

Abstract
Previously, we found that elastin-like polypeptide (ELP), when dried above the lower critical solution temperature on top of a hydrophilic fused silica disk, exhibited a dynamic coalescence behavior. The ELP initially wet the silica, but over the next 12 h, dewett the surface and formed aggregates of precise sizes and shapes. Using Fourier-transform infrared (FT-IR) spectroscopy, the present study explores the role of secondary structures present in ELP during this progressive desiccation and their effect on aggregate size. The amide I peak (1600–1700 cm–1) in the ELP’s FT-IR spectrum was deconvoluted using the second derivative method into eight subpeaks (1616, 1624, 1635, 1647, 1657, 1666, 1680, 1695 cm–1). These peaks were identified to represent extended strands, β-turns, 3(10)-helix, polyproline I, and polyproline II using previous studies on ELP and molecules similar in peptide composition. Positive correlations were established between the various subpeaks, water content, and aggregate size to understand the contributions of the secondary structures in particle formation. The positive correlations suggest that type II β-turns, independent of the water content, contributed to the growth of the aggregates at earlier time points (1–3.5 h). At later time points (6–12 h), the aggregate growth was attributed to the formation of 3(10)-helices that relied on a decrease in water content. Understanding these relationships gives greater control in creating precisely sized aggregates and surface coatings with varying roughness.
1. Introduction
Elastin-like polypeptides (ELPs) are a class of biopolymers that are known to exhibit an inverse phase transition behavior that allows them to coalesce from a solvated polymer to a coacervated state above a specific lower critical solution temperature (LCST). This transition temperature (Tt) can be influenced by changing ELP concentration, solution conditions (salt concentration, pH), and structural factors (guest residue, molecular weight (MW)).1−4 ELPs are considered an artificial, intrinsically disordered protein (IDP) that is predominately random coil, β-turns, and polyproline II (PPII) below the Tt. Above the Tt, the fraction of β-turn can increase in a temperature- and solution-condition manner.5 This may be especially important when undergoing interactions with a surface. Because ELP secondary structure is an ensemble of many statistical coil structures,6−8 ELPs have been used as an IDP model to provide a more fundamental understanding of other more complex IDPs.9 Typical applications for ELPs include the formation of amyloid-like structures,10 modeling phase separation behavior of biological systems,11,12 and more widely as drug delivery vectors13−15 and biological scaffolds.16−18 Thorough characterization and understanding of the prevalence of the types of secondary structures will provide enhanced predictability of ELPs’ behavior and pave the way for more advanced uses such as biological circuits and sensors that will rely on ELP’s interactions with the substrate surface.
Analysis of ELP secondary structures is limited to the use of specific spectroscopic methods due to their size. The most common methods of analysis are nuclear magnetic resonance spectroscopy (NMR) and circular dichroism (CD).19,20 While NMR is a powerful tool for the identification of structures present in proteins and polypeptides, the information it provides for polypeptides is challenging due to the repetitive nature of the constructs and the large ensemble of secondary structures.19 CD, while not as precise as NMR, can give a simplified overview of the secondary structures present and how they change over time. This information is compared to a spectral library of basis vectors to provide the interpretation of the peaks.20 CD can give information on strongly absorbing secondary structures but can also introduce errors in the interpretation of spectrally diverse β-sheet structures.20 Therefore, more than one technique is often used to help cross-identify spectra generated for the polypeptides to overcome the limitations of individual techniques.
Fourier-transform infrared (FT-IR) spectroscopy has been a popular method to identify the secondary structures in proteins due to its ease of use. Traditionally for polypeptides, deconvoluted peak positions were identified using protein structural assignments. These peak positions were previously identified using proteins with a single type of secondary structure.21,22 These preidentified peak positions have become invaluable starting references, but lack the specificity needed for more complex ensembles of secondary structures seen in polypeptides. The repeat units present in polypeptides contribute to distributions and overlapping interactions of secondary structures that are not found in stable proteins. Therefore, new peak assignments based on the individual polypeptides are needed.23 Serrano et al. used density functional theory (DFT) calculations and second derivative deconvolution of a small model ELP construct (VPGVG)3 (where V = valine, P = proline, and G = glycine) to identify secondary structures. They believed β-spirals to be a potential structure present during ELP coacervation based on previous studies.24 It was also interpreted that the ELP secondary structure shifted from a loosely packed β-spiral to a tighter packed β-helix when the NaCl concentration was increased to 1.5 M in the ELP solution. However, other studies have shown that a dynamic interchange of secondary structures prevents the VPGVG peptide from adopting a stable β-spiral conformation.10,25−27
The principal goal of this study was to identify the secondary structures present in ELP during desiccation on a silica surface and to examine the role that secondary structures play in maintaining the coalesced aggregate size. To this end, we explored the evolution of the various SynB1-ELP secondary structures using FT-IR spectroscopy by allowing peak assignments based on previous FT-IR studies on traditional proteins as well as NMR, CD, and Monte Carlo computer simulations on ELP.6,7 Statistical analysis was used to confirm peak assignments based on correlations between peaks in the deconvoluted amide I band. This work capitalized on the relatively water-free environments created by incubating the samples at 50 °C for extended periods on a silica surface and is expected to give more accurate peak shapes than traditional samples in water. Understanding these relationships may give greater control for applications where ELP will experience significant surface interactions such as creating coatings with varying roughness.28
2. Results
2.1. Surface Water Content
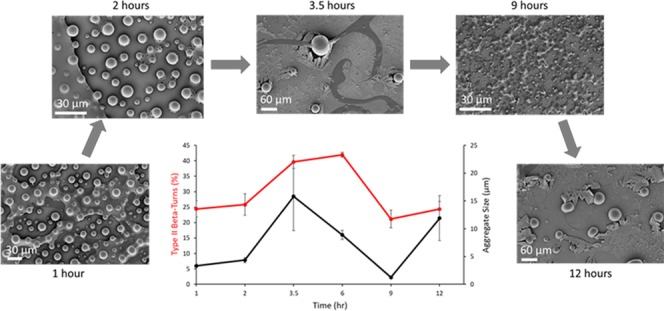
FT-IR spectra were taken at discrete time points during the incubation of ELP solution on silica at 50 °C to observe the effect that water removal has on the secondary structures. Previously, we used scanning electron microscopy to demonstrate that as water evaporates from the ELP solution, a layer of salt forms on top of the silica before the deposition of ELP.29 The strong peak at 3300 cm–1 is representative of the O–H vibrations of water and was used to measure the amount of water present on the surface without the addition of ELP. Above its LCST, ELP excludes itself from the bulk water by phase separation into aggregates.6,7 In contrast, the salts from phosphate-buffered saline (PBS) solution deposited atop silica retain small amounts of water for extended periods. A graph of the water content of the PBS solution deposited atop silica over time was constructed from the FT-IR spectra to visualize this behavior (Figure 1).29 This graph demonstrates that from 1 to 3.5 h, the bulk water loss occurs from the PBS–silica at a faster rate, while from 6 h until the 12 h time point, the water loss was significantly slower. This water retention by the deposited salts is thought to be one of the primary sources of the ELP’s observed ability to dynamically rearrange itself in various secondary structures.
Figure 1.

Water content decreases rapidly from 1 to 3.5 h due to evaporation; from 6 to 12 h, the water content remains stable, most likely from secondary interactions between the water, salts, and silica.
2.2. FT-IR Peak Assignments
After deconvolution, multiple peak positions were identified, as can be seen in Table 1. Identification of these peaks using traditional protein assignments proved difficult for most of the peaks because the traditional peak positions are for individual proteins exhibiting a single secondary structure and do not account for resonance, hydrogen bond strength, or the number of amino acid residues participating in the structure. Therefore, assignments were made using past data on SynB1-ELP generated from NMR, CD, and Monte Carlo simulations and utilizing other studies on similar sequenced peptides and polypeptides.21−27,30−32
Table 1. Deconvoluted Peak Positions with Traditional Protein Assignments and Those of SynB1-ELP.
| average position (cm–1) | traditional protein assignment | ELP assignment (time dependent) | average composition (%) |
|---|---|---|---|
| 1616 ± 0.4 | β-sheet aggregates24/extended strands25,26,30−32 | extended strands/γ-turn or bifurcated H-bond27 | 21.8 |
| 1624 ± 0.5 | β-sheet21−24 | PPI33/γ-turn or bifurcated H-bond27 | 2.8 |
| 1635 ± 0.4 | β-sheet21−24 | type II/type I(III) β-turn27 | 14.0 |
| 1647 ± 0.3 | Random coil21,23 | PPI/PPII-helix34−37 | 16.7 |
| 1657 ± 0.3 | α-helix21,23 | type II/type I(III) β-turn27 | 9.2 |
| 1666 ± 0.3 | 3(10)-helix, β-turn21−24 | type I(III) β-turn27/3(10)-helix21/PPII34 | 14.0 |
| 1680 ± 0.5 | β-turn21,23,27 | PPII-helix34,36 | 17.0 |
| 1695 ± 0.4 | β-sheet21/β-turn23,27 | type II β-turn27 | 4.8 |
2.3. FT-IR Peak Correlations
Two discrete time intervals were selected to determine correlations between secondary structures based on the rate of change of surface water content, as seen in Figure 1, and change in aggregate diameters, as seen in Figure 2. The time points from 1 to 3.5 h (Figure 3A) were selected as they represent both the largest amount of water present during desiccation and the largest drop in the amount of water on the surface from 31.5 to 8.6% (Figure 1). Strong positive correlations were found between the peaks at 1635 and 1657 cm–1 (R2 = 0.70), 1657 and 1695 cm–1 (R2 = 0.89), and 1635 and 1695 cm–1 (R2 = 0.94) (Figure 3A). A strong correlation was also found between the peak at 1616 and 1624 cm–1 (R2 = 0.80). Weak positive correlations were found between the peak at 1635 and 1666 cm–1 (R2 = 0.50), 1666 and 1680 cm–1 (R2 = 0.49), and 1647 and 1680 cm–1 (R2 = 0.34).
Figure 2.

Particle diameter measurements were taken at discrete time intervals from scanning microscopy images. Adapted or reprinted in part with permission from Cobb et al.29
Figure 3.
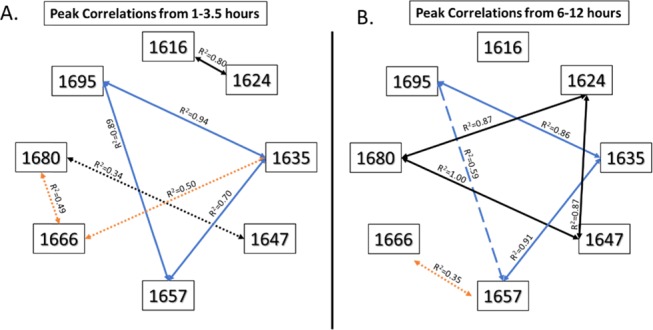
Positive correlations between peaks at three-time intervals that show a prevailing association between the peaks at 1635, 1657, and 1695 cm–1 (blue lines), which suggests that these peaks rely on each other’s presence to maintain their stability. A dynamic relationship is seen between these three peaks and the one at 1666 cm–1, which indicates a shift in the type of secondary structures being formed. Weak correlations are denoted by dotted lines, moderate correlations by dashed lines, and strong correlations by solid lines. The blue lines indicate the relationship between 1635, 1657, and 1695 cm–1; black lines show the relationship between 1616, 1624, 1647, and 1680 cm–1; orange lines indicate relationships to the peak at 1666 cm–1.
The time points from 6 to 12 h (Figure 3B) were analyzed as these points represent the lowest amount of water present on the surface (Figure 1) and the point at which the larger aggregates collapse and regrow on the silica (Figure 2).26 Again, the same positive correlations exist between the peaks at 1635, 1657, and 1695 cm–1, as well as between 1647 and 1680 cm–1.
Strong negative correlations were observed between the groups shown in Figure 4. A strong negative correlation (R2 = 0.91) exists between the β-turns and polyproline (PP) I and II structures and the extended strands (Figure 4A). A strong negative correlation (R2 = 0.90) exists between the polyproline I and II (PPI/PPII) and the type I(III) and II β-turns peaks at 1635 and 1647 cm–1 (Figure 4B). A strong negative correlation (R2 = 0.72) was observed between the type I(III) and II β-turns and the PPI/PPII peaks at 1647 and 1657 cm–1 (Figure 4C).
Figure 4.

Strong negative correlations were observed between (A) extended strands and PPI/PPII-helix, β-turns; (B) PPI/PPII-helix at 1646 cm–1 and β-turns at 1633 cm–1; (C) β-turns at 1656 cm–1 and PPI/PPII-helix at 1646 cm–1.
2.4. Amide II/Amide I Peak Ratio Comparison
The amide II to amide I peak ratio decreased from 1 to 3.5 h, while it increased at 5 h and remained level until the 9 h time point and drastically decreased at 12 h (Figure 5A). The decrease in the peak ratio was found to be weakly correlated (R2 = 0.47) to an increase in the percentage of β-turns present in the ELP (Figure 5B), while an increase in the peak ratio was found to be moderately correlated (R2 = 0.67) to an increase in the percentage of extended chains (Figure 5C).
Figure 5.

(A) Change in the amide II to amide I peak area ratio is associated with a change in secondary structure. (B) Correlations between the amide peak area ratio and secondary structures indicate that an increase in the amide peak area ratio is weakly (R2 = 0.47) associated with a decrease in the total number of β-turns. (C) Increase in the peak area ratio is moderately (R2 = 0.67) correlated to an increase in extended chains.
2.5. Correlations of Secondary Structures to Water Content and Coalesced Aggregate Size
Table 2 shows the correlations of the ELP aggregate size (Figure 2)26 and surface water content (Figure 1) to the secondary structures of ELP seen in FT-IR spectroscopy. For the 1–3.5 h time points, we observed that an increase in aggregate size correlated with the formation of β-turns, predominantly type II β-turns, as indicated by the positive correlations between aggregate size and the β-turn peaks at 1635, 1657, 1666, and 1695 cm–1 (Table 2A and TOC Figure). Negative correlations and thus a decrease in aggregate size were found to come from extended chains, γ-turns, and PPII-helix. A strong negative correlation was found between water content and the γ-turn peak at 1624 cm–1, while a weak positive correlation was found between water content and the β-turn peak at 1635 cm–1 (Table 2B).
Table 2. Correlations of the ELP Aggregate Size26 and Surface Water Content to the Secondary Structures of ELP Seen in FT-IR Spectroscopya.

(A) Large aggregate sizes rely on an increase in β-turns from 1 to 3.5 h and switch to the 3(10)-helix from 6 to 12 h to maintain their stability, as shown by the green shaded boxes. (B) A decrease in water content from 1 to 3.5 h shows no positive moderate or strong correlations between secondary structures, but the decrease in water content positively affects the formation of type II β-turns and 3(10)-helixes from 6 to 12 h, as shown by the green shaded boxes.
For the 6–12 h time points, the increase in aggregate size was found to strongly correlate with the type I (III) β-turn/3(10)-helix peak at 1666 cm–1 and weakly with that of type II β-turn peak at 1657 cm–1 and a decrease in the extended chain peak at 1616 cm–1 (Table 2A and TOC Figure). Contributions to an increase in aggregate size for the entire duration of 1–12 h were found to come from type II β-turns and a decrease in PPI structures. The decrease in water content for the entire duration of 1–12 h also showed positive correlations with the formation of type II β-turns and negative correlations with PPI/PPII-helix structures (Table 2B). Based on these observations, it appears that to increase aggregate size, the number of β-turns must be maximized and for this to occur, the water content on the surface must be less than 10% (Figure 1).
3. Discussion
3.1. FT-IR Peak Assignments
Both the device and the methodology matter for the precise quantification of the peak locations and area, which is why all samples were treated the same, and the same settings were used on the device. FT-IR measurements are very precise so long as enough scans are taken (usually ≥8) and the spectral resolution is reasonable for precise peak locations (≤4 cm–1). Second derivative analysis is subject to sensitivity in measurements and thus requires a device that is capable of outputting high-quality spectra. Prior knowledge of potential secondary structures and a priori determined criteria for noise elimination are also needed to interpret the deconvoluted peaks indicated by second derivative analysis. For our analysis, samples were secured on the attenuated total reflection (ATR) accessory with a clamping pressure of 110 psi, and 16 scans were taken in the spectral range of 4000–650 cm–1 with a spectral resolution of 2 cm–1. Measurements were performed on three separate samples to ensure reproducibility. Our method for eliminating peaks that may have been caused by noise in the spectra was to calculate the mean absolute percent error. The mean absolute percent error was calculated for each region in the amide I containing an underlying peak. Since we performed more than enough scans at a high resolution for each sample (n = 3), we did not have to eliminate any deconvoluted peaks as the noise in the spectra was minimal.
Table 1 summarizes the secondary structure assignments made in FT-IR for SynB1-ELP; the following discussion explains why the specific assignments were chosen. SynB1-ELP has been previously studied to elucidate its aggregation kinetics using dynamic light scattering, turbidity measurements, and analytical ultracentrifugation. NMR, CD, and computer simulations were used to find the secondary structures responsible for the coalescence of SynB1-ELP into phase-separated domains.5−7 The individual polypeptide repeat sequences in SynB1-ELP have a propensity to form type II β-turns, extended chains, type I(III) β-turns, PPI, PPII, and 3(10)-helixes as detailed below. The NMR delta-2D analysis showed a low probability for α-helix structures to form and a high probability for random coils to form. However, this study did not utilize relaxation times to determine whether NMR peaks were from β-turns.6 Another NMR study of the (VPGVG)3 peptide showed that β-turn content was between 20 and 40% of the structure at any given time.8 Other studies on the VPGVG peptide with a three-repeat units and in polymeric form showed its structural preference for forming type II β-turns.7,31,37,38 The overall hydrophobic structure of the VPGVG-based peptides has been thought to allow the formation of the β-turn. Molecular dynamics studies have shown similar results and give an estimated type II β-turn content at 10–20% for VPGVG.33 This is not surprising given the evidence that the PGX sequence is known to favor the type II β-turn. The X residue determines how stable the β-turn is versus forming a PPII structure. For PGV in VPGVG, valine is likely to form more PPII than leucine or alanine in the same structural position, which tend to favor β-turns.39,40
CD spectra from previous studies with SynB1-ELP show a negative peak at 195 nm, a positive peak at 210 nm, and a second negative peak at 225 nm. This combination has been shown to occur in VPGVG polypeptides with extended chain and type II-β-turns when assigning them to a class B β-turn spectra.5,7,41 Other studies have shown that class C′ assignments are more relevant when dealing with combinations of type II β-turns, extended chains, and type I(III) β-turns as the change in spectra can be dealt with as a combination of Gaussian bands rather than by individual peaks and valleys.42,43 This is especially pertinent given that other major structural components exist in SynB1-ELP, namely, VPGGG and VPGAG, in which similar peptide sequences have been shown to exhibit structures other than type II β-turns and extended chains (Table 1).34,44−53
The peptide VGGG has been shown to form type I(III) β-turns when heated above its transition temperature, which is expected given that the GGG and PGGG structures were known to form a 3(10)-helix (type III β-turns are one turn of a 3(10)-helix) often next to isolated β-strands or α-helixes depending on the surrounding amino acid sequence.44,45 Polyglycine has also been shown to form a type of PPII-helix known as the polyglycine II helix, which has the same bond angles as the 3(10)-helix but exhibits a lower degree of hydrogen bonding for stabilization.46 The higher degree of forming PPII helixes has been established in repeating elastin-like domains containing proline.34,38 While proline is regarded as a helix-breaker because of its restricted bond angles and limited ability to participate in hydrogen bonding,47 sequences containing repeats of proline have been shown to act as stabilizers for β-turns and PPII helixes, especially in the VPG and PG amino acid sequences.34,38,44,48
Similar to VPGGG, VPGAG is entirely hydrophobic, and the CH3 side group in alanine has been shown to stabilize the helical structures in VPGAG.49−51 Human tropoelastin peptide segments (exon 3, 7, 30) that are entirely hydrophobic and contain G, A, L, and P amino acids have been shown to take on the form of a PPII-helix. The exon 30 sequence also contains a small amount of type I(III) β-turns.52 Polypeptides that have nonpolar side chains such as alanine and valine have been shown to take the form of a 3(10)-helix as long as the hydrophobic sequence is at least three to eight amino acids long.49,53
We have interpreted the FT-IR spectra for SynB1-ELP (Table 1) considering that type II β-turns, extended chains, type I(III) β-turns, PPI, PPII, and 3(10)-helixes can be formed as detailed above. Extended strands have been shown to occur predominantly in proteins that have a high number of proline groups and in mobile amino acid segments that are typically four to five amino acid residues in length.54 The peak at 1616 cm–1 in the SynB1-ELP FT-IR spectrum is likely to be that of an extended strand.25,26,31,32 γ Turns and bifurcated hydrogen bonds have been shown to occur at wavenumbers lower than 1630 cm–1 due to the strong hydrogen bonding.27 We attribute the peak at 1624 cm–1 to these secondary structures. Figure 3A shows a strong positive correlation between the peak at 1616 and 1624 cm–1 from 1 to 3.5 h that could indicate the presence of extended strands, γ turns, and bifurcated hydrogen bonds in SynB1-ELP (Table 1). This relationship goes away after 5 h of incubation time, indicating the disappearance of these secondary structures.
The peak at 1657 cm–1 is traditionally assigned to a highly ordered structure in the form of an α-helix; however, we assign this peak to β-turns (Table 1).21,22 The likelihood that an ELP molecule forms an α-helix structure is extremely low because of the regular proline repeat groups along the polypeptide backbone that prevent a repeating α-helix structure.24,47 In addition, a previous study analyzed an ELP with 40 repeat units using 1H–15N heteronuclear single quantum coherence (HSQC) NMR and found a low probability of forming α-helix structures.6 CD measurements of SynB1-ELP indicate the presence of β-turns.7,35 β-turns are not expected to have constant amide I frequencies in FT-IR as the peaks are expected to shift heavily based on the polypeptide amino acid sequence.27 β-Turns have been shown to occur at wavenumbers as low as 1628 cm–1 and as high as 1694 cm–1. The lower frequencies arise from the introduction of proline residues into the peptide sequence and from a strongly hydrogen-bonded amide carbonyl group. The higher frequency peaks occur from amide carbonyl groups with almost no hydrogen bonding.27 Above its transition temperature at 65 °C, the type I(III) β-turns and 3(10)-helixes decrease to 10–20%, type II β-turns increase to 40–45%, and extended chains decrease to 40–50%.42
In FT-IR spectra of polypeptides and peptide sequences, similar to β-turns, the PPI and PPII amide I bands typically appear anywhere from 1620 to 1689 cm–1. The peak assignments in this article for SynB1-ELP focus on FT-IR assignments made to similar peptides and polypeptides (Table 1). These structures include GGG, APG, poly(PG), poly(P), and ALGGGALG.34−36,46 These peptides typically exhibit one to three peaks based on the amino acid residue diversity of the molecule and the variation in hydrogen bonding throughout the structure due to inter- and intramolecular interactions. The central peak locations for PPI and PPII are 1620–1630 cm–1 when the sample has a large amount of hydrogen bonding (usually from the presence of solvent), 1640–1653 or 1661–1668 cm–1 for moderate hydrogen bonding and some bound solvent, and 1685–1689 cm–1 when composed of weaker intermolecular hydrogen bonding. The CD spectra for SynB1-ELP has been shown to contain a negative band at 197 nm and a positive band at 212 nm for the PPI helix and a negative band at 195 nm and a positive band at 218 nm for the PPII-helix.52,55,56 This presence of PPI and PPII has been used to explain the shift in the SynB1-ELP CD spectral peaks from 196 and 214 nm at 5 °C to 199 and 210 nm at 45 °C, respectively, as the disappearance of PPII in favor of type II β-turns and PPI.7 These observations indicate that PPII may play a more primary role in keeping SynB1-ELP solvated below its transition temperature and may remain present to a lesser extent above the transition temperature.35,57 PPII structures in elastin-based peptides have been shown to exist when higher amounts of proline and glycine are present. These structures have been shown to contribute to an increase in backbone hydration of the polypeptide, which in turn is necessary to form PPII helixes and to confer ELP’s solubility at lower temperatures.44 Hydrogen-bonded turns have been shown to occur more often with decreasing backbone hydration and thus with a decrease in the amount of proline and glycine present in the polypeptide.44 The PPII dissolution is thermally driven and reversible in polyproline and short elastin-based peptide sequences.35,52,55,57 CD measurements have shown that for polyproline, the PPI structures give way to an increase in the amount of PPII-helix as the temperature is increased.35,56,57 Peptide sequences of elastin have been shown to have the opposite behavior: with an increase in temperature, the amount of PPII decreases52,55 and can lead to an increase in the amount of PPI rather than just from the appearance of type II β-turns for SynB1-ELP. The structure and degree of hydrogen bonding are too similar for separate detection using FT-IR. CD measurements are currently the only detection method that has been used to differentiate them completely.52,55,56 Therefore, we have used previous CD measurements of SynB1-ELP and peak correlations to justify our peak assignments.
If the β-spiral exists for SynB1-ELP, its characteristic peaks are likely to be observed at 1616 and 1656 cm–1 as predicted by the calculations performed by Serrano et al. for VPGVG, as these peaks are associated with type II β-turns and extended chain conformations.24 These two structures are thought to contribute to the formation of the β-spiral, but their presence does not mean the two structures cooperate for it to occur, especially since the two structures have negative correlations in FT-IR (Figure 4A). An NMR study on the (VPGVG)3 peptide showed that there was a constant interchange between the formation of β-turns and other structures that occurred too rapidly to form a stable environment for the formation of the β-spiral, thus precluding it from existing as one of the significant structures.8
3.2. FT-IR Peak Correlations
Prevailing correlations between the peaks at 1635, 1657, and 1695 cm–1 can be seen at all time points (Figure 3). Two peptide sequences, GPLG and GPGG, exhibit three peaks near these peaks seen in our study.58,59 These peptide sequences have type II β-turns, a proline residue, and a glycine in the C3 position.58,59 A previous study using NMR measurements of an ELP containing VPGVG found that the average number of β-turns present in the molecule to be 20–40%, and molecular dynamics simulations place that number between 10 and 20%.8,33 As can be calculated from Table 1, the average percentage of total β-turns (peaks 1635, 1657, 1666, and 1695 cm–1) that were present from 1 to 12 h using FT-IR was ∼42%, which approximates the NMR study.
The peaks at 1647, 1666, and 1680 cm–1 that exhibited positive correlations from 1 to 3.5 h were then assigned to PPII. During this time, the highest amount of water was present (Figure 1), which is needed to stabilize the PPII-helix. As the water evaporates and the aggregates mature over the extended incubation period, the PPII correlations go away, and new stronger correlations are established between 1624, 1647, and 1680 cm–1. Based on the previous data35,36,44,46 and the positive correlations between the peaks at 1624, 1647, and 1680 cm–1 from 3.5 to 12 h, we assigned these peaks to the PPI and PPII structures. These correlations indicate the disappearance of PPII and the formation of PPI. This structural transition could be one of the driving forces behind the LCST behavior of ELP, as PPII structures must maintain a hydrated state at lower temperatures to stabilize themselves in solution and thus makes PPII soluble. PPI is not as hydrated and is known to precipitate and form aggregates during the PPII to PPI transition.35,52,55,57
From 1 to 3.5 h, the 1666 cm–1 peak is weakly correlated to both 1635 and 1688 cm–1 peaks, the first being the intramolecular hydrogen bonding peak of type I(III) β-turns and the second being PPII.27,34−36,46 At 3.5–6 h, there are still two subpeaks present: one demonstrates a positive correlation between 1666 and 1635 cm–1, indicating type I(III) β-turns, and the other shows a strong positive correlation between 1666 and 1656 cm–1, indicating a 3(10)-helix with a low number of turns.21,27 Peak splitting is known to occur in helices when there are only a few amino acid residues present in the helix due to resonance frequency splitting from dipole coupling;60 the most common example of this is from the α-helix, which contains 3.6 amino acid residues per turn instead of 3 for the 3(10)-helix.
3.3. Amide II/I Peak Area Ratio
The amide I peak in the FT-IR spectrum represents the stretching vibrations of the carbonyl and carbon–nitrogen bonds and allows secondary interaction measurements. Its position is located between the spectral range of 1600 and 1700 cm–1 and is a combination of Gaussian bands.19−22 The amide II peak is a more complex peak to interpret as it is comprised of many bending and stretching vibrations.42 More recent studies using density functional theory calculations for peptides indicate that the amide II peak is primarily composed of hydrogen bonding that occurs with the NH group in the peptide residues.61 While deconvolution of the amide I peak is the most successful way to interpret the secondary structure, it has been shown that the ratio of the amide II peak area to the amide I peak area is related to a change in the secondary structure of a protein.62 More importantly, the amide II to amide I peak area ratio is proportional to the amount of hydrogen bonding that occurs in the carbonyl to the secondary amine structures of the peptide repeats.61 While the amide II to amide I peak area ratio does not provide a direct interpretation of the secondary structures, it can indicate if the structure truly changes over time by providing an internal reference from both peaks.
Through correlations to specific deconvoluted subpeaks in the amide I peak, secondary structures can be identified that significantly contribute to a change in the amide II to amide I peak area ratio (Figure 5A). Two secondary structures were identified that have an impact on the change in the amide II to amide I peak area ratio. β-Turns showed a weak negative correlation to the amide II to amide I peak area ratio (Figure 5B): as the number of β-turns increase, the peak area ratio decreases. The amide II to amide I peak area ratio decreases from 1 to 3.5 h, and at the same time, the aggregates increase in size (Figures 2 and 5A). This is not surprising given the reliance on β-turns for the aggregates to grow in size (Table 2). Extended strands were found to have the opposite effect and showed a moderate positive correlation to the amide II to amide I peak area ratio (Figure 5C). This inherently makes sense as the extended strands are present in isolated chains. As the chains become isolated, they interact more with solvent molecules; the area of the amide II peak should increase from an increase in the secondary interactions leading to an increase in the amide II to amide I peak area ratio. The amide II to amide I peak area ratio is seen to increase from 6 to 9 h when the aggregates collapse onto the surface of the silica (Figures 2 and 5A) and is also seen to increase when an increase in aggregate size begins to rely on 3(10)-helices instead of β-turns (Table 2). Figure 4A shows that a strong negative relationship exists between β-turns and extended strands and demonstrates that, for one structure to increase in percentage, the other must decrease. These relationships establish a pattern in secondary structures in the dynamic behavior of SynB1-ELP when desiccated on silica. From 1 to 3.5 h, the aggregates grow in size from β-turns, from 6 to 9 h the aggregates decrease in size due to extended chains, and finally, the aggregates regrow at 12 h from 3(10) helices.
3.4. Correlations of Secondary Structures to Water Content and Aggregate Size
Type II β-turns were shown to be the primary driving force for the formation of larger aggregates from 1 to 3.5 h; however, the increase in the number of β-turns was not correlated to a decrease in the amount of water present on the sample surface. This could mean that the early formation of β-turns is temperature driven, with the temperature for this experiment held constant above the LCST of the polypeptide. From 6 to 12 h, the aggregate size increase shifts from relying on type II β-turns to the 3(10)-helix and was shown to be a direct correlation with the decrease in water content of the sample. This shift in secondary structure reliance on aggregate size could explain why the aggregates grow in size for the initial period from 1 to 3.5 h before they collapse from 6 to 9 h and reform at 12 h. Overall, the main contributor to a larger aggregate size is the type II β-turn, which is thought to occur from the hydrophobicity of the polypeptide chain as the temperature of the solution is raised to its LCST. As the turns form, they begin to associate with nearby ELP molecules through hydrophobic turn–turn interactions that stabilize the coalesced aggregates in solution.5−7 This process likely breaks down around 6 h as there may not be enough water present to support the formation of turns, thus giving way to stabilization from the 3(10)-helix. Peptide hydrogen bonds, especially in α-helixes, are known to increase or maintain the strength of their hydrogen bonds as water is removed from the system, which could be happening to the type I(III) β-turns to cause them to form 3(10)-helices.63,64
4. Conclusions
The amide I peak was successfully deconvoluted using the second derivative method and eight underlying peaks were found at 1616, 1624, 1635, 1647, 1657, 1666, 1680, and 1695 cm–1. Analysis of past SynB1-ELP and similar peptide sequences showed the presence of not only type II β-turns and extended chains but also of PPI, PPII-helix, and type I(III) β-turns/3(10)-helix. The positive correlation between peaks linked them together and aided in their identification. Correlations analysis indicated that the primary driver of the increase in aggregate size was the formation of type II β-turns at earlier time points. As the water content decreased, reliance on 3(10)-helices for aggregate formation occurred at later time points. This study examines the role secondary structures play during desiccation of SynB1-ELP and can be used to predictably control the formation of ELP coatings.
5. Materials and Methods
5.1. Synthesis of SynB1-ELP
The methods for SynB1-ELP expression and purification have been previously described.29 Briefly, the SynB1-ELP was synthesized using a pET25b vector encoded to produce a repeating structure of MRGGPLSYSRRRFSTSTGR-GPGVG(VPG[V5G3A2]G)150WPGSGGC (MW = 61 739 g/mol) and transformed into BLR (DE3) Escherichia coli. Purification was achieved by repeated thermocycling of the cell lysate to isolate the ELP.
5.2. Aggregation of ELP on Silica
ELP was dissolved in phosphate-buffered saline (PBS) at a concentration of 3 mg/mL, and 10 μL of the sample was placed onto a fused silica disk (1.5 in. diameter; Thorlabs, Newton, New Jersey). The silica disk was then enclosed with a 35 mm Petri dish to prevent the collection of any dust and placed into an oven at 50 °C until the desired time point was reached. We described these methods in detail in our previous work.29
5.3. Fourier-Transform Infrared-Attenuated Total Reflectance Spectroscopy
A PerkinElmer Spectrum 100 instrument was used to analyze the samples. FT-IR spectroscopy was performed using a single reflectance attenuated total reflectance accessory utilizing a diamond crystal. Samples were secured on the ATR accessory with a clamping pressure of 110 psi, and 16 scans were taken in the spectral range of 4000–650 cm–1 with a spectral resolution of 2 cm–1. Measurements were performed on three separate samples. A background spectral series was obtained to cancel out any effects from the presence of atmospheric gases and water. Automatic baseline correction was performed using PerkinElmer Spectrum 9. Water content of the silica substrate was measured by placing a 10 μL drop of PBS solution onto the silica disc. The disc was placed into a 60 mm cell culture dish to prevent contamination from dust and placed into a Thermo Lindberg/Blue M digital oven (Thermo Scientific, Waltham, MA) set at 50 °C. The disc was removed from the oven at specified time points and a spectral measurement was obtained using FT-IR ATR as described above. Percent water content was calculated by isolating the hydroxyl peak from 2600 to 3700 cm–1 and normalizing the baseline for all spectra to that of the silica disc. Peak height using absorbance at 3200 cm–1 was used to calculate the percent water content, with the bare silica disk taken as 0% and the 0 h time point measurement as 100%.
5.4. Amide I Peak Deconvolution
Figure 6A shows the full FT-IR spectrum of the SynB1-ELP aggregated on the silica surface at 9 h. PeakFit version 4.12 (Systat Software Inc.) was used to deconvolute the amide I peak isolated in the spectral range of 1590–1715 cm–1, and a linear baseline was automatically fit. The automatic second derivative method in the PeakFit software was used to identify peak positions. Gaussian bands were fit to the peak positions in PeakFit to determine the relative abundance of secondary structures present. All amide I peak fits had R2 values of 0.99 (Figure 6B). The average composition was calculated by dividing the individual deconvoluted peak area by the total amide I peak area multiplied by 100. The mean absolute percent error was calculated for each region in amide I containing an underlying peak to demonstrate that the selected subpeaks did not occur from noise in the spectra.
Figure 6.

(A) Full FT-IR spectra taken at the 9 h time point. The amide I and amide II peaks located at 1590–1715 and 1510–1580 cm–1, respectively. (B) The deconvoluted amide I peak at 9 h demonstrating a good agreement between the original and deconvoluted peaks. The mean absolute percent error calculated for each region using the average of three spectra from three independent samples demonstrates that the selected subpeaks did not occur from noise in the spectra.
5.5. Statistical Analyses
Linear fits were used to determine the Pearson correlation between two variables. Correlation strengths were grouped into four bins based on the R2 values. Values of 0–0.29 were indicative of no correlation, 0.3–0.49 were deemed weak correlations, 0.5–0.69 were deemed moderate correlations, and 0.7–1 indicated a strong correlation. Results are reported as the mean ± 95% confidence interval.
Acknowledgments
This work made use of instruments in the Department of Biomedical Materials Science Shared Equipment Facility.
This work was funded by the National Institutes of Health (NIH; R01EB020006).
The authors declare no competing financial interest.
References
- Cho Y.; Zhang Y.; Christensen T.; Sagle L. B.; Chilkoti A.; Cremer P. S. Effects of Hofmeister Anions on the Phase Transition Temperature of Elastin-like Polypeptides. J. Phys. Chem. B 2008, 112, 13765–13771. 10.1021/jp8062977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay J. A.; Callahan D. J.; Fitzgerald K. N.; Chilkoti A. Quantitative Model of the Phase Behavior of Recombinant pH-responsive Elastin-like Polypeptides. Biomacromolecules 2010, 11, 2873–2879. 10.1021/bm100571j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreher M.; Raucher D.; Balu N.; Michael O.; Chilkoti A. Evaluation of an Elastin-like Polypeptide–Doxorubicin Conjugate for Cancer Therapy. J. Controlled Release 2003, 91, 31–43. 10.1016/s0168-3659(03)00216-5. [DOI] [PubMed] [Google Scholar]
- Meyer D. E.; Shin B. C.; Kong G. A.; Dewhirst M. W.; Chilkoti A. Drug Targeting Using Thermally Responsive Polymers and Local Hyperthermia. J. Controlled Release 2001, 74, 213–224. 10.1016/s0168-3659(01)00319-4. [DOI] [PubMed] [Google Scholar]
- Lyons D.; Bidwell G. L.; Kramer W. H.; Raucher D.; Correia J. J. Biophysical Characterization of an Elastin-Like-Polypeptide. Biophys. J. 2011, 100, 540a 10.1016/j.bpj.2010.12.3148. [DOI] [Google Scholar]
- Zhang Y.; Zai-Rose V.; Price C. J.; Bidwell G. L.; Correia J. J.; Fitzkee N. C. Modeling the Early Stages of Aggregation in Disordered Elastin-Like Proteins. Biophys. J. 2017, 112, 481a 10.1016/j.bpj.2016.11.2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zai-Rose V.; West S. J.; Kramer W. H.; Bishop G. R.; Lewis E. A.; Correia J. J. Effects of Doxorubicin on the Liquid-Liquid Phase Change Properties of Elastin-Like Polypeptides. Biophys. J. 2018, 115, 1431–1444. 10.1016/j.bpj.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichheld S. E.; Muiznieks L. D.; Keeley F. W.; Sharpe S. Direct Observation of Structure and Dynamics during Phase Separation of an Elastomeric Protein. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, E4408–E4415. 10.1073/pnas.1701877114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts S.; Dzuricky M.; Chilkoti A. Elastin-like Polypeptides as Models of Intrinsically Disordered Proteins. FEBS Lett. 2015, 589, 2477–2486. 10.1016/j.febslet.2015.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauscher S.; Baud S.; Miao M.; Keeley F. W.; Pomès R. Proline and Glycine Control Protein Self-Organization into Elastomeric or Amyloid Fibrils. Structure 2006, 14, 1667–1676. 10.1016/j.str.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Huber M. C.; Schreiber A.; von Olshausen P.; Varga B. R.; Kretz O.; Joch B.; Barnert S.; Schubert R.; Eimer S.; Kele P.; et al. Designer Amphiphilic Proteins as Building Blocks for the Intracellular Formation of Organelle-like Compartments. Nat. Mater. 2015, 14, 125–132. 10.1038/nmat4118. [DOI] [PubMed] [Google Scholar]
- Pastuszka M. K.; Okamoto C. T.; Hamm-Alvarez S. F.; MacKay J. A. Flipping the Switch on Clathrin-Mediated Endocytosis Using Thermally Responsive Protein Microdomains. Adv. Funct. Mater. 2014, 24, 5340–5347. 10.1002/adfm.201400715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew MacKay J.; Chen M.; McDaniel J. R.; Liu W.; Simnick A. J.; Chilkoti A. Self-Assembling Chimeric Polypeptide–Doxorubicin Conjugate Nanoaggregates That Abolish Tumours after a Single Injection. Nat. Mater. 2009, 8, 993–999. 10.1038/nmat2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidwell G. L. III; Davis A. N.; Fokt I.; Priebe W.; Raucher D. A Thermally Targeted Elastin-like Polypeptide-Doxorubicin Conjugate Overcomes Drug Resistance. Invest. New Drugs 2007, 25, 313–326. 10.1007/s10637-007-9053-8. [DOI] [PubMed] [Google Scholar]
- Massodi I.; Bidwell G. L.; Raucher D. Evaluation of Cell Penetrating Peptides Fused to Elastin-like Polypeptide for Drug Delivery. J. Controlled Release 2005, 108, 396–408. 10.1016/j.jconrel.2005.08.007. [DOI] [PubMed] [Google Scholar]
- McHale M. K.; Setton L. A.; Chilkoti A. Synthesis and in Vitro Evaluation of Enzymatically Cross-Linked Elastin-Like Polypeptide Gels for Cartilaginous Tissue Repair. Tissue Eng. 2005, 11, 1768–1779. 10.1089/ten.2005.11.1768. [DOI] [PubMed] [Google Scholar]
- Amruthwar S. S.; Janorkar A. V. In Vitro Evaluation of Elastin-like Polypeptide–Collagen Composite Scaffold for Bone Tissue Engineering. Dent. Mater. 2013, 29, 211–220. 10.1016/j.dental.2012.10.003. [DOI] [PubMed] [Google Scholar]
- Amruthwar S. S.; Janorkar A. V. Preparation and Characterization of Elastin-like Polypeptide Scaffolds for Local Delivery of Antibiotics and Proteins. J. Mater. Sci.: Mater. Med. 2012, 23, 2903–2912. 10.1007/s10856-012-4749-5. [DOI] [PubMed] [Google Scholar]
- Newton R. P. Methods in Molecular Biology, Volume 17: Spectroscopic Methods and Analyses NMR, Mass Spectrometry, and Metalloprotein. Rapid Commun. Mass Spectrom. 1994, 8, 224. 10.1002/rcm.1290080220. [DOI] [Google Scholar]
- Micsonai A.; Wien F.; Kernya L.; Lee Y.-H.; Goto Y.; Réfrégiers M.; Kardos J. Accurate Secondary Structure Prediction and Fold Recognition for Circular Dichroism Spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, E3095–E3103. 10.1073/pnas.1500851112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong J.; Yu S. Fourier Transform Infrared Spectroscopic Analysis of Protein Secondary Structures. Acta Biochim. Biophys. Sin. 2007, 39, 549–559. 10.1111/j.1745-7270.2007.00320.x. [DOI] [PubMed] [Google Scholar]
- Singh B.Infrared Analysis of Peptides and Proteins: Principles and Applications; American Chemical Society: Washington, 2000; pp 2–37. [Google Scholar]
- Adochitei A.; Drochioiu G. Rapid Characterization of Peptide Secondary Structure by FT-IR Spectroscopy. Rev. Roum. Chim. 2011, 56, 783–791. [Google Scholar]
- Serrano V.; Liu W.; Franzen S. An Infrared Spectroscopic Study of the Conformational Transition of Elastin-Like Polypeptides. Biophys. J. 2007, 93, 2429–2435. 10.1529/biophysj.106.100594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauerer A.; Lee G. Changes in the Amide I FT-IR Bands of Poly-l-Lysine on Spray-Drying from α-Helix, β-Sheet or Random Coil Conformations. Eur. J. Pharm. Biopharm. 2006, 62, 131–142. 10.1016/j.ejpb.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Jackson M.; Mantsch H. H. The Use and Misuse of FTIR Spectroscopy in the Determination of Protein Structure. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 95–120. 10.3109/10409239509085140. [DOI] [PubMed] [Google Scholar]
- Vass E.; Hollósi M.; Besson F.; Buchet R. Vibrational Spectroscopic Detection of Beta- and Gamma-Turns in Synthetic and Natural Peptides and Proteins. Chem. Rev. 2003, 103, 1917–1954. 10.1021/cr000100n. [DOI] [PubMed] [Google Scholar]
- Wenzel R. N. Surface Roughness and Contact Angle. J. Phys. Chem. A 1949, 53, 1466–1467. 10.1021/j150474a015. [DOI] [Google Scholar]
- Cobb J. S.; Zai-Rose V.; Correia J. J.; Janorkar A. V. Visualization of the Temperature Dependent Rearrangement of SynB1 Elastin-like Polypeptide on Silica Using Scanning Electron Microscopy. Anal. Biochem. 2018, 558, 41–49. 10.1016/j.ab.2018.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N.; Ramakrishnan C.; Srinivasan N. Stranded in Isolation: Structural Role of Isolated Extended Strands in Proteins. Protein Eng., Des. Sel. 2003, 16, 331–339. 10.1093/protein/gzg046. [DOI] [PubMed] [Google Scholar]
- Shivu B.; Seshadri S.; Li J.; Oberg K. A.; Uversky V. N.; Fink A. L. Distinct β-Sheet Structure in Protein Aggregates Determined by ATR–FTIR Spectroscopy. Biochemistry 2013, 52, 5176–5183. 10.1021/bi400625v. [DOI] [PubMed] [Google Scholar]
- Sen-Britain S.; Hicks W.; Hard R.; Gardella J. Jr.. The Mechanism of Secondary Structural Changes in Keratinocyte Growth Factor During Uptake and Release from a Hydroxyethyl(Methacrylate) Hydrogel Revealed by 2D Correlation Spectroscopy. 2018, arXiv:1808.03670. arXiv.org e-Print archive. https://arxiv.org/abs/1808.03670.
- Rauscher S.; Pomès R. The Liquid-Like Structure of Elastin. Biophys. J. 2018, 114, 369a 10.1016/j.bpj.2017.11.2043. [DOI] [Google Scholar]
- Martino M.; Bavoso A.; Guantieri V.; Coviello A.; Tamburro A. On the Occurrence of Polyproline II Structure in Elastin. J. Mol. Struct. 2000, 519, 173–189. 10.1016/s0022-2860(99)00299-9. [DOI] [Google Scholar]
- Dukor R. K.; Keiderling T. A. Mutarotation Studies of Poly-L-Proline Using FTIR, Electronic and Vibrational Circular Dichroism. Biospectroscopy 1998, 2, 83–100. . [DOI] [Google Scholar]
- Blout E. R.; Doyle B. B.; Traub W.; Lorenzi G. P. Conformational Investigations on the Polypeptide and Oligopeptides with the Repeating Sequence L-Alanyl-L-Prolylglycine. Biochemistry 1971, 10, 3052–3060. 10.1021/bi00792a011. [DOI] [PubMed] [Google Scholar]
- Muiznieks L.; Weiss A.; Keeley F. Structural Disorder and Dynamics of Elastin. Biochem. Cell Biol. 2010, 88, 239–250. 10.1139/O09-161. [DOI] [PubMed] [Google Scholar]
- Tamburro A. M.; Bochicchio B.; Pepe A. Dissection of Human Tropoelastin: Exon-By-Exon Chemical Synthesis and Related Conformational Studies. Biochemistry 2003, 42, 13347–13362. 10.1021/bi034837t. [DOI] [PubMed] [Google Scholar]
- Brahmachari S. K.; Ananthanarayanan V. S. Beta-Turns in Nascent Procollagen Are Sites of Posttranslational Enzymatic Hydroxylation of Proline. Proc. Natl. Acad. Sci. U.S.A. 1979, 76, 5119–5123. 10.1073/pnas.76.10.5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamburro A.; Guantieri V. Folded, B-Turns and Collagenlike Conformations of -Gly-Pro-and -Pro-Gly-Sequences in Synthetic Polytripeptides. Biopolymers 1984, 617–621. 10.1002/bip.360230403. [DOI] [Google Scholar]
- Sauer K.Methods in Enzymology: Biochemical Spectroscopy; London Academic Press, 1995; Vol. 246, pp 34–71. [Google Scholar]
- Perczel A.; Hollosi M.; Sandor P.; Fasman G. D. The Evaluation of Type I and Type II β-Turn Mixtures. Int. J. Pept. Protein Res. 2009, 41, 223–236. 10.1111/j.1399-3011.1993.tb00330.x. [DOI] [PubMed] [Google Scholar]
- Reiersen H.; Clarke A. R.; Rees A. R. Short Elastin-like Peptides Exhibit the Same Temperature-Induced Structural Transitions as Elastin Polymers: Implications for Protein Engineering. J. Mol. Biol. 1998, 283, 255–264. 10.1006/jmbi.1998.2067. [DOI] [PubMed] [Google Scholar]
- Rauscher S.; Baud S.; Miao M.; Keeley F. W.; Pomès R. Proline and Glycine Control Protein Self-Organization into Elastomeric or Amyloid Fibrils. Structure 2006, 14, 1667–1676. 10.1016/j.str.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Pal L.; Dasgupta B.; Chakrabarti P. 310-Helix Adjoining α-Helix and β-Strand: Sequence and Structural Features and Their Conservation. Biopolymers 2005, 78, 147–162. 10.1002/bip.20266. [DOI] [PubMed] [Google Scholar]
- Taga K.; Sowa M. G.; Wang J.; Etori H.; Yoshida T.; Okabayashi H.; Mantsch H. H. FT-IR Spectra of Glycine Oligomers. Vib. Spectrosc. 1997, 14, 143–146. 10.1016/s0924-2031(96)00061-6. [DOI] [Google Scholar]
- Li S. C.; Goto N. K.; Williams K. A.; Deber C. M. Alpha-Helical, but Not Beta-Sheet, Propensity of Proline Is Determined by Peptide Environment. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 6676–6681. 10.1073/pnas.93.13.6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H.; Grimsley G. R.; Razvi A.; Scholtz J. M.; Pace C. N. Increasing Protein Stability by Improving Beta-Turns. Proteins: Struct., Funct., Bioinf. 2009, 77, 491–498. 10.1002/prot.22509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khrustalev V. V.; Barkovsky E. V.; Khrustaleva T. A. The Influence of Flanking Secondary Structures on Amino Acid Content and Typical Lengths of 3/10 Helices. Int. J. Proteomics 2014, 2014, 360230 10.1155/2014/360230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoret L. M.; Sauer R. T. Tolerance of a Protein Helix to Multiple Alanine and Valine Substitutions. Folding Des. 1998, 3, 119–126. 10.1016/s1359-0278(98)00017-0. [DOI] [PubMed] [Google Scholar]
- Heitmann B.; Job G. E.; Kennedy R. J.; Walker S. M.; Kemp D. S. Water-Solubilized, Cap-Stabilized, Helical Polyalanines: Calibration Standards for NMR and CD Analyses. J. Am. Chem. Soc. 2005, 127, 1690–1704. 10.1021/ja0457462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochicchio B.; Aït-Ali A.; Tamburro A. M.; Alix A. J. P. Spectroscopic Evidence Revealing Polyproline II Structure in Hydrophobic, Putatively Elastomeric Sequences Encoded by Specific Exons of Human Tropoelastin. Biopolymers 2004, 73, 484–493. 10.1002/bip.10552. [DOI] [PubMed] [Google Scholar]
- Yoder G.; Polese A.; Silva R. A. G. D.; Formaggio F.; Crisma M.; Broxterman Q. B.; Kamphuis J.; Toniolo C.; Keiderling T. A. Conformational Characterization of Terminally Blockedl-(AMe)Val Homopeptides Using Vibrational and Electronic Circular Dichroism. 310-Helical Stabilization by Peptide–Peptide Interaction. J. Am. Chem. Soc. 1997, 119, 10278–10285. 10.1021/ja971392l. [DOI] [Google Scholar]
- Vass E.; Hollosi M.; Besson F.; Buchet R. Vibrational Spectroscopic Detection of Beta- and Gamma-Turns in Synthetic and Natural Peptides and Proteins. Chem. Rev. 2003, 103, 1917. 10.1021/cr000100n. [DOI] [PubMed] [Google Scholar]
- Tamburro A. M.; Bochicchio B.; Pepe A. Dissection of Human Tropoelastin: Exon-By-Exon Chemical Synthesis and Related Conformational Studies. Biochemistry 2003, 42, 13347–13362. 10.1021/bi034837t. [DOI] [PubMed] [Google Scholar]
- Kuemin M.; Engel J.; Wennemers H. Temperature-Induced Transition between Polyproline I and II Helices: Quantitative Fitting of Hysteresis Effects. J. Pept. Sci. 2010, 16, 596–600. 10.1002/psc.1245. [DOI] [PubMed] [Google Scholar]
- Harrington W. F.; Sela M. Studies on the Structure of Poly-l-Proline in Solution. Biochim. Biophys. Acta 1958, 27, 24–41. 10.1016/0006-3002(58)90289-0. [DOI] [PubMed] [Google Scholar]
- Lagant P.; Vergoten G.; FLeury G.; Loucheux-Lefebvre M.-H. Raman Spectroscopy and Normal Vibrations of Peptides Characteristic Normal Modes of a Type II B-Turn. Eur. J. Biochem. 1984, 139, 137–148. 10.1111/j.1432-1033.1984.tb07987.x. [DOI] [PubMed] [Google Scholar]
- Lagant P.; Vergoten G.; Fleury G.; Loucheux-Lefebvre M. Vibrational Normal Modes of Folded Prolyl-Containing Peptides Application to Turns. Eur. J. Biochem. 1984, 139, 149–154. 10.1111/j.1432-1033.1984.tb07988.x. [DOI] [PubMed] [Google Scholar]
- Nevskaya N. A.; Chirgadze Y. N. Infrared Spectra and Resonance Interactions of Amide-I and II Vibrations of Alpha-Helix. Biopolymers 1976, 15, 637–648. 10.1002/bip.1976.360150404. [DOI] [PubMed] [Google Scholar]
- Myshakina N.; Ahmed Z.; Asher S. Dependence of Amide Vibrations on Hydrogen Bonding. J. Phys. Chem. B 2008, 112, 11873–11877. 10.1021/jp8057355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georget D. M. R.; Belton P. S. Effects of Temperature and Water Content on the Secondary Structure of Wheat Gluten Studied by FTIR Spectroscopy. Biomacromolecules 2006, 7, 469–475. 10.1021/bm050667j. [DOI] [PubMed] [Google Scholar]
- Sheu S.-Y.; Yang D.-Y.; Selzle H. L.; Schlag E. W. Energetics of Hydrogen Bonds in Peptides. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 12683–12687. 10.1073/pnas.2133366100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon D. A.; Dobbs K. D.; Valentini J. J. Amide-Water and Amide-Amide Hydrogen Bond Strengths. J. Phys. Chem. C 1994, 98, 13435–13439. 10.1021/j100102a001. [DOI] [Google Scholar]