

Abstract
Heparan sulfate (HS) mediates a wide range of protein binding interactions key to normal and pathological physiology. Though liquid chromatography coupled with mass spectrometry (LC-MS) based disaccharide composition analysis is able to profile changes in HS composition, the heterogeneity of modifications and the labile sulfate group present major challenges for liquid chromatography tandem mass spectrometry (LC-MS/MS) sequencing of the HS oligosaccharides that represent protein binding determinants. Here, we report online LC-MS/MS sequencing of HS oligosaccharides using hydrophilic interaction liquid chromatography (HILIC) and negative electron transfer dissociation (NETD). A series of synthetic HS oligosaccharides varying in chain length (tetramers and hexamers), number of sulfate groups (3–7), sulfate patterns (sulfate positional isomers), and uronic acid epimerization (epimers) were separated and sequenced. The LC elution order of isomeric compounds was associated with their fine structure. The application of an online cation exchange device (ion suppressor) enhanced the precursor charge states, and the subsequent NETD produced abundant glycosidic fragments, allowing the characterization of both lowly sulfated and highly sulfated HS oligosaccharides. Furthermore, the diagnostic cross-ring ions differentiated the 6-O sulfation and 3-O sulfation, allowing unambiguous structural assignment. Collectively, this LC-NETD-MS/MS method is a powerful tool for sequencing of heterogeneous HS mixtures and is applicable for the differentiation of both isomers and epimers, for the characterization of saccharide mixtures with a varying extent of sulfation and even for the determination of both predominant and rare modification motifs. Thus, LC-NETD-MS/MS has great potential for further application to biological studies.
Graphical Abstract
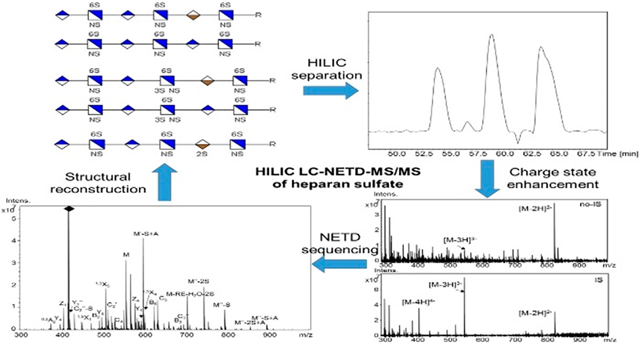
Heparan sulfate (HS) is a linear polysaccharide composed of alternating glucosamine (GlcN) and uronic acid (HexA) residues.1 As a kind of protein post-translational modification, HS mediates a wide range of key biochemical and developmental processes.2 Although a few function-specific HS motifs have been elucidated,3 HS sequences responsible for numerous cellular functions remain unknown. The need to better understand the structure/function relationships of HS has driven the development of advanced analytical methods for detailed structural characterization of these biomolecules. However, the heterogeneous nature of HS, such as O-sulfation positions, amine group modifications and uronic acid epimerization, caused by the postpolymerization modifications during biosynthesis, has made their structural determination a highly challenging task.
Liquid chromatography–mass spectrometry (LC-MS) methods, such as disaccharide analysis and oligosaccharide profiling, are able to profile the biological changes of HS at the compositional level.4 However, compelling research has shown that the interactions of HS and proteins are affected by the fine structure of HS, but not the average composition.5 For the fine structure analysis of HS, tandem mass spectrometry (MS/MS) dissociation methods employed include collision-induced dissociation (CID) and electron-activated dissociation (ExD).6 Although new methods and applications of HS analysis using LC-MS or MS/MS are reported every year, there are few reports demonstrating the online sequencing of HS using liquid chromatography tandem mass spectrometry (LC-MS/MS). A reliable system would greatly enhance understanding of structure/function relationship of HS. The first LC-MS/MS analysis of a mixture of HS tetrasaccharides was achieved in 2016, with a precolumn chemical derivatization scheme, which is not suitable for high-throughput biological samples.7 Subsequently, a porous graphitized carbon-MS/MS (PGC-MS/MS) approach showed effective separation of underived HS oligosaccharides.8 However, the method cannot differentiate the predominant sulfation motifs and rare cases (6-O-sulfate vs 3-O-sulfate, N-sulfate vs free amine), due to lack of informative cross-ring fragments from MS/MS, nor can it differentiate the epimerization (glucuronic acid, GlcA, vs iduronic acid, IdoA). Additionally, the characterized structures in such a workflow typically had one or less sulfate per disaccharide moiety, which does not cover the more highly sulfated saccharide motifs that mediate many protein binding interactions in HS chains.
Three challenges have made the online LC-MS/MS sequencing HS hard to achieve. The first is the separation of isomeric oligosaccharides using MS-compatible LC methods. Among LC separation methods, strong anion exchange (SAX) and reverse phase ion pairing (RP-IP) chromatography have the greatest resolution for HS oligosaccharides.9 However, both SAX and RP-IP have mobile phases that may result in signal suppression and source contamination. Size exclusion chromatography (SEC) and hydrophilic interaction liquid chromatography (HILIC) are MS compatible. However, both approaches have been used primarily for disaccharide analysis or oligosaccharide profiling; their capacity to resolve isomeric HS oligosaccharides is limited. While PGC has been used for decades for the analysis of N and O-linked glycans, and HS disaccharides,10 its application to HS oligosaccharides has only recently been reported and further investigation is needed.8 In addition, retention time drift remains a problem with PGC.
The second challenge is the fragility of sulfate groups. It was found that, in collision-induced dissociation, CID-MS/MS, protons on sulfate groups facilitate sulfate losses that diminish the abundances of informative backbone fragments and render unambiguous interpretation of the tandem mass spectrum a challenge.6a Since the past decade, electron activated dissociation, ExD, has been used for HS oligosaccharide sequencing and showed more tolerance to the free protons.6i Among dissociation methods, negative electron transfer dissociation (NETD) has been proved the most useful for HS sequencing as it produces informative fragmentation with fewer sulfate loss.6j,k Despite this, to date, NETD, or any other ExD, has never been used as a dissociation method for online LC-MS/MS analysis of HS oligosaccharides.
The third challenge is the precursor ion charge state. Although a mobile proton-tolerant MS/MS dissociation method helps to hedge the fragility of sulfate groups and to produce more informative fragments, careful precursor selection/treatment is also required to generate informative tandem mass spectra of HS oligosaccharides. In MS/MS experiments with direct sample infusion, charge state enhancement and sodium–hydrogen exchange can be used to reduce the number of free protons,6h,11 and precursors of highest charge state or cation adducted can be selected to optimize dissociation. However, for online LC-MS/MS analysis, the use of sodium salts for sodium–hydrogen exchange is not recommended because the presence of sodium adducts multiplies the number of possible fragment ion compositions so that confident spectral assignment becomes unfeasible. Precursor charge state enhancement using chemical reagents is not practical for LC-MS/MS, due to source contamination. Additionally, although ammonium buffers in LC system are volatile during MS ionization, proton transfer reactions lower the observed charge states of HS precursors, leading to increased mobile protons on the precursor ions and deleterious sulfate losses. This phenomenon poses a serious problem for online sequencing of HS oligosaccharides, especially those with a relatively high number of sulfate groups per disaccharide unit. This challenge has been largely neglected in the published literature.
Here, we describe the separation and sequencing of a wide range of HS oligosaccharides, varying in chain length (tetramers and hexamers), number of sulfate groups (3–7), sulfation pattern (sulfate positional isomers), and uronic acid stereochemical configuration (epimers), by HILIC LC-MS/MS with NETD as the dissociation method. A cation exchange device (ion suppressor) was used online, prior to MS detector, to remove ammonium ions and enhance the charge state of precursors. Our data highlight the advantages of HILIC LC-NETD-MS/MS based HS oligosaccharides separation and sequencing and the potential of this methodology as a valuable tool for further application on biological samples.
EXPERIMENTAL SECTION
Materials.
GAG standards (Figure 1) were chemically synthesized and purified as previously described.12 Reducing end modification R was confirmed to have a single conformation (α-linkage). The detailed synthetic methodology for tetrasaccharides without reducing end modification and hexasaccharides will be reported elsewhere. The number in the name of each compound indicates the number of sulfate groups. Lower case a, b, and c, in the name, are used to differentiate the isomeric compounds, including sulfate positional isomers and uronic acid epimers.
Figure 1.
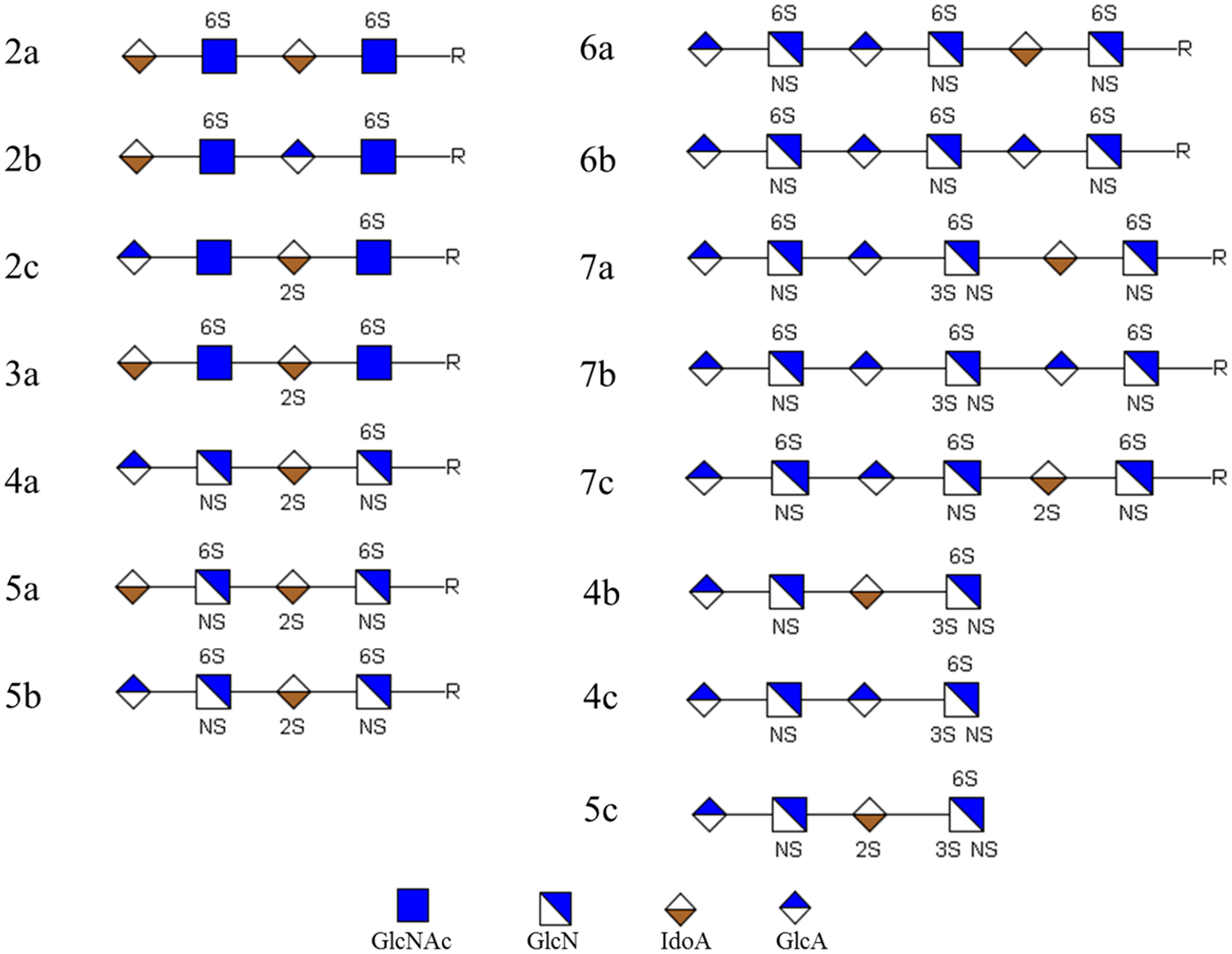
Structures of synthetic HS oligosaccharides used in this study; R = O(CH2)5NH2.
HILIC LC with Ion Suppressor.
An Accucore 150 Amide HILIC column (2.1 mm × 250 mm, Thermo Fisher Scientific, San Jose, CA) was used for the separation. Eluent A was 50 mM ammonium formate (pH 4.4) and Eluent B was 100% acetonitrile. For the separation of tetrasaccharides with reducing end modification, a linear gradient of 80–60% B over 80 min at a flow rate of 150 μL/min was used, followed by additional 40 min elution with 60% B and 30 min re-equilibration with 80% B (method 1). For the separation of tetrasaccharides with no modification and hexasaccharides, a linear gradient 60–50% B over 80 min at a flow rate of 150 μL/min was used, followed by 30 min re-equilibration with 60% B (method 2). All of HILIC experiments were carried at 25 °C, except the separation of tetramers without reducing end modification, which was carried at both 25 and 55 °C.
A chemically regenerated ion suppressor (ACRS 500, 2 mm, Thermo Fisher Scientific, San Jose, CA) was used after the column for online desalting as pervious described,13 prior to MS detector. The ion suppressor exchanged ammonium salts to their corresponding organic acids, thereby eliminating the proton-transfer reactions that diminished the magnitude of saccharide ion charge states. A solution of 100 mM sulfuric acid was used to regenerate the suppressor. Due to the natural features of the ion suppressor, a product designed to work with low percentage of organic solvent (≤40%), a postcolumn makeup flow of 100% water with a flow rate of 150 μL/min was applied to the HILIC eluent (80–50% acetonitrile) to make ion suppressor function correctly. The LC flow path and the scheme of ion exchange in ion suppressor are shown in Figure S1. The influence of the use of the ion suppressor on LC peak width and signal sensitivity was examined using LC method 2.
Offline MS/MS.
All MS and MS/MS experiments were carried out on a 12-T solariX hybrid Qh-FTICR mass spectrometer (Bruker Daltonics, Bremen, Germany) in the negative ionization mode. Prior to online LC-MS/MS sequencing, each compound was directly infused into the mass spectrometer using a nanoelectrospray source. NETD was performed to identify the optimal MS/MS conditions for each compound as previously described.6k The drying gas flow rate was 4 L/min. The resulting fragments of each compound were annotated and used as references for further online LC-MS/MS sequencing. CID-MS/MS was also performed for selected compounds to examine the difference of fragmentations between NETD and CID. External calibration using sodium-TFA clusters was implemented prior to MS/MS and LC-MS/MS experiments.
Online LC-MS/MS.
The online LC-MS/MS sequencing was carried out using a normal ESI source. As elution into the ESI electrospray source after ion suppressor was achieved at a flow rate of 300 μL/min, the drying gas flow rate were accordingly increased to 7 L/min. The molecular ions of interest with high charge state were selected in the preferred precursor list with a window of 100 ppm for LC-MS/MS, while the others were added in the exclusive list. The optimized NETD condition was set as a 300 ms reagent accumulation time and a 100 ms reaction time according to the preliminary Offline MS/MS experiment.
Data Analysis.
All spectra were processed by DataAnalysis 4.4 (Bruker, Bremen, Germany). Offline MS/MS spectra were interpreted using GAGfinder with a mass accuracy cutoff of 5 ppm or better.14 LC-MS/MS spectra were interpreted manually by checking the major glycosidic and cross-ring fragments using the fragment list generated from the corresponding Offline MS/MS results. Fragments were annotated according to the Domon and Costello nomenclature with an extension developed by Wolff-Amster.6c,15
RESULTS AND DISCUSSION
Precursor Charge State Enhancement Using an Ion Suppressor.
Due to the presence of ammonium cations in the LC mobile phase, which suppressed the ionization of the compounds containing negatively charged sulfate groups, the precursor charge states of HS oligosaccharides from LC-MS were always low.8 A low charge state resulted in more abundant sulfate losses and less abundant glycosidic bond and cross-ring fragmentations in both CID-MS/MS and ExD-MS/MS.6c,16 Moreover, as shown in Table S1, the precursor charge states did not increase for the highly sulfated compounds compared to those with a low degree of sulfation, indicating that for highly sulfated HS oligosaccharides, there were more free protons remaining on sulfate groups and it was even harder to produce informative MS/MS fragmentation. Thus, charge state enhancement was required.
Previously, an ion suppressor was applied to remove ammonium adduction for low molecular weight heparin profiling using SEC-MS. We found that the use of ion suppressor removed the ammonium salt, thus, increased the abundance and enhanced the charge state of the precursor of highly sulfated HS pentasaccharide, Arixtra.13 The ion abundance increase and precursor charge state enhancement perfectly met the requirement of LC-NETD-MS/MS. Because the ion suppressor was designed for the use in aqueous environment (<40% organic solvent), a postcolumn makeup flow was used to reduce the percentage of organic solvent in the effuent entering the ion suppressor to below 40% in this study. Over the entire LC gradient, the percentage of organic solvent that ran though the ion suppressor averaged below 40%, which allowed the ion suppressor to function correctly.
Consistent with previous work with SEC, the use of the ion suppressor in HILIC, as shown in Figure S2, greatly increased the signal intensities, which may improve the limit of detection. The increase in charge state of highly sulfated oligosaccharide was also observed (Table S1), which decreased the number of remaining free protons. The increased peak abundances and precursor charge states made the further LC-NETD-MS/MS sequencing of highly sulfated HS oligosaccharides feasible. The use of the ion suppressor widened the chromatographic peaks slightly. The broadening was due to both the void volume of the ion suppressor (<15 μL) and the greatly increased signal sensitivity. Since the influences of the use of the ion suppressor were largely positive, we then applied the ion suppressor to all LC separations.
LC Separation of HS Oligosaccharides.
The LC separation of tetramers is shown in Figure S3. The elution order of the tetramers corresponded to the number of sulfate groups, from 2 to 5, consistent with results observed from other HILIC experiments.17 Unlike other HILIC columns, the separation of isomeric compounds was achieved. Compound 2a was separated from its epimer 2b and sulfate positional isomer 2c, while 5a was separated from its epimer 5b. Additionally, we found that the C5 epimerization contributed to the separation of these epimers. In each pair of epimers, in which the compounds have the same sulfation patterns and the only difference is C5 epimerization, the one containing IdoA always eluted earlier than the ones containing GlcA, for example, 2a (IdoA–IdoA, for the pattern of epimerization, same below) eluted earlier than 2b (IdoA-GlcA), while 5a (IdoA–IdoA) eluted earlier than 5b (GlcA-IdoA). Compounds 2b and 2c, which had different sulfation patterns and epimerization, overlapped, indicating that the sulfation pattern also affects the retention time.
We observed similar elution patterns for separation of the hexamers (Figure S4). The compounds containing six sulfate groups eluted earlier than those containing seven. Two pairs of epimers were separated and the elution order of epimers was related to the C5 epimerization of uronic acid. Again, the isomer containing IdoA eluted earlier. Hexamer 6a (GlcA-GlcA-IdoA) eluted earlier than 6b (GlcA-GlcA-GlcA), while 7a (GlcA-GlcA-IdoA) eluted earlier than 7b (GlcA-GlcA-GlcA). This feature may allow the differentiation of epimers using LC instead of MS/MS since there is no universal method to characterize C5 epimerization of uronic acid in HS using MS/MS, albeit many efforts have been reported.6b,g,18 Additionally, sulfate positional isomer pairs, 7a and 7c, in which the only difference is the position of sulfate group, were separated, indicating that this LC method could reveal differences in sulfation patterns of similar compounds. However, compounds 7b and 7c overlapped in retention time, which reflected the effects of sulfation pattern and C5 epimerization. Although the pairs of 2b and 2c and 7b and 7c were not fully resolved by LC, the differentiation of these pairs was achieved by online LC-NETD-MS/MS.
We also observed good separation of HS oligosaccharides without reducing end modification, of which the structures were similar to the products from enzymatic digestion of heparan sulfate. As shown in Figure S5, three tetrasaccharides were separated. Consistent with the elution order of compounds with reducing end modification, the tetrasaccharide containing IdoA, 4b, eluted earlier than the one containing GlcA, 4c, indicating that the good separation did not rely on the reducing end modification. Thus, this HILIC method can be applied for native HS oligosaccharides. Additionally, we noticed that the anomers (or conformers) were separated at 25 °C (Figure S5a) and the anomer peaks merged when temperature was increased to 55 °C (Figure S5b), which was consistent with a previous report.17a
Optimization of HILIC Separations for HS Saccha-rides.
For polysaccharide lyase digests of HS, consisting primarily of disaccharides, 2.1 mm internal diameter capillary amide-HILIC columns perform well.17a For analysis of HS disaccharides released enzymatically from tissue slides, we use in-house packed nanocapillary columns. For such reduced scale chromatography, amide-HILIC shows poor retention of the unsulfated ΔHexA-GlcNAc disaccharide. We therefore use a HILIC-weak anion exchange (HILIC-WAX) resin that retains the range disaccharides containing 0–3 sulfate groups for nanocapillary LC-MS applications.19.20 For highly sulfated oligosaccharides, however, the HILIC-WAX stationary phase shows poor recovery of highly sulfated HS oligosaccharides. In the present work, we therefore used amide-HILIC for separation of such oligosaccharides. It is essential that the column hardware contain no titanium frits or filters because acidic saccharides bind to these surfaces.
In this work, we observed amide-HILIC resolution of saccharides that differ only by uronic acid epimerization at one position (2a vs 2b, 5a vs 5b, Figure S3; 6a vs 6b, 7a vs 7b, Figure S4; 4b vs 4c, Figure S5). In each of these examples, the saccharide with the IdoA positional isomer eluted earlier than did that with GlcA. A similar trend was observed for separation nitrous acid depolymerized disaccharides using amide-HILIC.17a The amide-HILIC retention mechanism is believed to reflect a combination of hydrophilic partitioning and secondary hydrogen binding and electrostatic interactions.21 The IdoA residue in oligosaccharides are present in two conformations, 1C4 and 2S0, in approximately 60:40 ratio, meaning that the presence of IdoA confers conformational flexibility.22 The pattern whereby IdoA-containing positional isomers elute earlier is consistent with weaker hydrogen bonding and dipole–dipole interactions than for the GlcA-containing positional isomer. It is known that, as the mobile phase buffer salt concentration increases, retention of hydrophilic molecules increases.23 Therefore, for future studies, ion suppressor should allow flexibility with regard to mobile phase salt concentration and additives to optimize the ability to separate saccharides that differ by uronic acid epimer positions.
LC-MS/MS Sequencing and Differentiation of Lowly Sulfated Tetramers.
NETD method produces tens to hundreds of fragment ions in every scan for each HS oligosaccharide,6k including glycosidic and cross-ring fragments, of which both normal type and neutral-loss type, for example, sulfate-loss type and hydrogen-loss type, can be observed. As a result, manual interpretation of LC-MS/MS of HS oligosaccharides is time-consuming. We therefore generated a fragment list of each compound using Offline NETD-MS/MS, of which the data were automatically interpreted by the software GAGfinder. The fragment lists were used as references for further online LC-MS/MS analysis (Tables S2−S6).
The sequencing of the lowly sulfated HS oligosaccharides, in terms of the ones containing one or fewer sulfate group per disaccharide, was straightforward. NETD was able to completely sequencing compounds 2a, 2b, and 2c, since there is no free proton left on the sulfate groups of the precursor [M − 2H]2− (Figure 2a). A full set of glycosidic cleavages was observed for each compound, allowing the assignment of the number of sulfate groups on each residue. For compounds 2a and 2b, each of the two sulfate groups were localized to one of the acetylated GlcN residues (GlcNAc), whereas for compound 2c, one sulfate group was localized to the acetylated GlcN residue at the reducing end and the other one to the adjacent IdoA residue. Additionally, there was no need to confirm the position of these sulfate groups, since the sulfate group can only locate at the 6-O position of the GlcNAc residue and the 2-O position of IdoA residue based on the well-known HS biosynthesis pathway. Thus, these disulfated tetramers were completely sequenced, as shown in Figure 2.
Figure 2.
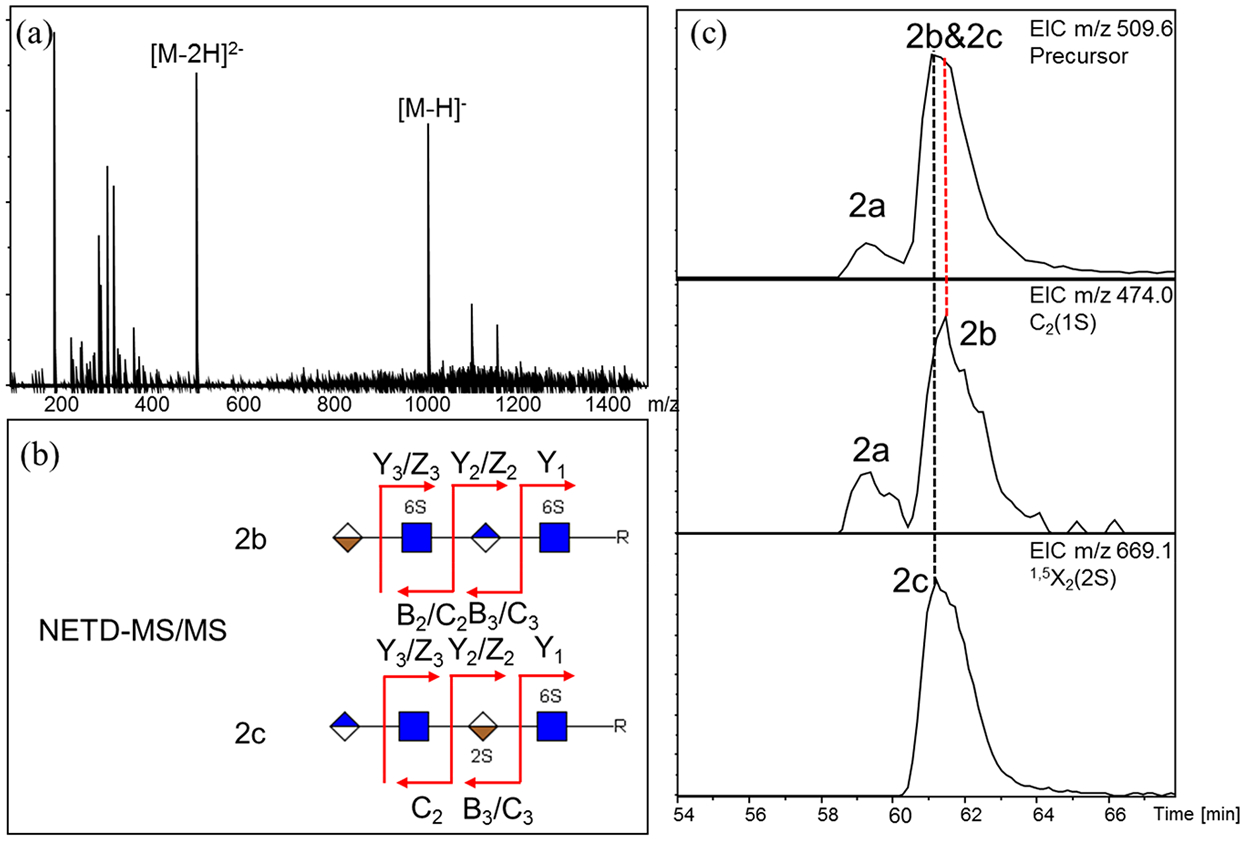
LC-NETD-MS/MS sequencing and differentiation of disulfated tetramers: (a) extracted mass spectrum at 61.50 min; (b) cleavage maps of compounds 2b and 2c generated by LC-NETD-MS/MS; and (c) differentiation of partially overlapped compounds using extracted ion chromatograms (EICs) of their corresponding diagnostic ions.
The different sulfation pattern in these isomeric compounds produced diagnostic ions for the further differentiation. For sulfate positional isomers 2b and 2c, B2 and C2 ions with one sulfate group, was exclusively produced by 2b, while Y2 and Z2 ions with two sulfate groups was exclusively produced by 2c (Tables S2 and S3). Additionally, cross-ring fragment 1,5X2 contained the same number of sulfate groups as did glycosidic fragments Y2 and Z2. Considering the intensities of these fragments, C2 was used as the diagnostic ion for compound 2b while 1,5X2 was used as the diagnostic ion for compound 2c. Although elution of compounds 2b and 2c partially overlapped, with 2c eluting slightly earlier than 2b, differentiation and identification of each isomer were achieved by examining the extracted ion chromatograms (EICs) of their corresponding diagnostic ions (Figure 2c). This feature makes further quantitative analysis feasible.
Although the pair of epimers, 2a and 2b, were separated by the HILIC column, no significant difference was found in their NETD-MS/MS spectra. Results from other epimer pairs showed the same problem, indicating that the differentiation of epimers by NETD MS/MS remains a challenge. Thus, in this study, epimers were differentiated using retention times.
LC-MS/MS Sequencing and Differentiation of Highly Sulfated Tetramers and Hexamers.
In the absence of the ion suppressor, [M − 3H]3− ions were not observed for pentasulfated saccharides, as shown for compound 5b (Figure 3a). By using the precursor of highest charge state, [M − 2H]2−, NETD-MS/MS failed to produce enough fragments for structural reconstruction (Figure S6). With the ion suppressor, however, abundant [M − 3H]3− precursor ions were observed, for which two free protons remained (Figure 3b). By using this triply charged precursor, NETD produced an informative tandem mass spectrum with a full set of glycosidic cleavages (Figure 3c and Table S4), allowing the determination of the number of sulfate groups on each residue. Additionally, we previously demonstrated that the diagnostic cross-ring fragments from NETD could be used to differentiate the predominant 6-O-sulfation and the rare 3-O-sulfation.6k Here, by using singly charged 0,2A2 ion (m/z 375.0) and 3,5A2 ion (m/z 329.0), and doubly charged 0,2A4 ion (m/z 475.5) and 3,5A4 ion (m/z 452.5), the sulfation pattern on both GlcN residues were assigned as NS6S but not NS3S, without any ambiguity. Meanwhile, cross-ring fragments 2,4A3 (m/z 556.0) and 3,5A3 (m/z 584.0) were used to assign the 2-O sulfation on the internal uronic acid albeit that kind of assignment can be assumed based on knowledge of the HS biosynthesis pathway. Thus, the pentasulfated tetramers were sequenced completely and all sulfate positions were assigned.
Figure 3.
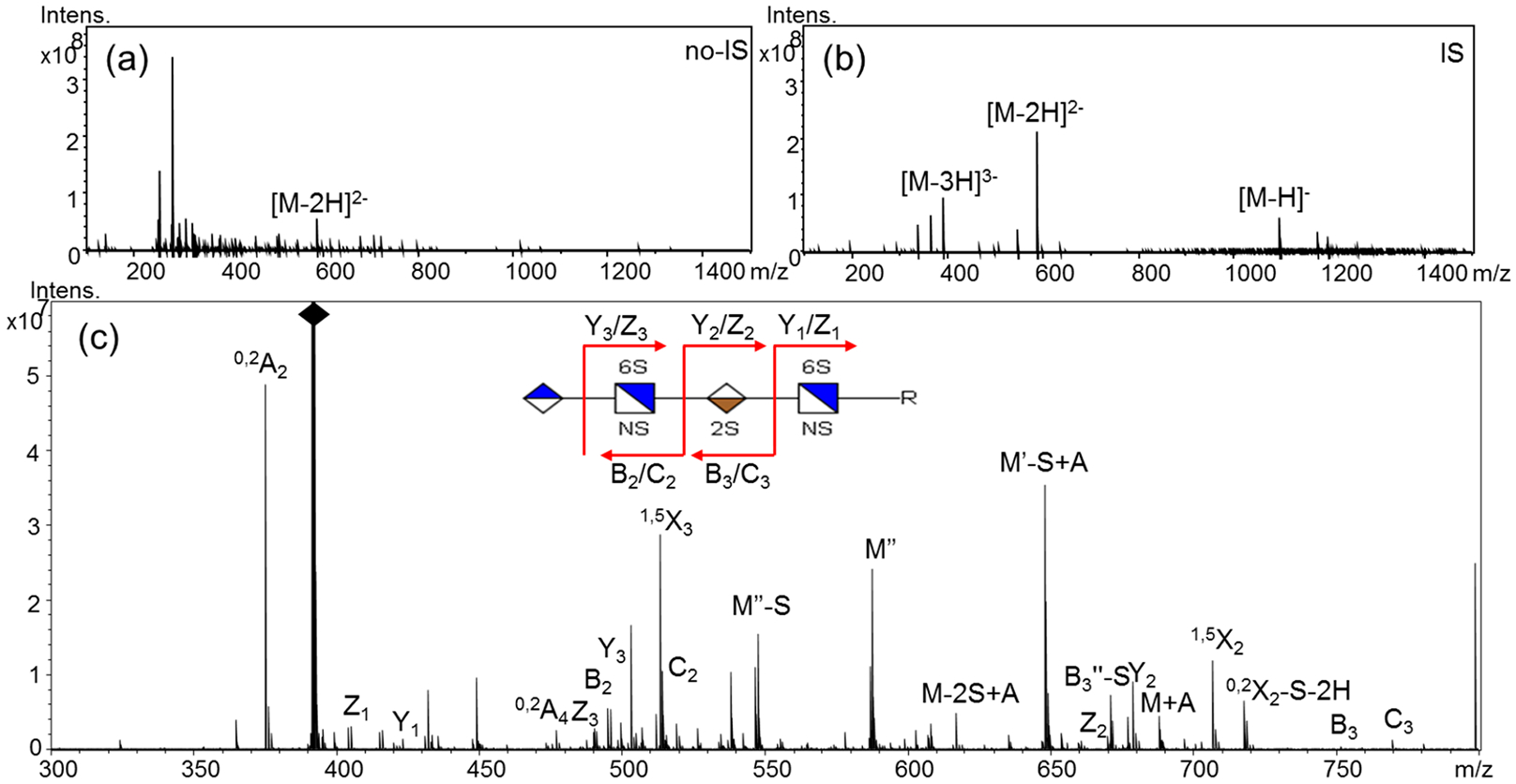
LC-NETD-MS/MS sequencing of pentasulfated tetramer 5b: (a) precursors of 5b from LC-MS without the ion suppressor; (b) precursors of 5b from LC-MS with the ion suppressor; and (c) annotated LC-NETD-MS/MS spectrum of 5b acquired at the retention time of 100.62 min; S, sulfate; A, NETD reagent.
The issue of low precursor charge state also impeded the analysis of highly sulfated hexamers when the ion suppressor was not applied. As shown in Table S1, for the hexamer 6a with six sulfate groups, [M-2H]2− was found to be highest charge state of the precursor while for the hexamer 7a with seven sulfate groups, [M-3H]3− was observed in a very low abundance (Figure 4a). Both precursors had four free protons on sulfate groups, resulting in insufficient NETD fragmentation for structural reconstruction (Figure S7). When the ion suppressor was used, [M − 3H]3− was the most abundant molecular ion of the heptasulfated hexasaccharides while [M − 4H]4− was also abundant (Figure 4b). By using the [M − 4H]4− precursors, hexamers with 7 sulfate groups were completely sequenced by NETD-MS/MS. As shown in Figure 4c, a full set of glycosidic fragments was observed for compound 7a, as well as 7b, indicating both structures contained two disulfated GlcN residues and one internal trisulfated GlcN residue. Sulfate groups on disulfated GlcN residues were confirmed as NS6S using diagnostic cross-ring fragments 3,5A2 and 3,5A6 (singly charged, m/z 329.0 and triply charged, m/z 467.0, respectively, Table S5). The internal trisulfated GlcN residue was automatically assigned as GlcNS3S6S. Hexamer 7c was confirmed to have three disulfated GlcN residues and one sulfated uronic acid next to the reducing end. Additionally, the same diagnostic cross-ring fragments 3,5A2 and 3,5A6 (singly charged, m/z 329.0 and triply charged, m/z 467.0, respectively) were used to confirm two GlcNS6S residues at both sides (Table S6). The sulfate groups on the internal disulfated GlcN residue were assigned as NS6S using the doubly charged 3,5A4 ion at m/z 412.5 in the Offline NETD-MS/MS experiment. Unfortunately, this ion was not observed in the online LC-MS/MS analysis due to its low relative abundance.
Figure 4.
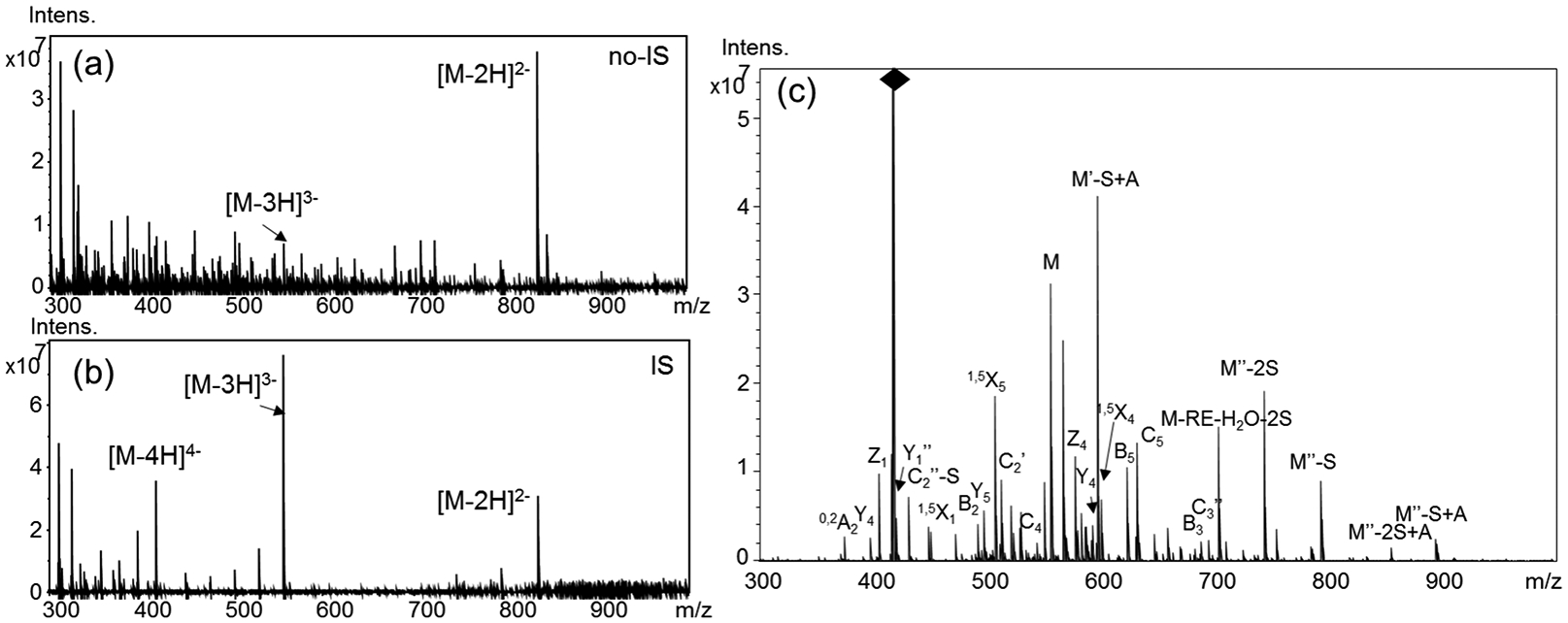
LC-NETD-MS/MS sequencing of heptasulfated hexamer 7a: (a) precursors of 7a from LC-MS without the ion suppressor; (b) precursors of 7a from LC-MS with the ion suppressor; and (c) annotated LC-NETD-MS/MS spectrum of 7a acquired at the retention time of59.54 min; RE, O(CH2)5NH2.
Aside from the full sequencing of these heptasulfated hexamers (shown in Figure S8 and Figure 5a), differentiation of the partially overlapped compounds 7b and 7c was achieved by examining the EICs of their corresponding diagnostic cross-ring fragments, C4 with five sulfate groups for compound 7b and 1,5X2 with three sulfate group for compound 7c (Figure S9). As shown in Figure 5b, compound 7b eluted earlier than compound 7c. Again, this feature made the present method feasible for quantitative analysis of biological samples.
Figure 5.
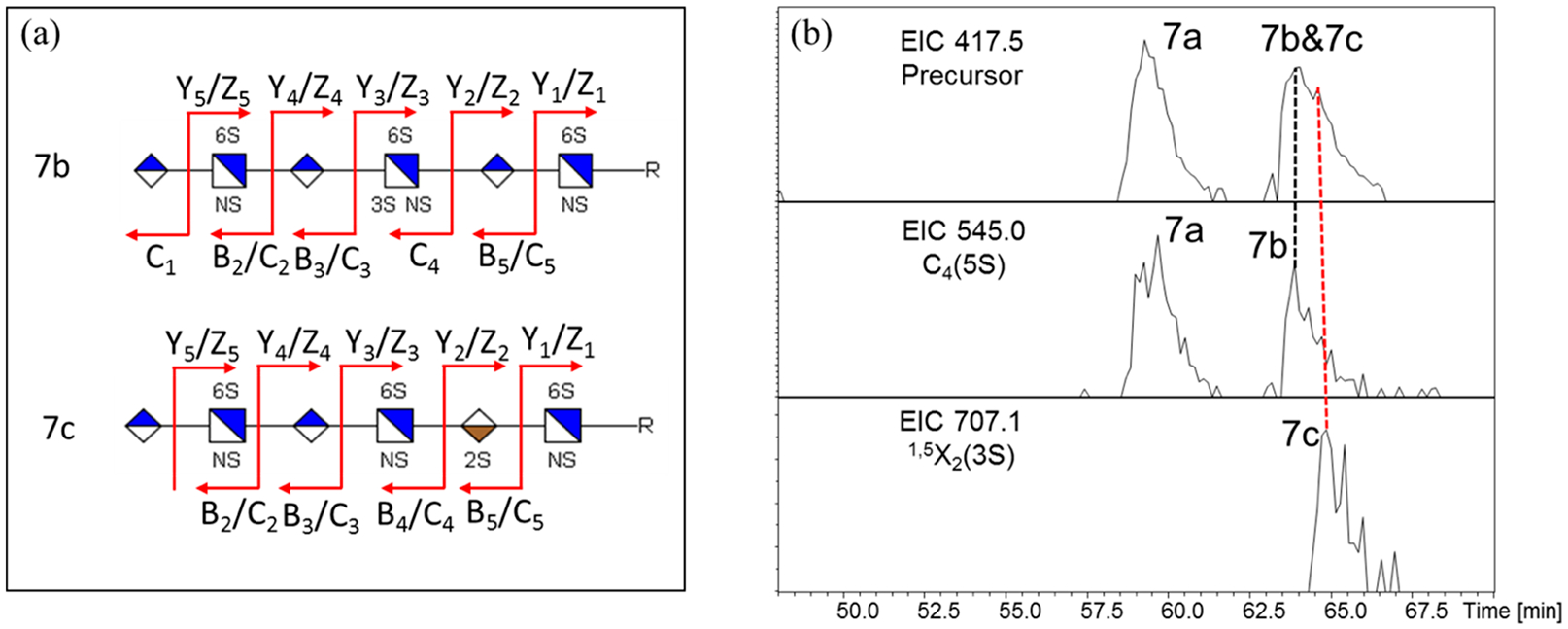
LC-MS/MS sequencing and differentiation of heptasulfated hexamers: (a) cleavage maps of compounds 7b and 7c generated by LC-NETD-MS/MS; and (b) differentiation of partially overlapped compounds using EICs of their corresponding diagnostic ions.
Comparison of CID and NETD-MS/MS Fragmentation.
Although the comparison of the fragmentation of CID and NETD has been reported,7 it remains a concern that whether CID-MS/MS can also produce informative fragmentation for structural reconstruction of HS oligosaccharides using the enhanced precursors. As shown in Figure S10 and Table S7, for the pentasulfated tetramer 5b, CID produced an informative tandem mass spectrum using the triply charged precursor, allowing the determination of the number of sulfate groups on each residue. However, compared with fragments in NETD, some glycosidic fragments were missing in CID, for example, all Z ions. Moreover, the observed glycosidic fragments were generally of relative low abundance and out-numbered by their sulfate-loss counterparts, which may lead to incorrect structural assignments. Additionally, the absence of cross-ring fragments made the assignments of sulfate positions impossible.
We also did the comparison using the hexamer 7a using [M − 4H]4− precursor. As shown in Figure S11 and Table S8, the absence of B1 and Y5 ions in the CID-tandem mass spectrum, resulted in the failure to identify the number of sulfate groups on GlcA and GlcN residues at the nonreducing end. Again, the sulfate-loss fragments were generally predominant and cross-ring fragments were absent using CID, leading to a failure of sequencing. These examples illustrate the advantages of NETD in comparison with CID as a tool of online LC-MS fragmentation.
CONCLUSIONS
Here we demonstrate that HILIC LC-NETD-MS/MS can be used for online sequencing of a wide range of HS oligosaccharides varying in chain length as well as degree of sulfation, and differentiation of isomeric compounds, including both sulfate positional isomers and epimers.
For the first time, we observed LC separation of epimers without the need of permethylation. The observation that for each epimeric pair, compounds containing IdoA eluted earlier than those containing GlcA, making it possible to identify the C5 epimerization of HS using LC retention time. The well-designed isomeric compounds allowed the determination of relationship between fine modifications and the chromatographic behaviors. The chain length, degree of sulfation, position of sulfation and C5 epimerization of uronic acid all affect the chromatographic selectivity. Synthesis and test of additional HS saccharides are in progress to validate this feature and to make it applicable to biological samples. Recently, ion mobility mass spectrometry (IM-MS) has been successfully applied for the analysis of HS oligosaccharides by our group.24 The combination of HILIC and IM for a two-dimentional separation of HS may provide better resolution of this class of compounds and may be more promising for detailed GAGome mapping and sequencing.
While MS/MS analysis of HS (and, by extension, heparin) oligosaccharides has been studied over several decades, the LC-MS/MS sequencing of these compounds remains a challenge to biomedicine. Here, with the use of the ion suppressor, which enhances the precursor charge state, and with the use of NETD, which is most tolerant to the lower charge states, we demonstrated online sequencing of both lowly sulfated and highly sulfated HS oligosaccharides. Notably, the ion suppressor is compatible with other LC systems, including PGC and SEC, therefore, this online charge state enhancing method for HS analysis can be widely applied.
The successful online sequencing of HS oligosaccharides provides a major opportunity to understand the structure/function relationship of this class of protein post-translational modification. However, the heterogeneous nature and the complex tandem mass spectra of HS made the manual data interpretation difficult. A software for peak finding and elemental composition assignment for glycosaminoglycan analysis by MS/MS has been developed recently by our lab.14 A software algorithm for automated annotation of HS and for LC-MS/MS sequencing is under development in our group.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grants P41GM104603, R21HL131554, and U01CA221234.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.9b02313.
Figure S1: The LC flow path and the scheme of ion exchange in the ion suppressor. Figure S2: Influence of the use of the ion suppressor on LC peak width and signal intensity. The comparison of LC chromatograms were shown in panels. Red, without the ion suppressor; black, with the ion suppressor. Figure S3: HILIC separation of the tetramers with reducing end modification. 2a was not shown in the BPC due to low abundance. Figure S4: HILIC separation of the hexamers. Figure S5: HILIC separation of tetramers without reducing end modification at different temperatures. Left panels, 25 °C; Right panels, 55 °C. Small annomeric peaks were observed and indicated with arrows in panels b and c. Figure S6: NETD-MS/MS spectrum of 5b using the [M − 2H]2− precursor. Figure S7: NETD-MS/MS spectrum of 7a using the [M − 3H]3− precursor. Figure S8: Annotated LC-MS/MS spectra of overlapped 7b and 7c at 64.03 and 65.71 min. Figure S9: Zoom-in LC-MS/MS spectra of overlapped 7b and 7c at 64.03 and 65.71 min for selected diagnostic ions: (a) C4(5S) at m/z 545.0; (b) 1,5X2(3S) at m/z 707.1. Figure S10: CID-MS/MS spectrum of 5b using the [M − 3H]3− precursor. Figure S11: CID-MS/MS spectrum of 7a using the [M − 4H]4− precursor. Table S1: Observed precursor of highest charge state of each compound in LC-MS. Compounds of charge state enhanced were marked in bold. Table S2: List of assigned peaks in the NETD-MS/MS spectrum of 2b, [M − 2H]2−. Relative intensities were normalized to the total intensity in the spectrum, same below. RE, reducing end modification; A, NETD reagent. Table S3: List of assigned peaks in the NETD-MS/MS spectrum of 2c, [M − 2H]2−. Table S4: List of assigned peaks in the NETD-MS/MS spectrum of 5b, [M − 3H]3−. Table S5: List of assigned peaks in the NETD-MS/MS spectrum of 7a, [M − 4H]4−. Table S6: List of assigned peaks in the NETD-MS/MS spectrum of 7c, [M − 4H]4−. Table S7: List of assigned peaks in the CID-MS/MS spectrum of 5b, [M − 3H]3−. Table S8: List of assigned peaks in the CID-MS/MS spectrum of 7a, [M − 4H]4− (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Esko JD; Lindahl UJ Clin. Invest 2001, 108 (2), 169–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Esko JD; Selleck SB Annu. Rev. Biochem 2002, 71, 435–71. [DOI] [PubMed] [Google Scholar]; (b) Chen YH; Narimatsu Y; Clausen TM; Gomes C; Karlsson R; Steentoft C; Spliid CB; Gustavsson T; Salanti A; Persson A; Malmstrom A; Willen D; Ellervik U; Bennett EP; Mao Y; Clausen H; Yang Z Nat. Methods 2018, 15 (11), 881–888. [DOI] [PubMed] [Google Scholar]; (c) Qiu H; Shi S; Yue J; Xin M; Nairn AV; Lin L; Liu X; Li G; Archer-Hartmann SA; Dela Rosa M; Galizzi M; Wang S; Zhang F; Azadi P; van Kuppevelt TH; Cardoso WV; Kimata K; Ai X; Moremen KW; Esko JD; Linhardt RJ; Wang L Nat. Methods 2018, 15 (11), 889–899. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Knelson EH; Nee JC; Blobe GC Trends Biochem. Sci 2014, 39 (6), 277–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Shukla D; Liu J; Blaiklock P; Shworak NW; Bai X; Esko JD; Cohen GH; Eisenberg RJ; Rosenberg RD; Spear PG Cell 1999, 99 (1), 13–22. [DOI] [PubMed] [Google Scholar]; (b) Pejler G; Danielsson A; Bjork I; Lindahl U; Nader HB; Dietrich CP J. Biol. Chem 1987, 262(24), 11413–21. [PubMed] [Google Scholar]
- (4).(a) Li G; Steppich J; Wang Z; Sun Y; Xue C; Linhardt RJ; Li L Anal. Chem 2014, 86 (13), 6626–32. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mao Y; Huang Y; Buczek-Thomas JA; Ethen CM; Nugent MA; Wu ZL; Zaia J J. Biol. Chem 2014, 289 (49), 34141–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Schultz V; Suflita M; Liu X; Zhang X; Yu Y; Li L; Green DE; Xu Y; Zhang F; DeAngelis PL; Liu J; Linhardt RJ J. Biol. Chem 2017, 292 (6), 2495–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Zaia J; Costello CE Anal. Chem 2003, 75 (10), 2445–55. [DOI] [PubMed] [Google Scholar]; (b) Wolff JJ; Chi L; Linhardt RJ; Amster IJ Anal. Chem 2007, 79 (5), 2015–22. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wolff JJ; Laremore TN; Busch AM; Linhardt RJ; Amster IJ J. Am. Soc. Mass Spectrom 2008, 19 (6), 790–8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wolff JJ; Leach FE 3rd; Laremore TN; Kaplan DA; Easterling ML; Linhardt RJ; Amster IJ Anal. Chem 2010, 82 (9), 3460–6. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Leach FE 3rd; Wolff JJ; Xiao Z; Ly M; Laremore TN; Arungundram S; Al-Mafraji K; Venot A; Boons GJ; Linhardt RJ; Amster IJ Eur. J. Mass Spectrom 2011, 17 (2), 167–76. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Leach FE 3rd; Xiao Z; Laremore TN; Linhardt RJ; Amster IJ Int. J. Mass Spectrom 2011, 308 (2–3), 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Leach FE 3rd; Arungundram S; Al-Mafraji K; Venot A; Boons GJ; Amster IJ Int. J. Mass Spectrom 2012, 330–332, 152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Shi X; Huang Y; Mao Y; Naimy H; Zaia JJ Am. Soc. Mass Spectrom 2012, 23 (9), 1498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Huang Y; Yu X; Mao Y; Costello CE; Zaia J; Lin C Anal. Chem 2013, 85 (24), 11979–86. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Leach FE; Riley NM; Westphall MS; Coon JJ; Amster IJ J. Am. Soc. Mass Spectrom 2017, 28 (9), 1844–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Wu J; Wei J; Hogan JD; Chopra P; Joshi A; Lu W; Klein J; Boons GJ; Lin C; Zaia JJ Am. Soc. Mass Spectrom 2018, 29(6), 1262–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Huang R; Zong C; Venot A; Chiu Y; Zhou D; Boons GJ; Sharp JS Anal. Chem 2016, 88 (10), 5299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Miller RL; Guimond SE; Prescott M; Turnbull JE; Karlsson N Anal. Chem 2017, 89 (17), 8942–8950. [DOI] [PubMed] [Google Scholar]
- (9).(a) Henriksen J; Roepstorff P; Ringborg LH Carbohydr. Res 2006, 341 (3), 382–7. [DOI] [PubMed] [Google Scholar]; (b) Wang Z; Zhang T; Xie S; Liu X; Li H; Linhardt RJ; Chi L Carbohydr. Polym 2018, 183, 81–90. [DOI] [PubMed] [Google Scholar]
- (10).(a) Karlsson NG; Schulz BL; Packer NH J. Am. Soc. Mass Spectrom 2004, 15 (5), 659–72. [DOI] [PubMed] [Google Scholar]; (b) Karlsson NG; Schulz BL; Packer NH; Whitelock JM J. Chromatogr. B: Anal. Technol. Biomed. Life Sci 2005, 824 (1–2), 139–47. [DOI] [PubMed] [Google Scholar]
- (11).Kailemia MJ; Li L; Ly M; Linhardt RJ; Amster IJ Anal. Chem 2012, 84 (13), 5475–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Arungundram S; Al-Mafraji K; Asong J; Leach FE 3rd; Amster IJ; Venot A; Turnbull JE; Boons GJ J. Am. Chem. Soc 2009, 131 (47), 17394–405. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zong C; Venot A; Li X; Lu W; Xiao W; Wilkes JL; Salanga CL; Handel TM; Wang L; Wolfert MA; Boons GJ J. Am. Chem. Soc 2017, 139 (28), 9534–9543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zaia J; Khatri K; Klein J; Shao C; Sheng Y; Viner R Anal. Chem 2016, 88 (21), 10654–10660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hogan JD; Klein JA; Wu J; Chopra P; Boons GJ; Carvalho L; Lin C; Zaia J Mol. Cell. Proteomics 2018, 17 (7), 1448–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Domon B; Costello CE Glycoconjugate J. 1988, 5 (4), 397–409. [Google Scholar]
- (16).Huang Y; Shi X; Yu X; Leymarie N; Staples GO; Yin H; Killeen K; Zaia J Anal. Chem 2011, 83 (21), 8222–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).(a) Gill VL; Aich U; Rao S; Pohl C; Zaia J Anal. Chem 2013, 85 (2), 1138–45. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Turiak L; Toth G; Ozohanics O; Revesz A; Acs A; Vekey K; Zaia J; Drahos LJ Chromatogr A 2018, 1544, 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).(a) Agyekum I; Patel AB; Zong C; Boons GJ; Amster J Int. J. Mass Spectrom 2015, 390, 163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Agyekum I; Zong C; Boons GJ; Amster IJ J. Am. Soc. Mass Spectrom 2017, 28 (9), 1741–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Raghunathan R; Polinski NK; Klein JA; Hogan JD;Shao C; Khatri K; Leon D; McComb ME; Manfredsson FP; Sortwell CE; Zaia J Mol. Cell. Proteomics 2018, 17 (9), 1778–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chen J; Kawamura T; Sethi MK; Zaia J; Repunte-Canonigo V; Sanna PP Sci. Rep 2017, 7 (1), 13931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).(a) Dulci M Hydrophilic Interaction Liquid Chromatography:Some Aspects of Solvent and Column Selectivity. https://assets.thermofisher.com/TFS-Assets/CMD/Technical-Notes/TN-20544-HILIC-Solvent-Column-Selectivity-TN20544-EN.pdf.; (b) Buszewski B; Noga S Anal. Bioanal. Chem 2012, 402 (1), 231–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Casu B; Petitou M; Provasoli M; Sinay P Trends Biochem. Sci 1988, 13 (6), 221–5. [DOI] [PubMed] [Google Scholar]
- (23).Guo Y; Gaiki SJ Chromatogr A 2005, 1074 (1–2), 71–80. [DOI] [PubMed] [Google Scholar]
- (24).Wei J; Wu J; Tang Y; Ridgeway ME; Park MA;Costello CE; Zaia J; Lin C Anal. Chem 2019, 91 (4), 2994–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.