

Abstract
Background
Anti-TNF treatment may be useful for the treatment of patients with refractory juvenile dermatomyositis (JDM). The aim of this study was to describe the use of infliximab and adalimumab therapy in juvenile dermatomyositis as an adjunctive treatment.
Methods
Sixty children recruited to the UK JDM Cohort and Biomarker Study that had received at least 3 months of anti-TNF treatment (infliximab or adalimumab) were studied. Childhood Myositis Assessment Scale (CMAS), Manual Muscle Testing (MMT8) and physician’s global assessment (PGA) were recorded. Skin disease was assessed using the modified skin disease activity score (DAS). Data were analysed using Friedman’s test for repeated measures analysis of variance.
Results
Compared to baseline, there were improvements at 6 and 12 months in skin disease (χ2(2) = 15.52, p = 0.00043), global disease (χ2(2) = 8.14, p = 0.017) and muscle disease (CMAS χ2(2) = 17.02, p = 0.0002 and MMT χ2(2) = 10.56, p = 0.005) in infliximab patients. For patients who switched from infliximab to adalimumab, there was improvement in global disease activity (χ2(2) = 6.73, p = 0.03), and trends towards improvement in CMAS, MMT8 and modified DAS. The median initial prednisolone dose was 6 [0–10] mg, and final was 2.5 [0–7.5] mg (p < 0.0001). Fifty-four per cent of patients had a reduction in the number and/or size of calcinosis lesions. Twenty-five per cent switched their anti-TNF treatment from infliximab to adalimumab. 66.7%of the switches were to improve disease control, 26.7% due to adverse events and 6.6% due to patient preference. A total of 13.9 adverse reactions occurred in 100 patient-years, of which 5.7 were considered serious.
Conclusion
Reductions in muscle and skin disease, including calcinosis, were seen following treatment with infliximab and adalimumab.
Keywords: Infliximab, Adalimumab, Juvenile dermatomyositis, Biologic therapy, P rheumatology
Background
Juvenile dermatomyositis (JDM) is a rare inflammatory disease of childhood that predominantly affects muscles and skin but is also a systemic multi-organ disease [1]. It is the most common idiopathic inflammatory myopathy (IIM) of childhood: incidence of 2–3 new cases per million/children/year [2]. The treatment of JDM has been challenging. The reasons include the rarity of the disease, its heterogeneous clinical phenotype and the small number of randomized, double-blind controlled clinical trials. Traditional treatment includes glucocorticoids and conventional immunosuppressive or immunomodulatory agents [3]. As treatment of refractory disease has been difficult, there is growing interest in evaluating novel therapies including newer biologics that target various pathways implicated in the pathogenesis of myositis [4]..
Anti-TNF biologics have been successful in treating various chronic inflammatory disorders [5, 6]. TNF has been identified in high levels in JDM patients who have a long disease course and calcinosis, which can be a debilitating complication [7–9]. There is some evidence that prolonged active disease is related to this complication and that its incidence can be reduced by earlier disease control [10]. A previous case series of 5 patients with refractory JDM treated with infliximab showed improvement in all 5 cases in core set measures [11]. Therefore, TNF may be a good potential therapeutic target for the treatment of JDM: however, evidence for efficacy of TNF blockade is limited. The goal of this study was to describe the efficacy and safety of anti-TNF treatment in patients recruited to the UK JDM Cohort and Biomarker Study (JDCBS).
Methods
Patients
Data from JDM patients treated with anti-TNF agents were analysed from the JDCBS [2]. Informed written parental consent and age-appropriate assent were obtained. This research was approved by the UK Northern & Yorkshire Medical Research and Ethics Committee. All patients had a diagnosis of definite or probable JDM according to the Bohan and Peter criteria [12, 13]. The new classification criteria were not employed as they are not yet used in routine clinical practice. Patients were included in the analysis if they had ever received anti-TNF therapy lasting for at least 3 months. Patients were excluded from the analysis if they were treated with etanercept or had no recorded outcome variables or dates of starting or ending anti-TNF. Indications for starting anti-TNF were active skin disease, calcinosis, muscle disease or general disease activity that the clinician considered to be refractory to conventional treatment.
Sixty patients treated with anti-TNF were identified, and data on the demographics, concomitant disease-modifying anti-rheumatic drugs (DMARDs) and adverse events are presented for this combined cohort in order to analyse treatments targeting this mechanism as a whole. Patients were excluded from subsequent efficacy analyses if they had allergic reactions to the first infusion and did not receive another anti-TNF (n = 2). For efficacy analysis, patients treated with infliximab alone (6 mg/kg every 4 weeks; n = 39) and patients treated with infliximab (6 mg/kg every 4 weeks) then adalimumab (24 mg/m2 every other week; n = 15) were analysed as two separate groups. Changes in levels of disease activity at 6 and 12 months after infliximab and adalimumab start were analysed for the respective groups. Finally, patients treated with adalimumab alone (24 mg/m2 every other week; n = 4) were not analysed statistically due to low numbers, but their clinical scores are described. A flow diagram outlining which patients were included in each analysis is presented Figure S1 in Supplementary Methods. Although patients who received etanercept were excluded from analysis, a description of their clinical scores is presented in Supplementary Results.
Data collection
Patient clinical data at 0, 6 and 12 months after infliximab/adalimumab start are described. For the safety analysis, patient clinical data were collected until the present date. Core outcome variables for JDM were collected including the Childhood Myositis Assessment Scale (CMAS, score range 0–53, with high scores indicating minimal disease) [14] and Manual Muscle Testing of 8 groups (MMT8, score range 0–80, with high scores indicating minimal disease) [15] that exist to standardize the muscle assessment. Both are part of the JDM Paediatric Rheumatology International Trials Organisation (PRINTO) [16] and International Myositis Assessment and Clinical Studies Group (IMACS) [17] core disease activity measures. The physician’s global assessment of disease activity (PGA, score range 0–10, with low scores indicating minimal disease) and the modified Disease Activity Score as a measure of skin disease activity (DAS, score range 0–5, with low scores indicating minimal disease) [18] were collected. Modified DAS includes the 4 skin components of a simplified version of the original DAS tool, which are Gottron’s papules (1 point), heliotrope rash (1 point), vasculitis (1 point) and erythema (2 points). The presence of calcinosis was also recorded based on a combination of physician’s assessment and imaging such as X-rays. As objective measures of calcinosis were not collected for this cohort, analyses of whether calcinosis improved were based on the physician’s assessment at the date of visit, which may have incorporated X-rays. Inter-observer variation was not recorded. Data on other medications received prior to the start, at start and at 12 months of anti-TNF therapy were recorded, including the dose of steroid treatment. Adverse events that occurred from the start of anti-TNF treatment until the end of follow-up were recorded. Severe adverse reactions were defined as the occurrence of death, hospitalization or any event that caused permanent damage. For patients who switched between anti-TNF therapies (infliximab to adalimumab), the reason for the switch and the duration of prior infliximab treatment at the time of the switch were noted.
Statistical analysis
The non-parametric Friedman’s test for repeated measures analysis of variance (ANOVA) was performed to identify significant main effects of time on clinical measures of disease activity. Separate analyses were performed for patients who received infliximab alone and for patients who received infliximab and then adalimumab. In the latter analysis, the date of switch from infliximab to adalimumab was taken as a new treatment start time. Post hoc tests to identify time-points at which clinical scores differed significantly from each other were performed using Wilcoxon signed-rank tests. The Bonferroni correction was used to adjust for multiple hypothesis testing, such that p values below 0.017 were considered statistically significant in the post hoc tests. Steroid doses at the start and 12 months after the start of anti-TNF therapy were compared using the Wilcoxon signed-rank test. Statistical analysis was performed using R version 3.5.1 and plots were generated using Graphpad Prism 5.
Results
Patient demographics, clinical features and medication
Of 60 patients assessed, 72% were female and 77% were Caucasian (Table 1). Most had been diagnosed with definite JDM (87%), with a minority diagnosed with probable JDM (3%), JDM overlap with scleroderma (7%) or JDM overlap with chronic arthritis (2%) The median age at disease onset was 5.2 [3.3–9.7] years. Median disease duration at the beginning of anti-TNF treatment was 3.1 [1.7–4.9] years, and median duration on anti-TNF therapy was of 2.5 [1.5–4] years. Of these patients, 59 had an autoantibody result: 19 (32%) had anti-TIF1γ, 7 (12%) had anti-NXP2, 1 (2%) had anti-MDA5, 1 (2%) had anti-Mi2, 1 (2%) had anti-SRP, 1 (2%) had anti-PL-7 and 1 (2%) had anti-HMGCR myositis-specific autoantibodies. A further 2 (3%) had anti-PMScl, and 1 (2%) had anti-Topo myositis-associated autoantibodies. One patient (2%) had both anti-U1RNP and anti-TIF1γ autoantibodies, 13 (22%) had unidentified autoantibodies and 11 (19%) had no-detectable autoantibodies.
Table 1.
Demographic and serological features of patients who received anti-TNF therapy (n = 60)
| Feature | Number (%) or median [IQR] |
|---|---|
| Sex | |
| Male | 17 (28%) |
| Female | 43 (72%) |
| Ethnicity | |
| Caucasian | 46 (77%) |
| Non-Caucasian | 14 (23%) |
| Diagnosis | |
| Definite JDM | 52 (87%) |
| Probable JDM | 2 (3%) |
| JDM overlap with scleroderma | 4 (7%) |
| JDM overlap with chronic arthritis | 1 (2%) |
| Age at disease onset | 5.2 [3.3–9.7] |
| Disease duration at anti-TNF start | 3.1 [1.7–4.9] |
| Duration on anti-TNF therapy | 2.5 [1.5–4] |
| Autoantibody | |
| Anti-TIF1γ | 19 (32%) |
| Anti-NXP2 | 7 (12%) |
| Anti-MDA5 | 1 (2%) |
| Anti-Mi2 | 1 (2%) |
| Anti-SRP | 1 (2%) |
| Anti-PL-7 | 1 (2%) |
| Anti-HMGCR | 1 (2%) |
| Anti-PMScl | 2 (3%) |
| Anti-Topo | 1 (2%) |
| Anti-U1RNP and anti-TIF1γ | 1 (2%) |
| Unidentified autoantibodies | 13 (22%) |
| No-detectable autoantibodies | 11 (19%) |
Regarding prior treatment, 98% of the patients were on methotrexate, azathioprine or hydroxychloroquine in monotherapy or in combination before starting anti-TNF (Table 2). Steroids were given prior to anti-TNF treatment in 92% of patients; of those with available data, 68% were still on steroids at anti-TNF start consistent with recalcitrant disease. Intravenous immunoglobulin (IVIg) treatment had been received by 11% of the patients prior to anti-TNF treatment, whilst 13% were still on IVIg at the start of the anti-TNF treatment.
Table 2.
Treatment at time of the first assessment of the 60 patients identified
| Treatment | Previously | At start of anti-TNF | After 12 months on anti-TNF |
|---|---|---|---|
| MTX/AZA/HQL1 | 48 (98%), n = 49 | 43 (80%), n = 56 | 41 (89%), n = 46 |
| Cyclophosphamide | 26 (43%), n = 60 | 3 (5%), n = 60 | 0, n = 43 |
| Immunoglobulin | 5 (11%), n = 47 | 6 (13%), n = 46 | 1 (2.5%), n = 40 |
| Oral steroids | 47 (92%), n = 51 | 26 (68%), n = 38 | 26 (58%), n = 45 |
| 2 or more DMARDs | 48 (98%), n = 49 | 30 (64%), n = 47 |
MTX methotrexate, AZA azathioprine, HQL hydroxychloroquine, n (%) absolute numbers (percentages) from the number of patients with available data
1Mycophenylate mofetil (MMF) was not used in the patients in this study
43% of the patients had finished treatment with cyclophosphamide (typically 6–7 doses, administered intravenously) before receiving anti-TNF. Five per cent (n = 3) of the patients started anti-TNF whilst they were still on cyclophosphamide (2 patients started anti-TNF 2 to 3 weeks before finishing the last cyclophosphamide dose of the course, and only 1 patient started anti-TNF before cyclophosphamide). After 12 months of anti-TNF therapy, none were still on cyclophosphamide. The median prednisolone daily dose at anti-TNF start was 6 [0–10] mg, and after 12 months of anti-TNF treatment was 2.5 [0–7.5] mg (p < 0.0001; Fig. 1, complete data available on 43 patients).
Fig. 1.
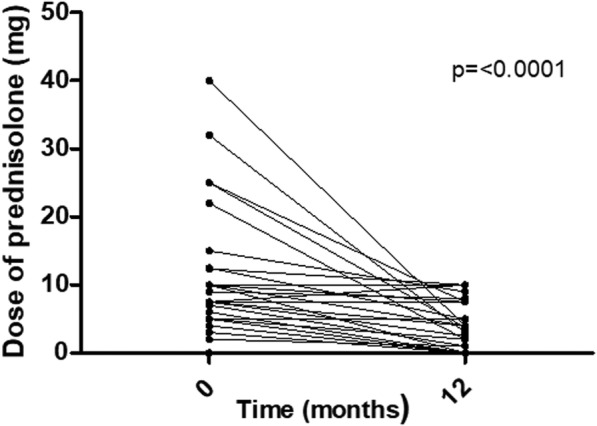
Steroid-sparing effect of use of anti-TNF therapy. Dose of prednisolone (mg/day) at anti-TNF start and 12 months of anti-TNF treatment. n = 43 patients (number of patients with complete data available)
Efficacy on infliximab therapy
In the 39 patients that received infliximab alone, global disease activity improved (χ2(2) = 8.14, p = 0.017; Fig. 2a). PGA decreased from 3.2 [1.8–5] at anti-TNF initiation to 0.9 [0.5–2.4] (p = 0.005) at 6 months and to 0.5 [0.3–1.3] at (p = 0.0003) 12 months. Skin involvement improved (χ2(2) = 15.52, p = 0.00043; Fig. 2b). Modified DAS decreased from 4 [1.5–5] at anti-TNF initiation to 2 [0–4] at 6 months (p = 0.002) and to 1 [0–3.3] at 12 months (p = 0.0006). Muscle involvement also improved in terms of CMAS and MMT 80 (χ2(2) = 17.02, p = 0.0002—Fig. 2c and χ2(2) = 10.56, p = 0.005—Fig. 2d, respectively). Median CMAS increased from 42 [37.8–49] at anti-TNF initiation to 50 [47–53] at 6 months (p = 0.03; not considered statistically significant following Bonferroni correction) and to 52 [50–53] (p = 0.0008) at 12 months. Median MMT 80 increased from 72 [59.8–78.3] to 77 [73–80] at 6 months (0.02; not significant) and to 80 [78–80] at 12 months (0.003).
Fig. 2.

Clinical measures over time in patients treated with infliximab (total of 43 patients). Clinical outcome measures of patients on infliximab are shown at baseline (time of starting infliximab) and at 6 and 12 months of infliximab treatment. a PGA, b Modified DAS, c CMAS and d MMT. n, number of patients with available data; PGA, Physician Global Assessment; DAS, Disease Activity Score; CMAS, Childhood Myositis Assessment Scale; MMT, Manual Muscle Testing
Of the 39 patients treated with infliximab alone, 15 patients were identified who had been treated with cyclophosphamide 1.9 [0.8–2.2] years prior to starting infliximab. Indications for cyclophosphamide include severe skin disease, severe muscle weakness, severe calcinosis, widespread vasculitis and failure to respond to first-line treatment. When these patients were excluded and the remaining patients analysed (n = 24), improvements in disease activity were observed in the remaining patients treated with infliximab alone (n = 24) for skin disease activity (χ2(2) = 6.08, p = 0.048 for modified DAS) and muscle disease activity (χ2(2) = 10.17, p = 0.006 for CMAS). Modified DAS reduced from 4 [1–4.3] at infliximab start to 2 [0–3] at 6 months (p = 0.018, not considered significant following Bonferroni correction) and 1 [0–3] at 12 months (p = 0.013). CMAS increased from 44 [38.8–50.5] at anti-TNF start to 52.5 [50–53] at 6 months (p = 0.11) and 52 [50–53] at 12 months (p = 0.03, not significant).
Efficacy after switching to adalimumab
Fifteen patients (25%) switched their anti-TNF treatment from infliximab to adalimumab. The median time of switching from infliximab to adalimumab was 2.3 months [1–3.8]. Ten (66.7%) of the switches were due to treatment inefficacy, 1 (6.6%) related to patient preference for subcutaneous administration and 4 (26.7%) were due to adverse events such as hypersensitivity reactions. From those 10 patients that switched due to treatment inefficacy, 8 were mainly due to active skin disease (5 had calcinosis lesions progressing). Only 3 of those 10 switches happened before 1 year on infliximab; all the others happened after 2 to 3 years on the drug.
For the patients who switched from infliximab to adalimumab (n = 15 patients), there was improvement in global disease activity (χ2(2) = 6.73, p = 0.03; Fig. 3a). PGA decreased from 1.2 [1–2.7] at adalimumab initiation to 0.5 [0.1–1.4] (p = 0.017; borderline significant) at 12 months. There were trends towards improvement in Modified DAS, CMAS and MMT8 (Fig. 3b–d).
Fig. 3.

Clinical measures in patients who switched from infliximab to adalimumab (total of 16 patients). Score shown at 0 (time of switch), 6 and 12 months of Adalimumab treatment. a PGA, b Modified DAS, c CMAS and d MMT. n, number of patients with available data; PGA, Physician Global Assessment; DAS, Disease Activity Score; CMAS, Childhood Myositis Assessment Scale; MMT, Manual Muscle Testing
For the limited number of patients on adalimumab alone (n = 4), median physician’s VAS at anti-TNF start was 2.6 (IQR 1.8–3.4), was 1.0 (IQR 0.5–1.25) at 6 months after anti-TNF start and was 1.5 (IQR 1.38–1.8) at 12 months after anti-TNF start. Median Modified DAS at anti-TNF start was 2 (IQR 1.5–2.3), was 3 (IQR 1.5–4) at 6 months after anti-TNF start and was 1 (IQR 0–2.5) at 12 months after anti-TNF start. Median CMAS at anti-TNF start was 52 (IQR 45–52), and there was no change over the year.
Resolution of calcinosis
Within the cohort of 60 patients, 28 patients (47%) with calcinosis were identified during their disease course up to the time of analysis. From the data recorded within the JDCBS [2] dataset, from the 28 patients, 15 (54%) had a reduction in the number and/or size of calcinotic lesions and calcinosis completely resolved in 8 (29%). Three out the 28 patients had fewer than 3 lesions which remained stable, and 3 out of the 28 patients had widespread lesions which remained stable. In 7, there were not sufficient data to evaluate changes. From the 15 patients that had a reduction in the number and/or size of calcinotic changes, we then further sub-analysed the 11 cases in which we had access to more detailed clinical information. This showed that the median time to improve was 2.75 [0.9–4] years, with a minimum of 0.25 years (3 months) and a maximum of 10 years. From those 11 patients, the calcinosis completely resolved in 4 of them in a median time of 2.8 [0.73–6.9] years and anti-TNF treatment was suspended in 3 of them with a median time of 4.6 [3–9] years after the beginning of the drug.
Adverse events
A total of 29 adverse events were reported, of which 12 were severe adverse events: 9 were allergic reactions to infliximab (HACA levels not routinely available in the UK), and there were 3 hospital admissions of Infliximab patients (sepsis in one and pneumonia in 2). One patient died due to small bowel perforation (probably secondary to disease damage) thought to be due to calcinosis in the intestinal walls. The patient was on infliximab for 6 years and then swapped to adalimumab for nearly 1 year before time of death. The remaining adverse reactions (n = 17) were not severe: 14 (82.4%) were due to infectious causes (5 bacterial upper respiratory infections, 4 viral infections, 2 skin infections, 2 episodes of tonsillitis, 1 episode of chicken-pox) and 3 (17.6%%) were due to local injection site reaction (n = 2) and 1 episode of skin rash. In 4 of the mild to moderate adverse reactions the drug had to be discontinued (2 patients on adalimumab had injection site reactions and 2 patients on infliximab had recurrent skin infections) whilst in the remaining patients temporarily withholding the drug proved sufficient. No tuberculosis or malignancy was recorded. According to the exposure time, a total of 13.9 adverse reactions occurred by 100 patient-years. 5.7 serious adverse reactions by 100 patient-years were reported.
Discussion
Biologic drugs have been used off-label since 2000 for JDM and other inflammatory myositis diseases with encouraging results [19]. High levels of TNF-α have been reported in JDM patients with a long disease course suggesting that it may play a significant role in refractory disease [7]. A recent study published by Spencer et al. [19], the results of a survey on CARRA members’ experience of using biologics in JDM, showed that survey responders considered that use of biologics significantly reduced complications in JDM (such as calcinosis, muscle atrophy, lipodystrophy) and were a logical therapeutic step after failure of corticosteroid and other immunosuppressive therapy, in JDM patients with resistant disease.
In our study, those who received infliximab and who switched from infliximab to adalimumab showed improvement in terms of global disease and improvement compared to their own baseline, but we cannot exclude the effects of other concomitant medications due to the lack of a control group. Muscle and skin disease appeared to reduce with infliximab treatment, with improvement in CMAS and MMT8 and the modified DAS. Importantly, we observed a reduction in steroid dose after 12 months of treatment with anti-TNF, which may suggest a possible steroid-sparing effect.
Major limitations of this study include missing data (commonly encountered in multi-site cohort studies) and the lack of a control group. We recognize that it is a limitation of our study that controls were not able to be included in the analysis. We have previously used observational data to model efficacy of cyclophosphamide, another second-line treatment for JDM, using the MSM method which allowed us to compare disease activity in patients treated with the drug to those who were not treated with the drug [20]. However, this method was not feasible in this study because we included two different drugs (infliximab and adalimumab) and treatment durations were heterogeneous. Obtaining evidence for second-line treatments in this rare disease is challenging, and there is an important role for observational studies and large case series like this study. Therefore, we cannot exclude effects of concomitant medications etc.
Infliximab was switched to adalimumab in 15 patients (25%). Ten (66.7%) of the switches were due to incomplete control of disease, mainly due to lack of skin disease control. The remaining causes for switching were adverse effects and patient preference.
Calcinosis remains a significant source of morbidity for many JDM patients, yet it is poorly understood and lacks uniform treatment approaches compared to other aspects of JDM. A recent study published by the CARRA group [21] emphasizes the inconsistency in the published literature regarding therapeutic effectiveness. The authors suggest that this can be explained by the relative inexperience of physicians and the multitude of different treatments and treatment scenarios [21]. In our study, there was a reduction in the number and/or size of calcinotic lesions in 54% of the patients that were on infliximab/adalimumab and calcinosis completely resolved in 29% of them, although we cannot attribute these effects to anti-TNF treatment alone. It was also noted that the reduction in number and size of calcinosis needed nearly 2 years on anti-TNF treatment. Although we had a patient that showed evident reduction on calcinosis lesions only after 3 months on anti-TNF treatment, the majority needed a much longer time and in one case it was necessary to continue 10 years of anti-TNF treatment until the calcinosis completely resolved. This highlights that an immediate improvement on the calcinosis should not be expected, and the anti-TNF treatment should not be stopped if an immediate improvement is not seen.
Regarding safety, the majority of the adverse events were mild to moderate and mostly due to infections causes. However, we had 12 serious adverse events, mostly (n = 9) related with allergic reactions to infliximab. Administration of infliximab is associated with a well-recognized risk of infusion-related adverse events. The exact aetiology and pathogenesis of those infusion reactions are often unclear, and findings regarding their allergic/immune nature are inconsistent [22]. Overall the anti-TNF drugs seemed to be well-tolerated in our population with an incidence of 5.7 serious adverse reactions by 100 patient-years, which is consistent with the literature on adult patients [23]. Importantly, the majority of patients had no adverse effects and tolerated the infliximab/adalimumab well, and in the context of patients needing long-term treatment, this could be of benefit.
This study is one of the largest to describe the use of infliximab and adalimumab in a large national cohort of JDM patients. Further clinical studies are required to assess the efficacy of anti-TNF treatment in JDM, controlling for the effects of other DMARDS, and to ascertain the optimum timing for treatment initiation and cessation. Stratified analysis by autoantibody subgroups could be addressed in future studies with greater numbers.
Conclusions
Reductions in muscle and skin disease, including calcinosis, were seen following treatment with infliximab and adalimumab. Infliximab and adalimumab were well-tolerated and the majority of patients had no adverse effects.
Supplementary information
Additional file 1: Supplementary Figure S1. Flow diagram to outline which patients were included or excluded in the study and which analyses they were included in. Supplementary Results.
Acknowledgements
We would like to thank all patients and their families who have contributed to the JDCBS. We also thank all members of the JDRG who contribute to the JDCBS. The JDRG members were as follows:
Dr. Kate Armon, Mr. Joe Ellis-Gage, Ms. Holly Roper, Ms. Vanja Briggs and Ms. Joanna Watts (Norfolk and Norwich University Hospitals), Dr. Liza McCann, Mr. Ian Roberts, Dr. Eileen Baildam, Ms. Louise Hanna, Ms. Olivia Lloyd, Susan Wadeson and Michelle Andrews (The Royal Liverpool Children’s Hospital, Alder Hey, Liverpool), Dr. Phil Riley, Ms. Ann McGovern, Verna Cuthbert (Royal Manchester Children’s Hospital, Manchester), Dr. Clive Ryder, Mrs. Janis Scott, Mrs. Beverley Thomas, Professor Taunton Southwood, Dr. Eslam Al-Abadi and Ruth Howman (Birmingham Children’s Hospital, Birmingham), Dr. Sue Wyatt, Mrs. Gillian Jackson, Dr. Mark Wood, Dr. Tania Amin, Dr. Vanessa VanRooyen and Ms. Deborah Burton, Louise Turner & Sarah Hanson (Leeds General Infirmary, Leeds), Dr. Joyce Davidson, Dr. Janet Gardner-Medwin, Dr. Neil Martin, Ms. Sue Ferguson, Ms. Liz Waxman and Mr. Michael Browne, Ms. Roisin Boyle, Ms. Emily Blyth (The Royal Hospital for Sick Children, Yorkhill, Glasgow), Dr. Mark Friswell, Professor Helen Foster, Mrs. Alison Swift, Dr. Sharmila Jandial, Ms. Vicky Stevenson, Ms. Debbie Wade, Dr. Ethan Sen, Dr. Eve Smith, Ms. Lisa Qiao, Mr. Stuart Watson and Ms. Claire Duong (Great North Children’s Hospital, Newcastle), Dr. Helen Venning, Dr. Rangaraj Satyapal, Mrs. Elizabeth Stretton, Ms. Mary Jordan, Dr. Ellen Mosley, Ms. Anna Frost, Ms. Lindsay Crate, Dr. Kishore Warrier, Stefanie Stafford, Kelly Sandhu & Tracey Dandy (Queens Medical Centre, Nottingham), Professor Lucy Wedderburn, Dr. Clarissa Pilkington, Dr. Nathan Hasson, Dr. Muthana Al-Obadi, Dr. Giulia Varnier, Dr. Sandrine Lacassagne, Mrs. Sue Maillard, Mrs. Lauren Stone, Ms. Elizabeth Halkon, Ms. Virginia Brown, Ms. Audrey Juggins, Dr. Sally Smith, Mrs. Sian Lunt, Ms. Elli Enayat, Mrs. Hemlata Varsani, Miss Laura Kassoumeri, Miss Laura Beard, Miss Katie Arnold, Mrs. Yvonne Glackin, Ms. Stephanie Simou, Dr. Beverley Almeida, Dr. Kiran Nistala, Dr. Raquel Marques, Dr. Claire Deakin, Dr. Parichat Khaosut, Ms. Stefanie Dowle, Dr. Charalampia Papadopoulou, Dr. Shireena Yasin, Dr. Christina Boros, Dr. Meredyth Wilkinson, Dr. Chris Piper, Mrs. Cerise Johnson-Moore, Ms. Lucy Marshall, Ms. Kathryn O’Brien, Ms. Emily Robinson (Great Ormond Street Hospital, London), Dr. Kevin Murray (Princess Margaret Hospital, Perth, Western Australia) Dr. Coziana Ciurtin, Dr. John Ioannou, Mrs. Caitlin Clifford and Ms. Linda Suffield (University College London Hospital, London) Ms. Helen Lee, Ms. Sam Leach, Ms. Helen Smith, Dr. Anne-Marie McMahon, Ms. Heather Chisem, Jeanette Hall, Ruth Kingshott and Maxine Mutten (Sheffield’s Children’s Hospital, Sheffield); Dr. Nick Wilkinson, Ms. Emma Inness, Ms. Eunice Kendall, Mr. David Mayers, Ruth Etherton, Danielle Miller and Dr. Kathryn Bailey (Oxford University Hospitals, Oxford); Dr. Jacqui Clinch, Ms. Natalie Fineman, Ms. Helen Pluess-Hall and Suzanne Sketchley (Bristol Royal Hospital for Children, Bristol); Dr. Joyce Davidson, Margaret Connon and Ms. Lindsay Vallance (Royal Aberdeen Children’s Hospital); Dr. Kirsty Haslam, Charlene Bass-Woodcock, Trudy Booth and Ms. Louise Akeroyd (Bradford Teaching Hospitals); Dr. Alice Leahy, Amy Collier, Rebecca Cutts, Emma Macleod, Dr. Hans De Graaf, Dr. Brian Davidson, Sarah Hartfree, Danny Pratt, Elizabeth Fofana and Lorena Caruana (University Hospital Southampton) and all the Children, Young people and their families who have contributed to this research.
Abbreviations
- CMAS
Childhood Myositis Assessment Scale
- JDRG
Juvenile Dermatomyositis Research Group
- MMT8
Manual Muscle Testing
- PGA
Physician’s global assessment
- DAS
Disease activity score
- JDM
Juvenile dermatomyositis
- IIM
Idiopathic inflammatory myopathy
- IVIg
Intravenous immunoglobulin
- JDCBS
Juvenile Dermatomyositis Cohort and Biomarker Study
- DMARD
Disease-modifying anti-rheumatic drug
- PRINTO
Paediatric Rheumatology International Trials Organisation
- IMACS
International Myositis Assessment and Clinical Studies Group
- ANOVA
Analysis of variance
- HACA
Human anti-chimeric antibody
Authors’ contributions
Author contributions are as follows: R C-M acquired, analysed and interpreted data and drafted the manuscript. CTD acquired, analysed and interpreted data and drafted the manuscript. SS acquired and analysed data. CP acquired and analysed data. LRW conceived and designed the study, interpreted data and drafted the manuscript. CAP conceived and designed the study, interpreted data and drafted the manuscript. The authors read and approved the final manuscript.
Authors’ information
Not applicable
Funding
Funding for the UK JDM Cohort and Biomarker study has been provided by generous grants from the Wellcome Trust UK [085860], Action Medical Research UK [SP4252], The Myositis Support Group UK, Arthritis Research UK [14518, 20164], The Henry Smith Charity, Great Ormond Street Children’s Charity [V1268], The Myositis Association, Cure JM and the National Institute for Health Research (NIHR). This research was supported by the NIHR Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust. The JDM Cohort study is adopted onto the NIHR Comprehensive Research Network. The Centre for Adolescent Rheumatology Versus Arthritis at UCL, UCL Hospital and GOSH is supported by grants from Versus Arthritis [20164] and Great Ormond Street Children’s Charity. Dr. Deakin was supported by a fellowship from The Myositis Association. This is a summary of independent research funded by the NIHR Rare Diseases Translational Research Collaboration and other funders. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health. None of these funding bodies has had a role in the design of the study or the collection, analysis and interpretation of data.
Availability of data and materials
Not applicable
Ethics approval and consent to participate
Informed written parental consent and age-appropriate assent were obtained. This research was approved by the UK Northern & Yorkshire Medical Research and Ethics Committee. This manuscript does not contain data from any individual person.
Consent for publication
Not applicable
Competing interests
The authors declare they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Raquel Campanilho-Marques and Claire T. Deakin contributed equally to this work.
Contributor Information
Clarissa A. Pilkington, Email: clarissa.pilkington@gosh.nhs.uk
for the Juvenile Dermatomyositis Research Group (JDRG):
Kate Armon, Joe Ellis-Gage, Holly Roper, Vanja Briggs, Joanna Watts, Liza McCann, Ian Roberts, Eileen Baildam, Louise Hanna, Olivia Lloyd, Susan Wadeson, Michelle Andrews, Phil Riley, Ann McGovern, Verna Cuthbert, Clive Ryder, Janis Scott, Beverley Thomas, Taunton Southwood, Eslam Al-Abadi, Ruth Howman, Sue Wyatt, Gillian Jackson, Mark Wood, Tania Amin, Vanessa VanRooyen, Deborah Burton, Louise Turner, Sarah Hanson, Joyce Davidson, Janet Gardner-Medwin, Neil Martin, Sue Ferguson, Liz Waxman, Michael Browne, Roisin Boyle, Emily Blyth, Mark Friswell, Helen Foster, Alison Swift, Sharmila Jandial, Vicky Stevenson, Debbie Wade, Ethan Sen, Eve Smith, Lisa Qiao, Stuart Watson, Claire Duong, Helen Venning, Rangaraj Satyapal, Elizabeth Stretton, Mary Jordan, Ellen Mosley, Anna Frost, Lindsay Crate, Kishore Warrier, Stefanie Stafford, Kelly Sandhu, Tracey Dandy, Lucy Wedderburn, Clarissa Pilkington, Nathan Hasson, Muthana Al-Obadi, Giulia Varnier, Sandrine Lacassagne, Sue Maillard, Lauren Stone, Elizabeth Halkon, Virginia Brown, Audrey Juggins, Sally Smith, Sian Lunt, Elli Enayat, Hemlata Varsani, Laura Kassoumeri, Laura Beard, Katie Arnold, Yvonne Glackin, Stephanie Simou, Beverley Almeida, Kiran Nistala, Raquel Marques, Claire Deakin, Parichat Khaosut, Stefanie Dowle, Charalampia Papadopoulou, Shireena Yasin, Christina Boros, Meredyth Wilkinson, Chris Piper, Cerise Johnson-Moore, Lucy Marshall, Kathryn O’Brien, Emily Robinson, Kevin Murray, Coziana Ciurtin, John Ioannou, Caitlin Clifford, Linda Suffield, Helen Lee, Sam Leach, Helen Smith, Anne-Marie McMahon, Heather Chisem, Jeanette Hall, Ruth Kingshott, Maxine Mutten, Nick Wilkinson, Emma Inness, Eunice Kendall, David Mayers, Ruth Etherton, Danielle Miller, Kathryn Bailey, Jacqui Clinch, Natalie Fineman, Helen Pluess-Hall, Suzanne Sketchley, Joyce Davidson, Margaret Connon, Lindsay Vallance, Kirsty Haslam, Charlene Bass-Woodcock, Trudy Booth, Louise Akeroyd, Alice Leahy, Amy Collier, Rebecca Cutts, Emma Macleod, Hans De Graaf, Brian Davidson, Sarah Hartfree, Danny Pratt, Elizabeth Fofana, and Lorena Caruana
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13075-020-02164-5.
References
- 1.Feldman BM, Rider LG, Reed AM, Pachman LM. Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet. 2008;371(9631):2201–2212. doi: 10.1016/S0140-6736(08)60955-1. [DOI] [PubMed] [Google Scholar]
- 2.Martin N, Krol P, Smith S, Murray K, Pilkington CA, Davidson JE, et al. A national registry for juvenile dermatomyositis and other paediatric idiopathic inflammatory myopathies: 10 years’ experience; the Juvenile Dermatomyositis National (UK and Ireland) Cohort Biomarker Study and Repository for Idiopathic Inflammatory Myopathies. Rheumatology (Oxford) 2011;50(1):137–145. doi: 10.1093/rheumatology/keq261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Enders FB, Bader-Meunier B, Baildam E, Constantin T, Dolezalova P, Feldman BM, et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis. 2017;76(2):329–340. doi: 10.1136/annrheumdis-2016-209247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moghadam-Kia S, Oddis CV, Aggarwal R. Modern therapies for idiopathic inflammatory myopathies (IIMs): role of biologics. Clin Rev Allergy Immunol. 2017;52(1):81–87. doi: 10.1007/s12016-016-8530-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maini R, St Clair EW, Breedveld F, Furst D, Kalden J, Weisman M, et al. Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. ATTRACT Study Group. Lancet. 1999;354(9194):1932–1939. doi: 10.1016/S0140-6736(99)05246-0. [DOI] [PubMed] [Google Scholar]
- 6.Brandt J, Sieper J, Braun J. Infliximab in the treatment of active and severe ankylosing spondylitis. Clin Exp Rheumatol. 2002;20(6 Suppl 28):S106–S110. [PubMed] [Google Scholar]
- 7.Pachman LM, Liotta-Davis MR, Hong DK, Kinsella TR, Mendez EP, Kinder JM, et al. TNFalpha-308A allele in juvenile dermatomyositis: association with increased production of tumor necrosis factor alpha, disease duration, and pathologic calcifications. Arthritis Rheum. 2000;43(10):2368–2377. doi: 10.1002/1529-0131(200010)43:10<2368::AID-ANR26>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 8.TO F, Lutz J, Pachman LM. Expression of TNFalpha by muscle fibers in biopsies from children with untreated juvenile dermatomyositis: association with the TNFalpha-308A allele. Clin Immunol. 2001;100(2):236–239. doi: 10.1006/clim.2001.5063. [DOI] [PubMed] [Google Scholar]
- 9.De Paepe B, Creus KK, De Bleecker JL. The tumor necrosis factor superfamily of cytokines in the inflammatory myopathies: potential targets for therapy. Clin Dev Immunol. 2012;2012:369432. doi: 10.1155/2012/369432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fisler RE, Liang MG, Fuhlbrigge RC, Yalcindag A, Sundel RP. Aggressive management of juvenile dermatomyositis results in improved outcome and decreased incidence of calcinosis. J Am Acad Dermatol. 2002;47(4):505–511. doi: 10.1067/mjd.2002.122196. [DOI] [PubMed] [Google Scholar]
- 11.Riley P, McCann LJ, Maillard SM, Woo P, Murray KJ, Pilkington CA. Effectiveness of infliximab in the treatment of refractory juvenile dermatomyositis with calcinosis. Rheumatology (Oxford) 2008;47(6):877–880. doi: 10.1093/rheumatology/ken074. [DOI] [PubMed] [Google Scholar]
- 12.Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts) N Engl J Med. 1975;292(7):344–347. doi: 10.1056/NEJM197502132920706. [DOI] [PubMed] [Google Scholar]
- 13.Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts) N Engl J Med. 1975;292(8):403–407. doi: 10.1056/NEJM197502202920807. [DOI] [PubMed] [Google Scholar]
- 14.Huber AM, Feldman BM, Rennebohm RM, Hicks JE, Lindsley CB, Perez MD, et al. Validation and clinical significance of the Childhood Myositis Assessment Scale for assessment of muscle function in the juvenile idiopathic inflammatory myopathies. Arthritis Rheum. 2004;50(5):1595–1603. doi: 10.1002/art.20179. [DOI] [PubMed] [Google Scholar]
- 15.Rider LG, Koziol D, Giannini EH, Jain MS, Smith MR, Whitney-Mahoney K, et al. Validation of manual muscle testing and a subset of eight muscles for adult and juvenile idiopathic inflammatory myopathies. Arthritis Care Res. 2010;62(4):465–472. doi: 10.1002/acr.20035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruperto N, Ravelli A, Pistorio A, Ferriani V, Calvo I, Ganser G, et al. The provisional Paediatric Rheumatology International Trials Organisation/American College of Rheumatology/European League Against Rheumatism Disease activity core set for the evaluation of response to therapy in juvenile dermatomyositis: a prospective validation study. Arthritis Rheum. 2008;59(1):4–13. doi: 10.1002/art.23248. [DOI] [PubMed] [Google Scholar]
- 17.Rider LG, Giannini EH, Brunner HI, Ruperto N, James-Newton L, Reed AM, et al. International consensus on preliminary definitions of improvement in adult and juvenile myositis. Arthritis Rheum. 2004;50(7):2281–2290. doi: 10.1002/art.20349. [DOI] [PubMed] [Google Scholar]
- 18.Lam CG, Manlhiot C, Pullenayegum EM, Feldman BM. Efficacy of intravenous Ig therapy in juvenile dermatomyositis. Ann Rheum Dis. 2011;70(12):2089–2094. doi: 10.1136/ard.2011.153718. [DOI] [PubMed] [Google Scholar]
- 19.Spencer CH, Rouster-Stevens K, Gewanter H, Syverson G, Modica R, Schmidt K, et al. Biologic therapies for refractory juvenile dermatomyositis: five years of experience of the Childhood Arthritis and Rheumatology Research Alliance in North America. Pediatr Rheumatol Online J. 2017;15(1):50. doi: 10.1186/s12969-017-0174-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deakin CT, Campanilho-Marques R, Simou S, Moraitis E, Wedderburn LR, Pullenayegum E, Pilkington CA, Juvenile Dermatomyositis Research Group Efficacy and safety of cyclophosphamide in severe juvenile dermatomyositis shown by marginal structural modeling. Arthritis Rheumatol. 2018;70(5):785–793. doi: 10.1002/art.40418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orandi AB, Baszis KW, Dharnidharka VR, Huber AM, Hoeltzel MF. Assessment, classification and treatment of calcinosis as a complication of juvenile dermatomyositis: a survey of pediatric rheumatologists by the childhood arthritis and rheumatology research alliance (CARRA) Pediatr Rheumatol Online J. 2017;15(1):71. doi: 10.1186/s12969-017-0199-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lichtenstein L, Ron Y, Kivity S, Ben-Horin S, Israeli E, Fraser GM, et al. Infliximab-related infusion reactions: systematic review. J Crohn’s Colitis. 2015;9(9):806–815. doi: 10.1093/ecco-jcc/jjv096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strand V, Ahadieh S, French J, Geier J, Krishnaswami S, Menon S, et al. Systematic review and meta-analysis of serious infections with tofacitinib and biologic disease-modifying antirheumatic drug treatment in rheumatoid arthritis clinical trials. Arthritis Res Ther. 2015;17:362. doi: 10.1186/s13075-015-0880-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Supplementary Figure S1. Flow diagram to outline which patients were included or excluded in the study and which analyses they were included in. Supplementary Results.
Data Availability Statement
Not applicable