

Abstract
Background
The enrichment of cancer stem cell-like cells (CSCs) has been considered to be responsible for tumor progression after an initial response to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (EGFR-TKIs) in patients with non-small cell lung adenocarcinoma (NSCLC/ADC). CSCs with ALDH1A1bright /CD44high expression contribute to the TKIs resistance in NSCLC/ADC cells. All-trans retinoic acid (ATRA) has been shown to be a potential targeted therapy against CSCs due to its ability to inhibit ALDH1A1 activity. We therefore investigated whether ATRA could circumvent the resistance to improve the response to gefitinib in NSCLC/ADC cells.
Methods
Treatment of NSCLC/ADC A549 and H1650 cells with gefitinib enriched the gefitinib surviving cells (GSCs). The expression of ALDH1A1 and CD44 and the IC50 values for gefitinib were determined by flow cytometry (FCM) and crystal violet assay in GSCs and ATRA-treated GSCs, respectively. Using DEAB as the positive control, direct inhibitory effect of ATRA on ALDH1A1 activity was determined by ALDEFLUOR assay,
Results
GSCs showed higher expression of ALDH1A1 and CD44 and IC50 values for gefitinib than their respective parental cells, suggesting that gefitinib can lead to propagation of CSC-enriched gefitinib-resistant cells. Treatment with ATRA was found to significantly reduce the increased expression of ALDH1A1 and CD44 and the IC50 values for gefitinib in A549GSC and H1650GSC cells, and ATRA could directly inhibit active ALDH1A1 as compared to DEAB.
Conclusion
Our findings suggest that combination treatment with ATRA prevents gefitinib-induced enrichment of ALDH1A1bright/CD44high CSCs and enhances gefitinib-induced growth inhibition of NSCLC/ADC cells.
Keywords: All-trans retinoic acid, ALDH1A1, CD44, EGFR tyrosine kinase inhibitors, Non-small cell lung adenocarcinoma
Background
Non-small cell lung cancer (NSCLC) is the most common type of lung cancer, accounting for 84% of all lung cancer diagnoses [1]. For advanced NSCLC, median survival remains poor at 7.9 months and only approximately one third of patients survive for 1 year or more despite conventional combination chemotherapy [2]. About 10 and 30% of NSCLCs in Western and Asian populations, respectively, express an activated mutant epidermal growth factor receptor (EGFRm) and the majority of such patients respond to adenosine triphosphate (ATP)-competitive EGFR tyrosine kinase inhibitors (EGFR-TKIs) such as gefitinib [3–5].
Despite initial dramatic efficacy of EGFR-TKIs in NSCLC patients with or without EGFR mutation, emergence of acquired resistance is almost inevitable at a median of 9–13 months, thereby limiting the benefits of EGFR-TKIs to NSCLC patients’ outcomes [5–8]. EGFR-TKIs resistance has become a major clinical challenge in NSCLC.
EGFR T790M-mutation-mediated TKIs resistance account for about 50% in acquired resistance of NSCLC patients, and the remaining 50% of patients acquire resistance to EGFR-TKIs via a non-T790M mediated resistance mechanism [9–11]. Alterations including secondary EGFR T790M mutation, MET amplification, and appearance of EMT features were observed in NSCLC. Cancer drug resistance has recently been linked, at least in part, to a small sub-population of cells within the tumour that possess stem-like properties [12]. It has been demonstrated that stem cell-like properties are enriched in CD44high and ALDH1A1bright subpopulations of some NSCLC cell lines [13, 14]. However, the manner of EGFR-TKIs exposure influences the mechanism of acquired resistance and the appearance of stem cell–like property with EGFR-TKI treatment. After initial gefitinib treatment, the residual cancer cells can survive the initial cycles of treatment and are proved to be a subset of gefitinib-induced enrichment of cancer stem-like cells (CSCs) [12]. It has been reported that the high-concentration gefitinib-exposure methods revealed different resistance mechanisms and CSC properties with ALDH1A1 and CD44 overexpression [13, 14]. Cancer stemness induced via up-regulation of ALDH1A1 and CD44 expression contributes to the acquisition of gefitinib resistance in EGFR-TKI sensitive NSCLC [15]. CSCs with ALDH1A1bright and CD44high may play a significant role in acquired resistance to gefitinib [16, 17]. A study reported that treatment of lung cancer cell lines with TKIs enriches the ALDHbright stem-like cells through EGFR-dependent activation of Notch3 and inhibition of EGFR kinase activity leads to an increase in ALDHbright cells [18]. It has been documented that inhibition of ALDH1A1 activity reduces chemotherapy resistance in various cancers such as ovarian and breast cancers [19–22]. As is reported, ALDH1A is linked to retinoic acid signaling and all-trans retinoic acid (ATRA), as a potential targeted therapy against cancer stem cells, could inhibit ALDH1A1 activity to improve chemotherapeutic efficacy [23, 24]. However, the role of ATRA in NSCLC is less well understood, in part, since it has not been fully established which cell subpopulation express the retinoic acid receptors (RARs). Studies have confirmed that RAR and RXR are expressed in NSCLC CSCs that co-express ALDH1 [25–27]. Using therapy-induced enrichment of CSCs may, therefore, prove to be an extremely useful method for studying CSCs and provide new clues regarding potential therapeutic targets such as ALDH1A1 for their efficient elimination, which will undoubtedly play an indispensable role in improving NSCLC patients’ survival.
In this study, we explored whether ATRA reduces resistance of therapy-induced enrichment of NSCLC CSCs through inhibiting ALDH1A1 activity to circumvent gefitinib resistance in NSCLC/ADC.
Methods
Cell culture and reagents
The human NSCLC/ADC H1650 cell line with EGFRdelE746-A750 mutation and A549 cell line with EGFRwt were obtained from the Committee on Type Culture Collection of Chinese Academy of Sciences (CTCCCAS, Shanghai, China). H1650 and A549 cells were chosen because their response to gefitinib have been extensively characterized (primary resistance to gefitinib).
Cell lines were cultured in RPMI 1640 medium (GIBCO) that contained 10% fetal calf serum (FCS), 2 mmol/L L-glutamine and 100 units/mL penicillin and 40 IU/mL gentamycin were maintained at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. Subconfluent cells (80%) were passaged with a solution containing 0.25% trypsin-0.5 mmol/L Ethylenediaminetetraacetic acid (EDTA). Cell lines were tested for mycoplasma and confirmed to be negative.
Gefitinib (N-[3-Chloro-4-fluorophenyl]-7-methoxy-6-[3-morpholinopropoxy]quinazolin-4- amine, CAS 184475–35-2, MF C22H24ClFN4O3, MW 446.907 g/mol, HPLC> 98%) and all-trans retinoic acid (ATRA, [2E,4E,6E,8E]-3,7-dimethyl-9-[2,6,6-trimethylcyclohexen- 1-yl] nona-2,4,6,8-tetraenoic acid, CAS 302–79-4, MF C20H28O2, MW 300.442 g/mol, HPLC> 98%) were purchased from Sigma Chemical Co. (St. Louis, MO, USA); ALDEFLUOR™ Kit (Cat. No.01700) was purchased from STEMCELL Technologies Inc.; BD Pharmingen™ PE mouse anti-human CD44 monoclonal antibody (Clone 515 Cat No. 550988) and its isotype mouse BALB/c IgG1 were purchased from BD Biosciences (Lake Franklin, NJ, USA).
AmoyDx ARMS EGFR mutation detection kit was purchased from Amoy Diagnostics Co. LTD (Xiamen, China).
Cell viability assay
Cell viability was measured by a colorimetric assay using crystal violet. To a 96-well plate, 5 × 103 cells/well were pre-cultured for 24 h, and then exposed to varying concentrations of gefitinib and ATRA, and 0.1% DMSO was used as a vehicle in triplicate. After 72 h, the supernatant was discarded as much as possible, and 100 μL of crystal violet solution (0.5% crystal violet in 30% methanol) was added to each well for 30 min, and then rinsed with tap water and dried at 40 °C. 100 μL of 10% SDS solution was added to each well and fully dissolved for 30 min. The absorbance at 595 nm was measured spectrophotometrically using a microplate reader (Infinite M200 Pro TECAN-Reader, Switzerland).
EGFR mutation testing
Genomic DNA from A549 and H1650 cells was manually extracted using a TIANamp Genomic DNA Kit (DP304, TIANGEN, China.) according to the manufacturer’s protocol. DNA was isolated by elution with 50 μl of Tris/Acetate/EDTA (TAE). EGFR mutations were detected with the AmoyDx Human EGFR Gene 29 Mutations Detection kit with fluorescence polymerase chain reaction (PCR) (Amoy Diagnostics, Xiamen, China) and assays were performed on CFX96 Touch (Bio-Rad, USA) real-time fluorescence quantitative PCR instrument according to the manufacturer’s instructions. Positive results were defined as [Ct (sample)-Ct (control)] \ Ct (cut-off).
Gefitinib-induced enrichment of GSC and ATRA treatment
H1650 and A549 cells were passaged with 15 μmol/L of gefitinib twice weekly for three consecutive weeks, and the resultant gefitinib surviving cells (A549GSC cells and H1650GSC cells) were incubated with 5 μmol/L of ATRA for 1–5 days. These cells were respectively harvested to test the expression of ALDH1A1 and CD44 by flow cytometer (FCM). The GSCs with enhanced expression of ALDH1A1 and CD44 are defined as GSC-enriched gefitinib-resistant cells.
Flow cytometry for ALDH1A1 and CD44 expression
Expression of ALDH1A1 and CD44 by A549 and H1650 cells were determined using ALDEFLUOR™ kit (FITC) and CD44 mAb (PE), respectively according to the manufacturer’s protocols. Briefly, A549 and H1650 cells (1 × 106) were harvested and stained with ALDH (DEAB as the negative control) and PE anti-human CD44 mAb (mouse IgG1 as the isotype control) staining. The stained cells were resuspended in 1 ml of Assay Buffer and subjected respectively to flow cytometrical analysis on FACSCanto II Flow Cytometer (Becton–Dickinson).
Determination for inhibition of ATRA on ALDH1A1 activity
Active ALDH1A1 was determined using ALDEFLUOR assay according to the manufacturer’s protocol. A549 GSCs and H1650 GSCs with ALDH1A1bright (5 × 105 cells/tube) were respectively exposed to varying concentrations of ATRA and DEAB (diethylaminobenzaldehyde, an inhibitor of ALDH1A1 activity), and washed twice with 2 ml ALDEFLUOR buffer and eventually resuspended in 500 μl ALDEFLUOR buffer, and then subjected to flow cytometrical analysis to determine the FITC AUC (area under curve) on FACS Canto II Flow Cytometer (Becton–Dickinson).
Results
Growth inhibition of H1650 and A549 cells by gefitinib and ATRA
As shown in Table 1 and Fig. 1a-d, we showed that there was no significant difference between H1650 cells and A549 cells for the response to gefitinib (IC50 5.26 vs. 8.42 μmol/L), however the IC50 values of gefitinib for H1650GSC and A549GSC cells significantly increased by 5.15-fold (from 5.26 to 27.11 μmol/L) and 4.39-fold (from 8.42 to 36.97 μmol/L), respectively as compared to their untreated cells (both P < 0.01). We found that pre-incubation with ATRA significantly enhanced gefitinib-induced growth inhibition and decreased the IC50 values for gefitinib by up to 2.27-fold (from 27.11 to 11.94 μmol/L) (P < 0.01), and 2.04-fold (from 36.97 to 18.17 μmol/L) (P < 0.01) for H1650GSC and A549 GSC cells, respectively. Interestingly, we found that ATRA significantly inhibited the growth of H1650GSC and A549GSC cells but did not obviously impact the growth of H1650 and A549 cells. These results suggest that both H1650GSC and A549GSC cells have a higher resistance to gefitinib, and ATRA could improve the response of H1650GSC and A549GSC cells to gefitinib.
Table 1.
Gefitinib-induced growth inhibition in combination with ATRA in NSCLC/ADC cells
| Cell line | n | Gefitinib | Gefitinib plus ATRA treatment | ||
|---|---|---|---|---|---|
| IC50 (μmol/L) | P | IC50 (μmol/L) | P | ||
| A549 | 3 | 8.42 ± 0.312 | – | – | – |
| A549GSC | 3 | 36.97 ± 2.017 | < 0.01 | 18.17 ± 1.271 | < 0.01 |
| H1650 | 3 | 5.26 ± 0.204 | – | – | – |
| H1650GSC | 3 | 27.11 ± 1.891 | < 0.01 | 11.94 ± 1.153 | < 0.01 |
NSCLC/ADC cells were treated with vehicle or gefitinib and treated with ATRA (10 μmol/L) plus gefitinib for 72 hoursh. Relative number of viable cells was assayed using crystal violet assay. IC50 values were measured as a curvilinear regression equation for each survival curve. Data represent mean values ± S.D. from three independent experiments. *P values, compared with gefitinib alone
Fig. 1.

Growth inhibition of gefitinib and ATRA on lung cancer cells. a. Cell viability (%) of A549 and A549GSC cells treated with gefitinib alone or combined with 10 μmol/L ATRA for 72 h; B. Cell viability (%) of H1650 and H1650GSC cells treated with gefitinib alone or combined with 10 μmol/L ATRA as described in Methods. c and d. Cell viability (%) of A549 and H1650 cells treated with ATRA alone, respectively. All experiments were performed in triplicate, and data are expressed as means±s.d. (n = 3). Error bars represent s.d. of replicate data points. *P < 0.05, compared to the control
GSC-enriched cell population mediates gefitinib resistance
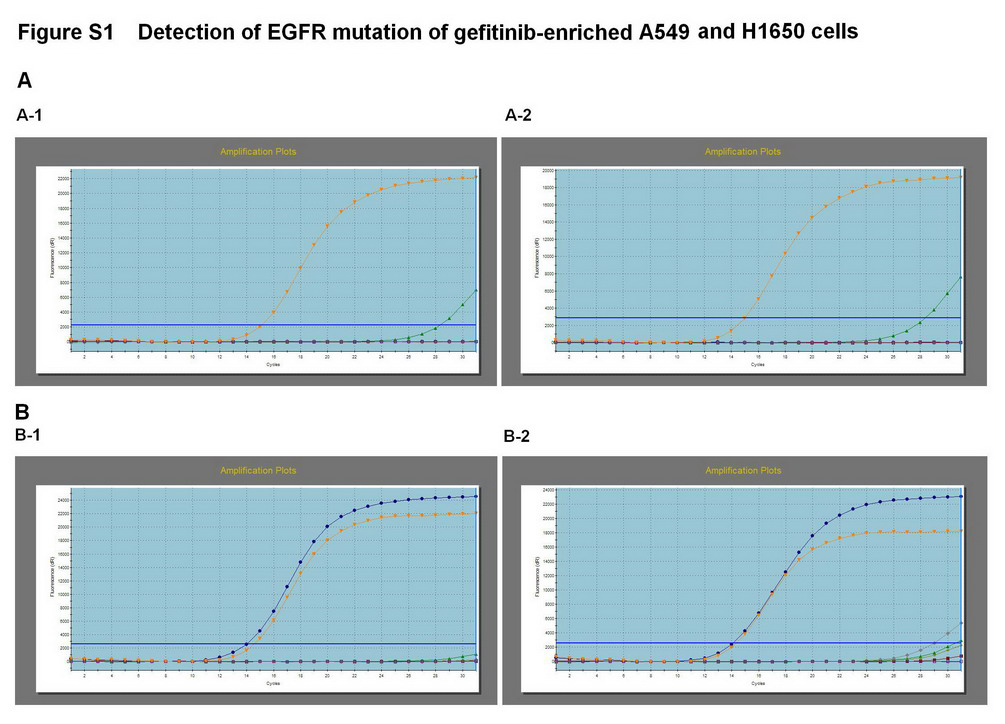
As shown in Fig. 2, H1650GSC and A549GSC cells showed increased expression of ALDH1A1 and CD44, compared to the untreated cells (P < 0.05). The expression of ALDH1A1 and CD44 in A549GSC cells increased from 2.8 to 4.8% and from 55.2 to 73.9%, respectively (Fig. 2a); the expression of ALDH1A1 and CD44 in H1650GSC cells increased from 3.1 to 11.2% and from 40.3 to 70.2%, respectively (Fig. 2b). However, no EGFR T790M mutation in A549GSC and H1650GSC cells was detected using quantitative fluorescence PCR as described in methods (Supplementary Figure 1). These results indicate that resistance of A549GSC and H1650GSC cells to gefitinib could be associated with ALDH1A1bright/CD44high but not associated with EGFR T790M mutation.
Fig. 2.

Detection of ALDH1A1 and CD44 expression of lung cancer cells treated with gefitinib by FCM as described in Methods. a. expression of ALDH1A1 (a-1 and a-2) and CD44 (a-3 and a-4) of A549 and A549GSC cells; b. expression of ALDH1A1 (b-1 and b-2) and CD44 (b-3 and b-4) of H1650 and H1650GSC cells. All experiments were performed in triplicate, and data are expressed as means±s.d. (n = 3). Error bars represent s.d. of replicate data points. *P < 0.05, compared to the control
ATRA reduces increased ALDH1A1 and CD44 expression of GSC cells
As shown in Figs. 3 and 4, following incubation with ATRA for a different time, the expression of ALDH1A1 and CD44 of A549GSC and H1650GSC cells showed a significant decrease in a time-dependent manner. Approximate 12-fold and 2.6-fold decrease in expression of ALDH1A1 and CD44 (from 4.8 to 0.4% and from 73.9 to 28.5%) (Fig. 3a and b) and 7-fold and 2.5-fold (from 11.2 to 1.6% and 70.2 to 28.4%) (Fig. 4a and b) were observed in A549GSC and H1650GSC cells, respectively, compared to the control (both P < 0.01), and as shown in Supplementary Figure 2, ATRA could directly inhibit ALDH1A1 activity as compared to DEAB, suggesting that ATRA reduces propagation of A549GSC and H1650GSC cells showing ALDH1A1bright/CD44high.
Fig. 3.

Detection of ALDH1A1 and CD44 expression of A549GSC cells incubation with ATRA for Day 1, 3, 5. a and b. Expression of ALDH1A1 (A-1 and A-2) and CD44 (B-1 and B-2) of A549GSC cells, respectively. All experiments were performed in triplicate, and data are expressed as means±s.d. (n = 3). Error bars represent s.d. of replicate data points. *P < 0.05, compared to the control
Fig. 4.

Detection of ALDH1A1 and CD44 expression of H1650GSC cells incubation with ATRA for day 1, 3, 5. a and b. Expression ALDH1A1 (A-1 and A-2) and CD44 (B-1 and B-2) of A549GSC cells, respectively. All experiments were performed in triplicate, and data are expressed as means±s.d. (n = 3). Error bars represent s.d. of replicate data points. *P < 0.05, compared to the control
Discussion
CSC cell-mediated drug resistance of cancers is a major cause leading to the failure in cancer therapies. Resistance to molecular targeted drugs of NSCLC is a common characteristic of CSC cells. Among EGFR non-T790M mutation NSCLC cases, a small sub-population of cells within the tumor that possess stem-like properties come into being a more resistant phenotype during12 months after an initial response to the EGFR-TKIs in NSCLC [12]. Since potential relapse of NSCLC may occur due to the enrichment of CSCs following TKI initial therapy, the generation of more effective therapeutic interventions based on CSC cell-mediated resistance of NSCLC to EGFR-TKIs is an urgent requirement [17].
There is increasing evidence to show that ALDH1A1bright/CD44high CSC-mediated EGFR-TKI resistance may be a major obstacle for EGFR-TKIs maintenance therapy of NSCLC. Loss of responsiveness to EGFR-TKIs in NSCLC with non-T790M EGFR mutation can be explained in terms of EGFR-TKI-resistant ALDH1A1bright/CD44high CSC that evolutionally possesses drug resistance and is often referred to as a tumor-initiating cell and associated with EGFR-TKIs non-responder [12, 17, 28]. Recent studies showed that ALDH1A1bright CSCs promote EGFR-TKI resistance in NSCLC [29, 30]. With respect to CD44, it has been reported that modulation of CD44 is detrimental to CSCs self-renewal and differentiation and NSCLC cells expressing CD44 are enriched for stem-like properties, suggesting that ALDH1A1bright/CD44high CSCs linked to tumor progression and EGFR-TKI resistance is associated with a significantly poor prognosis factor in NSCLC [14, 31].
Differentiating CSCs may provide such an approach to modulating or converting the phenotypes of CSCs for sustained treatment response of NSCLC to EGFR-TKIs, although the mechanisms underlying CSCs contribution to resistance of NSCLC to TKIs remain unclear [32].
The ALDH-retinoic acid pathway plays an important role in differentiation of CSCs. It has been shown that treatment of lung adenocarcinoma A549 cell with ATRA led to the downregulation of ALDH1A1 [33].
Retinoic acid (RA) can reduce the ALDH activity and CD44 expression, thus affecting cell proliferation, cancer invasiveness and sensitivity to various chemotherapy drugs [24, 34, 35]. ALDH1A1 has been shown to convert/oxidize retinaldehyde into RA in several tissues, and to be one of the target proteins of ATRA [24]. Treatment with ATRA increased the C/EBP homologous protein (GADD153) and GADD153-CCAAT-enhancing binding protein-β (C/EBP-β) interaction resulting in a decreased cellular availability of C/EBP-β for binding to the Raldh1 CCAAT box and high ATRA levels inhibit Raldh1 gene expression by sequestering C/EBP-β through its interaction to GADD153 [36, 37]. CD44 expression was highly responsive to ATRA as it was down regulated following treatment, and ATRA treatment also resulted in decreased migration and invasion of cancer cells and promoted tumor regression by inducing differentiation [34].
Retinoids prevent the development of several tumors and enhance the efficacy of cytotoxic drugs such as cisplatin and docetaxel [38, 39]. Retinoids bind to specific nuclear receptors, which function as transcriptional regulators controlling the expression of numerous genes. The retinoid X receptors (RXRs) and retinoic acid receptors (RARs) are selectively expressed in ALDHbright CSCs, indicating RA signaling mainly occurs via ALDHbright CSCs of lung cancer, which provides a potential mechanism to selectively target CSCs [26, 40]. As shown in Figure S3, RA signaling is modulated by two classes of nuclear retinoid receptors, RARs and RXRs. Both RXRs and RARs interact with multiple co-activator and co-repressor proteins to promote increased cell stemness or cell differentiation. Retinoic acid showed feedback inhibition of the ALDH1 gene through RARα and C/EBP-β [41]. Specifically, RA signaling regulates ALDH via the binding of ATRA to RXR and RAR that transcriptionally control ALDH gene expression [38].
Loss of retinoid receptors expression happens frequently in the development of carcino- genesis and induction of resistance to apoptosis. The known effect of ATRA on differentiation of cells is mediated through RARβ. RARβ belongs to the nuclear receptor (NR) superfamily of transcription factors. Upregulation of RARβ within the drug-resistant cancer cells, which exhibits loss of RARβ expression, has been shown to increase the susceptibility of cells to apoptosis induced by chemotherapeutic agents. Activation of RARs or RXRs contributes to induction of RARβ, growth inhibition and apoptosis by retinoids. It evidenced that the therapeutic anti-CSC and proapoptotic effects of ATRA are dependent on receptor class-selective retinoids and the expression of RARβ plays a role in mediating retinoid response in NSCLC cells [38, 41]. The loss of RARβ might contribute to enhanced cancer stemness and the apoptosis resistance of CSCs to gefitinib in NSCLC cells. ATRA can induce the apoptosis of NSCLC CSCs through activation of RARβ and its ability to down-regulate the CSCs markers in lung cancer cells [42, 43]. The expression of RARβ as well as RXRβ was reported to be downregulated in NSCLC, which enabled the cancer cell to evade apoptosis [44]. The RARβ is also known as tumor suppressor and the major target gene of retinoid action, and an enhanced level of RARβ protein exhibited its growth inhibitory action of lung cancer cells [44, 45]. RARβ can mediate retinoid action in lung cancer cells by promoting apoptosis. However, a fundamental question that remains unanswered is how ATRA and RARβ trigger apoptosis in lung cancer cells. Studies showed the overexpression of RARβ was accompanied by an increase in c-Myc and Bax but not TP53 protein expression and associated with an increase in the Bax/Bcl-2 ratio, and that ATRA enhanced G1 growth arrest, up-regulated p21and p27 and downregulated cyclin D1. These data suggest that the expression of RARβ is positively associated with ATRA-induced apoptosis and growth inhibition in lung cancer cells. It has been shown that RA inhibits EGFR expression at the transcriptional level by targeting the EGFR promoter leading to inhibition of lung cancer cell growth and arrests EGFR-TKI resistant NSCLC cells in the G0/G1 phase of the cell cycle by altering the expression of GATA-binding factor 6 (GATA6) and inhibits the activation of two important pathways involved in lung cancer progression namely EGFR and Wnt signaling to overcome TKI resistance [46]. Combinatorial treatment of retinoids with EGFR-TKIs drugs in EGFR-TKIs resistance lung cancer cells promotes the activation of GATA6 and then inhibits the activation of EGFR/Wnt signaling pathways and favors the association of RXR, RARβ, and cellular retinoic acid binding protein-2 (CRABP2). This complex inhibits the proliferation and promotes the differentiation of lung tumor cells via inhibiting activating protein-2 (AP-2), which result in re-sensitization of EGFR-TKIs resistant lung cancer cells [47].
In this study, a short-term gefitinib treatment was used to enrich A549GSCs and H1650GSCs. FCM assay showed that A549GSCs and H1650GSCs have a significant increase in proportions of ALDH1A1bright/CD44high cells (Fig. 2), and we further confirmed that these ALDH1A1bright/CD44high GSCs exhibit increased IC50 values for gefitinib compared to that of their respective parental cells (Table 1 and Fig. 1a and b), and are involved in CSCs but not in EGFR T790M-mediated gefitinib resistance (Supplementary Figure 1), suggesting that ALDH1A1bright/CD44high CSCs in NSCLC/ADC contribute to resistance to gefitinib. Interestingly, treatment with ATRA significantly reduced ALDH1A1 and CD44 expression of A549GSCs and H1650GSCs, and their IC50 values for gefitinib, thus returning to sensitization to gefitinib (Table 1 and Fig. 1a and b). ALDEFLUOR assay also showed that ATRA can directly inhibit ALDH1A1 activity in a concentration dependent manner (Supplementary Figure 2). These results showed that in contrast to the known tendency of EGFR-TKIs, such as gefitinib, to target the non-stem-like ALDH1A1-negative cell population, ATRA can modulate the ALDH1A1bright/CD44high cell population in NSCLC/ADC. Therefore, the synergistic antitumor effect of ATRA in combination with gefitinib might be a promising therapeutic strategy to circumvent CSC-mediated resistant NSCLC/ADC.
Conclusions
Our findings indicate that combinatorial treatment of ATRA with gefitinib could reduce CSC-mediated resistance by down-regulating expression of ALDH1A1 and CD44 and potentiate the anti-tumor effect of gefitinib in NSCLC/ADC.
Supplementary information
{kind=link}
Additional file 1 : Figure S1. Detection of EGFR mutation of A549GSC and H1650GSC cells by ARMS-qPCR as described in Methods.A. EGFR mutation of A549 cells before (A-1) and after (A-2) treatment with gefitinib; B. EGFR mutation of H1650 cells before(B-1) and after(B-2) treatment with gefitinib.
{kind=link}
Additional file 2 : Figure S2. Direct inhibitory effect of ATRA on ALDH1A1 activity in GSC cells (FITC AUC) by ALDEFLUOR assay as described in Methods.A. ALDH1A1 Activity of A549GSC (A-1, A-2 and A3); B. ALDH1A1 Activity of H1650GSC (BA-1, B-2 and B3).
{kind=link}
Additional file 3 : Figure S3. A potential mechanism by which ATRA regulates the response of lung cancer stem cells to gefitinib. ATRA binds and activates RARβ complex and related signaling molecules. The interaction of C/EBP homologous protein (GADD153) with GADD153-CCAAT-enhancing binding protein-β (C/EBP-β) results in a decreased cellular availability of C/EBP-β for binding to the Raldh1 CCAAT box, and high ATRA levels can sequester interaction of C/EBP-β with GADD153 to suppress expression of Raldh1 gene.
Acknowledgements
Not applicable.
Abbreviations
- EGFR-TKD
Epidermal growth factor receptor- tyrosine kinase domain
- TKIs
Tyrosine kinase inhibitors
- ATRA
All-trans retinoic acid
- NSCLC/ADC
Non-small cell lung adenocarcinoma
- ALDH1A1
Aldehyde dehydrogenase 1 family member A1
- CD44
Cluster of differentiation 44
- FCM
Flow cytometry
- IC50
Half maximal inhibitory concentration
- NSCLC
Non-small cell lung cancer
- EGFRm
Mutant epidermal growth factor receptor
- ATP
Adenosine triphosphate
- EGFR-TKIs
EGFR tyrosine kinase inhibitors
- ADC
Adenocarcinoma
- EGFRWT
Wild-type EGFR
- SCLC
Small cell lung cancer
- MET
Cellular-mesenchymal to epithelial transition factor
- ERBB2
Erythroblastic leukemia viral oncogene homolog 2
- PIK3CA
Phosphatidylino-sitol 3-kinases, catalytic, alpha polypeptide
- EMT
Epithelial to mesenchymal transition
- CSC
Cancer stem cell-like cell
- RAs
Retinoic acids
- RR
Response rate
- PFS
Progression-free survival
- APL
Acute promyelocytic leukemia
- CTCCCAS
The committee on type culture collection of Chinese academy of sciences
- FBS
Fetal bovine serum
- EDTA
Ethylenediaminetetraacetic acid
- TAE
Tris/Acetate/EDTA
- PCR
Polymerase chain reaction
- GSC
Gefitinib surviving cell
- TME
Tumor microenvironment
- GADD153
C/EBP homologous protein, CHOP
- EBPb
CCAAT-enhancing binding protein-B
- RARs
Retinoic acid receptors
- GATA6
GATA-binding factor 6
- CRABP2
Cellular retinoic acidbinding protein-2
- DEAB
Diethylaminobenzaldehyde
- AP-2
Activating protein-2
Authors’ contributions
WY, JH and LW conceived and designed this study; HH, HL and CG performed the experiments; KM and JC contributed to the quality control of data and algorithms; LH, PW and XL analyze the data/results; HH and XL wrote the manuscript; JH improved and revised the manuscript. All authors read and approved the final manuscript.
Funding
This study was co-financed by Chengdu Science and Technology Bureau and Sichuan Provincial Science and Technology Department. W.Y. and L.W. was supported by a grant from Chengdu Science and Technology Bureau (2014-HM01–00134-SF). L.W. was supported by a grant from Sichuan Provincial Science and Technology Department (2018JY0300). The funding bodies had no role in study design, data collection and analysis, interpretation of data or in writing the manuscript.
Availability of data and materials
Please contact author for data requests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Wenxiu Yao and Liyang Wang contributed equally to this work.
Contributor Information
Wenxiu Yao, Email: ywxhlx@sina.com.
Liyang Wang, Email: kuaiwly@163.com.
Huan Huang, Email: 515597510@qq.com.
Xin Li, Email: 673362285@qq.com.
Pinjia Wang, Email: 396375446@qq.com.
Kun Mi, Email: mikun@126.com.
Jia Cheng, Email: 13547913187@163.com.
Huifen Liu, Email: 247212390@qq.com.
Cuirong Gu, Email: 1357180462@qq.com.
Lingxiao Huang, Email: hlx2015@yahoo.com.
Jianming Huang, Email: hjianming@yahoo.com.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12885-020-06818-0.
References
- 1.Lung Cancer-Non-Small Cell . Statistics Approved by the Cancer. Net Editorial Board. 2019. [Google Scholar]
- 2.Chan SK, Gullick WJ, Hill ME. Mutations of the epidermal growth factor receptor in non-small cell lung cancer-search and destroy. Eur J Cancer. 2006;42:17–23. doi: 10.1016/j.ejca.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 3.Mok TS, Wu YL, Yu CJ, Zhou C, Chen YM, Zhang L, et al. Randomized, placebo- controlled, phase II study of sequential erlotinib and chemotherapy as first-line treatment for advanced non-small-cell lung cancer. J Clin Oncol. 2009;27:5080–5087. doi: 10.1200/JCO.2008.21.5541. [DOI] [PubMed] [Google Scholar]
- 4.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 5.Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 6.Kosaka T, Yatabe Y, Endoh H, Yoshida K, Hida T, Tsuboi M, et al. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res. 2006;12:5764–5769. doi: 10.1158/1078-0432.CCR-06-0714. [DOI] [PubMed] [Google Scholar]
- 7.Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–128. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 8.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 9.Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 10.Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19(8):2240–2247. doi: 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao J, Li HR, Jin C, Jiang JH, Ding JY. Strategies to overcome acquired resistance to EGFR TKI in the treatment of non-small cell lung cancer. Clin Transl Oncol. 2019;21(10):1287–1301. doi: 10.1007/s12094-019-02075-1. [DOI] [PubMed] [Google Scholar]
- 12.Freitas DP, Teixeira CA, Santos-Silva F, Vasconcelos MH, Almeida GM. Therapy- induced enrichment of putative lung cancer stem-like cells. Int J Cancer. 2014;134(6):1270–1278. doi: 10.1002/ijc.28478. [DOI] [PubMed] [Google Scholar]
- 13.Akunuru S, James Zhai Q, Zheng Y. Non-small cell lung cancer stem/ progenitor cells are enriched in multiple distinct phenotypic subpopulations and exhibit plasticity. Cell Death Dis. 2012;3:e352. doi: 10.1038/cddis.2012.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leung EL, Fiscus RR, Tung JW, Tin VP, Cheng LC, Sihoe AD, et al. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS One. 2010;5(11):e14062. doi: 10.1371/journal.pone.0014062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shien K, Toyooka S, Yamamoto H, et al. Acquired resistance to EGFR inhibitors is associated with a manifestation of stem cell-like properties in cancer cells. Cancer Res. 2013;73(10):3051–3061. doi: 10.1158/0008-5472.CAN-12-4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Westover D, Zugazagoitia J, Cho BC, Lovly CM, Paz-Ares L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann Oncol. 2018;29:i10–i19. doi: 10.1093/annonc/mdx703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suda K, Rivard CJ, Mitsudomi T, Hirsch FR. Overcoming resistance to EGFR tyrosine kinase inhibitors in lung cancer, focusing on non-T790M mechanisms. Expert Rev Anticancer Ther. 2017;17(9):779–786. doi: 10.1080/14737140.2017.1355243. [DOI] [PubMed] [Google Scholar]
- 18.Arasada RR, Amann JM, Rahman MA, Huppert SS, Carbone DP. EGFR blockade enriches for lung cancer stem-like cells through Notch3-dependent signaling. Cancer Res. 2014;74:5572–5584. doi: 10.1158/0008-5472.CAN-13-3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meng E, Mitra A, Tripathi K, et al. ALDH1A1 maintains ovarian cancer stem cell-like properties by altered regulation of cell cycle checkpoint and DNA repair network signaling. PLoS One. 2014;9(9):e107142. doi: 10.1371/journal.pone.0107142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Januchowski R, Wojtowicz K, Sterzyſska K, Sosiſska P, Andrzejewska M, Zawierucha P, Nowicki M, Zabel M. Inhibition of ALDH1A1 activity decreases expression of drug transporters and reduces chemotherapy resistance in ovarian cancer cell lines. Int J Biochem Cell Biol. 2016;78:248–259. doi: 10.1016/j.biocel.2016.07.017. [DOI] [PubMed] [Google Scholar]
- 21.Croker AK, Allan AL. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiationresistance of stem-like ALDHhiCD44+ human breast cancer cells. Breast Cancer Res Treat. 2012;133(1):75–87. doi: 10.1007/s10549-011-1692-y. [DOI] [PubMed] [Google Scholar]
- 22.Dinavahi SS, Bazewicz CG, Gowda R, Robertson GP. Aldehyde dehydrogenase inhibitors for Cancer therapeutics. Trends Pharmacol Sci. 2019;40(10):774–789. doi: 10.1016/j.tips.2019.08.002. [DOI] [PubMed] [Google Scholar]
- 23.Kim D, Choi BH, Ryoo IG, Kwak MK. High NRF2 level mediates cancer stem cell-like properties of aldehyde dehydrogenase (ALDH)-high ovarian cancer cells: inhibitory role of all-trans retinoic acid in ALDH/NRF2 signaling. Cell Death Dis. 2018;9(9):896. doi: 10.1038/s41419-018-0903-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moreb JS, Ucar-Bilyeu DA, Khan A. Use of retinoic acid/aldehyde dehydrogenase pathway as potential targeted therapy against cancer stem cells. Cancer Chemother Pharmacol. 2017;79(2):295–301. doi: 10.1007/s00280-016-3213-5. [DOI] [PubMed] [Google Scholar]
- 25.Picard E, Seguin C, Monhoven N, Rochette-Egly C, Siat J, Borrelly J, Martinet Y, Martinet N, Vignaud JM. Expression of retinoid receptor genes and proteins in non-small-cell lung cancer. J Natl Cancer Inst. 1999;91(12):1059–1066. doi: 10.1093/jnci/91.12.1059. [DOI] [PubMed] [Google Scholar]
- 26.Li Y, Dawson MI, Agadir A, Lee MO, Jong L, Hobbs PD, Zhang XK. Regulation of RAR beta expression by RAR-and RXR-selective retinoids in human lung cancer cell lines: effect on growth inhibition and apoptosis induction. Int J Cancer. 1998;75(1):88–95. doi: 10.1002/(SICI)1097-0215(19980105)75:1<88::AID-IJC14>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 27.Muñiz-Hernández S, Huerta-Yepez S, Hernández-Pedro N, Ramírez-Tirado LA, Aviles-Salas A, Maldonado A, et al. Association between nuclear expression of retinoic acid receptor alpha and beta and clinicopathological features and prognosis of advanced non-small cell lung cancer. Int J Clin Oncol. 2016;21(6):1051–1061. doi: 10.1007/s10147-016-1002-0. [DOI] [PubMed] [Google Scholar]
- 28.Hashida S, Yamamoto H, Shien K, et al. Acquisition of cancer stem cell-like properties in non-small cell lung cancer with acquired resistance to afatinib. Cancer Sci. 2015;106:1377–1384. doi: 10.1111/cas.12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang F, Qiu Q, Khanna A, Todd NW, Deepak J, Xing L, et al. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol Cancer Res. 2009;7:330–338. doi: 10.1158/1541-7786.MCR-08-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang CP, Tsai MF, Chang TH, Tang WC, Chen SY, Lai HH, et al. ALDH-positive lung cancer stem cells confer resistance to epidermal growth factor receptor tyrosine kinase inhibitors. Cancer Lett. 2013;328:144–151. doi: 10.1016/j.canlet.2012.08.021. [DOI] [PubMed] [Google Scholar]
- 31.Raha D, Wilson TR, Peng J, Peterson D, Yue P, Evangelista M, Wilson C, Merchant M, Settleman J. The cancer cell marker aldehyde dehydrogenase is required to maintain a drug-tolerant tumor cell subpopulation. Cancer Res. 2014;74:3579–3590. doi: 10.1158/0008-5472.CAN-13-3456. [DOI] [PubMed] [Google Scholar]
- 32.Gudas LJ, Wagner JA. Retinoids regulate stem cell differentiation. J Cell Physiol. 2011;226:322–330. doi: 10.1002/jcp.22417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moreb JS, Gabr A, Vartikar GR, Gowda S, Zucali JR, Mohuczy D. Retinoic acid downregulates aldehyde dehydrogenase and increases cytotoxicity of 4-hydroperoxycyclophosphamide and acetaldehyde. J Pharmacol Exp Ther. 2005;312:339–345. doi: 10.1124/jpet.104.072496. [DOI] [PubMed] [Google Scholar]
- 34.Chen J, Cao X, An Q, Zhang Y, Li K, Yao W. Inhibition of cancer stem cell like cells by a synthetic retinoid. Nat Commun. 2018;9:1406. doi: 10.1038/s41467-018-03877-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kitadai R, Bednarczyk R, Tuli N, Tiwari RK. Modulation of breast cancer stem cell marker CD44 by all-trans-retinoic acid (ATRA) Cancer Res. 2014;74:Abstract nr 3889. [Google Scholar]
- 36.Elizondo G, Medina-Díaz IM, Cruz R, Gonzalez FJ, Vega L. Retinoic acid modulates retinaldehyde dehydrogenase 1 gene expression through the induction of GADD153-C /EBPbeta interaction. Biochem Pharmacol. 2009;77:248–257. doi: 10.1016/j.bcp.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elizondo G, Corchero J, Sterneck E, Gonzalez FJ. Feedback inhibition of the retinaldehyde dehydrogenase gene ALDH1 by retinoic acid through retinoic acid receptor alpha and CCAAT/enhancer-binding protein beta. J Biol Chem. 2000;275:39747–39753. doi: 10.1074/jbc.M004987200. [DOI] [PubMed] [Google Scholar]
- 38.Jozan S, Paute S, Courtade-Saïdi M, Julié S, Vidal S, Bugat R, Valette A. All trans retinoic acid enhances CDDP-induced apoptosis: modulation of the CDDP effect on cell cycle progression. Int J Oncol. 2002;20:1289–1295. [PubMed] [Google Scholar]
- 39.Wang Q, Wieder R. All-trans retinoic acid potentiates Taxotere induced cell death mediated by Jun N-terminal kinase in breast cancer cells. Oncogene. 2004;23:426–433. doi: 10.1038/sj.onc.1207040. [DOI] [PubMed] [Google Scholar]
- 40.di Masi A, Leboffe L, De Marinis E, Pagano F, Cicconi L, Rochette-Egly C, Lo-Coco F, Ascenzi P, Nervi C. Retinoic acid receptors: from molecular mechanisms to cancer therapy. Mol Asp Med. 2015;41:1–115. doi: 10.1016/j.mam.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 41.Singh AT, Evens AM, Anderson RJ, Beckstead JA, Sankar N, Sassano A, Bhalla S, Yang S, Platanias LC, Forte TM, Ryan RO, Gordon LI. All trans retinoic acid nanodisks enhance retinoic acid receptor mediated apoptosis and cell cycle arrest in mantle cell lymphoma. Br J Haematol. 2010;150:158–169. doi: 10.1111/j.1365-2141.2010.08209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee MO, Han SY, Jiang S, Park JH, Kim SJ. Differential effects of retinoic acid on growth and apoptosis in human colon cancer cell lines associated with the induction of retinoic acid receptor beta. Biochem Pharmacol. 2000;59:485–496. doi: 10.1016/S0006-2952(99)00355-X. [DOI] [PubMed] [Google Scholar]
- 43.Tripathi SK, Pandey K, Panda M, Spinella MJ, Rengasamy KR, Biswal BK. The potential of retinoids for combination therapy of lung cancer: updates and future directions. Pharmacol Res. 2019;147:104331. doi: 10.1016/j.phrs.2019.104331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brabender J, Metzger R, Salonga D, Danenberg KD, Danenberg PV, Hölscher AH, Schneider PM. Comprehensive expression analysis of retinoic acid receptors and retinoid X receptors in non-small cell lung cancer: implications for tumor development and prognosis. Carcinogenesis. 2005;26:525–530. doi: 10.1093/carcin/bgi006. [DOI] [PubMed] [Google Scholar]
- 45.Lei M, de Thé H. Retinoids and retinoic acid receptor in cancer. Eur J Cancer Suppl. 2003;1:13–18. doi: 10.1016/S1359-6349(03)00010-7. [DOI] [Google Scholar]
- 46.Xu XC, Sozzi G, Lee JS, Lee JJ, Pastorino U, Pilotti S, Kurie JM, Hong WK, Lotan R. Suppression of nuclear retinoic acid receptor β in non-small cell lung cancer in vivo: implications in lung cancer development. J Natl Cancer Inst. 1997;89:624–629. doi: 10.1093/jnci/89.9.624. [DOI] [PubMed] [Google Scholar]
- 47.Zito G, Naselli F, Saieva L, Raimondo S, Calabrese G, Guzzardo C, Forte S, Rolfo C, Parenti R, Alessandro R. Retinoic Acid affects Lung Adenocarcinoma growth by inducing differentiation via GATA6 activation and EGFR and Wnt inhibition. Sci Rep. 2017;7:4770. doi: 10.1038/s41598-017-05047-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1 : Figure S1. Detection of EGFR mutation of A549GSC and H1650GSC cells by ARMS-qPCR as described in Methods.A. EGFR mutation of A549 cells before (A-1) and after (A-2) treatment with gefitinib; B. EGFR mutation of H1650 cells before(B-1) and after(B-2) treatment with gefitinib.
Additional file 2 : Figure S2. Direct inhibitory effect of ATRA on ALDH1A1 activity in GSC cells (FITC AUC) by ALDEFLUOR assay as described in Methods.A. ALDH1A1 Activity of A549GSC (A-1, A-2 and A3); B. ALDH1A1 Activity of H1650GSC (BA-1, B-2 and B3).
Additional file 3 : Figure S3. A potential mechanism by which ATRA regulates the response of lung cancer stem cells to gefitinib. ATRA binds and activates RARβ complex and related signaling molecules. The interaction of C/EBP homologous protein (GADD153) with GADD153-CCAAT-enhancing binding protein-β (C/EBP-β) results in a decreased cellular availability of C/EBP-β for binding to the Raldh1 CCAAT box, and high ATRA levels can sequester interaction of C/EBP-β with GADD153 to suppress expression of Raldh1 gene.
Data Availability Statement
Please contact author for data requests.