

Este grupo de erupciones está constituido por lesiones de morfología variable y etiología diversa. Sin embargo, todas tienen como característica común la presencia de eritema, reflejo de su naturaleza inflamatoria. Varias alteraciones parecen representar reacciones de hipersensibilidad, pero la etiología de la mayoría es desconocida. El diagnóstico diferencial de las púrpuras en el recién nacido es extenso, incluyendo alteraciones hematológicas, infecciones, traumatismos y alteraciones yatrogénicas.
Eritemas anulares
Eritema anular es un término descriptivo que engloba varias entidades de etiología desconocida caracterizadas por lesiones policíclicas circinadas que se extienden de forma periférica a partir de un foco central1 , 2. Dadas las escasas diferencias en las características clínicas, la edad de inicio, la duración de las lesiones individuales y la duración total de las erupciones, se han utilizado términos descriptivos para explicar estas alteraciones (v. tabla 19-1 ). Por razones de pronóstico es útil subdividir los eritemas anulares en formas transitorias y persistentes3. Las formas transitorias incluyen el eritema anular de la lactancia y la entidad peor establecida de eritema giratorio atrofiante transitorio neonatal. Los eritemas anulares persistentes incluyen el eritema anular centrífugo, el eritema anular familiar y el eritema giratorio persistente. En este epígrafe no se consideran otras formas de eritema anular, que corresponden a manifestaciones de enfermedades bien establecidas (p. ej., lupus neonatal) o con características clínicas o histológicas bien diferenciadas (p. ej., eritema multiforme, eritema crónico migratorio, eritema marginado reumático y eritema giratorio repentino).
TABLA 19-1.
Eritemas anulares
| Edad de inicio | Características clínicas | Duración de las lesiones individuales | Duración de la erupción | Curación | Histopatología | |
|---|---|---|---|---|---|---|
|
Formas transitorias | ||||||
| Eritema anular de la lactancia4 |
Lactancia temprana |
Placas anulares Sin descamación o formación de vesículas |
Días |
Transitorio (5-6 semanas; evolución cíclica) |
No deja lesiones residuales |
Infiltrado perivascular de eosinófilos |
|
Formas persistentes | ||||||
| Eritema anular centrífugo11 |
Edad adulta, es posible en el período neonatal |
En los bordes puede verse descamación leve |
Semanas |
Persistente (meses o años, desarrollando nuevas lesiones continuamente) |
Hiperpigmentación residual |
Perivascular superficial y profundo, linfocitario |
| Eritema familiar anular22 |
Lactancia temprana hasta la pubertad | Vesiculación y descamación posible | Días | Persistente (toda la vida, remisiones cortas) | Hiperpigmentación transitoria | Perivascular, superficial, linfocitario |
| Autosómico dominante |
Puede llevar asociada lengua geográfica | Espongiosis y paraqueratosis | ||||
| Prurito |
||||||
| Eritema giratorio persistente12 | Lactancia temprana | Descamación, posible constante vesiculación | Semanas | Persistente (toda la vida) | Hiperpigmentación transitoria | Perivascular linfocitario |
| Atrofia central | Espongiosis e hiperqueratosis | |||||
Eritema anular de la lactancia
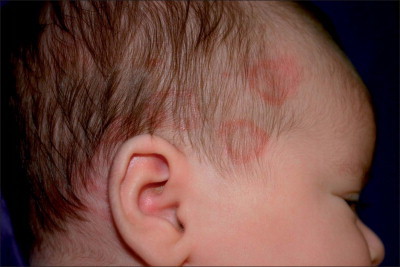
El eritema anular de la lactancia es una enfermedad benigna de la lactancia temprana caracterizada por pápulas urticariales que aumentan periféricamente, formando anillos de 2-3 cm o arcos, elevados, similares a cordones o con bordes urticariales4., 5., 6.. Las lesiones adyacentes confluyen formando lesiones arciformes y policíclicas (v. figura 19-1 ). En los bordes no hay vesiculación ni descamación. La erupción es asintomática. Las lesiones individuales se resuelven espontáneamente sin dejar rastro, pero siguen apareciendo lesiones nuevas de forma cíclica hasta la resolución completa, que tiene lugar durante el primer año de vida. Se han descrito algunos casos que duran varios años7., 8., 9..
FIGURA 19-1.

Eritema anular de la lactancia. La erupción era congénita.
La causa del eritema anular es desconocida y no se asocia a alteraciones sistémicas. Los estudios histológicos revelan un infiltrado superficial y profundo, denso y perivascular de células mononucleares y eosinófilos. No se observan figuras »en llama». La epidermis es normal o ligeramente espongiótica.
Los estudios de laboratorio son normales. La eosinofilia tisular no va acompañada de eosinofilia periférica. Los niveles de inmunoglobulinas, incluida la IgE, son normales. El diagnóstico diferencial debe incluir otras lesiones anulares de la infancia (v. la siguiente discusión). No se necesita tratamiento dada la naturaleza autolimitada de la erupción.
El eritema giratorio atrófico transitorio neonatal es una entidad peor definida10, caracterizada clínicamente por placas anulares con un borde eritematoso y un centro atrófico. Las lesiones aparecen en el período neonatal y desaparecen durante el primer año de vida. Los hallazgos histológicos incluyen atrofia epidérmica y un infiltrado mononuclear perivascular. Los estudios de inmunofluorescencia muestran depósitos granulares de IgG, C3 y C4 en la unión dermoepidérmica y alrededor de los capilares. El eritema giratorio atrofiante transitorio neonatal posiblemente representa una variedad neonatal de lupus eritematoso11.
Eritema anular centrífugo
El eritema anular centrífugo es una forma más persistente de eritema anular que generalmente afecta a los adultos12, si bien también puede ocurrir en niños y raramente en recién nacidos3 , 13., 14., 15., 16.. Las lesiones se parecen a nivel clínico a las del eritema anular de la infancia, aunque presentan vesículas y descamación en los bordes. La escamas se quedan detrás de los bordes que van progresando, los cuales, en contraste con el eritema anular del lactante, no está indurado. Las lesiones individuales se resuelven de forma espontánea al cabo de unas semanas; sin embargo, pueden ir apareciendo placas nuevas durante años o puede ser un trastorno que dura toda la vida. No se asocia a prurito.
El eritema giratorio persistente se engloba en el mismo espectro del eritema anular centrífugo16., 17., 18.. Algunos autores defienden la diferencia de esta forma de eritema y consideran que las características que los diferencian se encuentran en el inicio, la duración del proceso de más de 15 años, la presencia de un prurito ligero o grave y especialmente la presencia de vesiculación16.
Se cree que el eritema anular centrifugo representa una reacción de hipersensibilidad a diversos factores desencadenantes, incluyendo agentes infecciosos (Candida 19 , 20, virus de Epstein-Barr14 y Ascaris 21), fármacos o alimentos22 , 23 y neoplasias, especialmente en adultos. La inyección intradérmica de candidina o tricofitina puede reproducir las lesiones clínicas13 , 24.
Las características histológicas consisten en un infiltrado mononuclear denso, superficial, perivascular. Puede haber paraqueratosis o espongiosis epidérmica. No existe un tratamiento eficaz para todos los casos, Los agentes para el tratamiento incluyen la nistatina oral20, la anfotericina B19, los antifúngicos tópicos, antihistamínicos, cromoglicato disódico y el interferón α13.
Eritema anular familiar
Se han descrito casos hereditarios de eritema anular con una herencia autosómica dominante25 , 26. El cuadro debuta en la lactancia temprana. La descamación, la vesiculación y el prurito pueden ser más comunes que en el eritema anular centrífugo. Las lesiones desaparecen y dejan una hiperpigmentación residual. La cronicidad es la norma. Pueden asociarse a lengua geográfica26.
Diagnóstico diferencial del eritema anular
El diagnóstico diferencial incluye otras erupciones con lesiones similares a anillos, como son el lupus neonatal, el eritema multiforme, la urticaria, las lesiones urticariales del penfigoide, las infecciones fúngicas, el eritema crónico migratorio y la enfermedad de Lyme congénita7 , 27 , 28. Para descartar el lupus neonatal, se recomienda la determinación de los anticuerpos séricos (antinucleares SS-A y SS-B).
Lupus eritematoso neonatal29., 30., 31., 32., 33., 34., 35., 36.
El lupus eritematoso neonatal (LEN) es una enfermedad de recién nacidos producida por la transmisión de anticuerpos maternos. Las principales manifestaciones son dermatológicas y cardíacas. Los hallazgos cutáneos son transitorios. La enfermedad cardíaca, que es responsable de la mortalidad y morbilidad del LEN, se inicia en el útero y afecta permanentemente al sistema de conducción cardíaco. Otros hallazgos son las alteraciones hepáticas y hematológicas. Las madres de los lactantes con lupus eritematoso neonatal tienen en un 95% de los casos autoanticuerpos anti-Ro/SS-A. Los autoanticuerpos anti-La/SS-B y anti-U1RNP también se han visto implicados en la patogenia del LEN en una minoría de pacientes37 , 38.
Hallazgos cutáneos
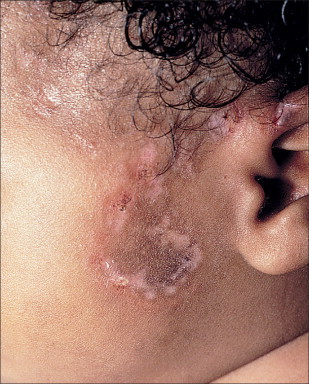
Un 50% de los lactantes con LEN tiene lesiones cutáneas y el bloqueo cardíaco está presente en el 10% de los casos29 , 36. Las lesiones generalmente se desarrollan a las pocas semanas de vida, si bien pueden estar presentes al nacer, lo que sugiere que la radiación ultravioleta (UV) no es esencial para el desarrollo de las lesiones cutáneas del LEN39. Clínicamente, la piel es análoga a la observada en el lupus cutáneo subagudo, en sus dos variantes: papuloescamosa y anular. Las lesiones papuloescamosas son más comunes y están caracterizadas por placas eritematosas, no induradas y descamativas (v. figura 19-2 ). En ocasiones las lesiones cutáneas tienen un aspecto atrófico (v. figura 19-3 ). Puede haber ulceraciones40. A diferencia del lupus discoide, no hay formación de cicatrices ni tapones foliculares. La variante anular, que ocurre casi de forma exclusiva en Japón, consiste en placas anulares e inflamatorias41. Entre las manifestaciones raras se encuentran el lupus profundo y la poiquilodermia generalizada, con erosiones y placas de alopecia42 , 43.
FIGURA 19-2.
Placas anulares de descamación del lupus eritematoso neonatal pareciendo una tiña corporal.
FIGURA 19-3.

Placas atróficas y ojos de mapache en la cara de dos gemelos con lupus neonatal.
Las lesiones del LEN pueden ser generalizadas, aunque son más comunes en la cara y en el cuero cabelludo, afectando de forma predominante las áreas periorbitarias y malares, lo que con frecuencia redetermina un aspecto de «ojos de mapache» (v. figuras 19-3 y 19-4 ). A menudo la aparición de la erupción se ve precipitada o agravada por la exposición al sol. Sin embargo, la exposición al sol no es estrictamente necesaria, dado que las lesiones pueden ocurrir en áreas protegidas, como son la región del pañal, las palmas y las plantas39 , 44 , 45. Las lesiones cutáneas son transitorias y dejan de aparecer alrededor de los 6 meses, cuando desaparecen los anticuerpos maternos. Las lesiones pueden provocar hipopigmentación y atrofia de la epidermis (v. figura 19-5 )36. Las telangiectasias son una secuela más permanente. Las telangiectasias pueden ser un signo inicial del LEN y aparecer sin venir precedidas de lesiones inflamatorias identificables; se han observado características de la piel marmórea telangiectásica congénita35 , 46. Se ha publicado un caso de LEN con un perfil serológico compatible en lupus inducido por fármacos en un recién nacido cuya madre había sido tratada con α-interferón durante el período de gestación47.
FIGURA 19-4.

«Ojos de mapache» y eritema facial prominente en un lactante con lupus neonatal.
Por cortesía de Dr. Joseph Lam.
FIGURA 19-5.
Cambios atróficos y pigmentarios en un lactante con lupus eritematoso neonatal. Este niño también tenía un bloqueo congénito A-V.
Hallazgos extracutáneos
La manifestación aislada más significativa es el bloqueo cardíaco completo congénito. Más de un 90% de estos casos son debidos al LEN. La mayoría de los pacientes tiene un bloqueo de tercergrado; sin embargo, se ha identificado la progresión a partir de un bloqueo de segundo grado48. El bloqueo cardíaco con frecuencia ya puede detectarse a las 20 semanas de gestación.
También puede ocurrir una enfermedad hepática transitoria, que se manifiesta por hepatomegalia (con un cuadro de colestasis) o elevación de las transaminasas hepáticas35 , 49., 50., 51., y trombocitopenia u otras citopenias aisladas52. Las petequias y la púrpura también se han descrito como un signo de presentación del LEN53. Otros hallazgos menos comunes incluyen trombosis asociada a anticuerpos anticardiolipina, hipocalcemia, paraparesia espástica, neumonitis y miastenia grave transitoria54., 55., 56.. La afectación del sistema nervioso central (SNC) se ha descrito en 10 de 11 lactantes consecutivos diagnosticados de LEN, en la forma de alteraciones en la ecografía o en la TC, que sin embargo no dieron lugar a manifestaciones neurológicas clínicas57. Esta afectación del SNC en el LEN probablemente sea un fenómeno transitorio.
Entre un 30% y un 50% de las madres de lactantes con LEN tiene una conectivopatía, con mayor frecuencia LES o síndrome de Sjögren. Sin embargo, la mayoría son asintomáticas. Se debate sobre el riesgo de estas madres de desarrollar de forma clara enfermedades del tejido conjuntivo, con una incidencia que oscila entre el 2% y más de un 70%36 , 58., 59., 60., 61., 62., 63..
Etiología/patogenia
Está universalmente aceptado que la transmisión placentaria de autoanticuerpos IgG maternos es necesaria para la patogenia del LEN, pero no suficiente64. Los autoanticuerpos implicados con más frecuencia han sido los anti-Ro/SS-A y anti-La/SS-B. Más de un 95% de los lactantes con LEN tienen anticuerpos anti-Ro y un 60%-80% tienen anticuerpos anti-La. Un pequeño subgrupo de lactantes afectados no tienen anticuerpos anti-Ro o anti-La, sino que presentan anti-U1RNP37 , 38. Las madres con títulos altos de anti-Ro y anti-La tienen un mayor riesgo de dar a luz a un lactante con LEN. A pesar de las observaciones iniciales basadas en técnicas de inmunotransferencia o ELISA de que los anticuerpos anti-52 kDa Ro se asociaban a un riesgo mayor de LEN que los anti-60 kDa Ro, las pruebas de laboratorio más precisas con inmunoensayo en línea han mostrado que los autoanticuerpos anti-Ro de 60 kDa son significativativamente más sensibles que los anticuerpos anti-Ro de 52 kDa y anti-La de 48 kDa65. Además, al comparar a las madres de niños con LEN que sólo desarrollan una erupción con los que sufren un bloqueo cardíaco congénito, no se han demostrado diferencias significativas en la prevalencia de ninguno de los tres anticuerpos entre los dos grupos65. Sin embargo, existe una diferencia significativa en el sentido que más madres de niños con bloqueo cardíaco congénito tienen anticuerpos anti-La que las madres con una enfermedad de características similares con niños no afectados65. Lo que es más importante, el nivel medio de anti-La parece ser mayor en las madres de lactantes con bloqueo cardíaco congénito que en las de niños con LEN cutáneo66.
No está claro por qué menos del 5% de las madres con anticuerpos anti-Ro y anti-La dan a luz a un niño afectado y por qué las madres de los niños afectados con frecuencia están asintomáticas a pesar de tener los mismos niveles de anticuerpos. Además, los gemelos son a menudo discordantes para el LEN y el LEN no se produce en todas las gestaciones. Posiblemente existan factores genéticos que faciliten el desarrollo del LEN en niños con anticuerpos maternos del lupus eritematoso. Se ha sugerido una conexión entre la erupción del LEN y el alelo HLA-DRB1*03, así como con el polimorfismo -308A en el gen TNF-α34. De manera alternativa, los microquimerismos maternos y/o de los hermanos pueden desempeñar un papel adicional, dado que se ha publicado que los niveles de microquimerismos se correlacionan con la actividad de la enfermedad67.
Pruebas de laboratorio e histopatología
Los estudios serológicos de los autoanticuerpos en la madre y el lactante demuestran anticuerpos anti-Ro, anti-La y/o anti-U1RNP. También puede haber anti-ADNn, anti-cardiolipina, antinucleares y factor reumatoide. El anticuerpo anti-Sm, muy específico para el lupus eritematoso sistémico, no se encuentra en el LEN. El titulo de anticuerpos en la madre generalmente es más alto que en el lactante, de forma que si sólo se utilizan técnicas de inmunodifusión el resultado en este último puede ser negativo. En estas condiciones deben utilizarse métodos más sensibles, como son el ELISA, la inmunotransferencia o el inmunoensayo en línea. La biopsia cutánea no suele resultar necesaria para el diagnóstico, pero muestra los cambios característicos del lupus eritematoso, que son atrofia epidérmica y vacuolización de la capa basal con escaso infiltrado linfohistiocitario en la unión dermoepidérmica y perianexial. En muchos casos, los cambios histopatológicos en los niños con LEN son mínimos. La inmunofluorescencia directa es positiva en un 50% de los casos, demostrando depósitos granulares de IgG, C3 e IgM en la unión dermoepidérmica. El examen histopatológico del corazón muestra sustitución del nódulo auriculoventricular por fibrosis o tejido graso. También pueden verse fibroelastosis endomiocárdica y conducto arterioso patente68 , 69, así como depósitos de IgG y de complemento70.
Diagnóstico diferencial
El diagnóstico diferencial incluye la rubéola congénita, la infección por citomegalovirus, el eritema anular de la lactancia, la tiña corporal y la dermatitis seborreica. También debe considerarse la sífilis congénita, aunque las lesiones de la mucosa no son una característica del LEN. Pueden encontrarse falsos positivos para la sífilis en el LEN. La telangiectasia y la fotosensibilidad pueden sugerir un síndrome de Bloom o un síndrome de Rothmund-Thomson. Los estudios serológicos en el lactante y en la madre ayudan a confirmar el diagnóstico. Raramente es necesaria la biopsia para realizar estudios histológicos o los estudios de inmunofluorescencia.
Evolución, manejo, tratamiento y pronóstico
A los neonatos con sospecha de LEN se les debe realizar una exploración física completa, electrocardiograma y hemograma completo que incluya plaquetas y pruebas de función hepática (v. cuadro 19-1 ).
Cuadro 19-1. Evaluación recomendada en niños con lupus eritematoso neonatal.
-
•
Exploración clínica de la piel
-
•
Exploración clínica completa
-
•
Biopsia cutánea (si no se logra un diagnóstico clínico)
-
•
Hemograma completo (incluyendo las plaquetas)
-
•
Detección selectiva de la coagulación (incluyendo el anticoagulante lúpico y los anticuerpos antifosfolipídicos)
-
•
Bioquímica sérica (incluyendo la función hepática)
-
•
Detección selectiva de autoanticuerpos: anti-nucleares, anti-Ro, anti-La, anti-RNP
-
•
Electrocardiograma y ecocardiograma. Derivar a un cardiólogo pediátrico
-
•
Ecografía del SNC. Considerar la TC o la RM del cerebro si la exploración clínica está alterada
Las lesiones cutáneas son transitorias. El tratamiento de la enfermedad cutánea consiste en protección solar y en la aplicación de esteroides tópicos. El tratamiento con pulsos de láser puede plantearse en las telangiectasias residuales. El bloqueo cardíaco congénito es permanente. En la mitad de los recién nacidos con bloqueo cardíaco completo es necesaria la implantación de un marcapasos durante el período neonatal32 , 61 , 71. La mortalidad media como consecuencia del bloqueo cardíaco completo en el período neonatal es del 15%; otro 10%-20% fallecen por complicaciones secundarias al marcapasos27 , 32. Algunos lactantes pueden desarrollar una cardiopatía de inicio tardío72., 73., 74..
Para las madres con anticuerpos anti-Ro y anti-La, el riesgo de dar a luz a un lactante con LEN oscila entre el 1%-20%, según si tienen un LES sintomático o asintomático29 , 32. El riesgo de recaída del bloqueo cardíaco completo en embarazos posteriores puede alcanzar un 25%61. Estos embarazos deben ser monitorizados de forma estrecha, con repetidas ecocardiografías fetales75. Si se detectan signos de insuficiencia cardíaca congestiva intrauterina, se puede administrar dexametasona, realizar una plasmaféresis o ambas32 , 76., 77., 78., 79..
A pesar de que el LEN generalmente es autolimitado, en un pequeño subgrupo de pacientes puede desarrollarse LES u otras enfermedades autoinmunitarias/reumatológicas en fases posteriores de la vida62 , 80 , 81. El riesgo exacto es desconocido.
Erupciones por fármacos82., 83., 84., 85.
Las reacciones cutáneas por fármacos son extraordinariamente raras durante el período neonatal dado que la capacidad de generar una respuesta inmunitaria inducida por fármacos parece ser baja en los lactantes86., 87., 88.. Además, la mayoría de las reacciones a fármacos precisan tiempo para la sensibilización, que puede ser de 1 a 4 semanas o incluso más, y una reexposición al fármaco causante de la misma. Finalmente, los recién nacidos y los lactantes jóvenes están menos expuestos a los fármacos que los adultos. Las reacciones adversas a fármacos pueden clasificarse de acuerdo con las características clínicas de la erupción (v. cuadro 19-2 ). Cuando se observe una lesión sospechosa, se debe obtener una historia detallada de las medicaciones, incluyendo los fármacos administrados a la madre, ya que pueden estar presentes en la leche. Las erupciones morbiliformes (v. figura 19-6 ) o maculopapulares (v. figura 19-7 ) son los dos tipos de reacciones más frecuentes en los neonatos, en las que suelen estar implicados los antibióticos (v. figura 19-8 ). Con frecuencia es difícil diferenciar una erupción por fármacos de un exantema viral.
Cuadro 19-2. Características clínicas de las reacciones cutáneas a fármacos.
Maculopapular
Urticarial
Similar a la enfermedad del suero
Síndrome de hipersensibilidad
Eritema multiforme
Necrólisis epidérmica tóxica
Fija medicamentosa
Purpúrica
Vasculítica
Liquenoide
Inducida por luz
FIGURA 19-6.

Erupción cutánea por fármacos debida a procainamida.
FIGURA 19-7.

Erupción maculopapular provocada por diazóxido.
FIGURA 19-8.
Erupción eritematosa extensa provocada por antibióticos sistémicos.
Se ha demostrado que la crema de EMLA, un anestésico local que suele utilizarse frecuentemente en las unidades neonatales, puede producir una erupción purpúrica localizada89 , 90. Este tipo de reacción se ve preferentemente en neonatos y la subsiguiente aplicación de la crema no siempre reproduce las lesiones purpúricas. La metahemoglobinemia es otra complicación del uso de EMLA en este grupo de edad90. Por tanto, la crema EMLA debe utilizarse con precaución en los lactantes que toman medicaciones que puedan inducir metahemoglobina, como son las sulfamidas, el paracetamol, la nitroglicerina, el nitroprusiato y la fenitoína, particularmente en niños con antecedentes de metahemoglobinemia.
La vancomicina, un antibiótico utilizado frecuentemente en los prematuros con infecciones nosocomiales por Staphylococcus epi dermidis, puede producir shock y erupción en los recién nacidos (síndrome del niño rojo)91., 92., 93.. Esta reacción se caracteriza por la aparición de una erupción intensa, macular, eritematosa sobre el cuello, cara y parte superior del tronco, poco tiempo después de terminar la infusión. Puede asociarse a hipotensión y shock. Esta reacción se resuelve espontáneamente en cuestión de horas. Frecuentemente se asocia a la infusión rápida; sin embargo, prolongar la infusión por encima de 1 hora no elimina por completo el riesgo92.
Los recién nacidos con SIDA tienen una mayor susceptibilidad a las reacciones por fármacos94 , 95. En los pacientes con SIDA las reacciones al trimetoprim/sulfametoxazol pueden ser graves, constituyendo una amenaza para la vida.96.
En la lactancia se ha descrito exantema fijo medicamentoso en el escroto y en el pene, con eritema y edema como consecuencia del clorhidrato de hidroxicina97. Sin embargo, el clorhidrato de hidroxicina raramente se administra en el período neonatal por el riesgo de efectos antimuscarínicos, como son nerviosismo y excitación.
Una reacción similar a la enfermedad de suero es una forma de reacción a los fármacos que tiene lugar predominantemente durante la infancia y se han publicado casos en niños de menos de 5 meses de edad98. Se caracteriza por fiebre, erupción urticarial y artralgias. También puede haber linfoadenopatias. A diferencia de la verdadera enfermedad del suero, no hay inmunocomplejos, vasculitis o afectación renal. El fármaco que se ha visto implicado con mayor frecuencia es el cefaclor98., 99., 100.. Este tipo de reacción puede afectar a lactantes con etiologías desconocidas o presumiblemente víricas (v. figura 19-9 ).
FIGURA 19-9.

Lactante con erupción similar a la enfermedad del suero.
El sindrome de reacción de hipersensibilidad es una grave reacción a los fármacos caracterizada por fiebre, erupción cutánea, linfadenopatías y afectación de los órganos internos, especialmente el hígado101 , 102. Los fármacos que se han visto implicados con mayor frecuencia son los antiepilépticos, por lo que no es raro en niños. Se ha publicado un caso mortal en un lactante de 3 meses y un caso en un lactante prematuro103 , 104.
La necrólisis epidérmica tóxica (NET) es extremadamente rara en los recién nacidos. Los casos de NET en los recién nacidos se han asociado a antibióticos y fenobarbital105. Todos los casos descritos han sido mortales.
El bromoderma vegetante es una reacción a los bromuros caracterizada por pápulas y pústulas coalescentes que forman lesiones inflamatorias vegetantes o seudotumores. Generalmente afecta al cuero cabelludo, la cara y las piernas. La mayoría de los casos de bromoderma vegetante han sido descritos en lactantes tras la ingestión de jarabes y de soluciones que contenían bromuro, de efectos sedantes y expectorantes, o con el espasmolítico bromuro de escopolamina106. La erupción desaparece tras suspender el tratamiento con bromuro. El riesgo de intoxicación sistémica, conocido como bromismo, hace aconsejable evitar los bromuros en los recién nacidos y en los lactantes.
Se han descrito otras publicaciones anecdóticas de toxicodermias en los lactantes muy jóvenes o en los recién nacidos, como son la erupción papulosa por el factor estimulador de colonias de granulocitos para la obtención de células pluripotenciales (v. figura 19-10 )107, una reacción liquenoide debida al tratamiento con ácido ursodesoxicólico para la hepatitis neonatal y un exantema maculopapuloso por el diazóxido utilizado para el tratamiento de la hiperglucemia neonatal (v. figura 19-7).
FIGURA 19-10.
Erupción papulopustulosa debida a CSF-G.
Urticaria
La urticaria (habones) ocurre a menudo en los niños, pero es rara antes de los 6 meses de edad y todavía lo es más en el período neonatal82 , 109., 110., 111., 112., 113., 114., 115., 116., 117.. La urticaria suele ser esporádica; sin embargo, se han descrito formas familiares con herencia autosómica dominante de muchas de las formas de urticaria, como son el dermografismo, la urticaria por calor, la urticaria por frío y la urticaria vibratoria.
La urticaria puede dividirse en dos tipos: aguda (dura menos de 6 semanas) y crónica (dura más de 6 semanas). Esta división arbitraria tiene significado pronóstico y etiopatogénico. En los lactantes, la urticaria es muy rara111 , 118. Las urticarias físicas representan un subgrupo especial de urticarias en las cuales los habones se inducen mediante diferentes estímulos físicos119. Entre ellos se incluyen los dermografismos, el frío, la presión, los colinérgicos, los acuagénicos, vibratorios y solares.
Hallazgos cutáneos
La urticaria se caracteriza por habones transitorios pruriginosos y edematosos (v. figura 19-11 ). Por definición, las lesiones duran menos de 24 horas. Las lesiones pueden ocurrir en la piel y en las mucosas. El angioedema o urticaria gigante es una entidad estrechamente relacionada, en la cual hay tumefacción de los tejidos subcutáneos profundos y edema difuso de los párpados, genitales, labios y lengua. Puede verse solo o más frecuentemente asociado a la urticaria «común». En el niño la urticaria muestra algunas características específicas. Los habones tienden a agruparse, adoptando curiosas formas policíclicas, serpiginosas o anulares (urticaria figurada, v. figura 19-12 ; o urticaria anular, v. figura 19-13 ) y puede transformarse en hemorrágica109 , 112. Con frecuencia el edema es pronunciado y doloroso. Estas características dan una imagen dramática a la erupción. En niños puede no aparecer prurito. La urticaria puede ser más frecuente y recidivante en pacientes atópicos112 , 115.
FIGURA 19-11.

Urticaria generalizada que siguió a la administración de la DPT y la polio.
FIGURA 19-12.

Lesiones urticariales policíclicas asociadas a la infusión de prostaglandinas E2.
FIGURA 19-13.

Urticaria anular de etiología desconocida.
Hallazgos extracutáneos
La urticaria aguda puede asociarse a signos de shock anafiláctico. En los casos de angioedema, también pueden aparecer dolor abdominal, diarrea, vómitos, dificultad respiratoria y dolor articular.
Etiología/patogenia82,113
La urticaria se desarrolla como consecuencia de un aumento en la permeabilidad de los capilares y de las pequeñas vénulas, lo que da lugar a extravasación del líquido hacia el espacio extravascular. Son responsables de estos cambios la activación de los mastocitos y la consiguiente liberación de los mediadores, entre los cuales la histamina es la mejor conocida. Son muchos los desencadenantes (secretagogos) que pueden iniciar la desgranulación de los mastocitos a través de los receptores de las membranas celulares, por un mecanismo dependiente de IgE o por la activación del complemento (secretagogos inmunológicos).
En niños, el agente que con mayor frecuencia provoca esta reacción son los fármacos y las infecciones, responsables en conjunto de un 40% de los casos de urticaria aguda112 , 115 , 117. Los antipiréticos (principalmente el ácido acetilsalicílico) y los antibióticos (amoxicilina, macrólidos y cefalosporinas) son los fármacos incriminados con mayor frecuencia. Las urticarias relacionadas con las comidas se asocian a la atopia112. La alergia a la leche de vaca es una de las principales causas de urticaria en lactantes, ya que aparece en el 6%-35% de los casos de intolerancia a la leche de vaca116 , 120 , 121.
Diagnóstico
El diagnóstico de urticaria se establece por la clínica. El examen histológico de una muestra de biopsia cutánea muestra dilatación vascular, edema e infiltrado inflamatorio perivascular compuesto por células linfohistiocitarias, células polimorfonucleares y, de forma más específica, eosinófilos. Pueden predominar los neutrófilos.
Para la evaluación de la urticaria generalmente no se necesitan pruebas de laboratorio. En algunos pacientes los niveles de IgE pueden estar elevados. No es necesaria una búsqueda exhaustiva de la causa subyacente si no se determina con la historia clínica. La velocidad de sedimentación eritrocitaria puede ser suficiente como prueba de cribado en las urticarias crónicas, dado que generalmente está elevada en las enfermedades asociadas a la urticaria crónica (p. ej., enfermedades del colágeno vascular). En el 10%-15% de los pacientes no se identifica la causa. No son fiables las pruebas cutáneas para descubrir los alérgenos sospechados.
Diagnóstico diferencial
La urticaria en los lactantes con frecuencia se diagnostica como eritema multiforme, edema hemorrágico agudo, eritema anular de la lactancia o enfermedad de Kawasaki. En un lactante con urticaria y dermografismo también debe considerarse la posibilidad de una mastocitosis cutánea sin lesiones cutáneas visibles122. Se debe plantear NOMID/CINCA (v. más adelante) en los lactantes jóvenes con urticaria. El predominio de los neutrófilos en las biopsias de piel en niños con NOMID puede ayudar en el diagnóstico diferencial, si bien no es específico al 100%. En caso de duda, en la actualidad hay pruebas genéticas para el NOMID.
Evolución, manejo, tratamiento y pronóstico
A pesar de sus síntomas alarmantes, en la lactancia temprana la urticaria generalmente es benigna. La excepción es el síndrome cutáneo, neurológico y articular infantil crónico (CINCA) (v. más adelante) y las urticarias físicas heredadas, que pueden persistir toda la vida. Si se necesita medicación, los antihistamínicos, como la difenhidramina o la hidroxicina, son los fármacos de elección. Sin embargo, los recién nacidos tienen mayor susceptibilidad a los efectos secundarios antimuscarínicos, como la excitación del sistema nervioso central que produce convulsiones. Los corticoesteroides sistémicos deben reservarse para casos de urticaria intratable.
Urticarias físicas familiares
Para muchas de las urticarias físicas se han descrito variantes autosómicas dominantes. Aunque son poco frecuentes, estos casos familiares empiezan pronto, incluso inmediatamente después de nacer, y persisten durante toda la vida, en general con una clínica cada vez más grave. La patogenia exacta de muchas de las urticarias físicas es desconocida.
La urticaria por frío familiar (UFF) es una alteración autosómica dominante caracterizada por el desarrollo de habones con sensación de quemadura y con frecuencia dolor e hinchazón de las articulaciones, rigidez, escalofríos e incluso fiebre después de la exposición al frío, especialmente en combinación con mal tiempo y clima de invierno123., 124., 125., 126.. Las lesiones aparecen en las áreas expuestas y luego se generalizan. Durante los ataques puede haber incluso leucocitosis. La reacción puede retrasarse hasta 6 horas después de la exposición al frío. A diferencia de lo que sucede en la urticaria por frío adquirida, la reacción no puede desencadenarse mediante la prueba del cubito de hielo, sino que el paciente debe verse sometido a temperaturas ambientales frías o a la inmersión en agua fría. En la biopsia cutánea predomina el infiltrado de neutrófilos. Los síntomas tienden a mejorar con la edad. La respuesta a los bloqueantes H1 y H2 y al ketotifeno es mala. El beneficio del tratamiento con estanozolol es limitado127. La UFF también se ha asociado a amiloidosis y sordera en el síndrome de Muckle-Wells (SMW)128. Recientemente se ha demostrado que tanto la UFF como el SMW son consecuencia de mutaciones en el gen CIAS1 (criopirina) y en realidad son la misma alteración y pueden compartir la misma mutación genética129. La UFF y el SMW también son enfermedades alélicas con el síndrome CINCA (v. más adelante), que también es consecuencia de una mutación en el gen CIAS1 130.
El dermografismo familiar (autosómico dominante) ha sido descrito en una sola familia extensa131. En los neonatos el dermografismo también puede ser una manifestación de una mastocitosis cutánea difusa «silente»122 , 132. La urticaria vibratoria también es una urticaria física autosómica dominante, la cual se desarrolla después de estímulos vibratorios repetidos o de estirar la piel133 , 134. La necesidad de traumatismos repetidos permite diferenciarla del dermografismo. La urticaria familiar acuagénica y la asociada a calor generalmente tienen su inicio durante la niñez135., 136., 137..
Síndrome crónico infantil neurológico cutáneo y articular (CINCA)138., 139., 140., 141.
El síndrome CINCA, también conocido como enfermedad inflamatoria sistémica de inicio neonatal (NOMID), es una enfermedad inflamatoria sistémica crónica de inicio neonatal caracterizada por una erupción cutánea, artropatía y manifestaciones del SNC. Los hallazgos cutáneos son los signos de presentación. La enfermedad sigue una evolución crónica con exacerbaciones febriles agudas, adenopatías y hepatoesplenomegalia. Dos tercios de los pacientes son prematuros.
Hallazgos cutáneos
Generalmente la erupción cutánea es la primera manifestación de la enfermedad y está presente al nacer o se desarrolla durante los 6 primeros meses de vida. Se caracteriza por máculas y pápulas urticariales generalizadas, evanescentes que en un mismo día emigran y aumentan y disminuyen de intensidad (v. figura 19-14 ). La erupción es persistente, si bien se nota una exacerbación de las lesiones cutáneas en los episodios de recrudescencia. Después de la exposición al sol las lesiones pueden ser pruriginosas, pero en general son asintomáticas140 , 141. En un paciente se ha descrito lengua geográfica y úlceras orales141.
FIGURA 19-14.

Enfermedad inflamatoria multisistémica de inicio neonatal (NOMID).
Hallazgos extracutáneos
La artropatía simétrica o asimétrica es otro hallazgo constante que puede ser grave en la mitad de los pacientes. Con frecuencia, en las primeras semanas está ausente, si bien generalmente se desarrolla durante el primer año140 , 142. La gravedad de la artropatía se correlaciona con el inicio temprano de los síntomas. Las rodillas son las articulaciones que se afectan con más frecuencia, seguidas de los tobillos y los pies, codos, muñecas y manos. La tumefacción de las articulaciones y el dolor son más intensos durante las recrudescencias. A la palpación es característica la consistencia ósea como consecuencia de la afectación de la epífisis y del cartílago de crecimiento y el sobrecremiento de la rótula. Se pueden producir contracturas articulares y graves deformidades.
Los síntomas y signos neurológicos, como son la cefalea, los vómitos y las convulsiones, se desarrollan a diferentes edades. También es común la afectación intelectual. Se han descrito tanto espasticidad como hipotonía. La afectación ocular es un hallazgo inconstante. La característica más común es el papiledema con o sin atrofia del nervio óptico. Las otras manifestaciones oculares incluyen uveítis, queratitis, conjuntivitis y coriorretinitis143. Estos cambios pueden dar lugar a ceguera total durante la edad adulta. También son comunes la sordera neurosensorial progresiva y la afonía.
Los niños afectados tienen un fenotipo característico. Hay un retraso de crecimiento progresivo y aumento del perímetro craneal con prominencia frontal. El cierre de las fontanelas está retrasado. Durante el período neonatal se puede desarrollar ictericia, especialmente en los pacientes con artropatía severa140.
Etiología/patogenia
Las mutaciones en el gen CIAS1 han sido identificadas en un 60% de los pacientes con el síndrome de CINCA144 , 145. El CIAS1 codifica una proteína, denominada criopirina, que está relacionada con la regulación de la apoptosis y de las vías de señalización inflamatorias145. Se ha propuesto que la urticaria familiar por frío y el CINCA representan los dos extremos de una misma enfermedad, definido por la magnitud de la expresión del fenotipo145. Existe una considerable superposición clínica entre ambas entidades.
Pruebas de laboratorio, hallazgos radiológicos e histopatología
Los hallazgos no específicos típicos de un proceso inflamatorio crónico incluyen la anemia microcítica; leucocitosis con elevación del recuento de los neutrófilos y de los eosinófilos, aumento del recuento de plaquetas y de la velocidad de sedimentación, y de los reactantes de fase aguda; e hiperglobulinemia G, A o M. Generalmente el factor reumatoide y los anticuerpos antinucleares son negativos. Puede haber una ligera elevación de las transaminasas. El examen del LCR muestra pleocitosis y elevación de las proteínas.
Los estudios radiológicos de las articulaciones afectadas muestran epífisis espiculadas irregulares y aumentadas de tamaño, con una apariencia trabecular engrosada140 , 142. Hay neoformación de hueso perióstico y son frecuentes las alteraciones en el cartílago. Con el tiempo, los huesos largos se abomban y se produce un acortamiento de la longitud de las diáfisis. La TC de la cabeza ha demostrado hidrocefalia y atrofia cerebral.
El examen histopatológico de la piel muestra neutrofilia intersticial y perivascular141 , 142. Se ha descrito una hidradenitis ecrina neutrofílica141. Las biopsias de los ganglios linfáticos, del hígado y de las sinoviales muestran signos inespecíficos de inflamación crónica.
Diagnóstico diferencial
El NOMID debe diferenciarse de la artritis reumatoide sistémica juvenil. Las principales diferencias son su inicio neonatal, la erupción persistente, la corta duración de los brotes de fiebre, la ausencia de rigidez matutina y la afectación del sistema nervioso central. La artropatía es deformante y los hallazgos radiológicos de epífisis agrandadas y desorganizadas es distintivo. Además, la respuesta a los antiinflamatorios no esteroideos es mala. También debe descartarse una urticaria y el predominio de eosinófilos en la biopsia cutánea puede ser un dato orientativo.
Evolución, manejo, tratamiento y pronóstico
La enfermedad sigue una evolución crónica con exacerbaciones agudas febriles. Ocasionalmente puede causar la muerte en la primera o segunda décadas de la vida. Para aliviar el dolor, pueden ser eficaces los fármacos antiinflamatorios, pero no alteran la evolución de la enfermedad. La prednisona ha sido paliativa en dosis que oscilan entre 0,5 y 2 mg/kg/día142. Se ha intentado tratamientos con clorambucil y penicilamina con un éxito modesto146 , 147. La talidomida ha mostrado efectos beneficiosos en un solo paciente148. Otras alternativas incluyen metrotrexato, el antagonista del receptor de IL-1 anakinra149., 150., 151., 152. y el agente anti-TNF-α etanercept153.
Eritema multiforme
El eritema multiforme (EM) es una enfermedad aguda, autolimitada de la piel y de las mucosas154., 155., 156.. Se ha considerado como un espectro de alteraciones, designadas como EM minor, que consiste en la afectación cutánea o de la piel y la boca, y EM major (síndrome de Stevens-Johnson; SSJ), que afecta por lo menos dos membranas mucosas con lesiones cutáneas variables. Algunos autores incluyen la necrólisis epidérmica tóxica dentro de este espectro, como una forma grave de SSJ. Las últimas evidencias sugieren que el EM y el SSJ tienen características diferentes y distintos factores precipitantes, por lo que tal vez es mejor abolir los términos EM major y EM minor 157 , 158. El EM es una enfermedad común en niños155, aunque es extremadamente rara en el período neonatal94 , 155 , 159., 160., 161., 162.. La necrólisis epidérmica tóxica se comenta en el capítulo 10.
Hallazgos cutáneos
El prototipo de las lesiones en la EM son pápulas eritematosas, edematosas de 1-3 cm que desarrollan unas vesículas oscuras, purpúricas o con centro necrótico. A menudo se ve un anillo edematoso pálido rodeado por un anillo externo eritematoso. Estos cambios de color concéntricos originan la típica lesión en diana o iris. En muchos casos sólo se ven dos zonas, con un simple anillo alrededor de la pápula central (lesión en diana atípica). Las lesiones se distribuyen simétricamente y en zonas acras sobre las superficies extensoras de las extremidades. Pueden extenderse al tronco, las superficies flexoras, palmas y plantas. En niños, son comunes las lesiones en la cara y oídos, y raras en el cuero cabelludo163 (v. figura 19-15 ).
FIGURA 19-15.
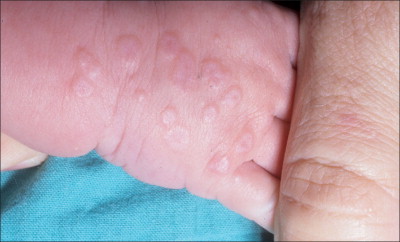
Lesiones diana en un recién nacido con eritema multiforme.
En el SSJ, las lesiones se localizan de forma más central, predominando en el tronco. Las dianas son atípicas y generalmente son planas. Las lesiones individuales tienden a fusionarse en grandes placas. Puede haber áreas de desprendimiento de la piel, si bien en general se afecta menos del 10% de la superficie corporal. Con frecuencia en la EM se observan lesiones en las mucosas y son un requisito para el diagnóstico de SSJ. La afectación de las mucosas se caracteriza por eritema o ampollas que rápidamente evolucionan hacia erosiones confluentes con formación de seudomembranas. La mucosa oral y las conjuntivas son las que se afectan con mayor frecuencia, aunque también se pueden afectar las mucosas genitales, anales, faríngea y del tracto respiratorio alto. El número de mucosas afectadas ha sido considerado como uno de los principales factores para diferenciar la EM y el SSJ.
Hallazgos extracutáneos
En la EM ocasionalmente puede haber leves signos prodrómicos inespecíficos como tos, rinitis y febrícula. En el SSJ es común la fiebre, las artralgias y la postración.
Etiología/patogenia
La EM ha sido considerada como un fenómeno de hipersensibilidad a múltiples factores precipitantes, como son los agentes infecciosos o los fármacos. Han sido identificados de forma bien documentada tres factores etiológicos: para el eritema multiforme las infecciones por herpes simple y por Mycoplasma y para el SSJ los fármacos. El herpes simple (VHS-1 o VHS-2) se considera responsable de más del 80% de los casos de EM en niños, incluso en aquellos casos en los que la infección no es aparente164. El EM asociado a VHS aparece 1-3 semanas después de las lesiones herpéticas y con frecuencia es recurrente. Sin embargo, no todos los episodios repetidos de herpes se siguen de EM. El ADN específico del VHS ha sido detectado por PCR e hibridación in situ en las lesiones de un gran número de niños con EM, independientemente de que este sea idiopático o claramente asociado a VHS164.
La intolerancia a la leche de vaca ha sido descrita como una causa de eritema multiforme en un neonato159. Los fármacos son las principales causas de SSJ y se ha relacionado con más frecuencia a las sulfonamidas, la fenilbutazona, la fenitoína y los derivados de la penicilina163. En un recién nacido y en dos lactantes, la única causa conocida posible de eritema multiforme fueron las vacunas162 , 165.
Pruebas de laboratorio e histopatología
En casos de mucha afectación puede verse una elevación de la velocidad de sedimentación, leucocitosis y ligero aumento de las transaminasas. En el SSJ pueden observarse alteraciones hidroelectrolíticas e hipoproteinemia. En los casos relacionados con fármacos puede verse eosinofilia.
El examen histopatológico de las lesiones tempranas muestra un infiltrado con una banda de linfocitos en la unión dermoepidérmica, con exocitosis y queratinocitos necróticos cerca de los linfocitos («necrosis de las células satélite»). Hay vacuolización de la capa basal con hendiduras focales en la unión dermoepidérmica. La parte superior de la dermis es edematosa. Con el tiempo, se produce una necrosis confluente más extensa de la epidermis con formación de ampollas subepidérmicas. En la EM predomina un infiltrado liquenoide, mientras que en el SSJ predomina la necrosis epidérmica.
Diagnóstico diferencial
Los casos típicos de EM o de SSJ raramente se confunden con otras entidades. En algunos casos hay que considerar la vasculitis urticarial. La enfermedad de Kawasaki también puede producir lesiones en diana; sin embargo, los hallazgos adicionales pueden permitir la diferenciación. Las reacciones parecidas a la enfermedad del suero, con frecuencia asociadas al uso de cefaclor u otros antibióticos, o incluso sin una etiología conocida, también puede producir lesiones en diana (v. figura 19-9).
Evolución, manejo, tratamiento y pronóstico
Generalmente el EM es autolimitado. Las lesiones individuales se curan en 1-2 semanas, con hiperpigmentación residual. La forma preferida de tratamiento es la de soporte. Se deben buscar las causas subyacentes. Está indicado el tratamiento de la infección subyacente y el cese de la administración de fármacos que no sean esenciales. Los corticoesteroides no son necesarios e incluso pueden empeorar una infección concomitante166 , 167. En el EM asociado a VHS puede ser beneficiosa la intervención temprana o incluso el tratamiento con aciclovir168.
El SSJ tiene un pronóstico menos favorable, con una tasa de mortalidad del 5%-15% si no se trata. En el SSJ el uso de corticoesteroides es más controvertido166 , 167 , 169 , 170. No hay ningún estudio controlado que haya probado su eficacia y en algunos estudios los pacientes tratados con corticoesteroides han tenido peor pronóstico171. Los corticoesteroides pueden predisponer a las infecciones secundarias y suprimir los signos de sepsis. El tratamiento de soporte es extremadamente importante.
Síndrome de sweet
El síndrome de Sweet, o dermatosis neutrofílica febril aguda, es una enfermedad benigna caracterizada por placas dolorosas y elevadas, con fiebre y leucocitosis periférica y hallazgo histológico de un infiltrado dérmico de leucocitos polimorfonucleares con una rápida respuesta a los corticoesteroides172., 173., 174., 175., 176.. Sólo se han descrito unos cuantos casos pediátricos177., 178., 179., 180., 181., 182., 183., 184., 185., 186., 187., 188., 189., 190., el más joven a las 5 semanas de vida180. Se han publicado dos hermanos de 2 semanas de vida con el síndrome de Sweet188.
Hallazgos cutáneos
Las lesiones del síndrome de Sweet tienen un inicio agudo, explosivo y se caracterizan por placas o nódulos eritematosos, indurados, dolorosos, con un tamaño variable entre 0,5 y 4 cm (v. figura 19-16 ). En fases más avanzadas pueden aparecer pequeñas pústulas. Los bordes pueden estar elevados, ser mamelonados o incluso vesiculosos. Algunas lesiones pueden mostrar un aclaramiento central, formando placas anulares (v. figura 19-17 ). Las lesiones son múltiples y están distribuidas por la cara y extremidades o, más raramente, por el tronco. Sin tratamiento, tienden a curar espontáneamente en unos meses. En algunos pacientes, especialmente en niños, las lesiones se curan con áreas de cutis laxa secundaria, lo que se conoce como síndrome de Marshall179 , 189 , 191.
FIGURA 19-16.

Lesiones nodulares del síndrome de Sweet con costras centrales.
FIGURA 19-17.

Progresión de las lesiones del síndrome de Sweet en el mismo paciente de la figura 19-16. Las placas y los nódulos se han aplanado y hay aclaramiento central.
Hallazgos extracutáneos
Es característica la fiebre alta con picos, pero puede estar ausente en un 50% de los casos176. Pueden aparecer artralgias o una artritis simétrica y en un tercio de los pacientes se puede ver conjuntivitis o iridociclitis176. También se ha descrito la afectación renal que se manifiesta por proteinuria y hematuria, así como afectación de los pulmones con infiltrados visibles en las radiografías. En raros casos puede haber afectación del sistema nervioso central, con cefalea, convulsiones o alteraciones en la conciencia. En estos casos el líquido cefalorraquídeo muestra pleocitosis con predominio de linfocitos192.
Etiología/patogenia
La patogenia es desconocida. Muchos de los pacientes publicados habían presentado antes del cuadro una infección del tracto respiratorio o títulos elevados de antiestreptolisina O176. Un 10% de los casos se han visto asociados a procesos malignos, especialmente leucemias mieloides o mielomonocíticas193. El síndrome de Sweet también se ha asociado a tumores sólidos, enfermedad inflamatoria intestinal, conectivopatías y enfermedad granulomatosa crónica o también puede ocurrir como un efecto adverso a fármacos194., 195., 196., especialmente con el factor estimulador de la formación de colonias de granulocitos197 o después de la vacunación198. Debido a estas asociaciones y a la rápida respuesta al tratamiento con corticoesteroides, se cree que el síndrome de Sweet representa una reacción de hipersensibilidad frente a los agentes infecciosos o antígenos tumorales.
Pruebas de laboratorio e histopatología
La elevación de la velocidad de sedimentación y la leucocitosis periférica son fenómenos acompañantes frecuentes. También pueden aparecer en ocasiones eosinofilia, anemia microcítica, elevación leve de las enzimas hepáticas y alteraciones en el análisis de orina. En algunos casos se han detectado anticuerpos anticitoplasma de neutrófilos176. En un caso de síndrome de Marshall se detecto un déficit de α1-antitripsina191.
Los cambios histopatológicos son diagnósticos173. Hay un infiltrado perivascular denso compuesto casi de forma exclusiva por neutrófilos. La dermis aparece edematosa y pueden formarse ampollas subepidérmicas. Puede verse espongiosis, exocitosis y vesiculación intraepitelial. Hay tumefacción endotelial y polvo nuclear, pero es típico que no exista una vasculitis verdadera.
Diagnóstico diferencial
Las lesiones del síndrome de Sweet inicialmente pueden parecerse a las de EM o del edema agudo hemorrágico. Las lesiones de las extremidades inferiores pueden recordar al eritema nudoso, pero las lesiones más típicas del síndrome de Sweet suelen afectar a otras localizaciones.
Evolución, manejo, tratamiento y pronóstico
El síndrome de Sweet es una entidad benigna, que sin embargo puede ser un marcador de una enfermedad maligna. Si se deja sin tratamiento, se resuelve de forma espontánea al cabo de unas semanas o meses. Las recaídas son comunes. El síndrome de Marshall tiene peor pronóstico, debido al desarrollo de elastólisis en los pulmones y por la afectación cardiovascular.
Los corticoesteroides orales son el tratamiento de elección y generalmente consiguen una pronta respuesta199. La administración de yoduro potásico ha sido exitosa en unos pocos casos200, igual que la administración de colchicina201, dapsona202, clofacimina189 e inmuglobulinas intravenosas184.
Enfermedad de kawasaki
La enfermedad de Kawasaki es una vasculitis aguda que afecta a las arterias musculares de tamaño pequeño y mediano, especialmente las arterias coronarias de los niños pequeños. En el pasado, muchos casos se denominaron arteritis nudosa. La enfermedad se caracteriza por fiebre que dura por lo menos 5 días, conjuntivitis no purulenta, un exantema polimorfo, eritema e hinchazón de las manos y de los pies, cambios inflamatorios en los labios y en la cavidad oral y una adenopatía cervical aguda no purulenta203., 204., 205., 206.. Los aneurismas o ectasia de las arterias coronarias se desarrollan en el 15%-25% de los niños no tratados y puede dar lugar a cardiopatía isquémica o muerte súbita.
La enfermedad de Kawasaki afecta de forma predominante a niños de menos de 5 años y tiene un pico de incidencia entre los 9 y los 11 meses207., 208., 209.. Es poco frecuente antes de los 6 meses de edad, si bien se han publicado casos en pacientes de menos de 2 semanas210., 211., 212.. Los niños se afectan 1,5 veces más que las niñas. La enfermedad de Kawasaki es una enfermedad endémica con agrupación epidémica y geográfica. Hay un predominio estacional al final del invierno y en la primavera, si bien esto puede diferir según los países213 , 214. Es más común en Japón, donde la incidencia anual es de 112 casos por 100.000 niños de menos de 5 años y está creciendo de forma continua215 , 216. Se han descrito casos familiares en las personas que habitan el mismo hogar217. La tasa de recaída es del 3% y algunos pacientes presentan dos o más recaídas.
Hallazgos cutáneos205,218
La piel se ve afectada en un 99% de los pacientes. El primer signo con frecuencia consiste en eritema difuso e induración dolorosa de las manos y de los pies. Entre 1 y 3 semanas después del inicio de la erupción esta empieza a descamarse en la parte distal de las uñas y la pérdida de piel puede extenderse hasta afectar toda la palma y las plantas. Puede presentarse una depresión horizontal de la placa ungueal (líneas de Beau).
El exantema polimorfo del tronco y de la parte proximal de las extremidades generalmente aparece a los 5 días del inicio de la fiebre (v. figura 19-18 ). Es una erupción inespecífica maculopapular o morbiliforme difusa, aunque también puede ser urticarial, escarlatiniforme o incluso pustulosa. No se han descrito erupciones ampollosas o vesiculosas. La erupción generalmente se localiza en el periné, lo que es una característica distintiva de esta fase precoz y se descama en 48 horas, precediendo a la descamación de la punta de los dedos de las manos y de los pies219 (v. figura 19-19 ). Se han descrito placas tipo pustuloso y psoriasis pustulosa, bien durante la fase aguda o durante la convalecencia de la enfermedad, lo que apoya que se trate de un proceso mediado por un superantígeno220., 221., 222., 223..
FIGURA 19-18.

Erupción morbiliforme en un lactante con enfermedad de Kawasaki.
FIGURA 19-19.

Erupción descamativa perineal precoz de la enfermedad de Kawasaki.
Los cambios en los labios y en la mucosa oral incluyen eritema, tumefacción y fisuras de los labios, lengua en frambuesa y eritema de la orofaringe. No se ven ulceraciones orales ni exudado faríngeo.
En los lactantes de menos de 6 meses se ha observado acrocianosis intermitente209, así como gangrena periférica224. Se han publicado casos de necrosis en el sitio donde previamente se había inyectado la BCG225., 226., 227., 228..
Hallazgos extracutáneos205,206,218,229
Una característica cardinal e inicial de la enfermedad es la fiebre de por lo menos 5 días de duración. Su inicio es abrupto y es alta, con picos de temperatura, generalmente >39 °C y en muchos casos >40 °C con varios picos diarios (fiebre remitente). En ausencia de un tratamiento apropiado, la fiebre persiste por término medio 11 días, si bien puede continuar durante 3-4 semanas y raramente más tiempo.
La inyección conjuntival bilateral no exudativa, que afecta principalmente a la conjuntiva bulbar, se inicia poco después del inicio de la enfermedad y ya puede haberse resuelto en el momento de la primera consulta. Mediante el examen con lámpara de hendidura es común observar una uveítis anterior, si bien raramente es sintomática.
La adenopatía cervical es el signo diagnóstico menos común, con una prevalencia del 65%. Generalmente es unilateral y se limita al triángulo cervical anterior. Los ganglios con frecuencia son duros, no fluctuantes y sólo ligeramente dolorosos.
Las alteraciones cardíacas son la principal causa de mortalidad y morbilidad. Puede afectarse el pericardio, miocardio, endocardio y las arterias coronarias. La miocarditis puede manifestarse en la fase aguda, pudiendo dar lugar a arritmias como consecuencia de la isquemia, insuficiencia cardíaca y afectación valvular, generalmente de la válvula mitral. Ocasionalmente puede haber un síndrome de bajo gasto cardíaco y shock. La auscultación cardíaca de un niño o un lactante con enfermedad de Kawasaki en la fase aguda muestra un precordio hiperdinámico, taquicardia, ritmo de galope y un soplo no patológico. También puede auscultarse un soplo pansistólico de insuficiencia mitral. La electrocardiografía puede mostrar arritmias, prolongación del intervalo PR y cambios inespecíficos del ST y de las ondas T. El ecocardiograma puede mostrar un derrame pericárdico en un 30% de los casos. Sin tratamiento, se desarrollan aneurismas coronarios en un 20% de los casos y se detectan con más frecuencia entre los 10 días y las 4 semanas del inicio de la enfermedad. Entre los factores de riesgo para el desarrollo de los aneurismas coronarios se incluye edad menor al año, sexo masculino, fiebre de más de 2 semanas, fiebre recurrente y retraso en el tratamiento. Los aneurismas también pueden desarrollarse en las arterias sistémicas de tamaño medio y dar lugar a gangrena periférica224.
En las primeras semanas de la enfermedad puede haber poliartritis y artralgias. Afecta a las articulaciones pequeñas y también a las grandes. La irritabilidad generalmente es prominente. También puede haber obnubilación y otros signos de meningitis aséptica. Son comunes los síntomas abdominales, como vómitos, diarrea y dolor. En casos raros puede haber dolor abdominal agudo, similar al del abdomen quirúrgico, como primer signo de la enfermedad. Con frecuencia hay una hepatitis leve, así como distensión aguda de la vesícula biliar (hidropesía vesical).
Raramente puede haber una parálisis facial periférica unilateral. Se han observado síntomas respiratorios como consecuencia de la formación de los nódulos pulmonares, infiltrados o el derrame pleural230., 231., 232.. Son hallazgos raros la hinchazón testicular, el síndrome hemofagocítico233 y una hipoacusia neurosensorial transitoria para las frecuencias altas (20-35 dB)234.
Etiología/patogenia235., 236., 237.
Los datos clínicos y epidemiológicos sugieren que la enfermedad de Kawasaki tiene características de una enfermedad infecciosa en un huésped susceptible inmunológicamente y de una vasculitis inmunitaria. Son muchos los agentes etiológicos, que van desde bacterias como las Propionibacterium, Staphylococcus, Streptococcus, Chlamydia y Yersinia, a virus como el Epstein-Barr, parvovirus, adenovirus, retrovirus y el nuevo coronavirus humano que se han asociado a la enfermedad de Kawasaki en diferentes focos geográficos; no obstante, ninguno ha mostrado una relación de forma consistente235. Gran parte del continuo debate en la literatura concierne al hecho de si la enfermedad de Kawasaki está relacionada con un superantígeno o un antígeno convencional235 , 237. Las evidencias sobre una enfermedad mediada por un superantígeno incluyen la identificación de organismos productores de superantígenos, aislamiento de superantígenos bacterianos o identificación de las características de la activación de un superantígeno, como son la expansión selectiva de los receptores de familias de linfocitos T Vβ2 y Vβ8, en los niños afectados. Pero la respuesta inmunitaria en la enfermedad de Kawasaki es más oligoclonal (ligada a antígenos, es decir, similar a la respuesta de un antígeno convencional) que policlonal (como las halladas típicamente en las respuestas ligadas a superantígenos), y las células plasmáticas secretoras de inmunoglobulinas A (IgA) juegan un papel central235 , 237.
Independientemente de la causa, la evidencia indica una activación inmunitaria generalizada con producción de varias citocinas proinflamatorias y a la activación endotelial que da lugar a la alteración de las arterias coronarias238., 239., 240., 241.. La citocina más estudiada ha sido el TNF-α, que generalmente está elevada en los niños con enfermedad de Kawasaki. Para la formación de las dilataciones aneurismáticas también pueden ser importantes las enzimas, como las metaloproteinasas de la matriz, que son capaces de dañar la integridad de la pared arterial. También pueden jugar un papel importante varias quimiotactinas que atraen neutrófilos y monocitos hacia las arterias coronarias242.
Existen determinantes genéticos del huésped que pueden desempeñar un papel en la susceptibilidad y en el resultado final de las arterias coronarias en la enfermedad de Kawasaki243. La tasa de incidencia en hermanos es 10 veces más alta que en la población general217 , 244 , 245. El riesgo de que se produzca en un gemelo es mayor que en un hermano normal. Los padres que han tenido enfermedad de Kawasaki en la infancia tienen más posibilidades de que sus hijos sufran la enfermedad y sus hijos tienen más riesgo de desarrollar una enfermedad recidivante246.
Pruebas de laboratorio e histopatología
En la fase aguda, los estudios de laboratorio muestran leucocitosis (>15.000/mm3) con una desviación hacia la izquierda, anemia normocrómica normocítica, aumento de la velocidad de sedimentación y de otros reactantes de fase aguda, disminución de la albúmina y niveles de IgE e IgM elevados. El grado de elevación de la VSG y de la proteína C reactiva puede mostrar discrepancias, debiéndose valorar ambas. Además, la elevación de la VSG puede ser secundaria al tratamiento con IGIV, por lo que no puede ser el único determinante del grado de actividad inflamatoria218. Los lípidos plasmáticos están alterados en la fase aguda, con una disminución del colesterol total y de HDL247 , 248. Puede haber una ligera elevación de las transaminasas e hipergammaglobulinemia policlonal. En la fase subaguda, en la semana segunda y tercera de la enfermedad, hay una trombocitosis importante, fenómeno que es casi universal y que se normaliza en 4-8 semanas. La trombocitopenia raramente se ve en la fase aguda y puede ser un signo de coagulación intravascular diseminada. Como fenómeno inespecífico se pueden detectar anticuerpos citoplasmáticos antineutrófilos. Puede haber piuria estéril con leve proteinuria. El líquido cefalorraquídeo muestra una pleocitosis mononuclear con proteínas y glucosa normales. Los hallazgos de la biopsia cutánea no son específicos. Hay edema en la dermis papilar, con un infiltrado de células mononucleares perivascular. Se ve vasculitis de las arterias medianas y grandes.
Diagnóstico218
No hay un prueba diagnóstica específica para la enfermedad de Kawasaki y por tanto se han establecido criterios clínicos para orientar las decisiones terapéuticas (v. cuadro 19-3 ). El diagnóstico clásico ha sido basado en la presencia de 5 días de fiebre y cuatro o cinco de las principales características clínicas. Las características clínicas generalmente aparecen de forma secuencial y no todas están presentes en un momento concreto, por lo que a veces es necesaria la vigilancia expectante antes de poder establecer el diagnóstico. Para evitar tener que retrasar el tratamiento hasta que hayan aparecido más de cuatro criterios y sabiendo que muchos pacientes con un Kawasaki «incompleto» desarrollarán la enfermedad en las arterias coronarias, es posible diagnosticar y tratar este proceso al cuarto día de enfermedad ante la presencia de cuatro de los principales criterios230 , 249. También se puede establecer el diagnóstico en pacientes con fiebre de 5 días y menos de cuatro características principales cuando se detecta la enfermedad de las arterias coronarias mediante la ecocardiografía bidimensional (2DE) o mediante angiografía coronaria. La enfermedad de Kawasaki debe considerarse en el diagnóstico diferencial de un niño pequeño, especialmente de menos de 1 año, con fiebre sin foco durante 5 días asociada a alguna de las principales características clínicas de la enfermedad o incluso ante la presencia de otros hallazgos clínicos y de laboratorio que no son criterios clásicos, pero que con frecuencia se encuentran en esta enfermedad (v. cuadro 19-3)218. Por ejemplo, la elevación de la PCR o de la VSG y la trombocitosis después de 7 días de enfermedad son comunes en las enfermedades víricas, pero se ven de forma universal en los niños con enfermedad de Kawasaki. La ecocardiografía puede ser útil en la evaluación de la «enfermedad de Kawasaki incompleta» y debe considerarse en cualquier lactante con menos de 6 meses con fiebre de más de 7 días de duración, evidencia en las pruebas de laboratorio de inflamación sistémica y sin otra explicación para la enfermedad febril218. A pesar de que los aneurismas raramente se forman antes del décimo día de la enfermedad, puede haber signos de arteritis coronaria, disminución en la contractilidad, insuficiencia mitral y derrame pericárdico. Con todas estas consideraciones se ha desarrollado un nuevo algoritmo para ayudar a decidir a qué paciente con enfermedad de Kawasaki incompleta se le debe realizar una ecocardiografía o administrar tratamiento con IGIV (v. figura 19-20 )218.
Cuadro 19-3. Criterios diagnósticos clásicos de la enfermedad de Kawasaki**Pacientes con fiebre de por lo menos 5 días y <4 criterios principales pueden diagnosticarse de enfermedad de Kawasaki cuando se detecten alteraciones de las arterias coronarias por ecografía en 2 dimensiones o angiografía.
-
1.
Fiebre de 5 días de duración y por lo menos cuatro de los siguientes criterios** :
-
2.
Cambios en las extremidades: eritema de las palmas, plantas; edema de las manos, pies; pérdida de piel periungueal en los dedos de las manos, dedos de los pies.
-
3.
Exantema polimorfo.
-
4.
Inyección conjuntiva bilateral sin exudado.
-
5.
Cambios en los labios y en la cavidad oral: fisuras, lengua en fresa, inyección difusa de la mucosa oral o faríngea.
-
6.
Linfadenopatía cervical de por lo menos 1,5 cm de diámetro.
-
7.
Exclusión de otras enfermedades con hallazgos similares.
Otros hallazgos clínicos/laboratorio en la enfermedad de Kawasaki
- Otros hallazgos clínicos y de laboratorio
- Hallazgos cardiovasculares
- Insuficiencia cardíaca congestiva, miocarditis, pericarditis, insuficiencia valvular
- Alteraciones en las arterias coronarias
- Aneurismas en las arterias de tamaño mediano
- Fenómeno de Raynaud
- Gangrena periférica
- Sistema musculoesquelético
- Artritis, artralgias
- Tubo digestivo
- Diarrea, vómitos, dolor abdominal
- Disfunción hepática
- Hidropesía de la vesicular biliar
- Sistema nervioso central
- Irritabilidad extrema
- Meningitis aséptica
- Hipoacusia neurosensorial
- Sistema genitourinario
- Uretritis/meatitis
- Otros hallazgos
- Eritema, induración en el sitio de inoculación del BCG
- Uveítis anterior (leve)
- Erupción descamativa en las ingles
- Hallazgos de laboratorio en la enfermedad de Kawasaki
- Leucocitosis con neutrofilia y formas inmaduras
- Elevación de la VSG
- Elevación de la PCR
- Anemia
- Alteraciones en los lípidos plasmáticos
- Hipoalbuminemia
- Hiponatremia
- Después de 1 semana con trombocitopenia
- Piuria estéril
- Elevación de las transaminasas séricas
- Elevación de la gammaglutamil transpeptidasa sérica
- Pleocitosis en el líquido cefalorraquídeo
- Leucocitosis en el líquido sinovial
Modificado de Pediatrics 2004; 114: 1708-1133.
FIGURA 19-20.

Evaluación de un caso de sospecha de enfermedad de Kawasaki incompleta. 1) Ante la ausencia de patrón de referencia para el diagnóstico, este algoritmo no puede estar basado en la evidencia, sino que representa la opinión informada del comité de un grupo de expertos. En cualquier momento en que se precise asistencia debe consultarse con un experto. 2) Lactantes de ≥6 meses de edad en el día ≥7 de fiebre sin otras explicaciones: deben llevarse a cabo pruebas de laboratorio; si hay evidencia de inflamación sistémica, debe realizarse un ecocardiograma, incluso si el lactante no tiene criterios clínicos. 3) Los pacientes con características sugerentes de enfermedad de Kawasaki se enumeran en el cuadro 19-3. Las características que sugieren otras enfermedades diferentes a la enfermedad de Kawasaki incluyen la conjuntivitis exudativa, faringitis exudativa, lesiones intraorales no confluyentes, erupción ampollosa o vesiculosa, linfadenopatía generalizada. Considerar diagnósticos alternativos. 4) Los criterios de laboratorio suplementarios incluyen albúmina ≥3 g/dL, anemia para su edad, elevación de la alanina aminotransferasa, plaquetas después de 7 días ≥450.000/mm3, recuento de leucocitos ≥15.000/mm3 y orina con ≥10 leucocitos por campo. 5) Pueden tratarse antes de llevar a cabo el ecocardiograma. 6) Para el propósito de este algoritmo el ecocardiograma se considera positivo si cumple cualquiera de las siguientes tres condiciones: puntuación del DAI o ACD ≥2,5, las arterias coronarias cumplen los criterios de aneurismas del Ministerio de Salud de Japón o ≥3 características sugerentes, incluyendo brillo perivascular, falta de estrechamiento, disminución de la función del ventrículo izquierdo, insuficiencia mitral, derrame pericárdico o puntuación en DAI o ACD de 2-2,5. 7) Si el ecocardiograma es positivo, se debe dar tratamiento a los niños dentro de los 10 días del inicio de la fiebre y después del día 10 en aquellos con signos clínicos y de laboratorio (PCR, VSG) o inflamación activa. 8) Pérdida de piel típica que empieza debajo del lecho ungueal de los dedos de las manos y después de los pies.
En ocasiones la enfermedad de Kawasaki puede ser «atípica» y debutar con unas características clínicas que no se ven de forma general, como son el dolor abdominal agudo, la afectación renal, la irritación meníngea, neumonía o abscesos retrofaríngeos.
Diagnóstico diferencial
Un gran número de enfermedades se parecen a la enfermedad de Kawasaki, incluyendo infecciones víricas, infecciones estreptocócicas, artritis reumatoide juvenil, eritema multiforme, síndrome de la escaldadura estafilocócica, síndrome del shock tóxico, reacciones de hipersensibilidad a los fármacos, fiebre de las Montañas Rocosas, leptospirosis, reacción de hipersensibilidad al mercurio (acrodinia) y adenitis cervical bacteriana. La leucopenia, la linfocitosis y la trombopenia pueden ser útiles para sugerir una infección vírica en lugar de una enfermedad de Kawasaki.
Evolución, manejo, tratamiento y pronóstico
La mortalidad y morbilidad de la enfermedad de Kawasaki dependen sobre todo de las lesiones en las arterias coronarias250., 251., 252., 253., 254.. Los aneurismas o ectasia de las arterias coronarias se desarrollan en un 15%-25% de los niños sin tratamiento y pueden causar una cardiopatía isquémica o muerte súbita. Con el tratamiento temprano el riesgo se reduce a casi un 5%-12%215 , 216. En un 30%-60% de los pacientes los aneurismas pequeños se resuelven en los 2 primeros años de la enfermedad255. Pero los aneurismas coronarios, sobre todo los gigantes (>8 mm), pueden persistir y complicarse por una oclusión trombótica o por una estenosis a la salida del aneurisma. Las lesiones estenóticas y la arteriosclerosis coronaria temprana pueden desarrollarse gradualmente al cabo de varios años, por lo que se necesita un seguimiento a largo plazo252 , 253 , 256 , 257.
Se han desarrollado varios sistemas de puntuación para predecir el riesgo de aneurismas de las arterias coronarias258. La puntuación de Harada es la que se utiliza en Japón259.
Dado que las secuelas principales de la enfermedad de Kawasaki se relacionan con el sistema de las arterias coronarias, los estudios radiológicos cardíacos son fundamentales para la evaluación de todos los pacientes con sospecha de enfermedad de Kawasaki, por lo que se recomiendan las ecocardiografías seriadas. La ecocardiografía debe centrarse en la visualización y la medición de las arterias coronarias y en la visualización y las medidas de la contractilidad del ventrículo izquierdo, función valvular, la presencia de derrame pericárdico y medidas de la raíz de la aorta260. El primer examen debe llevarse a cabo tan pronto como se sospeche el diagnóstico y sirve de base para los estudios posteriores. Por tanto, para los casos sin complicaciones debe repetirse a las 2 semanas y a las 6-8 semanas del inicio de la enfermedad218. En los pacientes que desarrollan aneurismas de las arterias coronarias u otras alteraciones cardíacas se recomienda una evaluación más frecuente. Otros estudios radiológicos no invasivos, como la RM, la ARM y la TC ultrarrápida, y las pruebas cardíacas de esfuerzo están siendo evaluadas para el manejo de la enfermedad de Kawasaki. Hasta que los aneurismas desaparezcan, pueden ser necesarias las pruebas de esfuerzo para orientar las recomendaciones sobre la realización de ejercicio y la necesidad de angiografía.
En la fase aguda de la enfermedad el tratamiento va dirigido a reducir la inflamación de la pared de las arterias coronarias y prevenir la trombosis de las coronarias, y el tratamiento a largo plazo de los individuos con aneurismas de las coronarias va dirigido a la prevención de la isquemia miocárdica y de los infartos. La administración de γ-globulinas (IGIV) combinadas con altas dosis de ácido acetilsalicílico es el tratamiento de elección en la fase aguda de la enfermedad229. El ácido acetilsalicílico solo no parece reducir la frecuencia del desarrollo de alteraciones coronarias, pero con las IGIV tiene un efecto antiinflamatorio aditivo. En la fase aguda el ácido acetilsalicílico se administra en dosis de 80-100 mg/kg cada 6 horas. La duración de las dosis altas de ácido acetilsalicílico varía según los centros. En muchas instituciones la dosis se reduce cuando el niño está afebril durante 48-72 horas, pero otros la mantienen hasta el día 14 de enfermedad y >48-72 horas después del cese de la fiebre. Tras esta fase aguda, se administra una dosis baja de ácido acetilsalicílico (3-5 mg/kg) como antiagregante hasta que no haya evidencia de cambios coronarios a las 6-8 semanas del inicio de la enfermedad. En los pacientes que desarrollan alteraciones en las coronarias se continúa la administración de dosis bajas de ácido acetilsalicílico hasta la regresión de los aneurismas de las arterias coronarias, pero algunos clínicos continúan su administración de forma indefinida. Se debe evitar la administración de ibuprofeno en niños que estén tomando ácido acetilsalicílico por sus efectos antiagregantes, dado que antagoniza la inhibición plaquetaria.
La administración de IGIV ha demostrado reducir la incidencia de aneurismas de las arterias coronarias desde el 20% al 3%-4%261., 262., 263., 264.. Una sola dosis de 2 mg/kg ha resultado mejor que la administración de dosis inferiores durante 4 días261 , 262. La IGIV debe iniciarse pronto, durante los 10 primeros días desde el inicio de la enfermedad y preferiblemente dentro de los 7 primeros. El tratamiento antes de los 5 días no parece evitar las secuelas cardíacas y puede ir asociado a una mayor necesidad de nuevos tratamientos con IGIV265 , 266. El tratamiento después de los 10 días debe considerarse si todavía hay signos de inflamación (elevación de la VSG o de la PCR) o persistencia de la fiebre267. No todos los pacientes responden a una dosis única de IGIV y pueden tener fiebre persistente o una recrudescencia más de 36 horas después de haber completado el tratamiento inicial, precisando una segunda dosis. Se ha observado que aquellos niños que reciben IGIV muy pronto en la evolución de la enfermedad están más predispuestos a recibir una segunda infusión. La vacunación con virus vivos u otras vacunas debe retrasarse al menos 11 meses después del tratamiento con una dosis alta de IGIV, tanto para asegurar una inmunización correcta como para evitar recidivas de la enfermedad de Kawasaki268.
El uso de corticoesteroides en combinación con IGIV como tratamiento inicial de la enfermedad de Kawasaki no está bien establecido. Los corticoesteroides acortan la duración de la fiebre y disminuyen la VSG y la PCR, pero no influyen en las complicaciones coronarias269 , 270. También se han usado para los fallos en el tratamiento con IGIV, pero su papel en la prevención o en la reversión de las alteraciones coronarias es incierto. Se ha recomendado restringirlos en niños con fiebre persistente tras dos infusiones de IGIV218. El régimen más común es la administración intravenosa de pulsos de metilprednisolona, 30 mg/kg durante 1-3 días.
La pentoxifilina puede tener un beneficio teórico en el tratamiento inicial de la enfermedad de Kawasaki dada la inhibición del ARN mensajero del TNF-α que produce, si bien los ensayos clínicos publicados son pequeños271. Es incierto el papel de los antagonistas del TNF-α, como el infliximab, del inhibidor de la glucoproteína receptora de plaquetas IIb/IIa abciximab, del intercambio de plasma y de los agentes citotóxicos para los pacientes con enfermedad de Kawasaki refractaria al tratamiento218 , 272.
Edema agudo hemorrágico
El edema agudo hemorrágico (EAH), púrpura en escarapela o enfermedad de Finkelstein es una vasculitis leucocitoclástica benigna que sólo afecta a la piel y ocurre en niños de menos de 2 años273., 274., 275., 276., 277., 278., 279., 280.. El EAH ha sido considerado una variante infantil de la púrpura de Schönlein-Henoch; sin embargo, debido a sus diferencias clínicas y pronosticas en ocasiones se considera una entidad diferente.
Hallazgos cutáneos
La enfermedad se caracteriza por un inicio abrupto de fiebre; edema doloroso en la cara, cejas, oídos, escroto y en las regiones acras de las extremidades, y una púrpura equimótica en la cara y en las extremidades. Por lo general el tronco no se afecta. Las lesiones individuales a menudo tienen un centro más oscuro y se expanden centrífugamente, dando una configuración en escarapela o similar a una diana. Las lesiones oscilan en tamaño entre 0,5 a 4 cm y pueden ser confluentes, formando placas policíclicas y anulares (v. figura 19-21 ). Pueden verse lesiones necróticas279 , 281 y ampollosas279 , 282. También se han descrito petequias en las mucosas283.
FIGURA 19-21.

Edema hemorrágico agudo.
Hallazgos extracutáneos
Salvo por la fiebre, no hay otras manifestaciones. En muchos pacientes el proceso viene precedido por una infección de vías respiratorias altas. Los espectaculares hallazgos cutáneos contrastan con el buen estado general del paciente.
Etiología/patogenia
La causa del EAH es desconocida. Se cree que representa una enfermedad mediada por inmunocomplejos precipitada por una infección precedente, especialmente del tracto respiratorio superior, por la ingesta de fármacos o por la inmunización. Los organismos que se han implicado con más frecuencia son las especies de Staphylococcus y Streptococcus y los virus (adenovirus, rotavirus).
Pruebas de laboratorio e histopatología
Puede haber leucocitosis (tanto linfocítica como granulocítica), trombosis, eosinofilia y una elevación de la VSG. Los análisis de orina, las pruebas para sangre oculta en heces, inmunoglobulinas y los niveles de complemento generalmente son normales. Ocasionalmente pueden hallarse inmunocomplejos en la circulación282.
El examen histopatológico de las muestras de biopsia cutánea demuestra una vasculitis leucocitoclástica de vasos pequeños. La inmunofluorescencia directa muestra depósitos de C3 y fibrinógeno en la pared de los vasos. En hasta un tercio de los casos se han notado depósitos de IgM, IgG, IgA e IgE282 , 284., 285., 286..
Diagnóstico diferencial
El diagnóstico diferencial incluye la púrpura de Schönlein-Henoch, los malos tratos físicos, la meningococemia y otras púrpuras, eritema multiforme, enfermedad de Kawasaki y el síndrome de Sweet273 , 283. La diferenciación con la púrpura de Schönlein-Henoch puede ser imposible280 (v. tabla 19-2 ). Los depósitos perivasculares de IgA no son útiles para diferenciar ambas entidades dado que pueden presentarse en ambas.
TABLA 19-2.
Diagnóstico diferencial entre el edema agudo hemorrágico y la púrpura de Henoch-Schönlein
| Edema agudo hemorrágico | Purpura de Schönlein-Henoch | |
|---|---|---|
| Inicio |
Agudo y espectacular |
Menos espectacular |
| Edad |
2 meses-2 años |
>2 años |
| Sexo |
Predominio en varones |
Predominio en varones |
| Localización |
Cara, oídos, extremidades |
Extremidades inferiores, nalgas |
| Morfología |
Púrpura grande, parecida a un medallón | Púrpura palpable |
| Edema facial |
Edema de las extremidades inferiores |
|
| Evolución |
Sin recidivas |
Recidivas frecuentes |
| Histopatología |
Vasculitis leucocitoclástica |
Vasculitis leucocitoclástica |
| Inmunodepósitos |
Variable IgA, IgM, C3 |
IgA |
| Afectación sistémica | No | Renal, digestivo, articulaciones, SNC |
Evolución, manejo y pronóstico
El pronóstico es excelente. La erupción desaparece de forma espontánea sin secuelas en 1-3 semanas. No es necesario el tratamiento con corticoesteroides, que pueden causar complicaciones y empeorar el pronóstico284. Durante la evolución clínica pueden observarse exacerbaciones, con la aparición de nuevos brotes de lesiones y fiebre281 , 285; sin embargo, las verdaderas recidivas semanas o meses después del primer episodio son raras280 , 283. Ha habido una sola publicación de un caso con invaginación ileoileal mortal en un lactante con lesiones cutáneas típicas de EHA281.
Porfirias287., 288., 289., 290., 291., 292., 293., 294., 295.
Las porfirias son un grupo de enfermedades caracterizadas por alteraciones en el metabolismo del grupo porfirina-hemo. Cada tipo es consecuencia de la actividad deficiente de una de las enzimas de la vía de síntesis del grupo hemo, lo que conduce a la acumulación de precursores del grupo hemo en plasma, glóbulos rojos, orina y heces295. Los genes de estas enzimas están bien caracterizados287 , 289 , 295. Las porfirias se heredan sobre todo de forma autosómica dominante con una penetrancia incompleta, aunque también pueden existir cuadros de herencia autosómica recesiva y patrones de herencia complejos. Las porfirias se clasifican como hepáticas o eritropoyéticas, según el órgano donde predomina el defecto en la síntesis del grupo hemo (v. tabla 19-3 ). Las manifestaciones cutáneas de las porfirias pueden ser clasificadas como fotosensibilidad aguda, con dolor-quemazón, edema y eritema poco después de la exposición al sol, o fotosensibilidad retardada que se manifiesta por fragilidad cutánea, ampollas subepidérmicas, acné miliar, alteraciones de la pigmentación y signos de esclerodermia. Las porfirias hepáticas generalmente se manifiestan por ataques agudos neuroviscerales y fotosensibilidad retardada y raramente se presentan antes de la pubertad salvo en las variantes homocigóticas. En la orina y heces puede detectarse una elevación de las porfirinas. Las porfirias eritropoyéticas se caracterizan por fotosensibilidad cutánea aguda desde la infancia temprana. Puede aparecer también una fotosensibilidad más retardada, aunque es menos característica de este tipo de porfiria293. En las porfirias eritropoyéticas, los niveles de porfirinas en los eritrocitos y en el plasma están elevados.
TABLA 19-3.
Clasificación de las porfirias, defecto enzimático, perfil de porfirinas y edad de inicio
|
Perfil de porfirinas |
|||||||
|---|---|---|---|---|---|---|---|
| Tejido de origen | Tipo | Déficit enzimático | Eritrocito | Plasma | Orina | Heces | Edad de inicio |
| Eritropoyética |
Porfiria eritropoyética congénita (PEC) | Uroporfirinógeno III sintetasa | URO-I, COPRO-I, PROTO | URO-1, COPRO-1, PROTO | URO-I > COPRO-I | COPRO-I | Nacimiento, lactancia |
| Protoporfiria eritropoyética (PEP) |
Ferroquelatasa |
PROTO |
PROTO |
Normal |
PROTO |
Lactancia, primera infancia |
|
| Hepática |
Porfiria aguda intermitente (PAI) | Porfobilinógeno deaminasa | Normal | Normal | ALA, PBG | Normal | Después de la pubertad, no tiene manifestaciones cutáneas |
| Porfiria variegata (PV) | Protoporfirinógeno oxidasa | Normal | COPRO, PROTO | COPRO > URO | PROTO > COPRO | Segunda década, variante homocigótica al nacer, con la lactancia o niñez temprana | |
| Coproporfiria hereditaria (CPH) | Oxidasa de coproporfirinógeno | Normal | COPRO | COPRO | COPRO | Tras la pubertad, variante homocigótica (harderoporfiria) al nacer, lactancia o niñez temprana | |
| Porfiria cutánea tardía (PCT) | Uroporfirinógeno descarboxilasa | Normal | URO | URO-I > III | ISOCOPRO | Tercera o cuarta década de vida | |
| Porfiria por ALA dehidratasa |
ALA dehidratasa |
PROTO |
ALA, COPRO, PROTO |
ALA, COPRO, URO |
COPRO, PROTO |
Cualquier edad, no tiene manifestaciones cutáneas |
|
| Hepatoeritropoyética | Porfiria hepatoeritropoyética (PHE) | Uroporfirinógeno decarboxilasa | Zn-PROTO | URO | URO-(I y III) | ISOCOPRO | Lactancia temprana |
ALA, 5-aminolevulinato; COPRO, coproporfirina; ISOCOPRO, isocoproporfirina; PBG, profobilinógeno; PROTO, portoporfirina; URO, uroporfirina.
La fotosensibilidad en las porfirias es máxima para las longitudes de onda ultravioletas entre 400 y 410 nm («banda de Soret »), el espectro de máxima absorción de las porfirinas.
El mecanismo fisiopatológico relacionado con las manifestaciones cutáneas de las porfirias es múltiple e implica la formación de especies reactivas del oxígeno296., 297., 298..
Las porfirias de los niños son relativamente raras, aunque su incidencia exacta es desconocida. Aquí sólo se revisarán las porfirias que tienen manifestaciones en la lactancia precoz.
Porfiria eritropoyética congénita
La porfiria eritropoyética congénita (PEC), también denominada enfermedad de Günther, es una alteración autosómica recesiva rara provocada por la deficiencia en la actividad de la uroporfirinógeno III (UROGEN III) sintetasa que da lugar a la conversión no enzimática del hidroximetilbilano a uroporfirinógeno I, un sustrato no fisiológico que se convierte en coproporfirinógeno I; estos isómeros del porfirinógeno I se oxidan entonces a uroporfirina I (URO-I) y coproporfirina I (COPRO-I), que son compuestos fototóxicos. Los niveles elevados de URO-I y COPRO-I en los eritrocitos dan lugar a una hemólisis masiva y a la liberación de las porfirinas acumuladas hacia la sangre periférica, piel, hueso y dientes y su excreción en grandes cantidades en la orina y en las heces.
Hallazgos cutáneos292,299,300
La PEC se presenta con fotosensibilidad grave desde el nacimiento o la lactancia temprana con formación de vesículas y ampollas en las áreas expuestas al sol, a los aparatos de fototerapia o incluso a la luz de ambiente301. También tiene una marcada fragilidad cutánea. Como consecuencia de la lesión fototóxica y del aumento de la fragilidad cutánea, hay graves mutilaciones, principalmente de los dedos de las manos y de la cara y en concreto de la nariz y las orejas, aunque también en las áreas protegidas del sol. La hipertricosis en la cara y las extremidades, la alopecia del cuero cabelludo y de las cejas como consecuencia de las cicatrices y los cambios pigmentarios (hiperpigmentación e hipopigmentación) son comunes. Con el tiempo, se produce una grave mutilación facial, con destrucción de los cartílagos nasales y auriculares, ectropión y eclabión, así como acortamiento y contracción de los dedos. Los fenotipos más leves pueden debutar en la niñez302.
Hallazgos extracutáneos
La acumulación de porfirinas en los dientes de leche y permanentes produce una coloración roja (eritrodoncia) y una fluorescencia roja en el examen con la luz de Wood, que es patognomónica. La orina también es rojiza, lo que provoca una coloración rosa fluorescente de los pañales, que es un signo precoz de diagnóstico. Las porfirinas pueden acumularse en el líquido amniótico, que adopta un tono marrón. Puede haber anemias hemolíticas graves y esplenomegalia secundaria. La anemia puede ser tan grave que ocasione una hidropesía fetal con muerte intraútero. Los pacientes que debutan de forma más tardía no desarrollan anemia hemolítica sino sólo trombocitopenia y mielodisplasia. Los cambios oculares incluyen ectropión, fotofobia y queratoconjuntivitis303. Otras manifestaciones pueden incluir osteodistrofia con aumento de la fragilidad ósea y cálculos en la vesícula ricos en porfirinas.
Genética
El gen de la UROGEN III sintetasa se localiza en el cromosoma 10. Se han identificado varias mutaciones304 y las diferentes mutaciones se correlacionan con el nivel de actividad residual de la enzima y con la gravedad de la enfermedad y de los genotipos302 , 305 , 306.
Pruebas de laboratorio e histopatología
Los exámenes histológicos de las biopsias de las ampollas muestran separación subepidérmica (dentro de la lámina lúcida) y un infiltrado inflamatorio mínimo. También puede verse una acumulación perivascular de un material hialino homogéneo PASpositivo y resistente a la diastasa (porfirinas), que se ve mejor con el microscopio de fluorescencia. En la tabla 19-3 se recoge el perfil de excreción de porfirina. Se puede medir la actividad de la URO III sintetasa. Es posible el diagnóstico prenatal midiendo la uroproporfirina I en el líquido amniótico o mediante el análisis de las mutaciones genéticas307., 308., 309., 310..
Diagnóstico diferencial
Deben considerarse otras enfermedades fotosensibles que se presentan en fases tempranas de la vida (v. cuadro 19-4 ) o enfermedades que se manifiestan con ampollas, como la epidermólisis ampollosa y el penfigoide ampolloso. La determinación de porfirinas es diagnóstica y permite la diferenciación de otras porfirias que se presentan en fases tempranas de la vida con fotosensibilidad.
Cuadro 19-4. Causas de fotosensibilidad en neonatos.

Pronóstico
La gravedad clínica de la PEC es altamente variable, oscilando desde hidropesía fetal, hepatoesplenomegalia y anemia grave intraútero a formas que se inician en la edad adulta con manifestaciones exclusivamente cutáneas311. Sin embargo, en la mayoría de los casos los pacientes sobreviven hasta la edad adulta, a pesar de graves mutilaciones o una gran desfiguración.
Tratamiento312
Es fundamental evitar la exposición al sol. Las cremas solares no consiguen una buena protección frente a la banda de radiación que produce heridas, por lo que se necesitan ropas protectoras y bloqueantes físicos de la luz solar. Se recomienda colocar filtros protectores para las longitudes de onda larga capaces de absorber UV en las ventanas de los coches y de las casas. Los niños con los fenotipos más graves se benefician de las repetidas transfusiones de eritrocitos y de la administración de hidroxiurea para suprimir la eritropoyesis. El hematocrito debe mantenerse por encima del 32%, con la administración apropiada de quelantes del hierro. La eficacia de las transfusiones repetidas de eritrocitos puede disminuir en la pubertad. Posteriormente, con frecuencia es necesaria la esplenectomía para controlar la anemia hemolítica313 , 314. También se han utilizado con resultados inconsistentes el carbón activado315., 316., 317., 318. y los β-carotenos319 , 320. El trasplante de médula ósea o de células pluripotenciales del cordón umbilical puede corregir la actividad enzimática321., 322., 323., 324., 325.. Se ha conseguido la terapia génica in vivo con reposición del gen alterado326 , 327.
Protoporfiria eritropoyética287,298,328
La protoporfiria eritropoyética (PPE) es la forma más común de porfiria cutánea, sin considerar la porfiria cutánea tardía (PCT). La PPE se produce por una deficiencia en la actividad de la ferroquelatasa, lo que conduce la acumulación de protoporfirina en los eritrocitos, plasma y heces. Los signos clínicos generalmente se inician en la lactancia o en la infancia temprana, con un pico de incidencia entre los 2 y 4 años de edad. La PPE generalmente se hereda de forma autosómica dominante, si bien la mayoría de los individuos que son heterocigóticos para la mutación son asintomáticos porque la actividad de la ferroquelatasa es seminormal329. Para que la protoporfirina se acumule en suficiente cantidad como para causar fotosensibilidad, se tiene que producir una reducción enzimática por debajo del umbral de alrededor del 35% de la actividad normal. Algunas familias pueden tener una herencia autosómica recesiva329., 330., 331..
Manifestaciones cutáneas
Las manifestaciones clínicas corresponden a una reacción fotosensible aguda, que desencadena un episodio de llanto al cabo de minutos de la exposición al sol debido al dolor de quemadura o a la sensación de pinchazos en las áreas expuestas (la cara y las partes dorsales de las manos). Algunos pacientes son fotosensibles a la luz fluorescente328. Se produce eritema, edema y lesiones urticariales, pero las vesículas y las ampollas son raras (v. figura 19-22 ). Pueden observarse pequeñas petequias tras la exposición prolongada al sol. Algunos pacientes sólo tienen síntomas subjetivos328. Con la exposición crónica se produce un característico engrosamiento con arrugas en los nudillos, arrugas alrededor de la boca (seudorrágades) y cicatrices superficiales elípticas en la nariz, mejillas y frente.
FIGURA 19-22.

Porfiria.
Por cortesía de Henry Lim y Tor Schwayder.
No se produce anemia hemolítica, aunque algunos pacientes pueden presentar una anemia microcítica e hipocrómica. Los cálculos de protoporfirina pueden aparecer en la niñez. Como consecuencia de la acumulación progresiva de protoporfirina en los hepatocitos, en un 2,5% de los pacientes se produce una insuficiencia hepática mortal, lo que modifica el pronóstico de esta enfermedad benigna. La herencia recesiva puede predisponer a una enfermedad hepática grave329 , 332.
Genética
El gen de la ferroquelatasa está en el cromosoma 18. En las familias con PPE se han identificado cerca de 70 mutaciones de este gen329 , 333. En la mayoría de los pacientes se precisa de la herencia de una segunda mutación para reducir la actividad enzimática por debajo del umbral crítico que produce síntomas. La herencia autosómica recesiva se ha demostrado en un 3% de los pacientes con PPE329.
Pruebas de laboratorio e histopatología
El examen histopatológico de las biopsias cutáneas en las áreas expuestas al sol muestra marcados depósitos concéntricos de material hialino alrededor de los vasos sanguíneos. Este material es PAS positivo y resistente a la diastasa.
En los pacientes con PPE se debe hacer un seguimiento estrecho de la función hepática y en los que presenten alteraciones de forma persistente se debe llevar a cabo una biopsia. En los niños con protoporfirinas eritrocitarias debe llevarse a cabo de forma periódica la determinación de porfirinas en sangre, orina y heces, dado que un aumento de la excreción de coproporfirinas y el elevado contenido de protoporfirinas eritrocitarias y una reducción de la excreción de protoporfirinas en las heces pueden ser predictores de la insuficiencia hepática334.
Diagnóstico
El diagnóstico de la PPE se establece con la determinación de niveles elevados de protoporfirina en los eritrocitos, plasma y heces. También pueden estar elevados los niveles de coproporfirina fecal y eritrocitaria. Para la detección selectiva puede utilizarse un ensayo microfluorimétrico rápido para la determinación de protoporfirinas en los eritrocitos de un frotis sanguíneo para visualizar eritrocitos fluorescentes335. El diagnóstico diferencial incluye otros tipos de porfiria, mientras que las causas de fotosensibilidad inmediata, como la EPML o la urticaria solar, no ocurren en los lactantes.
Tratamiento
La clave para el tratamiento de la protoporfiria eritropoyética es evitar el sol y el uso de protectores solares336 , 337. La dihidroxiacetona tópica puede ser útil en algunos pacientes al producir una pigmentación marrón287. La administración oral de β-carotenos (en niños, 30-90 mg/día) en estudios no controlados ha aumentado la tolerancia a la exposición solar dado que disminuye la formación de radicales libres334 , 338 , 339. Se ha propuesto fototerapia con una banda de luz ultravioleta estrecha, dado que esta longitud de onda no provoca fotosensibilidad340. También se ha utilizado la desensibilización mediante tratamiento con PUVA. Para prevenir la acumulación de protoporfirina en el hígado mediante la reducción de su producción se ha utilizado la administración de hierro oral, la hematina intravenosa, el tratamiento transfusional y una dieta rica en hidratos de carbono, pero su eficacia no se ha demostrado334. Para reducir la circulación enterohepática de protoporfirinas se han utilizado la colestiramina o el carbón activado341. También es esencial evitar el alcohol y los fármacos que interfieren con la función excretora del hígado. En algunos pacientes con insuficiencia hepática se ha realizado el trasplante hepático, si bien en estos casos el defecto enzimático no se corrige y por tanto el pronóstico a largo plazo es malo342., 343., 344., 345.. En el quirófano es necesario modificar la iluminación para evitar la fotoxicidad de los órganos expuestos.
Porfiria hepatoeritropoyética
La porfiria hepatoeritropoyética (PHE) es una enfermedad extraordinariamente rara provocada por una importante deficiencia de la uroporfirinógeno decarboxilasa como consecuencia de un estado homocigótico346., 347., 348., 349.. Las manifestaciones clínicas se inician en la lactancia, o de forma más común en la niñez temprana, y se parece a la porfiria cutánea tardía y a la enfermedad de Günther. La enfermedad generalmente se presenta con un oscurecimiento de la orina y fotosensibilidad cutánea retardada, con vesículas, fragilidad cutánea, acné miliar y cicatrices. Con el tiempo, pueden aparecer cambios como hipertricosis, cambios esclerodérmicos y mutilación similar a las manifestaciones de la enfermedad de Günther. También ocurren diferentes grados de anemia, hepatoesplenomegalia y alteraciones en la función hepática, aunque son menos frecuentes que en la porfiria eritropoyética congénita. El patrón de excreción de porfirinas se parece al de la porfiria cutánea tardía (PCT), con elevación urinaria de uroporfirinas y porfirinas 7-carboxiladas y una menor elevación de las coproporfirinas, porfirinas 6 y 5-carboxiladas. Es característica la elevación de las isocoproporfirinas en las heces. A diferencia de la PCT, el grupo proto está aumentado en los eritrocitos.
El tratamiento va dirigido a la protección solar. En la porfiria hepatoeritropoyética la hipertricosis se ha tratado con éxito con pulsos de alta intensidad de luz no coherente350.
Otras porfirias
Porfirias homocigóticas
Las otras porfirias cuyos síntomas aparecen en la lactancia o en la niñez temprana incluyen las variantes homocigóticas del déficit de aminolevulinato dehidratasa (ALAD), coproporfiria homocigótica (harderoporfiria), porfiria variegata homocigótica y porfiria aguda intermitente homocigótica351.
La deficiencia de ALAD es rara (se han publicado menos de 10 pacientes) y generalmente se manifiesta tarde en la infancia o en la edad adulta, si bien se han publicado casos de inicio neonatal352. Las manifestaciones clínicas desde el nacimiento son los ataques recurrentes de dolor, vómitos, hiponatremia y síntomas de polineuropatía que afectan a la función motora, incluyendo la respiración. Se encuentra una elevación en orina de los niveles de ácido 5-aminolevulínico y de coproporfirina. Es diagnóstico encontrar una actividad eritrocitaria muy baja de aminolevulinato dehidratasa. En los pacientes con inicio neonatal el trasplante de hígado tiene muy poco efecto353.
En la harderoporfiria, la ictericia neonatal, la anemia hemolítica y la hepatoesplenomegalia marcan el cuadro clínico354., 355., 356., 357.. Durante los tratamientos con fototerapia por la ictericia neonatal se pueden producir ampollas. El diagnóstico depende de la detección de niveles muy bajos de la actividad de la coproporfirinógeno oxidasa, elevación de la coproporfirina en la orina, importante elevación de la harderoporfirina y coproporfirina en las heces y de protoporfirina de cinc en los eritrocitos.
La porfiria variegata homocigótica puede presentarse poco después de nacer con importante fotosensibilidad o con más frecuencia con erosiones, ampollas y acné miliar después de pequeños traumatismos en las áreas expuestas al sol358., 359., 360.. No hay ataques agudos y se ha descrito retraso mental y del crecimiento, convulsiones, nistagmo y clinodactilia.
La variante homocigótica de la porfiria aguda intermitente puede presentarse en fases tempranas de la vida con ataxia, retraso mental, convulsiones, cataratas y hepatoesplenomegalia, pero los típicos ataques de la porfiria aguda intermitente no ocurren en estos niños361., 362., 363., 364.. No hay manifestaciones cutáneas. En la orina se encuentra elevación del porfobilinógeno y delta-ALA, cambios responsables del color naranja de la orina.
Porfirinemias transitorias365., 366., 367., 368.
La elevación transitoria en los niveles de porfirina se ha descrito en neonatos con enfermedad hemolítica del recién nacido y en neonatos con insuficiencia hepática severa debida a una tirosinemia del tipo I368. Estos lactantes desarrollan eritema, coloración violácea, púrpura, erosiones y ampollas en las áreas expuestas a fototerapia, con una brusca delimitación con las áreas fotoprotegidas. Puede haber sensibilidad a la luz solar.
Se observan niveles elevados en plasma/orina de porfirinas (especialmente coproporfirinas) y/o protoporfirina eritrocitaria, que se normaliza de forma espontánea durante los primeros meses de vida. La causa de esta porfirinemia transitoria es incierta, si bien probablemente sea debida a colestasis. Otros factores que probablemente también pueden verse implicados incluyen la transfusión de sangre y el uso de fármacos.
Púrpuras en el recién nacido
La púrpura en el neonato es prácticamente siempre una emergencia y debe ser motivo urgente de la búsqueda de sus causas. Aparte de los traumatismos, la púrpura en el recién nacido puede deberse a defectos en la coagulación, alteraciones plaquetarias o infecciones (v. cuadro 19-5 ). La eritropoyesis extramedular también causa lesiones purpúricas por mecanismos diferentes.
Cuadro 19-5. Diagnóstico diferencial de la púrpura neonatal.
-
1.
Eritropoyesis extramedular (lactante «en magdalena de arándanos»)
-
2.Alteraciones en la coagulación
- Déficit de proteína C y S (púrpura neonatal fulminante)
- Enfermedades hemorrágicas del recién nacido
- Deficiencias hereditarias de los factores de coagulación
-
3.Alteraciones plaquetarias
-
a.Destrucción inmune de plaquetas
- Trombocitopenia aloinmune neonatal
- Trombocitopenia materna autoinmunitaria (PTI, lupus)
- Trombocitopenia inmunitaria relacionada con fármacos
-
b.Alteración primaria en la producción/función de plaquetas
- Trombocitopenia con ausencia del síndrome del radio
- Síndrome de Wiskott-Aldrich
- Anemia de Fanconi
- Trombocitopenia megacariocítica congénita
- Trombocitopenia recesiva ligada al X
- Otras trombocitopenias hereditarias
- Síndrome de las plaquetas gigantes (Bernard-Soulier, May-Hegglin)
- Trisomía 13 o 18
- Variantes del síndrome de Alport
- Síndrome de las plaquetas grises
- Trombastenia de Glanzmann
-
c.Síndrome de Kasabach-Merritt*
-
a.
-
4.Infecciones†
- Congénitas (TORCH)
- Sepsis
- VIH
- Parvovirus B19
-
5.
Traumatismos
-
6.
Erupción purpúrica inducida por fototerapia.
Modificado de Baselga E, Drolet BA, Esterly NB. J Am Acad Dermatol 1997;37:673–705.
En la evaluación de un neonato con púrpura hay que obtener una historia materna y familiar de diátesis hemorrágicas y fenómenos tromboembólicos, ingesta de fármacos y síntomas de enfermedades infecciosas. Está indicada una exploración física general y descartar una sepsis. Los estudios de laboratorio deben incluir los valores de hemoglobina y hematocrito, recuento plaquetario, de células blancas, estudios de coagulación y serologías TORCH.
Eritropoyesis dérmica (bebé con aspecto de «magdalena de arándanos»)
La persistencia de la actividad eritropoyética del mesénquima dérmico fetal en el período neonatal da lugar a una erupción purpúrica característica que se denominó como niño con aspecto de «magdalena de arándanos». La erupción se notó por primera vez en recién nacidos con rubéola congénita (v. figura 19-23 ) y puede ser el resultado de otras infecciones intrauterinas (v. figura 19-24 ) y discrasias hematológicas369., 370., 371.. La erupción en forma de «magdalena de arándanos» también puede ser consecuencia de una infiltración metastásica de la dermis por tumores congénitos, sin una verdadera eritropoyesis extramedular (v. figura 19-25 ).
FIGURA 19-23.
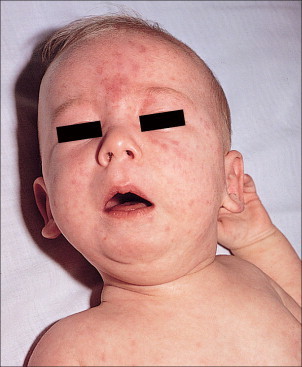
Lactante con rubéola congénita y lesiones «en magdalena de arándanos».
FIGURA 19-24.

Lesiones «en magdalena de arándanos» asociadas a la enfermedad de inclusión citomegálica.
FIGURA 19-25.

Infiltración de la piel por leucemia dando lugar a una piel con aspecto «en magdalena de arándanos».
Hallazgos cutáneos
Las lesiones cutáneas de estos niños consisten en pápulas redondas a ovales azul oscuras o rojizas, que no se blanquean con la presión, con un diámetro de 1 a 7 mm y que adoptan una distribución generalizada, aunque afectan sobre todo a la cabeza, cuello y tronco (v. figura 19-26 ). Las pápulas son firmes a la palpación, con un aspecto infiltrado que las diferencia de las petequias y de la púrpura, que en ocasiones también están presentes en el mismo paciente. Estas lesiones evolucionan a máculas oscuras, purpúricas-marronáceas que involucionan de forma espontánea en 2-6 semanas273. Las lesiones «en magdalena de arándanos» provocadas por un proceso infiltrativo generalmente son más grandes, más nodulares, menos hemorrágicas, en menor número y firmes a la palpación.
FIGURA 19-26.

Lesiones de eritropoyesis dérmica en un lactante con incompatibilidad Rh debido a fallo de la RhoGAM.
Hallazgos extracutáneos
Las alteraciones acompañantes varían según la causa subyacente.
Etiología/patogenia
En la era prevacunal la rubéola era la causa más común de eritropoyesis dérmica; sin embargo, en la actualidad, la principal causa es la infección congénita por citomegalovirus (CMV)369 , 370. La eritropoyesis dérmica se ha asociado a otras infecciones intrauterinas por Coxsackie B2369 y parvovirus B19372, así como discrasias hematológicas como la incompatibilidad del Rh (v. figura 19-24)373 , 374, incompatibilidad ABO materno fetal369, esferocitosis375 y con el síndrome de transfusión gemelar210 , 216 (v. cuadro 19-6 ). En raros casos puede ocurrir en niños sanos369 , 376.
Cuadro 19-6. Diagnóstico diferencial de las lesiones «en magdalena de arándanos».
- Eritropoyesis dérmica
- Infección congénita
- Rubéola
- Citomegalovirus
- Parvovirus B19
- Virus Coxsackie B2
- Enfermedades hemolíticas del recién nacido
- Incompatibilidad Rh
- Incompatibilidad del grupo sanguíneo
- Esferocitosis hereditaria
- Síndrome transfusional entre gemelos
- Enfermedades neoplásicas infiltrativas
- Neuroblastoma
- Rabdomiosarcoma
- Histiocitosis de células de Langerhans
Leucemia congénita
Modificado de Baselga E, Drolet BA, Esterly NB. J Am Acad Dermatol 1997;37:673–705.
Pruebas de laboratorio e histopatología
El examen histopatológico demuestra una colección mal delimitada de células eritroides nucleadas y no nucleadas, confinadas de forma predominante a la dermis reticular, que se extiende al tejido subcutáneo369., 370., 371.. Ocasionalmente puede haber entremezclados algunos precursores de la serie mieloide.
Los hallazgos de laboratorio dependen de la causa subyacente. En la evaluación del lactante con este cuadro están indicadas las siguientes pruebas: recuento de células sanguíneas, niveles de hemoglobina, serologías TORCH, cultivos virales y prueba de Coombs. La biopsia no siempre es necesaria para el diagnóstico, aunque puede ser de ayuda si se sospecha un proceso infiltrativo.
Diagnóstico diferencial
El diagnóstico diferencial incluye otras causas de púrpura neonatal, como son los defectos de coagulación, alteraciones plaquetarias e infecciones273. También deben considerarse las enfermedades neoplásicas que producen metástasis infiltrativas en el recién nacido, como los neuroblastomas377., 378., 379., rabdomiosarcomas380, leucemias mieloides381., 382., 383. y la histiocitosis de células de Langerhans, especialmente la variante reticulohistiocitosis congénita autoinvolutiva (Hashimoto-Pritzker)384 , 385.
Evolución, manejo, tratamiento y pronóstico
Las lesiones de verdadera eritropoyesis dérmica disminuyen y se resuelven de forma espontánea a las 3-6 semanas del nacimiento. El tratamiento corresponde al de la enfermedad subyacente.
Deficiencia de proteínas C y S (púrpura fulminante neonatal)
La púrpura fulminante neonatal es una enfermedad rara caracterizada por una necrosis hemorrágica masiva y progresiva de la piel acompañada en el período neonatal de trombosis de la vasculatura cutánea386., 387., 388., 389., 390., 391.. Ocasionalmente se afectan vasos mayores y otros órganos. El primer acontecimiento patológico es la trombosis diseminada, que es responsable del cuadro hematológico de coagulación intravascular diseminada (CID). En los neonatos, la púrpura fulminante generalmente es consecuencia de una alteración tromboembólica hereditaria que se atribuye a una deficiencia de proteína C, deficiencia de proteína S o resistencia a la activación de la proteína C como consecuencia de mutaciones en el factor V.
Hallazgos cutáneos
La púrpura fulminante neonatal se manifiesta a las 2-12 horas tras el nacimiento. En casos raros, se ha descrito un inicio tardío a los 6-10 meses de edad392. Las lesiones cutáneas consisten en equímosis extensas con una distribución difusa y con frecuencia simétrica que puede evolucionar espontáneamente a ampollas hemorrágicas y lesiones cutáneas necróticas de color púrpura-oscuro, que finalmente formarán una gruesa escara (v. figura 19-27 ). Las áreas equimóticas iniciales están muy bien definidas de la piel que las rodea y suelen mostrar un anillo rojo, inflamatorio que va avanzando. Son más comunes en los sitios de traumatismos o de presión, las nalgas, las extremidades, el tronco y el cuero cabelludo. Raramente se afectan las mucosas393. Si se instaura el tratamiento en las 1-3 primeras horas, antes de que se desarrolle la necrosis, las lesiones iniciales pueden ser reversibles390.
FIGURA 19-27.
Purpura fulminante neonatal debida a deficiencia de proteína C.
Hallazgos extracutáneos
También pueden afectarse otros órganos por la trombosis microvascular, destacando el SNC y el ojo, aunque también se puede afectar el riñón y el tubo digestivo. La afectación del seno cavernoso, que puede suceder en el útero, puede provocar hidrocefalia, convulsiones, hemorragias intracerebrales y retraso mental388 , 394 , 395. También puede verse microftalmía, cataratas y ceguera como consecuencia de hemorragias en el vítreo o en la retina388 , 389. También se han descrito trombosis venosa profunda y embolias pulmonares394.
Etiología/patogenia
La púrpura fulminante neonatal prácticamente siempre es debida a estados trombofílicos heredados, como son la deficiencia homocigótica de proteínas C y S o resistencia a la activación de la proteína C. Las infecciones bacterianas graves que llevan asociada una CID también pueden inducir una púrpura fulminante neonatal, si bien es más común en la lactancia o en la niñez temprana396 , 397. Las proteínas C y S son glucoproteínas dependientes de vitamina K con propiedades antitrombóticas398 , 399.
El déficit de proteína C es una enfermedad autosómica dominante con penetrancia incompleta399. Los pacientes homocigóticos o heterocigóticos compuestos tienen un fenotipo clínico grave y generalmente se presentan con una púrpura fulminante neonatal, si bien pueden estar asintomáticos o presentarse más tarde en la vida con trombosis de repetición400 , 401.
La deficiencia de proteína S también se transmite de forma autosómica dominante con penetrancia incompleta399. Los pacientes homocigóticos pueden desarrollar una púrpura neonatal fulminante, si bien el riesgo es menor que en los pacientes con déficit homocigótico de proteína C387 , 402.
La púrpura fulminante neonatal también puede estar provocada por resistencia de la proteína C a la activación debido a mutaciones en el gen del factor V393 , 403. La resistencia a la activación de la proteína C puede coexistir con deficiencias de las proteínas S y C, siendo un riesgo adicional para el desarrollo de una púrpura fulminante o complicaciones tromboembólicas, lo que explicaría en parte la incompleta penetrancia clínica de las enfermedades hereditarias tromboembólicas404.
Pruebas de laboratorio e histopatología
Los estudios de coagulación sanguínea muestran un cuadro de CID, incluyendo prolongación del tiempo de protrombina y de tromboplastina parcial, aumento de los productos de degradación de la fibrina, disminución del fibrinógeno y reducción de las plaquetas. Puede verse una anemia hemolítica microangiopática.
La biopsia de las lesiones cutáneas tempranas demuestra una oclusión de los vasos sanguíneos dérmicos por microtrombos. En las fases más avanzadas ya existe hemorragia y necrosis de la epidermis que cubre el área y ampollas subepidérmicas hemorrágicas. La necrosis fibrinoide secundaria de las paredes de los vasos dérmicos puede aparecer en las áreas de necrosis, pero no hay una vasculitis primaria390 , 405.
El diagnóstico definitivo del déficit de proteínas C y S se establece midiendo los valores de dichas proteínas en plasma399. La deficiencia de proteína C puede identificarse con ensayos inmunoenzimáticos midiendo la concentración de la proteína en plasma y dos ensayos funcionales miden la actividad enzimática y la actividad anticoagulante. Estas pruebas diferencian dos tipos de deficiencia de proteína C. En el tipo I, que es el más común, la menor síntesis de la proteína da lugar a una disminución en su concentración plasmática en los tres ensayos. En el tipo II hay una deficiencia cualitativa, de forma que los niveles son normales, mientras que los ensayos de función están alterados. Para la deficiencia de la proteína S están disponibles pruebas funcionales e inmunoenzimáticas y deben medirse tanto la forma libre como la inactiva que circula ligada a la proteína ligadora C4b399. La deficiencia del tipo I se caracteriza por un déficit de la proteína S total y libre, mientras que en el tipo II existe una proteína S normal, con baja actividad, y en el tipo III se observan unos niveles de proteína libre bajos con niveles totales normales.
La interpretación de los resultados de los ensayos puede ser difícil dado que los niveles de proteínas C y S están reducidos fisiológicamente en el período neonatal, y pueden ser indetectables en los recién nacidos enfermos con enfermedad hepática, síndrome de distrés respiratorio, CID o sepsis389 , 390 , 396 , 397 , 406. Por ello se recomienda un cribado completo de sepsis en cualquier caso de púrpura fulminante neonatal. Las determinaciones seriadas de los niveles de proteínas en los pacientes, así como en otros miembros de las familias, son necesarias para excluir una deficiencia transitoria y poder confirmar el diagnóstico de la forma congénita.
Diagnóstico diferencial
Las lesiones cutáneas de la púrpura fulminante son muy características y raramente se confunden con otras alteraciones. Deben considerarse otras causas de erupciones purpúricas en el neonato (v. cuadro 19-6).
Evolución, manejo, tratamiento y pronóstico
Sin tratamiento, la púrpura fulminante neonatal con frecuencia es mortal. Si se sospecha el diagnóstico debe iniciarse el tratamiento de forma inmediata, sin esperar los resultados de los valores de las proteínas C y S. El tratamiento temprano puede revertir por completo las lesiones cutáneas precoces. El tratamiento inicial consiste en la administración de plasma fresco congelado (10-15 mL/kg/12 h) o concentrado del complejo de protrombina, fuentes de proteína C, proteína S y proteína C activada389 , 390. Se ha desarrollado un concentrado de proteína C que tiene la ventaja de evitar la sobrecarga de volumen sanguíneo y no lleva consigo el riesgo de transmisión de enfermedades víricas407 , 408. Todavía no se dispone de un concentrado de proteína S para el uso clínico. El tratamiento sustitutorio debe continuarse hasta que se hayan curado todas las lesiones, generalmente unas 4-8 semanas. El tratamiento a largo plazo implica la administración cuidadosa de anticoagulantes orales, iniciándolos en dosis muy bajas y con el tratamiento protector para evitar la necrosis cutánea inducida por la cumarina. La experiencia a largo plazo con el uso de infusiones de proteína C es limitada. Hay unos pocos casos de trasplante hepático como tratamiento de la forma homocigótica de la deficiencia de proteína C409 , 410.
Erupción purpúrica inducida por la fototerapia
Esta púrpura benigna y transitoria en neonatos a los que se ha transfundido y que reciben fototerapia se caracteriza por lesiones de color frambuesa, que no blanquean, en los sitios expuestos, que respetan los sitios que no están expuestos a la luz (p. ej., sensores y sondas de temperatura)206 , 208. La erupción se desarrolla después de 1-4 días de fototerapia y desaparece espontáneamente tras la fototerapia. Histológicamente hay extravasación de eritrocitos en la dermis sin cambios en la epidermis. La patogenia de la enfermedad es desconocida, si bien en algunos pacientes se ha detectado una porfirinemia transitoria367 , 411. La naturaleza purpúrica de la erupción y la ausencia de «células de quemadura solar» diferencia esta erupción de las «quemaduras solares» provocadas por la exposición a lámparas UVA412. La porfiria eritropoyética congénita y la elevación transitoria de los niveles de porfirina en neonatos con enfermedad hemolítica pueden provocar fotosensibilidad411. También debe considerarse la fotosensibilidad inducida por fármacos en neonatos que hayan recibido agentes químicos fotosensibilizantes, como son los colorantes de fluoresceína, furosemida y azul de metileno413., 414., 415., 416..
Pacientes con fiebre de por lo menos 5 días y <4 criterios principales pueden diagnosticarse de enfermedad de Kawasaki cuando se detecten alteraciones de las arterias coronarias por ecografía en 2 dimensiones o angiografía.
Los pacientes con cuatro criterios principales pueden diagnosticarse al día 4 de fiebre.
La patogenia de esta entidad se relaciona tanto con la trombocitopenia como con una coagulopatía de consumo.
Las infecciones pueden provocar púrpura por varios mecanismos.
Bibliografía
- 1.Bressler GS, Jones RE., Jr Erythema annulare centrifugum. J Am Acad Dermatol. 1981;4:597–602. doi: 10.1016/s0190-9622(81)70063-x. [DOI] [PubMed] [Google Scholar]
- 2.Harrison PV. The annular erythemas. Int J Dermatol. 1979;18:282–290. doi: 10.1111/j.1365-4362.1979.tb01928.x. [DOI] [PubMed] [Google Scholar]
- 3.Toonstra J, de Wit RF. ‘Persistent’ annular erythema of infancy. Arch Dermatol. 1984;120:1069–1072. [PubMed] [Google Scholar]
- 4.Cox NH, McQueen A, Evans TJ, Morley WN. An annular erythema of infancy. Arch Dermatol. 1987;123:510–513. [PubMed] [Google Scholar]
- 5.Hebert AA, Esterly NB. Annular erythema of infancy. J Am Acad Dermatol. 1986;14:339–343. doi: 10.1016/s0190-9622(86)70039-x. [DOI] [PubMed] [Google Scholar]
- 6.Peterson AO, Jr, Jarratt M. Annular erythema of infancy. Arch Dermatol. 1981;117:145–148. [PubMed] [Google Scholar]
- 7.Wong LC, Kakakios A, Rogers M. Congenital annular erythema persisting in a 15-year-old girl. Australas J Dermatol. 2002;43:55–61. doi: 10.1046/j.1440-0960.2002.00556.x. [DOI] [PubMed] [Google Scholar]
- 8.Helm TN, Bass J, Chang LW, Bergfeld WF. Persistent annular erythema of infancy. Pediatr Dermatol. 1993;10:46–48. doi: 10.1111/j.1525-1470.1993.tb00013.x. [DOI] [PubMed] [Google Scholar]
- 9.Bottoni U, Innocenzi D, Bonaccorsi P. Erythema annulare centrifugum: report of a case with neonatal onset. J Eur Acad Dermatol Venereol. 2002;16:500–503. doi: 10.1046/j.1468-3083.2002.00546.x. [DOI] [PubMed] [Google Scholar]
- 10.Gianotti F, Ermacora E. Erythema gyratum atrophicans transiens neonatale. Arch Dermatol. 1975;111:615–616. [PubMed] [Google Scholar]
- 11.Puig L, Moreno A, Alomar A, de Moragas JM. Erythema gyratum atrophicans transiens neonatale: a variant of cutaneous neonatal lupus erythematosus. Pediatr Dermatol. 1988;5:112–116. doi: 10.1111/j.1525-1470.1988.tb01150.x. [DOI] [PubMed] [Google Scholar]
- 12.Tyring SK. Reactive erythemas: erythema annulare centrifugum and erythema gyratum repens. Clin Dermatol. 1993;11:135–139. doi: 10.1016/0738-081x(93)90110-x. [DOI] [PubMed] [Google Scholar]
- 13.Guillet MH, Dorval JC, Larregue M, Guillet G. Erytheme annulaire centrifuge de Darier à debut neonatal avec 15 ans de suivi. Efficacité de l’interféron et role de cytokines. Ann Dermatol Venereol. 1995;122:422–426. [PubMed] [Google Scholar]
- 14.Hammar H. Erythema annulare centrifugum coincident with Epstein–Barr virus infection in an infant. Acta Paediatr Scand. 1974;63:788–792. doi: 10.1111/j.1651-2227.1974.tb17006.x. [DOI] [PubMed] [Google Scholar]
- 15.Fried R, Litt JZ, Schonberg IL. Erythema annulare centrifugum (Darier) in a newborn infant. J Pediatr. 1957;50:66–67. doi: 10.1016/s0022-3476(57)80014-6. [DOI] [PubMed] [Google Scholar]
- 16.Klaber R. Erythema gyratum perstans (Colcott Fox): a case report, with discussion on relations with erythema centrifugum annulare (Darier) and dermatitis herpetiformis. Br J Dermatol. 1946;58:111–121. doi: 10.1111/j.1365-2133.1946.tb16284.x. [DOI] [PubMed] [Google Scholar]
- 17.Larregue M, Beuve-Mery M, Dupuy JM. Érythème annulaire centrifuge type Colcott–Fox (erythema gyratum perstans) Ann Dermatol Venereol. 1977;104:217–223. [PubMed] [Google Scholar]
- 18.Woederman MJ. Erythema gyratum repens. Dermatologica. 1964;128:391–392. doi: 10.1159/000254771. [DOI] [PubMed] [Google Scholar]
- 19.Schmid MH, Wollenberg A, Sander CA, Bieber T. Erythema annulare centrifugum and intestinal Candida albicans infection – coincidence or connection? Acta Dermatol Venereol. 1997;77:93–94. doi: 10.2340/00015555779394. [DOI] [PubMed] [Google Scholar]
- 20.Shelley WB. Erythema annulare centrifugum due to candida albicans. Br J Dermatol. 1965;77:383–384. doi: 10.1111/j.1365-2133.1965.tb14665.x. [DOI] [PubMed] [Google Scholar]
- 21.Hendricks AA, Lu C, Elfenbein GJ, Hussain R. Erythema annulare centrifugum associated with ascariasis. Arch Dermatol. 1981;117:582–585. doi: 10.1001/archderm.1981.01650090064030. [DOI] [PubMed] [Google Scholar]
- 22.Ashurst PJ. Erythema annulare centrifugum due to hydroxychloroquine sulfate and chloroquine sulfate. Arch Dermatol. 1967;95:37–39. doi: 10.1001/archderm.95.1.37. [DOI] [PubMed] [Google Scholar]
- 23.Shelley WB. Erythema annulare centrifugum. A case due to hypersensitivity to blue cheese penicillium. Arch Dermatol. 1964;90:54–58. doi: 10.1001/archderm.1964.01600010060012. [DOI] [PubMed] [Google Scholar]
- 24.Jillson OF, Hoekelman RA. Further amplification of the concept of dermatophytid. I. Erythema annulare centrifugum as a dermatophytid. AMA Arch Derm Syphilol. 1952;66:738–745. doi: 10.1001/archderm.1952.01530310076011. [DOI] [PubMed] [Google Scholar]
- 25.Watsky KL, Hansen T. Annular erythema in identical twins. Cutis. 1989;44:139–140. [PubMed] [Google Scholar]
- 26.Beare JM, Froggatt P, Jones JH, Neill DW. Familial annular erythema. An apparently new dominant mutation. Br J Dermatol. 1966;78:59–68. doi: 10.1111/j.1365-2133.1966.tb12176.x. [DOI] [PubMed] [Google Scholar]
- 27.Kikuchi I, Ogata K, Inoue S. Pityrosporum infection in an infant with lesions resembling erythema annulare centrifugum. Arch Dermatol. 1984;120:380–382. [PubMed] [Google Scholar]
- 28.Trevisan G, Stinco G, Cinco M. Neonatal skin lesions due to a spirochetal infection: a case of congenital Lyme borreliosis? Int J Dermatol. 1997;36:677–680. doi: 10.1046/j.1365-4362.1997.00217.x. [DOI] [PubMed] [Google Scholar]
- 29.Lee LA. Neonatal lupus erythematosus. J Invest Dermatol. 1993;100:9S–13S. doi: 10.1111/1523-1747.ep12355173. [DOI] [PubMed] [Google Scholar]
- 30.Lee LA. Neonatal lupus: clinical features, therapy, and pathogenesis. Curr Rheumatol Rep. 2001;3:391–395. doi: 10.1007/s11926-996-0009-3. [DOI] [PubMed] [Google Scholar]
- 31.Lee LA. Neonatal lupus erythematosus: clinical findings and pathogenesis. J Invest Dermatol Symp Proc. 2004;9:52–56. doi: 10.1111/j.1087-0024.2004.00827.x. [DOI] [PubMed] [Google Scholar]
- 32.Silverman ED, Laxer RM. Neonatal lupus erythematosus. Rheum Dis Clin North Am. 1997;23:599–618. doi: 10.1016/s0889-857x(05)70349-5. [DOI] [PubMed] [Google Scholar]
- 33.Watson RM, Lane AT, Barnett NK. Neonatal lupus erythematosus. A clinical, serological and immunogenetic study with review of the literature. Medicine (Baltimore) 1984;63:362–378. [PubMed] [Google Scholar]
- 34.Buyon JP, Clancy RM. Neonatal lupus: basic research and clinical perspectives. Rheum Dis Clin North Am. 2005;31:299–313. doi: 10.1016/j.rdc.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 35.Weston WL, Morelli JG, Lee LA. The clinical spectrum of anti-Ro-positive cutaneous neonatal lupus erythematosus. [Abstract] J Am Acad Dermatol. 1999;40:675–681. doi: 10.1016/s0190-9622(99)70146-5. [DOI] [PubMed] [Google Scholar]
- 36.Neiman AR, Lee LA, Weston WL, Buyon JP. Cutaneous manifestations of neonatal lupus without heart block: Characteristics of mothers and children enrolled in a national registry. [Abstract] J Pediatr. 2000;137:674–680. doi: 10.1067/mpd.2000.109108. [DOI] [PubMed] [Google Scholar]
- 37.Sheth AP, Esterly NB, Ratoosh SL. U1RNP positive neonatal lupus erythematosus: association with anti-La antibodies? Br J Dermatol. 1995;132:520–526. doi: 10.1111/j.1365-2133.1995.tb08705.x. [DOI] [PubMed] [Google Scholar]
- 38.Provost TT, Watson R, Gammon WR. The neonatal lupus syndrome associated with U1RNP (nRNP) antibodies. N Engl J Med. 1987;316:1135–1138. doi: 10.1056/NEJM198704303161807. [DOI] [PubMed] [Google Scholar]
- 39.Diociaiuti A, Paone C, Giraldi L. Congenital lupus erythematosus: case report and review of the literature. Pediatr Dermatol. 2005;22:240–242. doi: 10.1111/j.1525-1470.2005.22313.x. [DOI] [PubMed] [Google Scholar]
- 40.Elish D, Silverberg NB. Neonatal lupus erythematosus. Cutis. 2006;77:82–86. [PubMed] [Google Scholar]
- 41.Miyagawa S. Neonatal lupus erythematosus: a review of the racial differences and similarities in clinical, serological and immunogenetic features of Japanese versus Caucasian patients. J Dermatol. 2005;32:514–522. doi: 10.1111/j.1346-8138.2005.tb00793.x. [DOI] [PubMed] [Google Scholar]
- 42.Nitta Y. Lupus erythematosus profundus associated with neonatal lupus erythematosus. Br J Dermatol. 1997;136:112–114. [PubMed] [Google Scholar]
- 43.Crowley E, Frieden IJ. Neonatal lupus erythematosus: an unusual congenital presentation with cutaneous atrophy, erosions, alopecia, and pancytopenia. Pediatr Dermatol. 1998;15:38–42. doi: 10.1046/j.1525-1470.1998.1998015038.x. [DOI] [PubMed] [Google Scholar]
- 44.Cimaz R, Biggioggero M, Catelli L. Ultraviolet light exposure is not a requirement for the development of cutaneous neonatal lupus. Lupus. 2002;11:257–260. doi: 10.1191/0961203302lu171cr. [DOI] [PubMed] [Google Scholar]
- 45.See A, Wargon O, Lim A. Neonatal lupus erythematosus presenting as papules on the feet. Australas J Dermatol. 2005;46:172–176. doi: 10.1111/j.1440-0960.2005.00173.x. [DOI] [PubMed] [Google Scholar]
- 46.Thornton CM, Eichenfield LF, Shinall EA. Cutaneous telangiectases in neonatal lupus erythematosus. J Am Acad Dermatol. 1995;33:19–25. doi: 10.1016/0190-9622(95)90004-7. [DOI] [PubMed] [Google Scholar]
- 47.Fritz M, Vats K, Goyal RK. Neonatal lupus and IUGR following alpha-interferon therapy during pregnancy. J Perinatol. 2005;25:552–554. doi: 10.1038/sj.jp.7211342. [DOI] [PubMed] [Google Scholar]
- 48.Geggel RL, Tucker L, Szer I. Postnatal progression from second- to third-degree heart block in neonatal lupus syndrome. J Pediatr. 1988;113:1049–1052. doi: 10.1016/s0022-3476(88)80581-x. [DOI] [PubMed] [Google Scholar]
- 49.Lee LA, Sokol RJ, Buyon JP. Hepatobiliary disease in neonatal lupus: prevalence and clinical characteristics in cases enrolled in a national registry. Pediatrics. 2002;109:E11. doi: 10.1542/peds.109.1.e11. [DOI] [PubMed] [Google Scholar]
- 50.Cimaz R, Spence DL, Hornberger L, Silverman ED. Incidence and spectrum of neonatal lupus erythematosus: a prospective study of infants born to mothers with anti-Ro autoantibodies. J Pediatr. 2003;142:678–683. doi: 10.1067/mpd.2003.233. [DOI] [PubMed] [Google Scholar]
- 51.Scheker LE, Kasteler JS, Callen JP. Neonatal lupus erythematosus mimicking Langerhans’ cell histiocytosis. Pediatr Dermatol. 2003;20:164–166. doi: 10.1046/j.1525-1470.2003.20216.x. [DOI] [PubMed] [Google Scholar]
- 52.Kanagasegar S, Cimaz R, Kurien BT. Neonatal lupus manifests as isolated neutropenia and mildly abnormal liver functions. J Rheumatol. 2002;29:187–191. [PubMed] [Google Scholar]
- 53.Watson R, Kang JE, May M. Thrombocytopenia in the neonatal lupus syndrome. Arch Dermatol. 1988;124:560–563. [PubMed] [Google Scholar]
- 54.Bourke JF, Burns DA. Neonatal lupus erythematosus with persistent telangiectasia and spastic paraparesis. Clin Exp Dermatol. 1993;18:271–273. doi: 10.1111/j.1365-2230.1993.tb02186.x. [DOI] [PubMed] [Google Scholar]
- 55.Rider LG, Sherry DD, Glass ST. Neonatal lupus erythematosus simulating transient myasthenia gravis at presentation. J Pediatr. 1991;118:417–419. doi: 10.1016/s0022-3476(05)82160-2. [DOI] [PubMed] [Google Scholar]
- 56.Contractor S, Hiatt M, Kosmin M, Kim HC. Neonatal thrombosis with anticardiolipin antibody in baby and mother. Am J Perinatol. 1992;9:409–410. doi: 10.1055/s-2007-999276. [DOI] [PubMed] [Google Scholar]
- 57.Prendiville JS, Cabral DA, Poskitt KJ. Central nervous system involvement in neonatal lupus erythematosus. Pediatr Dermatol. 2003;20:60–67. doi: 10.1046/j.1525-1470.2003.03014.x. [DOI] [PubMed] [Google Scholar]
- 58.Lawrence S, Luy L, Laxer R. The health of mothers of children with cutaneous neonatal lupus erythematosus differs from that of mothers of children with congenital heart block. Am J Med. 2000;108:705–709. doi: 10.1016/s0002-9343(00)00408-3. [DOI] [PubMed] [Google Scholar]
- 59.Julkunen H, Eronen M. Long-term outcome of mothers of children with isolated heart block in Finland. Arthritis Rheum. 2001;44:647–652. doi: 10.1002/1529-0131(200103)44:3<647::AID-ANR113>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 60.Brucato A, Franceschini F, Gasparini M. Isolated congenital complete heart block: longterm outcome of mothers, maternal antibody specificity and immunogenetic background. J Rheumatol. 1995;22:533–540. [PubMed] [Google Scholar]
- 61.McCune AB, Weston WL, Lee LA. Maternal and fetal outcome in neonatal lupus erythematosus. Ann Intern Med. 1987;106:518–523. doi: 10.7326/0003-4819-106-4-518. [DOI] [PubMed] [Google Scholar]
- 62.Brucato A, Franceschini F, Buyon JP. Neonatal lupus: long-term outcomes of mothers and children and recurrence rate. Clin Exp Rheumatol. 1997;15:467–473. [PubMed] [Google Scholar]
- 63.Press J, Uziel Y, Laxer RM. Long-term outcome of mothers of children with complete congenital heart block. Am J Med. 1996;100:328–332. doi: 10.1016/S0002-9343(97)89492-2. [DOI] [PubMed] [Google Scholar]
- 64.Buyon JP, Kim MY, Copel JA, Friedman DM. Anti-Ro/SSA antibodies and congenital heart block: necessary but not sufficient. Arthritis Rheum. 2001;44:1723–1727. doi: 10.1002/1529-0131(200108)44:8<1723::AID-ART305>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 65.Gordon P, Khamashta MA, Rosenthal E. Anti-52 kDa Ro, anti-60 kDa Ro, and anti-La antibody profiles in neonatal lupus. J Rheumatol. 2004;31:2480–2487. [PubMed] [Google Scholar]
- 66.Silverman ED, Buyon J, Laxer RM. Autoantibody response to the Ro/La particle may predict outcome in neonatal lupus erythematosus. Clin Exp Immunol. 1995;100:499–505. doi: 10.1111/j.1365-2249.1995.tb03729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stevens AM, Hermes HM, Lambert NC. Maternal and sibling microchimerism in twins and triplets discordant for neonatal lupus syndrome-congenital heart block. Rheumatology (Oxford) 2005;44:187–191. doi: 10.1093/rheumatology/keh453. [DOI] [PubMed] [Google Scholar]
- 68.Costedoat-Chalumeau N, Amoura Z, Villain E. Anti-SSA/Ro antibodies and the heart: more than complete congenital heart block? A review of electrocardiographic and myocardial abnormalities and of treatment options. Arthritis Res Ther. 2005;7:69–73. doi: 10.1186/ar1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nield LE, Silverman ED, Smallhorn JF. Endocardial fibroelastosis associated with maternal anti-Ro and anti-La antibodies in the absence of atrioventricular block. J Am Coll Cardiol. 2002;40:796–802. doi: 10.1016/s0735-1097(02)02004-1. [DOI] [PubMed] [Google Scholar]
- 70.Lee LA, Coulter S, Erner S, Chu H. Cardiac immunoglobulin deposition in congenital heart block associated with maternal anti-Ro autoantibodies. Am J Med. 1987;83:793–796. doi: 10.1016/0002-9343(87)90918-1. [DOI] [PubMed] [Google Scholar]
- 71.Cruz RB, Viana VS, Nishioka SA. Is isolated congenital heart block associated to neonatal lupus requiring pacemaker a distinct cardiac syndrome? Pacing Clin Electrophysiol. 2004;27:615–620. doi: 10.1111/j.1540-8159.2004.00495.x. [DOI] [PubMed] [Google Scholar]
- 72.Costedoat-Chalumeau N, Amoura Z, Lupoglazoff JM. Outcome of pregnancies in patients with anti-SSA/Ro antibodies: a study of 165 pregnancies, with special focus on electrocardiographic variations in the children and comparison with a control group. Arthritis Rheum. 2004;50:3187–3194. doi: 10.1002/art.20554. [DOI] [PubMed] [Google Scholar]
- 73.Taylor-Albert E, Reichlin M, Toews WH. Delayed dilated cardiomyopathy as a manifestation of neonatal lupus: case reports, autoantibody analysis, and management. Pediatrics. 1997;99:733–735. doi: 10.1542/peds.99.5.733. [DOI] [PubMed] [Google Scholar]
- 74.Moak JP, Barron KS, Hougen TJ. Congenital heart block: development of late-onset cardiomyopathy, a previously underappreciated sequela. J Am Coll Cardiol. 2001;37:238–242. doi: 10.1016/s0735-1097(00)01048-2. [DOI] [PubMed] [Google Scholar]
- 75.Eronen M, Heikkila P, Teramo K. Congenital complete heart block in the fetus: hemodynamic features, antenatal treatment, and outcome in six cases. Pediatr Cardiol. 2001;22:385–392. doi: 10.1007/s002460010256. [DOI] [PubMed] [Google Scholar]
- 76.Saleeb S, Copel J, Friedman D, Buyon JP. Comparison of treatment with fluorinated glucocorticoids to the natural history of autoantibody-associated congenital heart block: retrospective review of the research registry for neonatal lupus. Arthritis Rheum. 1999;42:2335–2345. doi: 10.1002/1529-0131(199911)42:11<2335::AID-ANR12>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 77.Yang CH, Chen JY, Lee SC, Luo SF. Successful preventive treatment of congenital heart block during pregnancy in a woman with systemic lupus erythematosus and anti-Sjögren's syndrome A/Ro antibody. J Microbiol Immunol Infect. 2005;38:365–369. [PubMed] [Google Scholar]
- 78.Buyon JP, Swersky SH, Fox HE. Intrauterine therapy for presumptive fetal myocarditis with acquired heart block due to systemic lupus erythematosus. Experience in a mother with a predominance of SS-B (La) antibodies. Arthritis Rheum. 1987;30:44–49. doi: 10.1002/art.1780300106. [DOI] [PubMed] [Google Scholar]
- 79.Ishimaru S, Izaki S, Kitamura K, Morita Y. Neonatal lupus erythematosus: dissolution of atrioventricular block after administration of corticosteroid to the pregnant mother. Dermatology. 1994;189:92–94. doi: 10.1159/000246940. [DOI] [PubMed] [Google Scholar]
- 80.Martin V, Lee LA, Askanase AD. Long-term followup of children with neonatal lupus and their unaffected siblings. Arthritis Rheum. 2002;46:2377–2383. doi: 10.1002/art.10638. [DOI] [PubMed] [Google Scholar]
- 81.Buyon JP, Clancy RM. Neonatal lupus: review of proposed pathogenesis and clinical data from the US-based Research Registry for Neonatal Lupus. Autoimmunity. 2003;36:41–50. doi: 10.1080/0891693031000067340. [DOI] [PubMed] [Google Scholar]
- 82.Carder KR. Hypersensitivity reactions in neonates and infants. Dermatol Ther. 2005;18:160–175. doi: 10.1111/j.1529-8019.2005.05014.x. [DOI] [PubMed] [Google Scholar]
- 83.Wintroub BU, Stern R. Cutaneous drug reactions: pathogenesis and clinical classification. J Am Acad Dermatol. 1985;13:167–179. doi: 10.1016/s0190-9622(85)70156-9. [DOI] [PubMed] [Google Scholar]
- 84.Bonnetblanc JM. Réactions cutanées aux médicaments chez l’enfant. Ann Dermatol Venereol. 1997;124:339–345. [PubMed] [Google Scholar]
- 85.Knowles S, Shapiro L, Shear NH. Drug eruptions in children. Adv Dermatol. 1999;14:399–415. discussion 416. [PubMed] [Google Scholar]
- 86.Knowles S, Shapiro L, Shear NH. Serious dermatologic reactions in children. Curr Opin Pediatr. 1997;9:388–395. doi: 10.1097/00008480-199708000-00014. [DOI] [PubMed] [Google Scholar]
- 87.Kramer MS, Hutchinson TA, Flegel KM. Adverse drug reactions in general pediatric outpatients. J Pediatr. 1985;106:305–310. doi: 10.1016/s0022-3476(85)80314-0. [DOI] [PubMed] [Google Scholar]
- 88.Sharma VK, Dhar S. Clinical pattern of cutaneous drug eruption among children and adolescents in north India. Pediatr Dermatol. 1995;12:178–183. doi: 10.1111/j.1525-1470.1995.tb00149.x. [DOI] [PubMed] [Google Scholar]
- 89.Calobrisi SD, Drolet BA, Esterly NB. Petechial eruption after the application of EMLA cream. Pediatrics. 1998;101:471–473. doi: 10.1542/peds.101.3.471. [DOI] [PubMed] [Google Scholar]
- 90.Gourrier E, Karoubi P, el HA, Merbouche S. Use of EMLA cream in a department of neonatology. Pain. 1996;68:431–434. doi: 10.1016/s0304-3959(96)03208-3. [DOI] [PubMed] [Google Scholar]
- 91.Lacouture PG, Epstein MF, Mitchell AA. Vancomycin-associated shock and rash in newborn infants. J Pediatr. 1987;111:615–616. doi: 10.1016/s0022-3476(87)80133-6. [DOI] [PubMed] [Google Scholar]
- 92.Odio C, Mohs E, Sklar FH. Adverse reactions to vancomycin used as prophylaxis for CSF shunt procedures. Am J Dis Child. 1984;138:17–19. doi: 10.1001/archpedi.1984.02140390009004. [DOI] [PubMed] [Google Scholar]
- 93.Schaad UB, McCracken GH, Jr, Nelson JD. Clinical pharmacology and efficacy of vancomycin in pediatric patients. J Pediatr. 1980;96:119–126. doi: 10.1016/s0022-3476(80)80347-7. [DOI] [PubMed] [Google Scholar]
- 94.Salomon D, Saurat JH. Erythema multiforme major in a 2-month-old child with human immunodeficiency virus (HIV) infection. Br J Dermatol. 1990;123:797–800. doi: 10.1111/j.1365-2133.1990.tb04200.x. [DOI] [PubMed] [Google Scholar]
- 95.Prose NS. Cutaneous manifestations of pediatric HIV infection. Pediatr Dermatol. 1992;9:326–328. doi: 10.1111/j.1525-1470.1992.tb00618.x. [DOI] [PubMed] [Google Scholar]
- 96.Chanock SJ, Luginbuhl LM, McIntosh K, Lipshultz SE. Life-threatening reaction to trimethoprim/sulfamethoxazole in pediatric human immunodeficiency virus infection. Pediatrics. 1994;93:519–521. [PubMed] [Google Scholar]
- 97.Cohen HA, Cohen Z, Frydman M. Fixed drug eruption of the scrotum due to hydroxyzine hydrochloride (Atarax) Cutis. 1996;57:431–432. [PubMed] [Google Scholar]
- 98.King BA, Geelhoed GC. Adverse skin and joint reactions associated with oral antibiotics in children: The role of cefaclor in serum sickness-like reactions. J Paediatr Child Health. 2003;39:677–681. doi: 10.1046/j.1440-1754.2003.00267.x. [DOI] [PubMed] [Google Scholar]
- 99.Vial T, Pont J, Pham E. Cefaclor-associated serum sickness-like disease: eight cases and review of the literature. Ann Pharmacother. 1992;26:910–914. doi: 10.1177/106002809202600708. [DOI] [PubMed] [Google Scholar]
- 100.Hebert AA, Sigman ES, Levy ML. Serum sickness-like reactions from cefaclor in children. J Am Acad Dermatol. 1991;25:805–808. doi: 10.1016/s0190-9622(08)80973-5. [DOI] [PubMed] [Google Scholar]
- 101.Carroll MC, Yueng-Yue KA, Esterly NB, Drolet BA. Drug-induced hypersensitivity syndrome in pediatric patients. Pediatrics. 2001;108:485–492. doi: 10.1542/peds.108.2.485. [DOI] [PubMed] [Google Scholar]
- 102.Bessmertny O, Hatton RC, Gonzalez-Peralta RP. Antiepileptic hypersensitivity syndrome in children. Ann Pharmacother. 2001;35:533–538. doi: 10.1345/aph.10284. [DOI] [PubMed] [Google Scholar]
- 103.Yigit S, Korkmaz A, Sekerel B. Drug-induced hypersensitivity syndrome in a premature infant. Pediatr Dermatol. 2005;22:71–74. doi: 10.1111/j.1525-1470.2005.22116.x. [DOI] [PubMed] [Google Scholar]
- 104.Chen CJ, Huang YC, Wang CY, Lin TY. Fatal anticonvulsant hypersensitivity syndrome in an infant. Eur J Pediatr. 2003;162:893–894. doi: 10.1007/s00431-003-1190-2. [DOI] [PubMed] [Google Scholar]
- 105.Lohmeier K, Megahed M, Schulte KW. Toxic epidermal necrolysis in a premature infant of 27 weeks’ gestational age. Br J Dermatol. 2005;152:150–151. doi: 10.1111/j.1365-2133.2004.06258.x. [DOI] [PubMed] [Google Scholar]
- 106.Bel S, Bartralot R, Garcia D. Vegetant bromoderma in an infant. Pediatr Dermatol. 2001;18:336–338. doi: 10.1046/j.1525-1470.2001.01954.x. [DOI] [PubMed] [Google Scholar]
- 107.Torrelo A, Madero L, Mediero IG, Zambrano A. A cutaneous eruption from G-CSF in a healthy donor. Pediatr Dermatol. 2000;17:205–207. doi: 10.1046/j.1525-1470.2000.01753.x. [DOI] [PubMed] [Google Scholar]
- 108.Buyukgebiz B, Arslan N, Ozturk Y. Drug reaction to ursodeoxycholic acid: lichenoid drug eruption in an infant using ursodeoxycholic acid for neonatal hepatitis. J Pediatr Gastroenterol Nutr. 2002;35:384–386. doi: 10.1097/00005176-200209000-00030. [DOI] [PubMed] [Google Scholar]
- 109.Tamayo-Sanchez L, Ruiz-Maldonado R, Laterza A. Acute annular urticaria in infants and children. Pediatr Dermatol. 1997;14:231–234. doi: 10.1111/j.1525-1470.1997.tb00246.x. [DOI] [PubMed] [Google Scholar]
- 110.Haas N, Birkle-Berlinger W, Henz BM. Prognosis of acute urticaria in children. Acta Dermatol Venereol. 2005;85:74–75. doi: 10.1080/00015550410023545. [DOI] [PubMed] [Google Scholar]
- 111.Sackesen C, Sekerel BE, Orhan F. The etiology of different forms of urticaria in childhood. Pediatr Dermatol. 2004;21:102–108. doi: 10.1111/j.0736-8046.2004.21202.x. [DOI] [PubMed] [Google Scholar]
- 112.Mortureux P, Leaute-Labreze C, Legrain-Lifermann V. Acute urticaria in infancy and early childhood: a prospective study. Arch Dermatol. 1998;134:319–323. doi: 10.1001/archderm.134.3.319. [DOI] [PubMed] [Google Scholar]
- 113.Beltrani VS. Urticaria: reassessed. Allergy Asthma Proc. 2004;25:143–149. [PubMed] [Google Scholar]
- 114.Ghosh S, Kanwar AJ, Kaur S. Urticaria in children. Pediatr Dermatol. 1993;10:107–110. doi: 10.1111/j.1525-1470.1993.tb00032.x. [DOI] [PubMed] [Google Scholar]
- 115.Legrain V, Taieb A, Sage T, Maleville J. Urticaria in infants: a study of forty patients. Pediatr Dermatol. 1990;7:101–107. doi: 10.1111/j.1525-1470.1990.tb00663.x. [DOI] [PubMed] [Google Scholar]
- 116.Kauppinen K, Juntunen K, Lanki H. Urticaria in children. Retrospective evaluation and follow-up. Allergy. 1984;39:469–472. doi: 10.1111/j.1398-9995.1984.tb01970.x. [DOI] [PubMed] [Google Scholar]
- 117.Carter EL, Garzon MC. Neonatal urticaria due to prostaglandin E1. Pediatr Dermatol. 2000;17:58–61. doi: 10.1046/j.1525-1470.2000.01712.x. [DOI] [PubMed] [Google Scholar]
- 118.Hamel-Teillac D. Les urticaires chroniques de l’enfant. Ann Dermatol Venereol. 2003;130:1S69–11S. [PubMed] [Google Scholar]
- 119.Dice JP. Physical urticaria. Immunol Allergy Clin North Am. 2004;24:225–246. doi: 10.1016/j.iac.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 120.Heine RG, Elsayed S, Hosking CS, Hill DJ. Cow's milk allergy in infancy. Curr Opin Allergy Clin Immunol. 2002;2:217–225. doi: 10.1097/00130832-200206000-00011. [DOI] [PubMed] [Google Scholar]
- 121.Guillet MH, Guillet G. L’urticaire alimentaire de l’enfant. Revue de 51 observations. Allerg Immunol (Paris) 1993;25:333–338. [PubMed] [Google Scholar]
- 122.Ruiz-Maldonado R, Tamayo L, Ridaura C. Diffuse dermographic mastocytosis without visible skin lesions. Int J Dermatol. 1975;14:126–128. doi: 10.1111/j.1365-4362.1975.tb01429.x. [DOI] [PubMed] [Google Scholar]
- 123.Wanderer AA, Hoffman HM. The spectrum of acquired and familial cold-induced urticaria/urticaria-like syndromes. Immunol Allergy Clin North Am. 2004;24:259–286. doi: 10.1016/j.iac.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 124.Zip CM, Ross JB, Greaves MW. Familial cold urticaria. Clin Exp Dermatol. 1993;18:338–341. doi: 10.1111/j.1365-2230.1993.tb02212.x. [DOI] [PubMed] [Google Scholar]
- 125.Tindall JP, Beeker SK, Rosse WF. Familial cold urticaria. A generalized reaction involving leukocytosis. Arch Intern Med. 1969;124:129–134. [PubMed] [Google Scholar]
- 126.Doeglas HM, Bleumink E. Familial cold urticaria. Clinical findings. Arch Dermatol. 1974;110:382–388. [PubMed] [Google Scholar]
- 127.Ormerod AD, Smart L, Reid TM, Milford-Ward A. Familial cold urticaria. Investigation of a family and response to stanozolol. Arch Dermatol. 1993;129:343–346. doi: 10.1001/archderm.129.3.343. [DOI] [PubMed] [Google Scholar]
- 128.Haas N, Kuster W, Zuberbier T, Henz BM. Muckle–Wells syndrome: clinical and histological skin findings compatible with cold air urticaria in a large kindred. Br J Dermatol. 2004;151:99–104. doi: 10.1111/j.1365-2133.2004.06001.x. [DOI] [PubMed] [Google Scholar]
- 129.Dode C, Le DN, Cuisset L. New mutations of CIAS1 that are responsible for Muckle–Wells syndrome and familial cold urticaria: a novel mutation underlies both syndromes. Am J Hum Genet. 2002;70:1498–1506. doi: 10.1086/340786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pradalier A, Cauvain A, Oukachbi Z. Inherited autoinflammatory recurrent fevers. Allerg Immunol (Paris) 2006;38:5–9. [PubMed] [Google Scholar]
- 131.Jedele KB, Michels VV. Familial dermographism. Am J Med Genet. 1991;39:201–203. doi: 10.1002/ajmg.1320390216. [DOI] [PubMed] [Google Scholar]
- 132.Sahihi T, Esterly NB. Atypical diffuse cutaneous mastocytosis. Am J Dis Child. 1972;124:133–135. doi: 10.1001/archpedi.1972.02110130135022. [DOI] [PubMed] [Google Scholar]
- 133.Epstein PA, Kidd KK. Dermo-distortive urticaria: an autosomal dominant dermatologic disorder. Am J Med Genet. 1981;9:307–315. doi: 10.1002/ajmg.1320090407. [DOI] [PubMed] [Google Scholar]
- 134.Patterson R, Mellies CJ, Blankenship ML, Pruzansky JJ. Vibratory angioedema: a hereditary type of physical hypersensitivity. J Allergy Clin Immunol. 1972;50:174–182. doi: 10.1016/0091-6749(72)90048-6. [DOI] [PubMed] [Google Scholar]
- 135.Pitarch G, Torrijos A, Martinez-Menchon T. Familial aquagenic urticaria and Bernard–Soulier syndrome. Dermatology. 2006;212:96–97. doi: 10.1159/000089035. [DOI] [PubMed] [Google Scholar]
- 136.Bonnetblanc JM, Andrieu-Pfahl F, Meraud JP, Roux J. Familial aquagenic urticaria. Dermatologica. 1979;158:468–470. doi: 10.1159/000250799. [DOI] [PubMed] [Google Scholar]
- 137.Michaelsson G, Ros AM. Familial localized heat urticaria of delayed type. Acta Dermatol Venereol. 1971;51:279–283. [PubMed] [Google Scholar]
- 138.Kilcline C, Shinkai K, Bree A. Neonatal-onset multisystem inflammatory disorder: the emerging role of pyrin genes in autoinflammatory diseases. Arch Dermatol. 2005;141:248–253. doi: 10.1001/archderm.141.2.248. [DOI] [PubMed] [Google Scholar]
- 139.Hull KM, Shoham N, Chae JJ. The expanding spectrum of systemic autoinflammatory disorders and their rheumatic manifestations. Curr Opin Rheumatol. 2003;15:61–69. doi: 10.1097/00002281-200301000-00011. [DOI] [PubMed] [Google Scholar]
- 140.Prieur AM. A recently recognised chronic inflammatory disease of early onset characterised by the triad of rash, central nervous system involvement and arthropathy. Clin Exp Rheumatol. 2001;19:103–106. [PubMed] [Google Scholar]
- 141.Huttenlocher A, Frieden IJ, Emery H. Neonatal onset multisystem inflammatory disease. J Rheumatol. 1995;22:1171–1173. [PubMed] [Google Scholar]
- 142.Torbiak RP, Dent PB, Cockshott WP. NOMID – a neonatal syndrome of multisystem inflammation. Skeletal Radiol. 1989;18:359–364. doi: 10.1007/BF00361425. [DOI] [PubMed] [Google Scholar]
- 143.Dollfus H, Hafner R, Hofmann HM. Chronic infantile neurological cutaneous and articular/neonatal onset multisystem inflammatory disease syndrome: ocular manifestations in a recently recognized chronic inflammatory disease of childhood. Arch Ophthalmol. 2000;118:1386–1392. doi: 10.1001/archopht.118.10.1386. [DOI] [PubMed] [Google Scholar]
- 144.Feldmann J, Prieur AM, Quartier P. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet. 2002;71:198–203. doi: 10.1086/341357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Neven B, Callebaut I, Prieur AM. Molecular basis of the spectral expression of CIAS1 mutations associated with phagocytic cellmediated autoinflammatory disorders CINCA/NOMID, MWS, and FCU. Blood. 2004;103:2809–2815. doi: 10.1182/blood-2003-07-2531. [DOI] [PubMed] [Google Scholar]
- 146.Miura M, Okabe T, Tsubata S. Chronic infantile neurological cutaneous articular syndrome in a patient from Japan. Eur J Pediatr. 1997;156:624–626. doi: 10.1007/s004310050678. [DOI] [PubMed] [Google Scholar]
- 147.Hassink SG, Goldsmith DP. Neonatal onset multisystem inflammatory disease. Arthritis Rheum. 1983;26:668–673. doi: 10.1002/art.1780260515. [DOI] [PubMed] [Google Scholar]
- 148.Kallinich T, Hoffman HM, Roth J, Keitzer R. The clinical course of a child with CINCA/NOMID syndrome improved during and after treatment with thalidomide. Scand J Rheumatol. 2005;34:246–249. doi: 10.1080/03009740410010236. [DOI] [PubMed] [Google Scholar]
- 149.Hawkins PN, Lachmann HJ, Aganna E, McDermott MF. Spectrum of clinical features in Muckle–Wells syndrome and response to anakinra. Arthritis Rheum. 2004;50:607–612. doi: 10.1002/art.20033. [DOI] [PubMed] [Google Scholar]
- 150.Boschan C, Witt O, Lohse P. Neonatal-onset multisystem inflammatory disease (NOMID) due to a novel S331R mutation of the CIAS1 gene and response to interleukin-1 receptor antagonist treatment. Am J Med Genet A. 2006;140:883–886. doi: 10.1002/ajmg.a.31148. [DOI] [PubMed] [Google Scholar]
- 151.Lovell DJ, Bowyer SL, Solinger AM. Interleukin-1 blockade by anakinra improves clinical symptoms in patients with neonatal-onset multisystem inflammatory disease. Arthritis Rheum. 2005;52:1283–1286. doi: 10.1002/art.20953. [DOI] [PubMed] [Google Scholar]
- 152.Matsubayashi T, Sugiura H, Arai T. Anakinra therapy for CINCA syndrome with a novel mutation in exon 4 of the CIAS1 gene. Acta Paediatr. 2006;95:246–249. doi: 10.1080/08035250500341451. [DOI] [PubMed] [Google Scholar]
- 153.Federico G, Rigante D, Pugliese AL. Etanercept induces improvement of arthropathy in chronic infantile neurological cutaneous articular (CINCA) syndrome. Scand J Rheumatol. 2003;32:312–314. doi: 10.1080/03009740310003974. [DOI] [PubMed] [Google Scholar]
- 154.Brice SL, Huff JC, Weston WL. Erythema multiforme minor in children. Pediatrician. 1991;18:188–194. [PubMed] [Google Scholar]
- 155.Dikland WJ, Oranje AP, Stolz E, van JT. Erythema multiforme in childhood and early infancy. Pediatr Dermatol. 1986;3:135–139. doi: 10.1111/j.1525-1470.1986.tb00504.x. [DOI] [PubMed] [Google Scholar]
- 156.Hurwitz S. Erythema multiforme: a review of its characteristics, diagnostic criteria, and management. Pediatr Rev. 1990;11:217–222. doi: 10.1542/pir.11-7-217. [DOI] [PubMed] [Google Scholar]
- 157.Roujeau JC. Stevens–Johnson syndrome and toxic epidermal necrolysis are severity variants of the same disease which differs from erythema multiforme. J Dermatol. 1997;24:726–729. doi: 10.1111/j.1346-8138.1997.tb02524.x. [DOI] [PubMed] [Google Scholar]
- 158.Bastuji-Garin S, Rzany B, Stern RS. Clinical classification of cases of toxic epidermal necrolysis, Stevens–Johnson syndrome, and erythema multiforme. Arch Dermatol. 1993;129:92–96. [PubMed] [Google Scholar]
- 159.Ashkenazi S, Metzker A, Rachmel A, Nitzan M. Erythema multiforme as a single manifestation of cow's milk intolerance. Acta Paediatr. 1992;81:729–730. doi: 10.1111/j.1651-2227.1992.tb12349.x. [DOI] [PubMed] [Google Scholar]
- 160.Johnston GA, Ghura HS, Carter E, Graham-Brown RA. Neonatal erythema multiforme major. Clin Exp Dermatol. 2002;27:661–664. doi: 10.1046/j.1365-2230.2002.01098.x. [DOI] [PubMed] [Google Scholar]
- 161.Nanda S, Pandhi D, Reddy BS. Erythema multiforme in a 9-day-old neonate. Pediatr Dermatol. 2003;20:454–455. doi: 10.1046/j.1525-1470.2003.20332.x. [DOI] [PubMed] [Google Scholar]
- 162.Torrelo A, Moreno M, de P, I, Celma ML, Zambrano A. Erythema multiforme in a neonate. J Am Acad Dermatol 2003; 48: S78–S79. [DOI] [PubMed]
- 163.Sakurai M. Erythema multiforme in children: unusual clinical features with seasonal occurrence. J Dermatol. 1989;16:361–368. doi: 10.1111/j.1346-8138.1989.tb01281.x. [DOI] [PubMed] [Google Scholar]
- 164.Weston WL, Brice SL, Jester JD. Herpes simplex virus in childhood erythema multiforme. Pediatrics. 1992;89:32–34. [PubMed] [Google Scholar]
- 165.Frederiksen MS, Brenoe E, Trier J. Erythema multiforme minor following vaccination with paediatric vaccines. Scand J Infect Dis. 2004;36:154–155. doi: 10.1080/00365540410019200. [DOI] [PubMed] [Google Scholar]
- 166.Renfro L, Grant-Kels JM, Feder HM, Jr, Daman LA. Controversy: are systemic steroids indicated in the treatment of erythema multiforme? Pediatr Dermatol. 1989;6:43–50. doi: 10.1111/j.1525-1470.1989.tb00266.x. [DOI] [PubMed] [Google Scholar]
- 167.Yeung AK, Goldman RD. Use of steroids for erythema multiforme in children. Can Fam Phys. 2005;51:1481–1483. [PMC free article] [PubMed] [Google Scholar]
- 168.Tatnall FM, Schofield JK, Leigh IM. A double-blind, placebo-controlled trial of continuous acyclovir therapy in recurrent erythema multiforme. Br J Dermatol. 1995;132:267–270. doi: 10.1111/j.1365-2133.1995.tb05024.x. [DOI] [PubMed] [Google Scholar]
- 169.Samimi SS, Siegfried E. Stevens-Johnson syndrome developing in a girl with systemic lupus erythematosus on high-dose corticosteroid therapy. Pediatr Dermatol. 2002;19:52–55. doi: 10.1046/j.1525-1470.2002.00002.x. [DOI] [PubMed] [Google Scholar]
- 170.Kakourou T, Klontza D, Soteropoulou F, Kattamis C. Corticosteroid treatment of erythema multiforme major (Stevens–Johnson syndrome) in children. Eur J Pediatr. 1997;156:90–93. doi: 10.1007/s004310050561. [DOI] [PubMed] [Google Scholar]
- 171.Halebian PH, Corder VJ, Madden MR, Finklestein JL, Shires GT. Improved burn center survival of patients with toxic epidermal necrolysis managed without corticosteroids. Ann Surg. 1986;204:503–512. doi: 10.1097/00000658-198611000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cohen PR, Kurzrock R. Sweet's syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761–778. doi: 10.1046/j.1365-4362.2003.01891.x. [DOI] [PubMed] [Google Scholar]
- 173.Callen JP. Neutrophilic dermatoses. Dermatol Clin. 2002;20:409–419. doi: 10.1016/s0733-8635(02)00006-2. [DOI] [PubMed] [Google Scholar]
- 174.Fitzgerald RL, McBurney EI, Nesbitt LT., Jr Sweet's syndrome. Int J Dermatol. 1996;35:9–15. doi: 10.1111/j.1365-4362.1996.tb01607.x. [DOI] [PubMed] [Google Scholar]
- 175.von den DP. Sweet's syndrome (acute febrile neutrophilic dermatosis) J Am Acad Dermatol. 1994;31:535–556. doi: 10.1016/s0190-9622(94)70215-2. [DOI] [PubMed] [Google Scholar]
- 176.Kemmett D, Hunter JA. Sweet's syndrome: a clinicopathologic review of twenty-nine cases. J Am Acad Dermatol. 1990;23:503–507. doi: 10.1016/0190-9622(90)70250-l. [DOI] [PubMed] [Google Scholar]
- 177.Dunn TR, Saperstein HW, Biederman A, Kaplan RP. Sweet syndrome in a neonate with aseptic meningitis. Pediatr Dermatol. 1992;9:288–292. doi: 10.1111/j.1525-1470.1992.tb00350.x. [DOI] [PubMed] [Google Scholar]
- 178.Hassouna L, Nabulsi-Khalil M, Mroueh SM. Multiple erythematous tender papules and nodules in an 11-month-old boy. Sweet syndrome (SS) (acute febrile neutrophilic dermatosis) Arch Dermatol. 1996;132:1507. doi: 10.1001/archderm.1996.03890360099018. 1510. [DOI] [PubMed] [Google Scholar]
- 179.Levin DL, Esterly NB, Herman JJ, Boxall LB. The Sweet syndrome in children. J Pediatr. 1981;99:73–78. doi: 10.1016/s0022-3476(81)80960-2. [DOI] [PubMed] [Google Scholar]
- 180.Prasad PV, Ambujam S, Priya K. Sweet's syndrome in an infant – report of a rare case. Int J Dermatol. 2002;41:928–930. doi: 10.1046/j.1365-4362.2002.01681_1.x. [DOI] [PubMed] [Google Scholar]
- 181.Sedel D, Huguet P, Lebbe C. Sweet syndrome as the presenting manifestation of chronic granulomatous disease in an infant. Pediatr Dermatol. 1994;11:237–240. doi: 10.1111/j.1525-1470.1994.tb00593.x. [DOI] [PubMed] [Google Scholar]
- 182.Brady RC, Morris J, Connelly BL, Boiko S. Sweet's syndrome as an initial manifestation of pediatric human immunodeficiency virus infection. Pediatrics. 1999;104:1142–1144. doi: 10.1542/peds.104.5.1142. [DOI] [PubMed] [Google Scholar]
- 183.Collins P, Rogers S, Keenan P, McCabe M. Acute febrile neutrophilic dermatosis in childhood (Sweet's syndrome) Br J Dermatol. 1991;124:203–206. doi: 10.1111/j.1365-2133.1991.tb00435.x. [DOI] [PubMed] [Google Scholar]
- 184.Haliasos E, Soder B, Rubenstein DS. Pediatric Sweet syndrome and immunodeficiency successfully treated with intravenous immunoglobulin. Pediatr Dermatol. 2005;22:530–535. doi: 10.1111/j.1525-1470.2005.00133.x. [DOI] [PubMed] [Google Scholar]
- 185.Hazen PG, Kark EC, Davis BR. Acute febrile neutrophilic dermatosis in children. Report of two cases in male infants. Arch Dermatol. 1983;119:998–1002. [PubMed] [Google Scholar]
- 186.Itami S, Nishioka K. Sweet's syndrome in infancy. Br J Dermatol. 1980;103:449–451. doi: 10.1111/j.1365-2133.1980.tb07270.x. [DOI] [PubMed] [Google Scholar]
- 187.Lipp KE, Shenefelt PD, Nelson RP., Jr Persistent Sweet's syndrome occurring in a child with a primary immunodeficiency. J Am Acad Dermatol. 1999;40:838–841. doi: 10.1053/jd.1999.v40.a95650. [DOI] [PubMed] [Google Scholar]
- 188.Parsapour K, Reep MD, Gohar K. Familial Sweet's syndrome in 2 brothers, both seen in the first 2 weeks of life. J Am Acad Dermatol. 2003;49:132–138. doi: 10.1067/mjd.2003.328. [DOI] [PubMed] [Google Scholar]
- 189.Saxe N, Gordon W. Acute febrile neutrophilic dermatosis (Sweet's syndrome). Four case reports. S Afr Med J. 1978;53:253–256. [PubMed] [Google Scholar]
- 190.Tuerlinckx D, Bodart E, Despontin K. Sweet's syndrome with arthritis in an 8-month-old boy. J Rheumatol. 1999;26:440–442. [PubMed] [Google Scholar]
- 191.Hwang ST, Williams ML, McCalmont TH, Frieden IJ. Sweet's syndrome leading to acquired cutis laxa (Marshall's syndrome) in an infant with alpha 1-antitrypsin deficiency. Arch Dermatol. 1995;131:1175–1177. [PubMed] [Google Scholar]
- 192.Nobeyama Y, Kamide R. Sweet's syndrome with neurologic manifestation: case report and literature review. Int J Dermatol. 2003;42:438–443. doi: 10.1046/j.1365-4362.2003.01795.x. [DOI] [PubMed] [Google Scholar]
- 193.Cohen PR, Kurzrock R. Sweet's syndrome and cancer. Clin Dermatol. 1993;11:149–157. doi: 10.1016/0738-081x(93)90112-p. [DOI] [PubMed] [Google Scholar]
- 194.Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet's syndrome. J Am Acad Dermatol. 1996;34:918–923. doi: 10.1016/s0190-9622(96)90080-8. [DOI] [PubMed] [Google Scholar]
- 195.Govindarajan G, Bashir Q, Kuppuswamy S, Brooks C. Sweet syndrome associated with furosemide. South Med J. 2005;98:570–572. doi: 10.1097/01.SMJ.0000157530.63614.9B. [DOI] [PubMed] [Google Scholar]
- 196.Khan DB, Jappe U. Drug-induced Sweet's syndrome in acne caused by different tetracyclines: case report and review of the literature. Br J Dermatol. 2002;147:558–562. doi: 10.1046/j.1365-2133.2002.04817.x. [DOI] [PubMed] [Google Scholar]
- 197.Jain KK. Sweet's syndrome associated with granulocyte colony-stimulating factor. Cutis. 1996;57:107–110. [PubMed] [Google Scholar]
- 198.Jovanovic M, Poljacki M, Vujanovic L, Duran V. Acute febrile neutrophilic dermatosis (Sweet's syndrome) after influenza vaccination. J Am Acad Dermatol. 2005;52:367–369. doi: 10.1016/j.jaad.2004.07.061. [DOI] [PubMed] [Google Scholar]
- 199.Cohen PR, Kurzrock R. Sweet's syndrome: a review of current treatment options. Am J Clin Dermatol. 2002;3:117–131. doi: 10.2165/00128071-200203020-00005. [DOI] [PubMed] [Google Scholar]
- 200.Myatt AE, Baker DJ, Byfield DM. Sweet's syndrome: a report on the use of potassium iodide. Clin Exp Dermatol. 1987;12:345–349. doi: 10.1111/j.1365-2230.1987.tb02506.x. [DOI] [PubMed] [Google Scholar]
- 201.Suehisa S, Tagami H, Inoue F. Colchicine in the treatment of acute febrile neutrophilic dermatosis (Sweet's syndrome) Br J Dermatol. 1983;108:99–101. doi: 10.1111/j.1365-2133.1983.tb04584.x. [DOI] [PubMed] [Google Scholar]
- 202.Aram H. Acute febrile neutrophilic dermatosis (Sweet's syndrome). Response to dapsone. Arch Dermatol. 1984;120:245–247. [PubMed] [Google Scholar]
- 203.Kawasaki T, Kosaki F, Okawa S. A new infantile acute febrile mucocutaneous lymph node syndrome (MLNS) prevailing in Japan. Pediatrics. 1974;54:271–276. [PubMed] [Google Scholar]
- 204.Burns JC, Glode MP. Kawasaki syndrome. Lancet. 2004;364:533–544. doi: 10.1016/S0140-6736(04)16814-1. [DOI] [PubMed] [Google Scholar]
- 205.Newburger JW, Fulton DR. Kawasaki disease. Curr Opin Pediatr. 2004;16:508–514. doi: 10.1097/01.mop.0000137796.23813.64. [DOI] [PubMed] [Google Scholar]
- 206.Falcini F. Kawasaki disease. Curr Opin Rheumatol. 2006;18:33–38. doi: 10.1097/01.bor.0000197998.50450.f6. [DOI] [PubMed] [Google Scholar]
- 207.Levin M, Tizard EJ, Dillon MJ. Kawasaki disease: recent advances. Arch Dis Child. 1991;66:1369–1372. doi: 10.1136/adc.66.12.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Rosenfeld EA, Corydon KE, Shulman ST. Kawasaki disease in infants less than one year of age. J Pediatr. 1995;126:524–529. doi: 10.1016/s0022-3476(95)70344-6. [DOI] [PubMed] [Google Scholar]
- 209.Burns JC, Wiggins JW, Jr, Toews WH. Clinical spectrum of Kawasaki disease in infants younger than 6 months of age. J Pediatr. 1986;109:759–763. doi: 10.1016/s0022-3476(86)80689-8. [DOI] [PubMed] [Google Scholar]
- 210.Stanley TV, Grimwood K. Classical Kawasaki disease in a neonate. Arch Dis Child Fetal Neonatal Ed. 2002;86:F135–F136. doi: 10.1136/fn.86.2.F135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Krapf R, Zimmermann A, Stocker F. Lethal vasculitis of coronary arteries in a neonate and two infants: possible neonatal variant of the MLNS/IPN complex? Helv Paediatr Acta. 1981;36:589–598. [PubMed] [Google Scholar]
- 212.Bolz D, Arbenz U, Fanconi S, Bauersfeld U. Myocarditis and coronary dilatation in the 1st week of life: neonatal incomplete Kawasaki disease? Eur J Pediatr. 1998;157:589–591. doi: 10.1007/s004310050885. [DOI] [PubMed] [Google Scholar]
- 213.Burgner D, Harnden A. Kawasaki disease: what is the epidemiology telling us about the etiology? Int J Infect Dis. 2005;9:185–194. doi: 10.1016/j.ijid.2005.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Bell DM, Morens DM, Holman RC. Kawasaki syndrome in the United States 1976 to 1980. Am J Dis Child. 1960;137:211–214. doi: 10.1001/archpedi.1983.02140290003001. 1983. [DOI] [PubMed] [Google Scholar]
- 215.Yanagawa H, Nakamura Y, Yashiro M. Incidence survey of Kawasaki disease in 1997 and 1998 in Japan. Pediatrics. 2001;107:E33. doi: 10.1542/peds.107.3.e33. [DOI] [PubMed] [Google Scholar]
- 216.Yanagawa H, Nakamura Y, Yashiro M. Results of the nationwide epidemiologic survey of Kawasaki disease in 1995 and 1996 in Japan. Pediatrics. 1998;102:E65. doi: 10.1542/peds.102.6.e65. [DOI] [PubMed] [Google Scholar]
- 217.Fujita Y, Nakamura Y, Sakata K. Kawasaki disease in families. Pediatrics. 1989;84:666–669. [PubMed] [Google Scholar]
- 218.Newburger JW, Takahashi M, Gerber MA. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics. 2004;114:1708–1733. doi: 10.1542/peds.2004-2182. [DOI] [PubMed] [Google Scholar]
- 219.Friter BS, Lucky AW. The perineal eruption of Kawasaki syndrome. Arch Dermatol. 1988;124:1805–1810. [PubMed] [Google Scholar]
- 220.Eberhard BA, Sundel RP, Newburger JW. Psoriatic eruption in Kawasaki disease. J Pediatr. 2000;137:578–580. doi: 10.1067/mpd.2000.107840. [DOI] [PubMed] [Google Scholar]
- 221.Zvulunov A, Greenberg D, Cagnano E, Einhorn M. Development of psoriatic lesions during acute and convalescent phases of Kawasaki disease. J Paediatr Child Health. 2003;39:229–231. doi: 10.1046/j.1440-1754.2003.00117.x. [DOI] [PubMed] [Google Scholar]
- 222.Garty B, Mosseri R, Finkelstein Y. Guttate psoriasis following Kawasaki disease. Pediatr Dermatol. 2001;18:507–508. doi: 10.1046/j.1525-1470.2001.1862002.x. [DOI] [PubMed] [Google Scholar]
- 223.Tsai HJ, Wu WM, Chang YC. Annular pustules in Kawasaki disease: a further case indicating the association with psoriasis? Cutis. 2003;72:354–356. [PubMed] [Google Scholar]
- 224.Tomita S, Chung K, Mas M. Peripheral gangrene associated with Kawasaki disease. Clin Infect Dis. 1992;14:121–126. doi: 10.1093/clinids/14.1.121. [DOI] [PubMed] [Google Scholar]
- 225.Hsu YH, Wang YH, Hsu WY, Lee YP. Kawasaki disease characterized by erythema and induration at the Bacillus Calmette–Guérin and purified protein derivate inoculation sites. Pediatr Infect Dis J. 1987;6:576–578. [PubMed] [Google Scholar]
- 226.Bellet JS, Prose NS. Skin complications of Bacillus Calmette–Guérin immunization. Curr Opin Infect Dis. 2005;18:97–100. doi: 10.1097/01.qco.0000160895.97362.4f. [DOI] [PubMed] [Google Scholar]
- 227.Sinha R, Balakumar T. BCG reactivation: a useful diagnostic tool even for incomplete Kawasaki disease. Arch Dis Child. 2005;90:891. doi: 10.1136/adc.2004.071332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Weinstein M. Inflammation at a previous inoculation site: an unusual presentation of Kawasaki disease. CMAJ. 2006;174:459–460. doi: 10.1503/cmaj.051211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Dajani AS, Taubert KA, Gerber MA. Diagnosis and therapy of Kawasaki disease in children. Circulation. 1993;87:1776–1780. doi: 10.1161/01.cir.87.5.1776. [DOI] [PubMed] [Google Scholar]
- 230.Witt MT, Minich LL, Bohnsack JF, Young PC. Kawasaki disease: more patients are being diagnosed who do not meet American Heart Association criteria. Pediatrics. 1999;104:e10. doi: 10.1542/peds.104.1.e10. [DOI] [PubMed] [Google Scholar]
- 231.Alexandra FF. Inflammatory pulmonary nodules in Kawasaki disease. Pediatr Pulmonol. 2003;36:102–106. doi: 10.1002/ppul.10333. [DOI] [PubMed] [Google Scholar]
- 232.Palmer AL, Walker T, Smith JC. Acute respiratory distress syndrome in a child with Kawasaki disease. South Med J. 2005;98:1031–1033. doi: 10.1097/01.smj.0000182176.71475.a9. [DOI] [PubMed] [Google Scholar]
- 233.Palazzi DL, McClain KL, Kaplan SL. Hemophagocytic syndrome after Kawasaki disease. Pediatr Infect Dis J. 2003;22:663–666. [PubMed] [Google Scholar]
- 234.Knott PD, Orloff LA, Harris JP. Sensorineural hearing loss and Kawasaki disease: A prospective study. Am J Otolaryngol. 2001;22:343–348. doi: 10.1053/ajot.2001.26495. [DOI] [PubMed] [Google Scholar]
- 235.Yeung RS. Pathogenesis and treatment of Kawasaki's disease. Curr Opin Rheumatol. 2005;17:617–623. doi: 10.1097/01.bor.0000174184.15901.ee. [DOI] [PubMed] [Google Scholar]
- 236.Wang CL, Wu YT, Liu CA. Kawasaki disease: infection, immunity and genetics. Pediatr Infect Dis J. 2005;24:998–1004. doi: 10.1097/01.inf.0000183786.70519.fa. [DOI] [PubMed] [Google Scholar]
- 237.Burgner D, Harnden A. Kawasaki disease: what is the epidemiology telling us about the etiology? Int J Infect Dis. 2005;9:185–194. doi: 10.1016/j.ijid.2005.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Hirao J, Nagayoshi I. Development of coronary artery lesions in Kawasaki disease patients in relation to plasma interferon-gamma and interleukin-4 levels in the acute phase. Acta Paediatr Jpn. 1997;39:293–295. doi: 10.1111/j.1442-200x.1997.tb03603.x. [DOI] [PubMed] [Google Scholar]
- 239.Hirao J, Yamashita T. Circulating soluble CD23 levels in Kawasaki disease with coronary artery lesions. Acta Paediatr Jpn. 1997;39:397–399. doi: 10.1111/j.1442-200x.1997.tb03765.x. [DOI] [PubMed] [Google Scholar]
- 240.Hirao J, Hibi S, Andoh T, Ichimura T. High levels of circulating interleukin-4 and interleukin-10 in Kawasaki disease. Int Arch Allergy Immunol. 1997;112:152–156. doi: 10.1159/000237447. [DOI] [PubMed] [Google Scholar]
- 241.Sato N, Sagawa K, Sasaguri Y. Immunopathology and cytokine detection in the skin lesions of patients with Kawasaki disease. J Pediatr. 1993;122:198–203. doi: 10.1016/s0022-3476(06)80113-7. [DOI] [PubMed] [Google Scholar]
- 242.Hirao J, Yamashita T. Circulating soluble L-selectin levels in Kawasaki disease with coronary artery lesions. Acta Paediatr Jpn. 1997;39:290–292. doi: 10.1111/j.1442-200x.1997.tb03602.x. [DOI] [PubMed] [Google Scholar]
- 243.Burns JC, Shimizu C, Gonzalez E. Genetic variations in the receptor-ligand pair CCR5 and CCL3L1 are important determinants of susceptibility to Kawasaki disease. J Infect Dis. 2005;192:344–349. doi: 10.1086/430953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Hewitt M, Smith LJ, Joffe HS, Chambers TL. Kawasaki disease in siblings. Arch Dis Child. 1989;64:398–399. doi: 10.1136/adc.64.3.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Dergun M, Kao A, Hauger SB. Familial occurrence of Kawasaki syndrome in North America. Arch Pediatr Adolesc Med. 2005;159:876–881. doi: 10.1001/archpedi.159.9.876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Uehara R, Yashiro M, Nakamura Y, Yanagawa H. Clinical features of patients with Kawasaki disease whose parents had the same disease. Arch Pediatr Adolesc Med. 2004;158:1166–1169. doi: 10.1001/archpedi.158.12.1166. [DOI] [PubMed] [Google Scholar]
- 247.Salo E, Pesonen E, Viikari J. Serum cholesterol levels during and after Kawasaki disease. J Pediatr. 1991;119:557–561. doi: 10.1016/s0022-3476(05)82404-7. [DOI] [PubMed] [Google Scholar]
- 248.Newburger JW, Burns JC, Beiser AS, Loscalzo J. Altered lipid profile after Kawasaki syndrome. Circulation. 1991;84:625–631. doi: 10.1161/01.cir.84.2.625. [DOI] [PubMed] [Google Scholar]
- 249.Simonini G, Rose CD, Vierucci A. Diagnosing Kawasaki syndrome: the need for a new clinical tool. Rheumatology. 2005;44:959–961. doi: 10.1093/rheumatology/keh593. [DOI] [PubMed] [Google Scholar]
- 250.Singh-Grewal D, Wong M, Isaacs D. Diagnosis, treatment and outcome of Kawasaki disease in an Australian tertiary setting: a review of three years experience. J Paediatr Child Health. 2005;41:495–499. doi: 10.1111/j.1440-1754.2005.00690.x. [DOI] [PubMed] [Google Scholar]
- 251.Tsuda E, Kitamura S. National survey of coronary artery bypass grafting for coronary stenosis caused by Kawasaki disease in Japan. Circulation. 2004;110:II61–II66. doi: 10.1161/01.CIR.0000138194.61225.10. [DOI] [PubMed] [Google Scholar]
- 252.Nakamura Y, Aso E, Yashiro M. Mortality among persons with a history of Kawasaki disease in Japan: can paediatricians safely discontinue follow-up of children with a history of the disease but without cardiac sequelae? Acta Paediatr. 2005;94:429–434. doi: 10.1111/j.1651-2227.2005.tb01913.x. [DOI] [PubMed] [Google Scholar]
- 253.Nakamura Y, Yanagawa H, Harada K. Mortality among persons with a history of Kawasaki disease in Japan: the fifth look. Arch Pediatr Adolesc Med. 2002;156:162–165. doi: 10.1001/archpedi.156.2.162. [DOI] [PubMed] [Google Scholar]
- 254.Levy DM, Silverman ED, Massicotte MP. Longterm outcomes in patients with giant aneurysms secondary to Kawasaki disease. J Rheumatol. 2005;32:928–934. [PubMed] [Google Scholar]
- 255.Kato H, Sugimura T, Akagi T. Long-term consequences of Kawasaki disease. A 10- to 21-year follow-up study of 594 patients. Circulation. 1996;94:1379–1385. doi: 10.1161/01.cir.94.6.1379. [DOI] [PubMed] [Google Scholar]
- 256.Dajani AS, Taubert KA, Takahashi M. Guidelines for long-term management of patients with Kawasaki disease. Report from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation. 1994;89:916–922. doi: 10.1161/01.cir.89.2.916. [DOI] [PubMed] [Google Scholar]
- 257.Nakamura Y, Yanagawa H, Harada K. Mortality among persons with a history of Kawasaki disease in Japan: existence of cardiac sequelae elevated the mortality. J Epidemiol. 2000;10:372–375. doi: 10.2188/jea.10.372. [DOI] [PubMed] [Google Scholar]
- 258.Nakamura Y, Yashiro M, Uehara R. Use of laboratory data to identify risk factors of giant coronary aneurysms due to Kawasaki disease. Pediatr Int. 2004;46:33–38. doi: 10.1111/j.1442-200X.2004.01840.x. [DOI] [PubMed] [Google Scholar]
- 259.Harada K. Intravenous gammaglobulin treatment in Kawasaki disease. Acta Paediatr Jpn. 1991;33:805–810. doi: 10.1111/j.1442-200x.1991.tb02612.x. [DOI] [PubMed] [Google Scholar]
- 260.de ZA, Colan SD, Gauvreau K, Baker AL. Coronary artery dimensions may be misclassified as normal in Kawasaki disease. J Pediatr. 1998;133:254–258. doi: 10.1016/s0022-3476(98)70229-x. [DOI] [PubMed] [Google Scholar]
- 261.Terai M, Shulman ST. Prevalence of coronary artery abnormalities in Kawasaki disease is highly dependent on gamma globulin dose but independent of salicylate dose. J Pediatr. 1997;131:888–893. doi: 10.1016/s0022-3476(97)70038-6. [DOI] [PubMed] [Google Scholar]
- 262.Durongpisitkul K, Gururaj VJ, Park JM, Martin CF. The prevention of coronary artery aneurysm in Kawasaki disease: a meta- analysis on the efficacy of aspirin and immunoglobulin treatment. Pediatrics. 1995;96:1057–1061. [PubMed] [Google Scholar]
- 263.Newburger JW, Takahashi M, Burns JC. The treatment of Kawasaki syndrome with intravenous gamma globulin. N Engl J Med. 1986;315:341–347. doi: 10.1056/NEJM198608073150601. [DOI] [PubMed] [Google Scholar]
- 264.Furusho K, Kamiya T, Nakano H. High-dose intravenous gammaglobulin for Kawasaki disease. Lancet. 1984;2:1055–1058. doi: 10.1016/s0140-6736(84)91504-6. [DOI] [PubMed] [Google Scholar]
- 265.Fong NC, Hui YW, Li CK, Chiu MC. Evaluation of the efficacy of treatment of Kawasaki disease before day 5 of illness. Pediatr Cardiol. 2004;25:31–34. doi: 10.1007/s00246-003-0558-4. [DOI] [PubMed] [Google Scholar]
- 266.Muta H, Ishii M, Egami K. Early intravenous gamma-globulin treatment for Kawasaki disease: The nationwide surveys in Japan. J Pediatr. 2004;144:496–499. doi: 10.1016/j.jpeds.2003.12.033. [DOI] [PubMed] [Google Scholar]
- 267.Marasini M, Pongiglione G, Gazzolo D. Late intravenous gamma globulin treatment in infants and children with Kawasaki disease and coronary artery abnormalities. Am J Cardiol. 1991;68:796–797. doi: 10.1016/0002-9149(91)90658-8. [DOI] [PubMed] [Google Scholar]
- 268.Brogan PA, Bose A, Burgner D. Kawasaki disease: an evidence based approach to diagnosis, treatment, and proposals for future research. Arch Dis Child. 2002;86:286–290. doi: 10.1136/adc.86.4.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Sundel RP, Baker AL, Fulton DR, Newburger JW. Corticosteroids in the initial treatment of Kawasaki disease: Report of a randomized trial. J Pediatr. 2003;142:611–616. doi: 10.1067/mpd.2003.191. [DOI] [PubMed] [Google Scholar]
- 270.Okada Y, Shinohara M, Kobayashi T. Effect of corticosteroids in addition to intravenous gamma globulin therapy on serum cytokine levels in the acute phase of Kawasaki disease in children. J Pediatr. 2003;143:363–367. doi: 10.1067/s0022-3476(03)00387-1. [DOI] [PubMed] [Google Scholar]
- 271.Furukawa S, Matsubara T, Umezawa Y. Pentoxifylline and intravenous gamma globulin combination therapy for acute Kawasaki disease. Eur J Pediatr. 1994;153:663–667. doi: 10.1007/BF02190688. [DOI] [PubMed] [Google Scholar]
- 272.Burns JC, Mason WH, Hauger SB. Infliximab treatment for refractory Kawasaki syndrome. J Pediatr. 2005;146:662–667. doi: 10.1016/j.jpeds.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 273.Baselga E, Drolet BA, Esterly NB. Purpura in infants and children. J Am Acad Dermatol. 1997;37:673–705. doi: 10.1016/s0190-9622(97)70105-1. [DOI] [PubMed] [Google Scholar]
- 274.Caksen H, Odabas D, Kosem M. Report of eight infants with acute infantile hemorrhagic edema and review of the literature. J Dermatol. 2002;29:290–295. doi: 10.1111/j.1346-8138.2002.tb00265.x. [DOI] [PubMed] [Google Scholar]
- 275.Cox NH. Seidlmayer's syndrome: postinfectious cockade purpura of early childhood. J Am Acad Dermatol. 1992;26:275. doi: 10.1016/s0190-9622(08)80314-3. [DOI] [PubMed] [Google Scholar]
- 276.Cunningham BB, Caro WA, Eramo LR. Neonatal acute hemorrhagic edema of childhood: case report and review of the English-language literature. Pediatr Dermatol. 1996;13:39–44. doi: 10.1111/j.1525-1470.1996.tb01186.x. [DOI] [PubMed] [Google Scholar]
- 277.Cunningham BB, Eramo L, Caro W. Acute hemorrhagic edema of childhood present at birth. Pediatr Dermatol. 1999;16:68. doi: 10.1046/j.1525-1470.1999.016001068.x. [DOI] [PubMed] [Google Scholar]
- 278.Gonggryp LA, Todd G. Acute hemorrhagic edema of childhood (AHE) Pediatr Dermatol. 1998;15:91–96. doi: 10.1046/j.1525-1470.1998.1998015091.x. [DOI] [PubMed] [Google Scholar]
- 279.Ince E, Mumcu Y, Suskan E. Infantile acute hemorrhagic edema: a variant of leukocytoclastic vasculitis. Pediatr Dermatol. 1995;12:224–227. doi: 10.1111/j.1525-1470.1995.tb00163.x. [DOI] [PubMed] [Google Scholar]
- 280.Legrain V, Lejean S, Taieb A. Infantile acute hemorrhagic edema of the skin: study of ten cases. J Am Acad Dermatol. 1991;24:17–22. doi: 10.1016/0190-9622(91)70002-j. [DOI] [PubMed] [Google Scholar]
- 281.Larregue M, Lorette G, Prigent F, Canuel C. Oedème aigu hémorragique du nourrisson avec complication léthale digestive. Ann Dermatol Venereol. 1980;107:901–905. [PubMed] [Google Scholar]
- 282.Saraclar Y, Tinaztepe K, Adalioglu G, Tuncer A. Acute hemorrhagic edema of infancy (AHEI) – a variant of Henoch–Schonlein purpura or a distinct clinical entity? J Allergy Clin Immunol. 1990;86:473–483. doi: 10.1016/s0091-6749(05)80202-7. [DOI] [PubMed] [Google Scholar]
- 283.Dubin BA, Bronson DM, Eng AM. Acute hemorrhagic edema of childhood: an unusual variant of leukocytoclastic vasculitis. J Am Acad Dermatol. 1990;23:347–350. doi: 10.1016/0190-9622(90)70219-8. [DOI] [PubMed] [Google Scholar]
- 284.Amitai Y, Gillis D, Wasserman D, Kochman RH. Henoch–Schonlein purpura in infants. Pediatrics. 1993;92:865–867. [PubMed] [Google Scholar]
- 285.Lambert D, Laurent R, Bouilly D. Cedeme aigu hémorragique du nourrisson. Données immunologiques et ultrastructurales. Ann Dermatol Venereol. 1979;106:975–987. [PubMed] [Google Scholar]
- 286.Saraclar Y, Tinaztepe K. Infantile acute hemorrhagic edema of the skin. J Am Acad Dermatol. 1992;26:275–276. doi: 10.1016/s0190-9622(08)80315-5. [DOI] [PubMed] [Google Scholar]
- 287.Ahmed I. Childhood porphyrias. Mayo Clin Proc. 2002;77:825–836. doi: 10.4065/77.8.825. [DOI] [PubMed] [Google Scholar]
- 288.Gross U, Hoffmann GF, Doss MO. Erythropoietic and hepatic porphyrias. J Inherit Metab Dis. 2000;23:641–661. doi: 10.1023/a:1005645624262. [DOI] [PubMed] [Google Scholar]
- 289.Kauppinen R. Porphyrias. Lancet. 2005;365:241–252. doi: 10.1016/S0140-6736(05)17744-7. [DOI] [PubMed] [Google Scholar]
- 290.Mascaro JM. Porphyrias in children. Pediatr Dermatol. 1992;9:371–372. doi: 10.1111/j.1525-1470.1992.tb00634.x. [DOI] [PubMed] [Google Scholar]
- 291.Lim HW. Porphyria update. Pediatr Dermatol. 2000;17:75–83. [PubMed] [Google Scholar]
- 292.Lim HW, Cohen JL. The cutaneous porphyrias. Semin Cutan Med Surg. 1999;18:285–292. doi: 10.1016/s1085-5629(99)80027-3. [DOI] [PubMed] [Google Scholar]
- 293.Jensen JD, Resnick SD. Porphyria in childhood. Semin Dermatol. 1995;14:33–39. doi: 10.1016/s1085-5629(05)80037-9. [DOI] [PubMed] [Google Scholar]
- 294.Murphy GM. The cutaneous porphyrias: a review. British Photodermatology Group. Br J Dermatol. 1999;140:573–581. doi: 10.1046/j.1365-2133.1999.02754.x. [DOI] [PubMed] [Google Scholar]
- 295.Elder GH. Update on enzyme and molecular defects in porphyria. Photodermatol Photoimmunol Photomed. 1998;14:66–69. doi: 10.1111/j.1600-0781.1998.tb00014.x. [DOI] [PubMed] [Google Scholar]
- 296.Lim HW. Pathogenesis of photosensitivity in the cutaneous porphyrias. J Invest Dermatol. 2005;124:xvi–xvii. doi: 10.1111/j.0022-202X.2004.23531.x. [DOI] [PubMed] [Google Scholar]
- 297.Poh-Fitzpatrick MB. Molecular and cellular mechanisms of porphyrin photosensitization. Photo-Dermatology. 1986;3:148–157. [PubMed] [Google Scholar]
- 298.Poh-Fitzpatrick MB. Clinical features of the porphyrias. Clin Dermatol. 1998;16:251–264. doi: 10.1016/s0738-081x(97)00205-8. [DOI] [PubMed] [Google Scholar]
- 299.Lim HW, Murphy GM. The porphyrias. Clin Dermatol. 1996;14:375–387. doi: 10.1016/0738-081x(96)00067-3. [DOI] [PubMed] [Google Scholar]
- 300.Fritsch C, Bolsen K, Ruzicka T, Goerz G. Congenital erythropoietic porphyria. J Am Acad Dermatol. 1997;36:594–610. doi: 10.1016/s0190-9622(97)70249-4. [DOI] [PubMed] [Google Scholar]
- 301.Huang JL, Zaider E, Roth P. Congenital erythropoietic porphyria: clinical, biochemical, and enzymatic profile of a severely affected infant. J Am Acad Dermatol. 1996;34:924–927. doi: 10.1016/s0190-9622(96)90081-x. [DOI] [PubMed] [Google Scholar]
- 302.Berry AA, Desnick RJ, Astrin KH. Two brothers with mild congenital erythropoietic porphyria due to a novel genotype. Arch Dermatol. 2005;141:1575–1579. doi: 10.1001/archderm.141.12.1575. [DOI] [PubMed] [Google Scholar]
- 303.Venkatesh P, Garg SP, Kumaran E, Tewari HK. Congenital porphyria with necrotizing scleritis in a 9-year-old child. Clin Exp Ophthalmol. 2000;28:314–318. doi: 10.1046/j.1442-9071.2000.00330.x. [DOI] [PubMed] [Google Scholar]
- 304.Fontanellas A, Bensidhoum M, Enriquez de SR. A systematic analysis of the mutations of the uroporphyrinogen III synthase gene in congenital erythropoietic porphyria. Eur J Hum Genet. 1996;4:274–282. doi: 10.1159/000472214. [DOI] [PubMed] [Google Scholar]
- 305.Desnick RJ, Astrin KH. Congenital erythropoietic porphyria: advances in pathogenesis and treatment. Br J Haematol. 2002;117:779–795. doi: 10.1046/j.1365-2141.2002.03557.x. [DOI] [PubMed] [Google Scholar]
- 306.Xu W, Astrin KH, Desnick RJ. Molecular basis of congenital erythropoietic porphyria: mutations in the human uroporphyrinogen III synthase gene. Hum Mutat. 1996;7:187–192. doi: 10.1002/(SICI)1098-1004(1996)7:3<187::AID-HUMU1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 307.Kaiser IH. Brown amniotic fluid in congenital erythropoietic porphyria. Obstet Gynecol. 1980;56:383–384. [PubMed] [Google Scholar]
- 308.Lazebnik N, Lazebnik RS. The prenatal presentation of congenital erythropoietic porphyria: report of two siblings with elevated maternal serum alpha-fetoprotein. Prenat Diagn. 2004;24:282–286. doi: 10.1002/pd.852. [DOI] [PubMed] [Google Scholar]
- 309.Ikha-Dahmane F, Dommergues M, Narcy F. Congenital erythropoietic porphyria: prenatal diagnosis and autopsy findings in two sibling fetuses. Pediatr Dev Pathol. 2001;4:180–184. doi: 10.1007/s100240010143. [DOI] [PubMed] [Google Scholar]
- 310.Ged C, Moreau-Gaudry F, Taine L. Prenatal diagnosis in congenital erythropoietic porphyria by metabolic measurement and DNA mutation analysis. Prenat Diagn. 1996;16:83–86. doi: 10.1002/(SICI)1097-0223(199601)16:1<83::AID-PD812>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 311.Horiguchi Y, Horio T, Yamamoto M. Late onset erythropoietic porphyria. Br J Dermatol. 1989;121:255–262. doi: 10.1111/j.1365-2133.1989.tb01808.x. [DOI] [PubMed] [Google Scholar]
- 312.Dawe SA, Peters TJ, Du VA, Creamer JD. Congenital erythropoietic porphyria: dilemmas in present day management. Clin Exp Dermatol. 2002;27:680–683. doi: 10.1046/j.1365-2230.2002.01166.x. [DOI] [PubMed] [Google Scholar]
- 313.Guarini L, Piomelli S, Poh-Fitzpatrick MB. Hydroxyurea in congenital erythropoietic porphyria. N Engl J Med. 1994;330:1091–1092. doi: 10.1056/NEJM199404143301519. [DOI] [PubMed] [Google Scholar]
- 314.Piomelli S, Poh-Fitzpatrick MB, Seaman C. Complete suppression of the symptoms of congenital erythropoietic porphyria by long-term treatment with highlevel transfusions. N Engl J Med. 1986;314:1029–1031. doi: 10.1056/NEJM198604173141607. [DOI] [PubMed] [Google Scholar]
- 315.Gorchein A, Guo R, Lim CK. Porphyrins in urine, plasma, erythrocytes, bile and faeces in a case of congenital erythropoietic porphyria (Gunther's disease) treated with blood transfusion and iron chelation: lack of benefit from oral charcoal. Biomed Chromatogr. 1998;12:350–356. doi: 10.1002/(SICI)1099-0801(199811/12)12:6<350::AID-BMC761>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 316.Minder EI, Schneider-Yin X, Moll F. Lack of effect of oral charcoal in congenital erythropoietic porphyria. N Engl J Med. 1994;330:1092–1094. doi: 10.1056/NEJM199404143301520. [DOI] [PubMed] [Google Scholar]
- 317.Hift RJ, Meissner PN, Kirsch RE. The effect of oral activated charcoal on the course of congenital erythropoietic porphyria. Br J Dermatol. 1993;129:14–17. doi: 10.1111/j.1365-2133.1993.tb03304.x. [DOI] [PubMed] [Google Scholar]
- 318.Pimstone NR, Gandhi SN, Mukerji SK. Therapeutic efficacy of oral charcoal in congenital erythropoietic porphyria. N Engl J Med. 1987;316:390–393. doi: 10.1056/NEJM198702123160707. [DOI] [PubMed] [Google Scholar]
- 319.Maleville J, Babin JP, Mollard S. Porphyrie erythropoietique congenitale de Gunther et carotenoides. Essai therapeutique de 4 ans. Ann Dermatol Venereol. 1982;109:883–887. [PubMed] [Google Scholar]
- 320.Seip M, Thune PO, Eriksen L. Treatment of photosensitivity in congenital erythropoietic porphyria (CEP) with beta-carotene. Acta Dermatol Venereol. 1974;54:239–240. [PubMed] [Google Scholar]
- 321.Tezcan I, Xu W, Gurgey A. Congenital erythropoietic porphyria successfully treated by allogeneic bone marrow transplantation. Blood. 1998;92:4053–4058. [PubMed] [Google Scholar]
- 322.Thomas C, Ged C, Nordmann Y. Correction of congenital erythropoietic porphyria by bone marrow transplantation. J Pediatr. 1996;129:453–456. doi: 10.1016/s0022-3476(96)70082-3. [DOI] [PubMed] [Google Scholar]
- 323.Shaw PH, Mancini AJ, McConnell JP. Treatment of congenital erythropoietic porphyria in children by allogeneic stem cell transplantation: a case report and review of the literature. Bone Marrow Transplant. 2001;27:101–105. doi: 10.1038/sj.bmt.1702738. [DOI] [PubMed] [Google Scholar]
- 324.Harada FA, Shwayder TA, Desnick RJ, Lim HW. Treatment of severe congenital erythropoietic porphyria by bone marrow transplantation. J Am Acad Dermatol. 2001;45:279–282. doi: 10.1067/mjd.2001.114730. [DOI] [PubMed] [Google Scholar]
- 325.Dupuis-Girod S, Akkari V, Ged C. Successful match-unrelated donor bone marrow transplantation for congenital erythropoietic porphyria (Gunther disease) Eur J Pediatr. 2005;164:104–107. doi: 10.1007/s00431-004-1575-x. [DOI] [PubMed] [Google Scholar]
- 326.Geronimi F, Richard E, Lamrissi-Garcia I. Lentivirus-mediated gene transfer of uroporphyrinogen III synthase fully corrects the porphyric phenotype in human cells. J Mol Med. 2003;81:310–320. doi: 10.1007/s00109-003-0438-7. [DOI] [PubMed] [Google Scholar]
- 327.Kauppinen R, Glass IA, Aizencang G. Congenital erythropoietic porphyria: prolonged high-level expression and correction of the heme biosynthetic defect by retroviralmediated gene transfer into porphyric and erythroid cells. Mol Genet Metab. 1998;65:10–17. doi: 10.1006/mgme.1998.2739. [DOI] [PubMed] [Google Scholar]
- 328.DeLeo VA, Poh-Fitzpatrick M, Mathews-Roth M, Harber LC. Erythropoietic protoporphyria. 10 years experience. Am J Med. 1976;60:8–22. doi: 10.1016/0002-9343(76)90528-3. [DOI] [PubMed] [Google Scholar]
- 329.Whatley SD, Mason NG, Khan M. Autosomal recessive erythropoietic protoporphyria in the United Kingdom: prevalence and relationship to liver disease. J Med Genet. 2004;41:e105. doi: 10.1136/jmg.2003.016121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Goerz G, Bunselmeyer S, Bolsen K, Schurer NY. Ferrochelatase activities in patients with erythropoietic protoporphyria and their families. Br J Dermatol. 1996;134:880–885. [PubMed] [Google Scholar]
- 331.Norris PG, Nunn AV, Hawk JL, Cox TM. Genetic heterogeneity in erythropoietic protoporphyria: a study of the enzymatic defect in nine affected families. J Invest Dermatol. 1990;95:260–263. doi: 10.1111/1523-1747.ep12484876. [DOI] [PubMed] [Google Scholar]
- 332.Sarkany RP, Alexander GJ, Cox TM. Recessive inheritance of erythropoietic protoporphyria with liver failure. Lancet. 1994;344:958–959. doi: 10.1016/s0140-6736(94)92314-0. [DOI] [PubMed] [Google Scholar]
- 333.Frank J, Nelson J, Wang X. Erythropoietic protoporphyria: identification of novel mutations in the ferrochelatase gene and comparison of biochemical markers versus molecular analysis as diagnostic strategies. J Invest Med. 1999;47:278–284. [PubMed] [Google Scholar]
- 334.Murphy GM. Diagnosis and management of the erythropoietic porphyrias. Dermatol Ther. 2003;16:57–64. doi: 10.1046/j.1529-8019.2003.01609.x. [DOI] [PubMed] [Google Scholar]
- 335.Poh-Fitzpatrick MB, Piomelli S, Young P. Rapid quantitative assay for erythrocyte porphyrins. Arch Dermatol. 1974;110:225–230. [PubMed] [Google Scholar]
- 336.Mathews-Roth MM. The treatment of erythropoietic protoporphyria. Semin Liver Dis. 1998;18:425–426. [PubMed] [Google Scholar]
- 337.Todd DJ. Therapeutic options for erythropoietic protoporphyria. Br J Dermatol. 2000;142:826–827. doi: 10.1046/j.1365-2133.2000.03442.x. [DOI] [PubMed] [Google Scholar]
- 338.Miao LL, Mathews-Roth MM, Poh-Fitzpatrick MB. Beta carotene treatment and erythrocytic protoporphyrin levels. Arch Dermatol. 1979;115:818. [PubMed] [Google Scholar]
- 339.Mathews-Roth MM. Erythropoietic protoporphyria: treatment with antioxidants and potential cure with gene therapy. Meth Enzymol. 2000;319:479–484. doi: 10.1016/s0076-6879(00)19044-5. [DOI] [PubMed] [Google Scholar]
- 340.Warren LJ, George S. Erythropoietic protoporphyria treated with narrow-band (TL-01) UVB phototherapy. Australas J Dermatol. 1998;39:179–182. doi: 10.1111/j.1440-0960.1998.tb01278.x. [DOI] [PubMed] [Google Scholar]
- 341.Gorchein A, Foster GR. Liver failure in protoporphyria: long-term treatment with oral charcoal. Hepatology. 1999;29:995–996. doi: 10.1002/hep.510290314. [DOI] [PubMed] [Google Scholar]
- 342.Leone N, Marzano A, Cerutti E. Liver transplantation for erythropoietic protoporphyria: report of a case with medium-term follow-up. Dig Liver Dis. 2000;32:799–802. doi: 10.1016/s1590-8658(00)80359-x. [DOI] [PubMed] [Google Scholar]
- 343.McGuire BM, Bonkovsky HL, Carithers RL., Jr Liver transplantation for erythropoietic protoporphyria liver disease. Liver Transpl. 2005;11:1590–1596. doi: 10.1002/lt.20620. [DOI] [PubMed] [Google Scholar]
- 344.Meerman L, Haagsma EB, Gouw AS. Long-term follow-up after liver transplantation for erythropoietic protoporphyria. Eur J Gastroenterol Hepatol. 1999;11:431–438. doi: 10.1097/00042737-199904000-00012. [DOI] [PubMed] [Google Scholar]
- 345.Pimstone NR. Roles and pitfalls of transplantation in human porphyria. Liver Transpl. 2005;11:1460–1462. doi: 10.1002/lt.20621. [DOI] [PubMed] [Google Scholar]
- 346.Elder GH, Smith SG, Herrero C. Hepatoerythropoietic porphyria: a new uroporphyrinogen decarboxylase defect or homozygous porphyria cutanea tarda? Lancet. 1981;1:916–919. doi: 10.1016/s0140-6736(81)91615-9. [DOI] [PubMed] [Google Scholar]
- 347.Lim HW, Poh-Fitzpatrick MB. Hepatoerythropoietic porphyria: a variant of childhood-onset porphyria cutanea tarda. Porphyrin profiles and enzymatic studies of two cases in a family. J Am Acad Dermatol. 1984;11:1103–1111. doi: 10.1016/s0190-9622(84)70267-2. [DOI] [PubMed] [Google Scholar]
- 348.Piñol-Aguade J, Herrero C, Almeida J. Porphyrie hepato-erythroctaire, une nouvelle forme de porphyrie. Ann Dermatol Syphiligr. 1975;102:129–136. [PubMed] [Google Scholar]
- 349.Toback AC, Sassa S, Poh-Fitzpatrick MB. Hepatoerythropoietic porphyria: clinical, biochemical, and enzymatic studies in a three-generation family lineage. N Engl J Med. 1987;316:645–650. doi: 10.1056/NEJM198703123161101. [DOI] [PubMed] [Google Scholar]
- 350.Garcia-Bravo M, Lopez-Gomez S, Segurado-Rodriguez MA. Successful and safe treatment of hypertrichosis by high-intensity pulses of noncoherent light in a patient with hepatoerythropoietic porphyria. Arch Dermatol Res. 2004;296:139–140. doi: 10.1007/s00403-004-0490-3. [DOI] [PubMed] [Google Scholar]
- 351.Elder GH. Hepatic porphyrias in children. J Inherit Metab Dis. 1997;20:237–246. doi: 10.1023/a:1005313024076. [DOI] [PubMed] [Google Scholar]
- 352.Thunell S, Holmberg L, Lundgren J. Aminolaevulinate dehydratase porphyria in infancy. A clinical and biochemical study. J Clin Chem Clin Biochem. 1987;25:5–14. doi: 10.1515/cclm.1987.25.1.5. [DOI] [PubMed] [Google Scholar]
- 353.Thunell S, Henrichson A, Floderus Y. Liver transplantation in a boy with acute porphyria due to aminolaevulinate dehydratase deficiency. Eur J Clin Chem Clin Biochem. 1992;30:599–606. doi: 10.1515/cclm.1992.30.10.599. [DOI] [PubMed] [Google Scholar]
- 354.Lee DS, Flachsova E, Bodnarova M. Structural basis of hereditary coproporphyria. Proc Natl Acad Sci USA. 2005;102:14232–14237. doi: 10.1073/pnas.0506557102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Lamoril J, Puy H, Whatley SD. Characterization of mutations in the CPO gene in British patients demonstrates absence of genotype–phenotype correlation and identifies relationship between hereditary coproporphyria and harderoporphyria. Am J Hum Genet. 2001;68:1130–1138. doi: 10.1086/320118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Lamoril J, Martasek P, Deybach JC. A molecular defect in coproporphyrinogen oxidase gene causing harderoporphyria, a variant form of hereditary coproporphyria. Hum Mol Genet. 1995;4:275–278. doi: 10.1093/hmg/4.2.275. [DOI] [PubMed] [Google Scholar]
- 357.Nordmann Y, Grandchamp B, de VH. Harderoporphyria: a variant hereditary coproporphyria. J Clin Invest. 1983;72:1139–1149. doi: 10.1172/JCI111039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Palmer RA, Elder GH, Barrett D, Keohane SG. Homozygous variegate porphyria: a compound heterozygote with novel mutations in the protoporphyrinogen oxidase gene. Br J Dermatol. 2001;144:866–869. doi: 10.1046/j.1365-2133.2001.04147.x. [DOI] [PubMed] [Google Scholar]
- 359.Kauppinen R, Timonen K, von M. Homozygous variegate porphyria: 20 year follow-up and characterization of molecular defect. J Invest Dermatol. 2001;116:610–613. doi: 10.1046/j.1523-1747.2001.01293.x. [DOI] [PubMed] [Google Scholar]
- 360.Roberts AG, Puy H, Dailey TA. Molecular characterization of homozygous variegate porphyria. Hum Mol Genet. 1998;7:1921–1925. doi: 10.1093/hmg/7.12.1921. [DOI] [PubMed] [Google Scholar]
- 361.Beukeveld GJ, Wolthers BG, Nordmann Y. A retrospective study of a patient with homozygous form of acute intermittent porphyria. J Inherit Metab Dis. 1990;13:673–683. doi: 10.1007/BF01799566. [DOI] [PubMed] [Google Scholar]
- 362.Picat C, Delfau MH, de Rooij FW. Identification of the mutations in the parents of a patient with a putative compound heterozygosity for acute intermittent porphyria. J Inherit Metab Dis. 1990;13:684–686. doi: 10.1007/BF01799567. [DOI] [PubMed] [Google Scholar]
- 363.Llewellyn DH, Smyth SJ, Elder GH. Homozygous acute intermittent porphyria: compound heterozygosity for adjacent base transitions in the same codon of the porphobilinogen deaminase gene. Hum Genet. 1992;89:97–98. doi: 10.1007/BF00207051. [DOI] [PubMed] [Google Scholar]
- 364.Solis C, Martinez-Bermejo A, Naidich TP. Acute intermittent porphyria: studies of the severe homozygous dominant disease provides insights into the neurologic attacks in acute porphyrias. Arch Neurol. 2004;61:1764–1770. doi: 10.1001/archneur.61.11.1764. [DOI] [PubMed] [Google Scholar]
- 365.Crawford GH, Kim S, James WD. Skin signs of systemic disease: an update. Adv Dermatol. 2002;18:1–27. [PubMed] [Google Scholar]
- 366.Mallon E, Wojnarowska F, Hope P, Elder G. Neonatal bullous eruption as a result of transient porphyrinemia in a premature infant with hemolytic disease of the newborn. J Am Acad Dermatol. 1995;33:333–336. doi: 10.1016/0190-9622(95)91428-5. [DOI] [PubMed] [Google Scholar]
- 367.Paller AS, Eramo LR, Farrell EE. Purpuric phototherapy-induced eruption in transfused neonates: relation to transient porphyrinemia. Pediatrics. 1997;100:360–364. doi: 10.1542/peds.100.3.360. [DOI] [PubMed] [Google Scholar]
- 368.Vanden Eijnden S, Blum D, Clercx A. Cutaneous porphyria in a neonate with tyrosinaemia type 1. Eur J Pediatr. 2000;159:503–506. doi: 10.1007/s004310051319. [DOI] [PubMed] [Google Scholar]
- 369.Bowden JB, Hebert AA, Rapini RP. Dermal hematopoiesis in neonates: report of five cases. J Am Acad Dermatol. 1989;20:1104–1110. doi: 10.1016/s0190-9622(89)70141-9. [DOI] [PubMed] [Google Scholar]
- 370.Brough AJ, Jones D, Page RH. Dermal erythropoiesis in neonatal infants: a manifestation of intrauterine viral disease. Pediatrics. 1967;40:627–635. [PubMed] [Google Scholar]
- 371.Klein HZ, Markarian M. Dermal erythropoiesis in congenital rubella. Description of an infected newborn who had purpura associated with marked extramedullary erythropoiesis in the skin and elsewhere. Clin Pediatr (Phila) 1969;8:604–607. doi: 10.1177/000992286900801015. [DOI] [PubMed] [Google Scholar]
- 372.Silver MM, Hellmann J, Zielenska M. Anemia, blueberry-muffin rash, and hepatomegaly in a newborn infant. J Pediatr. 1996;128:579–586. doi: 10.1016/s0022-3476(96)70377-3. [DOI] [PubMed] [Google Scholar]
- 373.Hebert AA, Esterly NB, Gardner TH. Dermal erythropoiesis in Rh hemolytic disease of the newborn. J Pediatr. 1985;107:799–801. doi: 10.1016/s0022-3476(85)80422-4. [DOI] [PubMed] [Google Scholar]
- 374.Pizarro A, Elorza D, Gamallo C. Neonatal dermal erythropoiesis associated with severe rhesus immunization: amelioration by high-dose intravenous immunoglobulin. Br J Dermatol. 1995;133:334–336. doi: 10.1111/j.1365-2133.1995.tb02647.x. [DOI] [PubMed] [Google Scholar]
- 375.Argyle JC, Zone JJ. Dermal erythropoiesis in a neonate. Arch Dermatol. 1981;117:492–494. [PubMed] [Google Scholar]
- 376.Hendricks WM, Hu CH. Blueberry muffin syndrome: cutaneous erythropoiesis and possible intrauterine viral infection. Cutis. 1984;34:549–551. [PubMed] [Google Scholar]
- 377.Hawthorne HC, Jr, Nelson JS, Witzleben CL, Giangiacomo J. Blanching subcutaneous nodules in neonatal neuroblastoma. J Pediatr. 1970;77:297–300. doi: 10.1016/s0022-3476(70)80339-0. [DOI] [PubMed] [Google Scholar]
- 378.Shown TE, Durfee MF. Blueberry muffin baby: neonatal neuroblastoma with subcutaneous metastases. J Urol. 1970;104:193–195. doi: 10.1016/s0022-5347(17)61698-7. [DOI] [PubMed] [Google Scholar]
- 379.van EI. Cutaneous metastases in neuroblastoma. Dermatologica. 1968;136:265–269. doi: 10.1159/000254106. [DOI] [PubMed] [Google Scholar]
- 380.Kitagawa N, Arata J, Ohtsuki Y. Congenital alveolar rhabdomyosarcoma presenting as a blueberry muffin baby. J Dermatol. 1989;16:409–411. doi: 10.1111/j.1346-8138.1989.tb01291.x. [DOI] [PubMed] [Google Scholar]
- 381.Gottesfeld E, Silverman RA, Coccia PF. Transient blueberry muffin appearance of a newborn with congenital monoblastic leukemia. J Am Acad Dermatol. 1989;21:347–351. doi: 10.1016/s0190-9622(89)80032-5. [DOI] [PubMed] [Google Scholar]
- 382.Meuleman V, Degreef H. Acute myelomonocytic leukemia with skin localizations. Dermatology. 1995;190:346–348. doi: 10.1159/000246740. [DOI] [PubMed] [Google Scholar]
- 383.Resnik KS, Brod BB. Leukemia cutis in congenital leukemia. Analysis and review of the world literature with report of an additional case. Arch Dermatol. 1993;129:1301–1306. doi: 10.1001/archderm.129.10.1301. [DOI] [PubMed] [Google Scholar]
- 384.Enjolras O, Leibowitch M, Guillemette J. ‘Blueberry muffin baby’: hematopoiese extramedullaire neonatale? Leucemie monoblastique congenitale involutive? Ou histiocytose congenitale involutive? Ann Dermatol Venereol. 1990;117:810–812. [PubMed] [Google Scholar]
- 385.Enjolras O, Leibowitch M, Bonacini F. Histiocytoses langerhansiennes congenitales cutanees. A propos de 7 cas. Ann Dermatol Venereol. 1992;119:111–117. [PubMed] [Google Scholar]
- 386.Auletta MJ, Headington JT. Purpura fulminans. A cutaneous manifestation of severe protein C deficiency. Arch Dermatol. 1988;124:1387–1391. doi: 10.1001/archderm.124.9.1387. [DOI] [PubMed] [Google Scholar]
- 387.Mahasandana C, Suvatte V, Chuansumrit A. Homozygous protein S deficiency in an infant with purpura fulminans. J Pediatr. 1990;117:750–753. doi: 10.1016/s0022-3476(05)83335-9. [DOI] [PubMed] [Google Scholar]
- 388.Marciniak E, Wilson HD, Marlar RA. Neonatal purpura fulminans: a genetic disorder related to the absence of protein C in blood. Blood. 1985;65:15–20. [PubMed] [Google Scholar]
- 389.Marlar RA, Montgomery RR, Broekmans AW. Diagnosis and treatment of homozygous protein C deficiency. Report of the Working Party on Homozygous Protein C Deficiency of the Subcommittee on Protein C and Protein S, International Committee on Thrombosis and Haemostasis. J Pediatr. 1989;114:528–534. doi: 10.1016/s0022-3476(89)80688-2. [DOI] [PubMed] [Google Scholar]
- 390.Marlar RA, Neumann A. Neonatal purpura fulminans due to homozygous protein C or protein S deficiencies. Semin Thromb Hemost. 1990;16:299–309. doi: 10.1055/s-2007-1002683. [DOI] [PubMed] [Google Scholar]
- 391.van der Horst RL. Purpura fulminans in a newborn baby. Arch Dis Child. 1962;37:436–441. doi: 10.1136/adc.37.194.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Tuddenham EG, Takase T, Thomas AE. Homozygous protein C deficiency with delayed onset of symptoms at 7 to 10 months. Thromb Res. 1989;53:475–484. doi: 10.1016/0049-3848(89)90202-8. [DOI] [PubMed] [Google Scholar]
- 393.Pipe SW, Schmaier AH, Nichols WC. Neonatal purpura fulminans in association with factor V R506Q mutation. J Pediatr. 1996;128:706–709. doi: 10.1016/s0022-3476(96)80142-9. [DOI] [PubMed] [Google Scholar]
- 394.Seligsohn U, Berger A, Abend M. Homozygous protein C deficiency manifested by massive venous thrombosis in the newborn. N Engl J Med. 1984;310:559–562. doi: 10.1056/NEJM198403013100904. [DOI] [PubMed] [Google Scholar]
- 395.Sills RH, Marlar RA, Montgomery RR. Severe homozygous protein C deficiency. J Pediatr. 1984;105:409–413. doi: 10.1016/s0022-3476(84)80013-x. [DOI] [PubMed] [Google Scholar]
- 396.Chuansumrit A, Hotrakitya S, Kruavit A. Severe acquired neonatal purpura fulminans. Clin Pediatr (Phila) 1996;35:373–376. doi: 10.1177/000992289603500708. [DOI] [PubMed] [Google Scholar]
- 397.Gurses N, Islek I. Causes of purpura fulminans. Pediatr Infect Dis J. 1995;14:552–553. doi: 10.1097/00006454-199506000-00018. [DOI] [PubMed] [Google Scholar]
- 398.Petaja J, Manco-Johnson MJ, Protein C pathway in infants and children. Semin Thromb Hemost. 2003;29:349–362. doi: 10.1055/s-2003-42586. [DOI] [PubMed] [Google Scholar]
- 399.Aiach M, Borgel D, Gaussem P. Protein C and protein S deficiencies. Semin Hematol. 1997;34:205–216. [PubMed] [Google Scholar]
- 400.Grundy CB, Melissari E, Lindo V. Late-onset homozygous protein C deficiency. Lancet. 1991;338:575–576. doi: 10.1016/0140-6736(91)91144-j. [DOI] [PubMed] [Google Scholar]
- 401.Tripodi A, Franchi F, Krachmalnicoff A, Mannucci PM. Asymptomatic homozygous protein C deficiency. Acta Haematol. 1990;83:152–155. doi: 10.1159/000205194. [DOI] [PubMed] [Google Scholar]
- 402.Gomez E, Ledford MR, Pegelow CH. Homozygous protein S deficiency due to a one base pair deletion that leads to a stop codon in exon III of the protein S gene. Thromb Haemost. 1994;71:723–726. [PubMed] [Google Scholar]
- 403.Dahlback B. The discovery of activated protein C resistance. J Thromb Haemost. 2003;1:3–9. doi: 10.1046/j.1538-7836.2003.00016.x. [DOI] [PubMed] [Google Scholar]
- 404.Dahlback B. Blood coagulation and its regulation by anticoagulant pathways: genetic pathogenesis of bleeding and thrombotic diseases. J Intern Med. 2005;257:209–223. doi: 10.1111/j.1365-2796.2004.01444.x. [DOI] [PubMed] [Google Scholar]
- 405.Adcock DM, Hicks MJ. Dermatopathology of skin necrosis associated with purpura fulminans. Semin Thromb Hemost. 1990;16:283–292. doi: 10.1055/s-2007-1002681. [DOI] [PubMed] [Google Scholar]
- 406.Minutillo C, Pemberton PJ, Willoughby ML. Neonatal purpura fulminans and transient protein C deficiency. Arch Dis Child. 1990;65:561–562. doi: 10.1136/adc.65.5.561-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Baliga V, Thwaites R, Tillyer ML. Homozygous protein C deficiency – management with protein C concentrate. Eur J Pediatr. 1995;154:534–538. doi: 10.1007/BF02074829. [DOI] [PubMed] [Google Scholar]
- 408.Dreyfus M, Masterson M, David M. Replacement therapy with a monoclonal antibody purified protein C concentrate in newborns with severe congenital protein C deficiency. Semin Thromb Hemost. 1995;21:371–381. doi: 10.1055/s-2007-1000658. [DOI] [PubMed] [Google Scholar]
- 409.Angelis M, Pegelow CH, Khan FA. En bloc heterotopic auxiliary liver and bilateral renal transplant in a patient with homozygous protein C deficiency. J Pediatr. 2001;138:120–122. doi: 10.1067/mpd.2001.109199. [DOI] [PubMed] [Google Scholar]
- 410.Casella JF, Lewis JH, Bontempo FA. Successful treatment of homozygous protein C deficiency by hepatic transplantation. Lancet. 1988;1:435–438. doi: 10.1016/s0140-6736(88)91231-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Crawford RI, Lawlor ER, Wadsworth LD, Prendiville JS. Transient erythroporphyria of infancy. J Am Acad Dermatol. 1996;35:833–834. doi: 10.1016/s0190-9622(96)90096-1. [DOI] [PubMed] [Google Scholar]
- 412.Siegfried EC, Stone MS, Madison KC. Ultraviolet light burn: a cutaneous complication of visible light phototherapy of neonatal jaundice. Pediatr Dermatol. 1992;9:278–282. doi: 10.1111/j.1525-1470.1992.tb00348.x. [DOI] [PubMed] [Google Scholar]
- 413.George M. Methylene-blue-induced hyperbilirubinemia and phototoxicity in a neonate. Clin Pediatr (Phila) 2000;39:659–661. doi: 10.1177/000992280003901106. [DOI] [PubMed] [Google Scholar]
- 414.Porat R, Gilbert S, Magilner D. Methylene blue-induced phototoxicity: an unrecognized complication. Pediatrics. 1996;97:717–721. [PubMed] [Google Scholar]
- 415.Kearns GL, Williams BJ, Timmons OD. Fluorescein phototoxicity in a premature infant. J Pediatr. 1985;107:796–798. doi: 10.1016/s0022-3476(85)80421-2. [DOI] [PubMed] [Google Scholar]
- 416.Burry JN, Lawrence JR. Phototoxic blisters from high frusemide dosage. Br J Dermatol. 1976;94:495–499. doi: 10.1111/j.1365-2133.1976.tb05136.x. [DOI] [PubMed] [Google Scholar]