

Abstract
Background and Purpose
The mechanisms causing spontaneous epileptic seizures, including carbamazepine‐resistant/zonisamide ‐sensitive seizures and comorbidity in autosomal dominant sleep‐related hypermotor epilepsy (ADSHE) are unclear. This study investigated functional abnormalities in thalamocortical transmission in transgenic rats bearing rat S286L‐mutant Chrna4 (S286L‐TG) of α4 subunit of the nicotinic ACh receptor (nAChR) that corresponds to the human S284L‐mutant CHRNA4.
Experimental Approach
Effects of carbamazepine and zonisamide on epileptic discharges of S286L‐TG rat were measured using telemetry electrocorticogram. Transmission abnormalities of L‐glutamate and GABA in thalamocortical pathway of S286L‐TG rats were investigated using multiprobe microdialysis and ultra‐high‐performance liquid‐chromatography.
Key Results
Epileptic discharges in S286L‐TG rats were reduced by zonisamide but not by carbamazepine, similar to that of S284L‐ADSHE patients. Carbamazepine unaffected functional abnormality in transmission of S286L‐TG rats. However, zonisamide was able to compensate for the attenuated S286L‐mutant nAChR induced GABA release in frontal‐cortex, without affecting attenuated thalamocortical glutamatergic transmission. Excitatory effects of S286L‐mutant nAChR on thalamocortical transmission were attenuated compared with those of wild‐type nAChR. Loss‐of‐function of S286L‐nAChR enhanced transmission in thalamocortical motor pathway by predominantly attenuating GABAergic transmission. However, it attenuated transmission in thalamocortical cognitive pathway by reducing inhibitory GABAergic and excitatory glutamatergic transmission.
Conclusion and Implications
Our results suggest that functional abnormalities of S286L‐nAChR are associated with intra‐frontal and thalamocortical transmission, possibly contributing to the pathogenesis of ADSHE‐seizure and comorbidity of S284L‐ADSHE. Selective compensation of impaired GABAergic transmission by zonisamide (but not by carbamazepine) in frontal cortex may be involved, at least partially, in carbamazepine‐resistant ADSHE‐seizure of S284L‐ADSHE patients.
In the thalamocortical motor pathway, stimulatory effects of α4β2‐nAChR in the MoTN was eliminated, whereas inhibitory effects in the RTN were switched to stimulatory effects predominantly via impairment of GABAergic inhibition rather than glutamatergic excitation in the RTN‐MoTN pathway.
In contrast, the thalamocortical cognitive pathway showed that both the stimulatory and inhibitory effects of α4β2‐nAChR in respective MDTN and RTN were eliminated.
These functional abnormalities of the thalamocortical motor and cognitive pathways may contribute to pathogenesis of spontaneous S284L‐ADSHE seizure and its comorbidities (cognitive impairment).
Furthermore, selective compensation of impaired GABAergic transmission in the M2 by zonisamide (ZNS), but not carbamazepine (CBZ), probably plays important roles in the pathophysiology of carbamazepine‐resistant/zonisamide‐sensitive S284L‐ADSHE seizures.
Abbreviations
- ADSHE
autosomal dominant sleep‐related hypermotor epilepsy
- AMPA
amino‐3‐(3‐hydroxy‐5‐methyl‐isoxazol‐4‐yl)propanoic acid
- CBZ
carbamazepine
- HKMRS
50‐mM K+ composed modified Ringer's solution
- MDTN
mediodorsal thalamic nucleus
- MoTN
motor thalamic nuclei
- MRS
modified Ringer's solution
- M2
secondary motor cortex
- nAChR
nicotinic ACh receptor
- OFC
orbitofrontal cortex
- RJR2403
(E)‐N‐methyl‐4‐(3‐pyridinyl)‐3‐buten‐1‐amine
- RTN
reticular thalamic nucleus
- SHE
sporadic form sleep‐related hypermotor epilepsy
- S286L‐TG
transgenic rat bearing rat Chrna4 missense S286L mutation
- ZNS
zonisamide
What is already known
CHRNA4 gene mutations play important roles in autosomal dominant sleep‐related hypermotor epilepsy (ADSHE).
CHRNA4 genes encode for the α4 subunit of nicotinic ACh receptor (nAChR)
What this study adds
Loss‐of‐function of S286L‐nAChR produces paradoxical functional abnormalities in both the thalamocortical motor and cognitive pathways.
Zonisamide compensated the attenuated S286L‐mutant induced GABA release without affecting glutamate release in frontal‐cortex.
What is the clinical significance
Functional abnormalities of S284L‐mutant nAChR contribute to the pathogenesis of ADSHE‐seizures and cognitive impairment comorbidities.
1. INTRODUCTION
Comorbidities such as intellectual disability and autism are more frequently seen in epilepsy patients (up to 20‐fold) than in the general population. Both the epilepsy itself and the comorbidities negatively affect the prognosis and quality of life of these patients (Keezer, Sisodiya, & Sander, 2016; Strasser, Downes, Kung, Cross, & De Haan, 2018; van Ool et al., 2016). Various models have been proposed to account for the relationships between comorbid disorders (Keezer et al., 2016), but detailed understand of the mechanisms underlying the connections between epilepsy, intellectual disability and autism remain to be established.
Autosomal dominant sleep‐related hypermotor epilepsy (ADSHE; Tinuper et al., 2016) was first identified as distinct familial epilepsy (previously autosomal dominant nocturnal frontal lobe epilepsy: ADNFLE) in 1994 (Scheffer et al., 1995). ADSHE was initially considered to be a channelopathy, because relevant mutations were identified in genes encoding the https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=76 (nAChR) subunits, α2 (https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=463&familyId=76&familyType=IC), α4 (https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=465&familyId=76&familyType=IC) and β2 (https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=472&familyId=76&familyType=IC; Aridon et al., 2006; De Fusco et al., 2000; Hirose et al., 1999; Okada, Zhu, Yoshida, & Kaneko, 2010; Phillips et al., 2001; Steinlein et al., 1995). Seizures in ADSHE are symptomatically comparable to those seen in frontal lobe epilepsy and occur predominantly during the non‐rapid eye movement phase of sleep. These seizures are complex, stereotyped hyperkinetic seizures that consist of three types of motor seizures, nocturnal‐paroxysmal‐arousals, nocturnal‐paroxysmal‐dystonia and episodic‐nocturnal‐wandering (Okada et al., 2010; Provini et al., 1999). Interestingly, there is a sporadic form of sleep‐related hypermotor epilepsy (SHE; Tinuper et al., 2016; previously called nocturnal frontal lobe epilepsy [NFLE]), the clinical manifestations of which are indistinguishable from those of ADSHE (Okada et al., 2010; Provini et al., 1999). Therefore, any observed clinical phenotypes are considered to belong uniformly to ADSHE syndromes. There are, however, two clinical sub‐features related to a mutation in the nAChR gene that can be used to distinguish the two syndromes, cognitive impairment and sensitivity to anticonvulsants (Okada et al., 2010). https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5339 (CBZ) is a first‐choice anticonvulsant for ADSHE. Using relatively low doses leads to remission in approximately 60% of autosomal dominant sleep‐related hypermotor epilepsy/sleep‐related hypermotor epilepsy patients (Provini et al., 1999; Provini, Plazzi, Montagna, & Lugaresi, 2000; Scheffer et al., 1995). However, ADSHE patients carrying the S252L mutation in the CHRNA4 gene (currently termed as S284L mutation; Hirose et al., 1999; Ito et al., 2000; Okada et al., 2010; Zhu et al., 2008) and approximately 30% of other autosomal ADSHE patients are resistant to carbamazepine but responsive to other anticonvulsants such as https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6792, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6849 and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7047 (ZNS; Hirose et al., 1999; Ito et al., 2000; Okada et al., 2010; Rozycka, Skorupska, Kostyrko, & Trzeciak, 2003). Autosomal dominant sleep‐related hypermotor epilepsy patients with the S284L‐mutant CHRNA4 have also been known to be comorbid with cognitive disturbances such as autism and intellectual disabilities (Ito et al., 2000; Miyajima, Kumada, Saito, & Fujii, 2013).
To explore the pathogenesis of ADSHE/SHE, several animal models have been generated (Klaassen et al., 2006; Shiba et al., 2015; Teper et al., 2007; Zhu et al., 2008). In other respects, although functional abnormality in thalamocortical transmission is known to contribute to cognitive impairment (Alelu‐Paz & Gimenez‐Amaya, 2008; Fukuyama, Hasegawa, & Okada, 2018; Fukuyama, Kato, Murata, Shiroyama, & Okada, 2019; Okada et al., 2019; Okada, Fukuyama, Kawano, Shiroyama, & Ueda, 2019; Okada, Fukuyama, Nakano, & Ueda, 2019; Okada, Fukuyama, Shiroyama, & Ueda, 2019; Schuetze et al., 2016; Vertes, Linley, & Hoover, 2015) and propagation of epileptic discharge (Bertram, 2014), more intrinsic functional changes specific to these models remain to be elucidated. Some preliminary findings have been reported; for example, knockin mice carrying the S280F mutation in the Chrna4 have demonstrated enhanced cortical GABAergic inhibition in layers II/III of the frontal cortex (Klaassen et al., 2006), whereas S284L‐mutant Chrna4 transgenic rats have shown reduced synaptic and extra‐synaptic GABAergic transmission in layers II/III and V (Zhu et al., 2008). Nevertheless, there are no data from these models describing the pathogenesis of ADSHE‐seizures or the resistance of these animals to carbamazepine, nor are there data related to the comorbid cognitive impairment. Therefore, to understand the pathogenesis of ADSHE, the associated cognitive impairment and the pathophysiology of carbamazepine‐resistant seizures in S284L‐ADSHE, we generated genetic ADSHE model rat bearing the missense S286L mutation in the Chrna4 rat gene (S286L‐TG), which corresponds to the S284L mutation in the human CHRNA4 gene.
The α4β2‐nAChR is predominantly expressed in the frontal cortex and thalamus (Hillmer et al., 2012). Additionally, it has been well established that thalamocortical glutamatergic transmission regulates pathways associated with motor and cognitive functions (Asanuma, 1992; Beierlein, 2014; Fukuyama et al., 2018; Fukuyama et al., 2019; Okada, Fukuyama, Kawano, Shiroyama, Suzuki, & Ueda, 2019; Okada, Fukuyama, Kawano, Shiroyama, & Ueda, 2019; Okada, Fukuyama, Nakano, & Ueda, 2019; Okada, Fukuyama, Shiroyama, & Ueda, 2019). Based on these findings, the present study explores the functional abnormalities of thalamocortical transmission in S286L‐TG rats using multiprobe microdialysis. Specifically, to clarify the pathogenesis of S284L‐ADSHE seizures and the pathophysiology of carbamazepine‐resistant/zonisamide‐sensitive seizures, we determined functional abnormalities in the thalamocortical motor pathway comprising reticular thalamic nucleus (RTN), motor thalamic nuclei (MoTN) and secondary motor cortex (M2; Asanuma, 1992; Beierlein, 2014) of S286L‐TG. Furthermore, to clarify the mechanisms involved in cognitive impairment comorbidity in S284L‐ADSHE, functional abnormalities in the thalamocortical cognitive pathway comprising RTN, mediodorsal thalamic nuclei (MDTN) and orbitofrontal cortex (OFC; Fukuyama et al., 2018; Okada, Fukuyama, Kawano, Shiroyama, Suzuki, & Ueda, 2019; Okada, Fukuyama, Kawano, Shiroyama, & Ueda, 2019; Okada, Fukuyama, Nakano, & Ueda, 2019) were also determined. Finally, we assessed the effects of carbamazepine and zonisamide on the release of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1369 and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1067 in the thalamocortical motor pathways of S286L‐TG.
2. METHODS
2.1. Generation of S286L‐TG
Animal care along with the experimental procedures and protocols for animal experiments were approved by the Animal Research Ethics Committee of the Mie University School of Medicine (Nos. 24–37). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015) and with the recommendations made by the British Journal of Pharmacology. The cDNA of Chrna4 was cloned from a rat fetal brain cDNA panel (Clontech, Palo Alto, CA, USA) using PCR. The amplified cDNA fragment was subcloned in a pCRII‐TOPO vector (Invitrogen, Carlsbad, CA, USA). A rat S286L mutation in Chrna4, corresponding to the human S252L mutation of CHNRA4 (currently termed as S284L mutation), was introduced in the same construct using the QuickChange site‐direct mutagenesis kit (Stratagene Inc., La Jolla, CA, USA). The nucleotide exchange was c.856 T > C and c.857 C > T, which replaced a serine at 286th position with a leucine (S286L), according to the mRNA sequence of the rat Chrna4 (accession no. L31620). The probe sequence synthesised for quantitative PCR was also introduced at SmaI and the BlpI restriction sites within the cDNA sequence, with 36 silent nucleotide substitutions. To generate the S286L‐TG construct, the natural promoter region upstream of Chrna4 was amplified as a 1460 bp fragment and cloned into the BglII/IppoI sites of the pCI‐neo vector (Promega, Madison, WI), followed by the chimeric intron and the multiple cloning site. The Chrna4 (S286L) cDNA was then cloned into the EcoRI site to generate pCI‐neo‐S286L‐Chrna4 (https://www.addgene.org/plasmids/articles/28207545/). The microinjection of the transgene and generation of S286L‐TG animals was conducted at Japan SLC (Shizuoka, Japan), using standard procedures on Sprague–Dawley genetic background rats (https://scicrunch.org/resources/Any/search?q=RGD_12910483&l=RGD_12910483, SLC). Genomic DNA was prepared from the animal tails using a standard method and transformation was confirmed by PCR followed by direct sequencing using the primer pair, forward 5′‐CCAAGGACTCGCCGGC‐3′ and reverse 5′‐CGGTCCCAAAGACACAGACA‐3′.
2.2. Quantitative real‐time PCR (qRT‐PCR)
Brain tissues were preserved in RNAlater (Ambion, TX), and total RNA (1 μg) was isolated using the RNeasy Lipid Tissue Mini Kit (Qiagen, Venlo, Netherlands). Briefly, 1 μg of total RNA was purified and reverse‐transcribed into cDNA using ReverTra Ace qPCR RT Master Mix with gDNA Remover (Toyobo, Osaka, Japan), according to the manufacturer's instructions. For qRT‐PCR to determine expression of the S286L‐mutant Chrna4, wild‐type Chrna4 and total Chrna4 (S286L‐mutant plus wild‐type Chrna4), the cDNA was amplified using a set of custom sequence‐specific primers (Medical & Biological Laboratories, Nagoya, Japan) and detected with locked nucleic acids (LNA) probe (Medical & Biological Laboratories; Table 1) using CFX96 (RRID:SCR_017251, BioRad, Hercules, CA). The TaqMan primer‐probe sets for eukaryotic 18S rRNA (Hs99999901_s1, Applied Biosystem) for a common RNA mass normaliser endogenous control was purchased from Applied Biosystems (Austin, TX). The qRT‐PCR reactions were then performed in triplicate and normalised mRNA levels were determined using the comparative CT method (ΔΔCT) according to the manufacturer's (Applied Biosystems) protocol.
Table 1.
Probes and primers used for qRT‐PCR analysis
| Gene | Probes | Forward primer | Reverse primer |
|---|---|---|---|
| Wild Chrna4 | 5′‐TAGCCAATATCTCAGATGTGGTCCTCGT‐3′ | 5′‐CAGCCACATAGAGACCCG‐3′ | 5′‐GAGCAATGGACAAGCCAAAG‐3′ |
| S286L Chrna4 | 5′‐AATAAATGGTCCCGCCCCGTGG‐3′ | 5′‐GCTCAAAAGGCTGTTTTCTGG‐3′ | 5′‐ATAGATAATCCGAACCGCACC‐3′ |
| Total Chrna4 | 5′‐ACCCCCAGCCATCTA‐3′ | 5′‐CTATGACGGAAGGGTGCAGTG‐3′ | 5′‐ GTCGATGCTGCAGGAGCTCT‐3′ |
2.3. Simple Western analysis
Total plasma membrane proteins were extracted using Minute Plasma Membrane Protein Isolation Kit (Invent Biotechnologies, Plymouth, MN). Simple Western analyses were performed according to the ProteinSimple user manual. Lysates of frontal cortex and thalamus were mixed with a master mix (ProteinSimple, Santa Clara, CA) to a final concentration of 1X sample buffer, 1X fluorescent MW markers, and 40‐mM DTT and then heated at 95°C for 5 min. The samples, blocking reagent, primary antibodies, HRP‐conjugated second antibodies, chemiluminescent substrate, separation and stacking matrices were also dispensed into the designated wells of a 25‐well plate. After plate loading, the electrophoresis and immunodetection steps took place in the capillary system and were fully automated. Simple Western analysis was carried out at room temperature and instrument default settings were used. Capillaries were first filled with separation matrix, followed by stacking matrix and about 40 nl of sample. During electrophoresis, proteins were separated on the basis of MW through the stacking and separation matrices at 250 V for 40 min and then immobilised on the capillary wall using proprietary photoactivated capture chemistry. The matrices were then washed out and the capillaries incubated with a blocking reagent for 15 min. The target proteins were immunoprobed first with primary antibodies and with HRP‐conjugated secondary antibodies. Antibodies against GAPDH (NB300‐322SS, Novus Biologicals, Littleton, CO) and CHRNA4 (mAb299, Sigma‐Aldrich, St. Louis, MO) were diluted in antibody diluent II (ProteinSimple) to 1:100 and 1:50 dilutions, respectively. The antibody incubation time was 0–120 min with antibody diluents. Luminol and peroxide (ProteinSimple) were then added to generate chemiluminescence, which was captured by a CCD camera. The digital image was analysed with Compass software Ver4.1 (ProteinSimple), and quantified data of the detected protein were reported as MW and signal/peak intensities.
2.4. Electrocorticogram
Rats (10 weeks of age) were anaesthetized with 1.8% isoflurane and their heads were positioned in a stereotactic frame. Recording and reference screw electrodes were fastened to the skull over the frontal (A = +2.7 mm, L = +1.8 mm, relative to bregma) and occipital (A = −10.0 mm, L = +1.8 mm, relative to bregma) regions. Electrocorticograms of freely moving rats were telemetrically recorded (Unimec, Tokyo, Japan) and spike‐and‐ wave complexes were analysed using PowerLab (AD Instruments, Dunedin, New Zealand; RRID:SCR_001620). Wistar rats have been previously reported to occasionally show a spike‐and‐wave complex (Blumenfeld et al., 2008). In this study, we used male S286L‐TG and wild‐type littermates bred at our institution, which originated from the Sprague–Dawley strain in SLC. As seen in our previous study, we could not detect the spike‐and‐wave complex in wild‐type littermates (Zhu et al., 2008).
Six days after the telemetry implantation, the baseline electrocorticogram was recorded for 1 hr during light period (12:00–13:00). After baseline recordings, the rat was administered therapeutically relevant doses of zonisamide (30 mg·kg−1·day−1; n = 6) or carbamazepine (25 mg·kg−1·day−1; n = 6) for 7 days as a daily, single intraperitoneal injection (Zhu et al., 2008). Seven days after the administration of carbamazepine or zonisamide, the electrocorticogram was recorded for 1 hr during light period (12:00–13:00).
2.5. Microdialysis
A total of 182 rats were used in the experiments described. Male S286L‐TG and wild‐type littermates were anaesthetized with 1.8% isoflurane and then placed in a stereotactic frame. A concentric direct‐insertion type dialysis probe (0.22‐mm diameter, 3‐mm exposed membrane: Eicom, Kyoto, Japan) was implanted in the orbitofrontal cortex (OFC: A = +3.2 mm, L = +2.4 mm, V = −6.5 mm, relative to bregma). Another concentric direct‐insertion type probe with a shorter exposed membrane (0.22‐mm diameter, 2‐mm exposed membrane: Eicom) was then implanted in the secondary motor cortex (M2: A = +1.8 mm, L = +1.6 mm, V = −2.4 mm, relative to bregma), mediodorsal thalamic nucleus (MDTN: A = −3.0 mm, L = +0.9 mm, V = −6.2 mm, relative to bregma), MoTN comprising ventroanterior and ventrolateral thalamic nuclei (MoTN: A = −2.0 mm, L = +1.4 mm, V = −7.2 mm, relative to bregma) and reticular thalamic nucleus (RTN: A = −1.4 mm, L = +1.2 mm, V = −7.2 mm, relative to bregma; Paxinos & Watson, 2007).
Perfusion experiments were started 18 hr after recovery from isoflurane anaesthesia. The perfusion rate was set at 2 μl·min−1 in all experiments, using modified Ringer's solution (MRS) composed of the following (in mM): 145 Na+, 2.7 K+, 1.2 Ca2+, 1.0 Mg2+ and 154.4 Cl−, buffered with 2‐mM phosphate buffer and 1.1‐mM Tris buffer at pH 7.4. Extracellular levels of L‐glutamate and GABA were measured by ultra‐high‐performance liquid‐chromatography (UHPLC) 8 hr after starting the perfusion. When the coefficients of variation for L‐glutamate and GABA reached <5% over a period of 60 min (stabilisation), control data were obtained over another 60‐min period (pretreatment period). This was followed by perfusion with MRS containing target agents. To determine depolarisation‐induced release, the MRS perfusion medium was altered to include 50‐mM K+ and an equimolar decrease of Na+ (from 145 mM to 97.7 mM) to maintain isotonicity; the high‐potassium medium (HKMRS) was perfused for 20 min. Following the microdialysis experiments, the rats were killed under 1.8% isoflurane and their brains removed. The locations of the dialysis probes were verified by histological examination using 200‐μm‐thick tissue slices (Vibratome 1000, Technical Products International Inc, St. Louis, MO).
2.6. Microdialysis study designs and drug administration
All study designs used both S286L‐TG rats and their wild‐type littermates with the same probe configurations. The target pathways in each study design are described in Figure 1.
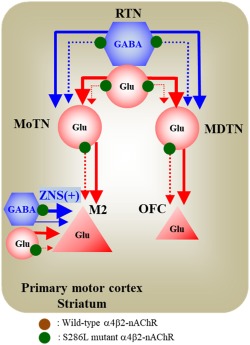
Figure 1.
Target thalamocortical pathways associated with S286L‐autosomal dominant sleep‐related hypermotor epilepsy (ADSHE). Based on the established thalamocortical pathways, the present study was designed to determine the functional abnormalities in the thalamocortical transmissions. Both the motor thalamic nuclei (MoTN) and mediodorsal thalamic nucleus (MDTN) are strongly regulated by GABAergic inhibition from the reticular thalamic nucleus (RTN;Asanuma, 1992; Fukuyama et al., 2018; Fukuyama et al., 2019; Okada, Fukuyama, Kawano, Shiroyama, Suzuki, & Ueda, 2019; Okada, Fukuyama, Kawano, Shiroyama, & Ueda, 2019; Okada, Fukuyama, Nakano, & Ueda, 2019). The pyramidal neurons in the frontal cortex, the secondary motor cortex (M2) and orbitofrontal cortex (OFC) receive glutamatergic projections from the thalamic nuclei (Fukuyama et al., 2018; Fukuyama et al., 2019; Kubota et al., 2013; Okada, Fukuyama, Kawano, Shiroyama, Suzuki, & Ueda, 2019; Okada, Fukuyama, Kawano, Shiroyama, & Ueda, 2019; Okada, Fukuyama, Nakano, & Ueda, 2019; Okada, Fukuyama, Shiroyama, & Ueda, 2019; Yamamura et al., 2009; Yamamura et al., 2009)
2.6.1. Study 1
To explore the pathogenesis and pathophysiology of epileptic seizure in S286L‐TG rats, the effects of therapeutically relevant concentration of zonisamide (500 μM: estimated concentration in extracellular fluid 98 μM; Yamamura, Ohoyama, Nagase, & Okada, 2009) and carbamazepine (100 μM: estimated concentration in extracellular fluid 22.2 μM; Yoshida, Okada, Zhu, & Kaneko, 2007) on 50 mM K+‐evoked releases of L‐glutamate and GABA in the focus region (M2) of S286L‐TG and wild type were determined. Perfusion was commenced with MRS with (experimental) or without (control) zonisamide and carbamazepine (Yamamura, Ohoyama, Nagase, & Okada, 2009; Yoshida et al., 2007) into the M2. After stabilisation of L‐glutamate and GABA in M2, the perfusion medium was switched to HKMRS containing the same agent for 20 min (Yoshida et al., 2007; Figure 4).
Figure 4.
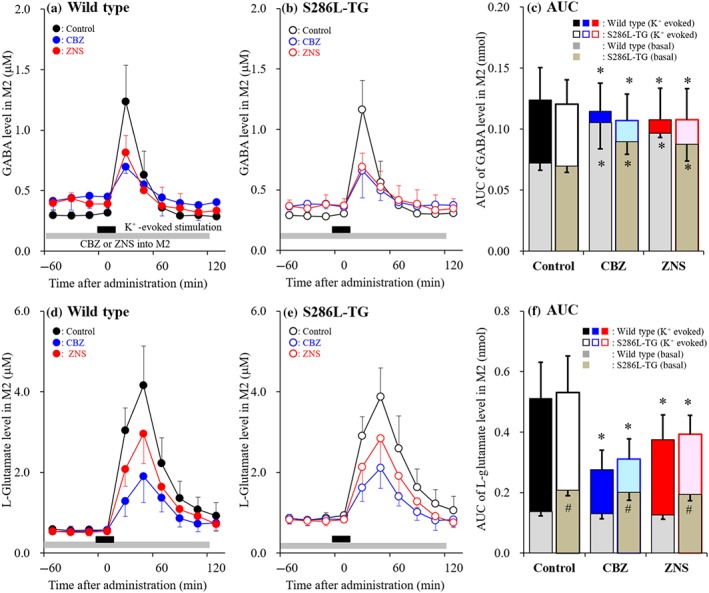
Effects of carbamazepine (CBZ) and zonisamide (ZNS) on K+‐evoked releases of GABA and L‐glutamate in the secondary motor cortex (M2; Study 1). Effects of perfusion with therapeutically relevant concentration of 500‐μM zonisamide (red circles) and 100 μM carbamazepine (blue circles) into the M2 on 50‐mM K+‐evoked releases of GABA (a, b) and L‐glutamate (d, e) in M2 of wild type (a, d) and S286L‐TG (b, e). Ordinates indicate mean extracellular levels of GABA and L‐glutamate (μM; N = 6), and abscissas indicate time after K+‐evoked stimulation (min). Grey bars indicate the perfusion with carbamazepine and zonisamide into M2. Black bars indicate the K+‐evoked stimulation (perfusion with HKMRS) into M2. (c, f) The AUC value of extracellular levels of GABA and L‐glutamate (nmol) after K+‐evoked stimulation (from 20 to 120 min) of panels (a), (b), (d) and (e), respectively. Grey columns in panels (c) and (d) indicate the AUC values of basal extracellular levels of GABA and L‐glutamate in panels (a), (b), (d) and (e). *P < .05 relative to control and # P < .05 relative to wild type by multivariate ANOVA with Tukey's post hoc test
2.6.2. Study 2
To explore the pathogenesis of epileptic seizure of S286L‐TG, the transmission abnormality associated with α4β2‐nAChR in the focus region (M2) of S286L‐TG was determined. Probes were implanted in the M2 and perfusion was commenced with MRS. After stabilisation of L‐glutamate and GABA in the M2, the perfusion medium was switched to MRS with (experimental) or without (control) 0, 10, 30 and 100μM RJR2403 for 120 min. https://www.guidetopharmacology.org/GRAC/DatabaseSearchForward?searchString=metanicotine&searchCategories=all&species=none&type=all&comments=includeComments&order=rank&submit=Search+Database (also known as metanicotine or rivanicline) is a selective α4β2‐nAChR agonist (Alexander et al., 2019; Figure 5).
Figure 5.
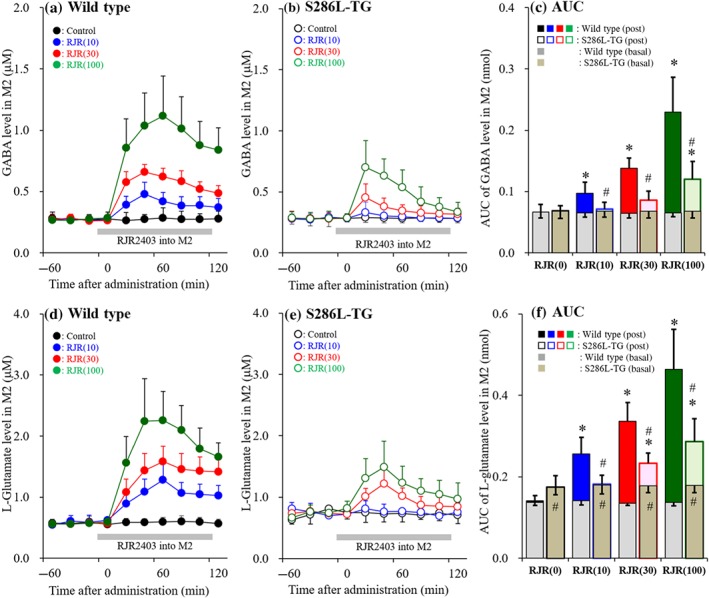
Concentration‐dependent effects of local administration of RJR2403 into the M2 on releases of GABA and L‐glutamate in the secondary motor cortex (M2; Study 2). Concentration‐dependent effects of perfusion with α4β2‐nAChR agonist, RJR2403 (10, 30 and 100 μM) into the M2 on releases of GABA (a, b) and L‐glutamate (d, e) in the M2 of wild type (a, d) and S286L‐TG (b, e). Ordinates indicate mean extracellular levels of GABA and L‐glutamate (μM; N = 6), and abscissas indicate time after RJR2403 administration (min). Grey bars indicate the perfusion with RJR2403 into the M2. (c, f) The AUC value of extracellular levels of GABA and L‐glutamate (nmol) during perfusion with RJR2403 (from 20 to 120 min) in panels (a), (b), (d) and (e), respectively. Grey columns in panels (c) and (f) indicate the AUC values of basal extracellular levels of GABA and L‐glutamate in panels (a), (b), (d) and (e). *P < .05 relative to control and # P < .05 relative to wild type by multivariate ANOVA with Tukey's post hoc test
2.6.3. Study 3
To explore the generator pathway associated with focus in S286L‐TG, the effects of α4β2‐nAChR on thalamocortical motor pathway (RTN‐MoTN‐M2) were determined. Probes were implanted in the RTN, MoTN, M2 and a series of experiments was conducted. The perfusion medium in the M2 was MRS at all times. Perfusion in MoTN and RTN was commenced with MRS with (experimental) or without (control) 100 μM RJR2403. After stabilisation of L‐glutamate release in the M2, the perfusion medium in the MoTN was switched to MRS containing the same agent, along with 100 μM https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4131 (AMPA) for 120 min (Okada, Fukuyama, Kawano, Shiroyama, Suzuki, & Ueda, 2019; Figure 6).
Figure 6.
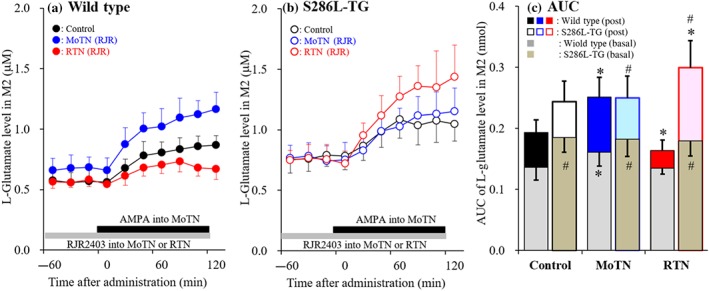
Effects of local administration of RJR2403 into the reticular thalamic nucleus (RTN) and motor thalamic nuclei (MoTN) on secondary motor cortex (M2) l‐glutamate release induced by AMPA‐evoked stimulation in the MoTN (Study 3). Effects of perfusion with or without (control) 100‐μM RJR2403 into MoTN (blue circles) and RTN (red circles) on M2 L‐glutamate release induced by perfusion with 100 μM AMPA into MoTN of wild type (a) and S286‐TG (b). Ordinates indicate mean extracellular L‐glutamate level (μM; N = 6), and abscissas indicate time after AMPA‐evoked stimulation (min). Grey bars indicate the perfusion with RJR2403 into MoTN or RTN. Black bars indicate the perfusion with AMPA into MoTN. (c) The AUC value of extracellular L‐glutamate level (nmol) during perfusion with AMPA (from 20 to 120 min) of panels (a) and (b). Grey columns in panel (c) indicate the AUC values of basal extracellular levels of L‐glutamate in panels (a) and (b). *P < .05 relative to control and # P < .05 relative to wild type by multivariate ANOVA with Tukey's post hoc test
2.6.4. Study 4
To clarify the functional abnormality of S286L‐nAChR in RTN, the effects of activation of α4β2‐nAChR in RTN on both glutamatergic and GABAergic transmissions in RTN‐motor thalamic nuclei pathway were determined. Probes were implanted in the RTN and MoTN. Perfusion in the RTN and MoTN was commenced with MRS. After stabilisation of L‐glutamate and GABA in MoTN, the perfusion medium in the RTN was switched to MRS containing with 0, 30 and 100 μM RJR2403 for 120 min. During perfusion with RJR2403 into the RTN, perfusion medium in MoTN was maintained as MRS (Figure 7).
Figure 7.
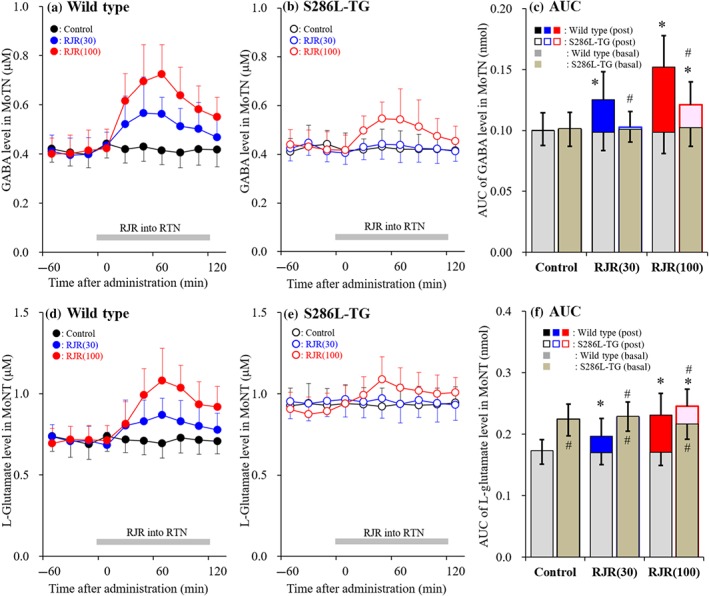
Concentration‐dependent effects of local administration of RJR2403 into the reticular thalamic nucleus (RTN) on releases of GABA and L‐glutamate in the motor thalamic nuclei (MoTN) (Study 4). Concentration‐dependent effects of perfusion with (0, 30 and 100 μM) RJR2403 into RTN on extracellular levels of GABA (a, b) and L‐glutamate (d, e) in MoTN of wild type (a, d) and S286L‐TG (b, e). Ordinates indicate mean extracellular levels of GABA and L‐glutamate (μM; N = 6), and abscissas indicate time after RJR2403 administration (min). Grey bars indicate perfusion with RJR2403 into RTN. (c, f) The AUC value of extracellular levels of GABA and L‐glutamate (nmol) during perfusion with RJR2403 (from 20 to 120 min) in panels (a), (b), (d) and (e), respectively. Grey columns in panels (c) and (f) indicate the AUC values of basal extracellular levels of GABA and L‐glutamate in panels (a), (b), (d) and (e). *P < .05 relative to control and # P < .05 relative to wild type by multivariate ANOVA with Tukey's post hoc test
2.6.5. Study 5
To clarify the pathophysiology of carbamazepine‐resistant/zonisamide‐sensitive epileptic seizures in S284L‐ADSHE, the effects of carbamazepine and zonisamide on α4β2‐nAChR‐induced regional release of L‐glutamate and GABA in the M2 were determined. Probes were implanted in the M2. Perfusion was commenced with MRS with (experimental) or without (control) zonisamide (500 μM) and carbamazepine (100 μM). After stabilisation of L‐glutamate and GABA in the M2, the perfusion medium was switched to MRS containing the same agent, along with 100 μM RJR2403 (Figure 8).
Figure 8.
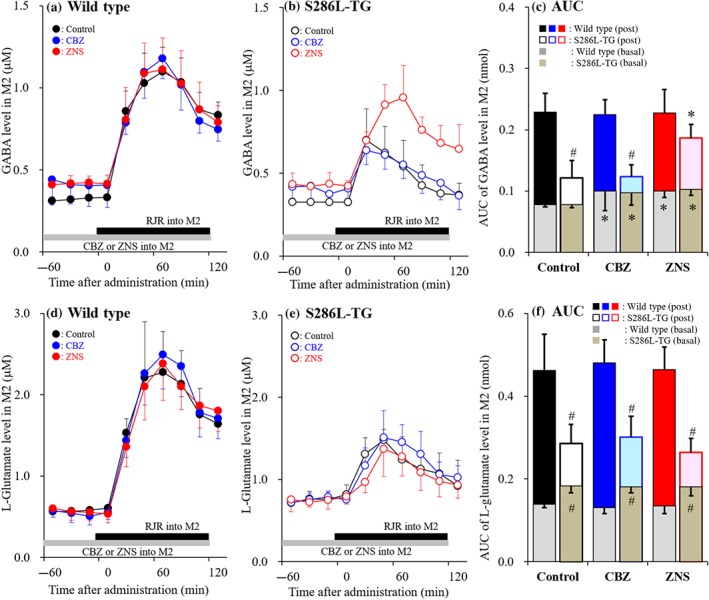
Effects of local administration of zonisamide (ZNS) and carbamazepine (CBZ) into the secondary motor cortex (M2) on RJR2403‐evoked release of GABA and L‐glutamate in the M2 (Study 5). Effects of perfusion with 100 μM carbamazepine (blue circles) and 500‐μM zonisamide (red circles) on 100‐μM RJR2403‐evoked releases of GABA (a, b) and L‐glutamate (d, e) in the M2 of wild‐type littermate (a, d) and S286L‐TG (b, e). Ordinates indicate mean extracellular levels of GABA and L‐glutamate (μM; N = 6), and abscissas indicate time after RJR2403‐evoked stimulation (min). Grey bars indicate the perfusion with carbamazepine and zonisamide into the M2. Black bars indicate the RJR2403‐evoked stimulation (perfusion with 100 μM RJR2403 into the M2). (c, f) The AUC value of extracellular levels of GABA and L‐glutamate (nmol) during perfusion with RJR2403 (from 20 to 120 min) in panels (a), (b), (d), and (e), respectively. Grey columns in panels (c) and (f) indicate the AUC values of basal extracellular levels of GABA and L‐glutamate in panels (a), (b), (d) and (e). *P < .05 relative to control and # P < .05 relative to wild type by multivariate ANOVA with Tukey's post hoc test
2.6.6. Study 6
To study the α4β2‐nAChR‐sensitive releases of L‐glutamate and GABA in the intra‐thalamic RTN‐MDTN pathway (Okada, Fukuyama, Kawano, Shiroyama, Suzuki, & Ueda, 2019), probes were implanted in both the RTN and MDTN. Perfusion was commenced with MRS in both the RTN and MDTN. After stabilising L‐glutamate and GABA in the MDTN, the perfusion medium in the RTN (but not MDTN) was switched to MRS containing 0, 30 and 100 μM RJR2403 for 120 min. During perfusion with RJR2403 into the RTN, perfusion medium in MDTN was maintained as MRS (Figure 9).
Figure 9.
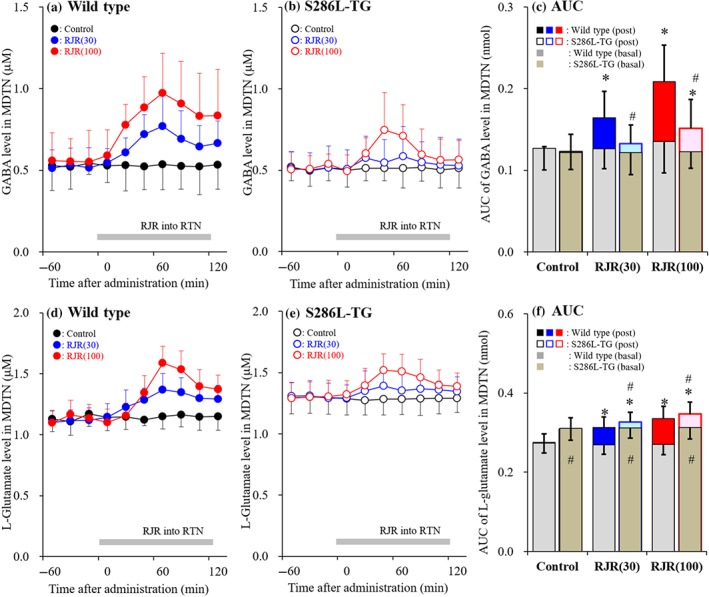
Concentration‐dependent effects of local administration of RJR2403 in the reticular thalamic nucleus (RTN) on releases of GABA and L‐glutamate in the mediodorsal thalamic nucleus (MDTN) (Study 6). Concentration‐dependent effects of perfusion with RJR2403 (0, 30 and 100 μM) into the RTN on extracellular levels of GABA (a, b) and L‐glutamate (d, e) in the MDTN of wild type (a, d) and S286L‐TG (b, e). Ordinates indicate mean extracellular levels of GABA and L‐glutamate (μM; N = 6), and abscissas indicate time after RJR2403 administration (min). Grey bars indicate the perfusion with RJR2403 into the RTN. (c, f) The AUC value of extracellular levels of GABA and L‐glutamate (nmol) during perfusion with RJR2403 (from 20 to 120 min) in panels (a), (b), (d), and (e), respectively. Grey columns in panels (c) and (f) indicate the AUC values of basal extracellular levels of GABA and L‐glutamate in panels (a), (b), (d) and (e). *P < .05 relative to control and # P < .05 relative to wild type by multivariate ANOVA with Tukey's post hoc test
2.6.7. Study 7
To study α4β2‐nAChR regulation in the RTN‐MDTN‐OFC thalamocortical pathway, probes were implanted in the RTN, MDTN and OFC, and a series of experiments was conducted. The perfusion medium in OFC was MRS at all times. Perfusion in the MDTN and RTN was commenced with MRS with (experimental) or without (control) 100 μM RJR2403. After stabilisation of L‐glutamate release in te OFC, the perfusion medium in the MDTN was switched to MRS containing the same agent, along with 100 μM AMPA for 120 min (Okada, Fukuyama, Kawano, Shiroyama, Suzuki, & Ueda, 2019; Figure 10).
Figure 10.
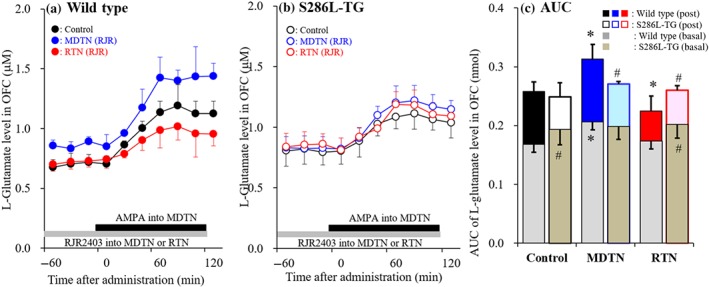
Effects of perfusion of RJR2403 into reticular thalamic nucleus (RTN) and mediodorsal thalamic nucleus (MDTN) on orbitofrontal cortex (OFC) L‐glutamate release induced by AMPA‐evoked stimulation in MDTN (Study 7). Effects of perfusion with or without (control) 100‐μM RJR2403 into the MDTN (blue circles) and RTN (red circles) on MDTN 100‐μM AMPA‐evoked L‐glutamate release in the OFC of wild type (a) and S286‐TG (b). Ordinates indicate mean extracellular L‐glutamate level (μM; N = 6), and abscissas indicate time after AMPA‐evoked stimulation (min). Grey bars indicate the perfusion with RJR2403 into MDTN or RTN. Black bars indicate the perfusion with AMPA into MDTN. (c) The AUC value of extracellular L‐glutamate level (nmol) during perfusion with AMPA (from 20 to 120 min) in panels (a) and (b). Grey columns in panel (c) indicate the AUC values of basal extracellular levels of L‐glutamate in panels (a) and (b). *P < .05 relative to control and # P < .05 relative to wild type by multivariate ANOVA with Tukey's post hoc test
2.7. Ultra‐high‐performance liquid‐chromatography (UHPLC)
L‐Glutamate and GABA levels were determined using UHPLC equipped with xLC3185PU (Jasco, Tokyo, Japan) and fluorescence detection (xLC3120FP, Jasco) following dual derivatisation with isobutyryl‐L‐cysteine and o‐phthalaldehyde (Karlsen, Korbo, Uylings, & Pakkenberg, 2014; Mtui, Gruener, & Dockery, 2015). Derivatisation solutions were prepared by dissolving isobutyryl‐L‐cysteine (2 mg) and o‐phthalaldehyde (2 mg) in 0.1‐ml ethanol, followed by the addition of 0.9‐ml sodium borate buffer (0.2 M, pH 9.0; Yamamura et al., 2013). Automated pre‐column derivatisation was performed by drawing 5‐μl aliquots of sample, standard, or blank solutions and 5 μl of derivatisation solution together into a reaction vial and incubating for 5 min before injection. The derivatised samples (5‐μl aliquots) were injected by an auto sampler (xLC3059AS, Jasco). The analytical column (YMC Triat C18, particle 1.8 μm, 50 × 2.1 mm, YMC, Kyoto, Japan) was maintained at 45°C, and flow rate was set at 500 μl·min−1. A linear gradient elution programme was performed over a period of 10 min with mobile phases A (0.05‐M citrate buffer, pH 5.0) and B (0.05‐M citrate buffer containing 30% acetonitrile and 30% methanol, pH 3.5). The excitation/emission wavelengths of the fluorescence detector were set at 280/455 nm.
2.8. Data analysis
All experiments in this study were designed with equally sized animal groups (N = 6) without carrying out a formal power analysis, in keeping with previous studies (Okada, Fukuyama, Kawano, Shiroyama, Suzuki, & Ueda, 2019; Okada, Fukuyama, Kawano, Shiroyama, & Ueda, 2019; Okada, Fukuyama, Shiroyama, & Ueda, 2019; Zhu et al., 2008). All values are expressed as mean ± SD and P < .05 (two‐tailed) was considered statistically significant for all tests. Drug doses in systemic and local administrations were selected based on values in previous studies (Okada, Fukuyama, Kawano, Shiroyama, Suzuki, & Ueda, 2019; Okada, Fukuyama, Kawano, Shiroyama, & Ueda, 2019; Okada, Fukuyama, Shiroyama, & Ueda, 2019; Zhu et al., 2008). Where possible, we sought to randomise and blind the data. In particular for the determination of extracellular transmitter levels, the sample order on the autosampler was determined by a random number table.
Regional transmitter concentrations were analysed by Mauchly's sphericity test followed by multivariate ANOVA using BellCurve for Excel ver. 3.2 (https://scicrunch.org/scicrunch/Resources/record/nlx_144509-1/SCR_017294/resolver; Social Survey Research Information Co., Ltd., Tokyo, Japan). When the data did not violate the assumption of sphericity (P > .05), the F value of multivariate ANOVA was analysed using sphericity assumed degrees of freedom. However, if the assumption of sphericity was violated (P < .05), the F value was analysed using Chi‐Muller's corrected degrees of freedom. When the F value for the drug/concentration/genotype factors of multivariate ANOVA was significant, the data were analysed by Tukey's post hoc test. Transmitter level was expressed as the AUC between 20 and 180 min (AUC20–180) after perfusion of target agent.
The effects of carbamazepine and zonisamide on spike‐and‐wave frequency in S286L‐TG, as well as mRNA expression of α4 subunit of nAChR (Chrna4), were analysed using two‐way ANOVA when there was no significant variance in homogeneity as determined by Levene's test. When the F value was significant (P < .05), data were subsequently analysed by Tukey's post hoc test. The protein expression of α4 subunit of nAChR was analysed by Student's t‐test because the F test was not significant. The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018).
2.9. Chemical agents
The selective α4β2‐nAChR agonist, (E)‐N‐methyl‐4‐(3‐pyridinyl)‐3‐buten‐1‐amine oxalate (RJR2403 oxalate) was obtained from Cosmo Bio (Tokyo, Japan; Alexander et al., 2019); carbamazepine and AMPA (Alexander et al., 2019) were obtained from Wako Chemicals (Osaka, Japan). Zonisamide was provided by Dainippon‐Sumitomo Pharma (Osaka, Japan). All compounds were prepared on the day of the experiment. RJR2403 and AMPA were dissolved in modified ringer solution (MRS). Both carbamazepine and zonisamide were initially made up as a 10‐mM stocks in DMSO and then diluted to 1 mM with MRS.
2.10. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
3. RESULTS
3.1. Validation of S286L‐TG
The expression of wild‐type versus the S286L‐mutant Chrna4 in the frontal cortex of S286L‐TG was 60% versus 40%, respectively (Figure 2a). In the thalamus, expression of wild‐type versus S286L‐mutant Chrna4 was 63% versus 37%, respectively (Figure 2b). The total expression of Chrna4 (wild‐type and S286L‐mutant Chrna4 combined) in the frontal cortex and thalamus of S286L‐TG was 119% and 139%, respectively of the expression level in wild‐type littermates (Figure 2a,b). Furthermore, protein expression of α4 subunit of nAChR in the frontal cortex and thalamus of S286L‐TG was also almost equal to the levels in wild‐type rats (Figure 2c,d). Clinical studies using PET have shown that the densities of α4β2‐nAChR distribution in the frontal cortex and thalamus of ADSHE patients are also equal to those of healthy volunteers (Picard et al., 2006). Therefore, transcript and protein expression levels of S286L‐mutant nAChR in S286L‐TG were similar to those seen in ADSHE patients.
Figure 2.
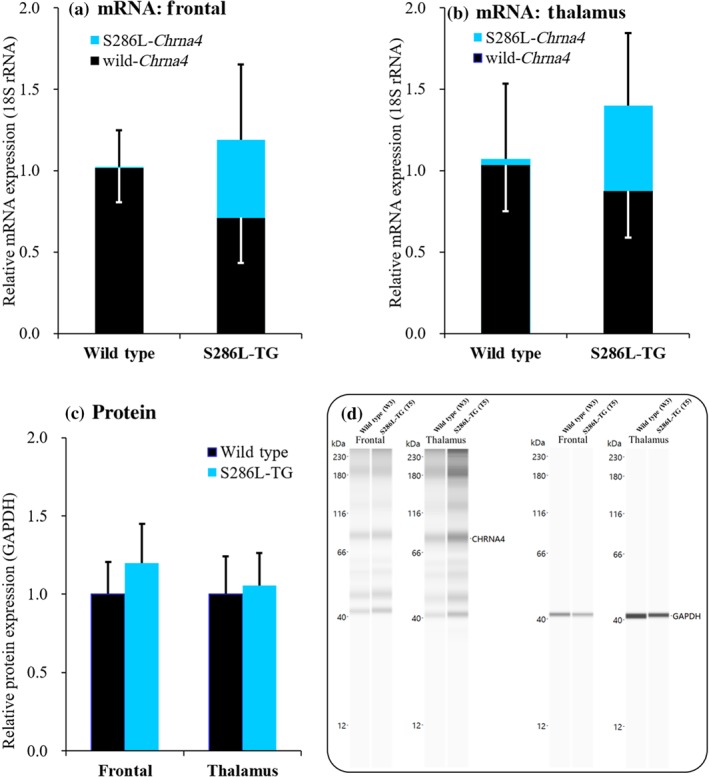
Construct validity of S286L‐TG. (a, b) mRNA expression of wild‐type Chrna4 (closed columns) and S286L‐mutant Chrna4 (blue columns) in frontal cortex and thalamus, respectively. Ordinates indicate the relative mRNA expression (%). (c) Protein levels of α4 subunit of nAChR in the frontal cortex and thalamus. Ordinates indicate the relative protein expression of α4 nAChR subunit (%). (d, e) Electropherogram and pseudo‐gel images using Simple Western results using anti‐GAPDH and anti‐CHRNA4 antibody for blotting of in frontal cortex and thalamus of wild‐type littermates and S286L‐TG
The typical electrocorticogram that is recorded during epileptic wandering is shown in Figure 3a. The typical spike‐and‐wave complex before and after sub‐chronic (7 days) administration of zonisamide and carbamazepine is indicated in Figure 3c. The spike‐and‐wave complex frequency of S286L‐TG rats was reduced by sub‐chronic administration of zonisamide, but not by carbamazepine (Figure 3b). The sensitivity of S286L‐TG rats is similar to that of ADSHE patients carrying the S286L‐mutation, who experience carbamazepine‐resistant but zonisamide‐sensitive seizures (Hirose et al., 1999; Ito et al., 2000; Okada et al., 2010; Rozycka et al., 2003).
Figure 3.
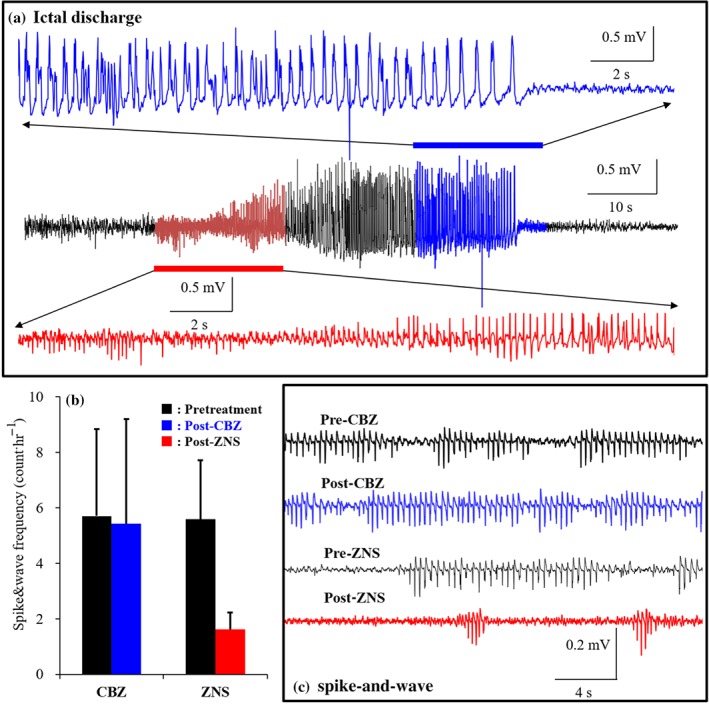
Face and predictive validities of S286L‐TG. (a) Typical electrocorticogram during epileptic wandering in S286L‐TG. (b) Effects of intraperitoneal administration of 25 mg·kg−1 carbamazepine (CBZ) or 30 mg·kg−1 zonisamide (ZNS) for 7 days on frequency of epileptic discharges (spike‐and‐wave complex) in S286L‐TG at before (pretreatment: closed columns) and after anticonvulsants treatment (post‐treatment of crbamazepine and zonisamide are blue and red columns, respectively). Ordinates indicate mean counts of spike‐and‐wave complex (counts·day−1; N = 6). *P < .05 versus pre‐administration with anticonvulsant, two‐way ANOVA with Tukey's post hoc test. (c) The typical spike‐and‐wave complex before (pre) and after (post) intraperitoneal administration of 25 mg·kg−1 carbamazepine and 30 mg·kg−1 zonisamide for 7 days
These results of spontaneous epileptic seizures, sensitivities to antiepileptic drugs, and expression of mRNA and protein of α4 subunit nAChR demonstrate that S286L‐mutation in the rat Chrna4 successfully recapitulates the effects of the equivalent S284L‐mutation in the human CHRNA4 gene. Hence, the S286L‐TG fulfil the validation criteria (face, construct and predictive validities) to present a viable genetic animal model for ADSHE (Okada et al., 2010).
3.2. Study 1: Effects of local administration of zonisamide or carbamazepine into secondary motor cortex on K+‐evoked GABA and L‐glutamate release
There was no differences in basal GABA release in the M2 between S286L‐TG and its wild‐type littermate (Figure 4a–c). Perfusion with 500‐μM zonisamide or 100 μM carbamazepine into the M2 increased basal GABA release, but significantly decreased K+‐evoked GABA release in both S286L‐TG and wild type (Figure 4a–c).
Basal L‐glutamate release in the M2 of S286L‐TG was significantly larger than that in wild type (Figure 4d–f). Perfusion with 500 μM zonisamide or 100 μM carbamazepine did not affect basal L‐glutamate release in the M2, but significantly decreased K+‐evoked L‐glutamate release in both S286L‐TG and wild type. We did not observe any differences between zonisamide and carbamazepine perfusion (Figure 4d–f).
The sensitivity of depolarisation‐induced release of these transmitters and sensitivity to anticonvulsants between were also the same between S286L‐TG and wild type. These results suggest that the transmission abnormality in M2 of S286L‐TG is probably not associated with pathogenesis or pathophysiology of epileptic seizure caused by S286L‐TG.
3.3. Study 2: Concentration‐dependent effects of local administration of RJR2403 into the secondary motor cortex on GABA and L‐glutamate release
Perfusion with RJR2403 (10, 30 and 100 μM) into the M2 caused a concentration‐dependent increase in the release of GABA in the M2 of both S286L‐TG and wild type (Figure 5a–c). The stimulation of GABA release by RJR2403 was significantly larger in wild type than in S286L‐TG (Figure 5c). All concentrations of RJR2403 significantly increased GABA release in wild type (peak time: 60 min), but only highest concentration of 100 μM RJR2403 increased GABA release in S286L‐TG (peak time: 20 min; Figure 5a–c). RJR2403 perfusion into the M2 also induced a concentration‐dependent increased in L‐glutamate release in both S286L‐TG (peak time: 40 min) and wild type (peak time: 60 min; Figure 5d–f). However, the stimulatory effect was significantly greater in wild type compared with S286L‐TG (Figure 5f). All concentrations of RJR2403 increased L‐glutamate release in wild type, but only the 30 and 100 μM concentrations caused an increase in S286L‐TG (Figure 5f).
Thus, we demonstrated that the sensitivity of glutamatergic and GABAergic transmission to α4β2‐nAChR is impaired in the focus region of S286L‐TG, whereas the desensitisation of α4β2‐nAChR is probably enhanced in S286L‐TG when compared to wild type.
3.4. Study 3: Effects of local administration of RJR2403 into the reticular thalamic nucleus and motor thalamic nuclei on secondary motor cortex L‐glutamate release, induced by AMPA‐evoked motor thalamic nuclei stimulation
The first two studies could not detect any hypersensitivity or hyperactivity in the M2 (ADSHE focus region). Therefore, these results suggest that M2 per se cannot produce epileptic discharges (ictal or interictal discharges) but probably triggers epileptic discharges by integrating external inputs from generator regions (epileptic focus generator). Typical symptoms of ADSHE are hyperactivities relevant to the M2, striatum and MoTN (Plazzi, Tinuper, Montagna, Provini, & Lugaresi, 1995; Provini et al., 1999). Given that the M2 receives thalamocortical glutamatergic input from MoTN (Berendse, Galis‐de Graaf, & Groenewegen, 1992; Conde, Maire‐Lepoivre, Audinat, & Crepel, 1995), the thalamocortical (MoTN‐M2) glutamatergic transmission associated with α4β2‐nAChR was therefore explored as a possible generator.
Perfusion with RJR2403 (100 μM) into the MoTN significantly increased basal L‐glutamate release in the M2 of wild type but not S286L‐TG. However, perfusion with RJR2403 into the RTN did not affect M2 L‐glutamate release in either genotype (Figure 5a–c). Perfusion with 100 μM AMPA into the MoTN increased L‐glutamate release in the M2 of both genotypes (Figure 6a–c), but the degree of increase remained equivalent between both wild type and S286L‐TG (Figure 6c). Perfusion with RJR2403 into the MoTN increased AMPA‐induced L‐glutamate release in wild type but not S286L‐TG (Figure 6a–c), whereas perfusion with RJR2403 into the RTN decreased AMPA‐induced L‐glutamate release in wild type, but conversely increased it in S286L‐TG (Figure 6a–c).
These results suggest that the stimulatory function of α4β2‐nAChR in the MoTN on thalamocortical glutamatergic transmission was attenuated in S286L‐TG, whereas its inhibitory function in the RTN converted to a stimulatory function in S286L‐TG. Therefore, S286L‐mutant nAChR generates disinhibition of thalamocortical glutamatergic transmission. This functional abnormality in the inhibitory regulation associated with S286L‐nAChR in RTN probably plays important roles in the pathogenesis of S284L‐ADSHE.
3.5. Study 4: Concentration‐dependent effects of local administration of RJR2403 into the reticular thalamic nucleus on GABA and L‐glutamate release in the motor thalamic nuclei
There was no difference in basal GABA release in the MoTN of S286L‐TG and wild type (Figure 7a–c). Perfusion with RJR2403 (30 and 100 μM) into the RTN induced a concentration‐dependent increase in GABA release in the MoTN of wild type, whereas only 100 μM significantly increased the release in S286L‐TG (Figure 7a–c).
Basal L‐glutamate release in the MoTN of S286L‐TG was significantly larger than that in wild type (Figure 7d–f). Perfusion with RJR2403 (30 and 100 μM) into the RTN caused a concentration‐dependent increase in L‐glutamate release in the MoTN of wild type, but this effect was only very weakly observed in S286L‐TG and only at the highest dose, 100 μM, of RJR2403 (Figure 7d–f).
Activation of α4β2‐nAChR in RTN stimulates both glutamatergic and GABAergic transmission in RTN‐motor thalamic nuclei pathway. However, the S286L‐nAChR produces disinhibition in MoTN, since perfusion with RJR2403 enhanced glutamatergic but did not affect GABAergic transmission in RTN‐motor thalamic nuclei pathway.
3.6. Study 5: Effects of local administration of zonisamide and carbamazepine into the secondary motor cortex on RJR2403‐evoked GABA and L‐glutamate release
Perfusion with therapeutic‐relevant concentration of carbamazepine or zonisamide into the M2 increased basal GABA release in the M2 of both S286L‐TG and wild type (Figure 8a–c). Perfusion with 100 μM RJR2403 into the M2 significantly increased basal GABA release in both genotypes, but the stimulatory effects were significantly greater in the wild type (Figure 8c). Neither carbamazepine nor zonisamide affected RJR2403‐evoked GABA release in the M2 of the wild type (Figure 8a,c). In contrast, carbamazepine perfusion did not affect RJR2403‐evoked GABA release in the M2 of S286L‐TG, but zonisamide enhanced this release (Figure 8b,c).
Perfusion with either carbamazepine or zonisamide into the M2 did not affected basal L‐glutamate release in either genotype (Figure 8d,f). Perfusion with 100 μM RJR2403 into the M2 increased L‐glutamate release in both S286L‐TG and wild type, but did so more effectively in the wild type (Figure 8d–f). This RJR2403‐induced increase in release was unaffected by carbamazepine or zonisamide perfusion in both genotypes (Figure 8d–f).
These results suggest that carbamazepine and zonisamide do not affect α4β2‐nAChR‐induced glutamatergic and GABAergic transmissions in the M2 of wild type. Zonisamide does, however, enhance α4β2‐nAChR‐associated GABAergic inhibition without affecting excitatory glutamatergic transmission in the M2 of S286L‐TG.
3.7. Study 6: Concentration‐dependent effects of local administration of RJR2403 in the reticular thalamic nucleus on GABA and L‐glutamate release in the mediodorsal thalamic nucleus
To explore the pathogenesis of the cognitive impairment comorbidity in S284L‐ADSHE, the transmission abnormality associated with α4β2‐nAChR in the thalamocortical cognitive pathway (Fukuyama et al., 2018; Okada, Fukuyama, Kawano, Shiroyama, Suzuki, & Ueda, 2019; Okada, Fukuyama, Kawano, Shiroyama, & Ueda, 2019) of S286L‐TG was investigated.
There was no difference in basal GABA release in MDTN between S286L‐TG and wild type (Figure 9a–c). Perfusion with RJR2403 (30 and 100 μM) into the RTN induced a concentration‐dependent increase in GABA release in the MDTN of wild type (Figure 9a,c), whereas only the highest dose (100 μM) of RJR2403 significantly increased the release in S286L‐TG (Figure 9b,c).
Basal L‐glutamate release in the MDTN of S286L‐TG was significantly higher than in wild type (Figure 9d–f). Perfusion with RJR2403 into the RTN significantly increased L‐glutamate release in the MDTN of both genotypes (Figure 9d–f), but the effect was significantly stronger in wild type (Figure 9f).
Similar to the thalamocortical motor pathway, the functional abnormalities seen in the thalamocortical cognitive pathway of S286L‐TG show a marked loss‐of‐function in the both glutamatergic and GABAergic transmissions.
3.8. Study 7: Effects of RJR2403 perfusion into reticular thalamic nucleus and mediodorsal thalamic nucleus on orbitofrontal cortex L‐glutamate release induced by AMPA‐evoked stimulation in mediodorsal thalamic nucleus
To explore the mechanisms behind the functional abnormality in glutamatergic transmission in the thalamocortical cognitive pathway of S286L‐TG, the effects of α4β2‐nAChR in the MDTN and RTN on glutamatergic transmission in the OFC were assessed.
Basal L‐glutamate release in the OFC of S286L‐TG animals was significantly higher than in wild type (Figure 10a–c). Perfusion with RJR2403 (100 μM) into the MDTN significantly increased L‐glutamate release in the OFC of wild type, but not in S286L‐TG (Figure 10a–c). Perfusion with RJR2403 into the RTN, however, did not affect basal L‐glutamate release in both genotypes (Figure 10a–c). Perfusion of 100 μM AMPA into the MDTN significantly increased L‐glutamate release in both S286L‐TG and wild type (Figure 10a–c). Perfusion of RJR2403 into the MDTN significantly increased AMPA‐evoked L‐glutamate release in wild type, but not in S286L‐TG (Figure 10a–c), whereas perfusion into the RTN significantly decreased the release in wild type but not in S286L‐TG (Figure 10a–c).
In wild‐type rats, the regulatory mechanisms associated with α4β2‐nAChR in the thalamocortical cognitive pathway is similar to that in thalamocortical motor pathways. The loss‐of‐function mutation of S286L causes similar effects in the MDTN in thalamocortical cognitive pathway and the MoTN in thalamocortical motor pathway. The functional abnormality of α4β2‐nAChR in the RTN, however, is different between thalamocortical cognitive and motor pathways. Indeed, in spite of a gain‐of‐function effect of S286L‐nAChR in the RTN of the thalamocortical motor pathway, activation of α4β2‐nAChR does not affect glutamatergic transmission in the thalamocortical cognitive pathway.
4. DISCUSSION
4.1. Candidate neural pathways for pathomechanism of autosomal dominant sleep‐related hypermotor epilepsy
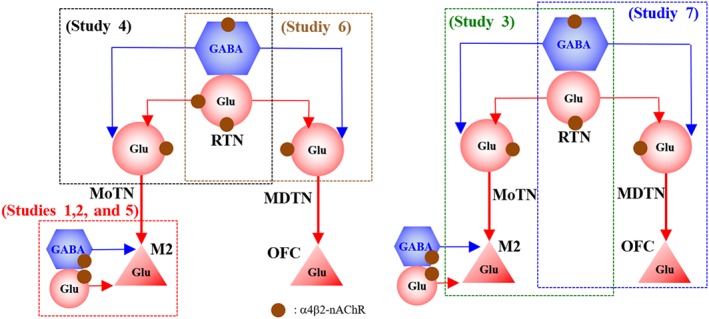
Typical symptoms of ADSHE are hyperactivities of the frontal cortex (premotor cortex and OFC), striatum and MoTN (Plazzi et al., 1995; Provini et al., 1999). M2 receives thalamocortical glutamatergic inputs from MoTN and sends glutamatergic projections to the primary motor cortex and striatum (Berendse et al., 1992; Conde et al., 1995), which is considered to generate nocturnal‐paroxysmal‐dystonia (Fedi et al., 2008). Several studies have indicated that disturbance of MDTN, which primarily projects to the OFC, is particularly relevant to cognitive dysfunctions (Alelu‐Paz & Gimenez‐Amaya, 2008; Dermon & Barbas, 1994; Fukuyama et al., 2018; Okada, Fukuyama, Kawano, Shiroyama, Suzuki, & Ueda, 2019; Okada, Fukuyama, Kawano, Shiroyama, & Ueda, 2019; Schuetze et al., 2016; Vertes et al., 2015) such as intellectual disability (Karlsen et al., 2014), autism (Schuetze et al., 2016), epileptic cognitive‐impairment and arousal (Leeman‐Markowski, Smart, Faught, Gross, & Meador, 2015). Additionally, various thalamic nuclei, including MDTN and MoTN, receive GABAergic inhibition from RTN (Asanuma, 1992; Beierlein, 2014; Fukuyama et al., 2019; Okada, Fukuyama, Kawano, Shiroyama, Suzuki, & Ueda, 2019; Okada, Fukuyama, Shiroyama, & Ueda, 2019). The RTN, in turn, receives excitatory cholinergic terminals from the brainstem and other thalamic nuclei (Beierlein, 2014). Several clinical and preclinical studies suggest that functional abnormalities in α4β2‐nAChR‐associated thalamocortical transmission might be involved in the pathogenesis or pathophysiology of ADSHE and/or its comorbidities (Figure 11). Based on these data and our hypothesis, we analysed thalamocortical glutamatergic transmission in the context of (a) pathogenesis of ADSHE, (b) pathophysiology of carbamazepine‐resistant/zonisamide‐sensitive ADSHE‐seizures and (c) pathogenesis of comorbidities of S284L‐ADSHE.
Figure 11.
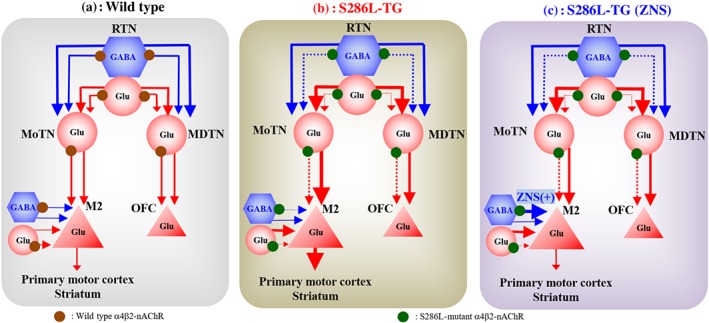
Proposed hypothesis for functional abnormalities of thalamocortical motor and cognitive pathways in S286L‐TG. Reticular thalamic nucleus (RTN) projects both glutamatergic and GABAergic terminal to motor thalamic nuclei (MoTN) and mediodorsal thalamic nucleus (MDTN). GABAergic inhibition in RTN‐MoTN and RTN‐MDTN is predominant rather than glutamatergic excitation in wild type (a). Activation of α4β2‐nAChR in the RTN enhances both glutamatergic and GABAergic transmission in the RTN‐MoTN and RTN‐MDTN pathways. The MoTN and MDTN project glutamatergic terminal to respective the secondary motor cortex (M2) and orbitofrontal cortex (OFC). In the MoTN and MDTN, α4β2‐nAChR activates glutamatergic transmission to the frontal cortex. The M2 glutamatergic neuron, which receives inhibitory GABAergic (from other cortical region) and excitatory glutamatergic (from thalamus and other cortical region) inputs, and projects to primary motor cortex and striatum. Loss‐of‐function of S286L‐mutant α4β2‐nAChRs in RTN generates GABAergic disinhibition in the both MoTN and MDTN, leading to enhancement of glutamatergic transmission in respective the M2 and OFC (b). Zonisamide (ZNS) compensates the attenuated GABAergic transmission in the M2, whereas carbamazepine (CBZ) does not affect (c)
Previous electrophysiological studies using two‐electrode voltage‐clamp of Xenopus oocytes, expressing mutant nAChR in a heterozygous manner, indicated that ADSHE‐nAChR increased ACh‐sensitivity and gain‐of‐function depending on experimental conditions. For the most part, however, ADSHE‐nAChR show enhanced desensitisation (Okada et al., 2010). Although patch‐clamp studies have also demonstrated several functional abnormalities of S284L‐α4β2‐nAChR, including enhanced desensitisation and ACh‐sensitivity without use‐dependent potentiation (Matsushima et al., 2002; Okada et al., 2010; Rodrigues‐Pinguet et al., 2003; Zhu et al., 2008). Our study shows a loss‐of‐function of S286L‐nAChR in both thalamocortical motor and cognitive pathways. Indeed, continuous perfusion with an α4β2‐nAChR agonist increased GABA and L‐glutamate release in M2 (Figure 5), MoTN (Figure 7) and MDTN (Figure 9) of both wild type and S286L‐TG, although the peak time differed between the two genotypes. Taken together with previous electrophysiological findings (Matsushima et al., 2002; Okada et al., 2010; Rodrigues‐Pinguet et al., 2003; Zhu et al., 2008), the hypersensitive functional abnormalities to ACh of ADSHE‐nAChR (S284L, S280F, V287M, V287L, and I279N; Okada et al., 2010) potentiate the functional abnormality of enhanced desensitisation of ADSHE‐nAChR. These reciprocal enhancement of function abnormalities of S284L‐nAChR between enhanced hypersensitivity and desensitisation probably contributes to the pathogenesis of ADSHE‐seizures.
Despite observable spontaneous seizures in S286L‐TG, no functional abnormalities in depolarisation‐induced L‐glutamate or GABA release in the M2 were noted. Although zonisamide reduced epileptic discharges in S286L‐TG, but carbamazepine did not, these anticonvulsants are voltage‐sensitive Na+ channel inhibitors which inhibit depolarisation‐induced L‐glutamate and GABA release in the M2 of both genotypes. We have previously demonstrated that S284L‐TG showed no hypersensitivity to maximal electroshock seizures as compared to wild type (Zhu et al., 2008). In summary, these data suggest that seizure‐susceptibility of S286L‐TG is unrelated to M2, per se its function (Nakatsu et al., 2004; Zhu et al., 2008).
4.2. Pathogenesis of autosomal dominant sleep‐related hypermotor epilepsy‐seizures
There are two hypothesised pathogeneses of ADSHE‐seizures. The first was proposed using S280F‐knockin mice (Klaassen et al., 2006), which suggests that ADSHE‐seizures are generated by inhibitory synchronisation in focus regions via activation of S280F‐α4β2‐nAChRs located on the presynaptic terminals and somato‐dendritic compartments of cortical GABAergic interneurons. The second hypothesis comes from our work in S284L‐TG rats, which indicates that the attenuation of both synaptic and extra‐synaptic GABAergic transmission produces relative hyperactivation of glutamatergic transmission in ADSHE focus regions (Okada et al., 2010; Zhu et al., 2008).
The present study highlights the regulatory mechanisms associated with α4β2‐nAChR in the thalamocortical motor pathway and functional abnormalities of S286L‐mutation (Figure 11). Our results indicate that α4β2‐nAChR stimulates transmission in thalamocortical motor pathway as excitatory system in each region. However, activation of α4β2‐nAChR in RTN inhibited L‐glutamate release in M2 (Figure 6) by enhancing GABAergic inhibition and glutamatergic excitation in MoTN (Figure 7). Therefore, α4β2‐nAChR activation in RTN enhances both excitatory glutamatergic and inhibitory GABAergic transmissions to MoTN, but typically exhibits suppression of MoTN activity in wild type (Figure 11; Asanuma, 1992; Beierlein, 2014).
Perfusion with RJR2403 (a selective α4β2‐nAChR agonist) into the M2 increased regional L‐glutamate and GABA releases in wild type, but this effect was attenuated in M2 of S286L‐TG (Figure 5). Furthermore, activation of S286L‐nAChR in MoTN did not affect MoTN‐M2 thalamocortical glutamatergic transmission (Figure 6). These results indicate that the S286L‐mutation induces loss of regulation associated with α4β2‐nAChR in the thalamocortical motor pathway. Remarkably, activation of α4β2‐nAChRs in RTN inhibited glutamatergic transmission in the thalamocortical motor pathways of wild type, but paradoxically enhanced it in S286L‐TG (Figures 6 and 11). Therefore, loss‐of‐function in S286L‐nAChR increases excitatory glutamatergic transmission in the thalamocortical motor pathways by switching from inhibitory to excitatory regulation in RTN‐motor thalamic nuclei pathway (Figure 11). This functional switching may contribute to S286L‐ADSHE seizures, which are electrophysiologically detected in the M2. Although the M2 itself cannot independently generate epileptic discharge, it can integrate external excitatory inputs from the thalamocortical motor pathway resulting in generation of epileptic discharges in the secondary motor cortex. Thus, secondary motor cortex plays an important role in the generation of ADSHE‐seizures as the focus region.
Basal extracellular L‐glutamate level in the MoTN, MDTN, M2 and OFC of S286L‐TG was higher than that in wild type. The basal extracellular GABA level, on the other hand, was equivalent for both genotypes. This imbalance between L‐glutamate and GABA levels in S286L‐TG is probably generated by synaptic and extra‐synaptic GABAergic disinhibition (Zhu et al., 2008) and shifts in epileptic imbalance transmission (Okada et al., 2002). To better understand the mechanisms behind this, further studies are required.
4.3. Pathophysiology of carbamazepine‐resistant/zonisamide‐sensitive seizures in S286L‐TG
Zonisamide affects various transmission systems (Okada & Kaneko, 2011) and has dual action against GABAergic transmission, GABA release and GABAA receptor (Okada & Kaneko, 2011). Zonisamide enhances basal GABA release but inhibits depolarisation‐induced GABA release via inhibition of voltage‐dependent Na+ channel, carbonic anhydrase, ryanodine, inositol phosphate and δ‐opioid receptors (Okada & Kaneko, 2011; Yamamura et al., 2009; Yamamura et al., 2009; Yamamura, Ohoyama, Nagase, & Okada, 2009; Yoshida, Okada, Zhu, & Kaneko, 2005). Interestingly, inhibition of carbonic anhydrase reduces intracellular HCO3− and enhances Na+‐independent Cl−/HCO3− exchange, resulting in enhanced GABAA receptor function (Okada & Kaneko, 2011; Yamamura, Hamaguchi, Ohoyama, Sugiura, et al., 2009). Based on established pharmacological zonisamide profiles and carbonic anhydrase inhibitor efficacy against carbamazepine‐resistant S284L‐ADSHE seizures (Okada & Kaneko, 2011; Yamamura, Hamaguchi, Ohoyama, Sugiura, et al., 2009), we hypothesised that zonisamide prevents ADSHE‐seizures by enhancing GABAergic inhibition via postsynaptic GABAA receptors (Hirose et al., 1999; Ito et al., 2000; Okada et al., 2010; Rozycka et al., 2003). Surprisingly, our study did not show this, but other possible mechanisms for its therapeutic action. We did not observe any differences between the effects of carbamazepine and zonisamide on RJR2403‐induced releases of L‐glutamate and GABA in the M2 of wild type. In the S286L‐TG, however, zonisamide enhanced RJR2403‐induced GABA release without affecting L‐glutamate release in the M2 (Figure 8). Although established zonisamide actions on GABAergic transmission may partially explain the compensation of attenuated GABA release in S286L‐TG, the present study cannot fully explain its mechanism of action.
We have previously reported that zonisamide enhances astroglial release of endogenous agonists of group‐II (xanthurenic acid) and group‐III (cinnabarinic acid) https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=40 (Fukuyama, Tanahashi, Hoshikawa, Shinagawa, & Okada, 2014). Preclinical studies have demonstrated that group‐II metabotropic glutamate receptor agonists decrease nicotine reinforcement and seeking (Li, Semenova, D'Souza, Stoker, & Markou, 2014). Indeed, a recent clinical trial has reported that zonisamide decreases nicotine withdrawal and craving during smoking cessation (Dunn et al., 2016). Therefore, zonisamide‐induced enhancement of astroglial release of endogenous metabotropic glutamate receptor agonists probably contributes to inhibition of carbamazepine‐resistant ADSHE‐seizures in S286L‐TG. Further studies are required to explain the detailed mechanisms behind this compensation of reduced α4β2‐nAChR‐induced GABA release in M2 of S286L‐TG by zonisamide.
4.4. Pathogenesis of comorbidities in S284L‐autosomal dominant sleep‐related hypermotor epilepsy
In analysing the regulation of thalamocortical cognitive transmission by α4β2‐nAChR and its functional abnormalities in S286L‐TG, we found that the regulatory mechanisms associated with α4β2‐nAChRs resembled those in thalamocortical motor pathway of wild type. There were different functional abnormalities in both the thalamocortical motor and cognitive pathways of S286L‐TG. Stimulatory effects of α4β2‐nAChR in RTN of S286L‐TG on L‐glutamate and GABA in MDTN were attenuated as compared to wild type (Figure 9), similar to the effect seen in the RTN‐motor thalamic nuclei pathway (Figure 7). However, contrary to thalamocortical motor pathway, activation of α4β2‐nAChR in the RTN inhibited MDTN‐OFC glutamatergic transmission in wild type but not in S286L‐TG (Figure 10). In other words, inhibitory regulation of α4β2‐nAChR in the RTN on MDTN‐OFC glutamatergic transmission was abolished in S286L‐TG (Figure 11).
Recent clinical reports have emphasised that MDTN disturbance is particularly relevant for cognitive dysfunction in psychosis (Alelu‐Paz & Gimenez‐Amaya, 2008; Fukuyama et al., 2018; Okada, Fukuyama, Kawano, Shiroyama, Suzuki, & Ueda, 2019; Okada, Fukuyama, Kawano, Shiroyama, & Ueda, 2019; Okada, Fukuyama, Nakano, & Ueda, 2019; Okada, Fukuyama, Shiroyama, & Ueda, 2019; Schuetze et al., 2016; Vertes et al., 2015). Functional abnormalities of MDTN have been observed in patients with intellectual disability (Karlsen et al., 2014), autism (Schuetze et al., 2016) and epileptic psychosis (Leeman‐Markowski et al., 2015). MDTN receives projections from various cortical and subcortical regions associated with learning, memory, emotion and perceptual integration (McCormick & Wang, 1991; Porrino, Crane, & Goldman‐Rakic, 1981; Russchen, Amaral, & Price, 1987). MDTN‐OFC glutamatergic transmission is considered to play important roles in maintaining flexible stimulus–reward associations (Okada, Fukuyama, Kawano, Shiroyama, Suzuki, & Ueda, 2019); MDTN‐lesioned monkeys continue to respond to stimuli after being satiated with associated food rewards (Mitchell, Browning, & Baxter, 2007; Okada, Fukuyama, Kawano, Shiroyama, Suzuki, & Ueda, 2019). Therefore, MDTN partially regulates the integration of emotional and sensory information sent to the OFC. In summary, impaired α4β2‐nAChR regulation on thalamocortical cognitive transmission might contribute to cognitive impairment seen in S284L‐ADSHE.
5. CONCLUSION
This study provided evidence for the pathomechanisms of S284L‐ADSHE seizure and the associated comorbidities, using S286L‐TG as a valid ADSHE model. Loss‐of‐function of S286L‐nAChR produces paradoxical functional abnormalities in both the thalamocortical motor and cognitive pathways. In the thalamocortical motor pathway of S286L‐TG, stimulatory effects of α4β2‐nAChR in the MoTN was eliminated, whereas inhibitory effects in the RTN were switched to stimulatory effects predominantly via impairment of GABAergic inhibition rather than glutamatergic excitation in the reticular thalamic nucleus‐motor thalamic nuclei pathway. In contrast, the thalamocortical cognitive pathway of S286L‐TG brains showed that both the stimulatory and inhibitory effects of α4β2‐nAChR in respective MDTN and RTN were eliminated. These functional abnormalities of the thalamocortical motor and cognitive pathways in S286L‐TG may contribute to pathogenesis of spontaneous S284L‐ADSHE seizure and its comorbidities (cognitive impairment). Furthermore, selective compensation of impaired GABAergic transmission in the M2 by zonisamide, but not carbamazepine, probably plays important roles in the pathophysiology of carbamazepine‐resistant/zonisamide‐sensitive S284L‐ADSHE.
AUTHOR CONTRIBUTIONS
M.O., K.F., and T.S. designed the research. M.O. and K.F. performed experiments. M.F. generated transgenic rats. All authors have given their final approval for the manuscript.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://bpspubs.onlinelibrary.wiley.com/doi/abs/10.1111/bph.14207, and https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14206, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
ACKNOWLEDGEMENT
This study was supported by Japan Society for the Promotion of Science (22390224 and 15H04892).
Fukuyama K, Fukuzawa M, Shiroyama T, Okada M. Pathogenesis and pathophysiology of autosomal dominant sleep‐related hypermotor epilepsy with S284L‐mutant α4 subunit of nicotinic ACh receptor. Br J Pharmacol. 2020;177:2143–2162. 10.1111/bph.14974
REFERENCES
- Alelu‐Paz, R. , & Gimenez‐Amaya, J. M. (2008). The mediodorsal thalamic nucleus and schizophrenia. Journal of Psychiatry & Neuroscience, 33, 489–498. [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Mathie, A. , Peters, J. A. , Veale, E. M. , Striessnig, J. , Kelly, E. , … CGTP collaboratories (2019). The Coincise Guide to PHARMACOLOGY 2019/20: Ion channels. British Journal of Pharmacology, 176, S142–S228. 10.1111/bph.14749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridon, P. , Marini, C. , Di Resta, C. , Brilli, E. , De Fusco, M. , Politi, F. , … Curia, G. (2006). Increased sensitivity of the neuronal nicotinic receptor α2 subunit causes familial epilepsy with nocturnal wandering and ictal fear. American Journal of Human Genetics, 79, 342–350. 10.1086/506459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asanuma, C. (1992). Noradrenergic innervation of the thalamic reticular nucleus: A light and electron microscopic immunohistochemical study in rats. The Journal of Comparative Neurology, 319, 299–311. 10.1002/cne.903190209 [DOI] [PubMed] [Google Scholar]
- Beierlein, M. (2014). Synaptic mechanisms underlying cholinergic control of thalamic reticular nucleus neurons. The Journal of Physiology, 592, 4137–4145. 10.1113/jphysiol.2014.277376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendse, H. W. , Galis‐de Graaf, Y. , & Groenewegen, H. J. (1992). Topographical organization and relationship with ventral striatal compartments of prefrontal corticostriatal projections in the rat. The Journal of Comparative Neurology, 316, 314–347. 10.1002/cne.903160305 [DOI] [PubMed] [Google Scholar]
- Bertram, E. H. (2014). Extratemporal lobe circuits in temporal lobe epilepsy. Epilepsy & Behavior, 38, 13–18. 10.1016/j.yebeh.2014.07.012 [DOI] [PubMed] [Google Scholar]
- Blumenfeld, H. , Klein, J. P. , Schridde, U. , Vestal, M. , Rice, T. , Khera, D. S. , … Levin, A. R. (2008). Early treatment suppresses the development of spike‐wave epilepsy in a rat model. Epilepsia, 49, 400–409. 10.1111/j.1528-1167.2007.01458.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde, F. , Maire‐Lepoivre, E. , Audinat, E. , & Crepel, F. (1995). Afferent connections of the medial frontal cortex of the rat. II. Cortical and subcortical afferents. The Journal of Comparative Neurology, 352, 567–593. 10.1002/cne.903520407 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Fusco, M. , Becchetti, A. , Patrignani, A. , Annesi, G. , Gambardella, A. , Quattrone, A. , … Casari, G. (2000). The nicotinic receptor β2 subunit is mutant in nocturnal frontal lobe epilepsy. Nature Genetics, 26, 275–276. 10.1038/81566 [DOI] [PubMed] [Google Scholar]
- Dermon, C. R. , & Barbas, H. (1994). Contralateral thalamic projections predominantly reach transitional cortices in the rhesus monkey. The Journal of Comparative Neurology, 344, 508–531. 10.1002/cne.903440403 [DOI] [PubMed] [Google Scholar]
- Dunn, K. E. , Marcus, T. F. , Kim, C. , Schroeder, J. R. , Vandrey, R. , & Umbricht, A. (2016). Zonisamide reduces withdrawal symptoms but does not enhance varenicline‐induced smoking cessation. Nicotine & Tobacco Research, 18, 1171–1179. 10.1093/ntr/ntv236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedi, M. , Berkovic, S. F. , Scheffer, I. E. , O'Keefe, G. , Marini, C. , Mulligan, R. , … Reutens, D. C. (2008). Reduced striatal D1 receptor binding in autosomal dominant nocturnal frontal lobe epilepsy. Neurology, 71, 795–798. 10.1212/01.wnl.0000316192.52731.77 [DOI] [PubMed] [Google Scholar]
- Fukuyama, K. , Hasegawa, T. , & Okada, M. (2018). Cystine/glutamate antiporter and aripiprazole compensate NMDA antagonist‐induced dysfunction of thalamocortical L‐glutamatergic transmission. International Journal of Molecular Sciences, 19, 3645 10.3390/ijms19113645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuyama, K. , Kato, R. , Murata, M. , Shiroyama, T. , & Okada, M. (2019). Clozapine normalizes a glutamatergic transmission abnormality induced by an impaired NMDA receptor in the thalamocortical pathway via the activation of a group III metabotropic glutamate receptor. Biomolecules, 9, 234 10.3390/biom9060234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuyama, K. , Tanahashi, S. , Hoshikawa, M. , Shinagawa, R. , & Okada, M. (2014). Zonisamide regulates basal ganglia transmission via astroglial kynurenine pathway. Neuropharmacology, 76(Pt A), 137–145. 10.1016/j.neuropharm.2013.08.002 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillmer, A. T. , Wooten, D. W. , Slesarev, M. S. , Ahlers, E. O. , Barnhart, T. E. , Murali, D. , … Christian, B. T. (2012). PET imaging of α4β2* nicotinic acetylcholine receptors: Quantitative analysis of 18F‐nifene kinetics in the nonhuman primate. Journal of Nuclear Medicine: Official Publication, Society of Nuclear Medicine, 53, 1471–1480. 10.2967/jnumed.112.103846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose, S. , Iwata, H. , Akiyoshi, H. , Kobayashi, K. , Ito, M. , Wada, K. , … Mitsudome, A. (1999). A novel mutation of CHRNA4 responsible for autosomal dominant nocturnal frontal lobe epilepsy. Neurology, 53, 1749–1753. 10.1212/wnl.53.8.1749 [DOI] [PubMed] [Google Scholar]
- Ito, M. , Kobayashi, K. , Fujii, T. , Okuno, T. , Hirose, S. , Iwata, H. , … Kaneko, S. (2000). Electroclinical picture of autosomal dominant nocturnal frontal lobe epilepsy in a Japanese family. Epilepsia, 41, 52–58. 10.1111/j.1528-1157.2000.tb01505.x [DOI] [PubMed] [Google Scholar]
- Karlsen, A. S. , Korbo, S. , Uylings, H. B. , & Pakkenberg, B. (2014). A stereological study of the mediodorsal thalamic nucleus in Down syndrome. Neuroscience, 279, 253–259. 10.1016/j.neuroscience.2014.08.046 [DOI] [PubMed] [Google Scholar]
- Keezer, M. R. , Sisodiya, S. M. , & Sander, J. W. (2016). Comorbidities of epilepsy: Current concepts and future perspectives. The Lancet Neurology, 15, 106–115. 10.1016/S1474-4422(15)00225-2 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160(7), 1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassen, A. , Glykys, J. , Maguire, J. , Labarca, C. , Mody, I. , & Boulter, J. (2006). Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy. Proceedings of the National Academy of Sciences of the United States of America, 103, 19152–19157. 10.1073/pnas.0608215103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota, M. , Miyata, J. , Sasamoto, A. , Sugihara, G. , Yoshida, H. , Kawada, R. , … Murai, T. (2013). Thalamocortical disconnection in the orbitofrontal region associated with cortical thinning in schizophrenia. JAMA Psychiatry, 70, 12–21. 10.1001/archgenpsychiatry.2012.1023 [DOI] [PubMed] [Google Scholar]
- Leeman‐Markowski, B. A. , Smart, O. L. , Faught, R. E. , Gross, R. E. , & Meador, K. J. (2015). Cessation of gamma activity in the dorsomedial nucleus associated with loss of consciousness during focal seizures. Epilepsy & Behavior, 51, 215–220. 10.1016/j.yebeh.2015.07.027 [DOI] [PubMed] [Google Scholar]
- Li, X. , Semenova, S. , D'Souza, M. S. , Stoker, A. K. , & Markou, A. (2014). Involvement of glutamatergic and GABAergic systems in nicotine dependence: Implications for novel pharmacotherapies for smoking cessation. Neuropharmacology, 76(Pt B), 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima, N. , Hirose, S. , Iwata, H. , Fukuma, G. , Yonetani, M. , Nagayama, C. , … Sugiyama, H. (2002). Mutation (Ser284Leu) of neuronal nicotinic acetylcholine receptor α4 subunit associated with frontal lobe epilepsy causes faster desensitization of the rat receptor expressed in oocytes. Epilepsy Research, 48, 181–186. 10.1016/s0920-1211(01)00336-9 [DOI] [PubMed] [Google Scholar]
- McCormick, D. A. , & Wang, Z. (1991). Serotonin and noradrenaline excite GABAergic neurones of the guinea‐pig and cat nucleus reticularis thalami. The Journal of Physiology, 442, 235–255. 10.1113/jphysiol.1991.sp018791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP . British Journal of Pharmacology, 172, 3189–3193. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, A. S. , Browning, P. G. , & Baxter, M. G. (2007). Neurotoxic lesions of the medial mediodorsal nucleus of the thalamus disrupt reinforcer devaluation effects in rhesus monkeys. The Journal of Neuroscience, 27, 11289–11295. 10.1523/JNEUROSCI.1914-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyajima, T. , Kumada, T. , Saito, K. , & Fujii, T. (2013). Autism in siblings with autosomal dominant nocturnal frontal lobe epilepsy. Brain & Development, 35, 155–157. 10.1016/j.braindev.2012.07.012 [DOI] [PubMed] [Google Scholar]
- Mtui, E. , Gruener, G. , & Dockery, P. (2015). Fitzgerald's clinical neuroanatomy and neuroscience (7th ed.). Elsevier: Philadelphia. [Google Scholar]
- Nakatsu, F. , Okada, M. , Mori, F. , Kumazawa, N. , Iwasa, H. , Zhu, G. , … Ohno, H. (2004). Defective function of GABA‐containing synaptic vesicles in mice lacking the AP‐3B clathrin adaptor. The Journal of Cell Biology, 167, 293–302. 10.1083/jcb.200405032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada, M. , Fukuyama, K. , Kawano, Y. , Shiroyama, T. , Suzuki, D. , & Ueda, Y. (2019). Effects of acute and sub‐chronic administrations of guanfacine on catecholaminergic transmissions in the orbitofrontal cortex. Neuropharmacology, 156, 107547 10.1016/j.neuropharm.2019.02.029 [DOI] [PubMed] [Google Scholar]
- Okada, M. , Fukuyama, K. , Kawano, Y. , Shiroyama, T. , & Ueda, Y. (2019). Memantine protects thalamocortical hyper‐glutamatergic transmission induced by NMDA receptor antagonism via activation of system xc− . Pharmacology Research & Perspectives, 7, e00457 10.1002/prp2.457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada, M. , Fukuyama, K. , Nakano, T. , & Ueda, Y. (2019). Pharmacological discrimination of effects of MK801 on thalamocortical, mesothalamic, and mesocortical transmissions. Biomolecules, 9, 746 10.3390/biom9110746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada, M. , Fukuyama, K. , Shiroyama, T. , & Ueda, Y. (2019). Lurasidone inhibits NMDA antagonist‐induced functional abnormality of thalamocortical glutamatergic transmission via 5‐HT7 receptor blockade. British Journal of Pharmacology, 176, 4002–4018. 10.1111/bph.14804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada, M. , & Kaneko, S. (2011). Different mechanisms underlying the antiepileptic and antiparkinsonian effects of zonisamide In Foyaca‐Sibat H. (Ed.), Novel treatment of epilepsy (pp. 23–36). Rijeka: InTech; 10.5772/19562 [DOI] [Google Scholar]
- Okada, M. , Zhu, G. , Yoshida, S. , Kanai, K. , Hirose, S. , & Kaneko, S. (2002). Exocytosis mechanism as a new targeting site for mechanisms of action of antiepileptic drugs. Life Sciences, 72, 465–473. 10.1016/s0024-3205(02)02283-x [DOI] [PubMed] [Google Scholar]
- Okada, M. , Zhu, G. , Yoshida, S. , & Kaneko, S. (2010). Validation criteria for genetic animal models of epilepsy. Epilepsy & Seizure, 3, 109–120. 10.3805/eands.3.109 [DOI] [Google Scholar]
- Paxinos, G. , & Watson, C. (2007). The rat brain: In stereotoxic coordinates (6th ed.). Academic Press: San Dieg. [Google Scholar]
- Phillips, H. A. , Favre, I. , Kirkpatrick, M. , Zuberi, S. M. , Goudie, D. , Heron, S. E. , … Mulley, J. C. (2001). CHRNB2 is the second acetylcholine receptor subunit associated with autosomal dominant nocturnal frontal lobe epilepsy. American Journal of Human Genetics, 68, 225–231. 10.1086/316946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard, F. , Bruel, D. , Servent, D. , Saba, W. , Fruchart‐Gaillard, C. , Schollhorn‐Peyronneau, M. A. , … Steinborn, B. (2006). Alteration of the in vivo nicotinic receptor density in ADNFLE patients: A PET study. Brain, 129, 2047–2060. 10.1093/brain/awl156 [DOI] [PubMed] [Google Scholar]
- Plazzi, G. , Tinuper, P. , Montagna, P. , Provini, F. , & Lugaresi, E. (1995). Epileptic nocturnal wanderings. Sleep, 18, 749–756. 10.1093/sleep/18.9.749 [DOI] [PubMed] [Google Scholar]
- Porrino, L. J. , Crane, A. M. , & Goldman‐Rakic, P. S. (1981). Direct and indirect pathways from the amygdala to the frontal lobe in rhesus monkeys. The Journal of Comparative Neurology, 198, 121–136. 10.1002/cne.901980111 [DOI] [PubMed] [Google Scholar]
- Provini, F. , Plazzi, G. , Montagna, P. , & Lugaresi, E. (2000). The wide clinical spectrum of nocturnal frontal lobe epilepsy. Sleep Medicine Reviews, 4, 375–386. 10.1053/smrv.2000.0109 [DOI] [PubMed] [Google Scholar]
- Provini, F. , Plazzi, G. , Tinuper, P. , Vandi, S. , Lugaresi, E. , & Montagna, P. (1999). Nocturnal frontal lobe epilepsy. A clinical and polygraphic overview of 100 consecutive cases. Brain, 122(Pt 6), 1017–1031. [DOI] [PubMed] [Google Scholar]
- Rodrigues‐Pinguet, N. , Jia, L. , Li, M. , Figl, A. , Klaassen, A. , Truong, A. , … Cohen, B. N. (2003). Five ADNFLE mutations reduce the Ca2+ dependence of the mammalian α4β2 acetylcholine response. The Journal of Physiology, 550, 11–26. 10.1113/jphysiol.2003.036681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozycka, A. , Skorupska, E. , Kostyrko, A. , & Trzeciak, W. H. (2003). Evidence for S284L mutation of the CHRNA4 in a white family with autosomal dominant nocturnal frontal lobe epilepsy. Epilepsia, 44, 1113–1117. 10.1046/j.1528-1157.2003.07603.x [DOI] [PubMed] [Google Scholar]
- Russchen, F. T. , Amaral, D. G. , & Price, J. L. (1987). The afferent input to the magnocellular division of the mediodorsal thalamic nucleus in the monkey, Macaca fascicularis . The Journal of Comparative Neurology, 256, 175–210. 10.1002/cne.902560202 [DOI] [PubMed] [Google Scholar]
- Scheffer, I. E. , Bhatia, K. P. , Lopes‐Cendes, I. , Fish, D. R. , Marsden, C. D. , Andermann, E. , … Berkovic, S. F. (1995). Autosomal dominant nocturnal frontal lobe epilepsy. A distinctive clinical disorder. Brain, 118(Pt 1), 61–73. 10.1093/brain/118.1.61 [DOI] [PubMed] [Google Scholar]
- Schuetze, M. , Park, M. T. , Cho, I. Y. , MacMaster, F. P. , Chakravarty, M. M. , & Bray, S. L. (2016). Morphological alterations in the thalamus, striatum, and pallidum in autism spectrum disorder. Neuropsychopharmacology, 41, 2627–2637. 10.1038/npp.2016.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba, Y. , Mori, F. , Yamada, J. , Migita, K. , Nikaido, Y. , Wakabayashi, K. , … Ueno, S. (2015). Spontaneous epileptic seizures in transgenic rats harboring a human ADNFLE missense mutation in the β2‐subunit of the nicotinic acetylcholine receptor. Neuroscience Research, 100, 46–54. 10.1016/j.neures.2015.06.003 [DOI] [PubMed] [Google Scholar]
- Steinlein, O. K. , Mulley, J. C. , Propping, P. , Wallace, R. H. , Phillips, H. A. , Sutherland, G. R. , … Berkovic, S. F. (1995). A missense mutation in the neuronal nicotinic acetylcholine receptor α4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nature Genetics, 11, 201–203. 10.1038/ng1095-201 [DOI] [PubMed] [Google Scholar]
- Strasser, L. , Downes, M. , Kung, J. , Cross, J. H. , & De Haan, M. (2018). Prevalence and risk factors for autism spectrum disorder in epilepsy: A systematic review and meta‐analysis. Developmental Medicine and Child Neurology, 60, 19–29. 10.1111/dmcn.13598 [DOI] [PubMed] [Google Scholar]
- Teper, Y. , Whyte, D. , Cahir, E. , Lester, H. A. , Grady, S. R. , Marks, M. J. , … Drago, J. (2007). Nicotine‐induced dystonic arousal complex in a mouse line harboring a human autosomal‐dominant nocturnal frontal lobe epilepsy mutation. The Journal of Neuroscience, 27, 10128–10142. 10.1523/JNEUROSCI.3042-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinuper, P. , Bisulli, F. , Cross, J. H. , Hesdorffer, D. , Kahane, P. , Nobili, L. , … Ottman, R. (2016). Definition and diagnostic criteria of sleep‐related hypermotor epilepsy. Neurology, 86, 1834–1842. 10.1212/WNL.0000000000002666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ool, J. S. , Snoeijen‐Schouwenaars, F. M. , Schelhaas, H. J. , Tan, I. Y. , Aldenkamp, A. P. , & Hendriksen, J. G. M. (2016). A systematic review of neuropsychiatric comorbidities in patients with both epilepsy and intellectual disability. Epilepsy & Behavior, 60, 130–137. 10.1016/j.yebeh.2016.04.018 [DOI] [PubMed] [Google Scholar]
- Vertes, R. P. , Linley, S. B. , & Hoover, W. B. (2015). Limbic circuitry of the midline thalamus. Neuroscience and Biobehavioral Reviews, 54, 89–107. 10.1016/j.neubiorev.2015.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamura, S. , Hamaguchi, T. , Ohoyama, K. , Sugiura, Y. , Suzuki, D. , Kanehara, S. , … Okada, M. (2009). Topiramate and zonisamide prevent paradoxical intoxication induced by carbamazepine and phenytoin. Epilepsy Research, 84, 172–186. 10.1016/j.eplepsyres.2009.01.015 [DOI] [PubMed] [Google Scholar]
- Yamamura, S. , Hoshikawa, M. , Dai, K. , Saito, H. , Suzuki, N. , Niwa, O. , & Okada, M. (2013). ONO‐2506 inhibits spike‐wave discharges in a genetic animal model without affecting traditional convulsive tests via gliotransmission regulation. British Journal of Pharmacology, 168, 1088–1100. 10.1111/j.1476-5381.2012.02132.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamura, S. , Ohoyama, K. , Hamaguchi, T. , Kashimoto, K. , Nakagawa, M. , Kanehara, S. , … Okada, M. (2009). Effects of quetiapine on monoamine, GABA, and glutamate release in rat prefrontal cortex. Psychopharmacology, 206, 243–258. 10.1007/s00213-009-1601-9 [DOI] [PubMed] [Google Scholar]
- Yamamura, S. , Ohoyama, K. , Hamaguchi, T. , Nakagawa, M. , Suzuki, D. , Matsumoto, T. , … Okada, M. (2009). Effects of zotepine on extracellular levels of monoamine, GABA and glutamate in rat prefrontal cortex. British Journal of Pharmacology, 157, 656–665. 10.1111/j.1476-5381.2009.00175.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamura, S. , Ohoyama, K. , Nagase, H. , & Okada, M. (2009). Zonisamide enhances delta receptor‐associated neurotransmitter release in striato‐pallidal pathway. Neuropharmacology, 57, 322–331. 10.1016/j.neuropharm.2009.05.005 [DOI] [PubMed] [Google Scholar]
- Yamamura, S. , Saito, H. , Suzuki, N. , Kashimoto, S. , Hamaguchi, T. , Ohoyama, K. , … Okada, M. (2009). Effects of zonisamide on neurotransmitter release associated with inositol triphosphate receptors. Neuroscience Letters, 454, 91–96. 10.1016/j.neulet.2009.02.065 [DOI] [PubMed] [Google Scholar]
- Yoshida, S. , Okada, M. , Zhu, G. , & Kaneko, S. (2005). Effects of zonisamide on neurotransmitter exocytosis associated with ryanodine receptors. Epilepsy Research, 67, 153–162. 10.1016/j.eplepsyres.2005.10.001 [DOI] [PubMed] [Google Scholar]
- Yoshida, S. , Okada, M. , Zhu, G. , & Kaneko, S. (2007). Carbamazepine prevents breakdown of neurotransmitter release induced by hyperactivation of ryanodine receptor. Neuropharmacology, 52, 1538–1546. 10.1016/j.neuropharm.2007.02.009 [DOI] [PubMed] [Google Scholar]
- Zhu, G. , Okada, M. , Yoshida, S. , Ueno, S. , Mori, F. , Takahara, T. , … Hirose, S. (2008). Rats harboring S284L Chrna4 mutation show attenuation of synaptic and extrasynaptic GABAergic transmission and exhibit the nocturnal frontal lobe epilepsy phenotype. The Journal of Neuroscience, 28, 12465–12476. 10.1523/JNEUROSCI.2961-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]