

Abstract
Cooperation and harmonisation of standards in the pharmaceutical domain is already a reality and, in fact, has been increasingly important in recent decades. This harmonisation seems natural and logical in the current environment of increased globalisation imposed by the current geo-economic-political situation. However, even though this is a reality, it is important to analyse whether or not all stakeholders benefit from this increased cooperation and harmonisation, in order to recommend future actions or potential improvements.
Cooperation, convergence and harmonisation is indeed not an end in itself. Elimination of differences, agreement on common standards and the increase of cooperation are needed to protect and promote global public health. It is therefore very important to thoroughly analyse the value and impact of the harmonisation and cooperation process, and to determine whether or not all the efforts made throughout the years have indeed met expectations. After many years of experience, it seems important to have a thorough evaluation to answer several important questions:
-
▸
What has been the benefit of the multiple harmonisation and cooperation initiatives so far?
-
▸
Should such processes continue? Are there any other alternatives to resolve current challenges?
-
▸
Should current initiatives and projects be improved? If so, how and what are the priorities?
-
▸
Would greater harmonisation and closer cooperation be beneficial? Is it possible? Is it necessary? What are the limits of this process?
-
▸
What are the important lessons from the past and the on-going harmonisation initiatives that should be considered for planning the next steps?
This book section’s goal is to answer these important questions by thoroughly evaluating the added value of cooperation, convergence and harmonisation. This book section also reviews the critical parameters and influencing factors for cooperation, convergence and harmonisation. This review/analysis of critical parameters and influencing factors is essential to understand the complexity of the situation and effectively discuss how to organise the next steps of global cooperation.
Keywords: Globalisation, Regulation, Medicine, Cooperation, Convergence, Harmonisation, International, Regional, Bilateral, Network, Pharmaceutical, Standard, Requirement, Best Practice, Regulatory capacity, resources, expertise, infrastructure, Communication, Planning, Organisation, Structure, Implementation, monitoring, Voluntary cooperation, Legal commitment, Influence, Politics, Political decisions, Economy, Cultures, Traditions, Ethnic Factors, Medical practices, legislations, Statutes, regulatory systems, cost effectiveness
Following the review of all major harmonization initiatives in the pharmaceutical sector (see Part I), two questions need to be answered before any action can be recommended:
-
▸
Do the ongoing and past harmonization initiatives have (or have they had) value, and if so, would further harmonization be possible and would it be beneficial?
-
▸
What are the important lessons from the past, and the ongoing harmonization initiatives that should be considered for planning the next steps?
II-1). Value of the Cooperation, Convergence, and Harmonization in the Pharmaceutical Domain
Differences in regulations between countries have been a problem in many areas (i.e., mobile phones, electrical equipment, etc.). In past decades, these differences were more problematic, but today it is common to travel and to buy and sell items on the Internet between people living throughout the world. To support this globalization of economic and social expectations, the harmonization of standards and regulations is necessary in many areas. Therefore, competent authorities began to collaborate, most of the time under the United Nations (UN) organization, to exchange information and to harmonize their regulations. These collaborations have been successful in certain domains (e.g., international air navigation), while challenging in others (e.g., climate change).
Cooperation and harmonization of standards in the pharmaceutical domain are already a reality, and have in fact been increasingly important in the past several decades (see Part I). This harmonization seems natural and logical in the current environment of increased globalization imposed by the current geo-economic-political situation. However, even though this is a reality, it is important to analyze whether all stakeholders benefit from this increased cooperation and harmonization in order to recommend future actions or potential improvements.
II-1.1). Value for Patients and Global Public Health
Even though there are differences between developed and developing countries or between regions, diseases go beyond borders and are present worldwide. It therefore seems logical that the development and manufacture of medicines against these diseases should be based on the same global standards, independent of where the sites of manufacture are or the clinical studies occur.
The harmonization of standards and cooperation between countries are beneficial to patients and global public health in several ways:
-
▸
Increase of Worldwide Access to Medicines: Pharmaceutical access is defined as the timely availability of quality medicines to those patients who need them. The unavailability of some medicines poses a real threat to public health and welfare. Many interrelated factors (i.e., availability of financial resources, government policies, infrastructure conditions, private and public sector insurance programs, appropriate use, supply management, manufacturing capacity, research and development decisions, etc.) determine the level of access. It is clear that the level of harmonization of pharmaceutical regulations and cooperation (regionally or globally) also influences pharmaceutical access in both developed and developing countries. These activities increase the availability of high-quality, safe, and effective medicines worldwide, and promote better access to a larger worldwide population. For example, the “drug lag” in Japan (i.e., delay of availability of new medicines vs. the US) has been partly attributed to specific local requirements requested by Japanese regulators. The same analysis partly attributed the recent reduction of this “drug lag” (from 2.4 years in 2006 to 1.1 years in 2010) to the implementation of PMDA measures to increase global cooperation [280].
By promoting the conduct of multinational clinical trials that meet international standards, the harmonization of regulation also facilitates faster access to innovative and quality medicines for worldwide patients. Indeed, harmonization of rules (i.e., implementation of ICH Good Clinical Practices [GCP]) and the introduction of a “mature” regulatory system (i.e., consistent and transparent) increase the interest of the global pharmaceutical industry in performing multinational studies, and therefore increase access to drugs in development for patients from these countries.
-
▸
Promotion of the Development and Implementation of High Standards: Collaboration facilitates dissemination, recognition, and adoption of best practices. Moreover, if the creation/harmonization of an international standard follows an appropriate and rigorous process (i.e., based on scientifically driven discussions), the quality, robustness, and relevance of such standards will increase as it integrates different expertise, experiences, and points of view from the best worldwide experts in the field. This will ultimately benefit the patients who will have the assurance that their medicines have been developed and manufactured according to the highest standards.
In certain domains, there are a limited number of experts worldwide (e.g., new technologies or specific diseases), therefore it is critical to gather these international experts together to develop high-quality standards.
-
▸
Reduction of Unnecessary Testing in Animals and Humans: The reduction of unnecessary testing in animals and humans should always be a priority for ethical reasons. The amount of human and animal experimentation is reduced when companies only have to produce one set of data for all regions.
-
▸
Promotion of Innovation and Development of Medicines for Unmet Medical Needs: One could question if the increase in harmonization of standards can decrease innovation and delay availability of a drug in certain markets. For example, it could be argued that if today Country A requires a one-year study to support the registration of a new product in a specific indication, while Country B requires only six months, the harmonization of requirements could potentially delay the availability of this product in Country B by six months. However, although there is indeed a risk of delay, which may in fact be beneficial for the patient if the harmonization discussions conclude that the risk/benefit assessment is more relevant at 12 months, harmonization and cooperation activities are in fact contributing to pharmaceutical innovation by promoting predictable and consistent requirements [281] and reducing regulatory uncertainty. Reducing the risk for industry (by releasing clear and harmonized technical guidelines and accepting foreign data), and increasing return on investment (by increasing global marketing opportunities), these harmonization activities stimulate investment in research and development (R&D). Moreover, the elimination of redundancy and duplication of work and testing to satisfy different requirements frees up resources for R&D. Finally, MedDRA and other harmonization of terminologies facilitate and increase communication between experts, which in turn increases cooperation and innovation.
Harmonization of regulations and requirements can also make development of certain types of products financially viable for industry. These development projects would indeed not be funded if different worldwide requirements needed different programs or clinical studies to support global registration. For example, before ICH recommendations on how to demonstrate comparability of biotechnological/biological products subject to changes in their manufacturing process [282], regional disparities created economic problems (i.e., wasted inventories, shelf-life, several parallel manufacturing processes) or implementation delays for industry, which discouraged process upgrades and improvements.
Finally, it is also important to note that harmonization of requirements is critical for the development and availability of orphan drugs that treat life-threatening conditions and rare diseases because it allows multinational clinical studies and obviates the development challenges due to the limited number of patients. The success of the European Union (EU) orphan drug regulation demonstrated how harmonization and cooperation can improve public health and increase availability of these types of medicines. Prior to this European legislation, a number of Member States had adopted specific measures to increase knowledge on rare diseases, but these initiatives did not lead to any significant progress in research on rare diseases. Following the implementation of EU Regulation (EC) No 141/2000, the number of orphan medicinal products authorized significantly increased. During the first five years of the implementation, 458 applications for orphan designation were submitted, resulting in 268 products being designated, relating to over 200 different rare conditions [283].
-
▸
Contribution to the Control of Medicine Quality: The harmonization of technical quality standards (e.g., Good Manufacturing Practices [GMP] domain) contributes to ensure that medicines, most of the time developed, manufactured, and tested in different countries, are of good quality.
Moreover, falsified medicines are a major threat to public health and safety. As falsifications become more sophisticated, the risk that falsified medicines reach patients in undeveloped (as well as developed) countries increases every year. This calls for a comprehensive strategy at the international level. International cooperation already exists through the International Medical Products Anti-Counterfeiting Taskforce (IMPACT) [284] created by WHO in 2006. International cooperation and harmonization of regulatory actions are critical to ensure success against falsified medicines.
-
▸
Contribution to the Evaluation and Monitoring of the Safety of Medicines: The establishment of a global collaboration framework and the harmonization of reporting (i.e., formats and processes) enhance the safety of products. It allows exchange of information and rapid communications between worldwide DRAs regarding new product safety problems via agreed-upon common tools (e.g., Electronic Individual Case Safety Reports, Periodic Safety Update Report [PSUR], and Development Safety Update Report [DSUR]). This is critical to better monitor the safety of medicines, and has a direct benefit for patients.
Such cooperation also allows pooling of information from different sources, regions, and countries. This increase of the quantity of safety information facilitates the early detection of possible safety signals and therefore improves the monitoring of product safety.
Pharmacovigilance would be even more effective if it was supported by a truly effective “global” system. Pharmaceutical companies already have global departments that assess the safety profile of a product based on safety reporting from different parts of the world where the product is registered and marketed. True harmonization of practices and cooperation between worldwide DRAs is also necessary to have a real safety profile of the product (especially for an orphan drug where the patient population is limited).
-
▸
Support to Developing Countries: Global cooperation and harmonization of standards support and assist achievement of the UN Millennium Development Goals (MDGs) as discussed in Part I-1.1. These activities improve health in developing regions by increasing access to safe and effective medicines of good quality. This is accomplished by strengthening the technical and administrative capacity via collaboration and the sharing of resources and expertise [285].
Technical and administrative assistance from the international community (i.e., ICH, WHO, or major DRAs) are indeed critical in supporting the development and enhancement of regulation in certain regions (e.g., Africa). This assistance (i.e., training, development of high quality standards, and technology transfer) helps developing countries to establish a mature regulatory system. It provides mentoring opportunities where more experienced DRAs from developed countries can share knowledge and experience with less advanced DRAs from developing countries. Without the help from international organizations, these countries/regions would not have the resources, expertise, or time to discuss, assess, and regulate new topics (i.e., biosimilars, gene therapies, etc.). If they apply “standard” rules to these types of state-of-the-art products and therapy, the patients may be exposed to risk.
Harmonization and cooperation also facilitate the economic development of low-income countries because they increase the attractiveness of their local facilities for the manufacture of medicines and the conduct of clinical studies. A 2010 Industry Survey [286] showed that better cooperation between African countries (to reduce country-specific requirements) would promote access to new medicines, encourage more companies to register medicines in Africa, and ensure a continuous supply of medicines. Indeed, specific country requirements increase the cost of medicines to African countries, and in some cases contribute to the discontinuation of medical supplies to these countries.
Finally, until recently, the focus of DRAs from developing countries was to exercise a regulatory oversight on products that were licensed and used in developed countries. However, some new products (i.e., new vaccines) are now being developed for use in the developing world, and sometimes exclusively in these markets. More clinical trials are being conducted in countries with weak regulatory systems, who are therefore confronted with new challenges which they are not in a position to address (i.e., assessing clinical trial applications and marketing applications not yet assessed by a developed country) [287]. Several initiatives, such as the WHO prequalification of products, the WHO regulatory pathways initiative, the WHO Initiative for Vaccine Research, or the EMA Scientific Opinion via the “Article 58” procedure, have already been established. International cooperation between WHO, developed countries, and developing countries is critical in this situation.
All the above benefits of harmonization and cooperation to patients and global public health are even more apparent if one reviews the public health risks that the lack of harmonization would generate. For example, in the absence of the ICH process, the DRAs of the three ICH regions may have continued to diverge in their practice and may have requested further local/regional focus and repetition in drug development activities. This increase of development time and efforts would have surely delayed the availability of key medicines to patients. Moreover, if there were no cooperation and harmonization, certain developing countries would not be able to develop and implement functioning regulatory systems and pharmaceutical regulation on their own. This lack of pharmaceutical control would alleviate unethical activities that would ultimately impact global public health with:
-
▸
Increased production and importation of substandard/counterfeit medicines [288]
-
▸
Conduct of unethical clinical trials
-
▸
Corruption
-
▸
Distribution of unregistered medicines
-
▸
Irrational prescribing practice
-
▸
Irrational dispensing practice
The case of biosimilar products also demonstrates how the lack of harmonized requirements (or lack of implementation of harmonized standards) can impact public health. Although these products have recently been an important global health topic of discussion and many countries prepared appropriate requirements over the past several years, other countries have not yet developed specific regulations (or not yet integrated the WHO recommendations) to cover the risk associated with the approval and use of these specific products. Due to the lack of appropriate regulation related to this specific type of product (and sometimes without having any regulations for biologics at all), these countries (i.e., Argentina) evaluate and approve these “biosimilar” products using the same requirements as those for standard generics. They do not take into consideration the specific risks associated with biosimilar products [289] not present in the case of standard generic (e.g., immunogenicity). In some countries, the need for less expensive medicines may also link to the approval of subpotent biological/biosimilar products. This lack of implementation of international requirements and standards has an obvious risk for patients and public health.
II-1.2). Value for Regulators
DRAs have the mandate to protect and enhance the health of their population by facilitating access to medicines of public health importance without compromising on quality, safety, and efficacy. However, as international markets expand and pharmaceutical companies operate more and more globally, the task of regulators to assess compliance with legislation and monitor the safety and quality of medicines becomes increasingly difficult and resource intensive. In addition, cuts in government budgets in recent years oblige the DRAs to rethink processes and utilization of resources. This is obviously a difficult exercise because they need to manage new challenges (i.e., increased globalization but also sophisticated products, new technologies, increased communication through the Internet, counterfeiting, etc.) with less budget and resources. In this context, maintaining the capacity and expertise to meet their obligations becomes a problem. As rightly stated by the Heads of Government of the Caribbean Community in the Nassau Declaration on Health in July 2001, “while the resources and absorptive capacity of no one single institution, country or nation are sufficient to reverse the negative trend, the evidence of ‘best practices’ and technological breakthroughs, the international, regional and national mechanisms and frameworks … provide hope of what can be achieved through a collective response” [290].
Cooperation with other countries seems indeed the only alternative to manage the situation and to address the specific challenges associated with the globalization of the development, manufacture, and distribution of medicines. This is not a choice anymore if these countries want to provide people with more effective and safer medicines more quickly [291]. Therefore, cooperation has been intensified at different levels and the networking of institutions in developing and developed countries is an important element in building regulatory capacity and trust. Development of common standards for scientific evaluation and inspection also facilitates regulatory communication and information sharing. Of course, each country will remain responsible for the final decision (e.g., to approve/suspend a drug), but exchange of evaluation/information (e.g., safety alerts) is a “must” and not a “nice to have” anymore. Worldwide DRAs are required to harmonize their activities with the international community. Numerous Memorandum of Agreements (MoA) or Memorandum of Understanding (MoU) have been put in place between DRAs to allow legal exchange of information.
However, even if this has been imposed on them, most regulators now agree that domestic and international measures complement each other, and therefore domestic and international operations should be carried out seamlessly with the understanding that they are inseparable in nature [292]. For example, the new international focus of the US FDA and the reorganization of its inspectional resources (i.e., the opening of overseas offices) helped to enforce compliance, and it is now easier for the US FDA to control and inspect these foreign manufacturing sites [293]. The ICH process also demonstrated that harmonization brings value and benefits to DRAs because it improves consistency, efficiency, and transparency of review, which in turn facilitates information sharing among regulators and ultimately promotes faster access to life-saving treatments to patients on a worldwide basis [294].
Moreover, harmonization and cooperation also allow regulators to learn from each other’s experiences, to leverage international expertise, and to keep up with international best practices and standards. For example, the decision by the EMA and US FDA to collaborate on biosimilar products will certainly help the US FDA to catch up with Europe in its development of the regulation of these products. Indeed, Europe has already gathered a lot of experience in this area (the first biosimilar product was approved in Europe in 2006), while the US has just recently passed their law on biosimilars. In Europe the regulatory framework for biosimilars is largely established, with both general guidelines and product-specific guidelines (e.g., human insulin, somatropin, erythropoietins, interferon-alpha, low-molecular-weight heparins, and monoclonal antibodies) having been put in place by the EMA. The EMA is also currently working on draft guidelines for a number of other product class-specific guidelines, including interferon-beta and follicle-stimulation hormone. The US, on the other hand, is lagging behind the EU because the legal pathway (Biologics Price Competition and Innovation [BPCI] Act) was only signed into law on March 23, 2010 by US President Barack Obama [295].
DRAs from developing countries also benefit from the experience, expertise, and resources from other countries. Regulators from these developing countries also see the value in cooperating with both developed and developing countries [296]. For instance, the cooperation project between Brazil and Mozambique demonstrates that cooperation and harmonization between developing countries can be very beneficial [297].
Finally, harmonization also decreases the duplication of activities and therefore facilitates work sharing and optimization of DRAs’ time and resources. It can help manage DRAs’ workloads more efficiently (critical due to the increase of complex and voluminous data to review in a resource-constrained environment), and ultimately improve overall regulatory performance. It is also a way to resolve some of the resource limitations and budget constraints in developing countries. However, although important, resource saving should not be the only objective to ensure success of cooperation. Even if it is expected that cooperation and harmonization can increase efficiency in the long term, such activities can in fact require more work at the beginning. Harmonization and cooperation can deliver much more than resource saving (e.g., better informed decisions, increased protection of public health, and facilitated availability of medicines in developing countries).
Even if the vast majority of worldwide regulators and national DRAs fully support and recognize the value of harmonization and cooperation, some resistance may sometimes arise. This sporadic resistance from some regulators and national authorities to increase cooperation and harmonization is inherent to the relative fear of losing “power” and sovereignty. These concerns need to be kept in mind when establishing new harmonization projects or collaboration as it could raise implementation challenges, even if this threat is not fully realized. Indeed, even if cooperation obviously implies recognition of other evaluations and expertise, regional or global cooperation does not replace national judgment/decisions and harmonization does not automatically mean a loss of national sovereignty or autonomy. It depends on the model selected (i.e., integration vs. cooperation) and the stage of the harmonization process. Collaborative mechanisms (i.e., joint assessments or inspections) does not imply common decision making. Close collaboration can occur even if a registration decision itself stays in the hands of sovereign nations. The European system also demonstrates that, even in a case of integration where Member States relinquished part of their sovereignty in favor of the Community and delegated some of their decision-making powers to shared institutions and supranational bodies, national DRAs remained crucial for the pharmaceutical network. For example, experts and regulators involved in harmonization activities come from national DRAs (i.e., EMA is a network of experts who are made available by the national DRAs of all EU Member States).
Regional and global collaboration does not replace national DRAs. This is an evolution of regulators’ activities and responsibilities. National DRAs will still be responsible for managing specific countries’ health needs and establishing public health priorities. Even if a global medicines evaluation system were established, national DRAs would remain the ultimate decision-making bodies. However, everyone can learn something from others! To fulfill their mandate to promote and protect public health in the current global environment, each DRA needs to ask itself the following questions:
-
▸
What are the strengths of counterpart DRAs?
-
▸
How do we leverage each other competencies and expertise?
II-1.3). Value for Industry
The cost of drug development increased considerably in the past decade due to more advanced and expensive technology and new requirements. Today, the average cost of bringing a new medicine to market is US $1.3 billion [298], it takes approximately 12 years (from the first toxicity dose to the first launch), and it has a success rate of 5% [299]. The combination of this strong increase in research and development (R&D) costs and the decrease of new molecular entity (NME) approvals has significantly impacted R&D productivity. Moreover, reduction in prices and reimbursement of medicines by worldwide governments (due to health budget restrictions), combined with low sales growth, has placed additional pressure on profit margins for pharmaceutical companies [300]. The only solution for innovative companies to keep up with this decrease of R&D productivity and to improve their return on investment is to market new medicines globally with minimal additional costs required to satisfy the regulatory requirements of different countries.
However, although entering global markets has significant advantages and facilitates return on investment, pharmaceutical companies face challenges in marketing their products in different countries/regions with diverse regulatory requirements and practices. These different, and sometimes conflicting or contradictory, requirements make this global effort difficult, time consuming, and costly. Pharmaceutical companies must integrate the challenge of complying with these disparate international requirements and regulatory systems. This means expending resources to develop knowledge on local specificities and markets that translate to operational complexity and additional costs and time. Resources that could best be used for increasing R&D productivity are often used to meet different regulatory requirements that may add little to the evaluation of the risk–benefit of new therapies. More importantly, these specific local requirements delay the availability of new medicines for the population of these countries.
It is of course essential to establish a sufficient body of clinical and scientific evidence to ensure that newly introduced innovative medicines are safe and effective. This work is needed whatever the cost and resources it requires. However, some of the existing local regulatory requirements that necessitate repetition of work already conducted are viewed as impeding the ability to rapidly bring innovative medicines to address unmet medical needs. Indeed, this regulatory variability across multiple countries brings an extreme complexity to development activities and ultimately impedes, rather than facilitates, patient access to meaningful, new, evidence-based medicines [301]. On the contrary, consistency and scientific quality of recommendations from DRAs (e.g., for study design) ease the development of new medicines and reduce the risk.
The ICH process demonstrated that harmonization brings value and benefits to pharmaceutical industries as it enables industry to reduce development times by removing the duplication of studies that was previously required to gain global market approval for a new medicine. This directly affects the bottom line through reduced development times and resources (that can be allocated to new additional development projects). This harmonization of standards also facilitates organization of companies and intracompany globalization [302].
Harmonization of quality specifications and analytical tests between countries would also mean an important cost savings for industry. It would reduce the need for several tests and batch releases of the same product batch. Moreover, some countries require in-country testing of each lot marketed in their territory (these analytical tests are performed by a government laboratory or a third-party laboratory contracted by the DRAs). If all countries had the same specifications and tests, and agreed to accept analytical results from other countries, in-country testing would not need to be repeated in several countries. This elimination of duplication of testing would obviously reduce the cost, time, and resources associated with multiple lot releases and in-country testing. This is also important for in vivo assays, to reduce unnecessary use of animals.
Harmonization of requirements also facilitates the development of new markets that are currently not profitable for global companies due to the cost of specific requirements. However, it is important to ensure that global standards are appropriately implemented in each region/country to avoid disadvantaging local drug manufacturers/industries. The implementation of global standards should not create a barrier to local companies and facilitate big global industry. It is therefore important to balance the necessary implementation of scientific global standards to support/improve public health in all countries while addressing the needs of each country. Overregulating the pharmaceutical market in low-income countries could indeed impact the local economy.
Some industry representatives may at first be opposed to the harmonization of regulation for several reasons. The first concern from industry is the unnecessary increase of requirements that harmonization and cooperation could generate. This is a legitimate concern because an easy way to harmonize a topic between countries is to combine all current requirements from different countries (i.e., if one country requests Study A and another country requests Study B for the registration of new medicines, a quick solution would be to ask for both Studies A and B). To avoid this unreasonable duplication of requirements (which would be against one of the main objectives of harmonization: to reduce duplication of activities), the harmonization process needs to be considered carefully. This harmonization needs to be based on scientific evidence and evaluation to allow for the development of high standards. This is not a quick compromise or addition of requirements, but a common requirement established following a thorough evaluation of the situation. Of course, the basis of this common scientific evaluation needs to take into account the existing worldwide requirements. The ICH process is an excellent example of how scientific-driven evaluation can produce common high-quality standards.
Second, one could argue that these new global requirements could reduce flexibility and a creative “development program.” They are also concerned that harmonization may impact regulator and sponsor interaction during the development of medicines and that certain regulators may apply these new harmonized rules to everyone without discerning the specificity of each product/program. Although these concerns need to be kept in mind when organizing and implementing the next steps of global harmonization and cooperation, they seem to be not fully relevant for the following reasons:
-
▸
For nonemerging topics: Pharmaceutical companies prefer to avoid surprises. The worst situation for a company is the unknown and its associated risks. Therefore, the two major benefits of harmonization (i.e., allowing for a global development plan supporting global registration and decreasing the unknown and its risks) outweigh the potential flexibility of having diverse requirements. Moreover, if there is a good scientific reason not to follow a guideline/recommendation, the company will always have the opportunity to discuss this deviation to general rules via scientific meetings during development.
-
▸
For new emerging topics and therapies: Regulator and innovative companies’ interactions during development will always be necessary. Development of a new class of compound or the first products to treat a new indication or disease will always need this type of interaction and sometimes a creative development program. Even if a proactive discussion is put in place between DRAs for new emerging topics, the role of this discussion will be to agree on terminology, endpoint, etc. The relevance and importance of the dialogue between the regulators and the first company developing such medicines will still be critical.
Finally, some industry resistance may also come from the fear of creating a global system that would deliver a common global decision that, in the case of a negative outcome, would mean refusal in all countries worldwide. However, it is important to remember that a global system would not mean a global marketing authorization. Unlike the European integrated system, a global system could provide a concerted/shared evaluation of data, but not a common decision regarding risk/benefit assessment. Following the shared evaluation of technical data (or recognition of the assessment from another country), each national DRA would remain the decision-making body. Each country would conduct their individual regulatory decision-making process regarding the risk/benefit evaluation following their own procedures and requirements. Each DRA would provide an independent decision, which could therefore still differ. This national decision-making process would also happen if a global mutual recognition procedure were to be established.
II-1.4). Would Further Cooperation and Harmonization Be Possible and Beneficial?
As presented in Part I, a substantial amount has already been done to harmonize pharmaceutical regulations. Many harmonization initiatives (i.e., bilateral, regional, and global) have already been established. The combination of this proactive collaboration with the natural convergence of issues and priorities on a worldwide basis (due to the globalization of trade and development/manufacture of pharmaceutical products) led to the convergence of requirements. However, there is still divergence between countries on many topics. More can be done to continue to protect and promote global public health (e.g., further harmonization and support for international clinical studies, better proactive support for the development of global standards related to emerging issues and new technologies, closer cooperation between DRAs, etc.). Moreover, the lack of international coordination of all harmonization initiatives is a problem. All of these ongoing initiatives remain segregated. ICH (via the GCG) and WHO initiated a type of coordination, but these efforts only promote good communication between the different players to share experiences and information on projects. Although such communication between global and regional initiatives is of course important, it is not sufficient. There is clearly a lack of leadership in the coordination of these initiatives and duplication of efforts. More proactive management of international actions needs to be established, and a global strategy (which would define clear objectives and responsibilities) needs to be agreed on. All current participants of the harmonization (at national, regional, and global levels) are critical, but the management of all these ongoing activities would certainly decrease duplication and would be more efficient and productive.
There is also an urgent need to increase access to priority essential medicines by reducing the time it takes for beneficial therapies to reach patients in need in developing countries/regions (such as Africa). However, lack of appropriate resources is a major and common problem in these countries (most of these countries have weak or nonexistent independent DRAs with experienced experts and reviewers). Harmonization of pharmaceutical regulations and cooperation at regional and international levels is an important element of the solution. Sharing resources between countries and use of international expertise (i.e., ICH) can resolve the problem if this cooperation is well structured and if the political support and legal framework are available in addition to funding. Moreover, the harmonization of regional regulation will develop new and bigger pharmaceutical market opportunities that will interest pharmaceutical manufacturers and therefore the development of medicinal products for the entire region. These opportunities would not be developed if each country has different standards and requirements for the registration of such products. Pharmaceutical companies usually focus their efforts first on major developed countries (i.e., the US and EU), as they are the major markets and the regulatory approval processes are more developed and transparent in these countries. They seek first approval for a new product in these regions even if the product is largely needed in developing countries. This practice can delay the entry of a new medicine in a developing country by 10 to 15 years. For example, for rotavirus infection, where the majority of between 352,000 and 592,000 children who die annually of its effects live in the developing world, this could mean thousands of deaths before medication is available [303]. The first rotavirus vaccine was initially available in the US where rotavirus is not as large of a health issue compared to developing countries, where a substantial number of children die due to this virus and where there is less access to medical treatments.
The vast majority of stakeholders support this continued harmonization effort and consider cooperation as the only solution to tackling the new challenges brought by the increased globalization of the pharmaceutical sector. Drug development and manufacture is indeed more and more global, therefore harmonization is more important than ever. In certain instances (e.g., regional integration process), harmonization and cooperation are critical, as divergences in national product standards often act as a barrier to trade and impede the creation of a free trade area or a single market.
The need for further cooperation and harmonization in the pharmaceutical sector (i.e., information exchange, joint assessments, and inspections) to reduce duplication has been clearly highlighted by worldwide regulators during the 14th International Conference of DRAs [304]. Major DRAs have also recognized the need to substantially change their operating models and to increase their global partnerships in order to address the challenges of the future [305]. Industry representatives have also strongly emphasized on numerous occasions that they support these activities and would welcome further worldwide harmonization of requirements.
Some resistance has, however, been raised, and these opponents to harmonization have criticized the significant costs, effort, resources, and time that such initiatives require. A 2005 article [306] even challenged the benefit of such projects. It argued that international harmonization is driven by industry and that there have been examples of less stringent requirements for drug approval at the international level than at the national level. Although additional efforts are necessary, the examples listed in this article are not fully representative of the overall situation. This negative position against harmonization is not shared by the majority of stakeholders, and numerous regulators have strongly repeated their support of international cooperation and harmonization. These criticisms against harmonization and cooperation will certainly fade away if global harmonization is developed in a coordinated and transparent fashion involving all stakeholders. However, it is important to take these concerns into account and continue to make sure that ICH and other international standards are developed based on appropriate and relevant scientific facts and discussions. Each harmonization project needs to be carefully evaluated in order to ensure that new proposed harmonized standards can be implemented and will bring benefit to global public health. The harmonization process demands money and many resources, so it cannot only be an intellectual exercise. The ultimate goal is not the harmonization of the technical requirements and processes themselves, but the improvement of public health and the increased timely access to quality, safe, and effective drugs on a worldwide basis. The harmonization process should clearly maintain or increase current levels of public health protection. In any case, it should not create “lower” standards. Also, harmonization and development of global standards should not imply reduction of medical alternatives. Although not beneficial for the entire population, certain traditional medicines can be valuable for certain subpopulations, regions, or countries. The global system therefore needs to allow for flexibility to develop these medicines and not restrict the choice to one global therapy.
If the harmonization process continues at the same pace as during recent decades, a global system of assessment and control of medicines (at least for specific products such as orphan drugs) will certainly be established as it presents advantages for all stakeholders. The multiplicity of registration procedures, and sometimes standards/requirements, obliged the pharmaceutical companies to set up priorities regarding worldwide submission plans. This can affect the availability of medicines in some small markets or countries. The establishment of a worldwide registration procedure, using international harmonized requirements, would be the only way to make sure that new therapies are available at the same time in both major and small markets. By improving the transparency, predictability, and efficiency of regulatory processes, the global system would also contribute to reducing unnecessary regulatory burden and promoting industry compliance. It would also facilitate the communication and coordination of pharmaceutical activities for emerging diseases and crisis management.
Today, this ultimate step of harmonization and cooperation seems far away and very difficult to implement. However, the European pharmaceutical regulatory system was also considered impossible to implement in 1939, but its success, in a relatively short period of time, demonstrated it was indeed possible! Although the European integration model cannot be applied to establish a global system, it demonstrates that the many challenges and resistance against harmonization/cooperation can be overcome if appropriate decisions are taken. More specifically, the European experience demonstrated that:
-
▸
Cooperation between (and even integration of) countries with different economic and societal/cultural environments and pharmaceutical markets is possible.
-
▸
Legal enforcement of harmonization (i.e., the creation of a mandatory implementation of harmonized topics) facilitates and accelerates harmonization.
-
▸
Political support is essential to achieve major harmonization objectives.
-
▸
Implementation of ICH guidelines in countries with less-developed regulatory systems is possible (e.g., EU enlargement to the Eastern European countries in 2004).
-
▸
Regional cooperation facilitates global harmonization and implementation of ICH guidelines.
-
▸
Partnership between central bodies (i.e., EMA, European Pharmacopoeia [EP], EU control laboratory network) and national DRAs is possible as long as roles and responsibilities are clearly defined.
-
▸
Countries can recognize the assessment performed by one rapporteur or another country (i.e., inspection or assessment of an application) if they agree on the standards used for evaluation.
Of course, implementing a global system may be more difficult than establishing the European system as there is no integration objective and therefore no economic/political pressures to create a single market, as was the case in Europe. However, it is possible if there is a political commitment. Scientists, regulators, and pharmaceutical companies have already shown, via their cooperation through ICH, WHO, and regional and bilateral collaborations, that they are ready for the next phase of harmonization.
Harmonization is a long process and the establishment of a global system will take time. The establishment of this global system can only be foreseen as a long-term stepwise project. It will require a lot of effort and commitment to progressively plan, organize, and implement several measures (at national, regional, and global levels) that will gradually structure international cooperation. Continuous political support will be crucial through the entire process. The politics should help in defining the scope and objectives of the cooperation, but the harmonization process should be left to technical and scientific experts. Without political support and a well-defined, science-driven, stepwise plan focused on public health, the establishment of a global system will remain elusive.
To ensure ultimate success, this harmonization process needs to evolve and take into consideration the economic, political, and other parameters (discussed in Part II-2). Indeed, these measures need to integrate the current political and economic context. Any short-term utopian measure or action that would not fulfill the current political and economic interests (e.g., developing a global supranational system that would give global approvals via a centralized procedure without consulting the national or regional DRAs) would be unrealistic, unreasonable, and therefore not viable. Long-term major changes will only be possible when organization is in place and based on the (geo)political evolution, as was the case in Europe.
Also, to be successful and beneficial for everyone, the new global system needs to be customized and take into account the specific and varying needs of all countries. Countries are indeed very different in terms of medical practice, legal obligations, level of development, etc. This diversity has to be taken into account when developing the global system. Moreover, it is important that this evaluation and the proposed improvements take into account the different regulatory capacity of all countries. Some countries do not have the expertise and resources to ensure that high standards are met. Is it therefore necessary to impose a costly investment to implement all global harmonized guidelines in all countries? The answer to this question is complex. Of course, certain global standards (e.g., GMP or GCP) need to be implemented similarly in all countries for ethical reasons, and also because medicines and data produced in one country will be used on a worldwide basis. However, it is not fair and/or practical to impose all new “high-level requirements” on developing countries. The cost of implementation can be too high for some requirements that may not be critical in a developing country as these requirements may require state-of-the-art science, equipment, expertise, and resources not available in these developing countries. A balance should therefore be found between the necessary implementation of high standards that not only support public health, but also the realities of developing countries. Increasing the requirements without benefit to public health could have major negative impact on the economy and development of these countries. Introducing high standards for all medicines without differentiation could lead to the failure of providing treatments for neglected diseases in developing countries [307]. The development and commercialization of these treatments are only interesting for small companies in developing countries (not multinational companies). Because they are relatively unprofitable for major pharmaceutical companies, R&D efforts aimed at new treatments for certain tropical diseases, as well as the availability of existing products, have decreased. If small pharmaceutical companies interested in developing and commercializing these medicines are limited by the need to comply to general high global standards (developed by ICH for all new medicines without distinction), such treatments will no longer be available. Therefore, careful evaluation of the benefit and potential negative impact of any new harmonization initiatives (especially global initiatives) needs to be conducted and mechanisms need to be in place to allow appropriate flexibility in the treatment of certain diseases.
In summary, the thorough evaluation of the current system outlined in previous sections makes it evident that harmonization of pharmaceutical regulations offers many direct benefits to both DRAs and the pharmaceutical industry, with beneficial impact for the protection of public health. Key benefits include better monitoring of medicines; preventing duplication of clinical trials in humans and minimizing the use of animal testing without compromising safety and effectiveness; streamlining the regulatory assessment process for new drug applications; allowing more informed decisions; and reducing development times and resources for drug development. However, although previous and ongoing harmonization and cooperation initiatives have already been beneficial, major improvements are still possible and would clearly provide many advantages to all stakeholders. It would help developing countries by providing expertise, experience, and support, but it would also help developed countries by ensuring better control of the medicines used in their countries that were developed and manufactured outside their own borders. Today, a population’s health suffers or benefits not just from its own domestic regulatory environment, but also from decisions made outside the country. All the positive initiatives and dedication seen in recent decades at the national, regional, and global levels need to be leveraged so that this collective knowledge and these resources continue to improve global public health.
The establishment of a coordinated global pharmaceutical system would have a significant added value on the promotion and protection of global public health. The recommendations presented in Part III have been developed with the following questions in mind: What is the best structure and process for this global pharmaceutical system, and how can this framework/network be realistically implemented?
II-2). Critical Parameters and Influencing Factors for Cooperation, Convergence, and Harmonization
Following the review of the status of all ongoing initiatives (Part I), we concluded that further harmonization would be beneficial for all stakeholders (Part II-1.4). However, to effectively discuss how to organize these next steps of harmonization, it is critical to first analyze the lessons of the past and the ongoing harmonization initiatives to understand what the critical parameters of harmonization and its influencing factors are. This analysis is essential to ensure success of the next necessary steps.
II-2.1). Critical Parameters for Cooperation, Convergence, and Harmonization
II-2.1.1). Regulatory Capacity (i.e., Resources, Expertise, Infrastructure)
A pharmaceutical regulatory system, supported by relevant legislation, is an essential component of a functioning healthcare scheme. This regulatory system includes, at the very least, the necessary legislation and regulations, and an authority that controls all pharmaceutical products through pre-marketing evaluation, marketing authorization, and post-marketing surveillance. It should also include an inspectorate, access to a medicine quality control laboratory, enforcement mechanisms, and safety monitoring [308].
Implementation of medicine policies has improved over the years [309], but there are still important problems. The level of effectiveness of national regulations varies widely across countries, as regulatory capacity is very different from one country to another. Some have mature, well-developed, and well-resourced systems, while others have weak or no regulatory system at all. For various reasons, many DRAs do not have the full capacity to perform all regulatory functions. In some countries, the inclusion of the regulatory functions as a department of the Ministry of Health also creates some challenges [310,311][310][311].
As presented in Figure 4 , there are important differences in regulatory capacity among all the WHO Member States [312].
FIGURE 4.
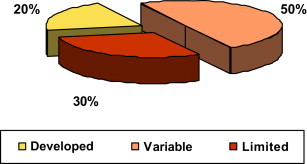
Differences in Regulatory Capacity Globally.
Source: Overview on medicines regulation: regulatory cooperation and harmonization in the focus, Presentation from Dr. Samvel Azatyan (WHO) at the WHO/UNICEF Technical Briefing Seminar on Essential Medicines Policies, 31 October – 4 November 2011, WHO Headquarters, Geneva, Switzerland.
Even in a given region, regulatory capacity can vary. For example, Figure 5 shows that in Africa, among 46 WHO Member States, there are major differences.
FIGURE 5.
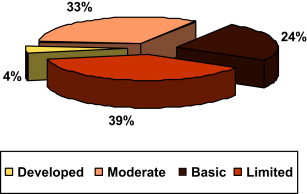
Differences in Regulatory Capacity in the African Region.
Source: Overview on medicines regulation: regulatory cooperation and harmonization in the focus, Presentation from Dr. Samvel Azatyan (WHO) at the WHO/UNICEF Technical Briefing Seminar on Essential Medicines Policies, 31 October – 4 November 2011, WHO Headquarters, Geneva, Switzerland.
Also, personnel resources of DRAs vary from one or two people in small countries [313] to thousands of people in developed countries. For example, the US FDA employs approximately 15,000 people, with approximately 4,500 dedicated to the assessment and control of biologics and drugs [314,315][314][315].
This worldwide variation needs to be taken into consideration when discussing the organization of international harmonization and cooperation because harmonization projects require time, money, resources, and expertise. Without regulatory capacity, it is indeed difficult for a country to be involved in harmonization initiatives. Moreover, due to these resource constraints, the requirements developed and successfully implemented in one country may not be equally successful in another.
Many governments do not seem to recognize the potential benefits of a strong medicine regulatory system, and do not make the necessary political and financial commitments to secure one [316]. However, in many instances, the issue is not a lack of political support, but a lack of appropriate mechanisms capable of translating the high degree of political commitment into concrete programs of community building and integration. In other words, the politicians are willing to initiate the activities, but they do not have the resources and infrastructure to support this ambition. Indeed, a lot of countries do not have the resources, capacity, and expertise to implement such a functioning regulatory system [317]. Their main challenges are the following:
-
▸
Costs associated with the development of a regulatory system, participation in harmonization, and implementation of agreed-upon common standards
-
▸
Limited human and financial resources and institutional capacity
-
▸
Lack of effective legislation
-
▸
Lack of technical expertise and a trained staff
-
▸
Lack of supportive environment (i.e., no policy framework and no legislation) and quality management systems
For example, according to Dr. José Luis Di Fabio (of the Area of Technology, Health Care and Research, Pan American Health Organization [PAHO]/WHO), 65% of the countries in the Americas have a regulatory authority, but most do not have the capacity to evaluate and regulate products [318]. The challenge in these countries is to develop the expertise and systems in order to raise the level of regulation and confidence and therefore allow recognition of this specific authority and system. Mutual recognition is indeed based on the confidence in all countries’ DRAs. This is why PAHO/WHO developed the “qualification procedure” for authorities.
Also, an assessment of medicines regulatory systems in 26 Sub-Saharan African countries over a period of eight years showed that, although structures for medicine regulation existed in all countries assessed (and the main regulatory functions were addressed), in practice, the measures were often inadequate and did not form a coherent regulatory system. Common weaknesses included a fragmented legal basis in need of consolidation, weak management structure and processes, and a severe lack of staff and resources. On the whole, countries did not have the capacity to control the quality, safety, and efficacy of the medicines circulating in their markets or passing through their territories [319].
This capacity issue is amplified by the fact that the sparse healthcare resources in these developing countries need to also manage other major public health issues. Most of the countries recognize the value of harmonization and cooperation (especially for developing countries), but the shortage of resources requires prioritization. Presently, harmonization is one of the priorities for developed countries (e.g., the EU or US are both involved in many global, regional, and bilateral initiatives), but it is not the first priority for less-developed countries. Their focus regarding the public health sector is, rightly, the prevention and control of certain emerging/resurging infectious and communicable diseases such as dengue, cholera, tuberculosis, Severe Acute Respiratory Syndrome (SARS), avian influenza, and typhoid fever, along with the prevention and control of HIV/AIDS. They also need to ensure access to quality essential medicines and low-priced generic products; in addition, the fight against substandard and counterfeit medicines is a growing issue.
In conclusion, it is evident that building regulatory capacity in developing countries is critical to allowing them to benefit from global harmonization, which in turn would benefit global public health. This support, with appropriate funding and resources, needs to be customized to their realities and needs. Complex systems and regulations from developed countries are not an appropriate response. The first focus should be to establish national and/or regional DRAs with appropriate powers in order to control the pharmaceutical market. Even with limited resources, this national or regional contact is necessary to liaise with other countries and develop a strategy and system based on a country’s individual needs. Many resources are available (e.g., via the WHO programs) to establish a pharmaceutical system and regulations appropriate to the level of development of the country [320]. To address the current issues in developing countries, it is also evident that national capacity building needs to be supported by regional and global cooperation and coordination.
II-2.1.2). Communication
Effective communication enhances cooperation, which in turn facilitates harmonization, especially for some cultures.
One of the initial steps of any harmonization initiative is to identify the main contacts (with the appropriate decision-making powers) from each party and build a strong communication channel between the identified parties. Understanding each other’s needs and challenges facilitates the building of trust and confidence in each other. This establishment of relationships between parties is key to a successful collaboration, as it will define the communication style throughout the entire process.
Good communication is key during all steps of the harmonization process, from initiation to implementation of the harmonized rules. But building strong communication based on mutual understanding and trust takes time, and many parameters can influence it (i.e., culture, distance between partners, fluency in English, etc.). Development and training of regulators on cultural differences and foreign languages (especially English) is obviously an important prerequisite to collaboration. For example, one of the first measures that the PMDA put in place to increase its international activities was to strengthen its foreign language training and daily educational activities in order to help relevant staff members improve their foreign language skills [321]. In addition, PMDA has also made efforts to improve and expand the English version of its website to provide the latest information in English [322].
Communication also involves the “human” factor. This human interaction should not be underestimated because good relationships and understanding of each other’s differences can facilitate the discussions/exchange and vice versa [323]. Indeed, harmonization does not only require infrastructure and resources, it requires willingness from the different players to communicate and exchange successfully. For example, during my research, regulators reported that some regulators from the US FDA or Health Canada have sometimes had more information and updates on activities happening in Europe than some small national European DRAs due to their close and regular communication with the EMA or certain national DRAs. Of course, as already mentioned in the previous section, Europe is a great example of harmonization. But even in this region that is mostly harmonized and integrated in terms of pharmaceutical regulation, there is some miscommunication occurring.
Changes in international trade and data protection [324,325][324][325], brought on by increased globalization, must also be considered. More specifically, the protection of commercially confidential informationa is an important issue because it impacts the communication and exchange of information between countries. It is obvious that it is important that DRAs exchange the maximum amount of information to achieve effective communication. Today, however, pharmaceutical companies and manufacturers provide different levels of information and data to different countries due to the risk associated with the dissemination of trade secrets. There is a concern that if major DRAs share information with “developing” DRAs, a leak could occur and proprietary/confidential information could become publicly available (e.g., proprietary information on a brand could be available to generic companies in developing countries). Consequently, most of the agreements between DRAs exclude the exchange of trade secret information and proprietary information and data (i.e., under these agreements the DRAs cannot exchange proprietary information/data on specific products). Regulators believe that these limitations have an important impact on cooperation as they limit communication and the exchange of information with other DRAs. This limitation would become even more problematic if a global framework for the evaluation of medicines (i.e., a EU Mutual Recognition Procedure [MRP]-type procedure) was established.
Pharmaceutical companies are already evaluating the pros and cons to registering a product in certain countries, especially where there is no (or limited) data exclusivity legislation in place, even if there are patent laws, because such laws are not always respected [326]. In this case, the harmonization of regulation between countries will obviously not be beneficial because medicines will not be registered and, therefore, available globally. To respond to this situation, several countries (including China and India) have started to issue “compulsory licenses” for local generic makers to produce drugs that are still within the patent life [327]. This amendment of legislation will certainly increase the concerns of pharmaceutical companies regarding trade secret information. In the case of China, the legislation amendment permits domestic firms that receive the licenses to be able to apply for permission to export their versions of the patented drugs for “reasons of public health,” which can cover a broad range of situations.
This problem of communication of trade secrets and confidential information therefore needs to be taken into consideration when developing global harmonization, worldwide cooperation mechanisms, and communication channels. Appropriate measures should be in place to balance the needs of developing countries to have access to essential medicines, but at the same time to avoid dissemination of confidential data that could impact innovation.
Finally, good communication between parties requires appropriate mechanisms and tools:
-
▸
Firstly, it is important before discussing harmonization to have a glossary to ensure that all parties use the same “language” because the different regulations impose conflicting definitions/terminology (e.g., definition of a medicinal, biotechnological, or biological product). The development of internationally recognized nomenclatures and classifications have been an important part of ICH and WHO work, and many tools have been developed (CTD, MedDRA, INN, ICD, ICF, ICHI, ATC, DDD, etc.). Moreover, the product classifications and/or nomenclatures need to be harmonized (especially in the case of an integration model) to facilitate communication and exchange of information between different national DRAs. Different brand/proprietary names of the same product in different countries can be confusing. Different nonproprietary names are a bigger problem. This issue has been partly resolved with the creation of the INN by the WHO. Indeed, before INN, the differences in nonproprietary names around the world were an issue (e.g., paracetamol vs. acetaminophen). These different names were given by different national bodies using different naming conventions. Today, INN is the worldwide standard for names. Unfortunately, in the US a USAN name is still a requirement (most other countries accept an INN-approved name). Moreover, USAN still has a different naming convention than the INN. Even if most of the new products have the same USAN and INN name, these different naming conventions can create some issues when a company wants to register a new name globally, creating conflicting feedback and positions that can take years to resolve.
-
▸
Secondly, harmonized and common training is very important between reviewers from different countries. This part of harmonization was recognized in Europe very early on in the process [328]. It facilitates the implementation of harmonized, high-quality performance standards, increases consistency in review/inspection, and decreases the discrepancies in the implementation of these standards.
-
▸
Thirdly, telecommunication and informatics infrastructures are important to ensure efficient communication. Appropriate communication tools supporting rapid and easy communication between parties (especially in the case of safety issues) need to be in place. In some countries, this communication (and exchange of information) is sometimes difficult due to the lack of infrastructure (e.g., computers). This lack of infrastructure has some impact on communication (i.e., connection to a safety/PhV database or clinical trial application). Basic communication tools such as communicating via e-mails or access to the Internet can still cause serious problems.
II-2.1.3). Definition of Clear Goals and Appropriate Planning
Harmonization is a long and stepwise process that requires huge effort and focus!
All successful harmonization arrangements to date (i.e., ICH, EU, etc.) have taken a significant amount of time to develop. They required numerous meetings, technical discussions, and complex negotiations to resolve scientific differences and/or legal issues. If the process had moved too fast, people may have been concerned about certain issues (e.g., losing their independence), and the success of the project may have been impacted. Trust and acceptance needs time, as evidenced by the establishment of relationships and cooperation between the ICH and non-ICH regions. The GCG was created in 1999, and it took almost 10 years for the GCG/ICH to create real partnerships with the Regional Harmonization Initiatives (RHIs). This time was necessary to get to know one another and to understand each other’s needs and challenges in order to establish the high level of trust, understanding, and respect necessary before setting up practical actions for the implementation of ICH guidelines in non-ICH regions (via training, shared meetings, etc.).
The most prevalent reasons for failure of alliance and cooperation projects are related to the lack of upfront clear specific goals and appropriate planning and structure (i.e., underestimation of time and the skills required) [329]. To be efficient and avoid any confusion, it is therefore important to clearly determine up front the scope and objective of the initiative. This alleviates future discussion and allows the focus to be on actions. It is also critical to establish a plan, with intermediate goals, in order to remain focused on the ultimate objectives. For example, ICH has worked since its creation towards harmonization of technical topics, while the EU developed a structure and system for harmonizing the laws and regulation of its member countries to promote both public health and the free circulation of pharmaceuticals within the European trade areas. Though these two organizations both work towards regulatory harmonization, they are very different as they have very different objectives.
True harmonization is much more than common documentation and standards. The objective is to have similar or collaborative approaches to medicine registration that ultimately allows for mutual recognition and/or centralized registration (if desired) in the long term. But this is a long process and it requires effective communication and collaboration (e.g., information sharing and working jointly) to understand similarities and differences and to build trust. The reality of resources and efforts needs to be taken into account when establishing this plan. Therefore, project planning and management is key.
Major steps can be distinguished as follows:
-
▸
A general exchange aimed at enhancing collaboration and mutual understanding.
-
▸
The formulation of a framework agreement as the formal basis to start the harmonization process.
-
▸
The development of harmonized nomenclatures and procedures.
-
▸
The adoption of harmonized product and qualification standards.
-
▸
The mutual recognition of the harmonized standards.
-
▸
Compliance to apply consistent practices in selected areas.
-
▸
Maintenance and update of a harmonized topic: Harmonization of the regulation is not a one-time activity. Science is not static, so harmonization of the technical requirements should not be static. Processes need to be put in place to ensure that when the fundamentals, assumptions, and science evolve, the agreement will be reassessed (and changed if necessary) to avoid countries evolving on their own.
Although the above sequence seems simple, in practice the tasks are tedious, and often initiate challenges that require systematic and thorough resolutions.
The African Medicines Registration Harmonization (AMRH) Initiative determined typical key milestones and timeframes for its regional medicines registration harmonization projects. This model, presented below in Figure 6 , can be generalized to any other worldwide harmonization project (i.e., international, regional, or subregional). Although each harmonization initiative can differ in its specific content and duration (due to the baseline level of harmonization and other legal, cultural, and political factors), the concept and process is always the same. This process takes time and the coordination of communication and the predetermination of a structured plan and process is critical.
FIGURE 6.
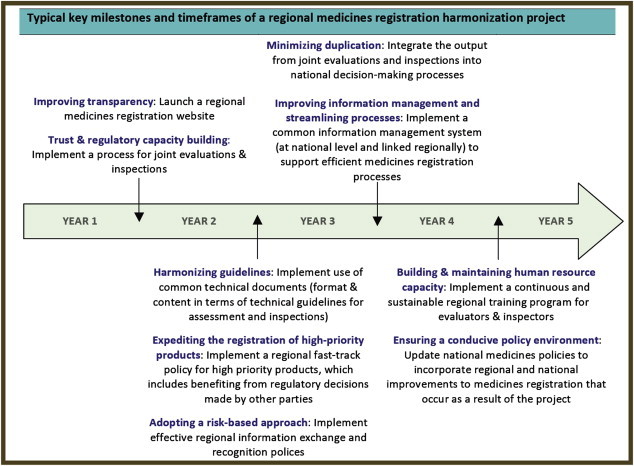
Model of Typical Key Milestones and Timeframes for Harmonization Projects.
Source: “African Medicines Registration Harmonization (AMRH) Initiative: Summary, Status and Future Plans,” NEPAD and WHO, November 2009.
II-2.1.4). Organization and Structure
The review of ongoing harmonization initiatives in Part I showed that there are different models of cooperation based on the scope and objectives of the project. They can range from a simple technical collaboration to full harmonization and integration of systems and regulations. Depending on the model, different options can be selected to enhance regulatory cooperation, each of these options requiring a different degree of collaboration and time to impact [329].
All successful organizations have an appropriate structure to coordinate efforts (ICH, WHO, and Europe are good examples). Harmonization is an intense process that requires dedicated resources and infrastructure to be successful. Responsibilities, duties, and functions need to be defined and distributed appropriately between the bodies.
At the very least, a harmonization initiative needs to be supported by:
-
▸
An oversight committee (called a “Steering Committee” or “Governance Committee”): The role of this committee is to set up priorities, coordinate actions and projects, and monitor those activities to ensure appropriate development towards predefined goals.
-
▸
A secretariat: The role of the secretariat is to support the administrative work and to provide project management. Transparency of the process requires publication of an agenda and meeting minutes, and dissemination of harmonized standards and decisions, etc.
Communication is also critical between the national institutions and regional or international initiatives. A designated contact needs to be established between national activities and regional/international discussions to avoid duplication of effort or even conflicting decisions. This contact is essential to (1) feed the regional and international discussion with specificities/challenges/etc. from the country; and (2) ensure good implementation of agreed-upon harmonized standards.
Finally, the structure and organization of harmonization initiatives also needs to evolve as the program evolves. A specific structure may be ideal for certain phases of harmonization/cooperation but not relevant for others. An early initiative needs a limited relationship at a high level, and the involvement of a finite number of people. However, the integration phase requires more stable and defined institutions. For example, SADC’s structure evolved over time. It started as a “Coordinating Conference” (with limited institutions and focused on coordination of national actions) before becoming a “Development Community” (with defined institutions and a focus on integration). To support this evolution, SADC’s structure changed over time, and is regularly evaluated to ensure that it adequately supports the initial objectives.
II-2.1.5). Implementation and Monitoring
After reaching agreement, the harmonized rules and standards need to be implemented, a key phase of any harmonization initiative. However, this ultimate step is not simple, and disharmony could still exist if the agreement is not adopted consistently in all regions.
Implementation of harmonized standards/requirements by each country can be more difficult than creating them. The results of such efforts depend on the willingness and commitment of all stakeholders. Many factors can affect the actual implementation of a standard, and therefore the overall harmonization process:
-
▸
The lack of resources, expertise, systems, or the organization necessary for the implementation.
-
▸
Inaccurate translation of a rule or technical recommendation.
-
▸
A different understanding of the rules due to cultural differences.
-
▸
The need for major modification of legislation not supported by the political system or legislators.
-
▸
A lack of pragmatism/practicality (i.e., if the rule is too vague, unclear, too theoretical, or leaves too much flexibility to the parties). For example, the texts relative to the arbitration process in Europe allow a country to refer a matter to the EMA/CHMP in case of disagreement during an MRP/Decentralized Procedure only if there is a potential risk to public health. However, this notion of public health risk is vague and has allowed countries to abuse this clause to trigger this procedure for any disagreement with the Reference Member State (RMS) evaluation.
All these factors can indeed impede the implementation phase or create diverse interpretations of recommendations/guidelines by the different parties (which is not the objective of a harmonization process). Diverse interpretation of a rule can lead to worsening of the situation and require additional efforts from industry to comply with a new standard.
There are many examples of harmonization initiatives that did not deliver the expected value due to misinterpretation of the rules/standards. For example, ICH has developed Guideline E5 to standardize the evaluation of ethnic factors and therefore avoid duplication of clinical studies. While industry and regulatory authorities have agreed that this guideline represents the best available model for developing clinical evidence of the safety and efficacy of new therapies across different ethnicities, its implementation has been problematic. Within the ICH countries, the challenges tend to be caused by ambiguities and lack of direction as to the sufficiency of data that would allow for extrapolation of findings to the new region. In emerging markets (i.e., non-ICH countries), the adoption, adaptation, and implementation of the principles outlined in ICH E5 are more problematic and tend to be much more systemic. For example, many of the emerging markets simply require that a bridging study always be performed [330].
To avoid these implementation problems, the strategy of implementation needs to be planned early. Goals and plans need to be agreed upon up front (before the harmonization process starts), and this ultimate goal needs to be kept in mind throughout the harmonization process. The plan needs to evaluate the benefit of the project (e.g., with regards to public health), but also assess its potential impact (on legislation and current rules) to anticipate potential limitations in its implementation. A great common standard will never be used if its implementation requires an expertise, a legal framework, an appropriate system, and/or resources not available in the countries/regions involved. In the case of harmonization of technical standards (i.e., ICH), the preparation of a successful implementation begins with the selection of the topics for harmonization. Technical guidelines must be value-added and able to be implemented (it should not remain a theoretical exercise). To facilitate implementation, it is also critical to build a scientific consensus among all stakeholders (consultation needs to happen early).
Several actions can facilitate the implementation phase:
-
▸
Adequate budget and time needs to be allocated to this last step of harmonization. For example, the Korean Ministry of Health invested in training, facilities, operation systems, etc., between $0.5 to $1 million/center/year for five years in nine clinical centers to support the implementation of GCP [331].
-
▸
Appropriate coordination and structure need to be established to support proper implementation. The success of ICH has been possible through effective and consistent guideline implementation (e.g., with the creation of IWG). Also, the SADC has implemented “National Committees” that are present in each Member State. This model is very interesting and may help other worldwide initiatives to implement the agreed-upon common rules. It creates a contact point at a more local level and therefore improves understanding, monitoring, and support for the implementation.
-
▸
Direct adoption of standards without regional/local “adaptation” needs to be favored. Sometimes a rule cannot be implemented in a country without modifications (e.g., due to a lack of resources/expertise). Some non-ICH regions/countries have been adopting and implementing the exact ICH guidelines. However, others have been adapting some ICH guidelines. These adaptations can clearly create some harmonization complications. It is therefore crucial to involve all regions early in the discussion to evaluate difficulties of future implementation. During the harmonization process, the working group needs to evaluate if the guidelines will not be applicable to certain regions/countries (or will require a certain level of adaptation).
-
▸
Legislative texts or agreements need to be quickly followed by detailed guidelines, standard operating procedures (SOPs), manuals, and/or questions and answers (Q&As), etc. These documents, clarifying the rules, reduce the risk of misinterpretation and/or conflicting interpretations of rules and agreements.
-
▸Transparency and appropriate communication needs to occur throughout the process involving all stakeholders early on (i.e., regulators, but also those affected by regulation such as industry, the public, etc.) to avoid major surprises at the time of implementation. The process should ensure the issuance of notice of a proposed regulation with a sufficient consultation period to [332]:
-
•Allow all stakeholders to have access to the draft proposals and to submit comments.
-
•Adequately consider and analyze those comments.
-
•Respond to significant points and explain the rationale for revisions when adopting the final regulation.
-
•
Openness and transparency in the preparation and application of regulations are also fundamental to ensure public confidence. Allowing people to review/comment and participate in the preparation of the harmonized rules and standards will clearly reduce surprises and help with implementation. In addition to promoting transparency, relevant consultation with all parties ensures that all perspectives on the issue have been considered, highlights alternative approaches and enhances awareness (which facilitates implementation and encourages compliance).
-
▸
Training needs to be organized because implementation is indeed much more important than publishing guidelines. The promotion of common understanding and training are key. All stakeholders need to become familiar with the new rules. Regulators and industry need to be trained, but also any other party affected by the rules (i.e., investigators, ethics committees, etc., in the case of GCP, for example)
-
▸
Implementation of new requirements needs to be monitored and corrective measures need to be taken if necessary. It is indeed crucial to ensure that everyone implement the rules and standards correctly, but also that all stakeholders continue to have a good understanding of the rules over time, especially when there are new versions and modifications of the rules.
PhRMA and the US FDA developed a simple methodology to assess the implementation, application, and utilization of ICH guidelines. This methodology, presented in Figure 7 , defines six steps (Topic Selection, Dissemination, Publication, Training, Implementation, and Management) and can be applied to any harmonization of technical standards. For each step, relevant processes and actions are defined.
FIGURE 7.

Implementation Process Flow.
Source: Presentation on Strategy of Implementation by CAPT Justina A. Molzon at the IV Pan American Conference on Drug Regulatory Harmonization, Dominican Republic, March 2005.
This methodology is an important tool to provide adequate assessment of implementation and resolution of potential problems that may occur during this implementation. The analysis also demonstrates that processes and systems need to be in place and resources allocated to ensure adequate implementation of agreed-upon standards.
II-2.1.6). Involvement of All Stakeholders
There are many individuals or organizations that are dependent on or are affected by the final product or output of a harmonization or cooperation project (i.e., national DRAs, regional and global bodies, industry, academia, patients, etc.). The early identification and involvement of all these stakeholders in the process increases the chance of success. Of course, involvement of stakeholders is tailored to meet the circumstances. Some stakeholders may be more crucial (i.e., DRAs) and therefore more involved (or involved at an earlier stage) than others, but this has to be clearly defined to avoid confusion. Roles and responsibilities of each stakeholderb need to be determined early and decision makers identified. Insufficient involvement and ineffective communication with stakeholders can lead to project failure. Involvement of the inappropriate stakeholders in a project may also reduce the value of the harmonization and may lead to criticism in the end.
Keeping all stakeholders informed and/or involved throughout the process has several practical advantages. First, it is important to take into consideration the different needs and challenges of all stakeholders when developing the objectives and scope of a harmonization project. This helps define practical and realistic goals. Second, keeping all stakeholders informed throughout the project builds trust and support in the process and decreases the risk of surprises at a late stage. Third, engaging stakeholders in the process through good management of their expectations develops their active interest and commitment to the project. This ensures that all parties will be actively engaged and ensures smooth implementation of harmonized standards.
It is also obvious that the degree of involvement and interaction between different stakeholders with varied backgrounds, experiences, and expertise leads to better standards that are more likely to be adopted and implemented. Also, in some countries, there is a lack of centralization and coordination of activities. In these countries, local authorities are not always closely collaborating with federal bodies, especially in the case of inspections [333]. It is therefore important to make sure that all the players in a system are involved and aware (not only the central DRAs) to ensure good implementation of common standards.
Today, there are cooperation and harmonization initiatives that involve only regulators (e.g., EMA, WHO–ICDRA) versus other initiatives that involve many players such as academic representatives and industry (e.g., ICH, APEC). Involvement of public interest groups (i.e., patient associations) is also very important to increase transparency and combat corruption in certain countries.
The ICH process is a good example of a harmonization initiative that understood early on that its activities necessitated the involvement of different stakeholders. The success of this unique initiative that includes both regulators and industry representatives has shown that the implementation of agreed-upon standards is indeed facilitated if both industry and DRAs share responsibility for decisions or actions. Moreover, the inclusion of observers from non-ICH regions and then RHIs and some individual DRAs (of countries which are a source of Active Pharmaceutical Ingredients [APIs], medicinal products, or clinical data for the ICH regions) also promote and facilitate the implementation of ICH standards. The same goals (developing and implementing harmonized standards and guidelines) would certainly be much more difficult to meet if this process had been managed by the DRAs of the three ICH regions only.
Finally, it is interesting to note that in the past decade many foundations and philanthropic organizations (i.e., the Bill and Melinda Gates Foundation, the Clinton Foundation, etc.) have been associated with harmonization work, especially in Africa. These new participants should continue to be further involved in collaboration initiatives as they bring additional resources and financial support. However, this growing complexity in the institutional landscape for global health, characterized by more partnerships, foundations, financial instruments, and bilateral and multilateral agencies that influence global health policy making, need to be coordinated. The challenge is to manage complexity and seek creative solutions that promote convergence around common goals. There are also opportunities to improve collaboration and use innovation at the national and international levels to fight inequities and to continue progress for better health [334].
II-2.1.7). Voluntary Cooperation versus Legal Commitment
Enforcement of harmonized standards and compliance to these standards are an important part of a harmonization initiative.
Today, most of the current harmonization processes are done on a voluntary basis (ICH, PANDRH, APEC, etc.). However, legal enforcement of harmonization (i.e., creating a mandatory implementation of a harmonized topic) facilitates and accelerates harmonization. The European experience has shown that having a legal basis for a cooperation initiative allows deeper harmonization and quicker implementation of common rules.
Of course it is more difficult to establish legal obligations in a nonintegration initiative. However, even in this situation, legal enforcement is possible if appropriate agreements are signed. MRAs can indeed be signed by one or more parties to mutually recognize or accept some or all aspects of each other’s requirements. Several examples demonstrated that the signature of such legal texts (especially when these agreements are comprehensive, technical, and easily implementable) promote further harmonization or cooperation. For instance, the signature of the Framework for Advancing Transatlantic Economic Integration between the European Union and the United States of America in 2007 by EC President José Manuel Barroso, German Chancellor Angela Merkel, and US President George W. Bush accelerated regulatory collaboration between the EU and US. Also, the implementation of the SADC pharmaceutical cooperation program was facilitated by the signature of the health protocol because this supportive legal text (defined in the SADC Treaty) was binding for the SADC Member States. Its Article 29 requires the Member States to cooperate in the harmonization of procedures of pharmaceuticals, quality assurance, and registration, and also in the production, procurement, and distribution of affordable essential drugs.
II-2.2). Factors Influencing Cooperation, Convergence, and Harmonization in the Pharmaceutical Domain
Intrinsic parameters reviewed in Part II-2.1 are very important to ensure success of a cooperation initiative. However, we will see in this section that other factors can also drastically influence the harmonization projects (including scientific debate on technical requirements). It is therefore important to take them into account when establishing cooperation and harmonization projects.
II-2.2.1). Globalization
Globalization is the primary factor that has accelerated and influenced convergence and harmonization of pharmaceutical regulations in past years.
Globalization is a fact of the 21st century. It brings many opportunities (e.g., access to new culture more easily) but also many challenges. The emergence of worldwide health threats such as the increasing number of counterfeit medicines or pandemic influenza (that can spread globally in a few days [335]) cannot be controlled at the local level. Also, the impact of the Internet on global access to information and markets has introduced significant new challenges and questions on the relevance of current regulatory systems. Borders count for very little in the light of these challenges.
Moreover, the globalization of the general economy (with increased travel of people and exchange of goods, finance, and information around the globe) and the modification of economic powers forced pharmaceutical companies to revise their strategy, processes, and management approach [336], which in turn triggered the globalization of the pharmaceutical market (i.e., development, manufacture, and distribution activities). This fundamental change in the global economic landscape requires countries to change their regulatory approaches to promote and protect the health of their population. For example, 24 million shipments of US FDA-regulated products were imported into the US in 2011 from 228 foreign jurisdictions. This represents a four-fold increase over past decade [337]. This trade activity should continue to increase as the most important consequences of the pressure to reduce costs and increase productivity will continue to move manufacturing activities to new locations, looking to global supply chains to reduce costs.c Just as cost pressures will only become more severe over time, companies will indeed continue the movement of manufacturing and production activities to lower-cost countries for the foreseeable future [338]. It is expected that eventually the growth in imported US FDA-regulated products will outpace the growth of US domestic production. According to some estimates, imports of US FDA-regulated products may triple by 2015 compared to 2007, corresponding to a 15% annual growth rate. Companies will not only be producing more products abroad, but these products will follow complex paths through multistep supply chains [339]. Today, it is not uncommon that raw materials, intermediates, excipients, and finished dosage forms are all manufactured in different countries.
Similarly, clinical studies are increasingly being conducted on a multiregional, if not global, basis [340]. About one quarter of all clinical trials in the EU are also performed in the third world [341], and approximately 60% of patients enrolled in pivotal trials included in European Marketing Authorization Applications (MAAs) come from outside Europe [342]. Also, the number of US Investigational New Drugs (INDs) and applications for marketing approval supported by foreign clinical trials has increased in recent years [343]. Even if North America and Europe have been the leading destinations in terms of trial numbers, a continuing shift towards other regions is expected [344,345][344][345]. Developing countries are increasingly more involved in these global studies [346]. Of the 149,672 studies listed on the US registry of clinical trials, a significant number have sites outside the US. Plate 7 shows the locations of these studies in 185 different countries (from all regions of the world).
In addition to the financial incentives, the developing countries offer a large pool of patients and the possibility of finding untreated or “naive” patients more easily.d Finally, many countries have been developing or enhancing their regulations (a majority of the time based on ICH recommendations) to attract clinical research and improve credibility of data and reported results [347,348][347][348]. For example, in Korea, the Korean Good Clinical Practice (KGCP) was established in December 1987, but has only been enforced since October 1995. In 2000, the KGCP was revised to harmonize with the ICH E6 guidance. Moreover, many other improvements have been implemented since 2000. All these improvements have translated to a significant increase of clinical trials (domestic and multinational) approved and conducted in Korea (from 31domestic studies in 1999 to 229 domestic plus 210 multinational studies in 2010 [349]).
Both India and China are the current leading destinations for pharmaceutical companies as they offer a number of advantages, most notably the lower cost of skilled labor. India in particular trains six times the number of chemists annually compared to the US, and companies can access this talent for 10% of the cost for the same in America. In total, estimates indicate that bulk manufacturing in India can reduce costs for US and EU companies by 30% to 40% [350]. Thanks to these major advantages, the Indian pharmaceutical industry has already established itself as a key player in the manufacturing arena, with almost half of the active pharmaceutical ingredients worldwide being produced there. Finished products, especially generics, are also very likely to come from countries with rapidly expanding pharmaceutical manufacturing, such as India [351]. Finally, the service industry from drug discovery to clinical development, data management, and statistical analysis, has also seen exponential growth with investment from practically all major global pharmaceutical companies [352]. India’s Phase 3 trials are growing seven times faster than the global average [353], supported by the attractiveness of its over 1 billion people, 15,000 hospitals, 900,000 hospital beds, and 700,000 physicians [354].
However, India’s attractiveness is today not only related to the fact that it is a location for cost-effective pharmaceutical product manufacturing and development. It has become a key market for finished products because the Indian middle class (estimated to be about 30% of the total population, making it more than 300 million, equivalent to the total US population) is now experiencing increased incidences of indications traditionally considered diseases of the West (e.g., hypertension, cardiovascular diseases, allergies, central nervous system diseases, etc.) and has the economic means to afford expensive Western therapies. Its growing economy, combined with the important regulatory changes in India over the past several years [355] and its desire to harmonize its regulation with ICH majors and other worldwide authorities, will certainly continue to position India as one of the new key countries for pharmaceutical companies [356]. Finally, it is also important to note that the presence of Indian generic firms in developed markets have quickly increased over the past several years [357].
The pharmaceutical market in China has already expanded over the past several decades with important investments from international companies [358,359][358][359]. From its 2003 ranking of ninth place, it represented the fifth largest pharmaceutical market in the world in 2008, and it is expected to continue to increase. It was also listed as the 13th country involved in clinical trials in 2009 (which represents an important increase compared to 2005 when it was listed as 29th) [360]. Also, Chinese pharmaceutical invention patent applications went from approximately 1,000 in 1999 to more than 9,000 in 2006 [361]. More cooperation is expected between the Chinese State Food & Drug Administration (SFDA) and foreign regulators with the international background and experience of the newly nominated SFDA Chief [362]. This will certainly continue to increase the influence of China on the global pharmaceutical market.
In addition to the impact of India and China on healthcare of developed countries, the low price of the medicines manufactured in these countries also impacts the public health of developing countries. The small developing countries are overly dependent on these low-price imported drugs. For example, about 85% of the generic antiretroviral products used in the SADC region are imported from India (and 15% are manufactured within the SADC region) [363]. With the global financial crisis affecting health budgets, it is expected that other countries will also increase their relationship with these emerging countries to reduce the prices of their imported medicines currently sourced from developed countries [364].
Finally, the increase of exchange between population/countries has created new needs and therefore new risks. For example, the growing demand for foreign traditional medicines in the Western environment is associated with new specific risks [365].
To summarize, globalization of the pharmaceutical market brings many opportunities to enhance global public health by forcing worldwide authorities to work together, but it also raises many specific concerns. The movement of clinical research to developing countries presents particular challenges, such as safeguarding the integrity of data, ensuring that equivalent ethical standards are met, the threat of double standards, and the need to have confidence in local regulatory and supervisory arrangements [366,367][366][367]. Acknowledging that the inspectional resources of the US FDA can inspect only 1% to 2% of clinical trial sites [368], it is clear that such challenges cannot be resolved by an increase of inspections.
Another area of growing concern relates to the increased manufacturing of products in developing countries, and in particular the potential for substandard material to enter the supply chain [369]. Several problems have indeed been associated with data generated from foreign countries or with imported products. It is therefore critical to ensure that products manufactured in foreign countries and data from clinical research conducted in third-world countries comply with international standards. Considering the huge number of foreign manufacturing sites or global studies, no DRA can adequately keep pace with the pressures of globalization alone. It is indeed impossible for them to rely on their own inspectional resources. For example, even the US FDA, which is the biggest DRA in the world with a total annual budget of approximately US $4.5 billion [370,371][370][371] and an Office of Regulatory Affairs [372] of approximately 5,000 employees [373], does not have the resources to inspect all foreign drug establishments. It has been estimated that at current rates it would take an estimated nine years for the US FDA to inspect every high-priority pharmaceutical facility just once [374].
In this context, the most effective way (and certainly the only way) to resolve these concerns brought on by globalization is to increase cooperation between DRAs and harmonization of standards between countries.
II-2.2.2). Evolution of the Communication between Worldwide Authorities
Communication between worldwide DRAs has increased tremendously over the last decade. Globalization has indeed encouraged regulators to work and communicate outside their country and region to fulfill their mission of public health protection, and DRAs have increasingly recognized the value of supporting their internal programs through international collaboration.
As explained in Part II-1, this increased communication and harmonization over the past several years has ultimately been beneficial for all players in the pharmaceutical sector (e.g., patients, regulators, and industry).
Initially, exchanges between worldwide DRAs were focused on major public health concerns (i.e., primarily safety issues) with approved products or with issues related to the development of new and innovative products. These communications were generally handled at a high level by the DRAs, did not occur frequently, and followed a formal/official process. With the implementation of specific Memorandums of Understanding (MoUs) between agencies, communication has improved considerably between major worldwide authorities (e.g., US FDA, EMA, Health Canada, MHLW, TGA, and AMSM [formerly AFSSAPS]), and this has expanded both formal and informal communication at many levels of the DRAs (including scientific and reviewer staff). In Europe, more open exchanges between national regulatory agencies were fostered through the formation of the EMA in 1995. Additionally, while they are not DRAs as such, bodies such as ICH and WHO have served as an international forum for the establishment of common regulatory principles as well as important international networking opportunities for regulatory scientists. Through these different levels of interaction, professional relationships and trust has been developed over time between key regulatory scientists and their respective agencies. Today, as a result, more regular formal and informal communications occur between worldwide DRAs and their respective staffs on many different topics. These topics include major concerns, but also other subjects related to specific products or their testing, registration, or post-marketing activities for all types of products. This type of interaction is now becoming integrated in the day-to-day activities of many of the major worldwide DRAs.
As an example, I have been involved in meetings with different worldwide DRAs for approximately 15 years. During that time, none of the agencies I have met indicated that they were exchanging information or communicated with other worldwide DRAs on the topics covered during our meetings (I acknowledge that they may have had these discussions without informing me). However, for the first time, during a Sponsor and Agency meeting in September 2009 with one of the major worldwide regulatory agencies, an Agency representative formally asked for authorization to communicate and exchange information with his counterpart from other major worldwide DRAs that were also involved with the topic covered during the meeting. Note that the subject of the meeting related to a manufacturing/testing change and not the development of a new or innovative product. This example clearly shows the evolution in the way of thinking concerning international cooperation, and the increase of communication and exchange between worldwide DRAs.
In addition to the natural but slow evolution of the communication between worldwide regulators described above, recent major events that required global coordination of public health and regulatory actions have tended to accelerate this evolution, such as:
-
▸
After the terrorist attack on the World Trade Center on September 11, 2001, and the subsequent US anthrax attacks and “copycat” bioterrorist pranks in Europe and North America, several major worldwide public health and regulatory authorities coordinated their actions against the perceived bioterrorist threats.
-
▸
In 2002/2003, the SARS crisis also required a worldwide coordination of efforts to limit the propagation of the virus and its consequences, which was greatly aided by the WHO network with the support of national public health and regulatory agencies.
-
▸
Between 2003 and 2005, an increased scientific and public awareness of the pandemic potential of H5N1 drove numerous exchanges between worldwide public health and regulatory authorities to deal with that potential threat.
-
▸
More recently, in 2009 the H1N1 outbreak resulted in one of the most urgent and widely coordinated international responses to a pandemic in recent decades. The global propagation of the virus again required globally coordinated actions and communication between worldwide DRAs. However, compared to previous crises, these actions and communications were not limited to the national management of the crises and the coordination and management of safety measures. For the first time on such an urgent and large scale, several worldwide DRAs (including the US FDA, EMA, national European DRAs, Health Canada, TGA, MHLW, and China’s SFDA) communicated in general terms with regard to the development of vaccines, discussed issues with international potency standards and testing, and exchanged updates on when products might come to the market and the status of clinical trials related to these vaccines. Note that product-specific trade secret information was specifically excluded from these multilateral discussions due to the lack of legal framework (however, such information may have been discussed during separate bilateral and/or trilateral discussions between DRAs that have specific MOUs and/or confidentiality agreements in place [375–377][375][376][377]). This multilateral activity was coordinated through WHO [378] and supported by the national agencies involved. The purpose of these important exchanges was to support national and international emergency preparedness and response, and individual jurisdictions proceeded with manufacturing and regulatory paths that they felt were most appropriate to their respective national needs. While different vaccine choices were made in various countries in the time leading up to the H1N1 pandemic with regard to the use of vaccine adjuvants, one result of international regulatory collaboration subsequent to the pandemic was the effort to develop international guidance through WHO on the use of vaccine adjuvants.
Even though cooperation has increased over the past decade, it is clear that the level of, and participation in, worldwide harmonization and cooperation is different for each country. Harmonization and cooperation is a long, step-by-step process requiring many prerequisites. The willingness of worldwide authorities to exchange and cooperate with their peers depends on the following factors:
-
▸
Memorandum of Understanding (MoU) signed with other countries
-
▸
National laws and regulations
-
▸
General level of comfort based on cultural history and previous experiences in cooperation (e.g., an EU regulatory representative would have had more experience with regulatory exchanges with comparable external DRAs than a US FDA representative, due to the former’s experience operating within the EU regulatory system)
-
▸
Individual behaviors (this is still important, but with enhanced international cooperation increasingly becoming an objective in key agencies, the culture in agencies is changing)
To conclude, it is obvious that communication between worldwide agencies will continue to increase. This continued evolution seems inevitable, and most of the players agree that such communication has been beneficial to public health in our increasingly global environment. Moreover, the above examples demonstrate that the rapid and extensive coordination that was achieved with the H1N1 outbreak and the more sustained efforts to develop international guidance through bodies such as ICH and WHO clearly illustrate the large-scale change that is taking place in international regulation. These types of technical and regulatory international cooperation are beneficial for everybody (especially the patients), and do not alter the ultimate responsibility of each country to render a decision for a specific product.
The H1N1 case demonstrated that while initial differences in opinion with regard to the risk/benefit evaluation for adjuvanted H1N1 vaccines resulted in different choices with respect to the use of these products at the time, collaboration in this area has driven the development of an international guidance on adjuvants.
II-2.2.3). Political Decisions and Commitments
Political stability is a prerequisite for the establishment of a developed pharmaceutical system. It is obvious that developing countries during war will have difficulties in developing and implementing appropriate regulations and structure to control their pharmaceutical market. These countries have other priorities and cannot devote adequate resources to the pharmaceutical sector and the harmonization initiatives such as ICH. For example, the Southern African Development Coordination Conference was created to focus on political liberation of the region and to lessen economic dependence on the then apartheid South Africa. It was only with the achievement of political independence by SADC Member States [379] and the end of the South African apartheid regime in 1994 that this organization became a “Development Community” (SADC). Economic integration (and all efforts towards harmonization) could then really begin in Southern Africa.
In some countries, corruption and conflict of interest can also weigh against harmonization and globalization of the pharmaceutical market. For example, it is expected that the recent major political changes in several Arab countries will ultimately have an impact on the pharmaceutical regulations and systems of these countries.
However, political stability in a country is not sufficient to adequately support the development of a pharmaceutical system. It is well established that no national DRAs will be successful in implementing pharmaceutical regulation if they do not have full and continuing government support [380]. To perform effectively, DRAs must have the necessary political support, legal power, human and financial resources, and independence in decision making [381].
Many events have demonstrated the critical influence of politics on the establishment of pharmaceutical regulations, including:
-
▸
The mediator event in France that triggered political decisions that significantly changed the French pharmaceutical regulations and system [382,383][382][383].
-
▸
The delay in the availability of biosimilars in the US, compared to many other countries, is due to a lack of political decision. In this case, the long political debate and multiple delays in passing laws delayed the registration of such products in the US for many years. It is important to note that this delay also delayed the harmonization of the requirements on a global basis as the lack of US framework prevented any global discussion on this subject.
-
▸
One important strategic change in the US was the approval of embryonic stem cell research that followed the election of President Barack Obama. Such research was forbidden under President George W. Bush’s administration. It was authorized three days after the start of President Obama’s administration, with the US FDA approval of a Phase 1 human study conducted by Geron Corporation to test the safety of a treatment derived from embryonic stem cells for patients with spinal cord damage [384].
Political decisions and commitments are also vital for the harmonization of regulations between countries.
Most of the harmonization initiatives started with political decisions. For example:
-
▸
European harmonization is a direct consequence of the political decision to establish the European Union.
-
▸
The establishment of WHO and its pharmaceutical programs was a political agreement between countries.
-
▸
The foundation for advanced technical and regulatory cooperation and harmonization between the EU and US came from the signature of the Framework for Advancing Transatlantic Economic Integration between the European Union and the United States of America by Commission President José Manuel Barroso, German Chancellor Angela Merkel, and US President George W. Bush (at the EU–US Summit on April 30, 2007).
This political support is also critical for the entire harmonization process, including the implementation of recommendations. It needs to define appropriate priorities, to devote appropriate resources, and to support the project administratively and financially. Harmonization is indeed not free. Even if it is believed that more efficient use of resources will reduce costs for the long term, cooperation and harmonization efforts require investments (i.e., time, resources, and budgets for communication and travel). A recurring theme in all harmonization initiatives is the enormous pressure on resources at every level. The creation of the European pharmaceutical system is a good example. The audit of this system in 2000 by Cameron McKenna and Andersen Consulting [385] has clearly shown this important need for resources. Authorities noted the scarcity of expert assessment resources and suitably qualified people for the large number of working groups and other demands that the pharmaceutical systems made of national DRAs. Even if the purpose of the creation of the EMA and the new regulatory procedures was to use resources more efficiently, in reality, pan-European regulation has seen no real dividends in terms of cost efficiencies through economy of scale. On the contrary, the political pressure for the involvement of a national authority in every policy and assessment activity that may affect its market and citizens, coupled with the increased complexity of the process of regulation, has increased the administrative burden of European regulation as well as its sophistication. One CPMP member expressed the view that the centralized system in the EU was capable of providing the best regulatory assessment in the world because it encouraged networking of experts across Member States, but it was admitted that the system was also very expensive to run as a result. National authorities were under significant resource pressure and the relative funding of the centralized system by the Community and indirectly by national agencies was an increasing cause for concern in some Member States.
Political support is always necessary and a driver of the harmonization, but it can have different objectives:
-
▸
Economic reasons: to create a free trade area or a single market with other countries.
-
▸
Increase communication with other countries: to cope with increased globalization of the pharmaceutical environment and enhance public health.
-
▸
Geopolitical reasons: developed countries support global harmonization to facilitate capacity building and control of the pharmaceutical market in developing countries. This support also benefits developed countries because it allows better control of products/data from these countries.
In Europe, the implementation of pediatric regulation is a good example of how political decisions can influence the regulation and harmonization/cooperation between countries to enhance public health. Prior to the introduction of the European Commission proposal (September 29, 2004), there were no adequate national measures to support the development of high-quality and adequate medicines for children. More than 50% of the medicines used to treat children had not been tested and were not authorized for that use [386]. Following a political decision, the EU regulation created these requirements and obligations. Following the implementation of this text on January 26, 2007, pharmaceutical companies started to invest in pediatric development and included this new component in their strategy. In 2009, the EMA received applications for Pediatric Investigation Plans (PIP) relating to 364 clinical indications.
However, although political support is clearly important, it is not automatic and easy to achieve.
Firstly, the priorities of ministers of health depend on the health issues in that country. In the developing countries where there is an important problem of HIV/AIDS (especially when this public health problem is coupled with an important poverty issue), health resources will be rightly dedicated to the prevention and management of this disease versus the harmonization of the pharmaceutical regulation with other countries and regions.
Secondly, national/regional protectionism can also delay or limit harmonization efforts. In this case, the lack of political commitment, sometimes exacerbated by lobbies (that benefit from loose regulation and lack of global harmonization), is a response to two political considerations:
-
▸
Loss of sovereignty: Any harmonization process raises questions about sovereignty as harmonization of standards implies a certain loss of control for the countries involved. It is obvious that the influence of politics is even more important in the case of MRAs or integration models. These models of harmonization raise practical and obvious concerns regarding loss of sovereignty. Resistance to relinquishing power can result in a clear obstacle to harmonization and cooperation. The joint Australia New Zealand Therapeutic Products Agency (ANZTPA) project is a good example where politics (and also economic factors) can interfere with harmonization [387].
-
▸
Industrial competitiveness: Some countries may see harmonization of regulation and cooperation with other countries as a concern for competitive advantage and want to protect the competitiveness of their own industry. For example, regulation on advanced therapy is one topic on which most developed countries in the world should fully collaborate because this is a new, important, and promising topic without historical differences that may be difficult to harmonize. However, due to the huge potential for industry, each country or region needs to balance cooperation/harmonization with competitiveness/stimulation of its own industry regarding new technologies. For example, in 2007, the EU institutions agreed on a Regulation on advanced therapies (Regulation (EC) No 1394/2007), designed to ensure the free movement of advanced therapy products within Europe, to facilitate access to the EU market, and to foster the competitiveness of European companies in the field. Indeed, the lack of an EU-wide regulatory framework for advanced therapy in the past led to divergent national approaches which hindered patients’ access to products, hampered the growth of this emerging industry, and ultimately affected EU competitiveness in a key biotechnology area [388]. This regulation has special incentives for small- and medium-sized enterprises.
To conclude, political support is clearly a key element to ensure a successful harmonization process. Therefore, a harmonization plan should evaluate, a priori, the potential political implications of the proposed projects to mitigate any negative impact and ensure appropriate support.
II-2.2.4). Economy
There is an important interdependence between economy and public health in general. Unfortunately, a bad economy can impact public health as demonstrated by the recent cuts in worldwide governmental health budgets due to the worldwide financial crisis. However, the opposite is also true. There is also a positive relationship between economic growth and improvement of public health. Studies have shown that the level of economy and public health of a country are indeed linked [389]. It seems obvious that when economies get richer, a greater proportion of diseases is treated and more sophisticated treatments are employed.e , f This improvement in public health in a specific country does not only benefit the patients who can enjoy the additional years of life without disability, but also benefits the country/community as a whole by decreasing the burden due to bad public health. Indeed, a combination of early death and an increase in disability in a younger active population (as quantified by the “disability adjusted life years lost” by the economists) has a clear impact on the economy of the country as it influences the increase/decrease of treatment costs, labor force, productivity, and hence the gross domestic product (GDP). For example, estimated losses in national income from heart disease, stroke, and diabetes in 2005 were US $18 billion in China, US $11 billion in the Russian Federation, US $9 billion in India, and US $43 billion in Brazil. These losses accumulate over time. It is estimated that, between 2005 and 2015, China will have lost US $558 billion in national income due to heart disease, stroke, and diabetes alone [390].
The relationship between economy and harmonization of pharmaceutical regulations is also important, but more complex.
First, economic decisions can influence harmonization initiatives, positively or negatively. Most of the regional initiatives related to the harmonization of pharmaceutical regulations are indeed a positive consequence of an intergovernmental decision to set up a trade bloc (e.g., Europe, GCC, ASEAN, MERCOSUR). The creation of these trade blocs requires a reduction of barriers to trade in all sectors (including the pharmaceutical sector) and the establishment of common requirements/standards. Depending on the level of economic integration, trade blocs can fall into different categories, such as preferential trading areas, free trade areas, customs unions, common markets, and economic and monetary unions. Of course, the higher the economic integration, the deeper the harmonization. However, economic considerations can also negatively impact harmonization efforts. Although globalization has created new opportunities, major pharmaceutical companies are still focusing their efforts of development to meet the needs of developed countries because these countries continue to represent an important part of their revenue [391]. World sales of pharmaceuticals are indeed highly skewed toward the developed world markets. Developing countries account for more than 80% of the world’s population, but they are responsible for only about 10% of global sales [392]. Companies therefore have no incentive to include developing countries and establish a global development plan if such a global plan would delay registration in Europe or the US.
Second, decisions in the pharmaceutical domain can also impact employment and the general economy. The pharmaceutical sector has experienced some important structural changes over the years (due mainly to technological innovations, increasingly stringent regulatory requirements, and globalization of activities) that have affected markets and increased the costs and risks of development of new products. Today, this sector is highly regulated and is very complex as it involves a large variety of players (i.e., multinational major pharmaceutical firms, but also start-up companies, a lot of contractors, and other research organizations such as universities, public and private research centers, financial institutions, DRAs, governments, health care systems, consumers, physicians, etc.). Many parameters can influence the competitiveness of this industry [393], including changes in regulation or integration/harmonization activities. However, the pharmaceutical market is clearly a “strategic” sector for all countries/regions in the world because it is large, high growth, high revenue, and globalized. For example, in Europe, the pharmaceutical sector employs more than 634,000 people and accounts for more than 17% of the EU R&D expenditure [394]. Therefore, actions in the pharmaceutical sector always have the dual objectives of safeguarding public health (by providing people with safe and effective medicines) while creating a business environment that stimulates research, boosts innovation, and supports the competitiveness of industry. This has to be kept in mind when working on harmonization initiatives to understand the resistance from some governments (especially global harmonization where the different regions have to consider this dual objective). For example, it is commonly agreed that the establishment of Europe and the creation of supra-national legislation in view of harmonization of practice and requirements was one of the factors (in addition to the increased globalization of the sector and other challenges) that impacted the competitiveness of Europe compared to the US and Asia. The implementation and interpretation of Community legislation by Member States created obstacles to the free movement of medicines. Overburdening requirements also affected competitiveness, especially for small- and medium-sized enterprises [395].
In summary, economic parameters and their potential consequences need to be considered when discussing harmonization activities. Of course, they should not be the major or unique factors, but they need to be considered along with other parameters because they may support or limit the harmonization process. If a harmonization project impacts the economy too much (by impacting the attractiveness and competitiveness of a country or region), there is a good chance that it will be difficult to implement as demonstrated by the introduction of the European Clinical Trials Directive. Notwithstanding the added value of this legislation, it has been recognized that this Directive has increased bureaucracy and the costs of clinical trials conducted in Europe [396], which in turn greatly reduced European competitiveness in the field of clinical research (especially in the context of globalization of these activities with emerging investigation sites in Eastern Europe and in the Asia-Pacific region). This was one of the reasons that convinced the European Commission to launch the “Impact on Clinical Research of European Legislation (ICREL) project to help determine the most relevant pathways for improvement of this new legislation. The legislative and regulatory framework is indeed one of the major determinants for the attractiveness of a given region for clinical research. This sector is critical to promoting public health, but is also crucial for the economy as it employs people and represents a substantial amount of money invested. It is, for example, a source of employment and of revenue for investigational sites and many subcontractors.
II-2.2.5). Differences in Cultures and Traditions
Cultural differences and diverse traditions have influenced the establishment of national regulations, requirements, and systems. For example, the importance and recognition of physicians and other stakeholders are different in each culture [397]. In addition, some products are sometimes used by a limited number of countries (i.e., the venotonic products primarily used in France or traditional therapies in certain countries). This difference of environment and the specific needs of local markets continue to require appropriate and specific local regulation and organization.
Cultural and traditional differences can also limit harmonization efforts on topics of common interest. For example, during the first meeting of the Pediatric Medicines Regulators’ Network in February 2010, participants observed that it may not be possible to reach a global consensus as to the ethics of clinical studies in children because opinions include cultural aspects. For example, current guidelines differ as to:
-
▸
Whether children should be included in research studies
-
▸
Whether research in children is beneficial to the participants themselves
-
▸
Whether healthy children may be subjected to clinical studies, and if so, under what circumstances
-
▸
The definition of the ages of “children”
Cooperation is still relevant to understanding differences and agreeing on technical topics (i.e., of clinical trials, terminology, etc.), but harmonization of ethical standards may not be possible due to these cultural differences [398].
Historical differences on how reviewers evaluate data and drug applications can also impact cooperation. For example, some review the summaries of information provided by the company first (this is the case in Europe, which used to require expert reports, now replaced by the CTD overviews). Others prefer starting with source data (e.g., the US). This type of historical difference needs to be taken into account when developing common Good Review Practices (GRevPs).
Finally, awareness of cultural differences is important in communication and should not be underestimated, as communication is critical to achieve cooperation and harmonization (see Part II-2.1.2). Communication codes (verbal and nonverbal) between French, Dutch, Italians, Americans, Japanese, Batswanans, Kuwaitis, or Brazilians are very different. It is therefore critical that people involved with harmonization and cooperation initiatives understand such differences to avoid problems of communication. Many courses and training on cultural differences are available. They are important tools to initiate and maintain respectful and strong relationships with counterparts.
The establishment of the EU (bringing together Member States with major historical, social, and cultural differences and 23 official languages) demonstrated that these differences can be overcome if there is a political commitment. However, it is important to keep these differences in mind when working on harmonization/cooperation initiatives to avoid communication problems and to understand approaches, systems, methods, and the interests of other parties.
II-2.2.6). Ethnic Factors
Ethnic factors are characteristics related to race or large populations grouped according to common traits and customs. This definition gives ethnicity, by virtue of its cultural as well as genetic implications, a broader meaning than racial. Ethnic factors may be classified as either intrinsic or extrinsic [399]:
-
▸
Intrinsic ethnic factors are factors that help to define and identify a subpopulation and may influence the ability to extrapolate clinical data between regions (e.g., genetic polymorphism, lean body mass, body composition, organ dysfunction, etc.).
-
▸
Extrinsic ethnic factors are factors associated with the environment and culture in which a person resides (e.g., diet, use of tobacco and alcohol, exposure to pollution and sunshine, socio-economic status, compliance with prescribed medications, etc.). They tend to be less dependent on genetics and more socially, culturally, and behaviorally determined.
Ethnic factors can limit global development of medicines and require duplication of clinical studies. These factors can indeed affect the medication’s safety, efficacy, dosage, and dose regimen, but also raise specific risks for a certain population [400].
Until now, DRAs have managed this risk by requesting that all, or much of, the foreign clinical data in support of registration be duplicated in a new population, and/or that bridging studies be performed to extrapolate data from another population.
The ICH E5 guideline [401] is an important tool to help characterize the influence of ethnic factors in order to facilitate extrapolation of data to different populations and therefore minimize the need for duplication of clinical studies. The recent increased cooperation between China, Korea, and Japan in the clinical domain is based on their ethnic similarities (genetic, but also cultural) [402].
Evaluation of the impact of ethnic factors on treatment effect is important to support global development and facilitate international clinical studies, but this evaluation is also key due to the changing demographics in most of the developed countries. For example, the US population is becoming increasingly ethnically and racially diverse. The Caucasian population is growing more slowly than other groups, but Caucasians continue to account for the majority of trial participants. The US FDA has expressed concern regarding the extrapolation of the results to others subgroups [403].
II-2.2.7). Differences in Medical Practices
Differences in medical practices and therapeutic approaches exist between countries and regions of the world. These differences (defined as extrinsic ethnic factors by ICH [401]) may be due to several reasons, including education, cultural/traditional differences, level of development of each country, specific and genetic diseases, etc. Different diets, importance of physical therapies, and environmental/climatic conditions also need to be considered.
These differences in medical practices can limit harmonization and complicate the design of global clinical studies:
-
▸
Choice of comparator products (due to different therapeutic arsenals and/or standard therapies in each country/region, but also due to confusion generated by different names of medicines)
-
▸
Selection of endpoint
-
▸
Definition of disease and clinical relevance and significance of results
-
▸
Monitoring of underlying illnesses
-
▸
Evaluation and limitation of concomitant therapies
It is interesting to see that ICH efficacy guidelines are not therapeutic class specific even when:
-
▸
There are, in some therapeutic classes, individual drug evaluation guidelines among the three regions.
-
▸
Differences between guidelines can result in obstacles to the mutual use and acceptance of clinical data.
At the ICH Steering Committee meeting in September 1998, it was agreed that this should be adopted as a new area of work for ICH, with the first such guideline being undertaken as a “pilot study” to assess the feasibility of extending work in this area. It was agreed to develop the first therapeutic class-specific guideline for anti-hypertensive drugs (E12). However, this pilot work showed how differences in medical practices can impact the global development of medicines. It was finally agreed that this document should be considered an “ICH Principle Document” rather than an “ICH Guideline” because there were differences in the requirements of the three regions that could not be harmonized (i.e., the need for studies comparing the new medicine with current standard therapy and the specific requirements regarding co-administration with other anti-hypertensives). No other guideline for clinical evaluation of specific therapeutic categories has been developed since this E12 document.
II-2.2.8). Difference in Legislation, Statutes, and Systems
While worldwide DRAs generally share similar scientific concerns, differences between legislations and regulatory systems/frameworks can present important obstacles to harmonization. Some countries have an independent agency (e.g., FDA in the US, Health Canada in Canada), some have semi-independent institutes, some have a multinational/supranational agency (EMA in Europe), and some do not have an independent agency (i.e., the system is driven by the Ministry of Health [MoH]). The difference in statutes of each DRA, when they exist, can sometimes impact the objectives of cooperation with their counterparts. In some countries, certain aspects of the pharmaceutical sector are governed by national laws, and other aspects are governed by state or provincial laws. This decentralization of roles and responsibilities provides another level of complexity for harmonization [404]. Although there has been significant progress during the last several decades, with strong participation and motivation of regulators, overcoming these obstacles will ultimately require unprecedented levels of international cooperation and profound modifications of legislations.
Most of the legislative texts have been voted in by national/regional legislators following major public health issues. Even if the legislators reacted to the same issues (e.g., the thalidomide problem in 1961), different actions have been taken and different fundamental laws have been passed in each country. This different legislative environment can impact harmonization efforts. For example, one of the reasons (among many others) for the delay of registration and use of biosimilar products in the US compared to other developed countries was the lack of a legal basis for such products (because most of the biologics were approved under the Public Health Service Act and not under the Federal Food, Drug and Cosmetic Act, which has allowed generic products since the Hatch–Waxman Amendment in 1984) [405]. Until US legislators defined this legal basis, it was impossible to have US regulation for this type of product and therefore harmonization of requirements with other countries.
The different status/legislation of borderline products in the world is another example demonstrating how legislation can complicate harmonization between countries. Some products can be considered “medicines” in certain countries and “medical devices” in other countries (e.g., products for dry eyes). Discussing the harmonization of regulations becomes difficult in this case because these products fall within different regulatory frameworks and also within the scope of different harmonization initiatives.
Finally, the legislative context also impacts the communication and exchange of information between DRAs. Indeed, some countries passed legislative texts to protect trade secrets and commercial information (e.g., the Uniform Trade Secrets Act in the US). Although government needs to protect sensitive information, these restrictions may impact/complicate harmonization and cooperation between countries [406].
The impact of differences in legislations (and the time and effort to overcome these differences and to change legislations) is well known and has already restricted or impacted certain harmonization initiatives. For example, the Transatlantic Administrative Simplification project between the EU and US (see Part I-3.2.5) was limited to actions that would not require changes in legislation. Also, one of the major issues of concern to PANDRH members is the recognition that serious limitations exist in some subregions, such as Central America, where no legal framework exists to authorize and implement the commitments made by technical groups [407].
Any new harmonization initiative needs to take this factor into account. It is important to avoid the need for legislation changes and to work within the current legislation boundaries to avoid delays. Regulation can be passed or revised more rapidly and simply. If a harmonization project requires legislative changes (in the case of an integration project or major harmonization efforts), this delay needs to be clearly factored in the plan since passing a new or revised law may require a lengthy process [408].
II-2.2.9). The Fourth Hurdle
In the past, three criteria were used to assess a new medicine and evaluate the risk/benefit ratio: quality, safety, and efficacy. A fourth hurdle has become more and more important in the last decade: the evaluation of the cost effectiveness of the medicine. In other words, it is not sufficient to demonstrate that a new drug is of high quality, safe, and efficacious, but it also has to have benefits over existing therapies. This new hurdle is obviously one of the consequences of the decrease of healthcare budgets worldwide. Escalating healthcare costs and the need to balance budgets have led payers in many jurisdictions to become more restrictive in their decisions to reimburse new expensive health technologies. These considerations mean that coverage has become a major roadblock for new medicines. Even if DRAs and payers have distinct roles and information needs, winning regulatory approval is of little use to industry or patients when the medicine is not reimbursed, as access to high-priced medicines will effectively be precluded for most patients. This new hurdle has led to a number of important consequences on the development of new products [409].
The increased importance of this new criteria (entirely based on national pharmaceutical needs and strategy) and the increased communication between DRAs and Health Technology Assessment (HTA) (e.g., as is the case in Europe [410]) will also influence the regulatory environment of new medicines [411]. This is an important change of environment that could impact the ongoing global harmonization of the regulation. For example, until now, the need to evaluate new medicines against existing therapies has been supported in Europe, but not by US regulators/legislators. Although this concept of comparative effectiveness has not yet become a new criterion to register a drug in the US, the change in US legislation will certainly harmonize the EU and US positions on this topic [412].
II-2.3). Conclusion
All the parameters and factors discussed above are critical to consider because they can influence the success of a harmonization/cooperation initiative. A good idea is not sufficient. All harmonization initiatives need to take these parameters into consideration to ensure success. Even the establishment of an appealing harmonization project, supported by all stakeholders, needs to integrate these factors and associated challenges.
The European harmonization of the rules for performing clinical trials is an excellent example to demonstrate that:
-
▸
All influencing factors and potential problems of implementation need to be taken into consideration very early (before the rule comes into effect).
-
▸
The choice of the legal framework for harmonization is critical.
-
▸
It is important to avoid any possible interpretation of the rules (rules and the implementation process should be as clear as possible).
-
▸
The implementation of harmonized rules is at least as important as the rules themselves.
-
▸
Monitoring of the implementation is required.
The harmonization of clinical trial requirements is one of the most critical harmonization topics for the development of new medicinal products. Ironically, the initial implementation of harmonized European requirements has been very difficult. Although the EU Clinical Trials Directive was an important step and everyone initially welcomed it (as it created a certain level of harmonization), it has been one of the most criticized pieces of the European legislation on medicines. As this harmonization has not been sufficiently far-reaching, its implementation has been very difficult and has led to an undesired increase of clinical trial complexity. The fact that the clinical trials legislation was established as a Directive (requiring transposition of its principles into national legislation) partly explains the problem. Because most of the EU countries already had their own legislation and practice before the adoption of the Directive, their interpretation of the Directive and the changes brought to the national legislation were highly dependent on this preexisting framework. As a result, the harmonization target was missed and these differences in interpretation of the modalities for the agreed-upon principles led to even higher complexity levels (especially for multinational clinical trials). Today, a sponsor of a clinical trial needs to have very detailed knowledge about every country’s national requirements for the clinical trial authorizations and has to integrate the different requirements to the protocol and the Investigational Medicinal Product Dossier (IMPD) resulting from parallel submission in multinational trials.
To better understand this problem of implementation, the European Commission launched a retrospective comparative study to measure and analyze the direct and indirect impact of the Clinical Trials Directive (and its related guidelines) on all categories of clinical research and on the different stakeholders. This project, called Impact on Clinical Research of European Legislation (ICREL), compared the mean differences for several parameters between 2003 (i.e., prior to the entry into force of the Clinical Trials Directive) and 2007.
According to the ICREL report [413]:
-
▸
There has been an indisputable increase in the administrative burden for DRAs (this is reflected by an increase in workforces and related costs, which is paralleled by a rise in fees).
-
▸
Performing a clinical trial has become considerably more difficult and costly. Staff needs in pharmaceutical companies for administrative work for submitting a request for authorization of a clinical trial has doubled compared to the circumstances prior to the entry into force of the Clinical Trials Directive.
-
▸
The timelines for the overall protocol and substantial amendment approval process were extended by approximately 30%.
In this report, some recommendations were also proposed, such as:
-
▸
Using a risk-based approach to regulation would result in a substantial reduction in workload and cost for both the Sponsor and DRAs, particularly for academic institutions that run a number of low-risk studies using marketed drugs. Not all clinical studies require the same level of requirements.
-
▸
Simplifying the Clinical Trial Authorization (CTA) process by the competent authorities through a single CTA for multinational trials would reduce duplication of efforts and also save time, costs, and expertise.
-
▸
Further harmonizing practices in ethics committee requirements.
Based on this ICREL report, the European Commission released a public consultation paper in October 2009 [414] that highlighted five key issues to be addressed:
-
▸
Multiple and divergent assessments of clinical trials: While the Clinical Trials Directive sets out common rules (that ensure harmonization of the concepts), experience shows that these requirements are applied very differently by the respective Member States, and conflicting points are brought up by the DRAs of these Member States.
-
▸
Inconsistent implementation of the Clinical Trials Directive (e.g., definition of a substantial amendment).
-
▸
The regulatory framework is not always adapted to the practical requirements.
-
▸
Adaptation to peculiarities in trial participants and trial design.
-
▸
Ensuring compliance with Good Clinical Practices in clinical trials performed in third-world countries. This is critical because about 25% of all clinical trials performed in the EU also involve at least one third-world country and 65% of all data/patients submitted in pivotal clinical studies in the framework of an application for an EU-wide marketing authorization are generated in third-world countries.
These key issues highlighted by the European Commission were shared by sponsors and other stakeholders involved in the conduct of clinical trials in Europe [415]. They confirmed that, although concepts promoted in these new texts were good and improved the situation (e.g., improvement of data reliability and increase of communication and cooperation), they did not achieve their aim. They stressed that it was very common to receive divergent assessments from Member States (certainly due to differences in culture, clinical practice, and health systems). They agreed that the new system created additional administrative costs/burdens and reduced the competitiveness of Europe on a global scale without improving the safety and rights of subjects. Moreover, it could affect the development of new and innovative treatments because clinical trials are sponsored not only by large multinational pharmaceutical companies, but also by small, local pharmaceutical companies or research organizations with limited resources.
The new revision of the EU legislation [416], based on an evaluation of failure that involved all stakeholders, will hopefully resolve most of the challenges in order to finally meet the predefined objective.
Learning from this EU case and recognizing the importance of the numerous and complex factors that can influence the outcomes of a harmonization/cooperation initiative, it seems important to follow a simple step-by-step approach when developing a harmonization initiative in order to integrate and better control all the parameters. Those steps are as follows:
-
▸
Step 1: Create an oversight committee with relevant decision-making power from each party. This committee will then need to meet on a regular basis to monitor progress of the initiative.
-
▸
Step 2: Define objectives and agree on clear and realistic goals.g
-
▸
Step 3: Put in place an appropriate structure and organization to support projects (i.e., secretariat, working parties, etc.). This structure needs to involve all relevant stakeholders, and at a minimum, continuous updates need to be provided.
-
▸
Step 4: Set up a plan to achieve intermediate goals and ultimate objectives. The plan needs to incorporate appropriate and realistic timelines and also define roles and responsibilities. The value of each funded project (i.e., development of a specific harmonized standard) needs to be evaluated by assessing its benefit against the current situation, the budget and resources available, the development, implementation, and maintenance it will require, and the potential implementation challenges.
-
▸
Step 5: Develop harmonized rules and standards. All stakeholders need to have the opportunity to review and comment on rules before they come into effect.
-
▸
Step 6: Implement new harmonized rules and standards.
-
▸
Step 7: Monitor implementation and use of each harmonized rule and standard and evaluate if this implementation achieves predefined goals and supports the ultimate objective.
-
▸
Step 8: Maintain and revise rules when needed.
Of course, this basic approach is only a starting point that needs to be customized to the specific situation, context, and objectives. Each project needs to be thoroughly detailed, but following this simple step-by-step approach provides a structured and logical process to integrate all critical parameters for success and limit the influence of external factors. It also gives time to partners for communication and the establishment of relationships, and ensures that political leaders are in agreement with the objectives, plan, and investment (i.e., resources, budget, etc.).
There is no unique and exhaustive legal interpretation of the concept of “commercially confidential information.” A 2007 EMA document (titled “Principles to Be Applied for the Deletion of Commercially Confidential Information for the Disclosure of EMEA Documents,” EMEA/45422/2006) considers that this information relates to either intellectual property “know-how” and trade secrets (such as formulae and processes) or commercial confidences (such as development plans).
Several methods allow clear and early definition of roles and responsibilities, such as the RACI model that defines who is “Responsible,” “Accountable,” “Consulted” or “Informed.”
For example, the cost of formulation of an Active Pharmaceutical Ingredient (API) can range from 15% to 40% less to produce in India as compared with the US.
A naive patient is a patient that has never been administered the investigational medicines.
There are several US studies that estimated the value of a year of life at approximately US $150,000, acknowledging that there are a lot of different numbers in the literature and that this value varies of course in the time period and is different in other countries (Murphy and Topel 2003; Norhaus 2003).
History has clearly demonstrated this relationship. During bad economic times (such as the financial crisis in 2008/2009), there is a decrease in start-up companies, which are mainly financed by venture capital (See 2009 BIO paper on “The Ongoing Financial Markets Crisis and Lack of Access to Capital Threatens America’s Biotechnology Industry and the Development of Innovative New Therapies for Patients”). Moreover, analysis has shown that R&D expenditure falls during recessions (see “Stress in the life sciences innovation model: Impact of the Global Financial Crisis, presented by Bruce Rasmussen during the APEC Life Sciences Innovation Forum in Singapore on 4 August 2009”). This decrease in start-up companies and R&D expenditure have an obvious impact on innovation and research/development of new therapies.
Each goal needs to be Specific, Measurable, Attainable, Relevant and Timely (also known as the “SMART” criteria).