

Abstract
Endogenous viral elements (EVEs) are found in many eukaryotic genomes. Despite considerable knowledge about genomic elements such as transposons (TEs) and retroviruses, we still lack information about nonretroviral EVEs. Aedes aegypti mosquitoes have a highly repetitive genome that is covered with EVEs. Here, we identified 129 nonretroviral EVEs in the AaegL5 version of the A. aegypti genome. These EVEs were significantly associated with TEs and preferentially located in repeat-rich clusters within intergenic regions. Genome-wide transcriptome analysis showed that most EVEs generated transcripts although only around 1.4% were sense RNAs. The majority of EVE transcription was antisense and correlated with the generation of EVE-derived small RNAs. A single genomic cluster of EVEs located in a 143 kb repetitive region in chromosome 2 contributed with 42% of antisense transcription and 45% of small RNAs derived from viral elements. This region was enriched for TE-EVE hybrids organized in the same coding strand. These generated a single long antisense transcript that correlated with the generation of phased primary PIWI-interacting RNAs (piRNAs). The putative promoter of this region had a conserved binding site for the transcription factor Cubitus interruptus, a key regulator of the flamenco locus in Drosophila melanogaster. Here, we have identified a single unidirectional piRNA cluster in the A. aegypti genome that is the major source of EVE transcription fueling the generation of antisense small RNAs in mosquitoes. We propose that this region is a flamenco-like locus in A. aegypti due to its relatedness to the major unidirectional piRNA cluster in Drosophila melanogaster.
Keywords: endogenous viral elements, EVE, A. aegypti, flamenco locus, RNA interference, piRNAs
INTRODUCTION
Endogenous viral elements (EVEs) are sequences derived from viruses integrated into eukaryotic genomes. EVEs can have either retroviral or nonretroviral origin. Retroviruses integrate into the host genome as part of their replication cycle using their own machinery that includes reverse transcriptase (RT) and integrase proteins (Wicker et al. 2007). Therefore, retroviral EVEs are autonomous elements. In contrast, integration of nonretroviral sequences is intriguing, since these do not usually integrate into host genomes (Katzourakis and Gifford 2010). This is especially the case with EVEs derived from RNA viruses that even lack DNA intermediates (referred to as nonretroviral integrated RNA virus sequences, NIRVS) (Palatini et al. 2017). Nonretroviral EVEs exist in different eukaryotic organisms including animals, plants, fungi, and even those that are unicellular, such as protists, although studies on integration mechanisms are mostly restricted to animals (Mette et al. 2002; Maori et al. 2007; Taylor and Bruenn 2009; Horie et al. 2010; Aiewsakun and Katzourakis 2015; Li et al. 2015; Parrish et al. 2015; Grybchuk et al. 2017). In mammals, integration of nonretroviral sequences is likely driven by autonomous transposable elements (TEs) (Zhdanov 1975; Geuking et al. 2009; Horie et al. 2010). In human and mouse cell lines, chimeras of viral and Alu elements are produced and integrated into the host genome during virus infection (Horie et al. 2010). Alu elements are nonautonomous and require long interspersed nucleotide elements (LINEs) for their mobilization and may also control the integration of viral sequences (Dewannieux et al. 2003).
Although EVEs are found in most animal genomes, it is unclear how these elements impact host biology. In some cases, EVEs might be beneficial to the host by retaining the ability to generate viral particles that compete with and inhibit related exogenous viruses (Fujino et al. 2014). EVEs may also be co-opted and give rise to new genes, thus impacting host genome evolution (Taylor and Bruenn 2009; Belyi et al. 2010). EVEs also generate small noncoding RNAs that feed into RNA interference (RNAi) pathways, although their functions remain unclear (Parrish et al. 2015; Whitfield et al. 2017). It has been proposed that EVE-derived small RNAs represent a mechanism of sequence-specific antiviral immune memory (Tassetto et al. 2019).
In invertebrates, current knowledge about EVEs is mostly restricted to retroviral elements (Feschotte and Gilbert 2012). This is likely due to the absence of nonretroviral EVEs in well-studied model organisms, such as D. melanogaster and C. elegans, which hampered characterization of mechanisms involved in the endogenization of these elements (Aiewsakun and Katzourakis 2015; Kryukov et al. 2018). Possible implications of nonretroviral EVEs for vertebrate antiviral immunity have increased the interest in studying their functions in invertebrates, especially in disease vectors such as ticks and mosquitoes (Fort et al. 2012; Fujino et al. 2014; Parrish et al. 2015; Honda and Tomonaga 2016; Palatini et al. 2017; Suzuki et al. 2017; Whitfield et al. 2017; Ter Horst et al. 2019).
A. aegypti mosquitoes are important vectors for human viruses and offer an interesting model to study EVEs since their genome contains over 60% of repetitive elements (Nene et al. 2007; Akbari et al. 2013; Li et al. 2015; Palatini et al. 2017; Whitfield et al. 2017; Matthews et al. 2018). Recent studies have started to uncover the diversity of viral families and explore possible functions of EVEs in A. aegypti (Katzourakis and Gifford 2010; Fort et al. 2012; Palatini et al. 2017; Suzuki et al. 2017; Whitfield et al. 2017). Nonretroviral EVEs identified in these mosquitoes are related to viruses from different families with RNA genomes (Palatini et al. 2017; Whitfield et al. 2017). A few mosquito EVEs correspond to coding regions within genes although most do not seem to be translated into proteins (Suzuki et al. 2017). Mosquito EVEs are frequently found in piRNA-generating clusters in association with TEs and generate abundant antisense small RNAs, similar to previous observations in vertebrates (Parrish et al. 2015; Palatini et al. 2017; Whitfield et al. 2017). EVE-derived piRNAs have been suggested to play a role in mosquito immunity against related exogenous viruses, although this still lacks supporting evidence in vivo (Girardi et al. 2017; Whitfield et al. 2017; Tassetto et al. 2019). Previous EVE studies in mosquitoes were based on incomplete versions of the A. aegypti genome (AaegL3 based on Nene et al. 2007 and the genome of the Aag2 cell line from Whitfield et al. 2017), which hamper inferences on abundance, integration preferences, and origin of EVEs. This could severely bias our interpretation of EVE biology. Indeed, overall mechanisms driving the integration of viral sequences and the possible functions of EVEs remain unclear.
Here, we carried out de novo identification of EVEs based on the AaegL5 version of the A. aegypti genome. This reference was assembled using long reads associated to a Hi-C strategy to achieve high coverage and generate a reference that is anchored end-to-end to the three A. aegypti chromosomes. This is extremely important since 40%–45% of contigs from the previous version could not be assigned to a single chromosome location (Juneja et al. 2014; Timoshevskiy et al. 2014). In addition, the AaegL5 version was able to “deduplicate” a number of sequences found in multiple copies in previous assemblies, thus providing a more concise reference (Matthews et al. 2018). Our EVE identification approach offered a unique advantage of consolidating fragmented elements and provided a more reliable reference for the assessment of their abundance and localization in the A. aegypti genome. EVEs in the AaegL5 version were associated with TEs and preferentially located in repeat-rich genomic clusters as observed in previous work (Palatini et al. 2017; Whitfield et al. 2017). In order to gain insights into possible functions, we investigated the transcriptional profile of EVEs. We observed that ∼99% of EVE transcription was antisense and a single ∼143 kb cluster in chromosome 2 generated around 42% of RNAs derived from viral elements in A. aegypti. This ∼143 kb cluster generated a single continuous antisense transcript that correlated with the accumulation of abundant EVE-derived small RNAs with a clear signature of phased primary piRNAs. These results indicate this is a unidirectional piRNA cluster that reveals a striking similarity with the flamenco locus from Drosophila melanogaster, including a conserved binding site for the transcription factor Cubitus interruptus. These observations have important implications for our understanding of EVE biology in mosquitoes.
RESULTS
Comprehensive de novo identification of EVEs in the AaegL5 genome
Characterization of EVEs remains challenging in most organisms. Here, we applied a strategy to identify and characterize EVEs using sequence similarity searches over the reference genome (Supplemental Fig. S1A). We applied our strategy to the vector mosquito A. aegypti whose previous genome versions has been shown to be covered with EVEs (Nene et al. 2007; Akbari et al. 2013; Palatini et al. 2017; Whitfield et al. 2017). In contrast, to previous work, we analyzed the AaegL5 version of the A. aegypti genome, which has been assembled with long reads providing significant improvement over older versions (Matthews et al. 2018). Using this reference, we ran a de novo prediction of open reading frames (ORFs) followed by sequence similarity comparisons against the Genbank database. A total of 8863 ORFs showed significant similarity (E-value <1 × 10−5) to viral sequences, which were reduced to 2168 after merging of adjacent ORFs. This was an essential step as we noted that adjacent EVEs showed similarity to the same virus and were likely a result of a single integration event (Supplemental Fig. S1B). We confirmed the relevance of this step by applying it to previous EVE data sets described in the genome of Aag2 cells (Whitfield et al. 2017). In this data set, merging of adjacent ORFs led to a reduction in the total number of EVEs from 472 to 352.
Next, 2168 EVEs were manually curated in order to separate retroviral elements and remove misidentified TEs and transpovirons. The latter are especially challenging since these are transposons integrated into the genome of DNA viruses (Desnues et al. 2012). The majority of EVEs (83%) were retroviral with only 129 non-retroviral elements remaining after curation (Supplemental Table S2). Our strategy yielded lower overall EVE numbers than expected based on recent reports using the genome of Aag2 cells and the AaegL3 version (Palatini et al. 2017; Whitfield et al. 2017). The use of a more concise and reliable reference genome contributed to the reduction in the number of EVEs. Indeed, applying our own strategy to the AaegL3 version of the A. aegypti genome resulted in the identification of 181 non-retroviral elements, an increase of more than 40% in the total number of EVEs compared to AaegL5 (Supplemental Table S3).
Correlation between diversity of EVEs and exogenous viruses in A. aegypti
We next analyzed the diversity of elements using this conservative set of 129 non-retroviral EVEs identified in the AaegL5 version of the A. aegypti genome. Using the closest viral sequence for classification, EVEs showed similarity to at least six viral families including Rhabdoviridae, Flaviviridae, and Phenuiviridae and numerous other unclassified viruses. Unclassified EVEs included sequences with similarity to many viruses recently described in insects such as Totivirus-like, Rhabdovirus-like, Partitivirus-like, and Virgavirus-like (Li et al. 2015; Lara Pinto et al. 2017).
We next investigated whether the diversity of viral families represented in EVEs correlated with viruses reported to be circulating in Aedes mosquitoes. Data mining in GenBank databases retrieved 116 unique viral sequences from 17 families and 40 more unclassified viruses previously described in Aedes mosquitoes. Using this data set, there was significant correlation between the abundance of viral groups in EVEs and exogenous viruses (r = 0.65, P = 0.003) (Fig. 1A). We also observed significant correlation when we restricted our analysis to sequences derived from unclassified viruses (r = 0.54, P = 0.04) (Supplemental Fig. S2).
FIGURE 1.

Characterization of nonretroviral EVEs in A. aegypti mosquitoes. (A) Scatter plot showing the correlation between the abundance of viral families assigned to EVEs and exogenous viruses circulating in A. aegypti mosquitoes. r (Pearson correlation) and P-values are indicated. Viral families represented among EVEs are highlighted with different colors. Only the most abundant viral families exclusively found among circulating viruses are also named. (B) Genomic origin of EVEs in A. aegypti. (C) Distance of EVEs to the closest feature annotated in the same or opposite strands of the A. aegypti genome. Significance of colocalization was computed using Fisher's exact test for genomic data. A bin of 500 nt was utilized. EVEs annotated as genes were not considered for this analysis. (D) Percentage of EVEs association with TEs in the A. aegypti genome. EVEs found within 500 nt of TEs were considered associated. (E) Number of EVEs per chromosome of the AaegL5 version of the A. aegypti genome. EVEs were grouped by bins of 20 kb separated by strand. The AaegL5 version of the A. aegypti genome was used for EVE identification.
EVEs only showed similarity to viruses with RNA genomes. Indeed, DNA viruses were significantly underrepresented in EVEs present in the A. aegypti genome compared to viruses circulating in mosquitoes (P = 0.0007) (Supplemental Fig. S3).
EVEs are often associated with TEs in genomic clusters
EVEs in the AaegL5 version of the A. aegypti genome were preferentially located in non-coding regions although a small percentage (15%) corresponded to exons (Fig. 1B). In the latter case, we note that a clear enrichment compared to the proportion of exons in the whole mosquito genome (2.17%) (Fig. 1B; Supplemental Fig. S4). This suggests frequent co-option of integrated EVEs by the host as previously proposed (Palatini et al. 2017; Suzuki et al. 2017; Whitfield et al. 2017). EVEs were often found in the vicinity of repeats and TEs (Fig. 1C). The Fisher enrichment test for genomic data indicated that these EVEs are significantly associated with TEs (P < 2 × 10−16) but not with repeats, as previously reported (Palatini et al. 2017; Suzuki et al. 2017; Whitfield et al. 2017). Most EVEs (52.17%) were within 500 nt of TEs (Fig. 1D).
Independently of TE association, EVEs were preferentially found in genomic clusters with at least two elements within a maximum 20 kb of each other. We identified a total of 21 clusters containing 91 EVEs compared to 38 single viral elements in the A. aegypti genome. These were distributed across the three chromosomes of A. aegypti with significant enrichment in Chr.2 (P = 0.046). This is noteworthy since we observed no specific enrichment for TEs in any of the A. aegypti chromosomes. EVE clusters and single elements were numbered according to the order they appear from the start of Chr.1 to the end of Chr.3 (Fig. 1E). The number of EVEs in clusters varied, with the largest one containing 17 elements (cluster 38) located on the end of the right arm of Chr.2 (Fig. 1E).
Disproportionate contribution of a single genomic cluster to EVE-derived transcripts and small RNAs in A. aegypti
Transcription of genomic elements is usually indicative of activity (Mills et al. 2007). We analyzed long RNA libraries from whole mosquitoes and observed EVE-derived transcription arising from several clusters and isolated elements in the A. aegypti genome (Fig. 2A). The vast majority (98.6%) of EVE-derived RNAs were antisense to the annotation. As expected, sense RNAs were preferentially derived from EVEs annotated as exons, whose expression levels were similar to other genes.
FIGURE 2.

EVEs are organized in genomic clusters that generate antisense RNAs. (A) Production of EVE-derived transcripts for each cluster or single EVE in the A. aegypti genome. (B) Scatter plot showing the relation between EVE transcription and the density of viral elements for each cluster or single EVE in the A. aegypti genome. (C) Contribution of each cluster or single EVE to total transcription and abundance of EVEs on reference genome. (D) Abundance of EVE-derived small RNAs for each cluster or single EVE in the A. aegypti genome. (E) Scatter plot showing the relation between EVE-derived small RNAs and the density of viral elements for each cluster or single EVE. (F) Contribution of each cluster or single EVE to total small RNA production and abundance of EVEs on reference genome. Fifty-nine EVE regions are defined, in which regions with more than one element are referred to as clusters. Each EVE region is numbered according to its location on AaegL5 version of the A. aegypti chromosomes, as indicated in Figure 1E. Fisher's exact test was applied. P-values are indicated for each comparison. RNA libraries from whole mosquitoes were used in this analysis.
Antisense EVE transcription occurred from different locations and there was poor correlation between transcription and EVE density (measured as the number of EVEs per kb) (Fig. 2B). We observed that a single location, cluster 38, accounted for 42% of EVE-derived antisense RNAs (Fig. 2C). Although cluster 38 had the largest number of EVEs, it had a significantly larger contribution to EVE-derived transcription when compared to its size (Fig. 2C). In addition to cluster 38, only single EVEs 10 and 59 contributed more than expected to EVE-derived transcription but with lower significance (Fig. 2C). Cluster 38 stood out in its capacity to generate transcripts even when the density of EVEs was weighed (Fig. 2B).
Antisense transcription is usually associated with mechanisms of regulation of gene expression such as RNA interference. We observed that several EVEs distributed across the A. aegypti genome generated small RNAs. Similar to our observation for transcription, EVE-derived small RNAs were mostly antisense (∼98%) (Fig. 2D). There was poor overall correlation between EVE density and the generation of antisense small RNAs (Fig. 2E). Here again cluster 38 disproportionally contributed with ∼45% of all EVE-derived small RNAs in A. aegypti (Fig. 2F). Cluster 38 had a significantly larger contribution to the generation of small RNAs compared to the number of viral elements (Fig. 2F). A larger contribution was also observed for a single EVE 56 but with lower significance.
Our data show that cluster 38 was unique in its disproportionate contribution to EVE-derived transcripts and small RNAs. The localization of EVEs within clusters and the generation of small RNAs has been noted in previous studies based on different reference genomes (Palatini et al. 2017; Suzuki et al. 2017; Ter Horst et al. 2019). However, the disproportionate contribution of a single EVE cluster for the generation of small RNAs has not been reported. In order to verify whether a similar EVE cluster was present in other genome references, we analyzed version AaegL3 using our own strategy. An EVE cluster located in supercontig 1.286 of this genome reference was similar to cluster 38 of the AaegL5 version (dotplot in Fig. 3A). In agreement with this similarity, supercontig 1.286 was localized to a close region in Chr.2 (2q44) using Restricted-site Associated DNA (RAD) sequencing by Juneja et al. (2014). Nevertheless, there were substantial differences in size and organization between the EVE cluster in the two versions of the genome (Fig. 3A). These may represent assembly errors in one of the references because this is a highly repetitive region that can be difficult to resolve. However, these differences could also reflect natural polymorphisms between strains. In order to further analyze these two possibilities, we took advantage of the long reads from the latest sequencing effort (Matthews et al. 2018). We observed homogeneous long read coverage along the cluster 38 in the AaegL5 version opposing to several gaps in the supercontig 1.286 of the AaegL3 version (Fig. 3B). For example, we observed a 5 kb block present in the AaegL3 assembly that is repeated four times spanning over 20 kb in the AaegL5 version (highlighted in the dotplot of Fig. 3A and shown in details in Fig. 3C). Each 5 kb block seems to represent a single copy of an LTR retrotransposon containing putative capsid and reverse transcriptase/integrase genes (Fig. 3C). Close analysis of local sequencing coverage of the 20 kb region in the AaegL5 version reveals individual long reads that contain many individual reads that spanned multiple copies of the 5 kb block (Fig. 3D). Together, these data give support to the structure of cluster 38 assembled in the AaegL5 version, which is an accurate reflection of the genome from the strain sequenced in Matthews et al. (2018). Although supercontig 1.286 of the AaegL3 version may represent natural variations found in other mosquito strains, previous available data suggests it contains assembly errors. Indeed, Timoshevskiy et al. (2014) placed supercontig 1.286 in two separate chromosomes (Chr.1 and Chr.2) by direct hybridization thus suggesting it is a misassembly.
FIGURE 3.

Comparative analysis of the largest EVE cluster in AaegL3 and AaegL5 versions of the A. aegypti genome. (A) Dotplot showing pairwise comparison of the largest EVE cluster in AaegL3 and AaegL5 versions of the mosquito genome. Shaded areas show a divergent region between the two genome versions that is further analyzed in C. (B) Graph showing coverage of the region spanning the largest EVE cluster identified in AaegL3 and AaegL5. (C) Zoom of a divergent region within the largest EVE cluster (shaded area in A) in the AaegL5 version of the mosquito genome showing the coverage of PacBio long reads. The structure of the region shows a repeated 5-kb block composed of a putative LTR retrotransposon containing capsid and a reverse transcriptase/integrase genes. (D) Single long reads containing the full repeated block give support for the assembly in the AaegL5 version of the A. aegypti genome.
Characterization of the largest EVE cluster in A. aegypti
EVE cluster 38 in Chr.2 covered a repetitive region of ∼143 kb containing three genes, 17 EVEs and 73 TEs, almost all oriented in the same coding strand (Fig. 4A). TEs identified in this region included LTR Retrotransposons (Pao/Bel and Ty3/Gypsy), Cut and Paste DNA transposon (Tc1) and mTA elements (MITE). Retrotransposons from the gypsy family were enriched and represented the majority of TEs within cluster 38 (∼59%) (TEs with black borders in Fig. 4A). Notably, the three genes in this cluster are all A. aegypti specific and correspond to EVEs that were annotated as exons. These genes showed no evidence for sense transcription and could be misannotations. The entire 143 kb region had very little coverage of sense RNAs but showed continuous coverage of antisense transcripts spanning both TEs and EVEs (Fig. 4A). There was significant correlation between transcription of adjacent EVEs and TEs inside cluster 38 suggesting the region is transcribed as a single unit (Supplemental Fig. S5). Genomic elements within cluster 38, both EVEs and TEs, also had high continuous coverage of antisense small RNAs (Fig. 4A). The generation of antisense transcription and small RNAs by TEs and EVEs within cluster 38 were significantly correlated (Fig. 4B). In addition, abundance of long and small RNAs were proportional to the length of the EVE or TE (Fig. 4B). These results are consistent with the hypothesis that this region generates a single long transcript that is continuously processed into small RNAs.
FIGURE 4.

A TE and EVE rich region in the A. aegypti genome is related to the flamenco locus. (A) Zoomed-in view of the shaded area in chromosome 2 corresponding to cluster 38 indicating EVE, gene and TE content per strand. Middle graphs show the density of transcripts and small RNAs considering single and multiple mapped reads. Bottom graph shows gap occupancy of small RNA coverage over the entire region. The dashed line represents a significance cutoff of P = 0.05 that the gap occurred by chance. Significant gaps are shown. (B) Correlation between production of long and small RNAs for each element within cluster 38. Each dot represents one element with the type (EVEs or TEs) and length indicated by colors and size of the symbol, respectively. (C,D) Size distribution (C) and base preferences (D) for EVEs and TEs within EVE cluster 38. (E) Relative frequency of distances between 3′ and 5′ ends of piRNAs derived from the same genomic strand within EVE cluster 38 in mosquitoes. (F) Nucleotide composition of the genomic neighborhood of small RNA 3′ ends. (G) Distance between 5′ ends of small RNAs in different strands for EVEs and TEs within EVE cluster 38. (H) Genomic context of promoter region of the flamenco locus and EVE cluster 38 in A. aegypti. Small RNAs between 24 and 29 nt were used for the analyses in B–F. RNA libraries from whole mosquitoes and flies were used in this analysis.
TE- and EVE-derived small RNAs from within cluster 38 had characteristics of canonical piRNAs. These were 24 to 29 nt in length and had significant enrichment for U at the first base (Fig. 4C,D). A similar profile was observed for small RNAs derived from EVEs and TEs within cluster 38 (Fig. 4C,D). There was significant 1-nt phasing between antisense small RNAs derived from TEs and EVEs within cluster 38, which occurs when a single transcript is processed continuously into primary piRNAs (Fig. 4E). We also observed that processing of the 3′ end of phased primary small RNAs derived from cluster 38 occurs preferentially at U as described for primary piRNAs in Drosophila (Fig. 4F; Gainetdinov et al. 2018). These data indicate that cluster 38 is transcribed as a long precursor transcript that is processed into primary piRNAs.
Sense small RNAs were rare in this region, but showed significant 10-nt overlap with antisense pairs and strong enrichment for A at the 10th base (Fig. 4D,G). These are signatures of the ping-pong amplification mechanism that occurs when piRNAs find complementary targets (Brennecke et al. 2007). These results reinforce that cluster 38 is a source of canonical primary piRNAs that can trigger targeting of complementary RNAs. We note that the ping-pong signal for TE-derived piRNAs (P < 1 × 10−5) was slightly more significant than for EVEs (P < 3 × 10−5), suggesting that targets for viral elements might be less available.
EVE cluster 38 in A. aegypti is similar to the flamenco locus of Drosophila melanogaster
The organization and characteristics of cluster 38 are reminiscent of unidirectional or uni-strand piRNA clusters such as the flamenco locus of D. melanogaster. The flamenco locus is a long (>100 kb) repeat-rich region composed of elements organized in the same coding strand that are transcribed into a single transcript (Brennecke et al. 2007). This non-coding transcript is processed into primary phased piRNAs that are preferentially processed at U nucleotides on the transcript (Han et al. 2015). Although the D. melanogaster genome does not contain EVEs, our results show important similarities between the overall organization and function of EVE cluster 38 from A. aegypti and the flamenco locus.
In Drosophila, transcription of the flamenco locus is mediated by the transcription factor (TF) Cubitus Interruptus (Ci), whose binding site is located at position −385 of the transcription start site (Goriaux et al. 2014). Analysis of the putative promoter region of cluster 38 revealed a highly conserved binding site for Ci at position −1772 from the predicted transcription start site (Fig 4H). As a control, our strategy was able to identify de novo the Ci binding site in the flamenco promoter. These results suggest that Ci could also play a role in the transcription of cluster 38 in A. aegypti mosquitoes.
Hence, genomic organization, transcription profile and production of small RNAs suggest that cluster 38 is a unidirectional piRNA cluster related to the flamenco locus despite a complete absence of sequence similarity between them. Hereafter, we refer to cluster 38 as the flamenco-like locus.
EVE- and TE-derived piRNAs from the flamenco-like locus associate with PIWI proteins
piRNAs derived from EVEs and TEs within the flamenco-like locus were resistant to oxidation, which indicates they are modified at the 3′ end (Fig. 5A). piRNAs are methylated at the 3′ end after they are loaded onto PIWI proteins (Horwich et al. 2007; Vodovar et al. 2012). Thus, we used immunoprecipitation data generated from Aag2 cells to confirm the association of piRNAs with mosquito PIWI proteins (Girardi et al. 2017). Although Aag2 cells do not express all PIWI proteins, they appear to have a fully functional piRNA pathway (Vodovar et al. 2012; Akbari et al. 2013; Girardi et al. 2017). In these cells, piRNAs derived from the flamenco-like locus were strongly associated with PIWI5 and PIWI6, but not PIWI4 and AGO3 (Fig. 5B). In Drosophila, piRNAs derived from dual-strand clusters associate with distinct PIWI proteins compared to uni-strand clusters, such as the flamenco locus (Ozata et al. 2019). In order to further look into the specificity of interactions between small RNAs and PIWI proteins in mosquitoes, we identified dual and uni-strand piRNA clusters in Aag2 cells (Supplemental Table S4). The flamenco-like locus was the major uni-strand piRNA cluster in mosquitoes as observed for the flamenco locus in Drosophila. The top five uni-strand clusters basically showed the same pattern as the flamenco-like locus and associated significantly with PIWI5 and PIWI6 (Fig. 5B). In contrast, piRNAs derived from the top five dual-strand clusters associated more significantly to AGO3 and only weakly with PIWI5 (Fig. 5B). As a control, microRNAs (miRNAs) did not show significant association with any PIWI protein since they represent another class of small RNAs (Fig. 5B). Together, these data support the idea that the flamenco-like locus is the major uni-strand piRNA cluster in the mosquito genome.
FIGURE 5.
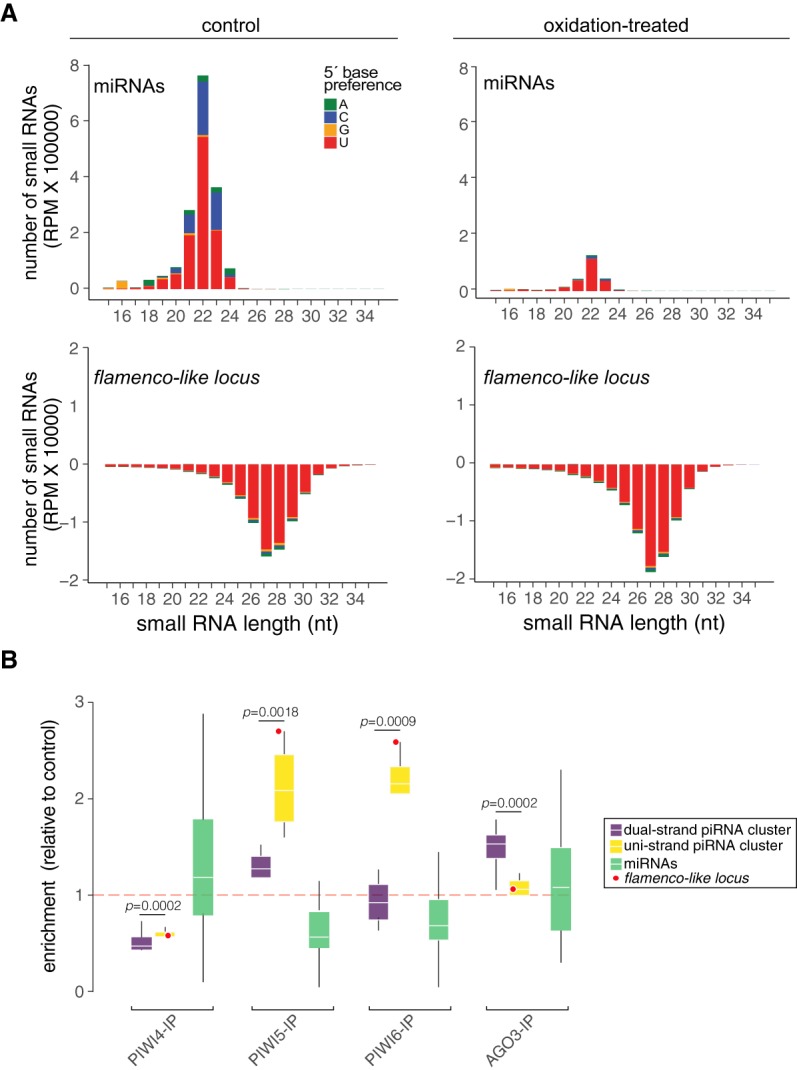
EVE-derived piRNAs from the flamenco-like locus associate with mosquito PIWI proteins. (A) Size profile of small RNA derived from miRNAs or the flamenco-like locus in control or oxidation-treated libraries. The nt at the 5′ end is indicated by color. (B) Association of piRNAs derived from the top five uni- and dual-strand clusters (highlighted in Supplemental Table S4) compared to microRNAs with different PIWI proteins based on immunoprecipitations from mosquito Aag2 cells. The flamenco-like locus belongs to the group of uni-strand piRNA clusters and is highlighted in red for comparison. Statistics were performed using Wilcoxon rank-sum test. Significant P-values (P < 0.01) are indicated. Small RNA libraries from mosquito Aag2 cells were used in these analyses.
TE-derived piRNAs from the flamenco-like locus showed complementarity to over 20% of all transcriptionally active transposons in the A. aegypti genome. Indeed, the presence of a ping-pong signal suggests active targeting of TEs by these piRNAs (Fig. 4D,G). EVE-derived piRNAs were also complementary to viral elements elsewhere in the mosquito genome although most of them did not generate sense RNAs. This lack of targets may explain the lower abundance of sense piRNAs derived from the flamenco-like locus (Fig. 4A). Other groups have also reported potential antiviral functions for EVE-derived piRNAs against exogenous viruses (Fort et al. 2012; Suzuki et al. 2017; Tassetto et al. 2019). We observed that few EVE-derived piRNAs (0.12%) from the flamenco-like locus showed high complementary (>90%) to genomes of 25 circulating viruses (Supplemental Table S5). However, we did not observe sense-derived piRNAs from these exogenous viruses, which would be indicative of active targeting.
DISCUSSION
Non-retroviral EVEs are repetitive elements that represent a new frontier in genome biology. These are found in many organisms but their functions remain unclear. Identification of EVEs in new genomes is still a challenge since they tend to follow mutation rates of the host genome thus accumulating changes that separate them from their original viral source (Preston 1996; Feschotte and Gilbert 2012; Whitfield et al. 2017). Aedes mosquitoes have recently been the focus of several EVE studies due the repetitive nature of their genome (Palatini et al. 2017; Suzuki et al. 2017; Whitfield et al. 2017; Tassetto et al. 2019). Here, we applied our own EVE identification strategy to the AaegL5 version of the A. aegypti genome, which is a significant improvement over previous references (Matthews et al. 2018). This new improved genome version coupled to our stringent strategy led to the identification of a highly reliable set of EVEs in the A. aegypti genome that could be explored to provide insights into the biology of these elements.
EVEs identified in A. aegypti were related to viruses with RNA genomes, which is consistent with recent reports (Palatini et al. 2017; Suzuki et al. 2017; Whitfield et al. 2017). However, several EVEs corresponded to viruses that were only recently described, suggesting that the analysis could still be limited by the availability of references (Li et al. 2015; Shi et al. 2016). EVEs were depleted for sequences derived from DNA viruses, even accounting for their underrepresentation in mosquitoes. This result suggests that integration is affected by the nature of the viral genome and its replication strategy. Since retrotranscription mediated by autonomous TEs has been shown to drive integration of viral sequences, the lower amounts of RNA produced by DNA viruses in comparison to RNA viruses could explain their underrepresentation (Weber et al. 2006; Whitfield et al. 2017). Similar to previous reports, our results suggested that TEs likely play a role on EVE integration (Palatini et al. 2017; Whitfield et al. 2017). Indeed, RT enzymes encoded by TEs are promiscuous in the recognition of target sites, and may bind viral RNA during infection thus producing TE-virus DNA hybrids (Preston 1996; Goic et al. 2016; Whitfield et al. 2017). This is probably the origin of non-retroviral EVEs that are integrated into the host genome in association with TEs. Mobilization of the associated TE can then lead to transposition of flanking viral sequences (Kidwell and Lisch 1997). Accordingly, EVEs tend to be clustered, which is consistent with the mechanism of TE mobilization (Tower et al. 1993). We also note that EVEs were preferentially found in the same coding direction as the closest TE (∼90%), reinforcing the idea of functional association.
A major frontier in EVE biology has been the assignment of possible functions for these elements. Our genome-wide analysis of EVE transcription indicated that the vast majority of EVEs only generated antisense transcripts, which would not be able to generate protein. Rather, EVE-derived transcripts correlated well with the generation of antisense small RNAs with characteristics of piRNAs. Although production of piRNAs by EVEs has been noted (Palatini et al. 2017; Suzuki et al. 2017; Whitfield et al. 2017; Ter Horst et al. 2019), the analysis of transcription and small RNAs derived from EVEs was not done before. In our work, this analysis prompted the identification of a single EVE cluster, number 38, covering a 143 kb region in Chr.2 of the AaegL5 version of the A. aegypti genome as the major source of transcripts and piRNAs derived from viral elements. Our data shows that EVE cluster 38 is the major uni-strand piRNA cluster in mosquitoes that is related to the flamenco locus from D. melanogaster despite any sequence similarity. The identification of genomic regions with similar organization and function have also been described in other Drosophila species and other organisms such as Arabidopsis (Malone et al. 2009; Grob et al. 2014). It is noteworthy that even within Drosophila species, there was significant sequence divergence and the locus was only conserved considering genomic organization and structure. In both Drosophila and Arabidopsis, these loci are heterochromatic islands showing elevated levels of methylation, enriched for remnants of RNA-mediated TEs and production of small RNAs (Zanni et al. 2013). These regions are preferred landing sites for TEs, which is presumably a host strategy to trap and promote silencing of these elements through the production of small RNAs (Grob et al. 2014). This has been well documented for TEs in the flamenco locus of Drosophila.
The identification of this unique cluster that we refer to as the flamenco-like locus has important implications for the possible functions of EVEs. Integrations of viral sequences into the flamenco-like locus are likely to be a byproduct of their association with TEs rather than driven by specific mechanisms directed at viruses. In Drosophila, TEs found in the flamenco locus are often non-functional (Zanni et al. 2013). Since TE-EVE hybrids are not likely to be functional, their integration within the flamenco-like locus could be favored in mosquitoes. Indeed, we observed EVE enrichment in Chr.2 although TEs are not concentrated in any specific chromosome of A. aegypti. Notably, EVE enrichment in Chr.2 is not significant if the flamenco-like locus is not taken into account, which suggests it is the only region that favors integration of EVEs in Chr.2. Importantly, the association between TEs and EVEs likely occurs by chance and TEs are homogeneously present throughout the A. aegypti genome. Thus, our results suggest that the enrichment of viral elements in the flamenco-like locus is based on the selection of non-functional TEs and not on the fact that EVE integrations may provide an advantage to the host. Hence, our work suggests that EVE-derived small RNAs are a byproduct of their association with non-functional TEs and do not have a direct function. Nevertheless, further work is still required to characterize possible functions of non-retroviral EVEs in mosquitoes and other animals. The identification of the flamenco-like locus as the major source of EVE-derived piRNAs in mosquitoes is an important step toward this goal. It will be of great interest to examine the structure and sequence of the flamenco-like locus in distinct strains of A. aegypti and other mosquito species to understand the evolution of this locus. Indeed, some differences in the flamenco-like locus between AaegL5 and AaegL3 versions of the A. aegypti genome could indicate some natural polymorphisms in this region.
MATERIALS AND METHODS
Reference genomes
We analyzed different versions of the A. aegypti genome (AaegL3 and AaegL5). Files were downloaded from VectorBase (www.vectorbase.org). References of transposable elements were downloaded from TEfam (https://tefam.biochem.vt.edu/tefam; accessed April 26, 2017) and VectorBase. Transposable elements in EVE Cluster 38 were manually curated in order to remove annotation redundancy.
Identification of EVEs
Genome-based identification
ORFs of at least 100 nucleotides in the A. aegypti genome were predicted using the getorf program from EMBOSS package (Rice et al. 2000). Sequence similarity searches were performed with DIAMOND software (Buchfink et al. 2015) using translations of predicted ORFs in all six frames as queries for comparison against the nonredundant protein sequence database (NR) in GenBank. The five best hits with maximum E-value threshold of 1.0 × 10−5 were considered.
ORF extension
Fragmented EVE sequences were consolidated in order to avoid misannotation and overestimations. Here, annotated viral sequences that were distant up to 150 nt to each other in the same genomic strand and showed similarity to the same exogenous virus were consolidated into a single EVE using the merge program from BEDTools package (Quinlan and Hall 2010).
Manual curation
After consolidation, putative EVEs were maintained if they did not show significant similarity (E-value < 10−5) by BLAST searches to: (i) retroviral sequences, (ii) sequences in other nonviral organisms, and (iii) or TEs.
Comparative genomics of EVE cluster
To examine read support for the flamenco-like region across reference genomes, all PacBio reads (NCBI SRA SRS2349110) generated as part of the AaegL5 reference genome assembly (Matthews et al. 2018) were aligned to the reference genome assembly regions. Alignments were performed using blasr (Chaisson and Tesler 2012) with the following flags: blasr –nproc 30 –bam –bestn 10 –minMatch 12 –maxMatch 30 –minSubreadLength 500 –minAlnLength 500 –minPctSimilarity 70 –minPctAccuracy 70 –hitPolicy randombest –randomSeed 1 –minPctSimilarity 70.0 –refineConcordant Alignments. PacBio alignments were filtered using samclip (https://github.com/tseemann/samclip) to remove reads with clippings of >100 bp. Depth of coverage from raw alignments or filtered reads was extracted with SAMtools “depth” (Li et al. 2009) and plotted using R and ggplot2. To examine read support for a repeat region located at EVE cluster 38 in AagL3 and AagL5 versions of the mosquito genome, individual alignments were visualized in IGV (Thorvaldsdóttir et al. 2013) to verify that there was read support comprised of individual reads that spanned multiple repeat copies.
Identification of circulating viruses based on NCBI databases
In order to assess viruses circulating in mosquitoes, we performed a comprehensive search in the NCBI Nucleotide database (http://www.ncbi.nlm.nih.gov/nuccore/) using esearch and efetch programs from the Entrez Programming Utilities (E-utilities) (Sayers 2009). The search was performed using keywords “Aedes OR mosquito” and txid10239 [Organism] as host and “viruses[filter]” in order to retrieve GenBank files in XML format. Organism name and family information of each viral sequence were retained based on the presence of “Viruses” as the upper taxa term in the taxonomy field and “Aedes aegypti” as the value of the host qualifier. Viruses that did not have a determined viral family by ICTV, referred to as unclassified, were characterized based on the literature (Li et al. 2015; Shi et al. 2016; Lara Pinto et al. 2017).
Genomic analysis
Comparisons between the genomic origin of EVEs and features annotated on the A. aegypti genome, including analysis of colocalization, were performed using Fisher's exact test, flank, merge, fisher, reldist, and closest programs built into the BEDTools package (Quinlan and Hall 2010). For analysis of association with the TE and repeats, regions up- and downstream from EVEs were analyzed for the presence of annotated repetitive elements in the A. aegypti genome (AaegL5), in which we considered association elements up to 500 nt distant in the same genomic strand.
Construction and sequencing of small RNA libraries
Total RNA was extracted from Aag2 cells using TRIzol (Invitrogen). RNA oxidation was performed according to protocols described by the Zamore Lab (April, 2014; http://www.umassmed.edu/zamore/resources/protocols/). Briefly, two portions of total RNA containing 20 µg each were dissolved in borate buffer (pH 8.6) and then mixed with 8 µL of freshly prepared 200 mM NaIO4 (oxidizing reaction) or 8 µL of water (control reaction) in a final volume of 40 µL. Reactions were incubated at room temperature for 30 min in the dark, and the RNA was size selected (18 to 30 nt) on a denaturing PAGE and eluted from the gel followed by ethanol precipitation in the presence of 300 mM NaCl and 5 µg of glycogen. The recovered small RNA fraction was entirely used for library construction utilizing the TruSeq Small RNA Library Prep Kit (Illumina). Samples were sequenced using the Illumina HiSeq 4000 platform at the IGBMC Microarray and Sequencing facility (Strasbourg, France).
Analysis of TE- and EVE-derived RNAs
Mapping of sequenced reads from long and small RNA libraries was performed using Bowtie (Langmead 2010) and Bowtie 2 (Langmead and Salzberg 2012), respectively. Counts were computed considering each strand separately and abundance of small and long RNAs was normalized by reads per million (RPM) or fragments per kilobase per million (FPKM), respectively. Two classes of reads were considered: (i) single or unique mappers representing reads that mapped only once to the reference genome; and (ii) multiple mappers encompassing reads that mapped two or more times to the reference genome. For analysis of the origin of small RNAs, only unique mappers were considered. For analysis of abundance, pondering quantification was used as previously described (Gainetdinov et al. 2018). Briefly, we considered the number of times that a small RNA sequence appears in the library divided by the number of mappings to the reference genome. Analysis of EVE-derived RNAs was restricted to those elements present in the current version of the A. aegypti genome (AaegL5).
Analysis of small RNAs’ characteristics
Distances between small RNAs were calculated as previously described (Han et al. 2015; Gainetdinov et al. 2018). Briefly, the frequency of 5′ to 5′ or 5′ to 3′ distances between 24- to 29-nt-long small RNAs in the same strand or 5′ to 5′ distances between 24- to 29-nt-long small RNAs in opposite strands was calculated and normalized by Z-score. Sequence preferences for the genomic neighborhoods of 3′ ends of piRNAs were generated with motifStack and plotted as weblogo (Ou et al. 2018). Gap occupancy analysis was performed as previously described (Marques et al. 2013). Briefly, we calculated the probability that each gap in the coverage of the reference did not occur by chance by considering the raw small RNA coverage and the abundance of reads in the boundaries of the gap.
Identification of transcription factor binding sites
Fasta files with transcription binding sites (TBS) motif sequences of Cubitus Interruptus (Ci), Br (Broad), BgB (Big Brother), and Dsx (Double sex) identified in Drosophila melanogaster (Goriaux et al. 2014) were obtained from Fly Factor Survey database (http://mccb.umassmed.edu/ffs/). We generated a PSP (position-specific priors) matrix for each TBS. We used the software FIMO (Grant et al. 2011) to calculate the background model and search for the target TF motif sequences in the promoter region of A. aegypti flamenco-like locus (1Kbp downstream and 2 Kbp upstream of the inferred transcription start site) in the AaegL5 reference genome with a P-value threshold of 0.001.
Identification of piRNA clusters
For the identification of piRNA clusters, small RNA libraries from Aag2 cells were mapped to the AaegL5 reference genome divided into 2 kb segments. Segments that presented high coverage were selected and classified based on the preferential size of mapped reads, as previously described (Brennecke et al. 2007). To avoid false-positives, regions that were annotated as miRNA genes were discarded. Genomic segments with an accumulation of a majority of small RNAs between 24 and 30 nt, coverage of at least 30% and a minimum of 500 reads were classified as piRNA clusters. Adjacent segments were consolidated as a single cluster. Clusters with piRNA coverage in both strands (ratio < 10:1) were classified as dual-strand. Information about the top 200 piRNA clusters identified in Aag2 cells are in Supplemental Table S4.
Evaluation of the impact of PIWI proteins on EVE-derived piRNAs
Small RNA libraries were compared to EVEs or TEs in the mosquito genome allowing for one mismatch using Bowtie. The pondered abundance of each element was calculated and normalized by RPM. A pseudocount of one was added to each EVE or TE before calculating fold changes. Changes in abundance of TEs and EVEs in immunoprecipitation (IP) libraries were calculated by normalizing each library by its control, GFP IP. Fold change was plotted as a boxplot. Differences in the association of piRNAs derived from uni- and dual-strand piRNA clusters in IPs of each PIWI protein were calculated using Wilcoxon rank-sum test.
Identification of potential piRNA targets
In order to identify viruses that could be potentially targeted by piRNAs originating from the flamenco-like locus, we first identified exogenous viral genomes closely related to EVEs in this region. Through sequence similarity searches against the NCBI NT database using BLAST software requiring E-value < 1 × 10−5, we selected candidate viral references. piRNAs derived the flamenco-like locus were aligned to these viral genomes up to two mismatches in total.
Statistics
Analyses of correlation were carried out using the Pearson correlation test. For analysis of EVE-TE association, we applied a Fisher's exact test modified to genomic data built into the BEDTools package. Enrichment for production of small and long RNAs derived from EVEs was computed using Fisher's exact test where P < 0.05 was shown.
DATA DEPOSITION
All libraries used in this work are publicly available in the SRA NCBI repository. Access numbers and descriptions are described in Supplemental Table S1.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Marques and Imler laboratories for discussion and the IGBMC core facility in Strasbourg for sequencing services. We also thank anonymous reviewers whose criticisms have contributed to this manuscript. This work was supported by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) to E.R.G.R.A. and J.T.M.; Fundação de Amparo a Pesquisa do Estado de Minas Gerais (FAPEMIG) and Instituto Nacional de Ciência e Tecnologia de Vacinas (INCTV) to J.T.M.; the Investissement d'Avenir Programs (ANR-10-LABX-0036 and ANR-11-EQPX-0022) to J.T.M. and J.L.I.; and Google Latin American Research Award (LARA 2019) to J.T.M. and J.P.P.A. This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brasil (CAPES), Finance Code 001. E.R.G.R.A., L.S.O., R.P.O., I.J.S.F., L.R.Q., and J.P.P.A. were supported with fellowships from CAPES.
Author contributions: J.T.M. and E.R.G.R.A. conceived the project. E.R.G.R.A., R.P.O., L.R.Q., I.J.S.F., J.P.P.A., L.S.O., A.G., B.J.M., J.L.I., and J.T.M. performed experiments and analyzed the data. J.L.I. and J.T.M. contributed reagents and materials. E.R.G.R.A., J.L., B.J.M., and J.T.M. wrote the paper. All authors read and contributed with suggestions to the latest version of the manuscript.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.073965.119.
REFERENCES
- Aiewsakun P, Katzourakis A. 2015. Endogenous viruses: connecting recent and ancient viral evolution. Virology 479–480: 26–37. 10.1016/j.virol.2015.02.011. [DOI] [PubMed] [Google Scholar]
- Akbari OS, Antoshechkin I, Amrhein H, Williams B, Diloreto R, Sandler J, Hay BA. 2013. The developmental transcriptome of the mosquito Aedes aegypti, an invasive species and major arbovirus vector. G3 3: 1493–1509. 10.1534/g3.113.006742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyi VA, Levine AJ, Skalka AM, Buchmeier MJ. 2010. Unexpected inheritance: multiple integrations of ancient bornavirus and ebolavirus/marburgvirus sequences in vertebrate genomes. PLoS Pathog 6: e1001030 10.1371/journal.ppat.1001030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ. 2007. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128: 1089–1103. 10.1016/j.cell.2007.01.043. [DOI] [PubMed] [Google Scholar]
- Buchfink B, Xie C, Huson DH. 2015. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12: 59–60. 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- Chaisson MJ, Tesler G. 2012. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desnues C, La Scola B, Yutin N, Fournous G, Robert C, Azza S, Jardot P, Monteil S, Campocasso A, Koonin EV, et al. 2012. Provirophages and transpovirons as the diverse mobilome of giant viruses. Proc Natl Acad Sci 109: 18078–18083. 10.1073/pnas.1208835109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewannieux M, Esnault C, Heidmann T. 2003. LINE-mediated retrotransposition of marked Alu sequences. Nat Genet 35: 41–48. 10.1038/ng1223. [DOI] [PubMed] [Google Scholar]
- Feschotte C, Gilbert C. 2012. Endogenous viruses: insights into viral evolution and impact on host biology. Nat Rev Genet 13: 283–296. 10.1038/nrg3199. [DOI] [PubMed] [Google Scholar]
- Fort P, Albertini A, Van-Hua A, Berthomieu A, Roche S, Delsuc F, Pasteur N, Capy P, Gaudin Y, Weill M. 2012. Fossil rhabdoviral sequences integrated into arthropod genomes: ontogeny, evolution, and potential functionality. Mol Biol Evol 29: 381–390. 10.1093/molbev/msr226. [DOI] [PubMed] [Google Scholar]
- Fujino K, Horie M, Honda T, Merriman DK, Tomonaga K. 2014. Inhibition of Borna disease virus replication by an endogenous bornavirus-like element in the ground squirrel genome. Proc Natl Acad Sci 111: 13175–13180. 10.1073/pnas.1407046111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov I, Colpan C, Arif A, Cecchini K, Zamore PD. 2018. A single mechanism of biogenesis, initiated and directed by PIWI proteins, explains piRNA production in most animals. Mol Cell 71: 775–790.e5. 10.1016/j.molcel.2018.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geuking MB, Weber J, Dewannieux M, Gorelik E, Heidmann T, Hengartner H, Zinkernagel RM, Hangartner L. 2009. Recombination of retrotransposon and exogenous RNA virus results in nonretroviral cDNA integration. Science 323: 393–396. 10.1126/science.1167375. [DOI] [PubMed] [Google Scholar]
- Girardi E, Miesen P, Pennings B, Frangeul L, Saleh MC, van Rij RP. 2017. Histone-derived piRNA biogenesis depends on the ping-pong partners Piwi5 and Ago3 in Aedes aegypti. Nucleic Acids Res 45: 4881–4892. 10.1093/nar/gkw1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goic B, Stapleford KA, Frangeul L, Doucet AJ, Gausson V, Blanc H, Schemmel-Jofre N, Cristofari G, Lambrechts L, Vignuzzi M, et al. 2016. Virus-derived DNA drives mosquito vector tolerance to arboviral infection. Nat Commun 7: 12410 10.1038/ncomms12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goriaux C, Desset S, Renaud Y, Vaury C, Brasset E. 2014. Transcriptional properties and splicing of the flamenco piRNA cluster. EMBO Rep 15: 411–418. 10.1002/embr.201337898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, Noble WS. 2011. FIMO: scanning for occurrences of a given motif. Bioinformatics 27: 1017–1018. 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grob S, Schmid MW, Grossniklaus U. 2014. Hi-C analysis in Arabidopsis identifies the KNOT, a structure with similarities to the flamenco locus of Drosophila. Mol Cell 55: 678–693. 10.1016/j.molcel.2014.07.009. [DOI] [PubMed] [Google Scholar]
- Grybchuk D, Akopyants NS, Kostygov AY, Konovalovas A, Lye LF, Dobson DE, Zangger H, Fasel N, Butenko A, Frolov AO, et al. 2017. Viral discovery and diversity in trypanosomatid protozoa with a focus on relatives of the human parasite Leishmania. Proc Natl Acad Sci 115: E506–E515. 10.1073/pnas.1717806115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han BW, Wang W, Li C, Weng Z, Zamore PD. 2015. Noncoding RNA. piRNA-guided transposon cleavage initiates Zucchini-dependent, phased piRNA production. Science 348: 817–821. 10.1126/science.aaa1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda T, Tomonaga K. 2016. Endogenous non-retroviral RNA virus elements evidence a novel type of antiviral immunity. Mob Genet Elements 6: e1165785 10.1080/2159256X.2016.1165785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie M, Honda T, Suzuki Y, Kobayashi Y, Daito T, Oshida T, Ikuta K, Jern P, Gojobori T, Coffin JM, et al. 2010. Endogenous non-retroviral RNA virus elements in mammalian genomes. Nature 463: 84–87. 10.1038/nature08695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwich MD, Li C, Matranga C, Vagin V, Farley G, Wang P, Zamore PD. 2007. The Drosophila RNA methyltransferase, DmHen1, modifies germline piRNAs and single-stranded siRNAs in RISC. Curr Biol 17: 1265–1272. [DOI] [PubMed] [Google Scholar]
- Juneja P, Osei-Poku J, Ho YS, Ariani CV, Palmer WJ, Pain A, Jiggins FM, Valenzuela JG. 2014. Assembly of the genome of the disease vector Aedes aegypti onto a genetic linkage map allows mapping of genes affecting disease transmission. PLoS Negl Trop Dis 8: e2652 10.1371/journal.pntd.0002652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzourakis A, Gifford RJ. 2010. Endogenous viral elements in animal genomes. PLoS Genet 6: e1001191 10.1371/journal.pgen.1001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwell MG, Lisch D. 1997. Transposable elements as sources of variation in animals and plants. Proc Natl Acad Sci 94: 7704–7711. 10.1073/pnas.94.15.7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryukov K, Ueda MT, Imanishi T, Nakagawa S. 2018. Systematic survey of non-retroviral virus-like elements in eukaryotic genomes. Virus Res 262: 30–36. 10.1016/j.virusres.2018.02.002. [DOI] [PubMed] [Google Scholar]
- Langmead B. 2010. Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics Chapter 11: Unit 11 7 10.1002/0471250953.bi1107s32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara Pinto AZd, Santos de Carvalho M, de Melo FL, Ribeiro ALM, Morais Ribeiro B, Dezengrini Slhessarenko R, Schneider BS. 2017. Novel viruses in salivary glands of mosquitoes from sylvatic Cerrado, Midwestern Brazil. PLoS One 12: e0187429 10.1371/journal.pone.0187429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CX, Shi M, Tian JH, Lin XD, Kang YJ, Chen LJ, Qin XC, Xu J, Holmes EC, Zhang YZ. 2015. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife 4: e05378 10.7554/eLife.05378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone CD, Brennecke J, Dus M, Stark A, McCombie WR, Sachidanandam R, Hannon GJ. 2009. Specialized piRNA pathways act in germline and somatic tissues of the Drosophila ovary. Cell 137: 522–535. 10.1016/j.cell.2009.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maori E, Tanne E, Sela I. 2007. Reciprocal sequence exchange between non-retro viruses and hosts leading to the appearance of new host phenotypes. Virology 362: 342–349. 10.1016/j.virol.2006.11.038. [DOI] [PubMed] [Google Scholar]
- Marques JT, Wang J-P, Wang X, de Oliveira KPV, Gao C, Aguiar ERGR, Jafari N, Carthew RW, Ding S-W. 2013. Functional specialization of the small interfering RNA pathway in response to virus infection. PLoS Pathog 9: e1003579 10.1371/journal.ppat.1003579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews BJ, Dudchenko O, Kingan SB, Koren S, Antoshechkin I, Crawford JE, Glassford WJ, Herre M, Redmond SN, Rose NH, et al. 2018. Improved reference genome of Aedes aegypti informs arbovirus vector control. Nature 563: 501–507. 10.1038/s41586-018-0692-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mette MF, Kanno T, Aufsatz W, Jakowitsch J, van der Winden J, Matzke MA, Matzke AJM. 2002. Endogenous viral sequences and their potential contribution to heritable virus resistance in plants. EMBO J 21: 461–469. 10.1093/emboj/21.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Bennett EA, Iskow RC, Devine SE. 2007. Which transposable elements are active in the human genome? Trends Genet 23: 183–191. [DOI] [PubMed] [Google Scholar]
- Nene V, Wortman JR, Lawson D, Haas B, Kodira C, Tu Z, Loftus B, Xi Z, Megy K, Grabherr M, et al. 2007. Genome sequence of Aedes aegypti, a major arbovirus vector. Science 316: 1718–1723. 10.1126/science.1138878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou J, Wolfe SA, Brodsky MH, Zhu LJ. 2018. motifStack for the analysis of transcription factor binding site evolution. Nat Methods 15: 8 10.1038/nmeth.4555. [DOI] [PubMed] [Google Scholar]
- Ozata DM, Gainetdinov I, Zoch A, O'Carroll D, Zamore PD. 2019. PIWI-interacting RNAs: small RNAs with big functions. Nat Rev Genet 20: 89–108. 10.1038/s41576-018-0073-3. [DOI] [PubMed] [Google Scholar]
- Palatini U, Miesen P, Carballar-Lejarazu R, Ometto L, Rizzo E, Tu Z, van Rij RP, Bonizzoni M. 2017. Comparative genomics shows that viral integrations are abundant and express piRNAs in the arboviral vectors Aedes aegypti and Aedes albopictus. BMC Genomics 18: 512 10.1186/s12864-017-3903-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish NF, Fujino K, Shiromoto Y, Iwasaki YW, Ha H, Xing J, Makino A, Kuramochi-Miyagawa S, Nakano T, Siomi H, et al. 2015. piRNAs derived from ancient viral processed pseudogenes as transgenerational sequence-specific immune memory in mammals. RNA 21: 1691–1703. 10.1261/rna.052092.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston BD. 1996. Error-prone retrotransposition: rime of the ancient mutators. Proc Natl Acad Sci 93: 7427–7431. 10.1073/pnas.93.15.7427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16: 276–277. 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Sayers E. 2009. Entrez programming utilities help. http://www.ncbi.nlm.nih.gov/books/NBK25499 [Google Scholar]
- Shi M, Lin X-D, Tian J-H, Chen L-J, Chen X, Li C-X, Qin X-C, Li J, Cao J-P, Eden J-S, et al. 2016. Redefining the invertebrate RNA virosphere. Nature 540: 539–543. 10.1038/nature20167. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Frangeul L, Dickson LB, Blanc H, Verdier Y, Vinh J, Lambrechts L, Saleh MC. 2017. Uncovering the repertoire of endogenous flaviviral elements in Aedes mosquito genomes. J Virol 91: e00571-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassetto M, Kunitomi M, Whitfield ZJ, Dolan PT, Sánchez-Vargas I, Garcia-Knight M, Ribiero I, Chen T, Olson KE, Andino R. 2019. Control of RNA viruses in mosquito cells through the acquisition of vDNA and endogenous viral elements. Elife 8: e41244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DJ, Bruenn J. 2009. The evolution of novel fungal genes from non-retroviral RNA viruses. BMC Biol 7: 88 10.1186/1741-7007-7-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter Horst AM, Nigg JC, Dekker FM, Falk BW. 2019. Endogenous viral elements are widespread in arthropod genomes and commonly give rise to PIWI-interacting RNAs. J Virol 93: e02124-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14: 178–192. 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timoshevskiy VA, Kinney NA, deBruyn BS, Mao C, Tu Z, Severson DW, Sharakhov IV, Sharakhova MV. 2014. Genomic composition and evolution of Aedes aegypti chromosomes revealed by the analysis of physically mapped supercontigs. BMC Biol 12: 27 10.1186/1741-7007-12-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tower J, Karpen GH, Craig N, Spradling AC. 1993. Preferential transposition of Drosophila P elements to nearby chromosomal sites. Genetics 133: 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodovar N, Bronkhorst AW, van Cleef KWR, Miesen P, Blanc H, van Rij RP, Saleh M-C, Pfeffer S. 2012. Arbovirus-derived piRNAs exhibit a ping-pong signature in mosquito cells. PLoS One 7: e30861 10.1371/journal.pone.0030861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. 2006. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol 80: 5059–5064. 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield ZJ, Dolan PT, Kunitomi M, Tassetto M, Seetin MG, Oh S, Heiner C, Paxinos E, Andino R. 2017. The diversity, structure, and function of heritable adaptive immunity sequences in the Aedes aegypti genome. Curr Biol 27: 3511–3519.e7. 10.1016/j.cub.2017.09.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, et al. 2007. A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8: 973–982. 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- Zanni V, Eymery A, Coiffet M, Zytnicki M, Luyten I, Quesneville H, Vaury C, Jensen S. 2013. Distribution, evolution, and diversity of retrotransposons at the flamenco locus reflect the regulatory properties of piRNA clusters. Proc Natl Acad Sci 110: 19842–19847. 10.1073/pnas.1313677110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhdanov VM. 1975. Integration of viral genomes. Nature 256: 471–473. 10.1038/256471a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.