

Abstract
Transposable elements (TEs) can damage genomes, thus organisms use a variety of mechanisms to repress TE expression. The PIWI–piRNA pathway is a small RNA pathway that represses TE expression in the germline of animals. Here we explore the function of the pathway in the somatic stem cells of Hydra, a long-lived freshwater cnidarian. Hydra have three stem cell populations, all of which express PIWI proteins; endodermal and ectodermal epithelial stem cells (ESCs) are somatic, whereas the interstitial stem cells have germline competence. To study somatic function of the pathway, we isolated piRNAs from Hydra that lack the interstitial lineage and found that these somatic piRNAs map predominantly to TE transcripts and display the conserved sequence signatures typical of germline piRNAs. Three lines of evidence suggest that the PIWI–piRNA pathway represses TEs in Hydra ESCs. First, epithelial knockdown of the Hydra piwi gene hywi resulted in up-regulation of TE expression. Second, degradome sequencing revealed evidence of PIWI-mediated cleavage of TE RNAs in epithelial cells using the ping-pong mechanism. Finally, we demonstrated a direct association between Hywi protein and TE transcripts in epithelial cells using RNA immunoprecipitation. Altogether, our data reveal that the PIWI–piRNA pathway represses TE expression in the somatic cell lineages of Hydra, which we propose contributes to the extreme longevity of the organism. Furthermore, our results, in combination with others, suggest that somatic TE repression is an ancestral function of the PIWI–piRNA pathway.
Keywords: PIWI, piRNA, Hydra, transposable elements, stem cells, aging
INTRODUCTION
The PIWI–piRNA pathway is best known for repressing transposable element (TE) expression in animal germlines (Siomi et al. 2011). The central effector complex of this pathway consists of PIWI proteins partnered with a class of small RNAs, the PIWI-interacting RNAs (piRNAs), which are typically 24–31 nucleotides (nt) in length (Aravin et al. 2006; Girard et al. 2006; Grivna 2006; Vagin 2006). TE repression is directed by cytoplasmic PIWI–piRNA complexes that target TE RNAs for degradation (Brennecke et al. 2007; Gunawardane et al. 2007). In some species, PIWI–piRNA complexes can also act in the nucleus to silence TE genomic loci (Watanabe et al. 2018; Batki et al. 2019). In addition to germline function, somatic expression and function for piwi genes and piRNAs has been demonstrated for many organisms, although in most cases the somatic targets have not been well explored (Mani and Juliano 2013; Ross et al. 2014; Ozata et al. 2019). In Drosophila, the piwi gene functions in several somatic tissues to repress TEs, including the ovary, fat body, and intestinal stem cells (Malone et al. 2009; Jones et al. 2016; Sousa-Victor et al. 2017). One possibility is that somatic TE repression is a derived function of the pathway in Drosophila, but two recent studies demonstrate widespread expression of PIWI family genes and TE-derived piRNAs in somatic tissues of arthropods and mollusks, suggesting that somatic repression of TEs is an ancestral function of the pathway in at least the protostome lineage (Jehn et al. 2018; Lewis et al. 2018).
To better understand the ancestral function of the PIWI–piRNA pathway in metazoans requires careful analysis of PIWI–piRNA pathway function in nonbilaterian taxa, such as cnidarians, which are the sister group to the bilaterians (Dunn et al. 2014). Recent work in the sea anemone Nematostella vectensis, a cnidarian, demonstrates that PIWI–piRNA pathway genes are expressed in both embryonic and adult somatic tissue and that a piwi gene is required for metamorphosis (Praher et al. 2017; Modepalli et al. 2018). Furthermore, degradome analysis demonstrated that the PIWI–piRNA pathway targets TEs in Nematostella, but the cell-type specificity of this targeting has not been characterized (Praher et al. 2017). Here we aim to identify somatic targets of the PIWI–piRNA pathway in Hydra. This cnidarian has several experimental advantages for such studies, including well-described somatic stem cell populations which express piwi genes, transgenic lines that allow for the collection of specific cell lineages for RNA analysis, and reagents for isolating PIWI–piRNA complexes (Juliano et al. 2014).
Hydra is composed of three cell lineages: ectodermal epithelial, endodermal epithelial, and interstitial; each lineage is supported by its own population of stem cells (Fig. 1A). The basic Hydra body plan consists of two epithelial monolayers (Fig. 1A). No fully undifferentiated stem cells exist to renew the epithelium. Instead, all body column epithelial cells are mitotically active, leading to the displacement of cells toward the extremities. At the oral and aboral ends, epithelial cells stop dividing and terminally differentiate to build the tentacles, hypostome, and basal disk. The mitotic ectodermal and endodermal body column cells therefore simultaneously function as epitheliomuscular cells and as epithelial stem cells (ESCs). Cells of the interstitial lineage include neurons, gland cells, the stinging nematocytes specific to cnidarians, and germ cells (Fig. 1A). This lineage is supported by an undifferentiated multipotent interstitial stem cell (ISC) population that continually produces neurons, gland cells, and nematocytes in a homeostatic adult animal. ISCs are also capable of producing germline stem cells (GSCs) when GSCs are experimentally depleted (Bosch and David 1987; Nishimiya-Fujisawa and Kobayashi 2012). Therefore, while ISCs have germline potential, ectodermal and endodermal ESCs are strictly somatic.
FIGURE 1.

Epithelial Hydra piRNAs have sequence signatures typical of ping-pong biogenesis. (A) Schematic of the Hydra body plan and cell types adapted from Siebert (2018). Hydra is comprised of three cell lineages. The two epithelial cell lineages, endoderm (end, green) and ectoderm (ect, blue), form two monolayers separated by a basal lamina called the mesoglea. The cells of the interstitial lineage (pink) are embedded among the epithelial cells; this lineage is supported by a multipotent ISC which gives rise to three somatic cell types: neurons, gland cells, and nematocytes. Interstitial cells shown: progenitors (prog), nematoblasts (nb), nematocyte (nem), ganglion neuron (gn), sensory neuron (sn), and zymogen gland cell (zmg). The ISCs are also able to produce GSCs (not shown). In contrast, the epithelial cell lineages are strictly somatic. The epithelial cells of the body column (shown in lighter colors) are mitotically active, unipotent stem cells that express PIWI proteins (Juliano et al. 2014). Due to mitotic divisions, these cells are translocated toward the oral and aboral ends of the animal. When epithelial cells arrive at the extremities (shown in darker colors), the cells exit the cell cycle, lose PIWI expression, and terminally differentiate to build the hypostome and tentacles at the oral end and the peduncle (ped) and basal disc (bd) at the aboral end. (B) Experimental design for Figures 1, 2: piRNAs were extracted from either untreated (wild-type, contains germline-competent interstitial lineage) or colchicine-treated (epithelial, does not contain interstitial cells) Hydra by immunoprecipitation (IP) using Hywi- and Hyli-specific antibodies (Juliano et al. 2014). piRNAs were isolated from immunoprecipitated complexes, sequenced, and mapped to the Hydra transcriptome (Siebert et al. 2019). (C–J) The number of occurrences of complementary overlap between the 5′ ends of Hywi- and Hyli-bound piRNAs; all four possible combinations were tested. “S” indicates piRNAs that map in a sense orientation to a transcript, and “AS” indicates piRNAs that map in an antisense orientation to a transcript. The high frequency of 10 bp overlap between Hywi- and Hyli-bound piRNAs is indicative of ping-pong biogenesis. This signal is present in piRNAs isolated from both wild-type (WT) (C–F) and epithelial (G–J) Hydra, indicating that the ping-pong biogenesis pathway is active in ESCs. The highest frequency of 10 bp overlaps occurs between antisense Hywi piRNAs and sense Hyli piRNAs in both WT (C) and epithelial Hydra (G). This is consistent with a previous study analyzing piRNAs isolated from WT Hydra vulgaris strain 105 (Juliano et al. 2014).
Hydra has two cytoplasmic PIWI proteins, Hywi and Hyli, which are located in perinuclear, cytoplasmic granules found in both ESCs and ISCs (Juliano et al. 2014; Lim et al. 2014). Hywi and Hyli participate in the conserved “ping-pong” piRNA biogenesis pathway that is well described in animal germlines. Analyzing piRNAs isolated from whole Hydra demonstrated that a large fraction of Hywi-bound piRNAs map to TEs in an antisense orientation, and a large fraction of Hyli-bound piRNAs map to TEs in a sense orientation. These piRNAs exhibit a typical “ping-pong” signature, which consists of a 10 bp overlap between the 5′ end of Hywi-bound piRNAs (initiator piRNAs) and the 5′ end of Hyli-bound piRNAs (responder piRNAs) (Juliano et al. 2014; Lim et al. 2014; Ozata et al. 2019). These data indicated that Hywi and Hyli likely participate in ping-pong mediated cleavage of TE RNAs, a conserved mechanism that occurs in the cytoplasmic granules of germ cells (Brennecke et al. 2007; Aravin et al. 2008). In addition, a recent study found evidence of phased piRNA production in Hydra, a conserved mechanism of piRNA biogenesis triggered by the initial ping-pong cleavage event (Gainetdinov et al. 2018). After cleavage, the remaining 3′ portion of the target RNA is fragmented into head-to-tail phased trailing piRNAs, which are sense with respect to the target RNA (Han et al. 2015; Homolka et al. 2015; Mohn et al. 2015; Gainetdinov et al. 2018).
Hywi knockdown in Hydra ESCs led to loss of epithelial integrity and death within 12 d, thus demonstrating a necessary function for the PIWI–piRNA pathway in Hydra somatic stem cells (Juliano et al. 2014). However, our previous study focused on piRNAs isolated from whole animals and did not directly test the identity of PIWI–piRNA pathway targets in ESCs. Therefore, the identity of PIWI–piRNA targets in the somatic ESCs was unknown and was the subject of this study. Here we report that a primary target of the PIWI–piRNA pathway in Hydra ESCs is TE RNAs. This targeting occurs through the conserved ping-pong mechanism, which couples piRNA biogenesis to TE destruction (i.e., the RNA transcripts of TEs are processed into piRNAs). This is supported by our findings that TE-derived piRNAs are prevalent in Hydra ESCs and that TE transcripts are up-regulated in response to epithelial hywi knockdown. Using degradome sequencing, we find evidence of PIWI-directed cleavage products of TE transcripts in somatic cells using the ping-pong mechanism, and we demonstrate a direct interaction between Hywi protein and target TE RNAs in ESCs. These observations demonstrate that the PIWI–piRNA pathway represses TEs in the somatic stem cells of Hydra, which further supports that TE repression is an ancestral function of the pathway in not only the germline, but also in somatic tissues.
RESULTS AND DISCUSSION
Hydra vulgaris AEP piRNAs have conserved sequence characteristics
In our previous study, we isolated and sequenced Hywi-bound and Hyli-bound piRNAs from Hydra vulgaris strain 105 and mapped these to genome and transcriptome assemblies generated from the same strain. These data suggested that the Hydra PIWI–piRNA pathway targets TEs through the conserved ping-pong mechanism (Juliano et al. 2014). However, we were not able to discern cell lineage-specific roles for the pathway because piRNAs were isolated from whole Hydra vulgaris strain 105. TE repression is a conserved function of the PIWI–piRNA pathway in germ cells (Aravin et al. 2007; Brennecke et al. 2007; Carmell et al. 2007; Houwing et al. 2007; Das et al. 2008; Kuramochi-Miyagawa et al. 2008), but targets in somatic cells are less well understood (Ross et al. 2014). Therefore, in this study we focused on identifying targets of the pathway specifically in the ESCs.
To understand lineage-specific functions of the PIWI–piRNA pathway in Hydra, we used transgenic lines with cell-lineage or cell-type specific expression of fluorescent proteins. These lines were generated using Hydra vulgaris strain AEP that produces eggs in the laboratory and is routinely used for the production of transgenic lines (Wittlieb et al. 2006). As this strain differs from the strain used in our previous study, Hydra vulgaris strain 105, which does not produce eggs in the laboratory and cannot be used to make transgenic lines, we began this study by testing if the two Hydra strains have the same piRNA characteristics. We first established that our Hywi and Hyli antibodies are able to immunoprecipitate PIWI–piRNA complexes from the AEP strain (Supplemental Fig. S1A–D). Next, we isolated and sequenced Hywi-bound and Hyli-bound piRNAs from the AEP strain, mapped these piRNAs to an AEP transcriptome (Fig. 1B; Siebert et al. 2019), and analyzed the sequences for evidence of ping-pong piRNA biogenesis. Similar to previous data from the 105 strain (Juliano et al. 2014; Lim et al. 2014), we found the following evidence of ping-pong in the AEP strain: (i) 86% of Hyli-bound piRNAs have an adenine at the 10th base from the 5′ end, indicating their likely identity as responder piRNAs (Supplemental Fig. S2; Ozata et al. 2019), and (ii) the presence of a 10 bp overlap between Hywi-bound and Hyli-bound piRNAs (Fig. 1C–F; Supplemental Fig. S3).
We used the piRNA mapping data to gain insights into possible targets. When mapping the piRNAs to the transcriptome, we considered both sense and antisense orientations with respect to the transcript (Supplemental Analysis 1). piRNAs mapping in an antisense orientation could indicate binding of PIWI–piRNA complexes to an RNA target, which could ultimately lead to cleavage of that target by the “slicer” activity of the PIWI domain (Saito et al. 2006; Gunawardane et al. 2007; Nishida et al. 2007). piRNAs mapping in a sense orientation could indicate that the RNA target has been directly processed into either responder piRNAs by the ping-pong mechanism or trailing piRNAs by the phasing mechanism (Brennecke et al. 2007; Han et al. 2015; Homolka et al. 2015; Iwasaki et al. 2015; Mohn et al. 2015; Ozata et al. 2019). Therefore, for antisense mapping we allowed three mismatches because cleavage of RNA targets can occur with imperfect base-pairing (Zhang et al. 2015), but for sense mapping, we did not allow any mismatches under the assumption that piRNAs derived directly from an RNA target should have the identical sequence. To understand piRNA mapping preferences we categorized transcripts as: (i) TE transcripts (“TE”), (ii) protein-coding transcripts with annotations (“gene”), (iii) unannotated transcripts with open reading frames of at least 100 amino acids (“uncharacterized”), and (iv) noncoding transcripts with open reading frames of less than 100 amino acids (“ncRNA”) (see Materials and Methods and Supplemental Analysis 1 for further details on transcript annotations). The “uncharacterized” transcripts could include TEs that we were unable to annotate, we therefore kept these transcripts as a separate category. Similar to previous data from the 105 strain (Juliano et al. 2014; Lim et al. 2014), we found that AEP piRNAs map to TE transcripts at a higher density than non-TE transcripts (Fig. 2A; Supplemental Table S1).
FIGURE 2.

Epithelial piRNAs map predominantly to TE transcripts. Hywi- and Hyli-bound piRNAs isolated from (A) wild-type (WT) Hydra and (B) epithelial Hydra were mapped to the transcriptome. The piRNA mapping density (mapped piRNA counts per kb per million counts) is shown on the y-axis of the box and whisker plots. The results for piRNA mapping are subdivided by: (i) Hywi- or Hyli-bound piRNAs, (ii) sense (S) or antisense (AS) orientation, and (iii) transcript class (see legend). In all cases, TEs (red) have a significantly higher mapping density than other transcript classes as determined using Tukey's range test (P < 0.001; Supplemental Analysis 1).
A previous study analyzing total small RNAs isolated from Hydra vulgaris strain 105 found evidence that sense-oriented piRNAs lie at regular intervals downstream from ping-pong cleavage sites; this is suggestive of phased piRNA biogenesis (Gainetdinov et al. 2018). In our mapping analysis of piRNAs isolated from Hydra vulgaris strain AEP we found Hywi-bound piRNAs adjacent to each other, such that the 3′ ends of sense-oriented piRNAs map directly adjacent to the 5′ ends of downstream mapped sense-oriented piRNAs; this occurred at a higher rate than was expected by chance (Z0 = 5.67, Z0 > 1.96 corresponds to P < 0.05) (Supplemental Fig. S4). This indicates that Hywi protein participates in phased piRNA biogenesis and also suggests that pre-piRNAs are not significantly trimmed at the 3′ end in Hydra. In contrast, we did not find a similar enrichment for Hyli-bound piRNAs (Z0 = 0.50) demonstrating that Hywi is primarily responsible for phased piRNA biogenesis in Hydra (Supplemental Fig. S4). In summary, we concluded that the PIWI–piRNA pathway functions similarly in the two commonly used Hydra strains, with both ping-pong and phased piRNA biogenesis occurring and with TE transcripts as the likely major target. Subsequent experiments in this study were done with Hydra vulgaris strain AEP, hereafter referred to simply as “Hydra”.
Hydra piRNAs isolated from epithelial stem cells are enriched for TE sequences
To gain insight into the function of the PIWI–piRNA pathway in Hydra ESCs, we first isolated and analyzed piRNAs from epithelial Hydra, which do not contain any cell types of the interstitial lineage. To obtain epithelial Hydra, we used an established colchicine treatment protocol to remove ISCs (Campbell 1976), which leaves ESCs as the only PIWI-expressing cell type. To confirm ISC depletion in our hands, we performed colchicine treatments using a transgenic line expressing GFP under the control of the cnnos1 promoter, which drives expression in ISCs (Hemmrich et al. 2012). Animals were treated with 0.4% colchicine for 8 h followed by a 10-d recovery period. Immunoblot analysis confirmed the loss of GFP-positive ISCs (Supplemental Fig. S1E). Given that stress could lead to increased expression of TEs (Horváth et al. 2017), we assayed TE abundance in colchicine-treated animals by qPCR. We found that neither hywi expression or TE expression is increased in colchicine-treated animals, therefore TE-derived piRNAs are not artificially abundant in the epithelial Hydra used in this study (Supplemental Fig. S1F). Epithelial Hydra obtained from colchicine treatment lack the interstitial lineage but can be cultured if fed by hand and show no deficits in asexual reproduction or regeneration (Marcum and Campbell 1978). In summary, the epithelial lineages are able to return to normal function after the colchicine treatment, thereby providing a suitable system for studying PIWI–piRNA pathway function in the ESCs.
To isolate the piRNAs expressed in epithelial cells, we performed Hywi and Hyli IPs from lysates prepared from colchicine-treated cnnos1::GFP Hydra (Fig. 1B). The isolated piRNAs were sequenced and the base distribution across the length of the piRNAs was analyzed. Similar to piRNAs isolated from WT animals, 90% of Hywi-bound piRNAs isolated from epithelial animals had a uridine at the 5′ end and 79% of Hyli-bound piRNAs had an adenine at the 10th base from the 5′ end (Supplemental Fig. S2). In addition, epithelial piRNAs showed the 10 bp overlap consistent with ping-pong piRNA biogenesis (Fig. 1G–J; Supplemental Fig. S3). The strongest 10 bp overlap was found between sense Hyli piRNAs and antisense Hywi piRNAs as expected. However, similar to piRNAs isolated from WT Hydra, other piRNA combinations showed some degree of 10 bp overlap. This included homotypic ping-pong, which also occurs in Drosophila, mice, and mollusks (Aravin et al. 2008; Zhang et al. 2012; Jehn et al. 2018). Overall, these data demonstrated that piRNAs expressed in the ESCs are processed by the ping-pong biogenesis pathway (Brennecke et al. 2007; Iwasaki et al. 2015). Furthermore, similar to our analysis for piRNAs isolated from Hydra that include the interstitial lineage, we found that Hywi-directed phased piRNA biogenesis occurs in ESCs (Z0 = 2.40, Supplemental Fig. S4).
To identify PIWI–piRNA pathway RNA targets in ESCs, we mapped epithelial piRNAs to the Hydra vulgaris AEP stranded transcriptome (Siebert et al. 2019) using the same strategy described above for piRNAs isolated from untreated animals (Fig. 1B). Using this mapping strategy, we found that for sense and antisense mapping, both Hywi-bound and Hyli-bound piRNAs map to TE transcripts at a significantly higher density than to non-TE transcripts (Fig. 2B; Supplemental Table S1; Supplemental Analysis 1). This was likely a reflection of TE RNAs being processed into piRNAs by the ping-pong mechanism, (Fig. 1G–J) or by a Hywi-mediated phasing (Supplemental Fig. S4; Han et al. 2015; Homolka et al. 2015; Mohn et al. 2015). For piRNAs that map in an antisense orientation, 68.0% of piRNAs isolated from WT animals and 85.1% of piRNAs isolated from epithelial animals mapped with at least one mismatch (Supplemental Analysis 1). This indicated that the majority of antisense mapping piRNAs originated from a locus distinct from the target locus. For example, many likely originated from piRNA cluster loci, which have been documented in Hydra vulgaris 105 strain using the genome assembly of that strain (Lim and Kai 2015). When considering antisense mapping events to the transcriptome, most non-TE transcripts had a low mapping density. However, for both the Hywi-bound and Hyli-bound antisense mapping, there were groups of gene transcripts with high antisense mapping density, which could indicate the existence of gene targets (Fig. 2). Sense mapping of Hywi-bound or Hyli-bound piRNAs to gene transcripts may indicate that these transcripts are processed into either responder or trailing piRNAs (Fig. 2). In summary, these data suggested that TEs are a major target of the PIWI–piRNA pathway in ESCs. In addition, this mapping strategy identified putative non-TE targets that we address further below.
To further test the lineage-specific functions of the PIWI–piRNA pathway, we analyzed total Hydra vulgaris AEP small RNAs that were isolated and sequenced from ectodermal, endodermal, and interstitial lineage cells sorted by FACS in our previous study (Juliano et al. 2014). We identified the piRNAs in the small RNA data sets by cross-referencing with the piRNAs sequenced from WT Hydra vulgaris AEP animals in this study (Supplemental Fig. S5A). Using this strategy, we produced a list of unique piRNA species and annotated the lineage(s) of origin for each sequence (Supplemental Fig. S5B–I). We next mapped the ectodermal and endodermal epithelial piRNAs to the Hydra transcriptome and, similar to our results using piRNAs isolated from epithelial animals, we found an enrichment for piRNAs mapping to TE transcripts as compared to other transcript categories (Supplemental Fig. S5J). In addition, this strategy allowed us to identify and map piRNAs specific to the interstitial lineage and we also found an enrichment for piRNAs mapping to TE transcripts as compared to other transcript categories (Supplemental Fig. S5K). An enrichment for transposon sequences in piRNAs expressed specifically in ISCs is expected because ISCs have germline potential.
RNAi knockdown of hywi in epithelial stem cells leads to the up-regulation of both TE and non-TE transcripts
Transcripts that are targeted by the PIWI–piRNA pathway should be up-regulated in response to down regulation of PIWI genes. To identify such transcripts, we used our previously validated hywi RNAi-1 transgenic line, which constitutively expresses a hywi hairpin under the control of an actin promoter that is not active in ISCs (Juliano et al. 2014). The hairpin construct also includes DsRed2, which is used to track transgenic tissue. The hairpin construct is integrated only in the interstitial lineage and is maintained through asexual propagation. In the asexually propagated hywi RNAi-1 transgenic line, the hairpin is not expressed in the PIWI-positive ISCs and therefore does not have a negative impact on the viability of the line. Integration of the hairpin construct in the interstitial lineage allows for germline propagation. After sexual reproduction, the transgene is now integrated into the genome of all cells in the offspring and the actin promoter drives expression in all epithelial cells and differentiated interstitial cells; the ESCs are the only piwi-positive cells that have RNAi transgene expression in the F1 generation. This allows us to analyze the effect of knocking down hywi in the ESCs using F1 animals. Using this strategy, we previously found that hywi knockdown in ESCs leads to death within 12 d of hatching (Juliano et al. 2014). In this study we performed RNA-seq and differential gene expression (DGE) analysis to compare gene expression between WT offspring (did not inherit the RNAi transgene) and hywi RNAi offspring (did inherit the transgene) 4 d after hatching. We identified 441 transcripts that were up-regulated in hywi RNAi animals as compared to WT siblings (Fig. 3A; Supplemental Tables S1, S2).
FIGURE 3.

Both TE and non-TE transcripts are up-regulated in response to epithelial hywi RNAi. (A) DGE analysis was performed to compare transcript expression levels between hywi RNAi animals and WT siblings; knockdown animals carry the hywi RNA-1 transgene (Juliano et al. 2014). Plot showing 458 differentially expressed transcripts (red); 441 transcripts are up-regulated and 17 transcripts are down-regulated (adjusted P-value <0.05). (B) Transcript class breakdown of the 441 transcripts up-regulated in response to hywi knockdown. (C,D) A second RNAi transgenic line (hywi RNAi-2) was used to validate the DGE results (Juliano et al. 2014). qPCR was used to test the levels of select (C) TE transcripts and (D) gene transcripts identified by the DGE performed on the hywi RNAi-1 transgenic line. Error bars reflect standard deviation. These results indicate that at least some transcripts up-regulated in the hywi RNAi-1 transgenic line are also up-regulated in the hywi RNAi-2 transgenic line, suggesting the specific effect of hywi knockdown.
Given that epithelial piRNAs were enriched for TE sequences, we predicted that TEs would be up-regulated upon RNAi knockdown of hywi in ESCs. However, among the 441 transcripts up-regulated in response to hywi knockdown only 7% (33 out of 441) were annotated as TEs (Fig. 3B). To gain insights into the identity of the 238 transcripts in the gene category that were up-regulated in response to hywi knockdown (either due to direct or indirect effects), we performed GO-term analysis. We found seven terms (biological process) significantly enriched, including “defense response” and “innate immune response” (adjusted P-value <0.05; Supplemental Analysis 2). This result could suggest that TE derepression triggers an innate immune response in the affected cells, a phenomenon which has been observed in other organisms (De Cecco et al. 2019; Simon et al. 2019).
To test the reproducibility of the DGE results, we used a second hywi RNAi line (hywi RNAi-2), which targets a different portion of the hywi gene. We generated this transgenic line using the same strategy as the hywi RNAi-1 line (Juliano et al. 2014). RNA was extracted from both WT and hywi RNAi hatchlings produced from the hywi RNAi-2 parental line. qPCR analysis of these samples was used to test the expression levels of four TE transcripts and five gene transcripts identified by the DGE analysis (Fig. 3A). These transcripts were also up-regulated in the hywi RNA-2 line, which suggests that the results we obtained in the DGE analysis were likely due to RNAi knockdown of hywi and not off target effects of the hywi hairpins (Fig. 3C,D).
Our DGE analysis raised the possibility that the PIWI–piRNA pathway has many non-TE targets in ESCs, but it was also possible that these were up-regulated due to secondary effects of TE up-regulation. To help discern between these two scenarios, we asked how many of the up-regulated transcripts had a high density of piRNAs mapping to them, which could indicate direct targeting by the PIWI–piRNA pathway. To identify transcripts with the highest density of mapped piRNAs, we ordered transcripts from the highest to lowest density of mapped epithelial Hywi piRNAs, and defined transcripts in the top 20% as “high mapping” (Supplemental Table S2). Of the 441 up-regulated transcripts, we found the following percentages to be “high mapping” in each category: (i) 97.0% (32 out of 33) for TE transcripts, (ii) 16.7% (six out of 36) for noncoding transcripts, (iii) 20.1% (27 out of 134) for uncharacterized transcripts, and (iv) 10.1% (24 out of 238) for gene transcripts. Therefore, we found that TE transcripts were overrepresented in the “high mapping” category. In addition, we identified 24 genes that may be direct targets of the PIWI–piRNA pathway in ESCs because they were both in the “high mapping” category and were up-regulated in response to hywi knockdown (Supplemental Table S3).
In the Hydra epithelial lineages, PIWI expression is largely restricted to the body column ESCs. We therefore predicted that direct targets of the PIWI–piRNA pathway would have lower expression in the ESCs as compared to the differentiated cells of the tentacles, hypostome, and basal disc where piwi genes are not expressed. To determine which of our 24 putative gene targets exhibited this expression pattern, we interrogated the available single-cell expression data (Siebert et al. 2019). In these visualizations we included genes that were found to be expressed in at least 1% of the ectodermal or endodermal epithelial cells (Siebert et al. 2019). We found epithelial expression for 16 of the 24 putative gene targets: (i) thirteen gene transcripts were expressed in both the endodermal and ectodermal epithelial lineages, (ii) one gene transcript was expressed only in the ectodermal epithelial lineage, and (iii) two gene transcripts were expressed only in the endodermal epithelial lineage. No epithelial expression was found for eight of the gene transcripts, which could indicate low expression in a homeostatic animal. This could be due to transcript degradation by the PIWI–piRNA pathway or an overall low level of transcription. For the 16 gene transcripts for which we had expression data, we generated plots to visualize expression profiles in epithelial cells along the oral-aboral axis (Supplemental Analysis 5). We inspected these plots for expression patterns that could suggest targeting by the piwi–piRNA pathway: higher expression in the piwi-negative cells at the extremities as compared to the piwi-positive cells in the body column. Of the 16 transcripts, the expression pattern of t14391 was the most suggestive of Hywi-mediated repression, with higher expression at the extremities in both the ectoderm and endoderm (Supplemental Fig. S6). Swissprot BLAST analysis of this transcript suggested similarity to PARP-12. Interestingly, PARP family genes are involved in stress response, including DNA damage repair, making this an interesting putative target for future study (Bai 2015).
PIWI–piRNA complexes cleave TE transcripts in epithelial stem cells
Our analysis of piRNA sequences isolated from ESCs strongly suggested that Hywi and Hyli repress TE RNAs by processing these RNAs into piRNAs using the ping-pong mechanism. To further test this, we looked for evidence of cleaved TE RNAs using degradome sequencing, a strategy used to isolate and sequence cleaved RNAs by selecting for uncapped RNAs with a poly(A) tail (Addo-Quaye et al. 2008; German et al. 2008). To determine the RNA targets cleaved by PIWI proteins in Hydra, we generated degradome sequencing libraries from both untreated and colchicine-treated cnnos1::GFP Hydra. Cleaved RNA targets of the PIWI–piRNA pathway display the ping-pong signature, which in Hydra most often manifests as a 10-bp overlap between the 5′ end of antisense-oriented Hywi-bound piRNAs with the 5′ end of sense-oriented Hyli-bound piRNAs and degradome fragments (Fig. 4A). We considered genes that have 10 or more of each of these species mapped and aligned in a “ping-pong” signature to be RNA targets of the PIWI–piRNA pathway. We identified 2047 such targets in WT animals and 254 such targets in epithelial animals (Supplemental Table S4). When considering the inverse of canonical ping-pong, Hywi oriented sense and Hyli oriented antisense, we identified 709 targets for WT and 74 targets for epithelial animals; therefore, this did occur, but at a lower rate than canonical ping-pong targeting. In libraries produced from both whole animals and epithelial animals, TEs comprised the highest percentage of transcripts displaying this signature (Fig. 4B; Supplemental Table S4). The second highest category was uncharacterized transcripts, which may include some TE transcripts that we were unable to annotate (Supplemental Table S4). Of the 441 transcripts up-regulated in response to hywi knockdown, 46 were identified as targets in the WT degradome library (Supplemental Table S4). The majority of these (38 out of 46) were either TEs (22) or uncharacterized transcripts (16). For gene transcripts, only three of the 24 that were up-regulated in response to hywi RNAi and had a high number of piRNAs mapping, were also identified as ping-pong targets in the degradome analysis (Supplemental Table S3). However, we found that 19 of these 24 gene transcripts were in the top 5% for Hywi piRNAs mapping in the sense orientation (Supplemental Table S3). This high number of sense mapped Hywi piRNAs could indicate that these transcripts are a target of phased piRNA processing, but that we were not able to detect the ping-pong cleavage site that initiated the phased piRNA production. In summary, the degradome results further supported that a major function of the PIWI–piRNA pathway in Hydra ESCs is to cleave TE transcripts, but also revealed some possible non-TE targets for future study.
FIGURE 4.
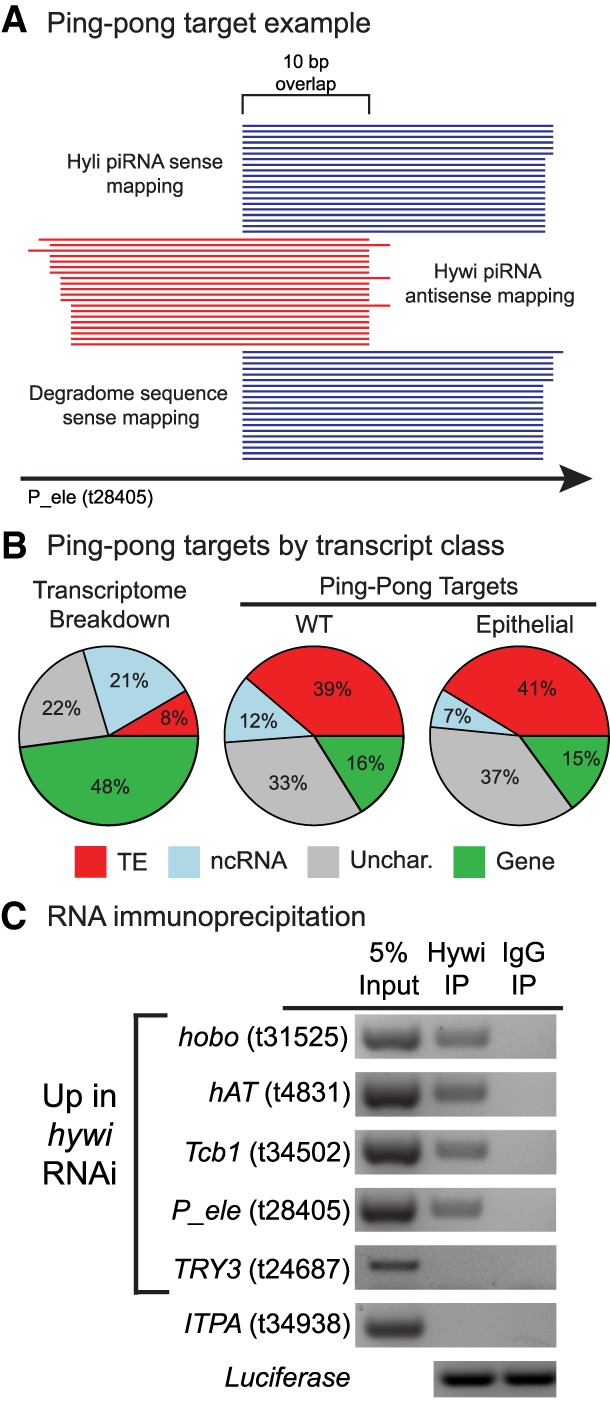
TE transcripts are cleaved by PIWI–piRNA complexes in Hydra ESCs. (A) Hywi-bound piRNAs, Hyli-bound piRNAs, and degradome reads were aligned to the transcriptome to identify likely direct targets of the PIWI–piRNA pathway. One example from the data is shown (t28405, P-element). The following pattern indicates cleavage: Antisense-oriented Hywi-bound piRNAs align with a 10-nt 5′ overlap with both sense-oriented Hyli-bound piRNAs and sense-oriented degradome reads. For a transcript to be considered a target of the PIWI–piRNA pathway, we require a minimum of 10 reads of each species to map in such an arrangement. (B) The degradome sequencing from WT Hydra identified 2047 transcripts (Supplemental Table S4) as targets of the PIWI–piRNA pathway, and the transcript class distribution of these targets is shown. The degradome sequencing from epithelial Hydra identified 254 transcripts (Supplemental Table S4) as targets of the PIWI–piRNA pathway, and the class distribution of these targets is shown. In contrast to the class distribution of the transcriptome as a whole, TE transcripts and uncharacterized transcripts comprise the majority of targets in both WT and epithelial animals. (C) RNA immunoprecipitation (RIP) was used to identify transcripts that directly interact with Hywi-piRNA complexes in ESCs (for complete results see Supplemental Fig. S7). Following RIP, luciferase mRNA was added to the washed beads to act as a positive control for RNA extraction and cDNA synthesis. RT-PCR was used to test for the presence of target transcripts in complexes immunoprecipitated with a Hywi antibody. The results for four TE transcripts are shown, which were amplified in three biological replicates (Supplemental Fig. S7). The following transcripts did not associate with Hywi protein as expected: (i) The TRY3 transcript (t24687) is up-regulated in response to hywi RNAi but has a low piRNA mapping density; and (ii) the ITPA transcript (t34938) did not change in response to hywi RNAi and has a low piRNA mapping density.
Finally, we tested whether Hywi-piRNA complexes physically associate with the somatic RNA targets identified by piRNA mapping, DGE analysis of hywi RNAi animals, and/or degradome sequencing. To this end, we performed RIP to isolate Hywi-piRNA-RNA ternary complexes from epithelial Hydra (Ilyin et al. 2017). We performed RT-PCR to test for the presence of the following transcripts in our immunoprecipitated complexes: (i) eight TE transcripts that were up-regulated in response to hywi knockdown, had a high number of piRNAs mapping, and were identified as targets in the degradome sequencing (Supplemental Table S3); (ii) seven gene transcripts that were up-regulated in response to hywi knockdown and had a high number of piRNAs mapping (one of these was also identified as a target in the degradome sequencing) (Supplemental Table S3); (iii) three gene transcripts that were up-regulated in response to hywi knockdown, but did not have a high number of piRNAs mapping; and (iv) six gene transcripts that were not affected by hywi knockdown and did not have a high number of piRNAs mapping to act as negative controls. We could reproducibly pull down four of the eight TE transcripts in all three biological replicates; the remaining four could be pulled down in at least one replicate. These data demonstrated a physical interaction between Hywi-piRNA complexes and TE RNAs (Fig. 4C; Supplemental Fig. S7; Supplemental Table S3). In contrast, we could not reproducibly pull-down transcripts from the remaining categories listed above (Supplemental Fig. S7). Three of the seven gene transcripts from the second category (t27033, t8142, and t5862) were pulled down in one or two of the three biological replicates (Supplemental Fig. S7; Supplemental Table S3). These three genes did not appear in the degradome sequencing, but do have a large number of sense mapping Hywi-bound piRNAs and therefore may be targeted by the phasing mechanism (Supplemental Table S3; Homolka et al. 2015; Pandey et al. 2017).
Conclusion
The mechanisms by which the PIWI–piRNA pathway repress TEs are best understood in the germline, but a large body of work demonstrates diverse functions for the pathway in somatic cells (for reviews, see Mani and Juliano 2013; Ross et al. 2014; Ozata et al. 2019). Here we find that the PIWI–piRNA pathway operates in the Hydra soma to degrade TE RNAs using the conserved ping-pong mechanism. We demonstrated this by isolating degraded RNAs and PIWI–piRNA complexes specifically from somatic cells. We found evidence of ping-pong mediated degradation of somatic TE RNAs and association of somatic PIWI–piRNA complexes with TE RNAs. Furthermore, loss of hywi function led to TE up-regulation in somatic cells. Recent studies surveying a wide variety of mollusk and arthropod species found evidence for widespread somatic ping-pong as a TE repression mechanism (Jehn et al. 2018; Lewis et al. 2018). Furthermore, the piwi genes have widespread somatic expression in Nematostella, another cnidarian in which the pathway likely represses TEs in the soma (Praher et al. 2017). Therefore, our data in combination with these previously published studies suggest that the PIWI–piRNA pathway has ancestral roles in both germline and somatic TE repression. Furthermore, the somatic function of the pathway may have been lost in some lineages, such as in mammals, leaving germline TE repression as the most prominent function.
TE expression is up-regulated in aging metazoan somatic tissues and in aging yeast, suggesting that TE derepression is a conserved feature of cellular aging (Maxwell et al. 2011; De Cecco et al. 2013; Li et al. 2013; Van Meter et al. 2014; Patterson et al. 2015; Wood et al. 2016). Some studies have shown that repressing TEs can extend lifespan, thus understanding the mechanisms by which long-lived organisms repress TEs is of interest (Wang et al. 2011; Jones et al. 2016; Wood et al. 2016; De Cecco et al. 2019; Simon et al. 2019). A longevity study demonstrated that mortality in Hydra does not increase with age, suggesting a lack of senescence (Schaible et al. 2015), but the mechanisms by which Hydra escapes aging are not understood. It has been hypothesized that the PIWI–piRNA pathway contributes to Hydra longevity through somatic TE repression (Sturm et al. 2017). Although this is a difficult hypothesis to test, our study demonstrates that the basic premise of this hypothesis is plausible because the PIWI–piRNA pathway does repress TE expression in Hydra somatic tissue. Interestingly, Drosophila piwi is required to repress TE expression in the fat body and in intestinal stem cells and this has been linked to longevity in these somatic tissues (Jones et al. 2016; Sousa-Victor et al. 2017). For example, in aging flies, TEs are up-regulated in intestinal stem cells and overexpression of piwi prevents the age-associated decline of intestinal stem cell function (Sousa-Victor et al. 2017). It is however not clear why piwi fails to be effective in the somatic tissues of aging Drosophila, whereas hywi and hyli appear to indefinitely protect somatic cells in Hydra. Comparative studies of somatic PIWI–piRNA pathway function in animals with a variety of life history strategies could reveal new insights into the role the pathway may play in protecting somatic cells from TE-induced damage and how this may influence the rate of aging.
MATERIALS AND METHODS
Code availability
Analysis code, scripts, and Supplemental Analysis files that are needed to reproduce the analyses are available in a git repository: https://github.com/cejuliano/bteefy_piwi_transposon
- 1_piRNA_and_Degradome_Counts.Rmd
- Generation of piRNA and degradome count files, transcript classification
- 2_Differential_Gene_Expression_GO_Analysis.Rmd
- hywi knockdown—Differential Gene Expression analysis, GO-term enrichment
- 3_Ping_Pong_Phasing.Rmd
- Ping-pong hit count files, ping-pong overlap frequency, ping-pong hit classification, piRNA phasing, Z-score analysis
- 4_Lineage_Sorted_piRNA_Mapping.Rmd
- Lineage-sorting and counting of piRNAs
- Assessment of homeostatic epithelial expression of putative hywi targets
Transgenic strains used in this study
Interstitial stem cell depletion was performed on cnnos1::GFP Hydra (Hemmrich et al. 2012). Differential gene expression (DGE) analysis was performed on our previously published hywi RNAi-1 animals (targeting bases 379–899 of the hywi gene) (Juliano et al. 2014). Validation was done by creating a second transgenic line using our previously published hywi RNAi-2 transgene (targeting bases 1557-2093 of the hywi gene) (Juliano et al. 2014). Hydra were cultured at 18°C and fed Artemia nauplii 1–3 times per week.
Colchicine treatment to remove the interstitial lineage
Colchicine treatment was performed as previously described (Campbell 1976). In brief, cnnos1::GFP Hydra (Hemmrich et al. 2012) were exposed to 0.4% colchicine dissolved in Hydra medium (HM) (0.3 mM CaCl2, 0.3 mM MgSO4, 0.5 mM NaHCO3, 0.08 mM K2CO3) for 8 h. After exposure, animals were rinsed with HM and transferred to fresh HM. Replicates were kept in separate dishes and HM was changed every day for 10 d after colchicine exposure while animals recovered. Ten animals were used per RIP experiment. To create degradome libraries, RNA was extracted from 10 Hydra per replicate using TRIzol (ThermoFisher Scientific) according to the manufacturer's instructions. Interstitial cell depletion was assayed using qPCR to detect levels of Cnnos1 and GFP transcripts using cDNA synthesized from 1 µg of TRIzol-extracted RNA. Interstitial cell depletion was also tested using an immunoblot to detect levels of GFP from three animals (see below for immunoblot details).
Immunoprecipitation of piRNAs with PIWI antibodies
Immunoprecipitation (IP) of PIWI proteins for piRNA isolation and sequencing was done using either 225 untreated Hydra vulgaris AEP or 225 colchicine-treated cnnos1::GFP Hydra. Hydra were centrifuged in a mini benchtop centrifuge (max speed 2000g), HM was removed, and animals were resuspended in 1 mL MCB buffer (50 mM HEPES, pH 7.5, 150 mM potassium acetate, 2 mM magnesium acetate, 10% glycerol, 0.1% Triton X-100, 0.1% NP-40, 1 mM DTT). The following were added fresh to the MCB buffer before use: (i) 1 tablet/10 mL of Pierce Protease Inhibitor Mini Tablets, EDTA-free (ThermoFisher Scientific); and (ii) 1 U/µL RNaseOUT Recombinant Ribonuclease Inhibitor (ThermoFisher Scientific). Hydra polyps in MCB buffer were homogenized on ice using a Dounce homogenizer. The resulting lysate was centrifuged at 20,000g for 10 min at 4°C and the supernatant was collected. Lysate was preincubated with 200 µL of rehydrated Protein A Sepharose CL-4B beads (GE Healthcare, Life Sciences) for 15 min at 4°C with rocking; to rehydrate beads, 100 mg of beads were washed and resuspended in 300 µL of MCB buffer. To remove beads, lysate was centrifuged at 20,000g for 10 min at 4°C and supernatant was collected. For each IP, 12 µg of purified Hywi or Hyli antibody (Juliano et al. 2014) was added to 500 µg of cleared protein lysate as measured by a NanoDrop ND-1000 Spectrophotometer 280 nM reading. The volume was then brought up to 1.2 mL per tube with MCB buffer. Next, the samples were incubated for 40 min with rocking at 4°C. Following antibody incubation, 60 µL of rehydrated beads were added to the lysate and rocked at 4°C for an additional 40 min. Beads were collected by centrifugation at 500g for 5 min at 4°C and then washed five times with 1 mL of MCB buffer. After the washes, the bead volume was brought up to 100 µL with MCB buffer and 10 µL was removed for immunoblot blot analysis (see below for immunoblot details). To isolate piRNAs, 1 mL of TRIzol was added to the remaining beads and RNA extraction was performed according to the manufacturer's instructions. The RNA pellet was resuspended in 20 µL of DEPC-treated water (Ambion). For size analysis, 8 µL was removed for 5′-end labeling with [γ-32P] ATP using polynucleotide kinase. Labeled RNAs were run on a TBE-Urea gel. The remaining 12 µL of RNA was used to prepare a sequencing library (see below for details on library preparation).
Immunoblotting
To detect GFP protein in cnnos1::GFP animals, 10 Hydra were homogenized in SDS-PAGE sample buffer for immunoblot analysis. Anti-GFP antibody was used at a 1:1000 dilution (Sigma, catalog #11814460001, Roche). All antibodies were diluted in blocking solution (3% w/v powdered milk dissolved in 0.1% TBS-Tween). For detection of Hywi and Hyli protein, 8 µg of purified anti-Hywi or anti-Hyli antibody in 50 mL of blocking solution was used for immunoblots. Anti-GFP was detected using goat anti-mouse IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 800 (ThermoFisher Scientific, A32730). Anti-Hywi was detected using goat anti-Rabbit IgG (H + L) Superclonal Secondary Antibody, Alexa Fluor 680 (Thermofisher Scientific, A27042). Anti-Hyli was detected using goat anti-guinea pig IgG (H + L) Cross Adsorbed Secondary Antibody, DyLight 800 conjugate (ThermoFisher Scientific, SA5-10100). All secondary antibodies were diluted 1:10,000 in blocking solution. Immunoblots were imaged using the LI-COR Odyssey platform.
piRNA and degradome sequencing library preparation
Sequencing libraries were prepared from immunoprecipitated piRNAs using the NEXTflex Small RNA-seq Kit v3 (PerkinElmer cat # NOVA-5132-05). The manufacturer's protocol was followed, but with modifications to 3′ and 5′ adapter ligations: 3′ adapter ligation was performed at 16°C overnight and 5′ adapter ligation was performed at 20°C for 2 h. Following adapter ligation, libraries were amplified for 18 cycles and selected for a size of 108–180 bp using BluePippin (Sage Science). For degradome sequencing, RNA was isolated using TRIzol from 20 untreated and colchicine-treated cnnos1::GFP Hydra. Starting with 2 µg of total RNA, poly(A) enrichment was performed to capture mRNAs, which was followed by 5′ adapter ligation. Ligation of the 5′ adaptor was done without removing the 5′ m7G cap so that only cleaved mRNAs were available for ligation. The RNA was then chemically fragmented and 3′ adapters were ligated. For ligations, 25% of the standard adapter mass was used. Libraries were prepared following the NEXTflex Small RNA-seq Kit v3 protocol. Libraries were amplified using 14 PCR cycles. Following amplification, the samples were sheared for 1 min at 94°C and size-selected for 125–200 bp using BluePippin. Degradome and piRNA libraries were pooled and sequenced using a HiSeq 4000 single end 50 bp run.
Analysis of sequenced piRNAs and degradome reads
Adapters were trimmed from piRNA and degradome sequencing reads using cutadapt (Martin 2011). After trimming, piRNAs and degradome reads were mapped to a previously published Hydra vulgaris AEP transcriptome (Siebert et al. 2019) using Bowtie v1.1.2 (Langmead et al. 2009). Mapping parameters were as follows: “-a –nofw -v 3 -S” was used for antisense mapping and “-a -k 40000 –norc -v 0 -S” was used for sense mapping. Degradome reads were mapped using the same parameters as sense piRNA reads. Multimappers were apportioned fractionally such that each piRNA mapping event was apportioned a single count according to the following formula:
where Ci is the count value for a transcript i, N is the number of times a particular piRNA maps to a unique location in the transcriptome, and Ri represents a piRNA mapping to transcript i.
Boxplots were generated in R using custom code (see Supplemental Analysis File “1_piRNA_and_Degradome_Counts.Rmd”). Ping-pong analysis was performed using custom scripts available in the accompanying git repository. Overlap plots, Z10, and Z0 scores were generated in R using custom code (see Supplemental Analysis file “3_Ping_Pong_Analysis.Rmd”). Mapping results were visually inspected using Integrative Genomics Viewer (IGV) (Robinson et al. 2011). The percentage of antisense piRNAs that map with mismatches was calculated using [(number of mapped piRNAs with up to three allowed mismatches) − (number of mapped piRNAs without allowed mismatches)]/(number of mapped piRNAs with up to three allowed mismatches).
Transcript annotation
Transposable element (TE): TEs were annotated in our transcriptome by using the Repbase, Swissprot, PFAM, and nr databases. First, BLAST against repeat class “Transposable element” available for Hydra vulgaris in Repbase (Bao et al. 2015) (https://www.girinst.org/repbase/) was used to identify transcripts with TE identity (e-value cutoff of 1 × 10−5). Next, BLAST against the Swissport database was used to add Uniprot protein descriptions to transcripts using the Uniprot Retrieve ID/mapping tool (https://www.uniprot.org/uploadlists/, cutoff e-value of 1 × 10−5). Transcripts with Uniprot protein descriptions containing the character strings “transpos”, “jerky”, or “mobile element” were classified as TEs. Finally, previously published PFAM annotations of our Hydra transcriptome (Siebert et al. 2019) were used to predict additional TEs. Transcripts predicted to encode domains containing “transposase”, “THAP”, or “_tnp_” in the domain descriptor were also classified as TEs. Gene: Transcripts with the following characteristics were placed in the “gene” category: (i) received a Swissprot or nr annotation and (ii) not classified as TE by the method described above. Uncharacterized: Transcripts with the following characteristics were placed in the “uncharacterized” category: (i) lack of Swissprot/nr/Pfam annotation, (ii) predicted ORF of 100 amino acids or greater, and (iii) not classified as a TE by the approach described above. This category contains taxonomically restricted genes. Noncoding (ncRNA): Transcripts with the following characteristics were placed in the “ncRNA” category: (i) lack of Swissprot hit, nr hit, or PFAM domain, (ii) ORF less than 100 amino acids, and (iii) not classified as a TE by the method described above. Annotation results described above are summarized in the Supplemental File “Table_S1”
GO-term enrichment analysis
GO-term enrichment analysis was performed on the 441 transcripts up-regulated in response to hywi RNAi-1 using GOATOOLS v0.6.10 (https://github.com/tanghaibao/goatools) (Klopfenstein et al. 2018). GO terms were considered enriched if the Bonferroni-corrected P-value was <0.05.
RNA immunoprecipitation (RIP) and RT-PCR
For each RIP experiment, 10 colchicine-treated cnnos1::GFP Hydra were used to prepare a lysate and perform an IP as described above for piRNA isolation. Prior to IP, 5% of the total cleared lysate volume was removed and saved as the “input” sample. After IP, 1 ng of Luciferase Control RNA (Promega L4561) was added to the washed beads as a positive control for subsequent RNA extraction and cDNA synthesis. RNA was extracted from the beads using TRIzol. cDNA was synthesized using M-MLV Reverse Transcriptase, RNase H Minus, Point Mutant (Promega, M3681), and random hexamers (ThermoFisher Scientific, N8080127). Three biological replicates were performed for each transcript tested (Supplemental Fig. S7). PCR was performed using primers spanning 75–150 bp (Supplemental Table S5) for each transcript using GoTaq Green Master Mix (Promega, M7122) for 35 cycles.
qRT-PCR
To test the effect of colchicine treatment on TE expression, RNA was extracted from 10 colchicine-treated cnnos1::GFP Hydra using TRIzol. To perform qPCR on hywi RNAi-2 hatchlings, RNA was extracted from 10 hatchlings using TRIzol. For all qPCR, cDNA was synthesized using M-MLV Reverse Transcriptase, RNase H Minus, Point Mutant (Promega, M3681), and random hexamers (ThermoFisher Scientific, N8080127). qRT-PCR was performed using SsoAdvanced Universal SYBR Green Supermix (BioRad,1725271) with primers spanning 75–150 bp for each transcript assayed (Supplemental Table S5). RP49 was used for normalization. Fold change was calculated using the ΔΔCt method.
Differential gene expression analysis
For DGE analysis, offspring from the hywi RNAi-1 transgenic line were collected 4 d post hatching. Offspring that did not inherit the transgene were considered “WT.” Three WT and three hywi RNAi replicates were collected consisting of 10 animals each and RNA was extracted using TRIzol. Total RNA libraries were prepared using the Tru-Seq stranded RNA Kit (Illumina RS-122-2201). Libraries were sequenced on one lane of an Illumina HiSeq 2000 using a single-end 50 bp sequencing strategy. Illumina TruSeq3 Adapters were trimmed using trimmomatic (2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36). RSEM v1.2.31 (Li and Dewey 2011), bowtie v1.1.2 (Langmead et al. 2009), and the transcriptome reference was used to estimate expression levels. Differential gene expression analysis was performed using edgeR, v 3.20.9 (Robinson et al. 2010). After expression normalization, replicates were contrasted for variance using function plotMDS. One replicate from each treatment was identified as an outlier and excluded from downstream analysis. Analysis code is available in file “2_Differential_Gene_Expression_GO_Analysis” in the accompanying git repository.
Small RNA and piRNA filtering
Small RNA libraries used for sorting piRNAs by lineage were generated in our previous study (Juliano et al. 2014); NCBI BioProject PRJNA213706. Analysis code is available in file “4_Lineage_Sorted_piRNA_Mapping” in the git repository.
Single cell data exploration
Single-cell sequencing data for epithelial cells from homeostatic Hydra were interrogated for expression of putative PIWI targets (Siebert et al. 2019). URD spline objects for endoderm and ectoderm are available from Dryad: https://doi.org/10.5061/dryad.v5r6077 (file Hydra_URD_analysis_objects). These objects contain expression data for genes that are expressed in at least 1% of the ectodermal or endodermal epithelial cells. Analysis code is available in file “5_Single_Cell_Data_Exploration” in the git repository.
DATA DEPOSITION
Raw reads from piRNA sequencing, degradome sequencing, and lineage-sorted mRNA sequencing have been submitted to the GEO repository GSE135440.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
The sequencing was carried out by the DNA Technologies and Expression Analysis Cores at the UC Davis Genome Center, supported by National Institutes of Health (NIH) Shared Instrumentation grant 1S10OD010786-01. We thank Lutz Froenicke and Siranoosh Ashtari for their assistance and expert advice in RNA library preparation and sequencing, Matt Settles and Nikhil Joshi of the UC Davis Bioinformatics Core for their help in building code for piRNA analysis, Rob Steele and Prashanth Rangan for their critical review and editing during manuscript preparation. We thank two anonymous reviewers for insightful and careful reviews that significantly improved the manuscript. B.B.T. was supported by an NIH Training Grant (5 T32 GM 7377-39). This study was supported by UC Davis start-up funds (C.E.J.) and by the NIH (C.E.J., 1K01AG044435-01A1).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.072835.119.
REFERENCES
- Addo-Quaye C, Eshoo TW, Bartel DP, Axtell MJ. 2008. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr Biol 18: 758–762. 10.1016/j.cub.2008.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P, Iovino N, Morris P, Brownstein MJ, Kuramochi-Miyagawa S, Nakano T, et al. 2006. A novel class of small RNAs bind to MILI protein in mouse testes. Nature 442: 203–207. 10.1038/nature04916 [DOI] [PubMed] [Google Scholar]
- Aravin AA, Sachidanandam R, Girard A, Fejes-Toth K, Hannon GJ. 2007. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science 316: 744–747. 10.1126/science.1142612 [DOI] [PubMed] [Google Scholar]
- Aravin AA, Sachidanandam R, Bourc'his D, Schaefer C, Pezic D, Toth KF, Bestor T, Hannon GJ. 2008. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell 31: 785–799. 10.1016/j.molcel.2008.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai P. 2015. Biology of poly(ADP-Ribose) polymerases: the factotums of cell maintenance. Mol Cell 58: 947–958. 10.1016/j.molcel.2015.01.034 [DOI] [PubMed] [Google Scholar]
- Bao W, Kojima KK, Kohany O. 2015. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6: 11 10.1186/s13100-015-0041-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batki J, Schnabl J, Wang J, Handler D, Andreev VI, Stieger CE, Novatchkova M, Lampersberger L, Kauneckaite K, Xie W, et al. 2019. The nascent RNA binding complex SFiNX licenses piRNA-guided heterochromatin formation. Nat Struct Mol Biol 26: 720–731. 10.1038/s41594-019-0270-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch TCG, David C. 1987. Stem cells of Hydra magnipapillata can differentiate into somatic cells and germ line cells. Dev Biol 121: 182–191. 10.1016/0012-1606(87)90151-5 [DOI] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ. 2007. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128: 1089–1103. 10.1016/j.cell.2007.01.043 [DOI] [PubMed] [Google Scholar]
- Campbell RD. 1976. Elimination of hydra interstitial and nerve cells by means of colchicine. J Cell Sci 21: 1–13. [DOI] [PubMed] [Google Scholar]
- Carmell MA, Girard A, van de Kant HJG, Bourc'his D, Bestor TH, de Rooij DG, Hannon GJ. 2007. MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev Cell 12: 503–514. 10.1016/j.devcel.2007.03.001 [DOI] [PubMed] [Google Scholar]
- Das PP, Bagijn MP, Goldstein LD, Woolford JR, Lehrbach NJ, Sapetschnig A, Buhecha HR, Gilchrist MJ, Howe KL, Stark R, et al. 2008. Piwi and piRNAs act upstream of an endogenous siRNA pathway to suppress Tc3 transposon mobility in the Caenorhabditis elegans germline. Mol Cell 31: 79–90. 10.1016/j.molcel.2008.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco M, Criscione SW, Peckham EJ, Hillenmeyer S, Hamm EA, Manivannan J, Peterson AL, Kreiling JA, Neretti N, Sedivy JM. 2013. Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell 12: 247–256. 10.1111/acel.12047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, Caligiana A, Brocculi G, Adney EM, Boeke JD, et al. 2019. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566: 1–33. 10.1038/s41586-018-0784-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn CW, Giribet G, Edgecombe GD, Hejnol A. 2014. Animal phylogeny and its evolutionary implications. Annu Rev Ecol Evol Syst 45: 371–395. 10.1146/annurev-ecolsys-120213-091627 [DOI] [Google Scholar]
- Gainetdinov I, Colpan C, Arif A, Cecchini K, Zamore PD. 2018. A single mechanism of biogenesis, initiated and directed by PIWI proteins, explains piRNA production in most animals. Mol Cell 71: 775–790.e5. 10.1016/j.molcel.2018.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- German MA, Pillay M, Jeong D-H, Hetawal A, Luo S, Janardhanan P, Kannan V, Rymarquis LA, Nobuta K, German R, et al. 2008. Global identification of microRNA–target RNA pairs by parallel analysis of RNA ends. Nat Biotechnol 26: 941–946. 10.1038/nbt1417 [DOI] [PubMed] [Google Scholar]
- Girard A, Sachidanandam R, Hannon GJ, Carmell MA. 2006. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 442: 199–202. 10.1038/nature04917 [DOI] [PubMed] [Google Scholar]
- Grivna ST. 2006. A novel class of small RNAs in mouse spermatogenic cells. Genes Dev 20: 1709–1714. 10.1101/gad.1434406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardane LS, Saito K, Nishida KM, Miyoshi K, Kawamura Y, Nagami T, Siomi H, Siomi MC. 2007. A slicer-mediated mechanism for repeat-associated siRNA 5’ end formation in Drosophila. Science 315: 1587–1590. 10.1126/science.1140494 [DOI] [PubMed] [Google Scholar]
- Han BW, Wang W, Li C, Weng Z, Zamore PD. 2015. Noncoding RNA. piRNA-guided transposon cleavage initiates Zucchini-dependent, phased piRNA production. Science 348: 817–821. 10.1126/science.aaa1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmrich G, Khalturin K, Boehm A-M, Puchert M, Anton-Erxleben F, Wittlieb J, Klostermeier UC, Rosenstiel P, Oberg H-H, Domazet-Lošo T, et al. 2012. Molecular signatures of the three stem cell lineages in hydra and the emergence of stem cell function at the base of multicellularity. Mol Biol Evol 29: 3267–3280. 10.1093/molbev/mss134 [DOI] [PubMed] [Google Scholar]
- Homolka D, Pandey RR, Goriaux C, Brasset E, Vaury C, Sachidanandam R, Fauvarque M-O, Pillai RS. 2015. PIWI slicing and RNA elements in precursors instruct directional primary piRNA biogenesis. Cell Rep 12: 418–428. 10.1016/j.celrep.2015.06.030 [DOI] [PubMed] [Google Scholar]
- Horváth V, Merenciano M, González J. 2017. Revisiting the relationship between transposable elements and the eukaryotic stress response. Trends Genet 33: 832–841. 10.1016/j.tig.2017.08.007 [DOI] [PubMed] [Google Scholar]
- Houwing S, Kamminga LM, Berezikov E, Cronembold D, Girard A, van den Elst H, Filippov DV, Blaser H, Raz E, Moens CB, et al. 2007. A role for Piwi and piRNAs in germ cell maintenance and transposon silencing in Zebrafish. Cell 129: 69–82. 10.1016/j.cell.2007.03.026 [DOI] [PubMed] [Google Scholar]
- Ilyin AA, Ryazansky SS, Doronin SA, Olenkina OM, Mikhaleva EA, Yakushev EY, Abramov YA, Belyakin SN, Ivankin AV, Pindyurin AV, et al. 2017. Piwi interacts with chromatin at nuclear pores and promiscuously binds nuclear transcripts in Drosophila ovarian somatic cells. Nucleic Acids Res 45: 7666–7680. 10.1093/nar/gkx355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki YW, Siomi MC, Siomi H. 2015. PIWI-interacting RNA: its biogenesis and functions. Annu Rev Biochem 84: 405–433. 10.1146/annurev-biochem-060614-034258 [DOI] [PubMed] [Google Scholar]
- Jehn J, Gebert D, Pipilescu F, Stern S, Kiefer JST, Hewel C, Rosenkranz D. 2018. PIWI genes and piRNAs are ubiquitously expressed in mollusks and show patterns of lineage-specific adaptation. Commun Biol 1: 137 10.1038/s42003-018-0141-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BC, Wood JG, Chang C, Tam AD, Franklin MJ, Siegel ER, Helfand SL. 2016. A somatic piRNA pathway in the Drosophila fat body ensures metabolic homeostasis and normal lifespan. Nat Commun 7: 13856 10.1038/ncomms13856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliano CE, Reich A, Liu N, Götzfried J, Zhong M, Uman S, Reenan RA, Wessel GM, Steele RE, Lin H. 2014. PIWI proteins and PIWI-interacting RNAs function in Hydra somatic stem cells. Proc Natl Acad Sci 111: 337–342. 10.1073/pnas.1320965111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopfenstein DV, Zhang L, Pedersen BS, Ramírez F, Warwick Vesztrocy A, Naldi A, Mungall CJ, Yunes JM, Botvinnik O, Weigel M, et al. 2018. GOATOOLS: a Python library for gene ontology analyses. Sci Rep 8: 10872 10.1038/s41598-018-28948-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramochi-Miyagawa S, Watanabe T, Gotoh K, Totoki Y, Toyoda A, Ikawa M, Asada N, Kojima K, Yamaguchi Y, Ijiri TW, et al. 2008. DNA methylation of retrotransposon genes is regulated by Piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev 22: 908–917. 10.1101/gad.1640708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SH, Quarles KA, Yang Y, Tanguy M, Frézal L, Smith SA, Sharma PP, Cordaux R, Gilbert C, Giraud I, et al. 2018. Pan-arthropod analysis reveals somatic piRNAs as an ancestral defence against transposable elements. Nat Ecol Evol 2: 174–181. 10.1038/s41559-017-0403-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12: 323 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Prazak L, Chatterjee N, Grüninger SGU, Krug L, Theodorou D, Dubnau J. 2013. Activation of transposable elements during aging and neuronal decline in Drosophila. Nat Neurosci 16: 529–531. 10.1038/nn.3368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim RSM, Kai T. 2015. A piece of the pi(e): the diverse roles of animal piRNAs and their PIWI partners. Semin Cell Dev Biol 47–48: 17–31. 10.1016/j.semcdb.2015.10.025 [DOI] [PubMed] [Google Scholar]
- Lim RSM, Anand A, Nishimiya-Fujisawa C, Kobayashi S, Kai T. 2014. Analysis of hydra PIWI proteins and piRNAs uncover early evolutionary origins of the piRNA pathway. Dev Biol 386: 237–251. 10.1016/j.ydbio.2013.12.007 [DOI] [PubMed] [Google Scholar]
- Malone CD, Brennecke J, Dus M, Stark A, McCombie WR, Sachidanandam R, Hannon GJ. 2009. Specialized piRNA pathways act in germline and somatic tissues of the Drosophila ovary. Cell 137: 522–535. 10.1016/j.cell.2009.03.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani SR, Juliano CE. 2013. Untangling the web: the diverse functions of the PIWI/piRNA pathway. Mol Reprod Dev 80: 632–664. 10.1002/mrd.22195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcum BA, Campbell RD. 1978. Development of Hydra lacking nerve and interstitial cells. J Cell Sci 29: 17–33. [DOI] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-througput sequencing reads. EMBnetjournal 17: 10–12. [Google Scholar]
- Maxwell PH, Burhans WC, Curcio MJ. 2011. Retrotransposition is associated with genome instability during chronological aging. Proc Natl Acad Sci 108: 20376–20381. 10.1073/pnas.1100271108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modepalli V, Fridrich A, Agron M, Moran Y. 2018. The methyltransferase HEN1 is required in Nematostella vectensis for microRNA and piRNA stability as well as larval metamorphosis. PLoS Genet 14: e1007590 10.1371/journal.pgen.1007590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn F, Handler D, Brennecke J. 2015. Noncoding RNA. piRNA-guided slicing specifies transcripts for Zucchini-dependent, phased piRNA biogenesis. Science 348: 812–817. 10.1126/science.aaa1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida KM, Saito K, Mori T, Kawamura Y, Nagami-Okada T, Inagaki S, Siomi H, Siomi MC. 2007. Gene silencing mechanisms mediated by Aubergine piRNA complexes in Drosophila male gonad. RNA 13: 1911–1922. 10.1261/rna.744307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimiya-Fujisawa C, Kobayashi S. 2012. Germline stem cells and sex determination in Hydra. Int J Dev Biol 56: 499–508. 10.1387/ijdb.123509cf [DOI] [PubMed] [Google Scholar]
- Ozata DM, Gainetdinov I, Zoch A, O'Carroll D, Zamore PD. 2019. PIWI-interacting RNAs: small RNAs with big functions. Nat Rev Genet 20: 89–108. 10.1038/s41576-018-0073-3 [DOI] [PubMed] [Google Scholar]
- Pandey RR, Homolka D, Chen K-M, Sachidanandam R, Fauvarque M-O, Pillai RS. 2017. Recruitment of Armitage and Yb to a transcript triggers its phased processing into primary piRNAs in Drosophila ovaries. PLoS Genet 13: e1006956 10.1371/journal.pgen.1006956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson MN, Scannapieco AE, Au PH, Dorsey S, Royer CA, Maxwell PH. 2015. Preferential retrotransposition in aging yeast mother cells is correlated with increased genome instability. DNA Repair (Amst) 34: 18–27. 10.1016/j.dnarep.2015.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praher D, Zimmermann B, Genikhovich G, Columbus-Shenkar Y, Modepalli V, Aharoni R, Moran Y, Technau U. 2017. Characterization of the piRNA pathway during development of the sea anemone Nematostella vectensis. RNA Biol 14: 1727–1741. 10.1080/15476286.2017.1349048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RJ, Weiner MM, Lin H. 2014. PIWI proteins and PIWI-interacting RNAs in the soma. Nature 505: 353–359. 10.1038/nature12987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Nishida KM, Mori T, Kawamura Y, Miyoshi K, Nagami T, Siomi H, Siomi MC. 2006. Specific association of Piwi with rasiRNAs derived from retrotransposon and heterochromatic regions in the Drosophila genome. Genes Dev 20: 2214–2222. 10.1101/gad.1454806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaible R, Scheuerlein A, Dańko MJ, Gampe J, Martínez DE, Vaupel JW. 2015. Constant mortality and fertility over age in Hydra. Proc Natl Acad Sci 112: 15701–15706. 10.1073/pnas.1521002112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebert S. 2018. Hydra cell lineages. https://commons.wikimedia.org/wiki/File:Hydra_cell_lineages.svg [Google Scholar]
- Siebert S, Farrell JA, Cazet JF, Abeykoon Y, Primack AS, Schnitzler CE, Juliano CE. 2019. Stem cell differentiation trajectories in Hydra resolved at single-cell resolution. Science 365: eaav9314 10.1126/science.aav9314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon M, Van Meter M, Ablaeva J, Ke Z, Gonzalez RS, Taguchi T, De Cecco M, Leonova KI, Kogan V, Helfand SL, et al. 2019. LINE1 derepression in aged wild-type and SIRT6-deficient mice drives inflammation. Cell Metab 29: 871–885. 10.1016/j.cmet.2019.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi MC, Sato K, Pezic D, Aravin AA. 2011. PIWI-interacting small RNAs: the vanguard of genome defence. Nat Rev Genet 12: 246–258. 10.1038/nrm3089 [DOI] [PubMed] [Google Scholar]
- Sousa-Victor P, Ayyaz A, Hayashi R, Qi Y, Madden DT, Lunyak VV, Jasper H. 2017. Piwi is required to limit exhaustion of aging somatic stem cells. Cell Rep 20: 2527–2537. 10.1016/j.celrep.2017.08.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm Á, Perczel A, Ivics Z, Vellai T. 2017. The Piwi-piRNA pathway: road to immortality. Aging Cell 16: 906–911. 10.1111/acel.12630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin VV. 2006. A distinct small RNA pathway silences selfish genetic elements in the germline. Science 313: 320–324. 10.1126/science.1129333 [DOI] [PubMed] [Google Scholar]
- Van Meter M, Kashyap M, Rezazadeh S, Geneva AJ, Morello TD, Seluanov A, Gorbunova V. 2014. SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat Commun 5: 1–10. 10.1038/ncomms6011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Geesman GJ, Hostikka SL, Atallah M, Blackwell B, Lee E, Cook PJ, Pasaniuc B, Shariat G, Halperin E, et al. 2011. Inhibition of activated pericentromeric SINE/Alu repeat transcription in senescent human adult stem cells reinstates self-renewal. Cell Cycle 10: 3016–3030. 10.4161/cc.10.17.17543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Cui X, Yuan Z, Qi H, Lin H. 2018. MIWI2 targets RNAs transcribed from piRNA-dependent regions to drive DNA methylation in mouse prospermatogonia. EMBO J 37: e95329 10.15252/embj.201695329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittlieb J, Khalturin K, Lohmann JU, Anton-Erxleben F, Bosch TCG. 2006. Transgenic Hydra allow in vivo tracking of individual stem cells during morphogenesis. Proc Natl Acad Sci 103: 6208–6211. 10.1073/pnas.0510163103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JG, Jones BC, Jiang N, Chang C, Hosier S, Wickremesinghe P, Garcia M, Hartnett DA, Burhenn L, Neretti N, et al. 2016. Chromatin-modifying genetic interventions suppress age-associated transposable element activation and extend life span in Drosophila. Proc Natl Acad Sci 113: 11277–11282. 10.1073/pnas.1604621113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Xu J, Koppetsch BS, Wang J, Tipping C, Ma S, Weng Z, Theurkauf WE, Zamore PD. 2012. Heterotypic piRNA Ping-Pong requires qin, a protein with both E3 ligase and tudor domains. Mol Cell 44: 572–584. 10.1016/j.molcel.2011.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Kang J-Y, Gou L-T, Wang J, Xue Y, Skogerboe G, Dai P, Huang D-W, Chen R, Fu X-D, et al. 2015. MIWI and piRNA-mediated cleavage of messenger RNAs in mouse testes. Cell Res 25: 193–207. 10.1038/cr.2015.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.