

Abstract
Peroxiredoxins (Prdxs) are a family of small (22–27 kDa) nonseleno peroxidases currently known to possess six mammalian isoforms. Although their individual roles in cellular redox regulation and antioxidant protection are quite distinct, they all catalyze peroxide reduction of H2O2, organic hydroperoxides and peroxynitrite.1,2 They are found to be expressed ubiquitously and in high levels,3 suggesting that they are both an ancient and important enzyme family. Prdxs can be divided into three major subclasses: typical 2-cysteine (2-Cys) Prdxs (Prdx1–4), atypical 2-Cys Prdx (Prdx 5) and 1-Cys Prdx (Prdx 6). Recent evidence suggests that 2-Cys peroxiredoxins are more than “just simple peroxidases”. This hypothesis has been discussed elegantly in recent review articles, considering “over”-oxidation of the protonated thiolate peroxidatic cysteine and post-translational modification of Prdxs as processes initiating a mechanistic switch from peroxidase to chaperon function.4–6 The process of over-oxidation of the peroxidatic cysteine (CP) occurs during catalysis in the presence of thioredoxin (Trx), thus rendering the sulfenic moiety to sulfinic acid,7 which can be reduced by sulfiredoxin (Srx).8,9 However, further oxidation to sulfonic acid is believed to promote Prdx degradation or, as recently shown, the formation of oligomeric peroxidase-inactive chaperones10 with questionable H2O2-scavenging capacity. In the light of this and given that Prdx1 has recently been shown by us11 and by others12–17 to interact directly with signaling molecules, we will explore the possibility that H2O2 regulates signaling in the cell in a temporal and spatial fashion via oxidizing Prdx1. Therefore, this review will focus on H2O2 modulating cell signaling via Prdxs by discussing: (1) the activity of Prdxs towards H2O2; (2) sub cellular localization and availability of other peroxidases, such as catalase or glutathione peroxidases; (3) the availability of Prdxs reducing systems, such as thioredoxin and sulfiredoxin and lastly, (4) Prdx1 interacting signaling molecules.
Keywords: oxidative stress, peroxiredoxins, cell signaling, phosphatases, peroxidases, transformation
Prdx1: “More than Just” a Peroxidase
Catalytic cycle.
All Prdxs share a conserved Cys residue, which corresponds to the N-terminal Cys51 (peroxidatic cysteine = CP) in mammalian Prdx1. The majority of peroxiredoxins (Prdxs 1–4) contain an additional conserved Cys residue in the C-terminal region that corresponds to Cys172 (resolving cysteine = CR) in mammalian Prdx1. The N-terminal Cys51 is oxidized by H2O2 to cysteine-sulfenic acid (Cys51-SOH) and reacts with Cys172-SH of the other subunit to produce an intermolecular disulfide (head to tail dimer), which can only be reduced by Trx, but not by GSH or glutaredoxin. Thus, the reducing equivalents stem from NADPH via thioredoxin reductase (TrxR) and Trx.18–20
Prdxs can become easily over-oxidized on their catalytically active cysteine from sulhydryl to sulfinic or sulfonic acid in vitro and in vivo.5,10 It has been postulated that this is due to the fact that during catalysis Cys51-SH exists as a thiolate anion (Cys51-S-), whereas the other cysteines (Cys17, Cys80 and Cys172) remain protonated at neutral pH. The thiolate Cys51 is very unstable and highly reactive with any accessible thiol to form either a disulfide or to further oxidize to sulfinic or sulfonic acid.21 Crystal structures of Prdx1, Prdx2 and their yeast or bacterial counterparts revealed that in reduced Prdxs, the sulfur atoms of the N- and C-terminal conserved cysteine residues are too far apart to react with each other. Thus, disulfide formation requires significant conformational changes, such as unwinding of the active site N-terminal helix and the movement of four loops. Surprisingly, Cys51 in crystallized Prdx2, isolated from aged erythrocytes, exists as a sulfinic moiety, buried within the active-site pocket with Cys171 partially exposed. There, sulfinic Cys51 forms a salt bridge with Arg127, similarly to as reduced Cys51 does.22 Based on this, it has been proposed that in a catalytic cycle, peroxidation of the catalytic cysteine in Prdx2 occurs in the fully folded active site, where the CP (Cys50 in Prdx2) is stabilized as a thiolate anion (Cys50-S-) by Thr52 and the opposing Arg127; Pro51 shield the active site from water. This active site environment lowers the pKa value of Cys50 to the range of 5–6, making the thiolate anion nucleophilic to attack the terminal oxygen of the peroxyl bond (RO-OH), generating Cys50-SOH and H2O.23 Cys50-SOH in turn, builds a disulfide bridge with the sulfhydryl group from the Cys171-SH concurrently releasing H2O. Lastly, the Trx systems reduces the disulfide bond by thiol-disulfide exchange to regenerate free thiols in Cys50 and Cys171.24
What causes the additional or “over”-oxidation of CP-SOH? It has been hypothesized that CP-SOH is also protected in the active-site pocket and similarly shielded from further oxidation residing in the active site pocket as the sulfinic Cys51 does by forming a salt bridge with Arg127.22 Yang at al. found that over-oxidation of Cys51 occurs during catalysis and correlates with increasing amounts of Trx.7 Thus, one could speculate that the binding of Trx to Prdx1-disulfides reduces Cys51-SOH, but then renders reduced Cys51-SH to de-stabilization and over-oxidation in the presence of H2O2, by inhibiting the sequestration of reduced Cys51 in the active-site pocket. Along those lines, Yang et al. further demonstrated that the rate of Prdx1 inactivation by H2O2 was increased in the range of 0.1–1 mM, despite an observed KM for H2O2 <20 μM. These observations indicate that the initial oxidation of Cys51-SH to sulfenic acid is achieved by H2O2 attracted to the active-site pocket with an affinity constant of <20 μM, but all subsequent oxidation to Cys-SO2H depends on increased collision with more H2O2 molecules.7 The above data illustrate one of the major differences between the peroxidase function of Prdxs and catalase: catalase decomposes H2O2 following an exponential decay, since its rate of H2O2 decomposition depends linearly on H2O2 concentration.25 This suggests that under high amounts of H2O2, catalase decomposes H2O2 fast and efficiently, whereas Prdxs become readily over-oxidized in conditions of high H2O2. Under conditions of low cellular H2O2 levels, Prdxs scavenge H2O2 more efficiently than catalase due to their high affinity towards H2O2. Therefore, one could propose that depending on the cellular H2O2 load, Prdxs and catalase function rather sequentially than synergistically as peroxidases (Fig. 1). When focusing on the efficiency of H2O2 elimination, Prdxs may seem limited as peroxidases compared to catalase. However, this limitation offers an efficient switch panel for controlling signaling via H2O2, since under increasing H2O2-stress Prdx1 exhibits less complex formation with signaling partners, including the kinases c-Abl13,26 and JNK14 and the phosphatase PTEN11 probably due to its own over-oxidation. Such loss of complex formation results in activation of c-Abl13,26 and JNK14 and inactivation of PTEN’s phosphatase activity.11
Figure 1.
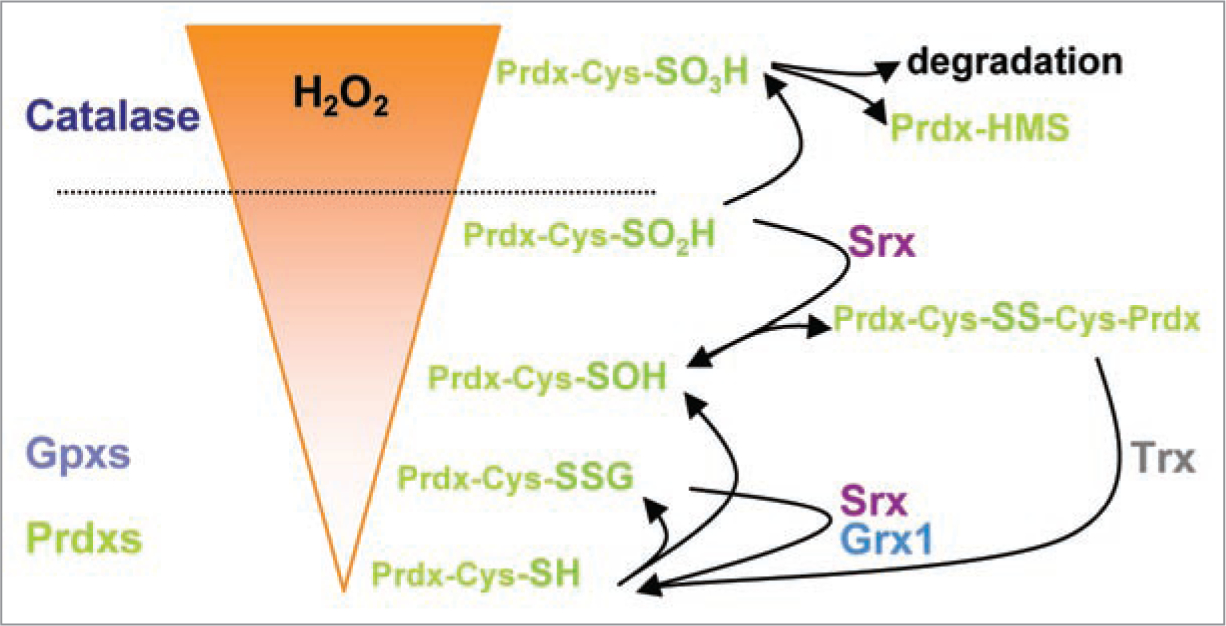
Cellular H2O2 levels are controlled sequentially by peroxidases. Prdxs and Gpxs scavenge smaller amounts of H2O2 whereas catalase catalyses higher amounts. Small amounts of H2O2 result in glutathionylation of Cys-thiols 51, 82 and 172 in Prdx1, which can be deglutathionylated by Srx and Grx1. The disulfide structure comprising sulfenic Prdx1 Cys51 and Cys172 from another Prdx1 protein can be reduced by Trx, Trx-reductase and NADPH. Further elevation of H2O2 promotes oxidation of Prdx1 Cys51 sulfenic acid to sulfinic acid. This process is reversible through an ATP and Mg2+ dependent reduction reaction induced by Srx. Oxidation of sulfinic Prdx1 Cys51 to sulfonic acid however is not reversible. Such over-oxidized Prdx1 proteins tend to form decamers with questionable peroxidase activity, but protein chaperone function. Such functional switch from peroxidase to chaperon elevates in turn cellular H2O2, which is then scavenged by catalase. Compared to Prdxs, catalase is not readily over-oxidized and decomposes H2O2 following an exponential decay, since its rate of H2O2 decomposition depends linearly on H2O2 concentration.
Regulation of 2-Cys Prdxs over-oxidation.
Recent studies have discovered that Prdxs have an additional function when over-oxidized, acting as molecular chaperones (reviewed most recently by Barranco-Medina et al.5). As discussed above, over-oxidation of 2-Cys Prdxs occurs only during catalysis in the presence of Trx, Trx-reductase and NADPH. It was previously believed that additional oxidation of CP-SOH initiated degradation of the Prdxs, since it was not known if a system reducing the Cys-sulfinic or the sulfonic moiety exists. However, studies measuring the protein half life of sulfinylated Prdx1 and Prdx2 indicated reversibility of the sulfinic protein product,27 suggesting a sulfinic acid reduction system may exist. Indeed, Biteau et al. identified in yeast a protein Srx, which reduced the sulfinic CP of 2-Cys yeast Prdx (Tsa1) to CP-SOH in a process which requires ATP hydolysis, Mg2+ and thiol as an electron donor.28
We now know that Srx regulates the chaperone function of 2-Cys Prdxs for the following reason: the molecular chaperone function for 2-Cys Prdxs is supposedly independent of its peroxidase activity and it correlates with the oligomerization status of the enzyme. Five homo-dimers of 2-Cys Prdxs form high molecular weight decamers, which can then further aggregate to form higher molecular complexes. It appears that a critical factor for determining the dimerdecamer equilibrium is the redox state of the catalytic cysteines, since the sulfinic enzymes favor decameric forms and the disulfide enzymes mainly prefer dimers. Under conditions of low H2O2 concentrations produced under normal cellular homeostasis, 2-Cys Prdxs form predominantly low-molecular weight oligomeric protein structures, which besides displaying peroxidase activity also protect proteins from degradation. However, following an unusual increase in H2O2, the 2-Cys Prdxs experience structural changes in which the low molecular weight complexes convert into high molecular complexes and act as chaperones.10,29 This provides the mechanistic basis for Srx to modulate H2O2-signaling in the cell by dissociating Prdxs high molecular weight complexes through reduction of Cys-sulfinic acid to Cys-sulfinic phosphoryl ester, which in turn becomes further reduced to a Cys-sulfenic acid by the Trx system.
H2O2 Modulates Cell Signaling through Oxidizing Prdx1
In order to better understand how H2O2 regulates signaling via Prdx1, we have to examine several aspects of H2O2 scavenging in the cell, by considering peroxidase activities of other peroxidases, localization of such peroxidases and availability of electron donors.
How Prdxs compare to other peroxidases in activity and localization.
Based on existing data, a direct biophysical and chemical comparison of H2O2 decomposition of each of the most common peroxidases (catalase, glutathione peroxidase (Gpx) and Prdx) in one study has not been done. So far each enzyme has been studied in individual studies making a direct comparison of Michaelis Menten kinetics difficult. However, reviewing the literature suggests that Prdx2 in erythrocytes possess a high affinity towards H2O2 since its oxidation occurred even in the presence of catalase, although its reaction rate with H2O2 was found comparable to catalase’s: 1.3 × 107 M−1 s−1. Catalase does not follow the Michaelis Menten constant since it decomposes H2O2 following an exponential decay, where its rate of H2O2 decomposition depends linearly on H2O2 concentration.25 This suggests then that under high amounts of H2O2, catalase decomposes H2O2 fast and efficiently, whereas in conditions of low H2O2 Prdxs may scavenge H2O2 more efficiently due to their higher affinity for H2O2. Next to catalase and glutathione peroxidase, Prdx1 and 2 are the third most abundant antioxidant proteins in erythrocytes and play a crucial role in erythrocytic H2O2 removal, since we and others have shown, in contrast to catalase or glutathione peroxidase,30,31 loss of Prdx1 or 2 resulted in hemolytic anemia in mice.32,33 While one study suggested that Prdx2 is easily over-oxidized in those cells when exposed to excess H2O2,25 another study found that erythrocytic Prdx2 appears mostly in a dimeric state. This was supported by the finding that TrxR activity is rather slow in red blood cells, compared to Jurkat cells where Prdx2 is easily found over-oxidized (sulfinic acid). The authors therefore suggested that Prdx2 is protected from over-oxidation since most of the protein is in a dimeric state and 2-Cys Prdxs function as non-catalytic scavengers of H2O2 in red blood cells.34 These data exemplify how cell signaling is modulated by H2O2 through the availability of Prdxs scavenging systems. Thus when Prdxs sulfenic acids are not properly reduced Prdxs over-oxidation with a functional switch from peroxidases to chaperones is promoted.
Similar to glutathione peroxidases, Prdxs are present in all subcellular compartments, however reports on the localization of Prdxs in the subcellular compartments are inconsistent, which is most likely due to the difference of species, tissues, cells and methods used in the studies. Electron micrographs of rat hepatocytes showed that Prdx1 is not only localized to the cytoplasm and nucleus, but also to the matrixes of mitochondria and peroxisomes. This is an interesting finding, since Prdx1 seems amongst the Prdx-family to possess the widest cellular distribution and also, to display the highest abundance in various tissues.35 Glutathione peroxidases are ubiquitiously expressed with high levels in erythrocytes, liver and kidney, in which the enzyme is found in the mitochondria and the cytosol.36 Catalase on the other hand is mainly found in the peroxisomes (together with Prdx1, GPX, MnSOD, Cu, Zn SOD, and other ROS decomposing enzymes)37 whereas in transgenic catalase-overexpressing mice, it could also be found extraperoxisomal in the cytosol and nucleus but not in the mitochondria.38 Taken together, these findings demonstrate a clear difference in cellular localization of the main peroxidases in the cell. Important to point out is that cytosolic H2O2 can only be scavenged by catalase after diffusing into the peroxisomes. This may also explain that H2O2 removal is a sequential event as mentioned above. Growth factor induced signaling increases H2O2 levels in the cell via mitochondrial and NADPH oxidase activity. Superoxide induced by growth factor signaling is either spontaneously or enzymatically dismutated to oxygen and H2O2. Such spontaneous rise in H2O2 is presumably low and localized if the stimulation is short and is controlled by peroxiredoxins and glutathione peroxidases. If however, H2O2 production continues due to prolonged receptor signaling, as found for example in cancer cells, Prdxs may shift to chaperone function and catalase activity scavenges more of the cellular H2O2 since H2O2 diffuses then into the peroxisomes. In general, catalase peroxidase activity probably works best under circumstances of higher cellular H2O2, as found in cancer cells or after acute high cellular exposure to H2O2.
Which factors modulate H2O2-induced signaling?
All three main peroxidases depend functionally on NADPH while glutathione peroxidase and Prdxs additionally require reductases and a suitable oxidant. Reduction of Trx by TrxR is needed in order to reduce disulfide-bridged Prdxs and glutathione reductase requires NADPH to reduced oxidized glutathione. Catalase utilizes NADPH differently by binding to it and thereby stabilizing catalase’s compound II. This in turn protects the enzyme from oxidation-induced inactivation,39 confirming that cellular catalase activity towards H2O2 correlates with intracellular concentrations of NADPH.40 Oxidation of glucose is the main source to generate either mitochondrial NADPH, as a product of the citric acid cycle in the mitochondrial matrix, or cytosolic and nuclear NADPH, as a product of the pentose phosphate pathway. Therefore, glucose availability is an important resource used to protect cells against H2O2-insult. This has been confirmed by experiments showing that glucose deprivation reduces the GSH/GSSG ratio through a decrease of NADPH levels.41
Several studies suggest that subcellular localization of reductant proteins influences cellular redox. For example, decreased protein expression of Trx, primarily in an oxidized, nuclear form, is found in exponentially growing cell cultures, compared to confluent quiescent cells in which Trx is primarily reduced and cytoplasmic.42 In addition, exponentially growing cells have higher overall H2O2-content and increased levels of GSSG. Along those lines, in proliferating cells (S + G2/M phase) glutathione was primarily found nuclear, whereas in resting cells (G1/G0 phase) it was found mostly cytoplasmic.43 Noh et al. have recently demonstrated that the Prdx sulfinyl reductase Srx translocates from the cytosol to the mitochondria in response to oxidative stress to reduce sulfinic acid of Prdx3,44 thereby most likely leaving part of the cytosolic sulfinic Prdxs in a sulfinic state and prone to further oxidation or formation of oligomeric structures.
Prdx1 was originally identified as a serum-induced gene that was three-fold overexpressed in Ras-transformed breast epithelial cells compared to non-transformed cells.45 It was subsequently found to have higher expression in S-phase of cells challenged with agents inducing ROS.46 Interestingly, phosphorylation of Prdx1 on Thr 90 by Cdc2 substantially decreases Prdx1 peroxidase activity, thereby elevating H2O2 towards the end of G2, allowing mitosis to occur.47 However at the same time, inactivation of Prdx1 presumably promotes H2O2-induced inactivation of the protein tyrosine phosphatases Cdc25.48 Oxidized Cdc25 phosphatases form intramolecular disulfides, which in turn are readily reduced by Trx.49 This emphasizes the interplay of H2O2 and antioxidants in normal cycling cells and appoints Prdx1 as an inhibitory regulator of the cell cycle. Since Ras transformed cells proliferate faster and have higher intracellular H2O2 levels due to induction of NADPH oxidases,50 the question arises to whether Prdx1 is functioning as a cell cycle promoter or inhibitor in cancer cells. Given all the above and considering that nuclear Prdx1 levels are increased in Ras-transformed fibroblasts compared to untransformed cells (Neumann CA, et al. unpublished data), it is plausible to conclude that in cancer cells, Prdx1 may be “immune” to Cdc2 induced phosphorylation due to conformational unavailability of Thr90, or that higher levels of Prdx1 expression may outcompete a possible inactivation by Cdc2. Moreover, the higher levels of ROS in cancer cells (recently reviewed by Trachootham et al.51) could result in Prdx oligomerization and chaperone activity. Since Cdc25 activity is preserved in cancer cells, it could be speculated that Cdc25 phosphatases are chaperoned by Prdxs in cancer cells. The exact biology of Prdxs chaperone function needs yet to be determined, not only in the context of cancer, where presumably cellular H2O2 is chronically elevated, but also in the context of an acute H2O2 insult. In cancer cells, elevated H2O2 is probably due to continuous signaling of either ligand binding-independent growth factor receptor signaling or mutations rendering kinases hyperactive or phosphatases inactive. The question is, if under such circumstances, Prdxs are mostly oligomerized and if such high molecular structures can then still be reduced by Srx, or if Srx itself is then mostly inactivated due to high H2O2 levels?
In normal cells mitogenesis did not cause hyperoxidation of either Prdx1 or Prdx2, whereas H2O2-dependent cell cycle arrest led to hyperoxidation of Prdx2 resulting in 66 and 140 Kda complexes, independent of the H2O2 dosage used. Alternatively, over-oxidation of Prdx1 was H2O2-dependent and was not detectable at higher concentrations of H2O2, but its formation of high oligomeric structures (around 600 KDa) was, suggesting a distinct difference of those two proteins, although they share a nearly 90% homology.52 Along those lines, Lee et al. demonstrated that the degree of H2O2-induced inactivation was higher for Prdx1 compared to Prdx2. However, replacing Cys83, which is only present in Prdx1 and not in Prdx2, with serine desensitized Prdx1 to H2O2-induced inactivation, abolished its chaperone function and supported the further finding that Cys83 is required to form disulfide structures in oligo-decameric structures at the dimerdimer interphase. This explains then why Prdx1 is mostly present as decamers while Prdx2 and Prdx1Cys83Ser are primarily found as dimers.53 If Prdx1 is more susceptible to over-oxidation, followed by a molecular switch to function as chaperone, the question arises if its reduction by Srx is slower or less efficient compared to Prdx2. Data from Chevallet and al. may have answered that question by showing that after over-oxidizing Prdx1 by using t-butylhydroperoxide treatment, Prdx1 recovered via retro-reduction far slower than Prdx2 did.54
Very recent evidence now adds a new layer of complexity to the regulation the peroxidase/chaperone function of Prdx1. Park and et al. showed that Srx catalyzes deglutathionylation of Prdx1. In this study Prdx1 was found glutathionylated on Cys 51, 83 and 172, only after small dosage of H2O2. The H2O2 dosages chosen were below the one inducing oligomerization. Interestingly, H2O2 dosages needed to induce glutathionylation varied between cell lines, correlating with the Trx levels expressed. Moreover, while Srx catalyzed the de-glutathionylation of Cys 83 and 172, glutaredoxin 1 (Grx1) catalyzed the de-glutathionylation of Cys 51, adding another level of control in Prdx1 function.44 These data suggest that regulation of Prdx1 activity is finely tuned and influenced by cellular H2O2 concentrations and the availability of its regulating enzymes such as Trx, Srx and Grx1 (summarized in Fig. 1).
How H2O2 and Prdx1 regulate cell signaling via interacting with signaling proteins.
Although over the past years all three major peroxidases have been found to interact with cellular signaling proteins, more binding partners have been identified for Prdx1 overall.11–17 More than 10 years ago, Prdx1 was identified by Wen et al. to regulate c-Abl tyrosine kinase activity by interacting with its SH-3 and kinase domains.13 Later, Prdx1 was also found to bind to JNK together with GSTpi, thereby suppressing radiation-induced JNK signaling.14 In both instances, it was evident that increased oxidative stress (H2O2 in the case of c-Abl and ionizing radiation in the case of JNK), inhibited these interactions and resulted in kinase activation. Although Kim and et al. speculated about a role of Prdx1 oxidation in regulating a loss of interaction with JNK, further evidence is still needed to understand exact details of the interaction, including stoichiometry and oxidation status of both binding partners. Prdx1, Gpx1,55 and catalase56 have also been found to associate directly with the c-Abl SH-3 domain. Since SH3 binding domains bind only one protein at a time, it would be interesting to know the biology determining the binding preference of c-Abl to Prdx1, Gpx1 or catalase. Of interest also is that Gpx1, like Prdx1, binds to c-Abl to inhibit its kinase activity and that this interaction is lost under H2O2-induced stress, resulting in activation of the c-Abl kinase.55 Catalase on the other hand, binds to c-Abl only under stress and is a substrate of c-Abl, whereas Prdx1 or Gpx are not. Phosphorylation of catalase by c-Abl has been shown on four of its tyrosine residues, which then either contribute to catalase activation56 or its degradation.57 Taken together, c-Abl activity seems well regulated by peroxidases, which reflects its important role in stress signaling in normal as well as in cancer cells.
An additional binding partner of Prdx1 is c-Myc.58 Prdx1 binds to the highly conserved Myc boxII, which is critically important for transformation and transcriptional activity. Prdx1-induced myc inhibition causes a broad but selective loss of c-Myc target gene regulation16 and Prdx1−/−MEFs show evidence of c-Myc activation.12 ASK-1, a kinase upstream of the JNK activating signaling cascade, interacts with Prdx1 after H2O2-induced stress.59 However, it is not clear from this study if such interaction is inhibiting ASK-1 activity. Considering all this, it becomes clear that Prdx1 is a promiscuous binding partner of stress signaling proteins, thereby regulating stress signaling cascades on multiple levels. An exciting prospect for future studies is the importance of the catalytic cysteines in Prdx1 in the regulatory process of modulating kinases activity given the differences in H2O2-induced binding or non-binding of Prdx1 with the kinases discussed above. We and others have taken first steps to investigate this problem in more detail. Park et al. showed that Prdx1 interacts with the androgen receptor (AR), enhancing its trans-activation. Interestingly, this process was independent of Prdx1 peroxidase activity suggesting that the AR is subject to Prdx1 chaperone function.15 The yeast protein Tsa1, corresponding to the human Prdx2, associates with ribosomes in an oxidation-dependent manner, since H2O2 treatment resulted in a loss of binding between Tsa1 WT, but not with an catalytically inactive Tsa1 (TsaC47S), suggesting that oxidation of Tsa1 results in (a) complex disruption of Tsa and the ribosomes and (b) a switch from the peroxidase to the chaperone function of Tsa1.60
Peroxides, are known to modify protein tyrosine phosphatases (PTPs) by oxidizing the catalytic cysteine in their active site. PTP catalytic property depends on a thiolate anion of a low pKa cysteine residue (pKa 4.7–5.4) located in the conserved motif of its active site.61,62 This highly nucleophilic group makes the initial attack on the phosphate group of the substrate, but renders the PTP extremely susceptible to oxidation, which results in its inactivation. The PTP and tumor suppressor PTEN exhibits phosphatase activity towards inositol and tyrosine phosphates and is known to be similarly inactivated through H2O2-mediated oxidation.63 A recent study showed that PTEN oxidation in stimulated macrophages resulted in the temporary inhibition of its phosphatase activity and in the downstream activation of Akt through its phosphorylation on Serine473.64 Analysis of human recombinant PTEN revealed that two of the five cysteines in its N-terminal phosphatase domain (Cys71 and Cys124) form a disulfide after oxidation, which resulted in the transient inhibition of its phosphatase activity.65 We have recently shown that Prdx1 associates with PTEN, protecting its lipid phosphatase from H2O2-induced inactivation.11 Our data demonstrated that Prdx1 most likely interacts with PTEN as a monomer, since adding Prdx1 to a PTEN lipid phosphatase reaction challenged by H2O2 preserved PTEN’s phosphatase activity fully when Prdx1 was added in a 1:1 ration and could not be further enhanced when additional Prdx was added. Treatment of cells with small amounts of H2O2 resulted in disruption of the complex and in oxidation of Prdx1. We think that Cys51 in Prdx1 regulates complex disruption for the following two reasons: (a) Testing the role of the catalytic Prdx1 cysteines showed that Prdx1C51S bound stronger to PTEN than Prdx1C172S or Prdx1C51,172S; (b) PTEN inhibited Prdx1 peroxidase activity in the presence of Trx, which may suggest that PTEN hinders Cys51-SH from folding back into the shielding pocket, thereby promoting its exposure to further oxidation and more importantly, the disruption of the Prdx1:PTEN complex. Yang et al. also demonstrated that the rate of Prdx1 inactivation by H2O2 was increased in the range of 0.1–1 mM, despite an observed KM for H2O2 <20 μM. These observations indicate that the initial oxidation of Cys51-SH to sulfenic acid is achieved by H2O2 attracted to the active-site pocket with an affinity constant <20 μM. All subsequent finding, since the amount of H2O2-inactivated PTEN in the presence of Prdx1 was independent of the H2O2 amount given (25 μM–500 μM H2O2).
Prdx1 is the first antioxidant protein reported protecting protein function from inactivation through interaction. Analysis of human recombinant PTEN revealed that two of the five cysteines in its N-terminal phosphatase domain (Cys71 and Cys124) form a disulfide bond after oxidation, which resulted in the transient inhibition of its phosphatase activity,63,65 which is essential for its tumor suppressive function.66,67 Heterozygous loss of germline PTEN, or temporal loss of somatic PTEN, in mice promotes tumor formation68,69 and is accompanied by enhanced cell proliferation, decreased cell sensitivity to apoptosis, and increased Akt kinase activity.69–71 Despite the resemblance of mouse PTEN deficiency with human Cowden’s disease,72 spontaneous forms of human breast cancer rarely exhibit loss of both PTEN alleles or other identifiable PTEN mutations.73 Since ROS are believed to be abnormally high in cancer cells,74 and present hyperactive Akt signaling,75 posttranslational modification such as oxidation may contribute to a loss of PTEN tumor suppressive function.72,73,76 It is known that PTEN negatively regulates Akt activity via dephosphorylating phosphatidylinositol (3,4,5) triphosphates (PIP3s), which are essential for the membrane recruitment and full activation of Akt.69,71 PTEN lipid phosphatase activity is at optimum after membrane binding via phosphatidylinositol (4,5) biphosphates (PIP2 s)77 and plays an important role in tumor suppression.66,67 Along those lines, Akt1 ablation protects (MMTV)-ErbB2/neu, and MMTV-v-H-Ras mice from breast cancer initiation.78,79 Lastly, loss of Prdx1 in H-Ras and ErbB-2 transformed PTEN positive MEFs increased transformation, whereas in transformed PTEN-negative MEFs it did not.11 This clearly demonstrated that Prdx1 tumor suppressive function is mainly achieved via PTEN regulation. Therefore it can be proposed that Prdx1 prevents Akt-driven transformation by protecting PTEN from oxidation-induced inactivation, since oxidation of Prdx1 dissociates the Prdx1:PTEN complex resulting in hyperactive Akt signaling and oncogenesis (Fig. 2).
Figure 2.
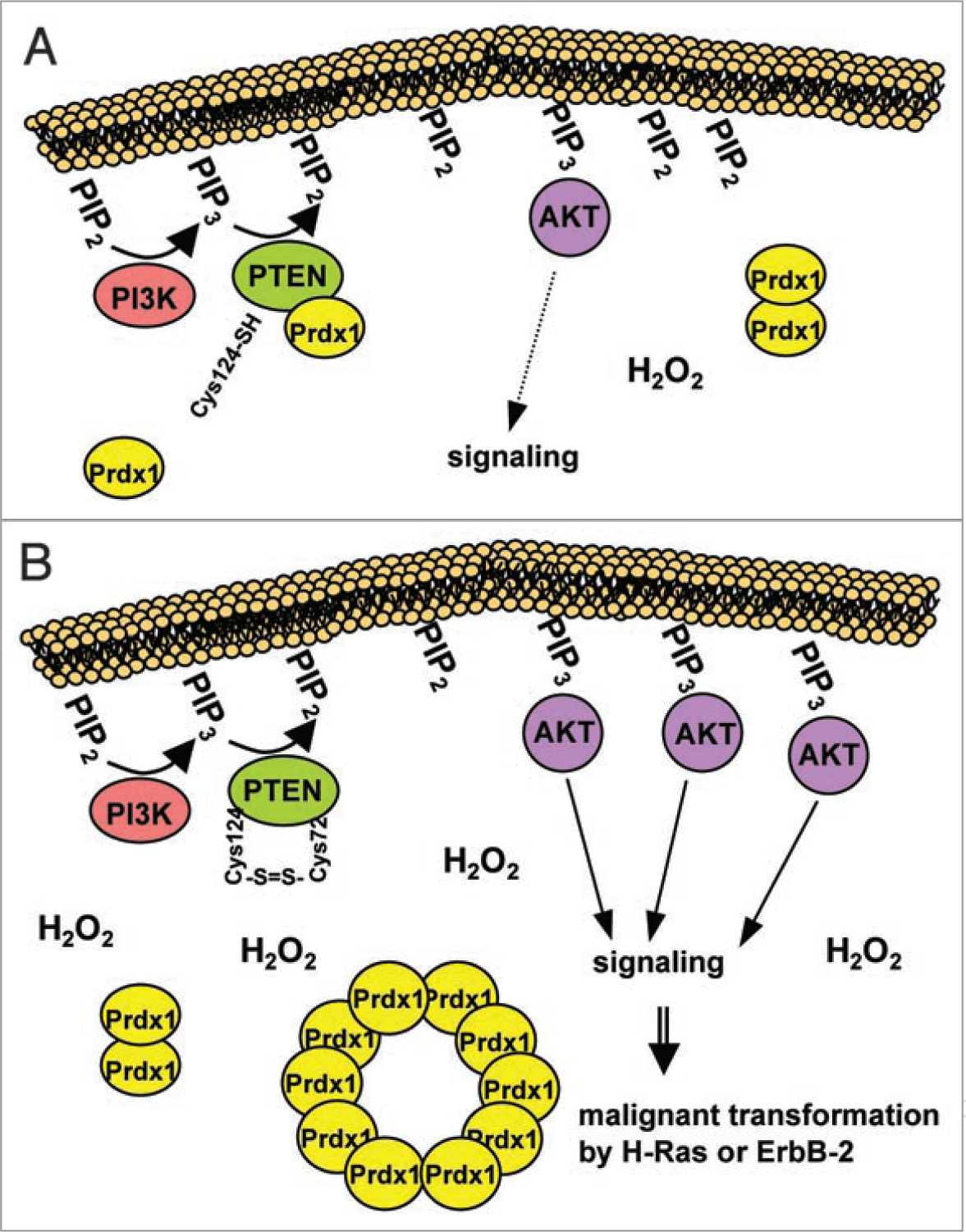
Prdx1 prevents Akt-driven tumorigenesis through protecting PTEN lipid phosphatase activity from oxidation-induced inactivation. (A) Prdx1 regulates PTEN phosphatase activity during oxidative stress, since binding of Prdx1 and PTEN occurs in conditions of mild or nil cellular stress. This constitutes a setting in which H2O2 is scavenged by Prdx1, which itself becomes in turn reversibly oxidized, in a controlled fashion. (B) However, under conditions of elevated oxidative stress, Prdxs are known to become irreversibly over-oxidized and dissociate from PTEN. Thereby, PTEN is inactivated by H2O2 resulting in hyperactivation of Akt. Hyperactive Akt then in turn can promote oncogenic signaling via ErbB-2- and Ras.
However, many questions still remain unanswered: for example, is Prdx1 reducing or preventing formation of the intramolecular disulfide build in PTEN following H2O2-induced stress? Or, is the interaction of Prdx1 and PTEN restricted to a certain cellular compartment?
Concluding Remarks
Over the recent years H2O2 has been recognized as a second messenger modifying cell signaling via protein oxidation.2 Yet, all aspects involved in regulating the production and elimination of H2O2 are still not fully understood. We acknowledge now that many different factors, such as sub cellular localization and expression levels of antioxidant systems can vary in different phase of the cell cycle and cell types, or that differences in individual peroxidase enzyme kinetics, as discussed here for catalase and Prdx1, can result rather in a sequential than synergistic H2O2-scavening. Since Prdxs are distributed throughout the cell and not like catalase only reduced to one cell organelle, they are capable to scavenge H2O2 where it is produced, for example, by NADPH oxidases and the mitochondria. Since NADPH oxidases are in close proximity to growth factor receptors and given that Prdxs have a high susceptibility to inactivation by oxidation, Prdxs function as “fine tuner” of cellular H2O2-signaling. By doing so, they dissociate from their binding partners, which in turn activates or inactivates the activity of the binding partner.
Acknowledgements
The authors wish to thank the Neumann lab and Drs. Yefim Manevich and Scott Eblen for fruit ful discussions. This work was supported by grants from the NIEHS-K22 ES012985, ACSIRG-97-219-05, Claudia Adams Barr Award-DFCI. All (C.A.N.) and Abney Research Foundation-MUSC (J.C.).
References
- 1.Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med 2005; 38:11–7. [DOI] [PubMed] [Google Scholar]
- 2.Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006; 312:1882–3. [DOI] [PubMed] [Google Scholar]
- 3.Wood ZA, Schroder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci 2003; 28:32–40. [DOI] [PubMed] [Google Scholar]
- 4.Hall A, Parsonage D, Horita D, Karplus PA, Poole LB, Barbar E. Redox-dependent dynamics of a dual thioredoxin fold protein: evolution of specialized folds. Biochemistry 2009; 48:5984–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barranco-Medina S, Lazaro JJ, Dietz KJ. The oligomeric conformation of peroxiredoxins links redox state to function. FEBS Lett 2009; 583:1809–16. [DOI] [PubMed] [Google Scholar]
- 6.Aran M, Ferrero DS, Pagano E, Wolosiuk RA. Typical 2-Cys peroxiredoxins—modulation by covalent transformations and noncovalent interactions. Febs J 2009; 276:2478–93. [DOI] [PubMed] [Google Scholar]
- 7.Yang KS, Kang SW, Woo HA, Hwang SC, Chae HZ, Kim K, et al. Inactivation of human peroxide 1 during catalysis as the result of the oxidation of the catalytic site to cysteine-sulfinic acid. J Biol Chem 2002; 277:38029–36. [DOI] [PubMed] [Google Scholar]
- 8.Woo HA, Jeong W, Chang TS, Park KJ, Park SJ, Yang JS, et al. Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-cys peroxiredoxins. J Biol Chem 2005; 280:3125–8. [DOI] [PubMed] [Google Scholar]
- 9.Chang TS, Jeong W, Woo HA, Lee SM, Park S, Rhee SG. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J Biol Chem 2004; 279:50994–1001. [DOI] [PubMed] [Google Scholar]
- 10.Lim JC, Choi HI, Park YS, Nam HW, Woo HA, Kwon KS, et al. Irreversible oxidation of the active-site cysteine of peroxiredoxin to cysteine sulfonic acid for enhanced molecular chaperone activity. J Biol Chem 2008; 283:28873–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao J, Schulte J, Knight A, Leslie NR, Zagozdzon A, Bronson R, et al. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Egler RA, Fernandes E, Rothermund K, Sereika S, de Souza-Pinto N, Jaruga P, et al. Regulation of reactive oxygen species, DNA damage, and c-Myc function by peroxiredoxin 1. Oncogene 2005; 24:8038–50. [DOI] [PubMed] [Google Scholar]
- 13.Wen S, VanEtten R. The Pag gene product, a stress-induced protein with antioxidant properties, is an Abl SH3-binding protein and a physiological inhibitor of c-Abl tyrosine kinase activity. Genes Dev 1997; 11:2456–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim YJ, Lee WS, Ip C, Chae HZ, Park EM, Park YM. Prx1 suppresses radiation-induced c-Jun NH2-terminal kinase signaling in lung cancer cells through interaction with the glutathione S-transferase Pi/c-Jun NH2-terminal kinase complex. Cancer Res 2006; 66:7136–42. [DOI] [PubMed] [Google Scholar]
- 15.Park SY, Yu X, Ip C, Mohler JL, Bogner PN, Park YM. Peroxiredoxin 1 interacts with androgen receptor and enhances its transactivation. Cancer Res 2007; 67:9294–303. [DOI] [PubMed] [Google Scholar]
- 16.Mu ZM, Yin XY, Prochownik EV. Pag, a putative tumor suppressor, interacts with the Myc Box II domain of c-Myc and selectively alters its biological function and target gene expression. J Biol Chem 2002; 277:43175–84. [DOI] [PubMed] [Google Scholar]
- 17.Chang R, Wang E. Mouse translation elongation factor eEF1A-2 interacts with Prdx-I to protect cells against apoptotic death induced by oxidative stress. J Cell Biochem 2007; 100:267–78. [DOI] [PubMed] [Google Scholar]
- 18.Chae HZ, Chung SJ, Rhee SG. Thioredoxin-dependent peroxide reductase from yeast. J Biol Chem 1994; 269:27670–8. [PubMed] [Google Scholar]
- 19.Chae HZ, Uhm TB, Rhee SG. Dimerization of a thiol specific antioxidant and the essential role of cystein 47. Proceedings National Academy of Sciences, USA 1994; 47:7022–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang SW, Chae HZ, Seo MS, Kim K, Baines IC, Rhee SG. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-alpa. J Biol Chem 1998; 273:6297–302. [DOI] [PubMed] [Google Scholar]
- 21.Claiborne A, Yeh JI, Mallett TC, Luba J, Crane EJ 3rd, Charrier V 3rd, et al. Protein-sulfenic acids: diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry 1999; 38:15407–16. [DOI] [PubMed] [Google Scholar]
- 22.Schroder E, Littlechild JA, Lebedev AA, Errington N, Vagin AA, Isupov MN. Crystal structure of decameric 2-Cys peroxiredoxin from human erythrocytes at 1.7 A resolution. Structure 2000; 8:605–15. [DOI] [PubMed] [Google Scholar]
- 23.Salsbury FR Jr, Knutson ST, Poole LB, Fetrow JS. Functional site profiling and electrostatic analysis of cysteines modifiable to cysteine sulfenic acid. Protein Sci 2008; 17:299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall A, Karplus PA, Poole LB. Typical 2-Cys peroxiredoxins—structures, mechanisms and functions. Febs J 2009; 276:2469–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manta B, Hugo M, Ortiz C, Ferrer-Sueta G, Trujillo M, Denicola A. The peroxidase and peroxynitrite reductase activity of human erythrocyte peroxiredoxin 2. Arch Biochem Biophys 2009; 484:146–54. [DOI] [PubMed] [Google Scholar]
- 26.Neumann CA, Wen ST, Van Etten RA. Role of the c-Abl tyrosine kinase in the cellular response to oxidative stress. Blood 1998; 92. [Google Scholar]
- 27.Woo HA, Chae HZ, Hwang SC, Yang KS, Kang SW, Kim K, et al. Reversing the inactivation of peroxiredoxins caused by cysteine sulfenic acid formation. Science 2003; 653–6. [DOI] [PubMed] [Google Scholar]
- 28.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 2003; 425:980–4. [DOI] [PubMed] [Google Scholar]
- 29.Rhee SG, Jeong W, Chang TS, Woo HA. Sulfiredoxin, the cysteine sulfinic acid reductase specific to 2-Cys peroxiredoxin: its discovery, mechanism of action, and biological significance. Kidney Int Suppl 2007; 3–8. [DOI] [PubMed] [Google Scholar]
- 30.Brigelius-Flohe R Glutathione peroxidases and redox-regulated transcription factors. Biol Chem 2006; 387:1329–35. [DOI] [PubMed] [Google Scholar]
- 31.Ho Y-S, M JL, Bronson R, Jin C, Gargano M, Sugawara M, et al. MIce deficient in cellular glutathione peroxidase develope normally and show no increased sensitivitty to hyperoxia. Journ of Biol Chem 1997; 272:16644–51. [DOI] [PubMed] [Google Scholar]
- 32.Neumann CA, Krause DS, Carman CV, Das S, Devendra D, Abraham JL, et al. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defense and tumor suppression. Nature 2003; 424:561–5. [DOI] [PubMed] [Google Scholar]
- 33.Lee TH, Kim SU, Yu SL, Kim SH, Park DS, Moon HB, et al. Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood 2003; 101:5033–8. [DOI] [PubMed] [Google Scholar]
- 34.Low FM, Hampton MB, Peskin AV, Winterbourn CC. Peroxiredoxin 2 functions as a noncatalytic scavenger of low-level hydrogen peroxide in the erythrocyte. Blood 2007; 109:2611–7. [DOI] [PubMed] [Google Scholar]
- 35.Immenschuh S, Baumgart-Vogt E. Peroxiredoxins, oxidative stress and cell proliferation. Antioxid Redox Signal 2005; 7:768–77. [DOI] [PubMed] [Google Scholar]
- 36.Esposito LA, Kokoszka JE, Waymire KG, Cottrell B, MacGregor GR, Wallace DC. Mitochondrial oxidative stress in mice lacking the glutathione peroxidase-1 gene. Free Radic Biol Med 2000; 28:754–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schrader M, Fahimi HD. Peroxisomes and oxidative stress. Biochim Biophys Acta 2006; 1763:1755–66 [DOI] [PubMed] [Google Scholar]
- 38.Zhou Z, Kang YJ. Cellular and subcellular localization of catalase in the heart of transgenic mice. J Histochem Cytochem 2000; 48:585–94. [DOI] [PubMed] [Google Scholar]
- 39.Kirkman HN, Gaetani GF. Mammalian catalase: a venerable enzyme with new mysteries. Trends Biochem Sci 2007; 32:44–50. [DOI] [PubMed] [Google Scholar]
- 40.Hoffschir F, Daya-Grosjean L, Petit PX, Nocentini S, Dutrillaux B, Sarasin A, et al. Low catalase activity in xeroderma pigmentosum fibroblasts and SV40-transformed human cell lines is directly related to decreased intracellular levels of the cofactor, NADPH. Free Radic Biol Med 1998; 24:809–16. [DOI] [PubMed] [Google Scholar]
- 41.Lord-Fontaine S, Averill-Bates DA. Heat shock inactivates cellular antioxidant defenses against hydrogen peroxide: protection by glucose. Free Radic Biol Med 2002; 32:752–65. [DOI] [PubMed] [Google Scholar]
- 42.Spielberger JC, Moody AD, Watson WH. Oxidation and nuclear localization of thioredoxin-1 in sparse cell cultures. J Cell Biochem 2008; 104:1879–89. [DOI] [PubMed] [Google Scholar]
- 43.Markovic J, Borras C, Ortega A, Sastre J, Vina J, Pallardo FV. Glutathione is recruited into the nucleus in early phases of cell proliferation. J Biol Chem 2007; 282:20416–24. [DOI] [PubMed] [Google Scholar]
- 44.Park JW, Mieyal JJ, Rhee SG, Chock PB. Deglutathionylation of 2-cys peroxiredoxin is specifically catalyzed by sulfiredoxin. J Biol Chem 2009; 284:23364–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prosperi MT, Ferbus D, Karczinski I, Goubin G. A human cDNA corresponding to a gene overexpressed during cell proliferation encodes a product sharing homology with amoebic and bacterial proteins. J Biol Chem 1993; 268:11050–6. [PubMed] [Google Scholar]
- 46.Prosperi MT, Ferbus D, Rouillard D, Goubin G. The pag gene product, a physiological inhibitor of c-abl tyrosine kinase, is overexpressed in cells entering S phase and by contact with agents inducing oxidative stress. FEBS Lett 1998; 423:39–44. [DOI] [PubMed] [Google Scholar]
- 47.Chang TS, Jeong W, Choi SY, Siquin Y, Kang SW, Rhee SG. Regulation of peroxiredoxin1 activity by cdc2-mediated phosphorylationj. Journal of Biological Chemistry 2002; 277:25370–6. [DOI] [PubMed] [Google Scholar]
- 48.Rhee SG, Yang KS, Kang SW, Woo HA, Chang TS. Controlled elimination of intracellular H(2)O(2): regulation of peroxiredoxin, catalase and glutathione peroxidase via post-translational modification. Antioxid Redox Signal 2005; 7:619–26. [DOI] [PubMed] [Google Scholar]
- 49.Rudolph J Redox regulation of the Cdc25 phosphatases. Antioxid Redox Signal 2005; 7:761–7. [DOI] [PubMed] [Google Scholar]
- 50.Mitsushita J, Lambeth J, Kamata T. The superoxide-generating oxidase Nox1 is functionally required for Ras oncogen transformation. Cancer Research 2004; 64:3580–5. [DOI] [PubMed] [Google Scholar]
- 51.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 2009; 8:579–91. [DOI] [PubMed] [Google Scholar]
- 52.Phalen TJ, Weirather K, Deming PB, Anathy V, Howe AK, van der Vliet A, et al. Oxidation state governs structural transitions in peroxiredoxin II that correlate with cell cycle arrest and recovery. J Cell Biol 2006; 175:779–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee W, Choi KS, Riddell J, Ip C, Ghosh D, Park JH, et al. Human peroxiredoxin 1 and 2 are not duplicate proteins: the unique presence of CYS83 in Prx1 underscores the structural and functional differences between Prx1 and Prx2. J Biol Chem 2007; 282:22011–22. [DOI] [PubMed] [Google Scholar]
- 54.Chevallet M, Wagner E, Luche S, van Dorsselaer A, Leize-Wagner E, Rabilloud T. Regeneration of peroxiredoxins during recovery after oxidative stress. J Biol Chem 2003; 278:37146–53. [DOI] [PubMed] [Google Scholar]
- 55.Cao C, Leng Y, Huang W, Liu X, Kufe D. Glutathione peroxidase 1 is regulated by the c-Abl and Arg tyrosine kinases. J Biol Chem 2003; 278:39609–14. [DOI] [PubMed] [Google Scholar]
- 56.Cao C, Leng Y, Kufe D. Catalase activity is regulated by c-Abl and Arg in the oxidative stress response. J Biol Chem 2003; 278:29667–75. [DOI] [PubMed] [Google Scholar]
- 57.Cao C, Leng Y, Liu X, Yi Y, Li P, Kufe D. Catalase is regulated by ubiquitination and proteosomal degradation. Role of the c-Abl and Arg tyrosine kinases. Biochemistry 2003; 42:10348–53. [DOI] [PubMed] [Google Scholar]
- 58.Sirvent A, Benistant C, Roche S. Cytoplasmic signalling by the c-Abl tyrosine kinase in normal and cancer cells. Biol Cell 2008; 100:617–31. [DOI] [PubMed] [Google Scholar]
- 59.Kim SY, Kim TJ, Lee KY. A novel function of peroxiredoxin 1 (Prx-1) in apoptosis signal-regulating kinase 1 (ASK1)-mediated signaling pathway. FEBS Lett 2008; 582:1913–8. [DOI] [PubMed] [Google Scholar]
- 60.Trotter EW, Rand JD, Vickerstaff J, Grant CM. The yeast Tsa1 peroxiredoxin is a ribosome-associated antioxidant. Biochem J 2008; 412:73–80. [DOI] [PubMed] [Google Scholar]
- 61.Peters GH, Frimurer TM, Olsen OH. Electrostatic evaluation of the signature motif (H/V)CX5R(S/T) in protein-tyrosine phosphatases. Biochemistry 1998; 37:5383–93. [DOI] [PubMed] [Google Scholar]
- 62.Zhang ZY, Dixon JE. Active site labeling of the Yersinia protein tyrosine phosphatase: the determination of the pKa of the active site cysteine and the function of the conserved histidine 402. Biochemistry 1993; 32:9340–5. [DOI] [PubMed] [Google Scholar]
- 63.Kwon J, Lee S, Yang K, Ahn Y, Kim Y, Stadtman ER, et al. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. PNAS 2004; 101:16419–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leslie N, Bennet D, Lindsay Y, Stewart H, Gray A, Downes C. Redox regulation of PI3-kinase signaling via inactivation of PTEN. EMBO J 2003; 22:5501–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. Journ of Biol Chem 2002; 20336–42. [DOI] [PubMed] [Google Scholar]
- 66.Koul D, Jasser SA, Lu Y, Davies MA, Shen R, Shi Y, et al. Motif analysis of the tumor suppressor gene MMAC/PTEN identifies tyrosines critical for tumor suppression and lipid phosphatase activity. Oncogene 2002; 21:2357–64. [DOI] [PubMed] [Google Scholar]
- 67.Tolkacheva T, Chan AM. Inhibition of H-Ras transformation by the PTEN/MMAC1/TEP1 tumor suppressor gene. Oncogene 2000; 19:680–9. [DOI] [PubMed] [Google Scholar]
- 68.Lu TL, Chang JL, Liang CC, You LR, Chen CM. Tumor spectrum, tumor latency and tumor incidence of the pten-deficient mice. PLoS ONE 2007; 2:1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998; 95:29–39. [DOI] [PubMed] [Google Scholar]
- 70.Backman SA, Ghazarian D, So K, Sanchez O, Wagner KU, Hennighausen L, et al. Early onset of neoplasia in the prostate and skin of mice with tissue-specific deletion of Pten. Proc Natl Acad Sci USA 2004; 101:1725–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stambolic V, Tsao MS, Macpherson D, Suzuki A, Chapman WB, Mak TW. High incidence of breast and endometrial neoplasia resembling human Cowden syndrome in pten+/− mice. Cancer Res 2000; 60:3605–11. [PubMed] [Google Scholar]
- 72.Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 1997; 16:64–7. [DOI] [PubMed] [Google Scholar]
- 73.Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer 2006; 6:184–92. [DOI] [PubMed] [Google Scholar]
- 74.Benhar M, Dalyot I, Engelberg D, Levitzki A. Enhanced ROS production in oncogenically transformed cells potentiates c-Jun N-Terminal kinase and p38 mitogen-activted protein kinase activation and sensitization to genotoxic stress. Mol Cell Biol 2001; 21:6913–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell 2003; 4:257–62. [DOI] [PubMed] [Google Scholar]
- 76.Ali IU, Schriml LM, Dean M. Mutational spectra of PTEN/MMAC1 gene: a tumor suppressor with lipid phosphatase activity. J Natl Cancer Inst 1999; 91:1922–32. [DOI] [PubMed] [Google Scholar]
- 77.Leslie NR, Downes CP. PTEN function: how normal cells control it and tumour cells lose it. Biochem J 2004; 382:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maroulakou IG, Oemler W, Naber SP, Tsichlis PN. Akt1 ablation inhibits, whereas Akt2 ablation accelerates, the development of mammary adenocarcinomas in mouse mammary tumor virus (MMTV)-ErbB2/neu and MMTV-polyoma middle T transgenic mice. Cancer Res 2007; 67:167–77. [DOI] [PubMed] [Google Scholar]
- 79.Skeen JE, Bhaskar PT, Chen CC, Chen WS, Peng XD, Nogueira V, et al. Akt deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and mTORC1-dependent manner. Cancer Cell 2006; 10:269–80. [DOI] [PubMed] [Google Scholar]