixture relative to reference standards of the respective products.
Characterization by 1H, 13C{1H}, and 19F{1H} NMR spectroscopy (when applicable).
While 11b and 13b could not be isolated, their presence was confirmed by HPLC analyses of the crude reaction mixture upon addition of a reference sample (see Figures S3 and S4).
In order to successfully translate a carboxylation method for use with [11C]CO2, the reaction must be compatible with the conditions that allow for a sub-stoichiometric quantity of [11C]CO2 to be delivered into solution and reasonably retained at elevated temperatures (80–120 °C) for the duration of the reaction. This entails the use of a high-boiling-point polar aprotic amide solvent, a Cu(I) source, an amine that serves as both a base and ligand for the copper species, and the organostannane precursor. Thus, we first carried out screening of the reaction conditions to maximize the RCY (non-isolated and decay-corrected) of [11C]benzoic acid ([11C]1b; see Table 2) using those adapted from our previous 11C-carboxylation method for [11C]bexarotene radiosynthesis as a starting point (substrate: 1a (0.1 M); solvent: N-methyl-2-pyrrolidone (NMP; 600 μL); temperature: 100 °C; time: 5 min; base/ligand: TMEDA (1.2 M); copper source: CuTC (8.7 mM)).16 RCYs were determined from radio-HPLC analyses of the product mixture, taking into account losses of volatile [11C]CO2 from the solution (i.e., during the transfer of activity into solution at room temperature, following heating, and upon acidifying the reaction mixture) using a sodium hydroxide coated silica (Ascarite II, 20–30 mesh) trap. Thus, the (decay-corrected) trapping efficiency (TE) was also quantified as the activity retained in the final product vial.
Table 2. Reaction Screening and Optimizationa.

| entry | precursor loading (μmol) | solvent | temperature (°C) | TEb (%) | RCYc (%) |
|---|---|---|---|---|---|
| 1 | 60 | NMP | 100 | 50 | 50 |
| 2 | 60 | DMA | 100 | 45 | 35 |
| 3d | 60 | DMF | 100 | 69 ± 3 | 67 ± 5 |
| 4e | 60 | DMF | 110 | 61 ± 12 | 58 ± 12 |
| 5 | 60 | DMF | 120 | 50 | 47 |
| 6 | 60 | NMP | 80 | 50 | 41 |
| 7f | 60 | NMP | 100 | 53 | 49 |
| 8g | 60 | DMF | 100 | 97 | 2 |
| 9h | 60 | DMF | 110 | 6 | 5 |
| 10i | 60 | DMF | 110 | 42 | 31 |
| 11j | 60 | DMF | 100 | 6 | 0.01 |
| 12 | 30 | DMF | 100 | 48 | 45 |
| 13 | 15 | DMF | 100 | 48 | 40 |
| 14d,k | 30 | DMF | 110 | 35 ± 3 | 33 ± 4 |
Unless otherwise stated, reactions were carried out using the following standard conditions and are n = 1: solvent (600 μL); 1a (19.6 μL, 60 μmol, 0.1 M); CuTC (5.3 μmol, 8.7 mM); TMEDA (0.8 mmol, 1.3 M); temperature: 100 °C; time: 5 min.
TE (trapping efficiency): percentage of total activity that remains in solution after acidifying the reaction mixture.
RCY (radiochemical yield): percentage of total activity converted to [11C]1b.
n = 3.
n = 2.
TMEDA (240 μL, 1.6 mmol, 2.6 M).
DMEDA as the base.
CuTC (0.1 mg, 0.52 μmol, 0.87 mM).
CuTC (5.4 mg, 28.6 μmol, 47.0 mM).
CuI as the Cu(I) source.
Half-quantity of all reagents: DMF (300 μL); 1a (9.8 μL, 30 μmol); CuTC (2.7 μmol); TMEDA (0.4 mmol).
Examination of three polar aprotic amide solvents (Table 2, entries 1–3), NMP, N,N-dimethylformamide (DMF), and N,N-dimethylacetamide (DMA), revealed the RCY to decrease sequentially in the following order: DMF (67 ± 5%) > NMP (50%) > DMA (35%). Variation of the reaction temperature between 80 and 120 °C suggests an optimum value of 100 °C (Table 2, entries 1, 3–6); increasing the temperature leads to a decrease in TE, while decreasing to 80 °C increases the percentage of activity that remains unreacted. Doubling the concentration of TMEDA had little effect on the RCY and TE. Exchanging the base for N,N′-dimethylethylenediamine (DMEDA) while dramatically improving the TE of the solution rendered the [11C]CO2 unreactive for the desired transformation (Table 2, entry 8). Riss et al. reported that a ca. 7-fold decrease in the quantity of the Cu(I) source (along with a reduction in fluoride source quantity) led to an increase in RCY for the carboxylation of boronic esters.25 A 10-fold decrease or 5-fold increase in CuTC concentration in our reaction, however, resulted in a detrimental effect on the RCY (Table 2, entries 9 and 10). Minimizing the quantity of CuTC is also desirable given the potential for complications that may arise from the formation of thiophene-2-carboxylic acid, which is generated after acidifying the reaction mixture, during the isolation and/or further derivatization of the desired product. Only trace amounts of [11C]1b were observed when CuI was used as the copper(I) source (Table 2, entry 11). This is expected given the dramatic effect of iodide for 2-thiophene carboxylate (TC) exchange for the activation of arylstannanes31,35 and is attributed to the exergonic differences of the initial transmetalation step (see Scheme 4). Finally, we systematically decreased the concentration of 1a in order to evaluate the quantity of the precursor that is required to carry out the transformation as it would be advantageous to decrease the required amount of precursor so as to simplify purification. Relative to the optimized reaction conditions ([1a] = 0.1 M, corresponding to ca. 60 μmol or 22 mg), decreasing the precursor loading by 50 and 75% (Table 2, entries 12–13) led to decreases in the resulting RCYs (i.e., 67 ± 5, 45, and 40%, respectively). While we tested the substrate scope (vide infra) using our optimized conditions, the results in Table 2 suggest that this reaction can tolerate a decrease in precursor loading. Attempts to decrease the precursor quantity by halving the amount of all reagents used once again led to a reduction in RCY (33 ± 4%, Table 2, entry 14), concurrent with a decrease in the TE of the solution. Systematic variation of the reaction parameters led to the following optimized conditions: temperature: 100 °C; time: 5 min; solvent: DMF (600 μL); [1a] = 0.1 M; [CuTC] = 8.7 mM; [TMEDA] = 1.3 M. A proof-of-concept reaction was carried out using the synthesis and isolation of [11C]1b as a representative 11C-labeled compound. Carbon-11-labeled 1b was isolated in a decay-corrected RCY of 45% relative to starting [11C]CO2, with molar activity Am = 9.3 GBq/μmol (250 mCi/μmol) and radiochemical purity >99%, with an overall synthesis and purification time of 29 min.
Scheme 4. Proposed Mechanism of the Carboxylation Reaction.
We next investigated the substrate scope of the reaction using the optimized conditions and tributyltin-substituted by para-substituted benzene derivatives (ArSnBu3; see Scheme 2), including those bearing electron-donating (pMe (2a), pMeO (3a)) and -withdrawing (pCF3 (4a), pCl (5a)) groups. The scope was then extended further to include N-heteroarenes (8a–11a) as well as tributyltin substituted by the electron-rich heteroarenes furan (6a) and thienyl (7a). A representative alkyne, trimethyl(phenylethynyl)tin (12a), was also investigated. The carboxylation of 1a–5a afforded moderate radiochemical yields (ca. 30–70%), with the exception of pCl-substituted 5a, which gave a lower yield. As the highest yield was obtained with the parent structure 1a, we postulate that the RCY is similarly impacted by the presence of electron-donating (pMe, pMeO) or -withdrawing (pCl, pCF3) groups. The reaction also facilitates the carboxylation of N-heteroarenes, with the formation of 11C-carboxylic acids bearing 3-pyridyl ([11C]8b) and 2-pyrazyl ([11C]11b) substituents being successfully generated with RCYs between 31 and 34%. On the other hand, the RCY is lower (4–6%) when carboxylation is carried out at both the 2- and 5-positions of a pyrimidyl ring (forming [11C]9b and [11C]10b, respectively). The yields appear to be most hindered with the transfer of electron-rich heteroarenes as the 2-furyl (6a)- and 2-thienyl (7a)-substituted tributyltin compounds afforded RCYs <3% in both instances. Alkynyl-substituted trimethyltin compound 12a did not yield the corresponding carboxylic acid [11C]12b. Radio-HPLC analysis of the crude reaction mixtures (following protonation) revealed that a relatively pure reaction had occurred in all cases; the only 11C-labeled impurity was attributed to [11C]CO2 and was common throughout all of the reactions.
Scheme 2. Substrate Scope of the 11C-Carboxylation Reaction. Non-isolated and Decay-Corrected RCYs Are in Parentheses and Are n = 2 Unless Otherwise Stated.

This new 11C-carboxylation method was successfully applied to the fully automated radiosynthesis and purification of [11C]bexarotene ([11C]13b; n = 3) using the TracerMaker (Scansys Laboratorieteknik, Denmark) synthesis module (Scheme 3). A decay-corrected RCY of 32 ± 5% was obtained relative to starting [11C]CO2, with molar activity Am = 38 ± 23 GBq/μmol (1.0 ± 0.6 Ci/μmol) and radiochemical purities >95% in all instances, from an overall synthesis and purification time of 41 ± 4 min. Isolated formulations (10% ethanol/90% saline v/v) contained up to 5.74 GBq (155 mCi) radioactivity and molar activities of up to Am = 63 GBq/μmol (1.7 Ci/μmol).39 These values are comparable to the decay-corrected RCY of 45% previously reported for [11C]13b16 using the corresponding boronic ester precursor.
Scheme 3. Radiosynthesis of [11C]Bexarotene.

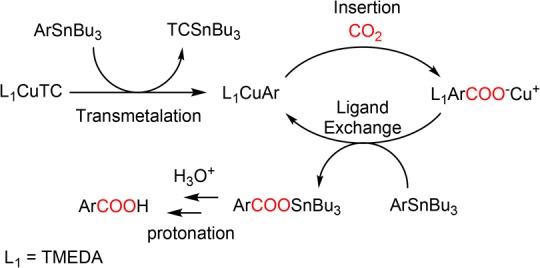
We propose the reaction mechanism illustrated in Scheme 4, wherein each step is supported by a literature precedent. The (hetero)arene (Ar) is first transferred from tributyltin to copper in the initial transmetalation step, generating the corresponding organocuprate (L1CuAr) along with Bu3SnTC.34,35 Experimental and theoretical studies have indicated that this process is reversible and close to ergoneutral.31,35 The organocuprate then readily undergoes CO2-insertion to form the carboxylate complex L1CuOOCAr.37,38 The large excess of arylstannane in solution then promotes a second ligand exchange process that simultaneously regenerates the organocuprate and forms the corresponding tributyltin carboxylate Bu3SnOOCAr, the latter of which is protonated to the carboxylic acid (ArCOOH) following acidic workup.40 TMEDA likely serves as the ligand (L1) for the active Cu(I) species in solution.25 As it is not mandatory for the reaction to be catalytic in the radiochemical experiments given the sub-stoichiometric quantities of [11C]CO2 being used, the mechanism in these cases may be viewed simply as a two-step transmetalation-insertion process. We envision that this methodology can be readily extended to reactions with 13CO2 and 14CO2.
In conclusion, we have developed a novel copper-mediated carboxylation methodology for the preparation of (hetero)aryl carboxylic acids from organostannanes using a stoichiometric quantity of CO2 under atmospheric pressure as well as sub-stoichiometric quantities of [11C]CO2. The successful radiosynthesis, purification, and formulation of [11C]bexarotene, using a fully automated commercial synthesis module, suggest that this methodology could be applied in radiopharmaceutical production.
Methods
General Methods and Instrumentation
1H and 13C{1H} NMR spectra were recorded at 400.22 (1H) and 100.64 MHz (13C{1H}) using a Bruker AV400 spectrometer and were referenced to the residual solvent proton and 13C signals, respectively. 19F{1H} NMR spectra were recorded at 376.58 MHz on a Bruker AV400 spectrometer and were referenced to an internal standard of 1,2-difluorobenzene (−140.53 ppm).30 High-resolution mass spectra and exact masses were determined on a Waters GCT Premier mass spectrometer using electron ionization, an Agilent 6538 Q-TOF mass spectrometer using electrospray ionization, or a JEOL AccuTOF model JMS-T1000LC mass spectrometer using DART ionization. Melting points were measured on a Mel-Temp Melting Point Apparatus (Barnstead International). HPLC analyses of reaction mixtures were carried out using a Phenomenex (Prodigy C18, 250 × 4.6 mm, 10 μm) analytical HPLC column coupled to a high-pressure isocratic pump (Shimadzu LC-20AT) and a variable-wavelength UV detector (Shimadzu SPD-20A; λ = 254 nm). Data were recorded and analyzed using PowerChrom chromatography software. Column chromatography was carried out using silica gel 40–60 Å (Oakwood Chemical) as the stationary phase.
Materials
Commercially available organostannanes (1a, 5a–12a) were used as received from Sigma-Aldrich and have ≥95% purity in all instances. Reference standards of the carboxylic acids 1b–13b were used as received from Sigma-Aldrich. Samples of TMEDA used for the synthesis of [11C]13b were stored under 4 Å molecular sieves, while CuTC (Sigma-Aldrich), CuI (Oakwood Chemical), LiCl (Oakwood Chemical), and Pd(PPh3)4 (Sigma-Aldrich) were handled in an argon-purged glove-bag. All other chemicals were obtained from Sigma-Aldrich, Oakwood Chemical, ACP Chemicals, Caledon, and Fisher Scientific and used as received without further purification.
Tributyl(p-tolyl)stannane (2a), tributyl(4-methoxyphenyl)stannane (3a), and tributyl(4-trifluoromethylphenyl)stannane (4a) were synthesized according to adapted literature procedures41,42 and exhibited 1H, 13C{1H}, 19F{1H} NMR (for 4a), and ESI-MS data that agree with those previously reported for the compounds.43,44 2,5-Dichloro-2,5-dimethylhexane (S1),45,46 1,1,4,4,6-pentamethyl-1,2,3,4-tetrahydronaphthalene (S2),47,48 4-iodophenyl-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)-methanone (S3),47 and 6-(1-(4-iodophenyl)vinyl)-1,1,4,4,7-pentamethyl-1,2,3,4-tetrahydronaphthalene (S4)16,49 were prepared according to previously published procedures and exhibited NMR spectral data that agree with those previously published for the compounds. Additional spectral data for S3: 13C{1H} NMR (CDCl3): 197.7, 148.0, 141.9, 137.61, 137.59, 134.9, 134.2, 131.6, 129.3, 127.9, 100.8, 34.93, 34.88, 34.3, 33.9, 31.7, 31.6, 19.9.
Tributyl(4-(1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)vinyl)phenyl)stannane (13a)
The title compound was prepared in 19% overall yield according to a five-step procedure starting from 2,5-dimethyl-2,5-hexanediol (with intermediate compounds S1–S4; see Scheme S1). In the final step, 13a was prepared from S4 using procedures adapted from those of Ritter and co-workers.50 An oven-dried 25 mL Schlenk tube was fitted with a stir-bar and cooled under an atmosphere of argon before the addition of S4 (0.50 g, 1.2 mmol) and anhydrous dioxane (ca. 10 mL). To the resulting mixture were added Pd(PPh3)4 (0.067 g, 0.058 mmol), LiCl (0.246 g, 5.81 mmol), and bis(tributyltin) (1.2 mL, 2.3 mmol) under a positive pressure of argon. The Schlenk tube was sealed and heated to 95 °C for ca. 20 h before being allowed to cool to room temperature. The resulting black solution was concentrated using a rotary evaporator. Purification of the crude residue by column chromatography on silica gel (eluent: hexanes/Et3N 99:1 (v/v)) afforded 13a as a colorless oil (0.422 g, 0.712 mmol, 61%, Rf = 0.36 in hexanes/Et3N 99:1 (v/v)). 13a was identified on the basis of its 1H, 13C{1H} NMR, ESI-MS, and HRMS data: 1H NMR (CDCl3): δ 7.38 (d, J = 7.9 Hz, JHSn = 37.9 Hz, 2 H), 7.23 (d, J = 7.9 Hz, 2 H), 7.13 (s, 1 H), 7.07 (s, 1 H), 5.74 (d, J = 1.5 Hz, 1 H), 5.18 (d, J = 1.5 Hz, 1 H), 1.98 (s, 3 H), 1.70 (s, 4 H), 1.53 (m, 6 H), 1.33 (sextet, J = 7.5 Hz, 6 H), 1.30 (s, 6 H), 1.27 (s, 6 H), 1.04 (m, JHSn = 50.4 Hz, 6 H), 0.88 (t, J = 7.3 Hz, 9 H). 13C{1H} NMR (CDCl3): δ 149.9, 143.8, 142.0, 141.3 (JCSn = 390.7, 370.3 Hz), 140.4 (JCSn = 10.3 Hz), 138.8, 136.4 (JCSn = 31.1 Hz), 132.8, 128.1, 127.8, 125.9 (JCSn = 41.2 Hz), 114.3, 35.28, 35.26, 34.0, 33.9, 31.93, 31.91, 29.1 (JCSn = 19.9 Hz), 27.4 (JCSn = 54.9 Hz), 19.9, 13.7, 9.6 (JCSn = 339.5, 324.2 Hz). EI-MS, m/z (relative intensity; Sn-containing isotopologic clusters are represented by the 120Sn isotopolog and are indicated with asterisks): 593.3* (1, M+), 537.2* (100, (M–C4H8)+), 481.2* (81, (M–2C4H8)+), 425.1* (85, (M–3C4H8)+), 367.0* (12, (M–C17H22)+), 289.2 (12, C22H25+), 103.1 (15, C8H7+). HRMS (EI-TOF+): [M]+ calcd for C35H53120Sn+ 593.3169; found 593.3185.
General Procedure for the Synthesis of Carboxylic Acids
Carboxylic acids 1b, 4b, 11b, and 13b were prepared according to procedures adapted from those of Takaya et al. for the copper(I)-catalyzed carboxylation of arylboronic esters.37 An oven-dried 10 mL v-vial, charged with a spin vane and sealed with a hole cap and septum, was allowed to cool in an atmosphere of argon. To the vial was added CuTC (1.8 mg, 9.4 μmol) followed by the addition of DMF (ca. 2.50 mL), TMEDA (ca. 0.25 mL), and the corresponding arylstannane (0.20 mmol) using microliter syringes. To the resulting pale-blue solution was introduced gaseous CO2, which was produced from dry ice (in a sealed vessel) and transferred to the solution using a hypodermic needle. Gaseous CO2 was bubbled through the reaction solution for ca. 25 min before the v-vial was sealed using a new hole cap and (unpunctured) septum. The sealed vial was then heated at 90 °C for 17 h before being allowed to cool to room temperature. An aliquot (150 μL) of the resulting solution was acidified with ca. 0.67 M aqueous formic acid (1.50 mL) and analyzed by HPLC (see Figures S1–S4). The bulk reaction mixture was transferred to an Erlenmeyer flask and quenched with ca. 0.67 M aqueous HCl solution (ca. 24 mL). The mixture was then transferred to a separatory funnel and extracted with Et2O (3 × 15 mL). The combined organic extracts were dried using MgSO4, filtered, and solvent-removed using a rotary evaporator. Pentane (ca. 1 mL) was added to the resulting crude oil to induce the precipitation of yellow crystals. The crystals were washed with additional portions of cold pentane (5 × 0.5 mL) before the solid was dried under vacuum (ca. 40 mmHg) for 1–2 h.
Benzoic Acid (1b)
The title compound was prepared using arylstannane 1a (73.4 mg, 0.200 mmol). 1b was isolated as yellow crystals (14.2 mg, 0.116 mmol, 58%, mp 118–120 °C (lit. 122 °C)51) and was identified on the basis of its 1H, 13C{1H} NMR, and HRMS data, which agree with those previously reported for the compound:52,531H NMR (CDCl3): δ 8.13 (d, J = 8.5 Hz, 2 H), 7.62 (t, J = 7.2 Hz, 1 H), 7.49 (t, J = 7.7 Hz, 2 H). 13C{1H} NMR (CDCl3): δ 171.7, 133.7, 130.2, 129.2, 128.5. HRMS (DART): [M + H]+ calcd for C7H7O2+ 123.0441; found 123.0446.
4-(Trifluoromethyl)benzoic Acid (4b)
The title compound was prepared using arylstannane 4a (90.8 mg, 0.200 mmol). 4b was isolated as pale-yellow crystals (13.1 mg, 0.0689 mmol, 33%) and was identified on the basis of its 1H, 13C{1H}, 19F{1H} NMR, and HRMS data, which agree with those previously reported for the compound:52,54,551H NMR (DMSO-d6): δ 13.4 (br, 1 H), 8.13 (d, J = 8.2 Hz, 2 H), 7.87 (d, J = 8.2 Hz, 2 H). 13C{1H} NMR (DMSO-d6): δ 166.2, 134.6, 132.5 (q, JCF = 32.2 Hz), 130.1, 125.6 (q, JCF = 3.7 Hz), 123.8 (q, JCF = 271.5 Hz). 19F{1H} NMR (DMSO-d6): -62.9. ESI-MS, m/z (relative intensity): 189.02 (100, (M–H)−), 145.03 (17, (M–CHO2)−). HRMS (ESI): [M–H]− calcd for C8H4F3O2– 189.0169; found 189.0171. Heating a sample of 4b in a melting point capillary tube led to sublimation at ca. 165 °C; a reference sample of 4b also sublimed at 170–175 °C.
Radiochemistry: General Remarks, Methods, and Instrumentation
No carrier added [11C]CO2 was produced using a MC17 cyclotron (Scanditronix) by the 14N(p,α)11C reaction and a gaseous target of N2 (+0.5% O2). The time at which [11C]CO2 was delivered to the synthesis module is defined as the start-of-synthesis, while the end-of-synthesis is defined as the time at which the activity of the isolated product formulation was measured.
Synthesis and Characterization of [11C]1b–[11C]12b
Reactions were carried out using 5 mL disposable centrifuge tubes (Kimble Glass Inc.) sealed with a hole cap and septum. Immediately prior to radiosynthesis, an oven-dried centrifuge tube was charged with the appropriate copper(I) salt in an argon-purged glove-bag followed by the sequential addition of solvent, base, and organostannane using microliter syringes. [11C]CO2 produced from the cyclotron was first transferred from the target to a molecular sieve trap at room temperature before being released (at 350 °C) and reconcentrated in a silica-gel trap cooled with liquid nitrogen. [11C]CO2 was then transferred to the reaction mixture by warming the trap to room temperature under a preset flow rate (ca. 10 mL/min) of gaseous He. [11C]CO2 was then transferred to the solution for 1 min and was followed by flushing the headspace with He (ca. 10 mL/min) for an additional minute. The reaction vessel was then sealed, and the solution was heated to the desired temperature for 5 min. Afterwards, the mixture was diluted with ca. 1 mL of water before being transferred to a septum-capped 20 mL glass vial (i.e., product vial). The solution was acidified with ca. 4 mL of aqueous 1 M formic acid solution and bubbled with nitrogen gas for an additional 2–3 min. The activity in the product vial was then quantified, and its radiochemical identity and purity were analyzed using radio-HPLC.
The vent-line of the reaction vessel was fitted with a sodium hydroxide coated silica (Ascarite II, 20–30 mesh) trap (denoted as AT1) to capture the activity lost during the transfer of [11C]CO2 into solution at room temperature. While the reaction vessel was sealed, AT1 was replaced with a second trap (AT2) to capture the activity that escapes the solution during heating and after the addition of ca. 1 mL of water. The vent-line of the product vial was also fitted with a third trap (AT3) to quantify the activity lost upon addition of the 1 M formic acid solution and bubbling the reaction mixture with nitrogen gas. All activity values were decay-corrected to the same point in time. The total activity for each reaction was estimated by adding the activities measured in the product vial and three traps (AT1–3). The decay-corrected radiochemical yield (RCY (%)) is defined as the amount of activity in the product vial multiplied by its radiochemical purity (as determined by radio-HPLC) and divided by the total activity. The decay-corrected trapping efficiency (TE (%)) is defined as one minus the ratio of activity in AT1–3 versus the total activity.
Procedure for the Synthesis and Isolation of [11C]1b
[11C]CO2 was produced from the cyclotron and transferred, as previously described, to a reaction mixture consisting of DMF (600 μL), CuTC (0.7 mg, 3.7 μmol), TMEDA (120 μL, 0.800 mmol), and 1a (19.6 μL, 60.1 μmol). The reaction vessel was then sealed and heated at 100 °C for 5 min. Subsequently, the reaction mixture was quenched/diluted with 1 M aqueous formic acid solution (ca. 4 mL), mixed, and transferred to a 5 mL HPLC loop before being injected to a semipreparative HPLC column (Phenomenex; Luna C18, 250 × 10 mm, 10 μm). The separation and isolation of [11C]1b were carried out using the Tracerlab FX2C module (General Electric) using a mobile phase consisting of acetonitrile/H2O/trifluoroacetic acid (TFA) (25:75:0.1; by volume) and a flow rate of 9 mL/min. The peak due to [11C]1b was then collected (retention time of ca. 9.5 min; see Figure S5) in a septum-capped 20 mL glass vial. An activity of 157 mCi was obtained at the end-of-synthesis from a total synthesis and purification time of ca. 29 min, corresponding to decay- and non-decay-corrected RCYs of 45 and 17%, respectively.
Procedure for the Synthesis and Isolation of [11C]13b
Immediately prior to radiosynthesis, an oven-dried 4 mL reaction vessel was charged with 13a (35.6 mg, 60.0 μmol). The vessel was then transferred to an argon-purged glove-bag where DMF (600 μL), TMEDA (120 μL, 0.800 mmol), and CuTC (0.7 mg, 3.7 μmol) were added, and the vessel was sealed with a hole cap and septum. The automated synthesis and isolation of [11C]13b were performed on a TracerMaker synthesis module (Scansys Laboratorieteknik, Denmark; see flowcharts in Figures S20 and S21). [11C]CO2 was produced from the cyclotron as previously described and transferred from the target to a HeySep D column (700 mg) cooled to −180 °C using liquid nitrogen. The HeySep D column was first gradually heated to −40 °C under helium flow (ca. 100 mL/min) to remove by-products that may have been produced during the 14N(p,α)11C reaction. [11C]CO2 was subsequently released by warming the trap to room temperature and transferred, under a preset flow rate (ca. 10 mL/min) of gaseous He, to the reaction vessel containing the solution mixture. The vessel was then sealed and heated at 100 °C for 5 min. Afterwards, the reaction mixture was diluted with ca. 2 mL of acetonitrile/H2O/TFA (80:20:0.1; by volume) and transferred to a 5 mL HPLC loop before being injected to a semipreparative HPLC column (Phenomenex; Gemini C18, 250 × 10 mm, 10 μm). The isolation of [11C]13b was carried out using a mobile phase consisting of acetonitrile/H2O/TFA (80:20:0.1; by volume) and a flow rate of 9 mL/min. The peak due to [11C]13b was then collected with a retention time of ca. 9.7 min (see Figure S6). After removal of the mobile phase, [11C]13b was then formulated in an ethanol/saline (10:90 v/v) mixture. The formulation at the end-of-synthesis contained 155 mCi of radioactivity from a synthesis and purification time of 37 min, corresponding to decay- and non-decay-corrected RCYs of 35 and 10%, respectively.
Analysis of Radiochemical Identity and Purity Using Radio-HPLC
Analysis of 11C-radiosynthesis reactions was carried out using a Phenomenex (Prodigy C18, 250 × 4.6 mm, 10 μm) analytical HPLC column coupled to a high-pressure isocratic pump (Shimadzu LC-20AT) and attached sequentially to a variable-wavelength UV detector (Shimadzu SPD-20A; λ = 254 nm) and radioactivity detector (Frisk-tech, Bicron; γ-ray). Data were recorded and analyzed using PowerChrom chromatography software. Seven different isocratic HPLC separation methods (denoted as methods A–G) were utilized over the course of this study: method A: Mobile phase: acetonitrile/H2O/TFA (25:75:0.1; by volume); flow rate: 2 mL/min. Method B: Mobile phase: acetonitrile/H2O/TFA (25:75:0.1; by volume); flow rate: 3 mL/min. Method C: Mobile phase: acetonitrile/H2O/TFA (40:60:0.1; by volume); flow rate: 2 mL/min. Method D: Mobile phase: acetonitrile/H2O/TFA (10:90:0.1; by volume); flow rate: 1 mL/min. Method E: Mobile phase: acetonitrile/H2O/TFA (10:90:0.1; by volume); flow rate: 2 mL/min. Method F: Mobile phase: acetonitrile/H2O/TFA (80:20:0.1; by volume); flow rate: 2 mL/min. Method G: Mobile phase: acetonitrile/0.1 M NH4CO2H (70:30; by volume); flow rate: 2 mL/min.
Acknowledgments
N.V. thanks National Institute on Ageing of the NIH (R01AG054473), the Azrieli Foundation, Canada Foundation for Innovation, Ontario Research Fund, and the Canada Research Chairs Program for support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c00524.
Analysis of reaction mixtures by HPLC for the synthesis of 1b, 4b, 11b, and 13b; HPLC chromatograms for the synthesis and isolation of [11C]1b and [11C]13b using semipreparative HPLC; analysis of radiochemical identity and purity for the synthesis of [11C]1b–[11C]13b; schematic flowcharts of the TracerMaker synthesis module; 1H, 13C{1H}, and 19F{1H} NMR spectra of synthesized compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Tortajada A.; Juliá-Hernández F.; Börjesson M.; Moragas T.; Martin R. Transition-Metal-Catalyzed Carboxylation Reactions with Carbon Dioxide. Angew. Chem., Int. Ed. 2018, 57, 15948–15982. 10.1002/anie.201803186. [DOI] [PubMed] [Google Scholar]
- Ahamed M.; Verbeek J.; Funke U.; Lecina J.; Verbruggen A.; Bormans G. Recent Progress in Metal Catalyzed Direct Carboxylation of Aryl Halides and Pseudo Halides Employing CO2: Opportunities for 11C Radiochemistry. ChemCatChem 2016, 8, 3692–3700. 10.1002/cctc.201600943. [DOI] [Google Scholar]
- Bragg R. A.; Sardana M.; Artelsmair M.; Elmore C. S. New trends and applications in carboxylation for isotope chemistry. J. Labelled Compd. Radiopharm. 2018, 61, 934–948. 10.1002/jlcr.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinsinger K.; Pieters G. The Emergence of Carbon Isotope Exchange. Angew. Chem., Int. Ed. 2019, 58, 9678–9680. 10.1002/anie.201905368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller P. W.; Long N. J.; Vilar R.; Gee A. D. Synthesis of 11C, 18F, 15O, and 13N Radiolabels for Positron Emission Tomography. Angew. Chem., Int. Ed. 2008, 47, 8998–9033. 10.1002/anie.200800222. [DOI] [PubMed] [Google Scholar]
- Rotstein B. H.; Liang S. H.; Holland J. P.; Collier T. L.; Hooker J. M.; Wilson A. A.; Vasdev N. 11CO2 fixation: a renaissance in PET radiochemistry. Chem. Commun. 2013, 49, 5621–5629. 10.1039/C3CC42236D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotstein B. H.; Liang S. H.; Placzek M. S.; Hooker J. M.; Gee A. D.; Dollé F.; Wilson A. A.; Vasdev N. 11C=O bonds made easily for positron emission tomography radiopharmaceuticals. Chem. Soc. Rev. 2016, 45, 4708–4726. 10.1039/C6CS00310A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X.; Rong J.; Wang L.; Vasdev N.; Zhang L.; Josephson L.; Liang S. H. Chemistry for Positron Emission Tomography: Recent Advances in 11C-, 18F-, 13N-, and 15O-Labeling Reactions. Angew. Chem., Int. Ed. 2019, 58, 2580–2605. 10.1002/anie.201805501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl K.; Halldin C.; Schou M. New methodologies for the preparation of carbon-11 labeled radiopharmaceuticals. Clin. Transl. Imaging 2017, 5, 275–289. 10.1007/s40336-017-0223-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei C.; Gee A. D. Recent progress in [11C]carbon dioxide ([11C]CO2) and [11C]carbon monoxide ([11C]CO) chemistry. J. Labelled Compd. Radiopharm. 2018, 61, 237–251. 10.1002/jlcr.3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl K.; Collier T. L.; Cheng R.; Zhang X.; Sadovski O.; Liang S. H.; Vasdev N. “In-loop” [11C]CO2 fixation: Prototype and proof of concept. J. Labelled Compd. Radiopharm. 2018, 61, 252–262. 10.1002/jlcr.3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A. A.; Garcia A.; Parkes J.; Houle S.; Tong J.; Vasdev N. [11C]CURB: Evaluation of a novel radiotracer for imaging fatty acid amide hydrolase by positron emission tomography. Nucl. Med. Biol. 2011, 38, 247–253. 10.1016/j.nucmedbio.2010.08.001. [DOI] [PubMed] [Google Scholar]
- Rusjan P. M.; Wilson A. A.; Mizrahi R.; Boileau I.; Chavez S. E.; Lobaugh N. J.; Kish S. J.; Houle S.; Tong J. Mapping Human Brain Fatty Acid Amide Hydrolase Activity with PET. J. Cereb. Blood Flow Metab. 2012, 33, 407–414. 10.1038/jcbfm.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasdev N.; Sadovski O.; Garcia A.; Dollé F.; Meyer J. H.; Houle S.; Wilson A. A. Radiosynthesis of [11C]SL25.1188 via [11C]CO2 fixation for imaging monoamine oxidase B. J. Labelled Compd. Radiopharm. 2011, 54, 678–680. 10.1002/jlcr.1908. [DOI] [Google Scholar]
- Rusjan P. M.; Wilson A. A.; Miler L.; Fan I.; Mizrahi R.; Houle S.; Vasdev N.; Meyer J. H. Kinetic Modeling of the Monoamine Oxidase B Radioligand [11C]SL25.1188 in Human Brain with High-Resolution Positron Emission Tomography. J. Cereb. Blood Flow Metab. 2014, 34, 883–889. 10.1038/jcbfm.2014.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotstein B. H.; Hooker J. M.; Woo J.; Collier T. L.; Brady T. J.; Liang S. H.; Vasdev N. Synthesis of [11C]Bexarotene by Cu-Mediated [11C]Carbon Dioxide Fixation and Preliminary PET Imaging. ACS Med. Chem. Lett. 2014, 5, 668–672. 10.1021/ml500065q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotstein B. H.; Placzek M. S.; Krishnan H. S.; Pekošak A.; Collier T. L.; Wang C.; Liang S. H.; Burstein E. S.; Hooker J. M.; Vasdev N. Preclinical PET Neuroimaging of [11C]Bexarotene. Mol. Imaging 2016, 15, 1536012116663054. 10.1177/1536012116663054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T.; Furusawa Y.; Yamada S.; Akehi M.; Takenaka F.; Sasaki T.; Akahoshi A.; Hanada T.; Matsuura E.; Hirano H.; Tai A.; Kakuta H. Positron Emission Tomography to Elucidate Pharmacokinetic Differences of Regioisomeric Retinoid X Receptor Agonists. ACS Med. Chem. Lett. 2015, 6, 334–338. 10.1021/ml500511m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen C. H.; Hansen H. D.; Bay T.; Vogensen S. B.; Lehel S.; Thiesen L.; Bundgaard C.; Clausen R. P.; Knudsen G. M.; Herth M. M.; Wellendorph P.; Frølund B. Radiosynthesis and Evaluation of [11C]3-Hydroxycyclopent-1-enecarboxylic Acid as Potential PET Ligand for the High-Affinity γ-Hydroxybutyric Acid Binding Sites. ACS Chem. Neurosci. 2017, 8, 22–27. 10.1021/acschemneuro.6b00335. [DOI] [PubMed] [Google Scholar]
- Shibahara O.; Watanabe M.; Yamada S.; Akehi M.; Sasaki T.; Akahoshi A.; Hanada T.; Hirano H.; Nakatani S.; Nishioka H.; Takeuchi Y.; Kakuta H. Synthesis of 11C-Labeled RXR Partial Agonist 1-[(3,5,5,8,8-Pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)amino]benzotriazole-5-carboxylic Acid (CBt-PMN) by Direct [11C]Carbon Dioxide Fixation via Organolithiation of Trialkyltin Precursor and PET Imaging Thereof. J. Med. Chem. 2017, 60, 7139–7145. 10.1021/acs.jmedchem.7b00817. [DOI] [PubMed] [Google Scholar]
- Fisher M. J.; McMurray L.; Lu S.; Morse C. L.; Liow J.-S.; Zoghbi S. S.; Kowalski A.; Tye G. L.; Innis R. B.; Aigbirhio F. I.; Pike V. W. [Carboxyl-11C]Labelling of Four High-Affinity cPLA2α Inhibitors and Their Evaluation as Radioligands in Mice by Positron Emission Tomography. ChemMedChem 2018, 13, 138–146. 10.1002/cmdc.201700697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida N. R.; Conda-Sheridan M. A review of the molecular design and biological activities of RXR agonists. Med. Res. Rev. 2019, 39, 1372–1397. 10.1002/med.21578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm M. F.; Zhang L.; Badea B. A.; White S. K.; Mais D. E.; Berger E.; Suto C. M.; Goldman M. E.; Heyman R. A. Synthesis and Structure-Activity Relationships of Novel Retinoid X Receptor-Selective Retinoids. J. Med. Chem. 1994, 37, 2930–2941. 10.1021/jm00044a014. [DOI] [PubMed] [Google Scholar]
- Tousi B. The emerging role of bexarotene in the treatment of Alzheimer’s disease: current evidence. Neuropsychiatr. Dis. Treat. 2015, 11, 311–315. 10.2147/NDT.S61309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riss P. J.; Lu S.; Telu S.; Aigbirhio F. I.; Pike V. W. CuI-Catalyzed 11C Carboxylation of Boronic Acid Esters: A Rapid and Convenient Entry to 11C-Labeled Carboxylic Acids, Esters, and Amides. Angew. Chem., Int. Ed. 2012, 51, 2698–2702. 10.1002/anie.201107263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafique W.; Khanapur S.; Spilhaug M. M.; Riss P. J. Reaching out for Sensitive Evaluation of the Mu Opioid Receptor in Vivo: Positron Emission Tomography Imaging of the Agonist [11C]AH7921. ACS Chem. Neurosci. 2017, 8, 1847–1852. 10.1021/acschemneuro.7b00075. [DOI] [PubMed] [Google Scholar]
- Wilson T. C.; Cailly T.; Gouverneur V. Boron reagents for divergent radiochemistry. Chem. Soc. Rev. 2018, 47, 6990–7005. 10.1039/C8CS00499D. [DOI] [PubMed] [Google Scholar]
- Pretze M.; Große-Gehling P.; Mamat C. Cross-Coupling Reactions as Valuable Tool for the Preparation of PET Radiotracers. Molecules 2011, 16, 1129–1165. 10.3390/molecules16021129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L.; Placzek M. S.; Hooker J. M.; Vasdev N.; Liang S. H. [11C]Cyanation of arylboronic acids in aqueous solutions. Chem. Commun. 2017, 53, 6597–6600. 10.1039/C7CC02886E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makaravage K. J.; Shao X.; Brooks A. F.; Yang L.; Sanford M. S.; Scott P. J. H. Copper(II)-Mediated [11C]Cyanation of Arylboronic Acids and Arylstannanes. Org. Lett. 2018, 20, 1530–1533. 10.1021/acs.orglett.8b00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allred G. D.; Liebeskind L. S. Copper-Mediated Cross-Coupling of Organostannanes with Organic Iodides at or below Room Temperature. J. Am. Chem. Soc. 1996, 118, 2748–2749. 10.1021/ja9541239. [DOI] [Google Scholar]
- Kang S.-K.; Kim J.-S.; Choi S.-C. Copper- and Manganese-Catalyzed Cross-Coupling of Organostannanes with Organic Iodides in the Presence of Sodium Chloride. J. Org. Chem. 1997, 62, 4208–4209. 10.1021/jo970656j. [DOI] [PubMed] [Google Scholar]
- Thapa S.; Shrestha B.; Gurung S. K.; Giri R. Copper-catalysed cross-coupling: an untapped potential. Org. Biomol. Chem. 2015, 13, 4816–4827. 10.1039/C5OB00200A. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Burton D. J. Copper(I)-Only Catalyzed Reactions of (E)-2,3-Difluoro-3-stannylacrylic Ester with Acid Chlorides and Mechanistic Studies of the “Copper Effect” in Stille Coupling Reactions. Org. Lett. 2004, 8, 1109–1111. 10.1021/ol053023x. [DOI] [PubMed] [Google Scholar]
- Wang M.; Lin Z. Stille Cross-Coupling Reactions of Alkenylstannanes with Alkenyl Iodides Mediated by Copper(I) Thiophene-2-carboxylate: A Density Functional Study. Organometallics 2010, 29, 3077–3084. 10.1021/om100304t. [DOI] [Google Scholar]
- Nayal O. S.; Hong J.; Yang Y.; Mo F. Cu-Catalysed carboxylation of aryl boronic acids with CO2. Org. Chem. Front. 2019, 6, 3673–3677. 10.1039/C9QO01023H. [DOI] [Google Scholar]
- Takaya J.; Tadami S.; Ukai K.; Iwasawa N. Copper(I)-Catalyzed Carboxylation of Aryl- and Alkenylboronic Esters. Org. Lett. 2008, 10, 2697–2700. 10.1021/ol800829q. [DOI] [PubMed] [Google Scholar]
- Ohishi T.; Nishiura M.; Hou Z. Carboxylation of Organoboronic Esters Catalyzed by N-Heterocyclic Carbene Copper(I) Complexes. Angew. Chem., Int. Ed. 2008, 47, 5792–5795. 10.1002/anie.200801857. [DOI] [PubMed] [Google Scholar]
- We have found that additional efforts to improve the molar activity, for instance by increasing the starting radioactivity of [11C]CO2 to ca. 64 GBq (from ca. 44.5 GBq), and storing the N,N,N’,N’-tetramethylethylenediamine (TMEDA) under 4Å molecular sieves for at least two weeks, improved the molar activity in that a value of Am = 63 GBq/μmol was obtained.
- Davies A., Organotin Carboxylates and Other Oxyesters. Stannylium Ions. In Organotin Chemistry; John Wiley & Sons: 2004; pp 203–213. [Google Scholar]
- Gligorich K. M.; Cummings S. A.; Sigman M. S. Palladium-Catalyzed Reductive Coupling of Styrenes and Organostannanes under Aerobic Conditions. J. Am. Chem. Soc. 2007, 129, 14193–14195. 10.1021/ja076746f. [DOI] [PubMed] [Google Scholar]
- Horino Y.; Sugata M.; Mutsuura I.; Tomohara K.; Abe H. Controllable Stereoselective Synthesis of (Z)- and (E)-Homoallylic Alcohols Using a Palladium-Catalyzed Three-Component Reaction. Org. Lett. 2017, 19, 5968–5971. 10.1021/acs.orglett.7b02979. [DOI] [PubMed] [Google Scholar]
- Oikawa A.; Kindaichi G.; Shimotori Y.; Okimoto M.; Hoshi M. Simple preparation of aryltributylstannanes and its application to one-pot synthesis of diaryl ketones. Tetrahedron 2015, 71, 1705–1711. 10.1016/j.tet.2015.01.048. [DOI] [Google Scholar]
- Komeyama K.; Asakura R.; Takaki K. A Sn atom-economical approach toward arylstannanes: Ni-catalysed stannylation of aryl halides using Bu3SnOMe. Org. Biomol. Chem. 2015, 13, 8713–8716. 10.1039/C5OB01096A. [DOI] [PubMed] [Google Scholar]
- Wang M.; Davis T.; Gao M.; Zheng Q.-H. Synthesis of a new fluorine-18-labeled bexarotene analogue for PET imaging of retinoid X receptor. Bioorg. Med. Chem. Lett. 2014, 24, 1742–1747. 10.1016/j.bmcl.2014.02.037. [DOI] [PubMed] [Google Scholar]
- Irwin L. J.; Zeits P. D.; Reibenspies J. H.; Miller S. A. Diffuse Diffraction from Parallel/Antiparallel Metallocene Pillars. Organometallics 2007, 26, 1129–1133. 10.1021/om060435g. [DOI] [Google Scholar]
- Sarshar S.Novel Therapeutic Agents for the Treatment of Cancer, Metabolic Diseases and Skin Disorders. US 200,802,342,29 (A1), September 25, 2008.
- Wagner C. E.; Jurutka P. W.; Marshall P. A.; Groy T. L.; van der Vaart A.; Ziller J. W.; Furmick J. K.; Graeber M. E.; Matro E.; Miguel B. V.; Tran I. T.; Kwon J.; Tedeschi J. N.; Moosavi S.; Danishyar A.; Philp J. S.; Khamees R. O.; Jackson J. N.; Grupe D. K.; Badshah S. L.; Hart J. W. Modeling, Synthesis and Biological Evaluation of Potential Retinoid X Receptor (RXR) Selective Agonists: Novel Analogues of 4-[1-(3,5,5,8,8-Pentamethyl-5,6,7,8-tetrahydro-2-naphthyl)ethynyl]benzoic Acid (Bexarotene). J. Med. Chem. 2009, 52, 5950–5966. 10.1021/jm900496b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. H.; Morandi B. Metathesis-active ligands enable a catalytic functional group metathesis between aroyl chlorides and aryl iodides. Nat. Chem. 2018, 10, 1016–1022. 10.1038/s41557-018-0078-8. [DOI] [PubMed] [Google Scholar]
- Tang P.; Furuya T.; Ritter T. Silver-Catalyzed Late-Stage Fluorination. J. Am. Chem. Soc. 2010, 132, 12150–12154. 10.1021/ja105834t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felpin F.-X.; Fouquet E. A Useful, Reliable and Safer Protocol for Hydrogenation and the Hydrogenolysis of O-Benzyl Groups: The In Situ Preparation of an Active Pd0/C Catalyst with Well-Defined Properties. Chem. – Eur. J. 2010, 16, 12440–12445. 10.1002/chem.201001377. [DOI] [PubMed] [Google Scholar]
- Ma C.; Zhao C.-Q.; Xu X.-T.; Li Z.-M.; Wang X.-Y.; Zhang K.; Mei T.-S. Nickel- Catalyzed Carboxylation of Aryl and Heteroaryl Fluorosulfates Using Carbon Dioxide. Org. Lett. 2019, 21, 2464–2467. 10.1021/acs.orglett.9b00836. [DOI] [PubMed] [Google Scholar]
- Charboneau D. J.; Brudvig G. W.; Hazari N.; Lant H. M. C.; Saydjari A. K. Development of an Improved System for the Carboxylation of Aryl Halides through Mechanistic Studies. ACS Catal. 2019, 9, 3228–3241. 10.1021/acscatal.9b00566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujihara T.; Nogi K.; Xu T.; Terao J.; Tsuji Y. Nickel-Catalyzed Carboxylation of Aryl and Vinyl Chlorides Employing Carbon Dioxide. J. Am. Chem. Soc. 2012, 134, 9106–9109. 10.1021/ja303514b. [DOI] [PubMed] [Google Scholar]
- Kremlev M. M.; Tyrra W.; Mushta A. I.; Naumann D.; Yagupolskii Y. L. The solid complex Zn(CF3)Br·2DMF as an alternative reagent for the preparation of both, trifluoromethyl and pentafluoroethyl copper, CuCF3 and CuC2F5. J. Fluorine Chem. 2010, 131, 212–216. 10.1016/j.jfluchem.2009.10.011. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.