

Abstract
Background and Purpose
In 2016, one person died and four others had mild‐to‐severe neurological symptoms during a phase I trial of the fatty acid amide hydrolase (FAAH) inhibitor BIA 10‐2474.
Experimental Approach
Pharmacodynamic and pharmacokinetic studies were performed with BIA 10‐2474, PF‐04457845 and JNJ‐42165279 using mice, rats and human FAAH expressed in COS cells. Selectivity was evaluated by activity‐based protein profiling (APBB) in rats. BIA 10‐2474 effect in stroke‐prone spontaneously hypertensive rats (SHRSP) was investigated.
Key Results
BIA 10‐2474 was 10‐fold less potent than PF‐04457845 in inhibiting human FAAH in situ but inhibited mouse brain and liver FAAH with ED50 values of 13.5 and 6.2 μg·kg−1, respectively. Plasma and brain BIA 10‐2474 levels were consistent with in situ potency and neither BIA 10‐2474 nor its metabolites accumulated following repeat administration. FAAH and α/β‐hydrolase domain containing 6 were the primary targets of BIA 10‐2474 and, at higher exposure levels, ABHD11, PNPLA6, PLA2G15, PLA2G6 and androgen‐induced protein 1. At 100 mg·kg−1 for 28 days, the level of several lipid species containing arachidonic acid increased. Daily treatment of SHRSP with BIA 10‐2474 did not affect mortality rate or increased the incidence of haemorrhage or oedema in surviving animals.
Conclusions and Implications
BIA 10‐2474 potently inhibits FAAH in vivo, similarly to PF‐04457845 and interacts with a number of lipid processing enzymes, some previously identified in human cells as off‐targets particularly at high levels of exposure. These interactions occurred at doses used in toxicology studies, but the implication of these off‐targets in the clinical trial accident remains unclear.
Abbreviations
- 2‐AG
2‐arachidonoylglycerol
- AA
arachidonic acid
- ABHD6
α/β‐hydrolase domain containing 6
- ABPP
activity‐based protein profiling
- AEA
anandamide
- AIG1
androgen‐induced protein 1
- BCRP
breast cancer resistance protein
- CES
carboxyl esterase
- CYP
cytochrome P450
- DAG
diacylglycerol
- FAAH
fatty acid amide hydrolase
- LIPE
lipase E
- MATE
multidrug and toxin extrusion
- MDR
multidrug resistance
- OAT
organic anion transporter
- OCT
organic cation transporter
- OEA
oleoylethanolamide
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PEA
palmitoylethanolamide
- P‐gp
P‐glycoprotein
- PLA2G15
PLA2 Group 15
- PLA2G6
PLA2 Group 6
- PNPLA6
patatin‐like phospholipase domain containing 6
- SHRSP
Spontaneously hypertensive rats stroke prone
- TAMRA‐FP
tetramethylrhodamine–fluorophosphonate
What is already known
BIA 10‐2474 is a FAAH inhibitor that was involved in a serious clinical trial accident.
An unknown off‐target effect of BIA 10‐2474 has been suggested to be the cause.
What this study adds
No evidence of accumulation or region‐specific enrichment of BIA 10‐2474 or metabolites was observed.
In rats, at high exposure, BIA 10‐2474 interacts with a number of lipid processing enzymes.
What is the clinical significance
The available information does not allow identification of the mechanism/s causing the clinical trial accident.
1. INTRODUCTION
In 2016, one subject died and four were hospitalized with neurological symptoms during a clinical trial with the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1400 (FAAH) inhibitor BIA 10‐2474, with reports that the subject who died, after receiving the 50‐mg dose for 5 days in the clinical trial, showed evidence of severe brain microhaemorrhage and several of the surviving subjects also showed evidence of mild to moderate microhaemorrhage as well as headache, cerebellar syndrome, memory impairment and altered consciousness. MRI showed bilateral and symmetric cerebral lesions, including microhaemorrhages and hyperintensities on fluid‐attenuated inversion recovery and diffusion‐weighted imaging sequences predominantly involving the pons and hippocampi. One patient became brain dead; the condition of two patients subsequently improved, but one patient had residual memory impairment and the other patient had a residual cerebellar syndrome. One patient remained asymptomatic (Kerbrat et al., 2016). The investigations by the French authorities concluded that it was an unexpected effect of the test drug, having ruled out other extraneous causes. Their conclusion was that the accident was likely to have been caused by an unknown off‐target effect of BIA 10‐2474 (Temporary Specialist Scientific Committee [TSSC], 2016).
The nature of this off‐target effect remains unknown, although it has been suggested that BIA 10‐2474 is not particularly selective for fatty acid amide hydrolase, while other compounds tested in the clinic, such as https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6694 (Johnson et al., 2011; Li et al., 2012) and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9012 (Keith et al., 2015), which are considered more selective and hence their lack of observed toxicity in man, despite being tested at exposure levels up to several hundred‐fold those required to fully inhibit fatty acid amide hydrolase (Li et al., 2012). A recent study (van Esbroeck et al., 2017) compared BIA 10‐2474 with PF‐04457845 at serine hydrolases using activity‐based protein profiling (ABPP) in human SW620 cells and identified some off‐targets specific to BIA 10‐2474, namely https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2592, CES2, CES3, α/β‐hydrolase domain containing 11 (ABHD11), https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2593, patatin‐like phospholipase domain containing 6 (PNPLA6) and α/β‐hydrolase domain containing 6 (https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2919), with the latter being inhibited with similar potency to fatty acid amide hydrolase. In human cortical neurons, the off‐targets identified were CES2, PNPLA6 and https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=275#2508 Group 15 (PLA2G15). An alteration in lipid profile was also shown in human cortical neurons. Although this study added important new information to the possible off‐target effects of BIA 10‐2474, such in situ experiments have limitations. In particular, it is not possible to reproduce the duration of exposure that the clinical subjects underwent nor the possible interplay between parent compound and metabolites. The in vivo studies in the rat reported here were designed to address this and also to provide comparative data at doses that were used in rat toxicology studies (Harris, Hardisty, Hayes, & Weber, 2019; Hayes, Hardisty, Harris, Okazaki, & Weber, 2019). The rat was used in these studies as the No‐observed‐adverse‐effect level (NOAEL) identified in rat toxicology was used to determine the clinical doses.
We have investigated the potency of BIA 10‐2474 on fatty acid amide hydrolase, its effects on brain fatty acid amides (FAAs) and exposure in plasma and the brain. We also studied BIA 10‐2474 selectivity using standard binding and enzymatic assays and activity‐based protein profiling against serine hydrolase enzymes in rat brain proteome in vitro and also ex vivo. As two other mechanism‐based fatty acid amide hydrolase inhibitors, PF‐04457845 and JNJ‐42165279, were considered safe in the clinic and in many of the published studies reported here, we have tested them in parallel with BIA 10‐2474 in order to help identify off‐targets specific to BIA 10‐2474. Much of the data reported here, namely, in vitro and in vivo pharmacodynamic results, were available to the authorities prior to the Clinical Trial Authorization. However, the broader off‐target screen at 50 μM performed at CEREP as well as the activity‐based protein profiling and lipid studies were carried out afterwards, as were the studies related to accumulation of the parent compound and its metabolites. In addition, in view of the MRI signs seen in the clinic (Kerbrat et al., 2016), to evaluate if BIA 10‐2474 might contribute to cerebral blood vessels rupturing, the effects of the compound were explored in stroke‐prone spontaneously hypertensive rats (SHRSP) in conjunction with high‐salt intake by evaluating mortality rates and examining cerebrovascular damage.
2. METHODS
Studies are reported following the editorial guidelines on experimental designs and analysis in pharmacology (Curtis et al., 2018).
2.1. Animal treatments
All animal procedures were conducted in accordance with the 2010/63/EU European Directive on the protection of animals used for scientific purposes, the Portuguese law on animal welfare (Decreto‐Lei 113/2013) and the Guide for the Care and Use of Laboratory Animals (eighth edition, 2011). The study carried out at Charles River (Edinburgh, UK) was conducted under U.K. regulations (license PBAD559F8, Toxicology of Pharmaceuticals, Protocol No. 1). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and the editorial on reporting animal studies (McGrath, & Lilley, 2015).
Male Crl:NMRI (Han) Mus musculus (IMSR Cat# CRL:605, https://scicrunch.org/resources/Any/search?q=Crl%3ANMRI(Han)%20Mus%20musculus&l=Crl%3ANMRI(Han)%20Mus%20musculus; 20–30 g, total number used was 190) and male Wistar (Crl:WI (Han) Rattus norvegicus (RGD Cat# 2308816, https://scicrunch.org/resources/Any/search?q=Crl%3AWI(Han)%20Rattus%20norvegicus&l=Crl%3AWI(Han)%20Rattus%20norvegicus; 200–250 g; total number used was 46) were purchased from Charles River (The Netherlands). Male SHRSP (A3NCrl stroke‐prone rats R. norvegicus; RGD Cat# 2311051, https://scicrunch.org/resources/Any/search?q=RGD_2311051&l=RGD_2311051; total number 70) were obtained from Charles River Laboratories (USA) and were 60 days old at the start of the study.
Mice were kept five per cage and rats were kept 3–8 per cage under controlled environmental conditions (12‐hr light–dark cycle, room temperature 22 ± 1°C). Food and tap water were allowed ad libitum and the experiments were carried out during daylight hours. Animals were socially housed for psychological/environmental enrichment and were provided with species‐appropriate environmental enrichment except during studies. As most of the studies reported here involving animals report ex vivo measurements, there was no blinding in the animal treatments. Animals were randomly distributed between treatment groups.
The strains chosen (Wistar rat and NMRI mice) are rodent species commonly used for pharmacological studies and accepted by regulatory agencies. The oral route was selected as it was the one used for human exposure.
2.1.1. For the evaluation of fatty acid amide hydrolase activity, fatty acid amides, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=729 and compound exposures
For the evaluation of fatty acid amide hydrolase activity (and determination of the median effective dose, ED50) and FAAs, overnight fasted NMRI mice (n = 4 per group) were given by gavage (p.o.) 0.001, 0.003, 0.01, 0.03, 0.1 and 0.3 mg·kg−1 (for BIA 10‐2474 and PF‐04457845) or 0.03, 0.1, 0.3, 1, 3, 10 and 30 mg·kg−1 (for JNJ‐42165279), formulated in 10% DMSO in water; volume administered was 8 ml·kg−1. At 8 hr after dosing, animals were anaesthetized with https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5480 60 mg·kg−1 i.p. and killed by decapitation after removal of a fragment of the liver and brain without the cerebellum. The liver and half of the brain were put into plastic vials containing fatty acid amide hydrolase membrane buffer (3 mM of MgCl2, 1 mM of EDTA, and 50 mM of Tris HCl, pH 7.4) and glass beads and were stored at −20°C. The remaining half brain was weighed, frozen in liquid nitrogen, and stored at −80°C until used for quantification of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2364 (AEA), https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1402 (PEA) and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2661 (OEA).
For the quantification of 2‐arachidonoylglycerol (2‐AG), overnight fasted NMRI mice (n = 4 per group) were given BIA 10‐2474 (10 mg·kg−1 in 0.2% hydroxypropyl methylcellulose [HPMC; Merck, Germany] prepared in saline) or vehicle orally. No food was given to animals after administrations except for the 24‐hr time point, which were allowed to have food access at the end of the administration day. At the specified time points 1, 4, 8 and 24 hr after dosing, mice were anaesthetized as described above, the blood was collected by cardiac puncture into heparinized tubes and the brain without the cerebellum was removed, split in half and put into pre‐weighed vials. Blood was processed for plasma isolation and collected samples were stored at −80°C until analysis.
For the quantification of BIA 10‐2474 and PF‐04457845 exposure, overnight fasted mice (n = 4 per group) were given BIA 10‐2474 (0.1 or 0.3 mg·kg−1 p.o.) or PF‐04457845 (0.03 and 0.1 mg·kg−1 p.o.) in 10% DMSO; volume of administration was 8 ml·kg−1. At the specified time points 1, 2, 4 and 8 hr after dosing, mice were anaesthetized as described above, the blood was collected by cardiac puncture into heparinized tubes, and the brain without the cerebellum was removed and put into pre‐weighed vials. Blood was processed for plasma isolation and collected samples were stored at −80°C until analysis.
For the quantification of BIA 10‐2474 and metabolites in brain sections, Wistar rats (n = 6 per group) fasted for 2 hr were given BIA 10‐2474 (10 mg·kg−1·day−1 in 0.2% HPMC prepared in saline, p.o.) or vehicle (n = 2). At the specified time points (Day 1: 1, 4 and 24 hr after dosing; Day 5: 4, 24, 48 and 72 hr after dosing) animals were anaesthetized as described above and perfused through the left ventricle with 20 ml of 0.9% NaCl. Brains were removed and dissected and the cerebellum, midbrain, pons, medulla oblongata, hippocampus and cerebral cortex were collected and stored at −80°C until analysis.
2.1.2. For the activity‐based protein profiling at serine hydrolases and lipidomics ex vivo
In a study carried out in Charles River (UK), male Han Wistar Crl:WI rats (190–290 g; total number 120) were orally dosed with vehicle (0.2% [w/v] HPMC) or BIA 10‐2474 (at 10, 30 or 100 mg·kg−1·day−1) for 1 to 28 days. Eight hours after dosing on Days 1, 7 and 28, 12 animals from each group were killed. The control group (n = 12) was killed on Day 28. Euthanasia was carried out by exposure of animals to rising levels of carbon dioxide and exsanguination. The brains of six animals from each group were split into two hemispheres: The right hemisphere was used for lipidomic analysis (see below) and the left was snap frozen and stored at −80°C until dispatch to Evotec (Toulouse, France) for activity‐based protein profiling analyses. For the remaining animals (six for each group), the entire brain, including the cerebrum, cerebellum and pons/medulla, was collected and preserved in 10% neutral buffered formalin. Representative samples of the brain, including the cerebrum, cerebellum and pons/medulla were embedded in paraffin, sectioned, mounted on glass slides and stained with haematoxylin and eosin. Two slides per animal were prepared, and histopathological evaluation was performed by a board‐certified veterinary pathologist with training and experience in laboratory animal pathology. Six sections were examined from each animal, with the cerebrum, cerebellum and pons/medulla included in these six sections.
2.2. Spontaneously hypertensive rats stroke prone (SHRSP)
All rats were first identified and weighed and then attributed iteratively to the treatment groups such that the mean and variance of body weight for each group was comparable. Ten SHRSP received standard rodent diet (2014 Teklad global rodent maintenance diet, Envigo, USA) and regular tap water (control group). The remaining 60 rats were placed on a stroke‐prone rodent diet (Zeigler Bros., Inc., USA) and given 1% NaCl to drink. Stroke‐prone diet has a lower potassium and protein content than standard diet and, in conjunction with high‐salt intake, accelerates the development of hypertensive end‐organ pathology (Stier, Chander, Gutstein, Levine, & Itskovitz, 1991). The rats on the stroke‐prone diet and high‐salt intake were divided into two groups receiving vehicle, 0.2% HPMC or BIA 10‐2474 (30 mg·kg−1 body weight, formulated in 0.2% HPMC, p.o.) at a volume of 4 ml·kg−1 body weight. The control rats also received an oral administration of vehicle. The various treatments were administered daily until approximately half the rats in either the vehicle or BIA 10‐2474 group had died. Mortality was registered twice daily except for weekends then it was once daily. Animals were killed with an overdose of sodium pentobarbital (120 mg·kg−1 body weight, i.p.) and if found moribund (i.e. lack of responsiveness to manual stimulation, immobility and/or an inability to eat or drink) and/or with a weight loss of ≥20% (an established humane end point, Morton & Griffiths, 1985). At the end of the study, surviving animals were killed and their brains were removed and fixed in 10% neutral buffered formalin. Brains were dispatched to AnaPath GmbH (Switzerland) where they were blindly processed and analysed. Brains were embedded in paraffin wax and seven cutting levels were taken from the brains, whenever possible, and stained by eosin and haematoxylin as previously described (Bolon et al., 2013). Histological changes were described according to distribution, severity and morphologic character. The lesion area was measured, whenever possible, for haemorrhage, oedema, inflammation and necrosis, either by circumference or using colour threshold measurements (UC30 camera and https://www.olympus-lifescience.com/en/software/cellsens/ V17, https://scicrunch.org/resources/Any/search?q=cellsens&l=cellsens, Japan).
2.3. Fatty acid amide hydrolase activity assay
2.3.1. In rat brain membranes in vitro
Fatty acid amide hydrolase activity was determined by measuring the formation of 3H‐ethanolamine from tritium (3H)‐labelled AEA in Wistar rat brain membranes prepared in house as described elsewhere (Kiss et al., 2011; Kiss et al., 2018). The reaction mixture (total volume of 200 μl) contained substrate solution (2 μM of AEA + 5 nM of 3H‐AEA + 0.1% fatty acid‐free BSA), 10 μg of protein, in fatty acid amide hydrolase incubation buffer (1 mM of EDTA and 10 mM of Tris, pH 7.6) and test compounds (BIA 10‐2474, PF‐04457845 or JNJ‐42165279) ranging from 1 nM to 10 μM. Stock solutions of compounds were prepared in 100% DMSO and the DMSO concentration in the assay was 0.1%. Reactions were started with the addition of the substrate solution and were carried out for 10 min before termination by the addition of 400 μl of activated charcoal suspension (8 g of charcoal in 32 ml of 0.5‐M HCl in continuous agitation). After 30 min of incubation at room temperature with agitation, charcoal was sedimented by centrifugation in microfuge (10 min at 12,100–16,060× g). Two hundred microlitres of the supernatant was added to 800 μl of OptiPhase SuperMix scintillation cocktail and counts per minute (cpm) was determined in a Microbeta TriLux scintillation counter. In each assay, blank samples (without protein) and controls (with vehicle and no compound) were prepared.
For the time‐dependent experiments, compounds were pre‐incubated with the enzyme for 5 to 60 min and reactions were started with the substrate as described above. For the no pre‐incubation period, reactions were started with the addition of the enzyme preparation.
For the determination of kinetic constants k obs and k inact/K I (Copeland, 2005), progression curves were performed where reactions were started with addition of 10‐ to 11‐μg enzyme in fatty acid amide hydrolase incubation buffer. Thereafter, reactions were carried out for 1, 3, 5, 7.5, 10, 12.5 and 15 min at 37°C before termination with activated charcoal suspension as described above. Disintegrations per minute was determined in a Microbeta TriLux scintillation counter. Data fitting and constant calculations are detailed in Section 2.15.
2.3.2. Ex vivo in the liver and brain
Fatty acid amide hydrolase activity in brain and liver homogenates was evaluated essentially as described (Beliaev et al., 2016) and as above. The amount of protein in the assays was 15 μg (brain) or 5 μg (liver) and there was no compound added. After a 15‐min pre‐incubation period at 37°C, the reaction was started with the substrate solution (cold AEA + radiolabelled AEA + BSA) and this was carried out for 10 min (brain) or 7 min (liver) before termination with 400 μl of activated charcoal suspension. Samples were processed and read in a Microbeta TriLux scintillation counter as described above for rat brain membranes assay. In each assay, blank samples (without protein) were prepared. The percentage of remaining enzymatic activity in the test samples was calculated in respect to the respective control samples after blank subtraction. Negative values were considered zero. Blank values varied between 73 and 171 cpm. The average cpm of controls was 3,952 ± 444 in the liver and 2,251 ± 223 in the brain (mean ± SEM, n = 4).
2.3.3. In COS‐human fatty acid amide hydrolase cells
Cell culture
COS‐7 cells (ATCC Cat# CRL‐1651, https://scicrunch.org/resources/Any/search?q=ATCC%20Cat%23%20CRL-1651&l=ATCC%20Cat%23%20CRL-1651) expressing human fatty acid amide hydrolase (COS‐hFAAH) were generated at Bial by transfecting the vector OmicsLink™ Expression clone EX‐M0033‐M02 (obtained from Genecopoeia, Rockville, MD) using PolyFect Transfection Reagent (Qiagen). Cells were cloned and clones expressing the active enzyme were propagated and stored. COS‐hFAAH were maintained in DMEM supplemented with 10% FBS, 4 mM of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=723, 25 mM of HEPES, 100 U·ml−1 of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4796, 100 μg·ml−1 of streptomycin, 0.25 μg·ml−1 of amphotericin B, and 0.5 mg·ml−1 of geneticin at 37°C in controlled humidified air with 5% CO2. Cells were seeded onto 24‐well plates at 50,000 cells per well the day before the experiments.
On the day after the seeding, medium was replaced by serum‐free medium and 3 hr later replaced by serum‐free medium containing the test compounds (BIA 10‐2474, PF‐04457845 or JNJ‐42165279) solutions (final concentration range 0.3 to 3,000 nM). After 1, 3 or 24 hr in the incubator, the solutions in the wells were aspirated and wells were washed twice with warm PBS buffer and added 150 μl of warm fatty acid amide hydrolase incubation buffer. The plates were transferred to a shaking water bath at 37°C and reaction was started with the substrate solution (as described for fatty acid amide hydrolase activity evaluation). Reactions were terminated after 5 min with 50 μl of 2‐M HCl. The contents of each well were transferred to 1.5‐ml microtubes with 400 μl of activated charcoal suspension and the procedure then followed the one described above for rat membranes. All conditions were initially tested in quadruplicate (i.e. four separate plates) and all values were normalized according to the control values for their particular plate. For JNJ‐42165279, additional concentrations were added in a separate experiment with repeats of some concentrations already tested, resulting in eight values for some concentrations.
Total protein was determined using the Bio‐Rad Protein Assay with a standard curve of BSA (50–250 μg·ml−1).
2.4. Quantification of BIA 10‐2474, PF‐04457845, and JNJ‐42165279 in COS‐human fatty acid amide hydrolase
On the day after the seeding, medium was replaced by serum‐free medium and 3 hr later replaced by 600 μl of test item solution (0.1 μM) or vehicle followed by the required incubation time (1, 3 or 24 hr). Immediately after the addition of compounds, 100 μl of supernatant was removed for compound quantification at Time 0. Uptake was stopped by removing the incubated solutions and washing twice with ice‐cold PBS pH 7.4 to remove any compound outside the cells. The incubation medium was retained and the cells were then lysed for 15 min by adding 300 μl of the internal standard solution in acetonitrile. The lysates and the cellular medium were transferred to a 96‐well plate and centrifuged for 20 min at 2,465× g. Five wells were tested for each condition. At each time point, 100 μl of internal standard working solution was added to 100 μl of supernatant sample for protein precipitation and 100 μl of DMEM without serum was added to 100 μl of cell lysates. The samples were vortex mixed, centrifuged for 10 min at 12,100–16,060× g (4°C) and then analysed using a LC/MS/MS TQ 6470 Series from Agilent Technologies with electrospray ionization with positive ion detection. Separation was performed on Poroshell 120 Phenyl‐Hexyl, 2.7 μm, 4.6 × 50 mm (Agilent), using a mobile phase A: Milli‐Q water and mobile phase B: acetonitrile with 0.1% of formic acid, with gradient conditions of 70% of A and 30% of B at 0 min, 30% of A and 70% of B at 4.0 min, and 70% of A and 30% of B at 4.1 min (BIA 10‐2474 and PF‐04457845) or 80% of A and 20% of B at 0 min, 20% of A and 80% of B at 4.0 min and 80% of A and 20% of B at 4.1 min (JNJ‐42165279).
2.5. Quantification of fatty acid amides and 2‐arachidonoylglycerol (2‐AG) in brain samples
Frozen brains were thawed at 4°C, and after weighing, 500 μl of chloroform:MeOH (2:1), 500 μl of phosphate buffer 0.1 M, pH 5.6, 1 μl of 125 μg·ml−1 internal standard (AEA‐d8, PEA‐d4, and OEA‐d2) spiking solution, or 5 μl of 100 μg·ml−1 internal standard (2‐AG‐d8) spiking solution was added immediately to each sample.
The samples were vortexed vigorously for 2 cycles of 5 s at 5,000 rpm and then centrifuged at 20,000× g for 10 min at 4°C. The organic layer was removed and collected into a tube. Five hundred microlitres of chloroform:MeOH (2:1) was added to the remaining aqueous layer and samples were again vortexed vigorously. The samples were centrifuged as described above, and organic layer was collected.
The extraction procedure with chloroform:MeOH (2:1; 500 μl) was repeated as described above. The collected organic layers were pooled and evaporated at 50°C until dry. The samples were reconstituted with 100 μl of acetonitrile and injected into a LC/MS/MS (TQ G6470A system from Agilent). The data were acquired with a MassHunter Workstation software for 6400 Series QQQ Version B.08.00 and analysed with MassHunter Workstation software for quantitative analysis, Version B.07.01 (Agilent Technologies). Fatty acid amide levels are expressed as % of control calculated using the control group killed at 8 hr after dosing on the same day for each experiment. The 2‐arachidonoylglycerol levels are expressed as μg·g−1 of tissue calculated by interpolation of a calibration curve.
2.6. Plasma and brain levels of BIA 10‐2474 and PF‐04457845 in mice
Plasma samples (100 μl) were thawed at 4°C and precipitated with 200 μl of acetonitrile and 0.1% formic acid containing the internal standard. The samples were centrifuged and the supernatant was injected into LC/MS/MS TQ from Agilent. BIA 10‐2474 and PF‐04457845 in the brain were quantified using the same method described in plasma, after the tissue homogenization.
To quantify BIA 10‐2474 and PF‐04457845, water:0.1% formic acid and acetonitrile:0.1% formic acid were used as mobile phase. Separation was performed using a Poroshell 120 Phenyl‐Hexyl, 2.7 μm, 4.6 × 50 mm (Agilent), with a column thermostat set at 40°C and 0.5 ml·min−1 flow. The separation started with 70% of water:0.1% formic acid and 30% of acetonitrile:0.1% formic acid; at 4.0 min, it was 30% of water:0.1% formic acid and 70% of acetonitrile:0.1% formic acid, and at 4.1 min, the gradient returned to the initial conditions. Two microlitres of sample that had been kept at 4°C in the autosampler was injected; the stop time was 6 min without post‐time. The sample was injected into a LC/MS/MS with electrospray ionization in positive mode ionization. The multiple reactions monitoring pair was m/z 301.4 → 145.1 for BIA 10‐2474 with a collision energy of 30 V; m/z 304.4 → 145.1 with a collision energy of 30 V for BIA 10‐4449 (internal standard); and m/z 456.2 → 335.1 with a collision energy of 14 V for PF‐04457845.
2.7. Brain concentrations of BIA 10‐2474 and its metabolites after acute and repeated administration to rats
After thawing at 4°C and weighing, water was added to tissues to give a tissue concentration of 0.1 g·ml−1. The samples were then homogenized and transferred to 1.5‐ml Eppendorf tubes. After centrifugation at 10,000× g for 20 min at 4°C, 100 μl of supernatant was added to 200 μl of internal standard working solution. The samples were vortexed and centrifuged for 10 min at 20,000× g. The supernatant was filtered and injected into LC/MS/MS. Data acquisition was done with MassHunter Workstation software for 6400 Series QQQ, Version B.08.00, and analysed with MassHunter Workstation software for quantitative analysis, Version B.07.01, Agilent Technologies. Samples were analysed for the presence of BIA 10‐2474, BIA 10‐2445, BIA 10‐2631, and BIA 10‐2639. Several values were obtained by extrapolation from the linear standard curves below the lower limit of quantification. Although caution should be exercised in interpreting these values, they have been presented for a better understanding of the distribution of BIA 10‐2474 and its metabolites.
2.8. Off‐target evaluation
All methods used for binding and enzymatic studies carried out by Eurofins are available on their website: http://www.cerep.fr/cerep/users/pages/catalog/profiles/catalog.asp.
2.9. Activity‐based protein profiling for activity at serine hydrolases in vitro
Activity‐based protein profiling in vitro was carried out at Evotec (Toulouse, France). Detailed methods are presented in the Supporting Information. In brief, membrane and soluble protein extracts were prepared from frozen male Wistar rat brains by homogenization in lysis buffer (50 mM of Tris, pH 7.9, and 0.32 M of sucrose). Total protein was quantified by BCA protein assay (Pierce). For the gel‐based competitive activity‐based protein profiling, 200 μg of protein extracts was incubated for 1 hr at room temperature with vehicle (DMSO), PMSF at 3 mM, BIA 10‐2474, JNJ‐42165279 or PF‐04457845 at 1 nM–100 μM. Active serine hydrolases were labelled with the ActivX TAMRA‐FP Serine Hydrolase Probe (Thermo Scientific). For the MS‐based competitive activity‐based protein profiling, 250 μg of protein extracts was incubated for 1 hr at room temperature with vehicle (DMSO), 3 mM of PMSF, 100 and 10 μM of BIA 10‐2474, 10 and 1 μM of JNJ‐42165279 and 0.1 and 0.01 μM of PF‐04457845. Active serine hydrolases were labelled with the ActivX Azido‐FP Serine Hydrolase Probe (Thermo Scientific), and samples were submitted either to in‐gel fluorescence visualization or to active serine hydrolase enrichment. For the latter, samples underwent a click chemistry reaction (Click‐iT Protein Enrichment Kit, Thermo Scientific). On‐bead enriched proteins were digested with 1 μg of trypsin/LysC mix, and the peptides obtained were analysed by nanoLC/MS/MS on a Q‐Exactive Plus (Thermo Fisher Scientific, Bremen) coupled either to an Ultimate 3500 RSLC (Dionex) or a nanoACQUITY UPLC system (Waters) operating in reverse phase (C18). Proteins quantified in fewer than two out of three replicates in at least one condition were removed, and missing values were replaced by a constant corresponding to the average of the lower level of the different conditions.
2.10. Activity‐based protein profiling for activity at serine hydrolases ex vivo
Activity‐based protein profiling ex vivo was carried out at Evotec (Toulouse, France). Detailed methods are presented in the Supporting Information. In brief, rat brain tissues stored frozen (from Charles River Study, see above) were homogenized in lysis buffer (100 mM of Tris, pH 7.9, and 0.32 M of sucrose) and active SerHs labelled with ActivX™ Azido‐FP Probe (Thermo Scientific). After precipitation, labelled proteins were reacted with Biotin Alkyne (Thermo Scientific) followed by enrichment with streptavidin magnetic beads (GE Healthcare). Samples were then either subject to Western blot analysis (to assess enrichment efficiency of biotin‐modified proteins) or proteomic analysis. For the latter, protein in beads was reduced, alkylated and digested with trypsin/LysC mix. Peptides were analysed by nanoLC/MS/MS. Protein identification and quantification were performed using MaxQuant software (Version 1.5.3.8; http://www.biochem.mpg.de/5111795/maxquant, https://scicrunch.org/resources/Tools/search?q=MaxQuant%20software&l=MaxQuant%20software) using a UniProt database (UniProtKB, https://scicrunch.org/resources/Tools/search?q=UniProt%20Knowledgebase%20(UniProtKB)%2C&l=UniProt%20Knowledgebase%20(UniProtKB)%2C; R. norvegicus, 36,126 sequences). The proteins quantified in fewer than two out of three replicates in at least one condition were removed, and missing values were replaced in each condition by a constant corresponding to the average of the Lower Inner Fence of the condition.
2.11. Lipid profiling
The brain right hemisphere (from Charles River Study, see above) was homogenized using a mechanical homogenization procedure (samples suspended in D‐PBS, without magnesium or calcium) with a final concentration of 5 mg·ml−1 (tissue wet weight per volume) and stored frozen at −80°C until dispatch to Lipotype GmbH (Dresden, Germany) where MS‐based lipid analysis was performed as described (Sampaio et al., 2011). Detailed methods are presented in the Supporting Information. Data analysis, including lipid identification, post‐processing, and normalization, was performed in a software based on LipidXplorer (Herzog et al., 2011). Only lipid identifications with a signal‐to‐noise ratio >5 and a signal intensity fivefold higher than in corresponding blank samples were considered for further data analysis.
2.12. Interactions with cytochromes
The rate of metabolite formation from selective cytochrome P450 (CYP) marker substrates by pooled human liver microsomes was measured in the presence of BIA 10‐2474 (0.1 to 30 μg·ml−1 equivalent to 0.33–99.2 μM) in Charles River (Edinburgh, UK) using internal protocols. For the assessment of time‐dependent inhibition, BIA 10‐2474 was pre‐incubated for a period of 30 min, in the presence and absence of co‐factor was included prior to co‐incubation with CYP probe substrates. The formation of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5239, 7‐hydroxycoumarin, hydroxybupropion, 6α‐hydroxypaclitaxel, 4‐hydroxytolbutamide, 4‐hydroxymephenytoin, 1‐hydroxybufuralol and 6‐hydroxychlorozoxazone was quantified as specific CYP markers for the activities of https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1319, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1321, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1324, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1325, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1326, CYP2C19, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1329, and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1330, respectively. As markers of https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1337 activity, the formation of 6β‐hydroxyltestosterone and 1′‐hydroxymidazolam was quantified. Incubations were also performed with probe substrate in the presence of enzyme selective inhibitors. Controls in the presence of solvent vehicle and in the presence of mechanism‐based inhibitors were also examined. Probe substrate incubates were analysed by HPLC with online radio‐detection or by LC/MS/MS.
2.13. Interactions with transporters
Transporter studies were carried out at Solvo (Szeged, Hungary) using their standard methods. BIA 10‐2474 was evaluated as inhibitor of human solute carrier (SLC) transporters organic anion transporter (https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1025 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1027), organic cation transporter (https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=196#1019 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1020), organic anion‐transporting polypeptide (OATP1B1 and OATP1B3), and https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=236#1216 and https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=236#1217 (MATE1 and MATE2‐K) and of human https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=136 https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=768 (P‐gp), https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=792 (BCRP) and bile salt export pump (BSEP). Drug transporters studies were performed using stably transfected cells generated in Solvo Biotechnology. CHO‐K1 cells (University of Zurich) were transfected with OAT1 and OCT1; HEK293 cells (Creative Cell Hungary) were transfected with OAT3, OATP1B1 and OATP1B3; and MDCKII cells (Life Technologies, Carlsbad, CA) were transfected with OCT2, P‐gp, BCRP, MATE1 and MATE2‐K. Vesicular transport assay was performed with HEK293 cells for BSEP. Test system transporter activities were confirmed using a positive control substrate or inhibitor for the individual drug transporter. The summary of conditions and materials used for the transporter assays is provided in Table S8.
To determine if BIA 10‐2474 is a multidrug resistance 1 (https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=768) substrate, the permeability of BIA 10‐2474 (10 μM) in the A–B and B–A directions was determined using MDCKII overexpressing MDR1 and parental cells. https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4726 (5 μM) was used as positive control and PSC833 (Valspodar; 10 μM) and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2406 (100 μM) were used as MDR1 inhibitor. The potential inhibition of MDR1 by BIA 10‐2474 was evaluated by measuring the efflux ratio of 3H‐digoxin in the presence of 33.3 μM of BIA 10‐2474. The positive control used was PSC833 (10 μM).
To determine if BIA 10‐2474 is a BCRP substrate, the permeability of BIA 10‐2474 (10 μM) in the A–B and B–A directions was determined using MDCKII overexpressing BCRP and parental cells. 3H‐Prazosin (1 μM) was used as positive control, and Ko134 (1 μM) was used as BCRP inhibitor. The potential inhibition of BCRP by BIA 10‐2474 was evaluated by measuring the apparent permeability of known BCRP substrate, 3H‐https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=503, in the presence of vehicle, known inhibitor Ko134 (1 μM) and BIA 10‐2474 (33.3 μM) for 1 hr.
2.14. Materials
BIA 10‐2474 (3‐(1‐(cyclohexyl (methyl)carbamoyl)‐1H‐imidazol‐4‐yl)pyridine 1‐oxide; Kiss et al., 2018), BIA 10‐2445 (N‐cyclohexyl‐N‐methyl‐4‐(pyridin‐3‐yl)‐1H‐imidazole‐1‐carboxamide), BIA 10‐2639 (3‐(1‐(((1R,4R)‐4‐hydroxycyclohexyl)(methyl)carbamoyl)‐1H‐imidazol‐4‐yl)pyridine 1‐oxide), and BIA 10‐2631 (N‐((1R,4R)‐4‐hydroxycyclohexyl)‐N‐methyl‐4‐(pyridin‐3‐yl)‐1H‐imidazole‐1‐carboxamide) were synthesized in the Chemistry Department at Bial. PF‐04457845 was obtained from AK Scientific (Union City, CA, USA), and JNJ‐42165279 was obtained from Hangzhou DayangChem (Hangzhou, China). Tritiated AEA [ethanolamine‐1‐3H] ([3H]‐AEA, 40–60 Ci·mmol−1) was obtained from American Radiochemicals (USA). All other materials and compounds were obtained from standard commercial vendors.
2.15. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology. For enzymatic assays, data are presented as mean ± SEM. IC50 and ED50 values are presented with 95% confidence intervals and were calculated using Prism 6 for Windows software (GraphPad Prism v6.05; https://scicrunch.org/resources/Tools/search?q=GraphPad%20Software&l=GraphPad%20Software) using the normalized sigmoidal dose–response curve for inhibition (variable slope).
For the calculation of IC50 values for JNJ‐42165279 inhibition of human recombinant fatty acid amide hydrolase in COS‐hfatty acid amide hydrolase, for the 1‐ and 3‐hr periods, the fitting of the experimental data included only data up to 30 nM as above this concentration, the inhibition level remained around 50%.
The k obs values derived for each inhibitor concentration [I] were obtained by non‐linear global fitting experimental data to the equation model below, where Y is fatty acid amide hydrolase activity (fmol·mg−1) and X is reaction time (min): . The ratio k inact/K I was determined as the slope of the linear fit of the obtained k obs values as function of the respective inhibitor concentration [I] according to the equation: .
For FAAs and compound quantifications, data are presented as mean ± SD. For compound quantification in rat brains, statistical significance (calculated using Prism 6 for Windows software) was evaluated using one‐way ANOVA. P < .05 was considered statistically significant.
For the activity‐based protein profiling studies, statistical differential analysis was performed using limma test (Kammers, Cole, Tiengwe, & Ruczinski, 2015; Smyth, 2004), and Benjamini–Hochberg false discovery rate correction was applied for multiple testing correction. Proteins with a false discovery rate <5% and a fold change of at least 2 were selected to be differentially modulated. Serine hydrolase proteins were annotated using a home‐made database referencing potential serine hydrolases. The database was built from two publications reviewing the human and mouse serine hydrolases (Bachovchin & Cravatt, 2012; Viader et al., 2016). In the in vitro study, a total of 244 human gene symbols were derived from the published data, and rat orthologues of those genes were found, and their UniProt ID (Swiss‐Prot or TrEMBL ID) was retrieved, representing the rat serine hydrolase database. In the ex vivo study, a total of 274 human gene symbols were derived from the published data, and rat orthologues of those genes were found, and their UniProt ID (Swiss‐Prot or TrEMBL ID) was retrieved, representing the rat serine hydrolase database.
For the lipid statistical analyses, an exploratory approach was taken using a non‐parametric two‐sided Wilcoxon tests (Mann–Whitney test) applied to every lipid feature. Lipid amounts were standardized to total lipid amount of each sample. P values were adjusted for multiple testing with the Benjamini and Hochberg method (Benjamini & Hochberg, 1995) and considered significant if P < .05 (Benjamini and Hochberg).
Survival in the SHRSP study was analysed by log‐rank (Mantel–Cox) test using Prism V6 (GraphPad, USA). A P value <.05 was regarded as statistically significant.
2.16. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
3. RESULTS
3.1. Inhibition of fatty acid amide hydrolase activity on rat brain membranes
All three compounds inhibited fatty acid amide hydrolase and IC50 values decreased with longer pre‐incubation times, although for PF‐04457845 this decrease was small (Table 1). The results are consistent with time‐dependent enzyme inhibition by all three inhibitors. PF‐04457845 was two orders of magnitude more potent at fatty acid amide hydrolase compared with BIA 10‐2474 and 1–2 orders of magnitude compared with JNJ‐42165279.
Table 1.
IC50 values determined for the inhibition of rat brain fatty acid amide hydrolase (FAAH) after various pre‐incubation periods of the enzyme with increasing concentrations of BIA 10‐2474, PF‐04457845 and JNJ‐42165279
| Pre‐incubation (min) | IC50 [95% CI] (nM) | ||
|---|---|---|---|
| BIA 10‐2474 | PF‐04457845 | JNJ‐42165279 | |
| 0 | 1,134.0 [745.2, 1,727.0] | 3.9 [3.1, 5.0] | 472.5 [359.7, 620.7] |
| 5 | 1,183.0 [954.8, 1,466.0] | 2.8 [2.0, 3.8] | 365.3 [266.6, 500.6] |
| 15 | 626.4 [522.6, 750.9] | 1.5 [1.3, 1.8] | 272.5 [243.2, 305.3] |
| 30 | 423.6 [300.1, 597.9] | 2.7 [2.4, 3.0] | 220.8 [181.0, 269.5] |
| 45 | 360.0 [265.4, 488.2] | 2.7 [2.4, 3.1] | 160.9 [129.3, 200.3] |
| 60 | 398.0 [338.7, 467.6] | 2.0 [1.6, 2.5] | 94.3 [73.8, 120.5] |
Note. IC50 values are presented with 95% confidence intervals (CIs; n = 4).
These potency differences were corroborated by the second‐order rate constant k inact /K I, for the three compounds at fatty acid amide hydrolase, determined from the plot of k obs values (obtained from fatty acid amide hydrolase progression curves at various inhibitor concentrations, Figure 1a) as a function of inhibitor concentration (Figure 1b). PF‐04457845 displayed the highest in vitro potency with a k inact/K I = 167,333 M·s−1 for inhibition of rat brain fatty acid amide hydrolase, followed by JNJ‐42165279 (k inact/K I = 5,092 M·s−1) and then BIA 10‐2474 (k inact/K I = 1,243 M·s−1).
Figure 1.
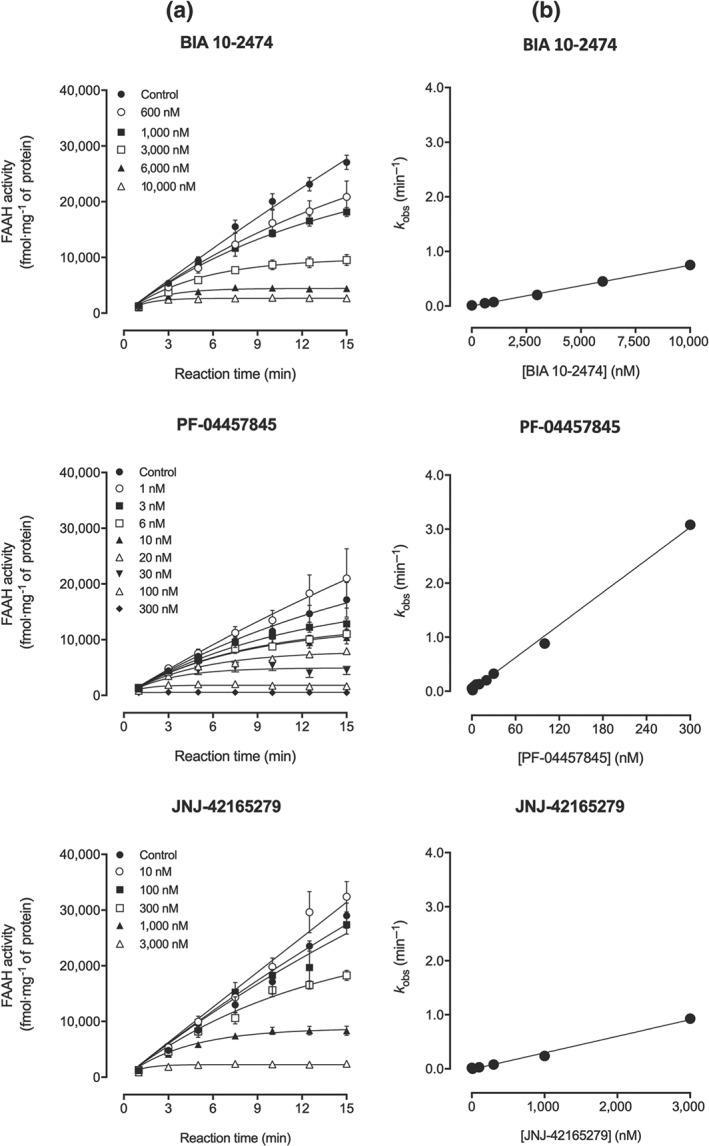
(a) Progression curves of rat brain fatty acid amide hydrolase( FAAH) activity determined in the presence of BIA 10‐2474 (600 nM to 10 μM) or PF‐04457845 (1 to 300 nM) or JNJ‐42165279 (10 nM to 3 μM). (b) From each curve was derived a k obs value that was plotted function of inhibitor concentrations. Results are means ± SEM (n = 4 except for BIA 10‐2474 1 μM, PF‐04457845 10 nM, and controls of these two compounds where n = 8) as obtained from GraphPad Prism 6 for Windows
3.2. Inhibition of fatty acid amide hydrolase activity on intact COS‐7 cells expressing human fatty acid amide hydrolase (COS‐human fatty acid amide hydrolase)
BIA 10‐2474, PF‐04457845 and JNJ‐42165279 concentration‐dependently inhibited human recombinant fatty acid amide hydrolase in COS‐human fatty acid amide hydrolase cells at all tested pre‐incubation periods (IC50 values are in Table 2). The IC50 values for PF‐04457845 were similar to those observed in vitro, in contrast to BIA 10‐2474 and JNJ‐42165279, which inhibited more potently fatty acid amide hydrolase in cells than in vitro.
Table 2.
IC50 values determined for the inhibition of human recombinant fatty acid amide hydrolase (FAAH) after various pre‐incubation periods of the COS‐hFAAH cells with increasing concentrations of BIA 10‐2474, PF‐04457845 and JNJ‐42165279
| Incubation (hr) | IC50 [95% CI] (nM) | ||
|---|---|---|---|
| BIA 10‐2474 | PF‐04457845 | JNJ‐42165279a | |
| 1 | 74.4 [62.2, 89.1] | 4.3 [3.0, 6.1] | 26.2 [17.3, 39.6] |
| 3 | 42.5 [32.4, 55.8] | 3.4 [2.5, 4.6] | 25.1 [14.2, 44.3] |
| 24 | 18.0 [13.8, 23.5] | n.c. | 41.5 [25.0, 69.0] |
Note. IC50 values are presented with 95% confidence intervals (CIs; n = 4 except for some concentrations of JNJ‐42165729 at 1 and 3 hr where n = 8). For PF‐04457845, it was not possible to obtain an IC50 for the 24‐hr time point since it was not possible to fit the experimental data to the sigmoidal dose–response equation.
Abbreviation: n.c., not calculated.
For the 1‐ and 3‐hr periods, the fitting of the experimental data to the dose–response inhibition curves included only data up to 30 nM. It should be noted that JNJ‐42165279 did not achieve full inhibition of FAAH at any of the pre‐incubation periods, even at the highest concentration (3 μM, for 1‐ and 3‐hr time points).
Levels of the compounds in the supernatant and in cell lysates, immediately after adding the compounds and after 1, 3, and 24 hr of incubation, showed that BIA 10‐2474 and JNJ‐42165279 were stable in cellular media over time and undetectable in the cell lysates. In contrast, PF‐04457845 was not stable in the cell medium but was detected in cell lysates and its concentration increased with longer incubation times (Table S1).
Both PF‐04457845 and JNJ‐42165279 showed higher levels of protein binding to cell homogenates, 28.0 ± 3.9 and 64.3 ± 4.0 (mean % free fraction ± SD), respectively at 500 μg·ml−1 of protein, compared with BIA 10‐2474 which was completely unbound (108.2 ± 1.9).
3.3. Effects of compounds on fatty acid amide hydrolase activity and endocannabinoid levels in mice
Oral administration of BIA 10‐2474 (0.1 mg·kg−1 p.o.) to mice resulted in >96.5% inhibition of liver fatty acid amide hydrolase over the first 8 hr (Figure 2a). Enzyme activity gradually recovered thereafter to 72.5% of control levels at 48 hr. In the brain, BIA 10‐2474 also inhibited fatty acid amide hydrolase, with maximal inhibition (91.4%) at 8 hr after administration and with the enzyme slowly recovering activity, returning to 41.8% of control levels at 48 hr. PF‐04457845 (0.1 mg·kg−1 p.o.) fully inhibited both hepatic and cerebral fatty acid amide hydrolase activity over 24 hr, with mean values above 88.7% inhibition activity. Afterwards, both liver fatty acid amide hydrolase activity and brain fatty acid amide hydrolase activity recovered reaching 72.3% and 43.8% of control levels, respectively at 48 hr after dosing. JNJ‐42165279 (0.1 mg·kg−1 p.o.) had virtually no inhibitory effect on either liver or brain fatty acid amide hydrolase activity over the tested 48 hr, with only a transient effect, at 1 hr after dosing, of 25.5% inhibition in the liver and 19.2% inhibition in the brain. Dose dependency of fatty acid amide hydrolase inhibition in the liver and brain was evaluated at 8 hr after oral administration of all three fatty acid amide hydrolase inhibitors. All showed dose‐dependent inhibition of fatty acid amide hydrolase (Figure 2b), and corresponding ED50 values at 8 hr are shown in Table 3.
Figure 2.
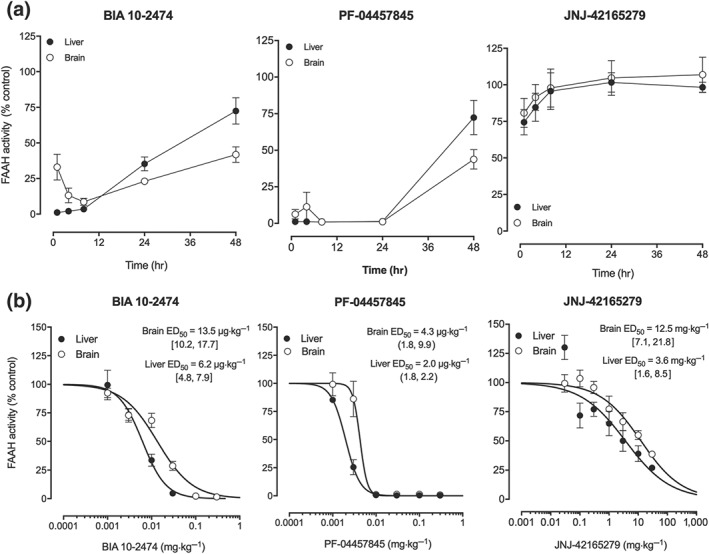
(a) Time course and (b) dose–response curves for inhibition of hepatic fatty acid amide hydrolase (FAAH; closed circles) and brain FAAH (open circles) in mice following oral administration of BIA 10‐2474, PF‐04457845 and JNJ‐42165279. A dose of 0.1 mg·kg−1 was used for the time course experiments. The dose–response was evaluated at 8 hr after administration. Results shown are means ± SEM (n = 4). The values shown near each curve are the ED50 values with 95% confidence intervals
Table 3.
ED50 values (with 95% CI, n = 4 except for one dose of PF‐04457845 where n = 3) determined for the inhibition of mouse liver and brain fatty acid amide hydrolase (FAAH) 8 hr after oral administration of increasing doses of BIA 10‐2474 and the reference compounds PF‐04457845 and JNJ‐42165279
| Compound | ED50 [95% CI] (μg·kg−1) | |
|---|---|---|
| Liver | Brain | |
| BIA 10‐2474 | 6.2 [4.8, 7.9] | 13.5 [10.2, 17.7] |
| PF‐04457845 | 2.0 [1.8, 2.2] | 4.3 [1.8, 9.9] |
| JNJ‐42165279 | 3, 650 [1574, 8461] | 12, 460 [7,110, 21,840] |
Abbreviation: CI, confidence interval.
The fatty acid amides, anandamide (AEA), palmitolyethanolamine (PEA) and oleoylethanolamide (OEA) were increased in mice 8 hr after administration of BIA 10‐2474 (0.1 and 0.3 mg·kg−1 p.o.) or PF‐04457845 (0.01 to 0.3 mg·kg−1 p.o.) when compared with vehicle‐treated mice (Table 4). In contrast, JNJ‐42165279 (1 to 30 mg·kg−1 p.o.) did not appear to affect anandamide, although both palmitolyethanolamine and oleoylethanolamide levels were increased at 10 and 30 mg·kg−1 p.o when compared with vehicle‐treated mice.
Table 4.
Mean concentrations of anandamide (AEA), palmitoylethanolamide (PEA) and oleoylethanolamide (OEA) in mouse brain 8 hr after oral administration of BIA 10‐2474 and the reference compounds PF‐04457845 and JNJ‐42165279
| Treatment (mg·kg−1 p.o.) | AEA | PEA | OEA |
|---|---|---|---|
| BIA 10‐2474 | |||
| 0.01 | 89.5 ± 9.3 | 107.2 ± 8.8 | 109.1 ± 8.6 |
| 0.03 | 87.0 ± 10.3 | 178.6 ± 62.7 | 143.1 ± 30.6 |
| 0.1 | 136.8 ± 16.4 | 1027.6 ± 148.0 | 676.5 ± 89.8 |
| 0.3 | 172.1 ± 29.5 | 1,223.3 ± 159.6 | 804.0 ± 107.6 |
| PF04457845 | |||
| 0.003 | 88.5 ± 7.5 | 100.6 ± 16.4 | 97.1 ± 8.0 |
| 0.01 | 220.2 ± 46.9 | 984.3 ± 107.1 | 837.5 ± 115.5 |
| 0.03 | 257.6 ± 16.9 | 1073.1 ± 85.1 | 886.1 ± 42.6 |
| 0.1 | 268.7 ± 10.8 | 1080.9 ± 62.0 | 933.4 ± 17.5 |
| 0.3 | 309.4 ± 16.6 | 1,141.9 ± 56.3 | 961.4 ± 48.2 |
| JNJ‐42165279 | |||
| 1 | 89.0 ± 20.6 | 123.4 ± 11.2 | 114.9 ± 13.36 |
| 3 | 83.6 ± 9.1 | 138.3 ± 19.4 | 121.7 ± 10.2 |
| 10 | 86.5 ± 4.8 | 205.6 ± 37.0 | 147.3 ± 13.8 |
| 30 | 95.5 ± 2.5 | 382.2 ± 103.5 | 234.9 ± 46.8 |
Note. Values are expressed as % of control values for n = 4 with SD.
BIA 10‐2474 (10 mg·kg−1 p.o.) had no clear effect on mouse brain 2‐arachidonoylglycerol (2‐AG) levels at 1, 4, 8 and 24 hr after administration (vehicle: 45.1 ± 7.9, 40.9 ± 3.4, 39.0 ± 13.2 and 38.0 ± 5.7 μg·g−1, respectively; BIA 10‐2474: 30.4 ± 11.7, 44.5 ± 4.9, 34.9 ± 7.7 and 31.4 ± 9.4 μg·g−1, respectively; all values mean ± SD, n = 4).
3.4. Plasma and brain levels of BIA 10‐2474 and PF‐04457845 in mice after oral administration
When administered orally at pharmacologically relevant doses, BIA 10‐2474 and PF‐04457845 were both detectable in plasma (Table 5). In the brain, BIA 10‐2474 was detected at both doses but with exposure values 5‐fold to 10‐fold lower than those in plasma, while PF‐04457845 was not detectable in the brain after administration of 0.03 mg·kg−1 p.o. but at 0.1 mg·kg−1 p.o. it was found, at approximately 10‐fold lower levels than in plasma (Table 5).
Table 5.
Pharmacokinetic parameters after oral administration for BIA 10‐2474 (0.1 and 0.3 mg·kg−1 p.o.) and PF‐04457845 (0.03 and 0.1 mg·kg−1 p.o.) in mouse plasma and brain (n = 4)
| Treatment (mg·kg−1) | Plasma | Brain | ||||||
|---|---|---|---|---|---|---|---|---|
| t ½ | t max | C max | AUClast | t ½ | t max | C max | AUClast | |
| (hr) | (hr) | (ng·ml−1) | (hr·ng·ml−1) | (hr) | (hr) | (ng·g−1) | (hr·ng·g−1) | |
| BIA 10‐2474 | ||||||||
| 0.1 | 2.2 | 1 | 33.0 | 109.7 | NR | 1 | 5.3 | 11.3 |
| 0.3 | 1.9 | 1 | 104.5 | 337.5 | 1.75 | 1 | 21.6 | 60.8 |
| PF‐04457845 | ||||||||
| 0.03 | NR | 2 | 1.3 | 4.3 | NR | NR | NR | NR |
| 0.1 | NR | 2 | 38.3 | 238.3 | NR | 2 | 3.7 | 22.9 |
Note. Concentrations of test substances were measured at 1, 2, 4, and 8 hr after administration. The limits of quantification were 0.1 ng·ml−1 for plasma and 1 ng·g−1 for the brain.
Abbreviation: NR, not reliable (insufficient data to calculate reliable values).
3.5. Brain levels of BIA 10‐2474 and its primary metabolites in rats following acute and repeated administration
To evaluate possible persistence of BIA 10‐2474 or its metabolites in the brain, concentrations of BIA 10‐2474 and its metabolites BIA 10‐2445, BIA 10‐2639 and BIA 10‐2631 were evaluated in rat cerebellum, midbrain, pons, medulla oblongata, hippocampus and cerebral cortex after oral BIA 10‐2474 (10 mg·kg−1 p.o.) at 1, 4 and 24 hr after acute administration and at 4, 24, 48 and 72 hr after repeat administration for five consecutive days. At 24 hr after acute administration and at 24, 48 and 72 hr after chronic administration, the levels of BIA 10‐2474 were below the limit of quantification (<50 ng·g−1 of tissue) in all brain sections. BIA 10‐2445 had a similar temporal pattern to BIA 10‐2474 but with much lower concentrations. Levels of BIA 10‐2631 and BIA 10‐2639 were below the limit of quantification at all time points measured and, apart from slightly higher levels of BIA 10‐2631 at 4 hr after administration of BIA 10‐2474, were not clearly different from background (Figure 3 and Table S2). There was only minor variation in levels of BIA 10‐2474 between brain regions and little variation between regions for the three measured metabolites (Table S2). Importantly, no significant differences in the levels of BIA 10‐2474 or its metabolites were observed between acute and repeated oral administration in any of the brain regions studied nor at any of the time points measured.
Figure 3.
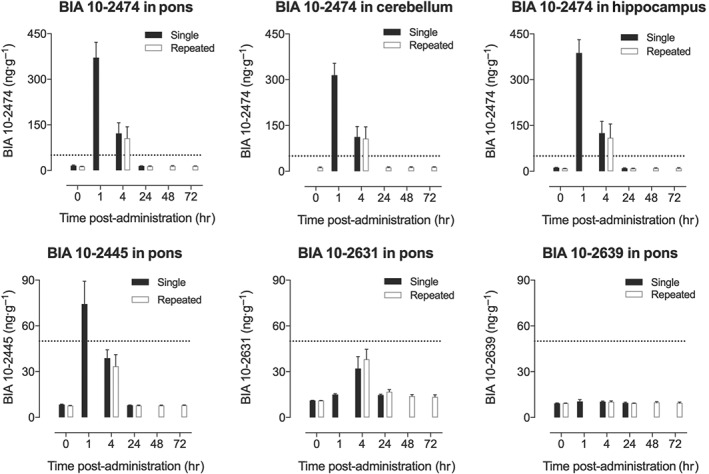
Regional brain levels of BIA 10‐2474 in the cerebellum, pons, and hippocampus (upper panels) and of its primary metabolites in the pons (lower panels) at various time points after acute (black bars) or 5 days repeated once daily (open bars) oral administration of BIA 10‐2474 at 10 mg·kg−1. The complete data set is available in Table S2. Values are mean and SD for n = 6 except vehicle where n = 2. The dotted line indicates the limit of quantification: Extrapolated values below this are shown to allow better comparison of acute and chronic treatments, but in these cases, the absolute values may be less reliable than those within the limits of quantification
3.6. Evaluation of off‐target activity in vitro
Potential off‐target activity was evaluated at Caliper for BIA 10‐2474 (10 μM) at 97 target assays and reported as part of the request for regulatory approval of the clinical trial. Subsequent studies at 10 and 50 μM were carried out for BIA 10‐2474, PF‐04457845 and JNJ‐42165279 at more than 220 targets at Eurofins. The complete data set for a total of 229 different sites is available as supplementary data (Table S3). BIA 10‐2474 had no significant activity at any of the sites studied. In contrast, PF‐04457845 was active at a number of sites some of which were consistent with published data (Li et al., 2012). JNJ‐42165279 showed some weak activity at just seven sites at 50 μM.
3.7. Interactions with cytochromes and transporters
In human hepatic microsomes, BIA 10‐2474 showed no or negligible inhibition of any of the CYP enzymes tested. Following co‐incubation, BIA 10‐2474 produced minor inhibition in CYP2D6 and CYP3A4 (testosterone as substrate), although there was no effect following pre‐incubation with BIA 10‐2474. Following co‐and pre‐incubation, BIA 10‐2474 (0.33–99.2 μM) did not exhibit any inhibitory effects on any of the other CYP isoforms examined in this study (CY1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2E1 and CYP3A4 [midazolam]).
In transporter inhibition assays, BIA 10‐2474 inhibited the MATE2‐K‐mediated probe substrate accumulation in a dose‐dependent manner with a calculated IC50 of 1.03 μg·ml−1 (3.4 μM) and maximum inhibition of 59%. Regarding OAT3‐, OCT1‐, OCT2‐ and MATE1‐mediated probe substrate accumulation, a slight inhibition was observed with BIA 10‐2474 at the highest concentration tested (10 μg·ml−1 or 33 μM) with a maximum inhibition of 34%, 26%, 25% and 24%, respectively. No IC50 was determined for these interactions. BIA 10‐2474 did not influence the OAT1‐, OATP1B1‐ and OATP1B3‐mediated probe substrate accumulation at the applied concentrations (0.033–33.3 μM). Accumulation of BIA 10‐2474 was similar in the transporter‐expressing and control cell (transporter specific fold accumulations were <2), indicating no active accumulation under all conditions tested.
In MDCKII‐BCRP and MDCKII‐MDR1 cells, BIA 10‐2474 showed higher permeability in the basolateral–apical direction than in the apical–basolateral direction, suggesting active transport of this compound. The highest observed net efflux ratio (ER; ERBCRP‐ERMDCKII or ERMDR1‐ERMDCKII) was 5.70 at 10 and 1 μM for BCRP and 2.24 at 10 μM for MDR1. The BCRP‐specific inhibitor (1 μM of Ko143) decreased net ER from 4.95 to 0.03, confirming the contribution of BCRP to the transport of BIA 10‐2474 across MDCKII‐BCRP monolayers. Similarly, the MDR1‐specific inhibitors PSC833 (10 μM) and verapamil (100 μM) decreased net ER from 1.45 to −0.11 and to −0.02, respectively, suggesting that BIA 10‐2474 is a weak MDR1 substrate in MDCKII‐MDR1 monolayers.
BIA 10‐2474 (33.3 μM) was evaluated on the BCRP‐ and MDR1‐mediated efflux of prazosin and digoxin in the monolayer inhibition assay. BIA 10‐2474 did not significantly change the BCRP‐mediated efflux of prazosin (net ER 10.8 and 9.41, without and with BIA 10‐2474) or the MDR1‐mediated efflux of digoxin (6.11 and 7.25, without and with BIA 10‐2474), indicating that BIA 10‐2474 is not an inhibitor of BCRP or MDR1 in the MDCKII monolayer assay.
3.8. Evaluation of off‐target activity using in vitro activity‐based protein profiling
BIA 10‐2474, PF‐04457845 and JNJ‐42165279 were first evaluated using a TAMRA‐FP (tetramethylrhodamine–fluorophosphonate) probe in both membrane and soluble rat brain fractions followed by studies with an azido‐FP probe to allow isolation and identification of serine hydrolases by MS. A band corresponding to the MW of fatty acid amide hydrolase was completely inhibited by 100 μM of BIA 10‐2474 and by 10 and 0.1 μM of JNJ‐42165279 and PF‐04457845, respectively. No other bands appeared to be affected by JNJ‐42165279 and PF‐04457845, whereas BIA 10‐2474 attenuated the band of another serine hydrolase (Figure S1). Studies with an azido‐FP probe confirmed that the band suppressed by all three inhibitors was fatty acid amide hydrolase and identified the additional band attenuated by BIA 10‐2474 as ABHD6 (Table S4). JNJ‐42165279 also inhibited the signal of ABHD11 at similar concentrations to its effects on fatty acid amide hydrolase. None of the other 32–36 serine hydrolase proteins identified in the membrane or soluble fractions were affected by the three compounds. The test substances also attenuated the signal for several non‐serine hydrolase proteins, but all were low abundance proteins and the effects were not always dose dependent (Table S5).
3.9. Evaluation of off‐target activity using ex vivo activity‐based protein profiling
The effect of BIA 10‐2474 on rat brain serine hydrolases was also evaluated after oral administration (10, 30, or 100 mg·kg−1·day−1 p.o.) to rats for 1, 7 or 28 days. Half of each brain was used for activity‐based protein profiling. The activity‐based protein profiling protocol used for whole‐brain homogenates was slightly modified and allowed the identification and quantification up to 72 serine hydrolases.
All doses of BIA 10‐2474 (10, 30 and 100 mg·kg−1 p.o. for 1, 7 or 28 days) resulted in almost complete inhibition of azido‐FP probe‐labelled fatty acid amide hydrolase in rat brain (Table 6). ABHD6 was also significantly inhibited at all doses from Day 1 but with some dose dependency, which did not clearly change with longer treatment. Several carboxyl esterase (CES) enzyme variants were also dose‐dependently inhibited without treatment duration having a clear impact. This was also true for ABHD11, although significant inhibition was only observed at 100 mg·kg−1.
Table 6.
Effect of BIA 10‐2474 treatment (10, 30, or 100 mg·kg−1 p.o.) for 1 to 28 days on serine hydrolase enzymes in rat brain
| Serine hydrolase | Protein ID | BIA 10‐2474 treatment condition (mg·kg−1·day−1 p.o.) | Protein ID and prevalence in controls | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 10 mg·kg−1 | 30 mg·kg−1 | 100 mg·kg−1 | Sequences for ID | Log2 intensity | ||||||||||||
| 1 day | 7 days | 28 days | 1 day | 7 days | 28 days | 1 day | 7 days | 28 days | 10 | 30 | 100 | 10 | 30 | 100 | ||
| FAAH1 | P97612 | 98 | 98 | 98 | 99 | 99 | 99 | 99 | 99 | 99 | 31 | 27 | 30 | 30.8 | 28.7 | 27.7 |
| Monoacylglycerol lipase (ABHD6) | Q5XI64 | 85 | 81 | 83 | 88 | 82 | 91 | 95 | 93 | 95 | 22 | 20 | 21 | 30.5 | 28.0 | 27.3 |
| Carboxyl esterase 1C | P10959 | 47 | 57 | 66 | 79 | 71 | 90 | 97 | 95 | 100 | 22 | 20 | 21 | 29.8 | 27.5 | 26.7 |
| Carboxyl ester hydrolase | D3ZGK7 | 69 | 73 | 79 | 98 | 67 | 99 | 99 | 100 | 100 | 3 | 3 | 3 | 24.9 | 22.9 | 22.9 |
| Carboxyl ester hydrolase | M0R9W7 | 12 | 25 | 12 | 9 | 71 | 94 | 81 | 97 | 98 | 9 | 6 | 7 | 24.5 | 21.9 | 21.4 |
| ABHD11 | D3ZXK4 | 3 | — | 11 | 10 | — | 39 | 64 | 64 | 82 | 16 | 13 | 14 | 30.9 | 28.6 | 27.9 |
| Signal peptidase complex catalytic subunit SEC11/11A | P42667; Q6P9X2 | 5 | 11 | 80 | 17 | 92 | 94 | — | — | — | 4 | 3 | 2 | 23.8 | 22.4 | 17.0 |
| Lysosomal thioesterase PPT2 | O70489 | — | — | 19 | 64 | 71 | 94 | nd | nd | nd | 5 | 3 | nd | 22.4 | 21.6 | nd |
| PLA2 Group 15 (PLA2G15) | Q675A5 | — | — | 24 | — | — | 43 | 29 | 58 | 71 | 9 | 10 | 8 | 25.6 | 23.3 | 22.5 |
| Patatin‐like phospholipase domain containing 6 (PNPLA6) | M0R715 | — | — | 12 | — | — | 30 | 17 | 44 | 63 | 61 | 47 | 46 | 29.1 | 26.6 | 25.8 |
| ABHD10 | Q5I0K5 | — | — | 6 | — | — | 12 | 9 | 38 | 63 | 18 | 16 | 16 | 30.7 | 28.1 | 27.3 |
| CaI‐PLA2 (PLA2G6) | P97570 | — | — | — | — | — | 42 | 7 | 81 | 69 | 9 | 6 | 4 | 22.7 | 20.2 | 19.7 |
| AIG1 protein | B2RZC0 | — | 9 | 9 | 11 | 10 | 53 | 19 | 62 | 72 | 6 | 6 | 6 | 29.6 | 26.2 | 26.7 |
Note. Values shown are % reduction of FP probe‐labelled serine hydrolase compared with vehicle as identified and quantified by LC/MS/MS. Each value is derived from six independent evaluations. Shaded cells with values in bold indicate proteins harbouring a fold change <−2 and an adjusted P value <.05 (Limma test and Benjamini–Hochberg correction for multiple testing). The experiment was analysed in three separate sub‐experiments, one for each of the dose levels. The protein ID and prevalence columns indicate the number of unique sequences used for identification of the protein in each sub‐experiment, and the log2 intensity of the protein by LC/MS/MS is given for the control group during each sub‐experiment. No value indicates no observed decrease in fold change.
Abbreviation: nd, not detected.
In contrast, a number of serine hydrolases showed a dose‐ and treatment duration‐related effect. These included in particular PLA2g15, PNPLA6, ABHD10 and androgen‐induced protein 1 (AIG1), which were unaffected on Day 1 but were significantly inhibited at 100 mg·kg−1 after 28 days of treatment. SEC11a and PPT2 were significantly inhibited at 30 mg·kg−1 after 28 days of treatment, but the effects at 100 mg·kg−1 could not be evaluated because of very low‐protein intensities. In addition, PLA2g6, although not reaching significance compared with vehicle‐treated rats, was completely inhibited in four out of six rats at 30 mg·kg−1 and three out of six rats at 100 mg·kg−1.
As in the in vitro study, non‐serine hydrolase proteins were identified including several that were significantly affected by BIA 10‐2474 treatment. Most were of low and variable abundance and none of the changes appeared to be dose related or consistent (Table S6).
3.10. Brain lipid profiling in rats given BIA 10‐2474 100 mg·kg−1·day−1 for 28 days
The half brain not used for the activity‐based protein profiling ex vivo study was used for a shotgun lipidomic analysis, and the lipid classes https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2796 (PE), plasmenyl PE, phosphatidylcholine (PC), phosphatidylinositol (PI) and DAG were evaluated in the brains of the rats dosed with vehicle or 100 mg·kg−1·day−1 for 28 days (Table S7).
In the PC class, all statistically altered diacyl species were increased compared with the control group. The other membrane phospholipid class evaluated, PE, was also significantly increased in species containing very‐long‐chain fatty acids, including arachidonic acid (AA—20:4) and docosahexaenoic acid (DHA—22:6). Plasmenyl PEs with AA and DHA were also statistically increased in the treated group. The signalling PI class presented just one lipid species with significant alterations, a phospholipid with AA that was increased by BIA 10‐2474. Two species of the DAG class, a source of FA/energy, were significantly decreased by BIA 10‐2474:DAG (16:0/18:1) and DAG (16:0/20:1).
3.11. Additional histological evaluations
Within the same study, the rat brains subjected to histopathology showed no abnormalities in any of the groups. Similarly, none of the animals used in this study showed any overt behavioural abnormalities during the course of treatment with BIA 10‐2474.
3.12. Effect of BIA 10‐2474 in SHRSP
The effects of the compound were explored in SHRSP by evaluating mortality rate and examining cerebrovascular damage. The study was terminated after 45 days when 53.3% (16 out of 30) of the animals receiving vehicle and 43.3% (13 out of 30) of animals receiving BIA 10‐2474 had died. No deaths were recorded in the control group. The mortality curves for the three treatment groups are shown in Figure 4. The mortality rate of SHRSP on a stroke‐prone diet in conjunction with high‐salt intake was significantly increased as compared with SHRSP on a standard rodent diet and tap water (control vs. vehicle, P = .0062). Log‐rank (Mantel–Cox) test comparison of the curves revealed no effect of BIA 10‐2474 treatment on mortality rate compared with vehicle in animals on stroke‐prone diet and high‐salt intake (P = .1882). In the rats surviving at the end of the study, histological lesions consisted of haemorrhage, oedema, and inflammation; necrosis was not observed. In the control group, five out of the 10 animals were affected by minor foci of haemorrhage in the dorsal cortex and pyramis distributed between Levels 2 and 5 that were deemed to represent preparation or agonal artefacts. In rats on high‐salt intake and receiving vehicle or BIA 10‐2474, the type of lesion, location, number and size were similar comparing affected animals. Three of the 14 surviving vehicle animals had haemorrhage and oedema in the corpus callosum and one of the 14 survivors had a focal inflammatory reaction in the dorsal cortex between Levels 2 and 4. Three of the 17 surviving BIA 10‐2474‐treated rats had haemorrhage in the hippocampus and in the dorsal cortex at Levels 2 and 3.
Figure 4.

Mortality rate of spontaneously hypertensive rats stroke‐prone (SHRSP) fed with a stroke‐prone diet and in conjunction with high‐salt intake (Groups 2 and 3). Group 1 animals were fed with normal rodent diet and given tap water. Animals from Groups 1 (n = 10) and 2 (n = 30) were daily given 0.2% HPMC, while animals from Group 3 (n = 30) were given BIA 10‐2474 (30 mg·kg−1·day−1), until termination of the study, when about 50% of animals of either Group 2 or 3 had died. Log‐rank (Mantel–Cox) test comparison of curves revealed a significant increase in mortality of animals fed with the stroke‐prone diet as compared with normal diet (Group 1 vs. Group 2, P = .0062) and no effect of BIA 10‐2474 treatment on mortality rate compared with vehicle in animals on stroke‐prone diet and high‐salt intake (Group 2 vs. Group 3, P = .1882)
4. DISCUSSION
The objectives of the studies reported here were several: firstly to characterize the pharmacology of BIA 10‐2474 using PF‐04457845 and JNJ‐42165279 as comparators, two fatty acid amide hydrolase inhibitors previously tested in humans and considered safe, and then to explore the selectivity of BIA 10‐2474 to identify potential off‐targets, both in vitro and in vivo. Our study concentrated on BIA 10‐2474 itself, as it represents the major species, >90% by AUC, found in plasma following its oral administration (Loureiro, Lopes, Bonifácio, Moser, & Soares‐da‐Silva, 2017) and secondly because the TSSC in its final report (TSSC, 2016) considered that the metabolites of BIA 10‐2474 were unlikely to play a role in the observed toxicity, because of their low circulating concentrations. This is reinforced by the present demonstration that brain levels of metabolites were substantially below those of the parent molecule. Furthermore, that the metabolite levels did not change between areas nor between acute and repeated administration. Finally, a study was designed to explore the potential effects of BIA 10‐2474 in an animal model known to be prone to have a high incidence of cerebral vascular stroke (Yang, Kimura‐Ohba, Thompson, & Rosenberg, 2016).
The present data characterizes BIA 10‐2474 as a time‐dependent mechanism‐based inhibitor of FAAH. In rat brain membranes, BIA 10‐2474 potency increased with pre‐incubation time, as expected for a time‐dependent inhibitor, similarly to that of PF‐04457845 and JNJ‐42165279. The IC50 values determined at each pre‐incubation time (from 360 to 1134 nM) were about two orders of magnitude higher than those of PF‐04457845 and 2–4‐fold higher than those for JNJ‐42165279. However, when using an intact cell preparation expressing human fatty acid amide hydrolase, the IC50 values determined for BIA 10‐2474 varied between 18 and 74 nM. This large variation in fatty acid amide hydrolase potency between membrane and whole‐cell preparations was not observed with either PF‐04457845 or JNJ‐42165279. Additionally, after oral administration the potency of BIA 10‐2474 to inhibit mouse brain or liver fatty acid amide hydrolase was similar to that of PF‐04457845. This higher potency in vivo has also been observed by other authors (Tong et al., 2017). The simplest explanation, as suggested by other authors (van Esbroeck et al., 2017), is that BIA 10‐2474 accumulates inside cells (Mateus, Matsson, & Artursson, 2013), but none of these compounds showed any evidence of such accumulation. Another possibility is that the effect was due to an active early metabolite instead of the parent BIA 10‐2474. However, metabolic studies performed in vitro with hepatocytes and in vivo in the rat with the radiolabelled BIA 10‐2474 (Loureiro, Fernandes‐Lopes, Moser, & Soares‐da‐Silva, 2018) showed no evidence for an early metabolite. Additionally, the only active metabolite identified had a similar in vitro potency to that of BIA 10‐2474. A differential interaction with the enzyme in its natural environment (in the membranes in vivo) or in the in vitro preparations could also be put forward. Although we do not have an explanation for this difference, the potency of BIA 10‐2474 determined in situ is more aligned with the concentrations that are active in vivo. Thus, at 0.1 mg·kg−1 p.o., a dose‐producing near‐maximal inhibition of fatty acid amide hydrolase, we obtained maximum levels in plasma and brain of 33.0 ng·ml−1 and 5.3 ng·g−1 (equivalent to 109 and 18 nM), respectively.
In a broad screen of 221 targets, BIA 10‐2474 had no significant activity at up to 50 μM. Additional testing at a further eight targets at 10 μM similarly revealed no significant activity. In contrast, PF‐04457845 showed significant activity at several receptors, transporters and ion channels, similar to data published by Johnson et al. (2011). In addition, the present study also shows that BIA 10‐2474 has relatively little impact on CYPs and transporters up to concentrations of at least 30 μM, apart from inhibition of MATE2‐K with an IC50 of 3.4 μM.
We identified an effect of BIA 10‐2474 against serine hydrolases both in vitro and ex vivo. The results obtained after chronic treatment in the ex vivo activity‐based protein profiling study confirmed most of the off‐targets previously identified by van Esbroeck et al. (2017), notably ABHD6 at exposure levels only slightly greater than those required for fatty acid amide hydrolase inhibition, and at higher exposures ABHD11 and PNPLA6. Although not detecting specifically activity against the rat orthologue of human CES2, we did identify activity against three members of the same https://www.uniprot.org/uniprot/?query=family:%22type-B+carboxylesterase%2Flipase+family%22 (CES 1C and two carboxyl ester hydrolases). We did not detect an effect against hormone‐sensitive lipase nor, as expected, fatty acid amide hydrolase2. However, we identified additional off‐target activity against the lipid metabolism‐related enzymes PLA2G15, lysosomal thioesterase PPT2, calcium‐independent PLA2 (PLA2G6) and ABHD10. Significant inhibition was also seen on the androgen‐induced protein 1 and signal peptidase complex catalytic subunit SEC11A. Possible reasons for the differences in off‐target identification include the interactions being specific to rodent enzymes (present study as opposed to human cells, van Esbroeck et al., 2017), and/or due to a metabolite or requiring very long exposure as they were primarily seen at 100 mg·kg−1 after 28 days. The differences between our in vitro and ex vivo activity‐based protein profiling data suggest that BIA 10‐2474 is less potent against all serine hydrolase enzymes in vitro, not just fatty acid amide hydrolase and ABHD6. Although we have not measured brain levels of BIA 10‐2474 at the doses used in our ex vivo study, data from GLP toxicology studies (Harris et al., 2019; Hayes et al., 2019) at comparable doses (10, 30, and 90 mg·kg−1 p.o.) and for comparable durations (28 and 30 days of treatment) provide plasma C max values of approximately 11, 47, and 101 μM at 10, 30 and 90 mg·kg−1 p.o. (irrespective of treatment duration), suggesting maximum brain concentrations around 20 μM at the 100 mg·kg−1 of dose used here (assuming the same ratio of 1:5 between brain and plasma as we see here for 0.1 and 0.3 mg·kg−1 in mice).
Lipid metabolism was altered in rat brain tissues in animals dosed with 100 mg·kg−1·day−1 as compared with the control group. All identified PEs and PCs containing AA were significantly increased in the BIA 10‐2474‐treated group reflecting accumulation of lipid species that are precursors of AEA, which, although not quantified in the lipid profiling study, is increased as a result of fatty acid amide hydrolase inhibition (as shown in mouse studies). Interestingly, phospholipids not only with AA but also with very‐long‐chain fatty acids were increased, representing an impact on downstream products of these lipid species. Our data are in part aligned with that previously reported (van Esbroeck et al., 2017), with increased long‐chain PC species and DAG. Further comparisons cannot be drawn since in our study, small lipids, such as fatty acids, N‐acetyl amines and small lysophospholipids, were not quantified due to method limitations. Although several of the serine hydrolases identified as BIA 10‐2474 off‐targets are involved in lipid metabolism, they metabolize classes of lipids, and a direct correlation between the lipid alterations observed and the activities of those enzymes cannot be made at present.
Some of these enzymes are associated with neurological syndromes in humans. Loss‐of‐function mutations in PLA2G6 (Arber, Li, Houlden, & Wray, 2016; Illingworth et al., 2014) and PNPLA6 (Synofzik et al., 2014; Topaloglu et al., 2014) have been identified as causing neurodevelopmental disorders and PPT2 knockout mice display a neurodegenerative phenotype, although PPT2 deficiency has not been described in humans (Gupta et al., 2003). Could any of the off‐targets identified here be responsible for the toxicity seen in the clinic with BIA 10‐2474? At the maximum plasma exposures seen in the clinic (around 2 μM at 50 mg and 6 μM at 100 mg, Rocha et al., 2017), it is almost certain that ABHD6 and CES would be inhibited. As far as we can ascertain, there are no reports of neurological consequences of either gene knockout or pharmacological inhibition of these targets. The other off‐targets mentioned above will probably have lower levels of inhibition at the measured clinical exposure, but we cannot determine at this stage to what extent any inhibition might be physiologically meaningful. It should be noted however that inhibitors of PNPLA6 can induce neurotoxicity in rodents if inhibition is greater than 80% (Wu & Casida, 1996) and animal models of the different point mutations and/or knockout mice of these targets reproduce the associated clinical symptoms (Long & Cravatt, 2011; Winrow et al., 2003).
However, the available data make it unlikely that the identified off‐targets were significantly occupied at the clinical doses, since other elements besides exposure to the inhibitor are involved such as the relative amounts of enzymes present in relation to both the inhibitor and substrate concentrations and the rate of recovery of enzyme activity. Nevertheless, the exposure levels of BIA 10‐2474 used in regulatory toxicology studies would have been sufficient to engage all of the off‐targets we have identified. However, neither the neurological syndromes nor the histopathological hallmarks of these human syndromes were seen in these toxicology studies. There was some mild axonal swelling in the medulla oblongata in the rat toxicology study at 90 mg·kg−1 after 13 weeks of treatment (in 3/10 male rats and 7/10 female rats) and vacuolation in the same region in about half the male rats at the same dose after 26 weeks of treatment. However, these signs were not observed in the 28‐day toxicology study (manuscript under editorial review), consistent with the absence of CNS histopathology in the present study.
There have been suggestions of other potential off‐targets such histone deacetylases, calcitoninhttps://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=48 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2363 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2359 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=590r (Molinski et al., 2017). However, the present data show that BIA 10‐2474 does not have affinity for any of these targets. Recently, Huang et al. (2019), using alkynylated analogues of BIA 10‐2474 and metabolites as activity‐based protein profiling probes showed that the two desmethyl metabolites were able to covalently bind catalytic cysteines of aldehyde dehydrogenases in human cells, namely, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2595, ALDH1B1 and https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=764#2467. These enzymes were not identified in the present study, although the data are not comparable, as the activity‐based protein profiling probes used in this study were designed to target specifically serine hydrolases.
Kerbrat et al. (2016) described several types of cerebral lesion in the affected subjects taking part in the clinical trial with BIA 10‐2474 as detected by MRI scans. They included lesions, microhaemorrhage and oedema, which were severe in the subject that died and mild to severe in two affected survivors. The SHRSP were established from a substrain of the spontaneously hypertensive rat (SHR) and shows a high incidence of cerebral stroke. We consider the SHRSP model that we have used here particularly relevant to the abnormalities described as the use of relatively young rats (60 days old) limits the role for hypertension but still results in cerebral haemorrhage in conjunction with high‐salt intake and low‐protein diet (Yang et al., 2016). Daily treatment with BIA 10‐2474 at the high dose of 30 mg·kg−1 p.o. did not affect mortality rate in this model and, in surviving animals, did not increase the incidence of haemorrhage or oedema. At this dose, the primary off‐targets identified by activity‐based protein profiling ex vivo, such as ABHD6 and CES, would have been inhibited as well as SEC11/11a, lysosomal thiophosphatase and androgen‐induced protein 1 (AIG1; see Table 6). However, the phospholipases identified by van Esbroek (van Esbroeck et al., 2017) and by us that have been associated clinically with neurological symptoms (i.e. PNPLA6 and PLA2g6) were probably unaffected at this dose.
It is important to point out some of the limitations of the present study in relation to identifying off‐target interactions that could be responsible for the clinical signs seen with BIA 10‐2474. The most obvious one is that we have used the rat for most of our studies. However, there is no other way to reproduce the duration of drug exposure that occurred in the clinical trial. It is nevertheless important that we have identified a very similar range of off‐targets to van Esbroeck et al. (2017) with what appears to be a similar relative potency compared with fatty acid amide hydrolase. However, relative potency of irreversible inhibitors such as BIA 10‐2474 and PF‐04457845 at serine hydrolase targets in vivo depends on several factors such as the inactivation constants, the quantity of enzyme present and the duration of exposure. None of the studies carried out so far with BIA 10‐2474 have done that. It would be important to characterize the interaction of the compounds to the identified off‐targets and to evaluate their in vivo off‐target occupancy at the different exposures to properly extrapolate our data to the clinical situation. It should also be noted that we, like others, have mostly considered CNS targets as being responsible for the clinical trial accident and serine hydrolases in particular. Although our study in SHRSP was carried out to explore possible mechanisms outside the CNS (albeit related to CNS vasculature), further studies outside the CNS and looking more broadly at protein targets using proteome‐wide screening techniques, such as surface plasmon resonance or MS imaging (Yang et al., 2016; Zhou, Liu, Rothschild, & Lim, 2016) could be of relevance. One other aspect that needs to be considered is that in this study and most of the non‐clinical studies with BIA 10‐2474, the rats were relatively young, although at the end of the 26‐week toxicology study, they would correspond to 25‐ to 30‐year‐old humans according to Sengupta (2013) and no pathology of the type seen in the clinic was observed. The subjects affected by toxicity of BIA 10‐2474 in the clinical trial were aged between 27 and 49 (Kerbrat et al., 2016) and so only slightly older than the rat‐equivalent age. Nevertheless, it will be useful in future studies to explore the effect of age on the sensitivity of the identified off‐targets to inhibition by mechanism‐based inhibitors such as BIA 10‐2474 as well as their rate of recovery.
The present study provides detailed information on the interaction of BIA 10‐2474 with fatty acid amide hydrolase and has explored potential off‐targets using traditional in vitro binding methods for non‐serine hydrolase targets and both in vitro and ex vivo activity‐based protein profiling for serine hydrolases. Although BIA 10‐2474 showed good selectivity in binding studies, the activity‐based protein profiling studies mostly confirmed the off‐targets already identified by van Esbroeck et al. (2017) and also identified several additional serine hydrolase off‐targets. Our data suggest that most of these interactions would have occurred at the doses used in preclinical toxicology studies. However, without further clinical information, such as the autopsy results and measured CNS exposure, it is not possible to determine if these off‐targets can be associated with the toxicity observed in the clinical trial with BIA 10‐2474. At present, the available data do not allow conclusions to be made about the specific mechanisms that could have caused the clinical trial accident.
AUTHOR CONTRIBUTIONS
M‐.J.B., F.S., and C.A. were responsible for planning and carrying out all the in vitro and in vivo assays evaluating fatty acid amide hydrolase activity and organizing external work with Eurofins. A.L. and C.F‐.L. carried out all PK assays, cell penetration studies, bioanalysis of FAA levels, and organizing external work with Solvo. P.N.P. was responsible for the SHRSP study. P.N.P. conceived and together with C.A. carried out data analysis for studies of kinetic constants. P.M. was involved in planning and conception of the studies and wrote the main manuscript text together with M‐.J.B. P.S‐.d‐.S. initiated the project and had overall responsibility for all studies reported here.
CONFLICT OF INTEREST
All authors were employed by Bial—Portela & Ca., S.A., during the work they performed in this manuscript and during its preparation.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://bpspubs.onlinelibrary.wiley.com/doi/abs/10.1111/bph.14207, and https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14206, and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
Supporting information
Table S1. Concentrations in nM of BIA 10‐2474, PF‐04457845 and JNJ‐42165279 in culture medium, supernatant and cell lysates after 1, 3 or 24 h incubation of COS‐hFAAH cells after application of 100 nM of test substance. The limit of quantification was 5 nM. All values are means ± SD (n = 5 except for 0 h that are pooled from the three incubation times and correspond to n = 15).
Table S2 Regional brain levels of BIA 10‐2474 and of its primary metabolites in various brain regions at various time points after acute or 5 days repeated once‐daily oral administration of BIA 10‐2474 at 10 mg kg−1. Values are mean and SD for n = 6 except vehicle where n = 2. The limit of quantification for all analytes was 50 ng g−1: extrapolated values below this value were used in compiling this table to allow better comparison of acute and chronic treatments for the later time‐points but in these cases the absolute values may be less reliable than those within the limits of quantification. nd: not determined.
Table S3. Comparison of BIA 10‐2474 with PF‐04457845 and JNJ‐42165279 at 229 assays for receptors, enzymes, transporters, ion channels, hormones, growth factors and inflammatory mediators. All results from both CROs for BIA 10‐2474 were considered non‐significant (ie below 50%) and so where data was available from both Caliper and Eurofins for BIA 10‐2474, only the Eurofins data has been retained. In addition, Caliper data for non‐selective ligands has been removed when individual receptor data was available from Eurofins. All values above 50% have been marked in bold.
Table S4. Effect of compound treatment (BIA 10‐2474, 10 and 100 μM; PF‐04457845, 10 and 100 nM; JNJ‐42165279 1 and 10 μM) on rat brain serine hydrolases (membrane fraction or soluble fraction). Values shown are % reduction of FP‐probe labelled serine hydrolase as compared to DMSO vehicle. Shaded cells with values in bold indicate proteins harbouring a fold‐change < −2 and an adjusted p‐value <0.05 (Limma test and Benjamini‐Hochberg correction for multiple testing). The protein ID and prevalence columns indicate the number of unique sequences used for identification of the protein in each sub‐experiment and the log2 intensity of the protein by LC–MS/MS is given for the DMSO group during each experiment. No value indicates no observed decrease in fold‐change.
Table S5. Non‐serine hydrolase proteins exhibiting reduced azido‐FP probe labelling in rat brain membrane fraction or soluble fraction by BIA 10‐2474 (10 and 100 μM), PF‐04457845 (10 and 100 nM) and JNJ‐42165279 (1 and 10 μM). Values in bold are significantly different from the corresponding DMSO control (p < 0.05, Limma test with Benjamini‐Hochberg correction). Note that apart from BAIAP2 all these proteins had relatively low log2 intensities and were detected inconsistently between experiments. Furthermore, many of the effects are not typically dose‐dependent. These observations suggest that these results may not be reliable and are presented here for completeness. nd – indicates not detected.
Table S6. Non‐serine hydrolase proteins exhibiting reduced or enhanced azido‐FP probe labelling in brains of rats treated with BIA 10‐2474 (10, 30 or 100 mg.kg−1 PO) for 1 to 28 days. Values shown are fold change; significantly reduced or enhanced labelling in at least one condition are highlighted in bold. The experiment was analysed in three separate sub‐experiments, one for each of the dose levels. The protein ID and prevalence columns indicate the number of unique sequences used for identification of the protein in each sub‐experiment and the log2 intensity of the protein by LC–MS/MS is given for the control group during each sub‐experiment.
Table S7. List of lipid species annotated with a significant change between rat brains dosed with 100 mg.kg−1.day−1 during 28 days of exposure (D100) compared to non‐dosed rat brains as control (D000). BH: p‐values adjusted for multiple testing with the Benjamini & Hochberg method (Benjamini Y & Hochberg Y, 1995, Journal of the Royal Statistical Society. Series B (Methodological):289–300).
Table S8. Summary of conditions used for the transporter assays
Figure S1. In‐gel fluorescence for serine hydrolases in the membrane fraction (top panel) or soluble fraction (lower panel) from rat brains labelled with ActivX™ TAMRA‐FP Serine Hydrolase Probe (2 μM final concentration) for 1 h min at room temperature following 1 h exposure to FAAH inhibitors at the indicated concentrations (PMSF tested at 3 mM). Labeling was stopped with 4x SDS‐PAGE loading buffer and heating at 60°C for 20 min. Labeled protein samples (50 μg protein.well−1) were separated by SDS‐PAGE on 4–12% Criterion™ XT Bis‐TrisGels. After electrophoresis, gels bands were visualized by exposure to a green LED light. Gels were documented with a SyngeneG‐BOX ChemiXT4 imaging system with an emission filter BP600/20 nm. The red arrow indicates the band corresponding to the molecular weight of fatty acid amide hydrolase which is inhibited in a concentration dependent manner by all the fatty acid amide hydrolase inhibitors. The green arrow shows another band inhibited only by BIA 10‐2474. No other bands appear to be inhibited. These gels were used to determine the concentrations to be used for the competitive mass spectrometry based activity‐based protein profiling studies described in the paper.
ACKNOWLEDGEMENT
We are particularly grateful for the support of Luisa Dória in the interpretation of the lipidomic studies and contributions to the manuscript.
Bonifácio M‐J, Sousa F, Aires C, et al. Preclinical pharmacological evaluation of the fatty acid amide hydrolase inhibitor BIA 10‐2474. Br J Pharmacol. 2020;177:2123–2142. 10.1111/bph.14973
DATA AVAILABILITY STATEMENT
The data sets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- Alexander, S. P. H. , Keely, E. , Mathie, A. , Peter, J. A. , Veale, E. L. , Armstrong, J. H. , … GTP collaborators (2019). The Concise Guide to pharmacology 2019/2020: Introduction and other protein target. British Journal of Pharmacology, 176, S1–S20. 10.1111/bph.14747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber, C. E. , Li, A. , Houlden, H. , & Wray, S. (2016). Review: Insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation: Unifying theories. Neuropathology and Applied Neurobiology, 42, 220–241. 10.1111/nan.12242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachovchin, D. A. , & Cravatt, B. F. (2012). The pharmacological landscape and therapeutic potential of serine hydrolases. Nature Reviews. Drug Discovery, 11, 52–68. 10.1038/nrd3620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beliaev, A. , Ferreira Humberto, S. , Learmonth David, A. , Bonifácio Maria, J. , Torrão, L. , Pires Nuno, M. , … Kiss, L. E. (2016). Synthesis and structure–activity relationships of ionizable 1,3,4‐oxadiazol‐2(3H)‐ones as peripherally selective FAAH inhibitors with improved aqueous solubility. Pure and Applied Chemistry, 88, 341–347. 10.1515/pac-2016-0104 [DOI] [Google Scholar]
- Benjamini, Y. , & Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B (Methodological), 57, 289–300. [Google Scholar]
- Bolon, B. , Garman, R. H. , Pardo, I. D. , Jensen, K. , Sills, R. C. , Roulois, A. , … Gumprecht, L. (2013). STP position paper: Recommended practices for sampling and processing the nervous system (brain, spinal cord, nerve, and eye) during nonclinical general toxicity studies. Toxicologic Pathology, 41, 1028–1048. 10.1177/0192623312474865 [DOI] [PubMed] [Google Scholar]
- Copeland, R. A. (2005). Evaluation of enzyme inhibitors in drug discovery In A guide for medicinal chemists and pharmacologists. New Jersey: Wiley Interscience. [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018) Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175: 987‐993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temporary Specialist Scientific Committee (2016). Report by the Temporary Specialist Scientific Committee (TSSC): “FAAH (Fatty Acid Amide Hydrolase)”, on the causes of the accident during a Phase 1 clinical trial in Rennes in January 2016.
- van Esbroeck, A. C. M. , Janssen, A. P. A. , Cognetta, A. B. 3rd , Ogasawara, D. , Shpak, G. , van der Kroeg, M. , … Allarà, M. (2017). Activity‐based protein profiling reveals off‐target proteins of the FAAH inhibitor BIA 10‐2474. Science, 356, 1084–1087. 10.1126/science.aaf7497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, P. , Soyombo, A. A. , Shelton, J. M. , Wilkofsky, I. G. , Wisniewski, K. E. , Richardson, J. A. , & Hofmann, S. L. (2003). Disruption of PPT2 in mice causes an unusual lysosomal storage disorder with neurovisceral features. Proceedings of the National Academy of Sciences, 100, 12325–12330. 10.1073/pnas.2033229100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–d1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, S. B. , Hardisty, J. F. , Hayes, A. W. , & Weber, K. (2019). Developmental and reproductive toxicity studies of BIA 10‐2474. Regulatory Toxicology & Pharmacology. 10.1016/j.yrtph.2019.104543 [DOI] [PubMed] [Google Scholar]
- Hayes, A. W. , Hardisty, J. F. , Harris, S. B. , Okazaki, Y. , & Weber, K. (2019). Oral repeated‐dose toxicity studies of BIA 10‐2474 in Wistar rat (p. 104540). Regulatory Toxicology & Pharmacology; in press. 10.1016/j.yrtph.2019.104540 [DOI] [PubMed] [Google Scholar]
- Herzog, R. , Schwudke, D. , Schuhmann, K. , Sampaio, J. L. , Bornstein, S. R. , Schroeder, M. , & Shevchenko, A. (2011). A novel informatics concept for high‐throughput shotgun lipidomics based on the molecular fragmentation query language. Genome Biology, 12, R8 10.1186/gb-2011-12-1-r8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Z. , Ogasawara, D. , Seneviratne, U. I. , Cognetta, A. B. , am Ende, C. W. , Nason, D. M. , … Cravatt, B. F. (2019). Global portrait of protein targets of metabolites of the neurotoxic compound BIA 10‐2474. ACS Chemical Biology, 14, 192‐197. 10.1021/acschembio.8b01097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illingworth, M. A. , Meyer, E. , Chong, W. K. , Manzur, A. Y. , Carr, L. J. , Younis, R. , … Kurian, M. A. (2014). PLA2G6‐associated neurodegeneration (PLAN): Further expansion of the clinical, radiological and mutation spectrum associated with infantile and atypical childhood‐onset disease. Molecular Genetics and Metabolism, 112, 183–189. 10.1016/j.ymgme.2014.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, D. S. , Stiff, C. , Lazerwith, S. E. , Kesten, S. R. , Fay, L. K. , Morris, M. , … Ahn, K. (2011). Discovery of PF‐04457845: A highly potent, orally bioavailable, and selective urea FAAH inhibitor. ACS Medicinal Chemistry Letters, 2, 91–96. 10.1021/ml100190t [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammers, K. , Cole, R. N. , Tiengwe, C. , & Ruczinski, I. (2015). Detecting significant changes in protein abundance. EuPA Open Proteomics, 7, 11–19. 10.1016/j.euprot.2015.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith, J. M. , Jones, W. M. , Tichenor, M. , Liu, J. , Seierstad, M. , Palmer, J. A. , … Breitenbucher, J. G. (2015). Preclinical characterization of the FAAH inhibitor JNJ‐42165279. ACS Medicinal Chemistry Letters, 6, 1204–1208. 10.1021/acsmedchemlett.5b00353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerbrat, A. , Ferre, J. C. , Fillatre, P. , Ronziere, T. , Vannier, S. , Carsin‐Nicol, B. , … Edan, G. (2016). Acute neurologic disorder from an inhibitor of fatty acid amide hydrolase. The New England Journal of Medicine, 375, 1717–1725. 10.1056/NEJMoa1604221 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss, L. E. , Beliaev, A. , Ferreira, H. S. , Rosa, C. P. , Bonifacio, M. J. , Loureiro, A. I. , … Soares‐da‐Silva, P. (2018). Discovery of a potent, long‐acting, and CNS‐active inhibitor (BIA 10‐2474) of fatty acid amide hydrolase. ChemMedChem, 13, 2177–2188. 10.1002/cmdc.201800393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss, L. E. , Ferreira, H. S. , Beliaev, A. , Torrao, L. , Bonifacio, M. J. , & Learmonth, D. A. (2011). Design, synthesis, and structure–activity relationships of 1,3,4‐oxadiazol‐2(3H)‐ones as novel FAAH inhibitors. Med Chem Comm, 2, 889–894. 10.1039/c1md00136a [DOI] [Google Scholar]
- Li, G. L. , Winter, H. , Arends, R. , Jay, G. W. , Le, V. , Young, T. , & Huggins, J. P. (2012). Assessment of the pharmacology and tolerability of PF‐04457845, an irreversible inhibitor of fatty acid amide hydrolase‐1, in healthy subjects. British Journal of Clinical Pharmacology, 73, 706–716. 10.1111/j.1365-2125.2011.04137.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, J. Z. , & Cravatt, B. F. (2011). The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chemical Reviews, 111, 6022–6063. 10.1021/cr200075y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loureiro, A. I. , Fernandes‐Lopes, C. , Moser, P. , & Soares‐da‐Silva, P. (2018). Distribution, metabolism and elimination of BIA 10‐2474 in the rat. The FASEB Journal, 32(1_supplement), 833.3. [Google Scholar]
- Loureiro, A. I. , Lopes, C. , Bonifácio, M. J. , Moser, P. , & Soares‐da‐Silva, P. (2017). Pharmacokinetics and pharmacodynamics of BIA 10‐2474, a novel FAAH inhibitor, following oral administration in the cynomolgus monkey. pA2 online 16: 212P.
- Mateus, A. , Matsson, P. , & Artursson, P. (2013). Rapid measurement of intracellular unbound drug concentrations. Molecular Pharmaceutics, 10, 2467–2478. 10.1021/mp4000822 [DOI] [PubMed] [Google Scholar]
- McGrath, J.C. & Lilley, E. (2015), Implementing guidelines on reporting research using animals (ARRIVE etc.). Br J Pharmacol, 172: 3189‐3193. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinski, S. V. , Shahani, V. M. , MacKinnon, S. S. , Morayniss, L. D. , Laforet, M. , Woollard, G. , … Windemuth, A. (2017). Computational proteome‐wide screening predicts neurotoxic drug–protein interactome for the investigational analgesic BIA 10‐2474. Biochemical and Biophysical Research Communications, 483, 502–508. 10.1016/j.bbrc.2016.12.115 [DOI] [PubMed] [Google Scholar]
- Morton, D. B. , & Griffiths, P. H. (1985). Guidelines on the recognition of pain, distress and discomfort in experimental animals and an hypothesis for assessment. The Veterinary Record, 116, 431–436. 10.1136/vr.116.16.431 [DOI] [PubMed] [Google Scholar]
- Rocha, F. , Santos, A. , Chassard, D. , Patat, A. , Astruc, A. , Falcão, A. , et al. (2017). Tolerability, pharmacokinetic and pharmacodynamic profile of BIA 10‐2474 in healthy volunteers following multiple ascending doses. Proceedings of the British Pharmacological Society at http://wwwpA2onlineorg/abstracts/Vol16Issue1abst178P.
- Sampaio, J. L. , Gerl, M. J. , Klose, C. , Ejsing, C. S. , Beug, H. , Simons, K. , & Shevchenko, A. (2011). Membrane lipidome of an epithelial cell line. Proceedings of the National Academy of Sciences of the United States of America, 108, 1903–1907. 10.1073/pnas.1019267108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta, P. (2013). The laboratory rat: Relating its age with human's. International Journal of Preventive Medicine, 4, 624–630. [PMC free article] [PubMed] [Google Scholar]
- Smyth, G. K. (2004). Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology, 3, 1–25. [DOI] [PubMed] [Google Scholar]
- Stier, C. T. , Chander, P. , Gutstein, W. H. , Levine, S. , & Itskovitz, H. D. (1991). Therapeutic benefit of captopril in salt‐loaded stroke‐prone spontaneously hypertensive rats is independent of hypotensive effect. American Journal of Hypertension, 4, 680–687. 10.1093/ajh/4.8.680 [DOI] [PubMed] [Google Scholar]
- Synofzik, M. , Gonzalez, M. A. , Lourenco, C. M. , Coutelier, M. , Haack, T. B. , Rebelo, A. , … Züchner, S. (2014). PNPLA6 mutations cause Boucher–Neuhäuser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain: A Journal of Neurology, 137, 69–77. 10.1093/brain/awt326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, J. , Mizrahi, R. , Houle, S. , Kish, S. J. , Boileau, I. , Nobrega, J. , … Wilson, A. A. (2017). Inhibition of fatty acid amide hydrolase by BIA 10‐2474 in rat brain. Journal of Cerebral Blood Flow and Metabolism, 37, 3635–3639. 10.1177/0271678X16668890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topaloglu, A. K. , Lomniczi, A. , Kretzschmar, D. , Dissen, G. A. , Kotan, L. D. , McArdle, C. A. , … Ojeda, S. R. (2014). Loss‐of‐function mutations in PNPLA6 encoding neuropathy target esterase underlie pubertal failure and neurological deficits in Gordon Holmes syndrome. The Journal of Clinical Endocrinology & Metabolism, 99, E2067–E2075. 10.1210/jc.2014-1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viader, A. , Ogasawara, D. , Joslyn, C. M. , Sanchez‐Alavez, M. , Mori, S. , Nguyen, W. , … Cravatt, B. F. (2016). A chemical proteomic atlas of brain serine hydrolases identifies cell type‐specific pathways regulating neuroinflammation. eLife, 5, e12345 10.7554/eLife.12345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winrow, C. J. , Hemming, M. L. , Allen, D. M. , Quistad, G. B. , Casida, J. E. , & Barlow, C. (2003). Loss of neuropathy target esterase in mice links organophosphate exposure to hyperactivity. Nature Genetics, 33, 477–485. 10.1038/ng1131 [DOI] [PubMed] [Google Scholar]
- Wu, S. Y. , & Casida, J. E. (1996). Subacute neurotoxicity induced in mice by potent organophosphorus neuropathy target esterase inhibitors. Toxicology and Applied Pharmacology, 139, 195–202. 10.1006/taap.1996.0158 [DOI] [PubMed] [Google Scholar]
- Yang, Y. , Kimura‐Ohba, S. , Thompson, J. , & Rosenberg, G. A. (2016). Rodent models of vascular cognitive impairment. Translational Stroke Research, 7, 407–414. 10.1007/s12975-016-0486-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y. , Liu, Z. , Rothschild, K. J. , & Lim, M. J. (2016). Proteome‐wide drug screening using mass spectrometric imaging of bead‐arrays. Scientific Reports, 6, 26125 10.1038/srep26125 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Concentrations in nM of BIA 10‐2474, PF‐04457845 and JNJ‐42165279 in culture medium, supernatant and cell lysates after 1, 3 or 24 h incubation of COS‐hFAAH cells after application of 100 nM of test substance. The limit of quantification was 5 nM. All values are means ± SD (n = 5 except for 0 h that are pooled from the three incubation times and correspond to n = 15).
Table S2 Regional brain levels of BIA 10‐2474 and of its primary metabolites in various brain regions at various time points after acute or 5 days repeated once‐daily oral administration of BIA 10‐2474 at 10 mg kg−1. Values are mean and SD for n = 6 except vehicle where n = 2. The limit of quantification for all analytes was 50 ng g−1: extrapolated values below this value were used in compiling this table to allow better comparison of acute and chronic treatments for the later time‐points but in these cases the absolute values may be less reliable than those within the limits of quantification. nd: not determined.
Table S3. Comparison of BIA 10‐2474 with PF‐04457845 and JNJ‐42165279 at 229 assays for receptors, enzymes, transporters, ion channels, hormones, growth factors and inflammatory mediators. All results from both CROs for BIA 10‐2474 were considered non‐significant (ie below 50%) and so where data was available from both Caliper and Eurofins for BIA 10‐2474, only the Eurofins data has been retained. In addition, Caliper data for non‐selective ligands has been removed when individual receptor data was available from Eurofins. All values above 50% have been marked in bold.
Table S4. Effect of compound treatment (BIA 10‐2474, 10 and 100 μM; PF‐04457845, 10 and 100 nM; JNJ‐42165279 1 and 10 μM) on rat brain serine hydrolases (membrane fraction or soluble fraction). Values shown are % reduction of FP‐probe labelled serine hydrolase as compared to DMSO vehicle. Shaded cells with values in bold indicate proteins harbouring a fold‐change < −2 and an adjusted p‐value <0.05 (Limma test and Benjamini‐Hochberg correction for multiple testing). The protein ID and prevalence columns indicate the number of unique sequences used for identification of the protein in each sub‐experiment and the log2 intensity of the protein by LC–MS/MS is given for the DMSO group during each experiment. No value indicates no observed decrease in fold‐change.
Table S5. Non‐serine hydrolase proteins exhibiting reduced azido‐FP probe labelling in rat brain membrane fraction or soluble fraction by BIA 10‐2474 (10 and 100 μM), PF‐04457845 (10 and 100 nM) and JNJ‐42165279 (1 and 10 μM). Values in bold are significantly different from the corresponding DMSO control (p < 0.05, Limma test with Benjamini‐Hochberg correction). Note that apart from BAIAP2 all these proteins had relatively low log2 intensities and were detected inconsistently between experiments. Furthermore, many of the effects are not typically dose‐dependent. These observations suggest that these results may not be reliable and are presented here for completeness. nd – indicates not detected.
Table S6. Non‐serine hydrolase proteins exhibiting reduced or enhanced azido‐FP probe labelling in brains of rats treated with BIA 10‐2474 (10, 30 or 100 mg.kg−1 PO) for 1 to 28 days. Values shown are fold change; significantly reduced or enhanced labelling in at least one condition are highlighted in bold. The experiment was analysed in three separate sub‐experiments, one for each of the dose levels. The protein ID and prevalence columns indicate the number of unique sequences used for identification of the protein in each sub‐experiment and the log2 intensity of the protein by LC–MS/MS is given for the control group during each sub‐experiment.
Table S7. List of lipid species annotated with a significant change between rat brains dosed with 100 mg.kg−1.day−1 during 28 days of exposure (D100) compared to non‐dosed rat brains as control (D000). BH: p‐values adjusted for multiple testing with the Benjamini & Hochberg method (Benjamini Y & Hochberg Y, 1995, Journal of the Royal Statistical Society. Series B (Methodological):289–300).
Table S8. Summary of conditions used for the transporter assays
Figure S1. In‐gel fluorescence for serine hydrolases in the membrane fraction (top panel) or soluble fraction (lower panel) from rat brains labelled with ActivX™ TAMRA‐FP Serine Hydrolase Probe (2 μM final concentration) for 1 h min at room temperature following 1 h exposure to FAAH inhibitors at the indicated concentrations (PMSF tested at 3 mM). Labeling was stopped with 4x SDS‐PAGE loading buffer and heating at 60°C for 20 min. Labeled protein samples (50 μg protein.well−1) were separated by SDS‐PAGE on 4–12% Criterion™ XT Bis‐TrisGels. After electrophoresis, gels bands were visualized by exposure to a green LED light. Gels were documented with a SyngeneG‐BOX ChemiXT4 imaging system with an emission filter BP600/20 nm. The red arrow indicates the band corresponding to the molecular weight of fatty acid amide hydrolase which is inhibited in a concentration dependent manner by all the fatty acid amide hydrolase inhibitors. The green arrow shows another band inhibited only by BIA 10‐2474. No other bands appear to be inhibited. These gels were used to determine the concentrations to be used for the competitive mass spectrometry based activity‐based protein profiling studies described in the paper.
Data Availability Statement
The data sets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.