

Abstract
Pancreatic cancer is one of the most aggressive cancers with poor prognosis and a low 5-year survival rate. The family of P21-activated kinases (PAKs) appears to modulate many signaling pathways that contribute to pancreatic carcinogenesis. In this work, we demonstrated that PAK1 is a critical regulator in pancreatic cancer cell growth. PAK1-targeted inhibition is therefore a new potential therapeutic strategy for pancreatic cancer. Our small molecule screening identified a relatively specific PAK1-targeted inhibitor, CP734. Pharmacological and biochemical studies indicated that CP734 targets residue V342 of PAK1 to inhibit its ATPase activity. Further in vitro and in vivo studies elucidated that CP734 suppresses pancreatic tumor growth through depleting PAK1 kinase activity and its downstream signaling pathways. Little toxicity of CP734 was observed in murine models. Combined with gemcitabine or 5-fluorouracil, CP734 also showed synergistic effects on the anti-proliferation of pancreatic cancer cells. All these favorable results indicated that CP734 is a new potential therapeutic candidate for pancreatic cancer.
Key words: PAK1, Pancreatic cancer, Inhibitor, Structure-based virtual screening, Synergistic effect
Abbreviations: ALP, alkaline phosphatase; ALT, alanine aminotransferase; ANOVA, analysis of variance; AST, aspartate aminotransferase; BCL-2, B-cell lymphoma-2; BUN, blood urea nitrogen; CCK-8, cell counting kit-8; CDC42, cell division cycle 42; DMEM, Dulbecco's modified Eagle's medium; DMSO, dimethylsulfoxide; ERK, extracellular regulated protein kinase; Gem, gemcitabine; 5-FU, 5-fluorouracil; GEPIA, gene expression profiling interactive analysis; GTEx, genotype-tissue expression; HEK293, human embryonic kidney 293; HTVS, high-throughput virtual screening; IMEM, improved minimum essential medium; IP, immunoprecipitation; MEK, mitogen-activated protein kinase kinase; MEM, modified Eagle's medium; NSCLC, non-small cell lung cancer; OS, overall survival; OHP, oxaliplatin; PAK, P21-activated kinase; PARP, poly(ADP-ribose) polymerase; PAX, paclitaxel; PSCs, pancreatic stellate cells; PUMA, P53 upregulated modulator of apoptosis; PVDF, polyvinylidene fluoride; RAC1, Rac family small GTPase 1; RIPA, radio immunoprecipitation assay; RPMI1640, Roswell Park Memorial Institute 1640 medium; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; SP, standard precision; TCGA, The Cancer Genome Atlas; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; XP, extra precision
Graphical abstract
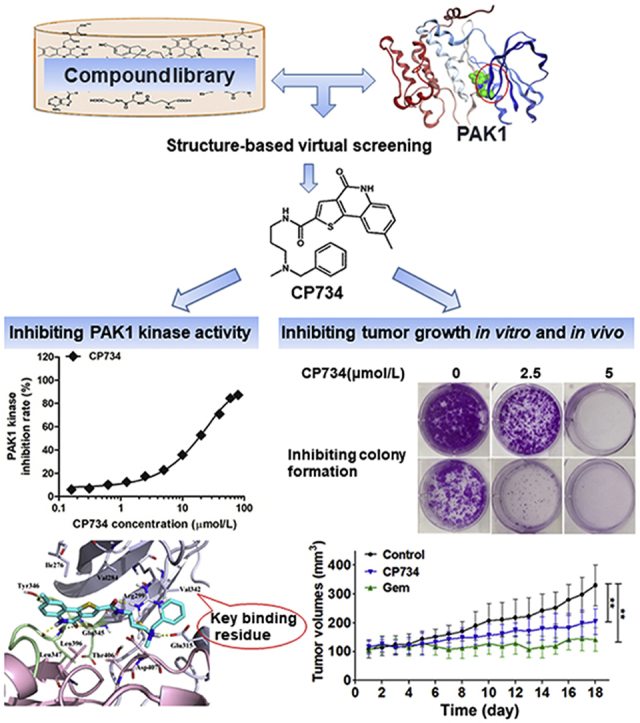
CP734 was identified as a relatively selective inhibitor of PAK1 by structure-based virtual screening. It significantly suppressed pancreatic cancer cell growth both in vitro and in xenograft models. This study will provide molecular insights into the development of novel PAK1 kinase inhibitors, which may benefit anti-cancer drug candidate development.
1. Introduction
Pancreatic cancer is the fourth leading cause of cancer-related deaths worldwide1. Although some advances were made recently in chemotherapeutics, the lack of early diagnosis and the limited number of effective therapeutic agents lead to a dismal 5-year survival rate of lower than 5%2,3. Thus, it is of great importance to seek new therapeutic agents for pancreatic cancer.
The P21-activated kinases (PAKs) belong to the non-receptor serine/threonine protein kinase family, and serve as important effector proteins for Rho GTPases, such as CDC42 and RAC14. PAKs are positioned at the intersection of several signaling pathways that mediate many different cellular processes, contributing to cancer development and progression5. The PAK family includes six isoforms that can be classified into two groups: group I (PAKs 1–3) and group II (PAKs 4–6)6. Of them, PAK1 and PAK4 are often up-regulated and/or hyper-activated in numerous malignant cell lines and various tumors7, 8, 9, 10. Previous studies showed that PAK1 plays an essential role in the proliferation of KRAS-driven colorectal carcinoma cells and also drives the transformation of KRAS-driven skin cancer11, 12, 13. Additionally, targeting PAK1 was shown to induce apoptosis in breast cancer cells and squamous NSCLCs cells14. Recent reports demonstrated that overexpression of PAK1 promotes the transforming activities of pancreatic cancer cells, while downregulation of PAK1 results in the lack of tumor formation in athymic mice15. Group I PAK inhibitor, FRAX597, improves the survival time in pancreatic cancer mouse model through suppressing pancreatic stellate cell activation16. Combination of PAK1 inhibitors with gemcitabine (Gem) delivers a synergistic inhibitory effect on pancreatic tumor growth17. Besides, elevated PAK1 activity confers Gem resistance in pancreatic cancer via modulation of multiple signaling crosstalks18. All these studies suggested that PAK1 could be a novel target for pancreatic cancer therapy.
To date, most of the reported PAKs inhibitors target pan-PAKs, which can be categorized into ATP-competitive inhibitors and allosteric inhibitors19, 20, 21, 22, 23, 24, 25. The poor druggability and selectivity limit their further development in preclinical/clinical trials. Herein, we identified a small molecule compound, CP734, as an ATP-competitive PAK1 inhibitor via structure-based virtual screening. Subsequently, we showed that CP734 blocked PAK1 activation not only in vitro but also in vivo, prohibiting the growth of pancreatic tumors. CP734 also showed synergistic effects in combination with Gem or 5-fluorouracil (5-FU) on the anti-proliferation of pancreatic cancer cells. These data supported the proposition that targeting PAK1 is a promising therapeutic strategy for pancreatic cancer and that CP734 has the potential application in managing tumorigenesis and progression of pancreatic cancer.
2. Materials and methods
2.1. Reagents
CP734 was purchased from Target Molecule Corp. (Boston, MA, USA); FRAX597 and G-5555 were from MedChemExpress (Shanghai, China); Z′-LYTETM biochemical assay kit from Invitrogen (Carlsbad, CA, USA); human recombinant protein kinase PAK1 from BPS Bioscience Inc. (San Diego, CA, United States); Primary antibodies against p-PAK1 (Lot. #2601), PAK1 (Lot. #2602), p-c-RAF (Lot. #9427), c-RAF (Lot. #9422), p-AKT: (Lot. #4060), AKT (Lot. #9272), cyclin D1 (Lot. #2922), c-MYC (Lot. #9402), p-P53 (Lot. #2521), P53 (Lot. #9282), P21 (Lot. #2946), PUMA (Lot. #4976), cyclin B1 (Lot. #4138), and β-actin (Lot. #3700) were purchased from Cell Signaling Technology (Danvers, MA, USA). The secondary antibodies were also from Cell Signaling Technology. All reagents for cell culture were purchased from Gibco (Thermofisher, Rockville, MA, USA).
2.2. Cell lines
The human pancreatic cancer cell lines (BxPC-3, PANC-1, CFPAC-1, and HPAF-II) were purchased from the Cell Bank of Shanghai Institute of Biochemistry and Cell Biology (Shanghai, China). The cell lines were authenticated by short-tandem repeat (STR) analysis. BxPC-3 was cultured in RPMI1640, PANC-1 in DMEM, CFPAC-1 in IMEM, and HPAF-II in MEM media supplemented with 10% FBS and 100 U penicillin/streptomycin.
2.3. Data mining
The mRNA level of PAK1 for pancreatic cancer was analyzed from the Gene Expression Profiling Interactive Analysis (GEPIA) online database (http://gepia.cancer-pku.cn/), a web-based server providing customizable functionalities based on The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression (GTEx) data26. Box plots with jitter was used for comparing the expression of PAK1 mRNA level in pancreatic cancer patients and the matched normal data.
2.4. Computational analysis of TCGA data
A TCGA dataset corresponding to 179 pancreatic cancer patients were obtained from the TCGA database. Patients were clustered into three groups according to PAK1 mRNA expression levels, namely high, medium and low PAK1 mRNA expression groups. Kaplan–Meier curves were plotted using the R software survival package, whereas the log-rank test and P value were calculated to determine significance.
2.5. Structure-based virtual screening of PAK1 inhibitors
Virtual screening of PAK1 inhibitors was performed using Glide in Schrodinger 2015-3 program package. The crystal structure of PAK1 (PDB ID: 4EQC) in complex with an ATP-competitive inhibitor was retrieved from the protein data bank. Hydrogen atoms were added by Maestro at pH 7.0. A restrained minimization was then performed with OPLS3 force field. The pocket for docking was defined around the original ligand at the active site for grid generation. The compound library of ChemDiv with more than 1.26 million diverse chemical structures was prepared in LigPrep panel using OPLS3 at pH 7.0 ± 2.0. Epik was adopted to generate ionization states. Three stages of virtual screening calculations (HTVS, SP and XP) were carried out, with increasing accuracy and computational cost. The retained ligands were analyzed and some of them were selected for experimental investigation.
2.6. Determination of enzymatic activity inhibition
The IC50 value of CP734 towards PAK1 activity was determined using the Z′-LYTE™ kinase assay kit Ser/Thr 19 peptide (Thermofisher). Briefly, PAK1 protein was premixed with 2 μmol/L of Z′-LYTE™ peptide substrate before adding the compound, after which they were thoroughly mixed and incubated at room temperature for 10 min. ATP was then added to initiate the reaction with a total volume of 10 μL. The kinase reaction was proceed for 1 h. Afterwards, the coumarin and fluorescein emission signals were measured by the Varioskan Flash (Thermofisher) microplate reader. The final kinase activity was calculated according to the kit manual.
2.7. Assessment of cell growth
Tumor cells were seeded in 96-well plates (5000 cells/well) the day before assays. A series of concentrations (between 0 and 40 μmol/L) of CP734 were added to the cells and incubated for 24 h. Cell growth was assessed by MTT assay in triplicate.
2.8. Colony formation assay
BxPC-3 or PANC-1 cells were pre-treated with dimethylsulfoxide (DMSO) or CP734 (10 or 20 μmol/L) for 24 h. Then, the cells were seeded in 6-well plates with a density of 1000 cells/well. Two weeks later, colonies were stained with 1% crystal violet and the colonies containing more than 50 cells were quantified.
2.9. Determination of intracellular enzyme activity
The intracellular PAK1 activity was determined by immunoprecipitation (IP) combined with Z′-LYTE™ kinase assay. BxPC-3 cells were incubated with indicated concentrations of CP734 for 6 h, then collected and lysed with cell lysis buffer. The lysate was pretreated with protein A/G agarose beads for 2 h, and 5 μg of anti-PAK1 antibody was added to immunoprecipitate PAK1. Finally, PAK1 protein was eluted with elution buffer (0.2 mmol/L glycine, pH 3.0), and used for kinase assay. The IP efficiency was confirmed by Western blot.
2.10. Cell cycle distribution
After treated with CP734 for 24 h, BxPC-3 or PANC-1 cells were collected and fixed in 75% ethanol overnight. Then, they were stained with 500 μL PI/RNase staining buffer. DNA content was analyzed using GuavaSoft 2.5 (Millipore, Billerica, CA, USA).
2.11. Western blot analysis
This assay was conducted as previously reported27. To extract proteins, pancreatic cancer cells treated with CP734 were lysed by RIPA (protease and phosphatase inhibitors added). Proteins with equal amounts were loaded on SDS-PAGE gel and separated by electrophoresis. Then the on-gel proteins were transferred to a PVDF membrane. The membranes were incubated with 5% non-fat milk for 1 h to block non-specific binding. Primary antibodies were added to probe at 4 °C overnight, followed by the addition of IRDye-conjugated secondary antibodies. Afterwards, images of the membranes were obtained by an Odyssey infrared fluorescent scanner (LI-COR Biosciences, Lincoln, NE, USA).
2.12. Cellular thermal shift assay
The cellular thermal shift assay was performed as previously reported28. HEK293 cells were first transfected with the Flag-tagged PAK1 expression plasmid. Twenty-four hours later, the transfected cells were incubated with DMSO or CP734 for 4 h. The cells were carefully collected and followed by heat treatment. A gradient of heat from 40 to 58 °C was implemented in Verti™ 96-well thermal cycler (Thermofisher). The remaining soluble PAK1 protein was detected by Western blot and the expression of each band was analyzed by ImageJ software(NIH, Bethesda, MD, USA). Then, data were normalized by setting the highest and lowest values in each set to 100% and 1%, respectively. Finally, the apparent Tagg values were obtained by fitting data to the Boltzmann Sigmoid equation with GraphPad Prism (La Jolla, CA, USA).
2.13. Animal models
All animal experiments were carried out following protocols approved by the Animal Care and Use Committee of Jiangsu Province, Academy of Traditional Chinese Medicine (Nanjing, China). Nude BALB/c mice (weight 13 ± 2 g) were raised in a pathogen-free and temperature-controlled environment. Subcutaneous injection of BxPC-3 cells (5 × 106) into the flank was then conducted to establish pancreatic carcinoma xenograft models. After identification of a palpable tumor (minimal volume of 100–150 mm3), mice were randomly divided into 3 groups (7 mice each). Vehicle and CP734 (10 mg/kg) groups were administered via intraperitoneal injection every day for 18 days. The mice of Gem (40 mg/kg) group were intraperitoneally injected twice a week. The dose of Gem was referred to the literature17. Body weights and tumor sizes for all mice were measured every 3 days. Calculations of tumor volumes were performed by Eq. (1):
| (1) |
where V represents the tumor volume, a stands for the minimum diameter of tumors, and b is the length of tumors perpendicular to a. After sacrificing all animals, the tumors were excised, weighed, snap-frozen in liquid nitrogen, and stored at −80 °C or fixed in 4% paraformaldehyde for further analyses.
Organ toxicity of CP734 was also evaluated. In brief, the aforementioned nude mice were treated with CP734 (10 mg/kg, once daily), Gem (40 mg/kg, twice a week), or vehicle control for 18 days (n = 7, per group). Afterwards, all animals were anesthetized by inhalation of isoflurane (3%). Blood samples were then collected from the retro-orbital plexus, and the main organs were excised, weighed, and fixed in 4% paraformaldehyde for further analysis. Complete blood counts were performed by Auto Hematology Analyzer BC-5380 (Mindray, Shenzhen, China), while plasma biochemical indices were analyzed by Cobas C311 (Roche Diagnostics, Basel, Switzerland).
2.14. Immunohistochemistry and TUNEL assay
Tumors or mouse tissues were fixed in 4% paraformaldehyde overnight, after which they were dehydrated and embedded in paraffin. Anti-Ki67 and anti-phospho-PAK1/PAK1 (CST) primary antibodies were employed for immunohistochemical assay. The One Step TUNEL Apoptosis Assay Kit (Beyotime, Shanghai, China) was used to assess the apoptosis in the tumor samples. The percentage of positive cells was determined by scanning the slides with an Aperio CS Scanscope scanner (Aperio, Vista, CA, USA) and quantified by Aperio's image viewer software (ImageScope, version 11.1.2.760, Aperio).
2.15. Drug combination studies
BxPC-3 cells were passaged at a density of 5000 cells/well in 96-well plates. Next day, cells were treated with the single or combining agents with fixed ratio simultaneously. After 48 h, cell viability was measure by MTT assay. Data were analyzed by CalcuSyn software (Biosoft, Cambridge, UK), using the Chou-Talalay method.
2.16. Statistical analysis
The difference between pairs of treatments was analyzed by unpaired Student t test using GraphPad Prism. Multiple treatment groups were compared with the vehicle control group via ANOVA assay. The intensity of the immune-reactive bands in Western blots was quantitated by ImageJ software. P < 0.05 was considered as statistically significant.
3. Results
3.1. PAK1 overexpression is associated with poor outcomes in patients with pancreatic cancer
To confirm the relationship between PAK1 and human pancreatic cancers, we analyzed PAK1 mRNA level in both tumor and normal tissues using the online server GEPIA, which is based on the database of TCGA. Compared with the normal tissue group, the PAK1 mRNA level is significantly up-regulated in human pancreatic cancer (*P < 0.05) (Fig. 1A). We next examined the correlation between the clinical prognoses of pancreatic cancer patients and the mRNA expression level of PAK1. Analysis of the TCGA dataset revealed a significant negative correlation between patients’ overall survival and PAK1 mRNA expression level (P = 0.015), highlighting the prognostic significance of PAK1 (Fig. 1B). These results suggested that PAK1 is dysregulated in pancreatic cancer and tightly related to its prognosis.
Figure 1.

PAK1 mRNA expression in patients with pancreatic cancer. PAK1 mRNA level (A) in human pancreatic cancer samples (n = 179) and normal tissues (n = 171). Data are represented as mean ± SD, *P < 0.05. (B) Kaplan–Meier analysis confirmed PAK1 mRNA expression level as an independent predictor of poor survival of pancreatic cancer. The above data were analyzed based on TCGA database.
3.2. PAK1 is a critical regulator in pancreatic cancer cell growth
To gain further insights into the role of PAK1 in the tumorigenic properties of pancreatic tumor cells, PAK1 was knocked down in BxPC3 and PANC-1 cells. The depletion efficiency of PAK1 was assessed by qPCR and Western blot (Fig. 2A and B). Compared with the nontarget shControl cells, the shPAK1-expressing cells showed an over 90% decrease in the PAK1 level. In order to disclose the possible link between PAK1 and the transforming activity of pancreatic cancer cells, we performed proliferation assays with the PAK1 knockdown models of BxPC3 and PANC-1 cells, respectively. It was observed that depleted expression of PAK1 exhibited decreased proliferation compared with the shControl in BxPC-3 cells (Fig. 2C). Data from shControl and shPAK1 expressed PANC-1 cells showed similar correlation. These results indicated that PAK1 is a critical regulator of cell growth in pancreatic cancer.
Figure 2.

Depletion of PAK1 decreased the proliferation of pancreatic cells. Knockdown of PAK1 in BxPC-3 and PANC-1 cells was confirmed by (A) Q-PCR and (B) Western blot. (C) PAK1 silencing retarded the proliferation of BxPC-3 and PANC-1 cells. shControl and shPAK1 transfected cells were subjected to MTT assay for 3 days after transfection. Data are represented as mean ± SD, n = 3. ***P < 0.001 compared with shControl cells.
3.3. Identification of CP734 as a potent inhibitor of PAK1
A structure-based virtual screening approach was adopted to discover PAK1 inhibitors. After three screening stages (HTVS, SP and XP), several candidate compounds from ChemDiv library were selected for enzyme inhibitory assay. Among them, CP734 was identified as a potent inhibitor of PAK1. The structure of CP734 was described according to the following nomenclature: N-[3-(benzyl-methylamino)propyl]-8-methyl-4-oxo-5H-thieno[4,5-c]quinoline-2-carboxamide (Fig. 3A). It inhibited PAK1 kinase activity in a dose-dependent manner, showing an IC50 value of 15.27 μmol/L in vitro (Fig. 3B). Yet it showed no significant inhibitory effect on PAK2, PAK3 or PAK6, as well as very weak inhibitory effects on PAK4 and PAK7 (also known as PAK5) with inhibition rates of 22% and 15% respectively at 100 μmol/L (Supporting Information Fig. S1). Further investigation demonstrated that CP734 stimulation obviously suppressed the intracellular PAK1 activity at 20 μmol/L in BxPC-3 cells (Fig. 3C and Supporting Information Fig. S2). As hyper-activation of PAK1 was closely linked to cell growth, we measured CP734's cytotoxic effect on pancreatic cancer cells. The results suggested that CP734 did retarded cell growth in both time and dose-dependent manners (Fig. 3D).
Figure 3.

Identification of CP734 as a potent PAK1-targeted inhibitor. (A) Schematic representation of hit discovery strategy. HTVS: high-throughput virtual screening mode; SP: standard-precision mode; XP: extra-precision mode. (B) In vitro inhibition profiles for CP734 against PAK1 in kinase reaction. (C) Inhibitory capacity of CP734 against intracellular PAK1 activity in BxPC-3 cells. (D) Cytotoxicity of CP734 towards BxPC-3 and PANC-1 cells. (E) The cellular thermal curve shift of PAK1 treated with CP734 (20 μmol/L). (F) Overall structure of PAK1 with CP734 bound in the ATP-binding site. The N-terminal lobe is shown in light gray, the hinge region is shown in light green, and the C-terminal lobe is shown in pink. (G) View of the PAK1 active site in complex with CP734. Residues that form the binding pocket are labeled. (H) Mutants V342F-PAK1 (M1) and V342K-PAK1 (M2) decreased the in vitro inhibition effect of CP734 compared with that of wide-type PAK1 (WT). Immunoprecipitation of the FLAG-tagged WT, M1 and M2 in HEK293 cells, and blotted with anti-FLAG and anti-PAK1 antibodies. (I) M1 and M2 attenuated the cell-growth inhibition activity of CP734 compared with that of WT. Overexpressed WT, M1 and M2 in BxPC-3 cells was detected by Western blot. All the data are represented as mean ± SD, n = 3. **P < 0.01, significantly different.
To further confirm CP734 as a PAK1-targeted inhibitor, we examined whether it binds with PAK1 protein directly by cellular thermal shift assay. CP734 showed a strong interaction with PAK1, illustrated by a shift of 2.85 °C in the apparent aggregation temperature (Tagg) of cellular PAK1 (Fig. 3E). As CP734 was initially identified with the ATP-binding pocket defined as the active site, we hypothesized that CP734 is an ATP-competitive inhibitor of PAK1. To unveil the structure basis for CP734 affinity towards PAK1, molecular docking was performed (Fig. 3F and G). The 4-oxo-5H-thieno[4,5-c]quinolone moiety of CP734 was docked in a similar position to the adenine base of ATP, probably dictating the orientation of the entire compound. The ring system is engaged in three hydrogen bonding interactions with the backbone of Glu345 and Leu347 at the hinge region. Meanwhile, the amide and ammonium groups form hydrogen bonds with Arg299 and Glu315 respectively at the N-lobe. CP734 is also involved in hydrophobic interactions with the hydrophobic pocket formed by Ile-276, Val-284, Val342 from the N-lobe, Tyr346, Leu347 from the hinge region and Leu396, Thr406 from the C-lobe.
To verify CP734 interacting with the ATP-binding pocket, a key amino acid Val342 in the pocket was mutated to Phe (V342F) or Lys (V342K). Mutant PAK1s maintained almost an equal kinase activity to wide-type PAK1 (Supporting Information Fig. S3). However, both the V342F and the V342K mutants were less sensitive to CP734 inhibition (Fig. 3H). Similarly, overexpression of V342F-PAK1 (M1) and V342K-PAK1 (M2) in BxPC-3 cells significantly abolished the cell-growth inhibitory activity of CP734 in comparison with that of wide-type PAK1 (Fig. 3I).
3.4. CP734 depleted downstream pathways of PAK1 in pancreatic cancer cells
PAK1 plays a vital role in the carcinogenensis of pancreatic cancer through regulating many important signaling pathways, such as RAS/ERK, PI3K/AKT, and WNT/β-catenin pathways29, 30, 31, 32. Therefore, CP734's effect on PAK1 and subsequent consequences were examined. As shown in Fig. 4A, the phosphorylation (T423) of PAK1 was evidently reduced by CP734 stimulation in a dose-dependent manner, followed by downregulation of phospho-c-RAF. Meanwhile, the inactivation of PAK1 decreased AKT phosphorylation and the expression of cyclin D1 and c-MYC (β-catenin pathway downstream proteins).
Figure 4.

CP734 depleted the downstream signaling pathways of PAK1 in pancreatic cancer cells. (A) CP734 inhibited PAK1 phosphorylation and its downstream signaling pathways. BxPC-3 and PANC-1 cells were incubated with CP734 for 24 h. The expression levels of the indicated proteins were examined by Western blot analysis. (B) CP734 influenced the expression of cell cycle related proteins modulated by PAK1. BxPC-3 and PANC-1 cells were incubated with CP734 for 24 h. The expression levels of the indicated proteins were examined by Western blot analysis. Data are representative of two independent experiments.
The hyper-phosphorylation of PAK1 regulates DNA damage signaling pathway and modulates cell cycle in cancer cells33, 34, 35. Here, CP734-inhibited PAK1 activity resulted in the activation of P53, upregulation of PUMA and P21, and reduction of cell cycle protein cyclin B1 (Fig. 4B), which contributed to the dysregulation of cell cycle. All these data suggested that CP734 stimulation inhibited PAK1 phosphorylation and subsequently regulated the downstream signaling pathways in pancreatic cells, ultimately impeding pancreatic cell growth.
3.5. CP734 inhibited the growth of pancreatic cancer cells
Four human pancreatic cancer cell lines, i.e., BxPC-3, PANC-1, CFPAC-1 and HPAF-II, were used to assess the anti-tumor potential of CP734. Firstly, PAK1 expression and activation was examined in these cells by Western blot assay. The results showed that PAK1 was expressed in all tested cells and especially hyper-activated in BxPC-3 and PANC-1 cells (Supporting Information Fig. S4), both of which have higher phosphorylation level at the central phosphorylation site T423 of PAK136. Next, the effect of CP734 on cell growth was assessed with two reported PAK1 inhibitors FRAX597 and G-5555 as controls. The results demonstrated that all the compounds suppressed the growth of the tested pancreatic cells dose-dependently (Fig. 5A). CP734 blocked the proliferation of BxPC-3, PANC-1, CFPAC-1 and HPAF-II cells with the IC50 values of 10.10, 16.84, 16.83 and 22.37 μmol/L, respectively. It exhibited a slightly stronger effect than G-5555 and weaker effect than FRAX597 on cell growth inhibition (Supporting Information Table S1). Subsequently, the cytotoxicity of these compounds towards normal cells was assessed by MTT assay. Unfortunately, all the compounds have cytotoxicity towards normal human pancreas duct cell hTERT-HPNE and normal human lung bronchus epithelial cell Beas-2B (Supporting Information Fig. S5).
Figure 5.

CP734 suppressed the growth of pancreatic cancer cells. (A) Anti-proliferation assay of CP734, FRAX597 and G-5555 for different human pancreatic cancer cells. (B) The percentage of cell cycle distribution for BxPC-3 and PANC-1 cells treated with indicated concentrations of CP734. (C) Representative plates of colony formation assay on: the response inhibition rate curve does not need to compare the P value, and the statistical analysis is not explained. BxPC-3 and PANC-1 cells treated with different concentrations of CP734. The values represent the means ± SD of triplicate experiments.
Futher results showed that CP734 caused G2/M cycle arrest in both BxPC-3 and PANC-1 cells in a dose-dependent manner (Fig. 5B). The colony formation activity was drastically reduced in CP734 treated cancer cells (Fig. 5C). Additionally, CP734 suppressed cell migration and caused apoptosis in pancreatic cancer cells (Supporting Information Figs. S6 and S7).
3.6. CP734 suppressed pancreatic cancer growth in murine model
To evaluate the in vivo antitumor efficacy of CP734, BxPC-3 cells were inoculated subcutaneously to establish xenograft models. Treatment with CP734 for 18 days had little effect on the body weight of the xenograft model mice, while the Gem treated group showed notable decline in the body weight (Fig. 6A). CP734 treatment led to considerable inhibition of tumor growth in BxPC-3 xenograft and lowered the tumor/body weight ratio compared with the control group (Fig. 6B). In situ TUNEL assays revealed an obvious increase in TUNEL-positive cells in CP734-treated xenografts (Fig. 6C). Moreover, the proportion of Ki67-positive nuclei was reduced to 38 ± 5% in CP734 group from 57 ± 15% in the control group (Fig. 6D), indicating that the anti-proliferative and apoptosis-inducing effects of CP734 in vivo were distinctive. Importantly, CP734 significantly suppressed the phosphorylation of PAK1 (control group: 42 ± 10%, CP734 group: 28 ± 7%, P < 0.01) in pancreatic tumors (Fig. 6D), corroborating our in vitro observations. Taken together, these results demonstrated that CP734 exhibited potent antitumor activity in pancreatic cancer mouse model.
Figure 6.

CP734 inhibited tumor growth in mouse xenograft model. (A) and (B) Mice implanted with BxPC-3 cells were administrated with CP734 or the vehicle control for 18 days. Body weight (A), tumor volume and tumor/body weight ratio (B) of CP734-treated or control-treated animals were monitored. Data are means ± SD relative to control group. **P < 0.01, *P < 0.05. (C) CP734 induced significant apoptosis in tumor xenograft model shown by in situ TUNEL assay. (D) Immunohistochemical analysis of Ki67, phospho-PAK1 and PAK1 were evaluated in xenograft tumor samples. Representative images are shown. Scale bar = 100 μm.
3.7. CP734 showed no obvious toxicity toward main organs in vivo
To assess the safety of CP734 in vivo, we determined its toxicity towards bone marrow, heart, liver, spleen and kidney in mice. All the blood cell indices were maintained within the normal ranges following CP734 treatment (Fig. 7A). Blood biochemical parameters (ALT, AST, ALP, BUN and creatinine) showed no significant changes after CP734 stimulation (Fig. 7B). The viscera weight indices suggested that CP734 had no significant toxicity toward main organs and Gem showed weak toxicity to the heart and liver (Fig. 7C). Finally, histopathologic evaluation of the main organs did not reveal any significant changes after CP734 stimulation (Fig. 7D). These data suggested that CP734 is safe at the treatment dose in vivo.
Figure 7.

CP734 shows little toxicity to main organs. Nude BALB/c mice (13 ± 2 g) were treated with CP734, Gem or vehicle control for 18 days (7 mice/group). All the animals were sacrificed after collecting blood samples from the retroorbital plexus. (A) Complete blood counts were done using AutoHematology Analyzer C-5380 (Mindray). Normal reference ranges for the tested parameters: WBC (2.6–12 × 103/mL), lymphocyte (1.3–9 × 103/mL), hemoglobin (10.1–16.1 g/dL), platelets (5.92–29.72 × 105/mL), and red blood cells (6.5–10.1 × 106/mL). (B) Results of plasma biochemical tests for ALT, AST, ALP, BUN, and creatinine in the control or drug-treated mice. (C) Viscera weight of all groups after mice sacrifice. The values represent the means ± SD of triplicate experiments. (D) H&E staining assay of main tissues after fixation.
3.8. Synergistic inhibitory effects of CP734 combined with chemotherapeutic drugs on the proliferation of pancreatic cancer cells
To extend the application of PAK1 inhibitors, we searched for combination drugs with CP734 against pancreatic cancer cells. Oxaliplatin (OHP), Gem, paclitaxel (PAX) and 5-FU are widely used chemotherapeutic drugs in cancer therapy. We measured the combined effect of CP734 with each of these drugs, respectively. As shown in Fig. 8, CP734 had strong synergistic effects with Gem (CI = 0.360) or 5-FU (CI = 0.420) and weak synergistic effect with OHP (CI = 0.810) against BxPC-3 pancreatic cancer cells. Yet it showed no synergistic effect with PAX (CI > 1). Therefore, the combination of CP734 with Gem or 5-FU imply a new strategy for pancreatic cancer therapy in the future.
Figure 8.

Synergistic effects of CP734 combined with chemotherapeutic drugs against pancreatic cancer cells. Anti-proliferation activity of CP734 combined with 5-FU (A), Gem (B), OHP (C) or PAX (D) against the BxPC-3 pancreatic cancer cells. Cells were treated with indicated doses of CP734, chemotherapeutic drugs, or their combinations for 48 h. Cell viability was measured by MTT assay. The values represent the means ± SD of triplicate experiments.
4. Discussion
Due to the poor prognosis, better biomarkers and potential druggable targets is in urgent need for pancreatic cancer. PAK1 is a group I PAK kinase which has been shown to be widely overexpressed and/or hyper-activated in a variety of human cancer tissues37. PAK1 regulates multiple complex signaling transduction networks that contribute to various cellular processes, e.g., cytoskeleton modeling, cell motility, survival and proliferation, and cell cycle progression38. To investigate the exact function of PAK1 in pancreatic cancer, we first sought the correlation between the expression levels of PAK1 and the clinical prognoses in pancreatic patients. The statistical analysis suggested that increased expression of PAK1 correlates to the poor outcome of patients with pancreatic cancer (Fig. 1B). Then we knocked down PAK1 expression in BxPC-3 and PANC-1 cells by shRNA and found that depletion of PAK1 decreased the proliferation of pancreatic cancer cells. Our results agree with the previous report17, illustrating a vital role of PAK1 in pancreatic cancer.
Subsequently, we turned to the discovery of small-molecule therapeutics targeting PAK1. Several types of PAK inhibitors have been reported, most of which are limited by the lack of target specificity. Due to the highly conservation of the ATP-binding sites in PAK family, it is not trivial to identify selective PAK1 inhibitors. In this study, we identified a relatively selective inhibitor of PAK1-CP734 through virtual screening, with no obvious inhibitory effect on the other PAK family members (Fig. 3 and Fig. S1). By molecular docking, we predicted that CP734 binds at the ATP-binding pocket of PAK1 (Fig. 3F and G). Mutagenesis of V342 in the ATP-binding pocket interfered with CP734's activity against PAK1, confirming that CP734 binds with PAK1 at the ATP-binding pocket and V342 may be one of the key sites for their interaction (Fig. 3H and I). Consequently, we have identified a new candidate compound CP734 which directly targets PAK1.
Although several small-molecular PAK1 inhibitors has been reported22, 23, 24, 25, their potential therapeutic efficacy on pancreatic cancer remains unclear. Herein, we evaluated the anti-tumor effect of CP734, demonstrating that it significantly inhibited the cell proliferation, attenuated cell cycle, and induced apoptosis of a panel of pancreatic cancer cells. We also revealed that CP734 suppressed cellular phosphorylation of PAK1, and disrupted downstream-signaling pathways, such as RAS–RAF signaling pathways. Moreover, CP734 increased the expression level of PUMA and P21 proteins, reduced cell cycle protein cyclin B1, and finally caused cell cycle arrest in G2/M phase. These results offered insights into the inhibitory molecular mechanisms of CP734 on pancreatic cancer cells.
Excitingly, CP734 also exhibited inspiring antitumor efficacy in human xenograft murine model. It suppressed the activity of PAK1 and significantly inhibited the tumor growth of BxPC-3 xenograft. Compared with the clinically used anti-pancreatic cancer drug Gem, CP734 showed little impact on the body weight and negligible toxicity to the crucial organs of the mice.
Although Gem remains a standard chemotherapeutic drug for the therapy of pancreatic cancer, combination medication with Gem to decrease chemotherapy-associated cytotoxicity and enhance therapeutic efficiency is desired. Previous studies have reported that PAK1 regulates NF-κB transcription, which plays a role in Gem resistance13,39. Therefore, PAK1 inhibition combined with Gem may achieve synergistic effect on the treatment of pancreatic cancer. In this study, we found that CP734 had strong synergistic effects with Gem or 5-FU on the anti-proliferation of BxPC-3 pancreatic cancer cells (Fig. 8), implying a new strategy for pancreatic cancer therapy in the future.
5. Conclusions
A small molecule compound CP734 was identified as a relatively selective inhibitor of PAK1. CP734 suppressed pancreatic cancer cell growth both in vitro and in xenograft models. Results from this study provide insights into the development of novel PAK1 kinase inhibitors, with CP734 being a potential anti-cancer drug candidate for further development.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81873057, 81973527), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (Integration of Chinese and Western Medicine) grant (China).
Author contributions
Peng Cao and Xiaoyan Sun designed research, analyzed data and supervised the project. Peng Cao, Jiao Chen, Xiaoyan Sun and Jiaqi Wang drafted the manuscript. Jiaqi Wang, Xiaoyan Sun, Lingxia Zhu and Yonghua Zhu conducted experiments and analyzed data. Jiao Chen screened the compound and did the molecular docking. Yuhan Yang performed the public data mining. Jiaqi Wang, Yonghua Zhu, Jiayu Zhao, and Chunping Hu conducted experiments. Rafael Rosell, Yang Yang and Xueting Cai analyzed data.
Conflicts of interest
The authors disclose no potential conflicts of interest.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2019.11.015.
Contributor Information
Xiaoyan Sun, Email: xiaoyande126@126.com.
Peng Cao, Email: cao_peng@njucm.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Suker M., Beumer B.R., Sadot E., Marthey L., Faris J.E., Mellon E.A. FOLFIRINOX for locally advanced pancreatic cancer: a systematic review and patient-level meta-analysis. Lancet Oncol. 2016;17:801–810. doi: 10.1016/S1470-2045(16)00172-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cao X., Hu Y., Luo S., Wang Y.J., Gong T., Sun X. Neutrophil-mimicking therapeutic nanoparticles for targeted chemotherapy of pancreatic carcinoma. Acta Pharm Sin B. 2019;9:575–589. doi: 10.1016/j.apsb.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Radu M., Semenova G., Kosoff R., Chernoff J. PAK signalling during the development and progression of cancer. Nat Rev Cancer. 2014;14:13–25. doi: 10.1038/nrc3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carroll S.L., Ratner N. How does the Schwann cell lineage form tumors in NF1?. GLIA. 2008;56:1590–1605. doi: 10.1002/glia.20776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Widemann B.C. Current status of sporadic and neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Curr Oncol Rep. 2009;11:322–328. doi: 10.1007/s11912-009-0045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yeo D., He H., Baldwin G., Nikfarjam M. The role of p21-activated kinases in pancreatic cancer. Pancreas. 2015;44:363–369. doi: 10.1097/MPA.0000000000000276. [DOI] [PubMed] [Google Scholar]
- 8.Rane C.K., Minden A. P21 activated kinase signaling in cancer. Semin Cancer Biol. 2019;54:40–49. doi: 10.1016/j.semcancer.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 9.Chen S., Auletta T., Dovirak O., Hutter C., Kuntz K., El-ftesi S. Copy number alterations in pancreatic cancer identify recurrent PAK4 amplification. Cancer Biol Ther. 2008;7:1793–1802. doi: 10.4161/cbt.7.11.6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kimmelman A.C., Hezel A.F., Aguirre A.J., Zheng H., Paik J.H., Ying H. Genomic alterations link Rho family of GTPases to the highly invasive phenotype of pancreas cancer. Proc Natl Acad Sci U S A. 2008;105:19372–19377. doi: 10.1073/pnas.0809966105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tabusa H., Brooks T., Massey A.J. Knockdown of PAK4 or PAK1 inhibits the proliferation of mutant KRAS colon cancer cells independently of RAF/MEK/ERK and PI3K/AKT signaling. Mol Cancer Res. 2013;11:109–121. doi: 10.1158/1541-7786.MCR-12-0466. [DOI] [PubMed] [Google Scholar]
- 12.Chow H.Y., Jubb A.M., Koch J.N., Jaffer Z.M., Stepanova D., Campbell D.A. p21-Activated kinase 1 is required for efficient tumor formation and progression in a Ras-mediated skin cancer model. Cancer Res. 2012;72:5966–5975. doi: 10.1158/0008-5472.CAN-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baker N.M., Yee Chow H., Chernoff J., Der C.J. Molecular pathways: targeting RAC-p21-activated serine-threonine kinase signaling in RAS-driven cancers. Clin Cancer Res. 2014;20:4740–4746. doi: 10.1158/1078-0432.CCR-13-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ong C.C., Jubb A.M., Haverty P.M., Zhou W., Tran V., Truong T. Targeting p21-activated kinase 1 (PAK1) to induce apoptosis of tumor cells. Proc Natl Acad Sci U S A. 2011;108:7177–7182. doi: 10.1073/pnas.1103350108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jagadeeshan S., Krishnamoorthy Y.R., Singhal M., Subramanian A., Mavuluri J., Lakshmi A. Transcriptional regulation of fibronectin by p21-activated kinase-1 modulates pancreatic tumorigenesis. Oncogene. 2015;34:455–464. doi: 10.1038/onc.2013.576. [DOI] [PubMed] [Google Scholar]
- 16.Yeo D., Phillips P., Baldwin G.S., He H., Nikfarjam M. Inhibition of group 1 p21-activated kinases suppresses pancreatic stellate cell activation and increases survival of mice with pancreatic cancer. Int J Cancer. 2017;140:2101–2111. doi: 10.1002/ijc.30615. [DOI] [PubMed] [Google Scholar]
- 17.Yeo D., He H., Patel O., Lowy A.M., Baldwin G.S., Nikfarjam M. FRAX597, a PAK1 inhibitor, synergistically reduces pancreatic cancer growth when combined with gemcitabine. BMC Canc. 2016;16:24. doi: 10.1186/s12885-016-2057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jagadeeshan S., Subramanian A., Tentu S., Beesetti S., Singhal M., Raghavan S. P21-activated kinase 1 (Pak1) signaling influences therapeutic outcome in pancreatic cancer. Ann Oncol. 2016;27:1546–1556. doi: 10.1093/annonc/mdw184. [DOI] [PubMed] [Google Scholar]
- 19.Rudolph J., Crawford J.J., Hoeflich K.P., Wang W. Inhibitors of p21-activated kinases (PAKs) J Med Chem. 2015;58:111–129. doi: 10.1021/jm501613q. [DOI] [PubMed] [Google Scholar]
- 20.Licciulli S., Maksimoska J., Zhou C., Troutman S., Kota S., Liu Q. FRAX597, a small molecule inhibitor of the p21-activated kinases, inhibits tumorigenesis of neurofibromatosis type 2 (NF2)-associated Schwannomas. J Biol Chem. 2013;288:29105–29114. doi: 10.1074/jbc.M113.510933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Viaud J., Peterson J.R. An allosteric kinase inhibitor binds the p21-activated kinase (Pak) autoregulatory domain covalently. Mol Cancer Ther. 2009;8:2559–2565. doi: 10.1158/1535-7163.MCT-09-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deacon S.W., Beeser A., Fukui J.A., Rennefahrt U.E., Myers C., Chernoff J. An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chem Biol. 2008;15:322–331. doi: 10.1016/j.chembiol.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim D.J., Choi C.K., Lee C.S., Park M.H., Tian X., Kim N.D. Small molecules that allosterically inhibit p21-activated kinase activity by binding to the regulatory p21-binding domain. Exp Mol Med. 2016;48 doi: 10.1038/emm.2016.13. e299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karpov A.S., Amiri P., Bellamacina C., Bellance M.H., Breitenstein W., Daniel D. Optimization of a dibenzodiazepine hit to a potent and selective allosteric PAK1 inhibitor. ACS Med Chem Lett. 2015;6:776–781. doi: 10.1021/acsmedchemlett.5b00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yi C., Maksimoska J., Marmorstein R., Kissil J.L. Development of small-molecule inhibitors of the group I p21-activated kinases, emerging therapeutic targets in cancer. Biochem Pharmacol. 2010;80:683–689. doi: 10.1016/j.bcp.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang Z., Li C., Kang B., Gao G., Li C., Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuliantie E., Dai X.C., Yang D., Crack P.J., Wang M.W. High-throughput screening for small molecule inhibitors of the type-I interferon signaling pathway. Acta Pharm Sin B. 2018;8:889–899. doi: 10.1016/j.apsb.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jafari R., Almqvist H., Axelsson H., Ignatushchenko M., Lundbäck T., Nordlund P. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc. 2014;9:2100–2122. doi: 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
- 29.Tonsgard J.H. Clinical manifestations and management of neurofibromatosis type 1. Semin Pediatr Neurol. 2006;13:2–7. doi: 10.1016/j.spen.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 30.Kolberg M., Høland M., Agesen T.H., Brekke H.R., Liestøl K., Hall K.S. Survival meta-analyses for 41,800 malignant peripheral nerve sheath tumor patients with and without neurofibromatosis type 1. Neuro Oncol. 2013;15:135–147. doi: 10.1093/neuonc/nos287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Packer R.J., Rosser T. Therapy for plexiform neurofibromas in children with neurofibromatosis 1: an overview. J Child Neurol. 2002;17:638–641. doi: 10.1177/088307380201700816. [DOI] [PubMed] [Google Scholar]
- 32.Carroll S.L. Molecular mechanisms promoting the pathogenesis of Schwann cell neoplasms. Acta Neuropathol. 2012;123:321–348. doi: 10.1007/s00401-011-0928-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y., Gu X., Li W., Zhang Q., Zhang C. PAK1 overexpression promotes cell proliferation in cutaneous T cell lymphoma via suppression of PUMA and p21. J Dermatol Sci. 2008;90:60–67. doi: 10.1016/j.jdermsci.2017.11.019. [DOI] [PubMed] [Google Scholar]
- 34.Ye D.Z., Jin S., Zhuo Y., Field J. p21-Activated kinase 1 (Pak1) phosphorylates BAD directly at serine 111 in vitro and indirectly through Raf-1 at serine 112. PLoS One. 2011;6 doi: 10.1371/journal.pone.0027637. e27637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Motwani M., Li D.Q., Horvath A., Kumar R. Identification of novel gene targets and functions of p21-activated kinase 1 during DNA damage by gene expression profiling. PLoS One. 2013;8 doi: 10.1371/journal.pone.0066585. e66585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zenke F.T., King C.C., Bohl B.P., Bokoch G.M. Identification of a central phosphorylation site in p21-activated kinase regulating auto-inhibition and kinase activity. J Biol Chem. 1999;274:32565–32573. doi: 10.1074/jbc.274.46.32565. [DOI] [PubMed] [Google Scholar]
- 37.Molli P.R., Li D.Q., Murray B.W., Rayala S.K., Kumar R. PAK signaling in oncogenesis. Oncogene. 2009;28:2545–2555. doi: 10.1038/onc.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dummler B., Ohshiro K., Kumar R., Field J. Pak protein kinases and their role in cancer. Cancer Metastasis Rev. 2009;28:51–63. doi: 10.1007/s10555-008-9168-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Sousa C.L., Monteiro G. Gemcitabine: metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur J Pharmacol. 2014;741:8–16. doi: 10.1016/j.ejphar.2014.07.041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.