

Abstract
Drug repurposing is an efficient strategy for new drug discovery. Our latest study found that nitazoxanide (NTZ), an approved anti-parasite drug, was an autophagy activator and could alleviate the symptom of Alzheimer's disease (AD). In order to further improve the efficacy and discover new chemical entities, a series of NTZ-based derivatives were designed, synthesized, and evaluated as autophagy activator against AD. All compounds were screened by the inhibition of phosphorylation of p70S6K, which was the direct substrate of mammalian target of rapamycin (mTOR) and its phosphorylation level could reflect the mTOR-dependent autophagy level. Among these analogs, compound 22 exhibited excellent potency in promoting β-amyloid (Aβ) clearance, inhibiting tau phosphorylation, as well as stimulating autophagy both in vitro and in vivo. What's more, 22 could effectively improve the memory and cognitive impairments in APP/PS1 transgenic AD model mice. These results demonstrated that 22 was a potential candidate for the treatment of AD.
Key words: Alzheimer's disease, Autophagy, Nitazoxanide, β-amyloid, Tau protein
Abbreviations: Aβ, β-amyloid; AChEIs, acetylcholinesterase inhibitors; AD, Alzheimer's disease; APP, amyloid precursor protein; BBB, blood–brain barrier; CNS, central nervous system; mTOR, mammalian target of rapamycin; MWM, Morris Water Maze; NCEs, new chemical entities; NFTs, neurofibrillary tangles; NMDA, N-methyl-d-aspartate; NTZ, nitazoxanide; PAMPA, parallel artificial membrane permeation assay; PBL, porcine brain lipid; SPs, senile plaques; WORT, wortmannin
Graphical abstract
Through the drug redevelopment of nitazoxanide, we acquired new chemical entities with potent anti-AD efficacy. The candidate derivative 22 could promote Aβ clearance, inhibit tau phosphorylation, as well as stimulate autophagy both in vitro and in vivo. In addition, 22 worked well in APP/PS1 transgenic AD model mice.

1. Introduction
Alzheimer's disease (AD) is the most common neurodegenerative disorder characterized by memory loss and cognitive dysfunction1. There are lots of hypotheses about its cause but the underlying pathogenesis has not been fully elucidated, which seriously hinders the development of new drugs for AD treatment. Although so many efforts have been devoted to the R&D of anti-AD agents in recent years, the 99.6% failure rate of clinical research is among the highest for any therapeutic area, and it has been 16 years since the last drug, memantine, was launched2. Currently, the available therapeutic options for AD are limited to several acetylcholinesterase inhibitors (AChEIs) and one N-methyl-d-aspartate (NMDA) receptor antagonists, which only provide limited symptomatic relief. Therefore, there are still lack effective drugs in the clinic that could slow down or halt the disease progression, and new drug discovery strategy with high efficiency and low risk should be encouraged.
The two most representative pathological hallmarks of AD are amyloid senile plaques (SPs) caused by accumulated β-amyloid (Aβ) peptides, and neurofibrillary tangles caused by abnormally hyperphosphorylated tau protein (NFTs)3, 4, 5. SPs and NFTs have been closely involved in the pathogenesis of AD, and simultaneous regulations of Aβ clearance and tau phosphorylation have been treated as promising strategies for drug discovery against AD6, 7, 8.
Aβ is a type of peptides, which is produced from proteolytic cleavage of the amyloid precursor protein (APP) by β- and γ-secretase4, 9, 10. The amyloid hypothesis believes that excessive production and accumulation of Aβ peptides can promote the formation of SPs and result in neuronal death6, 11, 12. Aβ-targeted therapies have played a central role in the treatment of AD in the last two decades. A large number of research projects focusing on this therapeutic strategy for decreasing Aβ level have been published, and some drug candidates have entered clinical trials, such as β-secretase (BACE1) inhibitor verubecestat (MK-8931)13, 14, γ-secretase inhibitor semagacestat (LY450139)15, 16, and anti-Aβ monoclonal antibody solanezumab (LY2062430)17, 18. Tau protein is kind of microtubule-associated protein that able to promote microtubules assembly and stabilize the integrity and function of microtubules in the neuronal cytoskeleton18, 19. In AD patients, hyperphosphorylated tau protein disengaged from microtubules, misfolded, and self-assembled into neurotoxic paired helical filaments (PHFs) which eventually aggregated to form the intracellular NFTs20, 21. Therapeutic strategies that target the tau pathway have received much attention for the potential treatment of AD, and some tau-based drug candidates also have reached to clinical research22. However, at present, the two hypotheses are still controversial and questionable, for the clinical drugs targeting these two pathological hallmarks can't cure AD. Based on the complexity of the pathogenesis of AD, more and more study showed that these two pathological symptoms are not independent but promote each other. Under pathological conditions, Aβ-induced neurotoxicity induces abnormal hyperphosphorylation of tau, which in turn directly or indirectly promotes Aβ production and inhibits Aβ clearance, leads to the inability to completely cure AD for compounds that target only a single pathology. Therefore, the compound nitazoxanide (NTZ) and its analogues able to break the cycle of Aβ and tau hyperphosphorylation may show potential in the treatment of AD.
Autophagy is the process of clearing abnormal protein aggregates and damaged organelles that affect normal functions in cells23, 24. Mounting evidence suggests that autophagy deficits might contribute to the pathogenesis of numerous neurodegenerative diseases, including AD25, 26, 27, 28. Aβ can be swallowed into autophagic vesicles and fuse with the lysosome, and then be degraded in the process of autophagy29. Autophagy has been confirmed to be a major pathway for Aβ clearance, along with Aβ degradation enzymes30. In addition, many studies indicated that autophagy could facilitate the degradation of a variety of forms of tau proteins31. For example, autophagy inducer, rapamycin, was beneficial to the degradation of insoluble forms of tau32. Thus, autophagy may be a promising therapeutic target for the development of anti-AD drugs.
Chemical genetics facilitates the discovery of new drug molecules, especially the forward chemical genetics, which screens the chemical libraries of small molecules for a interested phenotype, to discover positive molecules that could regulate the phenotype, and then uses the resulting molecules to treat the corresponding disease and identify potential drug targets. Therefore, this strategy can be used to discover new drug molecules and explore new mechanisms33. With a lab in-house FDA-approved drug library as the screening chemical library, and the phosphorylation level of p70S6K as the interested phenotype, NTZ (an anti-parasite drug) was screened out as an autophagy activator for the potential treatment of AD, this is the R&D strategy of drug repurposing. Our latest study has reported this finding34. NTZ was able to stimulate autophagy and promote Aβ clearance by inhibiting signaling pathway of both PI3K/AKT/mTOR/ULK1 and NQO1/mTOR/ULK1, and restrain tau hyperphosphorylation by inhibiting PI3K/AKT/GSK3β signaling pathway, as well as effectively ameliorate memory and cognitive impairments of APP/PS1 transgenic AD model mice34. In order to discover new chemical entities (NCEs) and further improve the anti-AD efficacy, structural modification based on NTZ has been carried out, this is the so called drug redevelopment (Fig. 1).
Figure 1.

Strategy of drug repurposing and drug redevelopment.
In this work, 52 NTZ-based compounds were designed and synthesized, and their abilities to promote autophagy were evaluated by phosphorylation level of p70S6K, which was the direct substrate of mTOR (mammalian target of rapamycin). The phosphorylation of p70S6K could reflect mTOR-dependent autophagy level35. At last, we found a candidate compound 22 with good blood–brain barrier (BBB) permeability and low hERG inhibition activity. What's more, 22 exhibited the most potent efficacy, even far more than NTZ, and it could not only significantly promote β-amyloid (Aβ) clearance and inhibit tau phosphorylation but also largely improve the memory and cognitive impairments of APP/PS1 AD model mice. These results demonstrated that compound 22 is a new chemical entity and promising agent for the treatment of AD.
2. Results and discussion
2.1. Design and synthesis of NTZ derivatives
In this study, NTZ was set as the lead compound and structural modification was carried out, aim to discover NCEs with improved efficacy and good BBB permeability that like central nervous system (CNS) drugs.
First, the 2-acetoxy group of NTZ was modified to obtain derivatives 2 (hydroxyl, tizoxanide) and 3 (hydrogen). Then we investigated the substituent effect of 5-position on the thiazole ring of NTZ. The 5-nitro group of NTZ was replaced by hydrogen (4), cyano (5), ester group (6 and 7), methylthio (8), methanesulfonyl (9), carboxyl (10), piperidyl (11), morpholinyl (12), pyrrolyl (13), and piperazinyl (14) to obtain derivatives 4–14.
Compound 2, the active metabolite of NTZ, was commercially available. The general synthetic routes of derivatives 3–10 were described in Scheme 1. The commercially available substituted 2-aminothiazoles (55–58) and previously prepared substituted 2-aminothiazoles (59 and 60, Supporting Information Sections 1, 2) were reacted with benzoyl chloride or 2-acetoxybenzoyl chloride with the Et3N as base to obtain target compounds 3–8. Compound 8 reacted with m-chloroperbenzoic acid and the S atom was oxidized to sulphone to afford compound 9. The ester group of compound 6 was hydrolyzed by LiOH to give intermediate 61, followed by reaction with acetic anhydride to afford target compound 10.
Scheme 1.

Syntheses of compounds 3−10. Reagents and conditions: (a) Et3N, THF, rt, 3 h; (b) mCPBA, DCM, rt, 30 min; (c) LiOH, THF, H2O, rt, overnight; (d) Ac2O, H2SO4, 75 °C, 20 min.
Scheme 2 depicted the synthetic route of derivatives 11–14. The starting material 2-amino-5-bromothiazole hydrobromide was reacted with corresponding secondary amine derivatives through nucleophilic substitution reaction to provide key intermediates 62 (piperidine), 63 (morpholine), 64 (pyrrole), and 65 (1-(tert-butoxycarbonyl)piperazine). The intermediates 62–65 were reacted with 2-acetoxybenzoyl chloride in the presence of Et3N to afford target compounds 11–13, and intermediate 62 respectively. The N-Boc protecting group of 62 was removed by utilizing silica gel in toluene at 80 °C to afford target compound 14.
Scheme 2.

Syntheses of compounds 11–14. Reagents and conditions: (a) corresponding secondary amine derivatives, morpholine, pyrrole, or 1-(tert-butoxycarbonyl)piperazine, K2CO3, DMF, 60 °C, 3 h; (b) Et3N, THF, rt, 3 h; (c) silica gel, toluene, 80 °C, 4 h.
Morpholinyl containing derivative 12 was identified to display excellent p70S6K phosphorylation inhibitory activity (Table 1), so the 5-morpholinothiazole moiety (12) was retained in the following structural modification. The 2-acetoxyphenyl fragment of 12 was treated as the next modifying part, replaced by different substituted 2-hydroxyphenyl (15–18), various substituted phenyl (19–29), naphthyl (30 and 31), heteroaryl (32–38), cycloalkyl (39–44), alkyl (45–49), fluoroalkyl (50), arylalkyl (51 and 52), oxocycloalkyl (53) and oxoalkyl (54), and 40 derivatives (15–54) were designed. The syntheses of derivatives 15–54 were outlined in Scheme 3. Derivatives 15–18, and 23–54 were obtained by acylation reaction of intermediate 63 with corresponding acyl chlorides 76–103 respectively. Some acyl chlorides were commercially available, and the others were prepared by corresponding carboxylic acids reacting with thionyl chloride or oxalyl chloride. The acetyl group of compounds 11, 22 and 23 were hydrolyzed by LiOH to obtain derivatives 19–21 respectively.
Table 1.
Chemical structures of compounds 1–14 and their inhibitory activities of p70S6K phosphorylation.
| Compd. |
R1 |
R2 |
Inhibition ratio of p70S6K phosphorylation at 20 μmol/L (%)a |
| NTZ (1) | NO2 | OAc | 50.21±3.11 |
| 2 | NO2 | OH | 43.09±2.98 |
| 3 | NO2 | H | 64.71±4.20 |
| 4 | H | OAc | 9.98±5.16 |
| 5 | CN | OAc | 70.85±3.56 |
| 6 | COOCH3 | OAc | 32.72±7.87 |
| 7 | COOBn | OAc | 18.88±5.27 |
| 8 | SCH3 | OAc | 44.74±9.98 |
| 9 | SO2CH3 | OAc | 10.67±10.51 |
| 10 | COOH | OAc | −4.51±19.40 |
| 11 |  |
OAc | 56.27±4.61 |
| 12 |  |
OAc | 85.20±1.76 |
| 13 |  |
OAc | 69.85±0.53 |
| 14 | 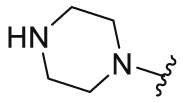 |
OAc | 28.21±6.44 |
Results are expressed as the mean ± SD (n ≥ 3).
Scheme 3.

Syntheses of compounds 15–54. Reagents and conditions: (a) SOCl2, toluene, DMF, 80 °C, 3 h; (b) oxalyl chloride, DCM, DMF, 0 °C to rt, 4 h; (c) NaOH, THF, H2O, 50 °C, overnight; (d) LiOH, THF, H2O, rt, overnight.
2.2. Inhibitory activity of p70S6K phosphorylation
For the primary assay, the inhibitory activities of tizoxanide (2, the active metabolite of NTZ) and derivatives 3–54 for the phosphorylation of p70S6K with concentration of 20 μmol/L were measured by α-screen assay in SH-SY5Y cell. NTZ and wortmannin (WORT) were the reference compounds. The results were summarized in Table 1, Table 2. First, since NTZ was a prodrug which could be rapidly hydrolyzed to its active metabolite tizoxanide (2) in vivo, we tested the inhibitory activity of tizoxanide against the phosphorylation of p70S6K, and found its activity was almost comparable to that of NTZ (Table 1). Then, we also replaced the 2-acetoxy with hydrogen to give compound 3, and its activity was slightly improved compared to NTZ. Subsequently, we modified the substituents on the 5-position of thiazole ring to afford compounds 3–14 to determine whether the activity may be enhanced. Among them, compound 12, with the nitro group replaced by morpholinyl group, exhibited the most potency with an inhibitory ratio of 85.20% at 20 μmol/L (Table 1). However, once the 5-nitro group was removed (4) or replaced by ester groups (6 and 7), carboxyl (9), methanesulfonyl (10), or piperazinyl (14), the inhibitory activities against the phosphorylation of p70S6K were decreased significantly (Table 1).
Table 2.
Chemical structures of compounds 15–54 and their inhibitory activities of phosphorylation of p70S6K.
| Compd. |
R3 |
Inhibition ratio of p70S6K phosphorylation at 20 μmol/L (%)a |
Compd. |
R3 |
Inhibition ratio of p70S6K phosphorylation at 20 μmol/L (%)a |
| 15 |  |
42.25±0.19 | 36 |  |
83.32±0.41 |
| 16 | 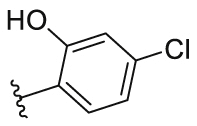 |
21.02±4.56 | 37 |  |
81.20±1.60 |
| 17 | 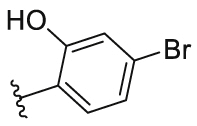 |
15.41±5.32 | 38 | 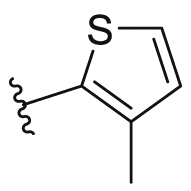 |
80.32±0.71 |
| 18 |  |
8.76±5.93 | 39 |  |
63.76±1.31 |
| 19 |  |
53.79±3.85 | 40 |  |
66.21±3.74 |
| 20 |  |
83.86±1.54 | 41 |  |
63.37±4.65 |
| 21 |  |
79.20±1.87 | 42 |  |
61.15±1.44 |
| 22 |  |
88.95±0.78 | 43 | 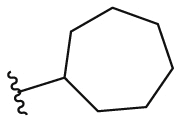 |
51.10±1.59 |
| 23 |  |
84.96±0.98 | 44 |  |
26.05±11.29 |
| 24 |  |
77.51±1.48 | 45 |  |
43.86±4.45 |
| 25 | 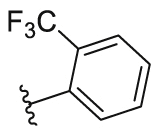 |
22.05±1.85 | 46 |  |
44.98±8.58 |
| 26 |  |
65.98±1.74 | 47 |  |
61.71±2.51 |
| 27 |  |
53.24±5.72 | 48 |  |
61.32±5.39 |
| 28 |  |
55.38±1.66 | 49 |  |
40.38±3.37 |
| 29 |  |
26.90±6.23 | 50 |  |
61.35±5.44 |
| 30 |  |
78.75±2.50 | 51 | 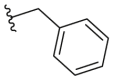 |
47.82±3.90 |
| 31 |  |
80.96±1.60 | 52 |  |
81.60±1.86 |
| 32 |  |
81.09±4.27 | 53 | 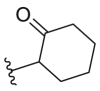 |
44.94±4.00 |
| 33 |  |
79.80±5.03 | 54 |  |
66.46±3.73 |
| 34 | 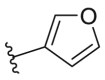 |
80.23±0.88 | WORT | – | 90.68±1.46 |
| 35 |  |
79.84±1.17 |
Results are expressed as the mean±SD (n ≥ 3). −Not applicable.
As shown in Table 2, replacing the 2-acetoxyphenyl group with various types of substituents remarkably affected the inhibitory activities of p70S6K phosphorylation. First of all, we replaced the 2-acetoxy on the phenyl of compound 12 with the hydroxyl (19), and also introduced a substituent on the 4-position of phenyl to afford compounds 15–18, which both lead to significant decline in activities. Then, we varied the position of hydroxyl and acetoxy on the phenyl or removed the 2-acetoxy, and compounds 20–24 with relatively effective activities were identified. Especially, compound 22 with acetoxy on the meta-position of the phenyl, exhibited the most inhibitory potency (88.95% at 20 μmol/L, Table 2). Furthermore, changing 2-acetoxy on the phenyl into other substituents (25–29) was not tolerated, leading to an obvious decline in potency. In addition, the 2-acetoxyphenyl was modified to naphthyl (30 and 31) and heteroaryl (32–38), and these compounds showed good inhibitory activities. However, replacing the 2-acetoxyphenyl with (cyclo)alkyl (39–49), fluoroalkyl (50), arylalkyl (51 and 52), oxocycloalkyl (53) and oxoalkyl (54) was not beneficial for the inhibitory potency.
2.3. In vitro blood−brain barrier permeation assay
One of the major requirements for the development of drugs targeting the CNS is the good BBB permeability. A parallel artificial membrane permeation assay (PAMPA)36, 37, 38, 39, 40, was performed to explore whether the optimized compounds 12, 20–24, 30–38, and 52 had the ability to penetrate into the brain. The in vitro membrane permeability (Pe) of the selected compounds were determined through the porcine brain lipid (PBL), and it had been concluded that the compounds with the Pe value more than 3.08 × 10−6 cm/s could penetrate BBB (CNS+), while the Pe values less than 1.13 × 10−6 cm/s could not penetrate BBB (CNS−). For the compounds with the Pe values in these boundary limits (1.13 × 10−6 cm/s < Pe < 3.08 × 10−6 cm/s), their BBB permeability were unsure (CNS+/−, Supporting Information Section 3, Fig. S1, Tables S1 and S2). As described in Table 3, NTZ displayed Pe value between the boundary limits, indicating its poor BBB permeability which conformed to the physicochemical properties of the anti-parasitic drug. Whereas, except for the compounds 20 and 21, the majority of the tested compounds (12, 22–24, 30–38, and 52) exhibited Pe values above 3.08 × 10−6 cm/s, demonstrating that they had the potency to penetrate BBB.
Table 3.
Permeability results (Pe, 10−6 cm/s) from the PAMPA–BBB assay for compounds 12, 20–24, 30–38, and 52 and their prediction of BBB penetration.
| Compd. | Pe (10−6 cm/s)a | Predictionb |
|---|---|---|
| NTZ | 1.26±0.21 | CNS+/− |
| 12 | 5.76±0.57 | CNS+ |
| 20 | 0.45±0.03 | CNS− |
| 21 | 0.09±0.01 | CNS− |
| 22 | 6.02±0.27 | CNS+ |
| 23 | 4.92±0.31 | CNS+ |
| 24 | 12.62±1.22 | CNS+ |
| 30 | 5.76±0.55 | CNS+ |
| 31 | 3.42±0.07 | CNS+ |
| 32 | 6.16±0.50 | CNS+ |
| 33 | 10.41±1.25 | CNS+ |
| 34 | 6.82±0.50 | CNS+ |
| 35 | 9.24±0.47 | CNS+ |
| 36 | 10.62±0.10 | CNS+ |
| 37 | 9.89±0.37 | CNS+ |
| 38 | 9.88±0.75 | CNS+ |
| 52 | 10.50±1.13 | CNS+ |
Values are expressed as the mean ± SD (n ≥ 3).
Compounds with permeabilities Pe > 3.08 × 10−6 cm/s could cross the BBB by passive diffusion (CNS+). Compounds with Pe < 1.13 × 10−6 cm/s show low BBB permeation (CNS−), and compounds with 1.13 × 10−6 cm/s < Pe < 3.08 × 10−6 cm/s show uncertain BBB permeation (CNS+/−).
2.4. Inhibitory activities of compounds 12, 22, and 36 against the phosphorylation of p70S6K at different concentrations
In consideration of the favorable inhibitory ratios of compounds 12, 22, and 36 against the phosphorylation of p70S6K at 20 μmol/L, as well as the good BBB permeation, we tested their inhibitory activities at different concentrations in SH-SY5Y cell additionally. As shown in Fig. 2, compounds 12, 22, and 36 could effectively suppress the phosphorylation of p70S6K at different concentrations (range 5–40 μmol/L), and the inhibitory activities of compounds 22 and 36 were significantly stronger than that of compound 12.
Figure 2.

Inhibitory activities of compounds 12, 22, 36 and NTZ against the phosphorylation of p70S6K at 5, 10, 20, and 40 μmol/L, with wortmannin (WORT) as control compound. All data were obtained from three independent experiments and presented as mean ± SEM (t-test, ***P < 0.001 vs. DMSO group).
2.5. The cell viability of compounds 12, 22, and 36
AD is a chronic disease, safety evaluation is essential. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to evaluate the cytotoxicity of compounds 12, 22, and 36 on the BV2 and SH-SY5Y cells. NTZ was tested as reference. As depicted in Fig. 3, compounds 22 and 36, exhibited no cytotoxicity at concentrations of 2, 5, 10, 20, 40 μmol/L after incubation with BV2 cells for 24 h. The cell viability decreased when increasing the concentration to a relatively high level of 80, 160 μmol/L, just like approved NTZ. Compound 12 began to decrease the viability of BV2 cells at concentration of 20 μmol/L, displayed relatively high cytotoxicity than 22 and 36. For SH-SY5Y cells, compounds 22 and 36 exhibited the similar low cytotoxicity, 12 decreased the viability at concentration of 5 μmol/L. Therefore, compounds 22 and 36 were proved to have low cytotoxicity at the BV2 and SH-SY5Y cells.
Figure 3.

Cell viability of BV2 and SH-SY5Y cells exposed to compounds NTZ, 12, 22, and 36 at different concentrations (range 0–160 μmol/L) for 24 h. (A) NTZ to BV2 cells; (B) 12 to BV2 cells; (C) 22 to BV2 cells; (D) 36 to BV2 cells; (E) NTZ to SH-SY5Y cells; (F) 12 to SH-SY5Y cells; (G) 22 to SH-SY5Y cells; (H) 36 to SH-SY5Y cells. The results are expressed as the percentage of viable cell observed after treatment with compounds vs. zero-treated cells (100%) and are shown as the means ± SEM; n = 3, *P < 0.05, **P < 0.01, ***P < 0.001 vs. zero-treated group.
2.6. Compound 22 stimulated autophagy via mTOR-dependent signaling pathway
Given that p70S6K is the direct substrate of mTOR35, we have measured p70S6K phosphorylation level to evaluate the derivatives-regulated mTOR activity before34. Next, we explored whether the optimized compounds 22 and 36 could regulate AKT/mTOR/ULK1 signaling pathways and enhance autophagy by Western blot assay. As shown in Fig. 4, compound 22 dose-dependently suppressed the phosphorylation of AKT, mTOR, p70S6K, and decreased ULK1 phosphorylation at Ser 757, a direct mTOR phosphorylation site. These results indicated that compound 22 could suppress the inactivation of ULK1 by inhibiting the phosphorylation of AKT/mTOR, thus promoting the activation of autophagy. As shown in Fig. 5, compound 36 could also effectively inhibit the phosphorylation of AKT, mTOR, and p70S6K, but it had no effect on the ULK1 phosphorylation. Therefore, it was uncertain that compound 36 could stimulate autophagy. In addition, the compounds-induced autophagy stimulation was assessed by evaluating the expressions of 3 classic autophagy marker proteins, microtubule-associated protein 1 light chain 3 II (LC3 II), Beclin 1 and p6241, 42. We found that compounds 22 and 36 both could upregulate the protein levels of Beclin 1 and LC3 II, and decrease the p62 protein level (Fig. 6B–E), which indicated the enhancement of autophagy.
Figure 4.

The Western blot results of compound 22 inhibited AKT/mTOR signaling pathway and promoted autophagy. (A) The BV2 cells were treated with different concentrations of 22 (20, 10, 5 and 0 μmol/L), NTZ (20 μmol/L) and WORT (2 μmol/L) for 24 h, and phosphorylation levels of AKT, mTOR, p70S6K and ULK1 were detected by Western blot assay, GAPDH was used as loading control in Western blot assays. (B) and (C) Quantitation results of (A). (One-way ANOVA, Dunnett's multiple comparison test. n = 3. *P < 0.05, **P < 0.01 and ***P < 0.001 vs. DMSO group).
Figure 5.

The Western blot results of compound 36 inhibited AKT/mTOR signaling pathway and promoted autophagy. (A) The BV2 cells were treated with different concentrations of 36 (20, 10, 5 and 0 μmol/L), NTZ (20 μmol/L) and WORT (2 μmol/L) for 24 h, and phosphorylation levels of AKT, mTOR, p70S6K and ULK1 were detected by western blot assay, GAPDH was used as loading control in Western blot assays. (B) and (C) Quantitation results of (A). (One-way ANOVA, Dunnett's multiple comparison test. n = 3. *P < 0.05, **P < 0.01 and ***P < 0.001 vs. DMSO group).
Figure 6.

(A) CLSM images of BV2 cells transiently expressing mTagRFP-mWasabi-LC3. Scale bar: 25 μm. The Western blot results of compound 22 and 36 promoted autophagy. (B) and (D) The BV2 cells were treated with different concentrations of 22 and 36 (20, 10, 5 and 0 mol/L), NTZ (20 mol/L) and WORT (2 mol/L) for 24 h, and the protein levels of LC3, Beclin 1 and p62 were detected by Western blot assay, GAPDH was used as loading control in Western blot assays. (C) and (E) Quantitation results of (B) and (D). (One-way ANOVA, Dunnett's multiple comparison test, n = 3. *P < 0.05, **P < 0.01 and ***P < 0.001 vs. DMSO group).
Moreover, an overexpression vector encoding mTagRFP-mWasabi-LC3 was used to verify the activation of 22 and 36 on autophagy. At early stage of autophagosomes biogenesis, puncta of mTagRFP-mWasabi-LC3 accumulated and displayed both green and red fluorescent signals (mRFP-GFP + yellow). Notably, when autophagosomes were fused with lysosomes, they formed acidic autolysosomes (mRFP-GFP + red), and the co-located mTagRFP-mWassbi-LC3 emitted only a red signal because the green signal quenched immediately under acidic conditions. As shown in Fig. 6A, both 22 and 36 increased the numbers of both yellow and red puncta compared with the control group, while autophagy inhibitor CQ efficiently abolished the effect induced by 22 and 36. At the same time, compared with NTZ, 22 and 36 have better autophagy agonistic. All the above results demonstrated that 22 and 36 could stimulate autophagy.
2.7. Compound 22 promoted Aβ clearance and decreased the phosphorylation of tau protein
The modulation of Aβ clearance by compound 22 was studied through an ELISA assay in BV2 cell. As depicted in Fig. 7, compound 22 dose-dependently decreased both Aβ1−40 and Aβ1−42 content in the cell. What's more, compound 22 exhibited stronger potency than NTZ both for Aβ1−40 (20 μmol/L) and Aβ1−42 (5 and 10 μmol/L). Next, we investigated whether compound 22 also exhibited the activity in suppressing the phosphorylation of tau protein by Western blot assay in the SH-SY5Y cell, and LiCl was used as positive control. The BV2 cell was more suitable for Aβ related assay43, but in this assay we chose SH-SY5Y cell which belonged to neuroblastoma cell line that closely resembles primary neurons. As shown in Fig. 8, compound 22 dose-dependently inhibited tau phosphorylation at multiple sites including Ser199, 396 and Tyr231. Therefore, it was proved that compound 22 was able to promote Aβ clearance and suppress the phosphorylation of tau protein. Hence, compound 22 exhibited the multiple properties as potential anti-AD agent, for both promoting Aβ clearance and decreasing the phosphorylation of tau protein.
Figure 7.

Compound 22 promoted Aβ40 (A) and Aβ42 (B) clearance. BV2 cells were treated with different concentrations of NTZ and 22 (5, 10 and 20 μmol/L) for 24 h, followed by the addition of 2 μg/mL soluble Aβ1-40 and Aβ1-42, then incubated for 3 h. The Aβ40 and Aβ42 level was detected by ELISA assay (t-test, *P < 0.05 and ***P < 0.001 vs. DMSO group; #P < 0.05 vs. NTZ group).
Figure 8.

Compound 22 reduced the phosphorylation level of tau protein. (A) SH-SY5Ycells were cultured with different concentrations of 22 (5, 10 and 20 μmol/L), NTZ (20 μmol/L) and LiCl (10 μmol/L) for 24 h, and the phosphorylation level of tau was detected by Western blot assay, GAPDH was used as loading control in Western blot assays. (B) Quantitation results of (A). (One-way ANOVA, Dunnett's multiple comparison test, n = 3. *P < 0.05, **P < 0.01 and ***P < 0.001 vs. DMSO group).
2.8. hERG inhibitory activity
The effect on hERG potassium channels is an important evaluation method for cardiotoxicity of preclinical drugs44. Therefore, the hERG inhibitory activities of compound 22 and NTZ were detected by patch clamp assay using cisapride as a positive control (Supporting Information Section 4). The results were shown in Table 4, and both compound 22 and NTZ showed very weak hERG inhibition compared to cisapride, indicating that they have no potential cardiac side effects.
Table 4.
The inhibitory activity against hERG potassium channels.a
| Compound | 22 | NTZ | Cisapride |
|---|---|---|---|
| IC50 (μmol/L) | >40 | >40 | 0.33 |
The results are presented as IC50 values for hERG inhibition.
2.9. Compound 22 improved memory and cognitive impairments in APP/PS1 transgenic mice
The effects of compound 22 on ameliorating memory and cognitive impairments were evaluated in APP/PS1 transgenic AD model mice by Morris Water Maze (MWM) assay and nesting test. The APP/PS1 transgenic mice [B6C3-Tg (APPswe, PS1dE9)], which expressed chimeric mouse/human Swedish mutant amyloid precursor protein (Mo/HuAPP695swe) and mutant human presenilin 1 protein (PS1-dE9), exhibited early AD symptoms at 6–7 months of age, and these symptoms became more serious after 9 months of age45. Herein, non-transgenic normal mice were orally administered vehicle (NV), while 9-month-old APP/PS1 transgenic mice were orally administered vehicle (TV). NTZ was at a dose of 30 mg/kg, and compound 22 was at two doses (10 and 30 mg/kg, respectively) over a period of 100 days before conducting MWM assay and nesting test.
During the last 7 days of the treatment, escape latencies to find the platform were recorded for the animals of different experimental groups in the MWM assay. Besides, we removed the platform on the 8th day, and recorded their crossing times over the hidden location of the platform. As shown in Fig. 9A and B, APP/PS1 mice (transgenic vehicle, TV) obtained longer escape latencies and less crossing time than normal mice (nontransgenic vehicle, NV). Whereas, NTZ (30 mg/kg) significantly shortened the escape latencies of APP/PS1 mice compared with that of the TV group. Corresponding to NTZ, compound 22 significantly reduced the escape latencies at an equivalent dose (30 mg/kg) and a lower dose (10 mg/kg). Even more, 22 significantly increased the crossing times at a dose of 30 mg/kg compared with the TV group.
Figure 9.

Compound 22 effectively improved learning and memory impairments of APP/PS1 transgenic mice. Morris Water Maze assay and nesting test were used to evaluate the effect of 22 on APP/PS1 transgenic mice. (A) The escape latency during platform trials (two-way ANOVA, n = 10. *P < 0.05, **P < 0.01, ***P < 0.001 compared with TV group). (B) The crossing times of the platform in the probe trials (one-way ANOVA, n = 10. *P < 0.05, **P < 0.01 compared with TV group). (C) The evaluation of the house building capacity by nesting test (one-way ANOVA, n = 10. *P < 0.05, **P < 0.01 compared with TV group).
APP/PS1 mice suffered from impaired the nest-building ability at 6 months of age. The nesting test was reported as a useful index of behavioral deficits, and widely used in the evaluation of anti-AD agents46. As shown in Fig. 9C, the nesting scores of TV mice were lower than NV mice, while APP/PS1 mice that were treated with NTZ (30 mg/kg) and compound 22 (30 mg/kg) exhibited an obvious improvement in nesting test compared with TV group.
These results above revealed that compound 22 effectively improved memory and cognitive impairments in APP/PS1 transgenic AD model mice.
2.10. Compound 22 reduced Aβ level and inhibited tau phosphorylation in APP/PS1 transgenic mice
Our above assays demonstrated compounds 22 could reduce Aβ level and inhibit tau phosphorylation level at cellular level, so what about the in vivo condition. We subsequently evaluated the inhibition of compound 22 against the Aβ40/42 level in both the cerebral cortex and hippocampus of the APP/PS1 transgenic mice using an ELISA assay. The results were shown in Fig. 10A–D, the Aβ40/42 level of TV group were expectedly higher than the NV group. The NTZ and 22 treating group displayed significantly reduced Aβ40/42 level both in the cerebral cortex and hippocampus, and 22 was more potent than the approved NTZ. A Western blot assay was subsequently conducted to detect the inhibition of 22 against p70S6K and tau phosphorylation in the cortex of the APP/PS1 transgenic mice. As depicted in Fig. 10E–J, compounds 22 administration with both 10 and 30 mg/kg doses, could significantly reduce the p70S6K phosphorylation level, also restrained tau protein phosphorylation at multiple sites, including Ser396, 199 and Tyr231. NTZ was treated as reference. These results demonstrated compounds 22 promote Aβ clearance and inhibit hyperphosphorylation of tau both in vitro and in vivo.
Figure 10.

Compound 22 reduced Aβ, tau and p70S6K phosphorylation level in APP/PS1 transgenic mice. (A–D) ELISA assay was used to evaluate Aβ40/42 levels in APP/PS1 transgenic mice (t-test, n = 4, *P < 0.01, **P < 0.005, ***P < 0.001 compared with TV, #P < 0.01 compared with NTZ-30 mg/kg group). Compound 22 inhibited p70S6K and tau phosphorylation level at 10 μmol/L (E) and 30 μmol/L (H) concentrations in APP/PS1 transgenic mice by Western blot assay. (F and G) quantitative results of (E). (I and J) Quantitative results of (H). GAPDH was used as a loading control in Western blot assays, t-test, n = 3, *P < 0.01, **P < 0.005, ***P < 0.001 compared with TV. NV, nontransgenic vehicle group; TV, transgenic vehicle group. Values indicate the mean ± SEM.
3. Conclusions
Given that the current drugs for the treatment of AD only provide limited symptomatic relief, it is of great significance to discover more effective therapies against AD based on new strategies. Prof. Chong and Sullivan claim that the most fruitful basis for the discovery of a new drug is to start with an old drug47, and in this work, we adopted the strategy of drug redevelopment. The FDA approved anti-parasitic drug NTZ was found to potentially treat AD by promoting Aβ clearance and inhibiting tau protein phosphorylation. The main purpose of redevelopment of NTZ is to acquire new chemical entities with more potent efficacy than NTZ. We designed and synthesized 52 NTZ-derived compounds and they were evaluated as autophagy activator against AD. 14 derivatives (12, 22–24, 30–38, and 52) were found to display significantly enhanced p70S6K phosphorylation inhibitory activities, as well as greatly improved BBB permeability compared to NTZ. Among them, compound 22 exhibited the most potent activity in promoting Aβ clearance, inhibiting tau phosphorylation, and stimulating autophagy. Further experiments demonstrated that compound 22 stimulated autophagy by suppressing the phosphorylation of AKT/mTOR/ULK1 signaling pathway. Compound 22 also displayed good BBB permeability, low cytotoxicity, and low hERG inhibitory activity. The in vivo pharmacological results revealed that compound 22 could reduce the escape latency, increase the crossing times in MWM assay, and improve nest-building behavior of APP/PS1 transgenic AD model mice, which indicated compound 22 could effectively improve memory and cognitive impairments in APP/PS1 mice even more potent than NTZ. In summary, our findings suggested that compound 22 could be considered as a potential drug candidate for further development of novel anti-AD agent.
4. Experimental
4.1. General methods of chemistry
Solvents and reagents were purchased from Acros, Adamas–beta, Alfa Aesar, Energy Chemical, J&K, Shanghai Chemical Reagent Co., and TCI, and were used without further purification. Analytical thin-layer chromatography (TLC) was performed on HSGF 254 (150–200 μm thickness; Yantai Huiyou Co., China). Melting points were determined on an SGW X-4 melting point apparatus (Shanghai, China) without correction. Nuclear magnetic resonance (NMR) spectra were recorded with a Bruker AMX-400 NMR (Bruker Company, Germany). Chemical shifts were reported in parts per million (ppm, δ) downfield from tetramethylsilane. Proton coupling patterns were described as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), and broad (br). Low- and high-resolution mass spectra (LR-MS and HR-MS) were obtained by electric ionization (EI) and electrospray ionization (ESI) using Waters GCT Premier and Waters LCT (Waters Corporation, Milford, NE, USA). HPLC data analysis of compounds 3–54 were performed on an Agilent 1100 (Agilent Technologies, CA, USA) with a quaternary pump and diode-array detector (DAD). The peak purity was verified with UV spectra. All analogs were confirmed to be ≥ 95% pure (Supporting Information Section 5, Table S4), and were identified with non-PAINS on the web at http://fafdrugs3.mti.univparis-diderot.fr/recommended by editors from the ACS (American Chemical Society).
4.2. The synthesis of target compounds and key intermediates
4.2.1. N-(5-nitrothiazol-2-yl)benzamide (3)
To a solution of 2-amino-5-nitrothiazole (516 mg, 3.56 mmol) in anhydrous THF (10 mL) was added triethylamine (989 μL, 7.12 mmol) and benzoyl chloride (413 μL, 3.56 mmol), and the mixture was stirred at room temperature for 3 h until complete, as indicated by TLC. The solvent was evaporated under reduced pressure, and the residue was purified on a silica gel column using mixtures of EtOAc/petroleum ether (1/1, v/v) as eluent, obtaining the target compound 3 as a white solid. Yield: 43%, m.p. 258–260 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.60 (s, 1H), 8.72 (s, 1H), 8.13 (d, J = 7.7 Hz, 2H), 7.70 (t, J = 7.3 Hz, 1H), 7.59 (t, J = 7.6 Hz, 2H). HR-MS (EI) m/z Calcd. C10H7N3O3S (M+) 249.0208, Found 249.0209.
4.2.2. 2-Acetyloxy-N-(thiazol-2-yl)benzamide (4)
4 was synthesized by the general procedure of 3 given above, 2-amino-5-nitrothiazole was replaced by 2-aminothiazole, and benzoyl chloride was replaced by 2-acetoxybenzoyl chloride, to afford 4 as a white solid. Yield: 52%, m.p. 126–131 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.60 (s, 1H), 7.78 (d, J = 7.6 Hz, 1H), 7.62 (t, J = 7.8 Hz, 1H), 7.54 (d, J = 3.6 Hz, 1H), 7.41 (t, J = 7.5 Hz, 1H), 7.31–7.25 (m, 2H), 2.22 (s, 3H). HR-MS (EI) m/z Calcd. C12H10N2O3S (M+) 262.0412, Found 262.0415.
4.2.3. 2-Acetyloxy-N-(5-cyanothiazol-2-yl)benzamide (5)
5 was synthesized by the general procedure of 3 given above, 2-amino-5-nitrothiazole was replaced by 2-amino-5-cyanothiazole, and benzoyl chloride was replaced by 2-acetoxybenzoyl chloride, to afford 5 as a white solid. Yield: 29%, m.p. 185–187 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.43 (s, 1H), 8.42 (s, 1H), 7.79 (dd, J = 7.7, 1.4 Hz, 1H), 7.64 (td, J = 7.9, 1.5 Hz, 1H), 7.40 (td, J = 7.6, 0.8 Hz, 1H), 7.28 (d, J = 7.6 Hz, 1H), 2.19 (s, 3H). HR-MS (EI) m/z Calcd. C13H9N3O3S (M+) 287.0365, Found 287.0364.
4.2.4. Methyl 2-(2-acetoxybenzamido)thiazole-5-carboxylate (6)
6 was synthesized by the general procedure of 3 given above, 2-amino-5-nitrothiazole was replaced by methyl 2-aminothiazole-5-carboxylate, and benzoyl chloride was replaced by 2-acetoxybenzoyl chloride, to afford 6 as a white solid. Yield: 62%, m.p. 162–165 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.14 (s, 1H), 8.24 (s, 1H), 7.82 (d, J = 7.4 Hz, 1H), 7.66 (t, J = 7.8 Hz, 1H), 7.43 (t, J = 7.5 Hz, 1H), 7.30 (d, J = 8.1 Hz, 1H), 3.83 (s, 3H), 2.23 (s, 3H). HR-MS (EI) m/z Calcd. C14H12N2O5S (M+) 320.0467, Found 320.0465.
4.2.5. Benzyl 2-(2-acetoxybenzamido)thiazole-5-carboxylate (7)
7 was synthesized by the general procedure of 3 given above, 2-amino-5-nitrothiazole was replaced by 59, and benzoyl chloride was replaced by 2-acetoxybenzoyl chloride, to afford 7 as a white solid. Yield: 63%, m.p. 159–163 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.15 (s, 1H), 8.28 (s, 1H), 7.81 (d, J = 7.1 Hz, 1H), 7.66 (t, J = 7.1 Hz, 1H), 7.47–7.37 (m, 6H), 7.30 (d, J = 8.1 Hz, 1H), 5.34 (s, 2H), 2.23 (s, 3H). HR-MS (EI) m/z Calcd. C20H16N2O5S (M+) 396.0780, Found 396.0777.
4.2.6. 2-Acetyloxy-N-(5-(methylthio)thiazol-2-yl)benzamide (8)
8 was synthesized by the general procedure of 3 given above, 2-amino-5-nitrothiazole was replaced by 60, and benzoyl chloride was replaced by 2-acetoxybenzoyl chloride, to afford 8 as a white solid. Yield: 69%, m.p. 142–146 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.73 (s, 1H), 7.77 (d, J = 6.6 Hz, 1H), 7.63 (td, J = 8.0, 1.6 Hz, 1H), 7.55 (s, 1H), 7.41 (td, J = 7.6, 1.0 Hz, 1H), 7.28 (d, J = 8.1 Hz, 1H), 2.46 (s, 3H), 2.23 (s, 3H). HR-MS (EI) m/z Calcd. C13H12N2O3S2 (M+) 308.0289, Found 308.0287.
4.2.7. 2-Acetyloxy-N-(5-(methylsulfonyl)thiazol-2-yl)benzamide (9)
To a solution of 8 (107 mg, 0.35 mmol) in DCM (5 mL) was added a solution of 85% 3-chloroperoxybenzoic acid (70 mg, 0.35 mmol) in DCM (2 mL). After stirring at room temperature for 30 min, another solution of 85% 3-chloroperoxybenzoic acid (70 mg, 0.35 mmol) in DCM (2 mL) was added to the mixture until complete. The reaction was quenched by saturated aqueous Na2S2O3 solution, and then the mixture was poured into water (50 mL) and extracted with DCM. The combined organic layers were dried over anhydrous Na2SO4, filtered, and condensed to afford 9 as a white solid. Yield: 89%, m.p. 179–181 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.34 (s, 1H), 8.18 (s, 1H), 7.83 (d, J = 7.5 Hz, 1H), 7.68 (t, J = 7.7 Hz, 1H), 7.44 (t, J = 7.5 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 3.40 (s, 3H), 2.24 (s, 3H). HR-MS (EI) m/z Calcd. C13H12N2O5S2 (M+) 340.0188, Found 340.0186.
4.2.8. 2-(2-Acetoxybenzamido)thiazole-5-carboxylic acid (10)
To a mixture of 61 (80 mg, 0.30 mmol) in acetic anhydride (1.5 mL) was added two drops of H2SO4 (98%), and then heated to 75 °C for 20 min. The mixture was poured into water (40 mL) and extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4, filtered, and condensed to afford 9 as a white solid. Yield: 94%, m.p. 216–219 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.06 (s, 2H), 8.13 (s, 1H), 7.81 (d, J = 7.8 Hz, 1H), 7.65 (t, J = 7.5 Hz, 1H), 7.42 (t, J = 7.4 Hz, 1H), 7.30 (d, J = 7.9 Hz, 1H), 2.23 (s, 3H). HR-MS (EI) m/z Calcd. C13H10N2O5S (M+) 306.0310, Found 306.0309.
4.2.9. 2-Acetyloxy-N-(5-(piperidin-1-yl)thiazol-2-yl)benzamide (11)
11 was synthesized by the general procedure of 3 given above, 2-amino-5-nitrothiazole was replaced by 62, and benzoyl chloride was replaced by 2-acetoxybenzoyl chloride, to afford 11 as a yellow solid. Yield: 67%, m.p. 160–164 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.16 (s, 1H), 7.73 (d, J = 7.6 Hz, 1H), 7.59 (t, J = 7.6 Hz, 1H), 7.38 (t, J = 7.6 Hz, 1H), 7.25 (d, J = 8.1 Hz, 1H), 6.67 (s, 1H), 3.07–2.99 (m, 4H), 2.22 (s, 3H), 1.68–1.59 (m, 4H), 1.56–1.42 (m, 2H). HR-MS (EI) m/z Calcd. C17H19N3O3S (M+) 345.1147, Found 345.1146.
4.2.10. 2-Acetyloxy-N-(5-morpholinothiazol-2-yl)benzamide (12)
12 was synthesized by the general procedure of 3 given above, 2-amino-5-nitrothiazole was replaced by 63, and benzoyl chloride was replaced by 2-acetoxybenzoyl chloride, to afford 12 as a yellow solid. Yield: 95%, m.p. 172–175 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.22 (s, 1H), 7.73 (d, J = 7.2 Hz, 1H), 7.60 (t, J = 7.3 Hz, 1H), 7.38 (t, J = 7.5 Hz, 1H), 7.25 (d, J = 8.0 Hz, 1H), 6.74 (s, 1H), 3.75–3.70 (m, 4H), 3.05–3.00 (m, 4H), 2.22 (s, 3H). HR-MS (EI) m/z Calcd. C16H17N3O4S (M+) 347.0940, Found 347.0939.
4.2.11. 2-Acetyloxy-N-(5-(1H-pyrrol-1-yl)thiazol-2-yl)benzamide (13)
13 was synthesized by the general procedure of 3 given above, 2-amino-5-nitrothiazole was replaced by 64, and benzoyl chloride was replaced by 2-acetoxybenzoyl chloride, to afford 13 as a gray solid. Yield: 58%, mp 218–221 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.56 (s, 1H), 11.38 (s, 1H), 7.79 (d, J = 7.4 Hz, 1H), 7.66–7.57 (m, 2H), 7.41 (t, J = 7.5 Hz, 1H), 7.28 (d, J = 8.1 Hz, 1H), 6.85 (s, 1H), 6.30 (s, 1H), 6.10 (s, 1H), 2.23 (s, 3H). EI-MS m/z 327.1 (M+); 165 (100%); HRMS (EI) m/z Calcd. C16H13N3O3S (M+) 327.0678, Found 327.0679.
4.2.12. 2-Acetyloxy-N-(5-(piperazin-1-yl)thiazol-2-yl)benzamide (14)
To a solution of 66 (100 mg, 0.22 mmol) in toluene (5 mL) was added dry silica gel (500 mg), and then the mixture was heated to 80 °C for 8 h. The solvent was evaporated under reduced pressure, and the residue was purified on a silica gel column using mixtures of MeOH/DCM (1/20, v/v) as eluent, obtaining the target compound 14 as a yellow solid. Yield: 51%, m.p. 216–218 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.89 (s, 2H), 7.96 (d, J = 7.7 Hz, 1H), 7.43 (t, J = 7.0 Hz, 1H), 7.08–6.87 (m, 2H), 6.79 (s, 1H), 3.59 (s, 4H), 3.14–2.93 (m, 4H), 2.04 (s, 3H). HR-MS (EI) m/z Calcd. C16H18N4O3S (M+) 346.1100, Found 346.1101.
4.2.13. 4-Fluoro-2-hydroxy-N-(5-morpholinothiazol-2-yl)benzamide (15)
To a solution of 4-fluoro-2-hydroxybenzoic acid (337 mg, 2.16 mmol) in toluene (5 mL) was added two drops of DMF and thionyl chloride (5 mL) under nitrogen, and the mixture was heated to 80 °C for 3 h. Then, the solvent was evaporated under reduced pressure to afford crude 67, which was used directly. To a solution of 63 (200 mg, 1.08 mmol) and triethylamine (0.45 mL, 3.23 mmol) in anhydrous THF (5 mL) was added a solution of 67 obtained above in anhydrous THF (5 mL), and the mixture was stirred at room temperature for 3 h until complete, as indicated by TLC. The solvent was evaporated to dryness under reduced pressure, and the residue was purified on a silica gel column using mixtures of EtOAc/petroleum ether (1/1, v/v) as eluent, obtaining the target compound 15 as a yellow solid. Yield: 41%, m.p. 255–257 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.99 (s, 2H), 7.73 (d, J = 7.9 Hz, 1H), 7.42–7.29 (m, 1H), 6.92–6.83 (m, 1H), 6.82 (s, 1H), 3.82–3.66 (m, 4H), 3.12–2.95 (m, 4H). HR-MS (EI) m/z Calcd. C14H14FN3O3S (M+) 323.0740, Found 323.0736.
4.2.14. 4-Chloro-2-hydroxy-N-(5-morpholinothiazol-2-yl)benzamide (16)
16 was synthesized by the general procedure of 15 given above, 4-fluoro-2-hydroxybenzoic acid was replaced by 4-choro-2-hydroxybenzoic acid, to afford 16 as a yellow solid. Yield: 45%, m.p. 249–253 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.77 (s, 2H), 7.92 (d, J = 8.4 Hz, 1H), 7.08–6.90 (m, 2H), 6.79 (s, 1H), 3.80–3.64 (m, 4H), 3.13–2.94 (m, 4H). HR-MS (EI) m/z Calcd. C14H14ClN3O3S (M+) 339.0444, Found 339.0443, Calcd. C14H14ClN3O3S (M+) 341.0415, Found 341.0420.
4.2.15. 4-Bromo-2-hydroxy-N-(5-morpholinothiazol-2-yl)benzamide (17)
17 was synthesized by the general procedure of 15 given above, 4-fluoro-2-hydroxybenzoic acid was replaced by 4-bromo-2-hydroxybenzoic acid, to afford 17 as a brown solid. Yield: 53%, m.p. 226–230 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.74 (s, 2H), 7.83 (d, J = 8.3 Hz, 1H), 7.14 (s, 1H), 7.09 (d, J = 8.3 Hz, 1H), 6.78 (s, 1H), 3.79–3.65 (m, 4H), 3.08–2.96 (m, 4H). HR-MS (EI) m/z Calcd. C14H14BrN3O3S (M+) 382.9939, Found 382.9934, Calcd. C14H14BrN3O3S (M+) 384.9919, Found 384.9922.
4.2.16. 2-Hydroxy-N-(5-morpholinothiazol-2-yl)-4-(trifluoromethyl)benzamide (18)
18 was synthesized by the general procedure of 15 given above, 4-fluoro-2-hydroxybenzoic acid was replaced by 2-hydroxy-4-(trifluoromethyl)benzoic acid, to afford 18 as a yellow solid. Yield: 21%, m.p. 240–243 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.92 (s, 2H), 8.08 (d, J = 8.3 Hz, 1H), 7.29–7.15 (m, 2H), 6.82 (s, 1H), 3.81–3.66 (m, 4H), 3.12–2.99 (m, 4H). HR-MS (EI) m/z Calcd. C15H14F3N3O3S (M+) 373.0708, Found 373.0710.
4.2.17. 2-Hydroxy-N-(5-morpholinothiazol-2-yl)benzamide (19)
To a solution of 11 (100 mg, 0.29 mmol) in THF (10 mL) was added a solution of LiOH (20 mg, 0.86 mmol) in water (5 mL), and the mixture was stirred at room temperature overnight. The pH was adjusted to about 5–6 with 1 mol/L HCl, and the mixture was poured into water and extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4, filtered, and condensed to afford 19 as a yellow solid. Yield: 87%, m.p. 220–222 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.09 (s, 2H), 7.96 (d, J = 6.8 Hz, 1H), 7.43 (t, J = 7.4 Hz, 1H), 7.07–6.87 (m, 2H), 6.77 (s, 1H), 3.82–3.66 (m, 4H), 3.15–2.93 (m, 4H). HR-MS (EI) m/z Calcd. C14H15N3O3S (M+) 305.0834, Found 305.0835.
4.2.18. 3-Hydroxy-N-(5-morpholinothiazol-2-yl)benzamide (20)
20 was synthesized by the general procedure of 19 given above, 11 was replaced by 22, to afford 20 as a yellow solid. Yield: 93%, m.p. 228–232 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.18 (s, 1H), 9.79 (s, 1H), 7.49 (d, J = 7.9 Hz, 1H), 7.42–7.37 (m, 1H), 7.30 (t, J = 7.9 Hz, 1H), 6.99 (dd, J = 8.0, 1.9 Hz, 1H), 6.75 (s, 1H), 3.77–3.69 (m, 4H), 3.08–2.99 (m, 4H). HR-MS (EI) m/z Calcd. C14H15N3O3S (M+) 305.0834, Found 305.0835.
4.2.19. 4-Hydroxy-N-(5-morpholinothiazol-2-yl)benzamide (21)
21 was synthesized by the general procedure of 19 given above, 11 was replaced by 23, to afford 21 as a yellow solid. Yield: 90%, m.p. 229–231 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.97 (s, 1H), 10.25 (s, 1H), 7.94 (d, J = 8.8 Hz, 2H), 6.84 (d, J = 8.8 Hz, 2H), 6.73 (s, 1H), 3.78–3.67 (m, 4H), 3.07–2.95 (m, 4H). HR-MS (EI) m/z Calcd. C14H15N3O3S (M+) 305.0834, Found 305.0833.
4.2.20. 3-Acetyloxy-N-(5-morpholinothiazol-2-yl)benzamide (22)
22 was synthesized by the general procedure of 15 given above, 4-fluoro-2-hydroxybenzoic acid was replaced by 3-acetoxybenzoic acid, to afford 22 as a yellow solid. Yield: 59%, m.p. 191–194 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.35 (s, 1H), 7.96 (d, J = 7.9 Hz, 1H), 7.82 (t, J = 1.9 Hz, 1H), 7.57 (t, J = 7.9 Hz, 1H), 7.39 (dd, J = 8.0, 1.6 Hz, 1H), 6.77 (s, 1H), 3.79–3.67 (m, 4H), 3.09–2.98 (m, 4H), 2.31 (s, 3H). HR-MS (EI) m/z Calcd. C16H17N3O4S (M+) 347.0940, Found 347.0942.
4.2.21. 4-Acetyloxy-N-(5-morpholinothiazol-2-yl)benzamide (23)
23 was synthesized by the general procedure of 15 given above, 4-fluoro-2-hydroxybenzoic acid was replaced by 4-acetoxybenzoic acid, to afford 23 as a yellow solid. Yield: 47%, m.p. 212–215 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.28 (s, 1H), 8.09 (d, J = 8.6 Hz, 2H), 7.29 (d, J = 8.6 Hz, 2H), 6.77 (s, 1H), 3.80–3.68 (m, 4H), 3.12–2.96 (m, 4H), 2.31 (s, 3H). HR-MS (EI) m/z Calcd. C16H17N3O4S (M+) 347.0940, Found 347.0938.
4.2.22. N-(5-Morpholinothiazol-2-yl)benzamide (24)
To a solution of 63 (100 mg, 0.54 mmol) in anhydrous THF (5 mL) was added triethylamine (225 μL, 1.62 mmol) and benzoyl chloride (75 μL, 0.65 mmol, commercially available), and the mixture was stirred at room temperature for 3 h until completed, as indicated by TLC. The solvent was evaporated to dryness under reduced pressure, and the residue was purified on a silica gel column using mixtures of EtOAc/petroleum ether (1/1, v/v) as eluent, obtaining the target compound 24 as a yellow solid. Yield: 68%, m.p. 211–213 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.28 (s, 1H), 8.05 (d, J = 7.3 Hz, 2H), 7.61 (t, J = 7.3 Hz, 1H), 7.53 (t, J = 7.5 Hz, 2H), 6.77 (s, 1H), 3.82–3.63 (m, 4H), 3.12–2.93 (m, 4H). HR-MS (EI) m/z Calcd. C14H15N3O2S (M+) 289.0885, Found 289.0887.
4.2.23. N-(5-Morpholinothiazol-2-yl)-2-(trifluoromethyl)benzamide (25)
25 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by 2-(trifluoromethyl)benzoyl chloride, to afford 25 as a white solid. Yield: 43%, m.p. 262–265 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.42 (s, 1H), 7.96–7.55 (m, 4H), 6.74 (s, 1H), 3.73 (s, 4H), 3.03 (s, 4H). HR-MS (EI) m/z Calcd. C15H14F3N3O2S (M+) 357.0759, Found 357.0760.
4.2.24. 2-Fluoro-N-(5-morpholinothiazol-2-yl)benzamide (26)
26 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by 2-fluorobenzoyl chloride, to afford 26 as a yellow solid. Yield: 73%, m.p. 204–207 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.23 (s, 1H), 7.69 (td, J = 7.6, 1.5 Hz, 1H), 7.61 (ddd, J = 9.3, 6.3, 1.7 Hz, 1H), 7.40–7.28 (m, 2H), 6.75 (s, 1H), 3.81–3.65 (m, 4H), 3.11–2.94 (m, 4H). HR-MS (EI) m/z Calcd. C14H14FN3O2S (M+) 307.0791, Found 307.0794.
4.2.25. N-(5-Morpholinothiazol-2-yl)-2-nitrobenzamide (27)
27 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by 2-nitrobenzoyl chloride, to afford 27 as a yellow solid. Yield: 38%, m.p. 245–248 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.51 (s, 1H), 8.15 (d, J = 8.2 Hz, 1H), 7.86 (t, J = 7.4 Hz, 1H), 7.82–7.71 (m, 2H), 6.75 (s, 1H), 3.80–3.67 (m, 4H), 3.13–2.95 (m, 4H). HR-MS (EI) m/z Calcd. C14H14N4O4S (M+) 334.0736, Found 334.0732.
4.2.26. 2-Methyl-N-(5-morpholinothiazol-2-yl)benzamide (28)
28 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by 2-methylbenzoyl chloride, to afford 28 as a yellow solidYield: 63%, m.p. 223–226 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.14 (s, 1H), 7.49 (d, J = 7.4 Hz, 1H), 7.45–7.37 (m, 1H), 7.34–7.25 (m, 2H), 6.73 (s, 1H), 3.79–3.66 (m, 4H), 3.08–2.96 (m, 4H), 2.38 (s, 3H). HR-MS (EI) m/z Calcd. C15H17N3O3S (M+) 303.1041, Found 303.1043.
4.2.27. 2-Methoxy-N-(5-morpholinothiazol-2-yl)benzamide (29)
29 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by 2-methoxybenzoyl chloride, to afford 29 as a yellow solidYield: 33%, m.p. 154–156 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.47 (s, 1H), 7.71 (dd, J = 7.6, 1.7 Hz, 1H), 7.60–7.51 (m, 1H), 7.21 (d, J = 8.4 Hz, 1H), 7.09 (t, J = 7.5 Hz, 1H), 6.73 (s, 1H), 3.92 (s, 3H), 3.79–3.67 (m, 4H), 3.08–2.95 (m, 4H). HR-MS (EI) m/z Calcd. C15H17N3O3S (M+) 319.0991, Found 319.0992.
4.2.28. N-(5-Morpholinothiazol-2-yl)-1-naphthamide (30)
30 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by 1-naphthoyl chloride, to afford 30 as a yellow solid. Yield: 52%, m.p. 248–250 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.44 (s, 1H), 8.26–8.17 (m, 1H), 8.11 (d, J = 8.3 Hz, 1H), 8.06–8.00 (m, 1H), 7.81 (dd, J = 7.1, 0.9 Hz, 1H), 7.68–7.54 (m, 3H), 6.78 (s, 1H), 3.83–3.68 (m, 4H), 3.15–2.97 (m, 4H). HR-MS (EI) m/z Calcd. C18H17N3O2S (M+) 339.1041, Found 339.1039.
4.2.29. N-(5-Morpholinothiazol-2-yl)-2-naphthamide (31)
31 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by 2-naphthoyl chloride, to afford 31 as a yellow solid. Yield: 38%, m.p. 221–225 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.43 (s, 1H), 8.73 (s, 1H), 8.13–7.98 (m, 4H), 7.71–7.60 (m, 2H), 6.79 (s, 1H), 3.81–3.69 (m, 4H), 3.10–2.99 (m, 4H). HR-MS (EI) m/z Calcd. C18H17N3O2S (M+) 339.1041, Found 339.1042.
4.2.30. N-(5-Morpholinothiazol-2-yl)furan-2-carboxamide (32)
32 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by 2-furoyl chloride, to afford 32 as a yellow solid. Yield: 63%, m.p. 199–201 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.22 (s, 1H), 7.97 (d, J = 0.8 Hz, 1H), 7.57 (d, J = 3.1 Hz, 1H), 6.75 (s, 1H), 6.71 (dd, J = 3.5, 1.7 Hz, 1H), 3.80–3.65 (m, 4H), 3.09–2.95 (m, 4H). HR-MS (EI) m/z Calcd. C12H13N3O3S (M+) 279.0678, Found 279.0679.
4.2.31. N-(5-Morpholinothiazol-2-yl)thiophene-2-carboxamide (33)
33 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by 2-thiophenecarbonyl chloride, to afford 33 as a yellow solid. Yield: 65%, m.p. 202–205 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.39 (s, 1H), 8.16 (s, 1H), 7.92 (d, J = 4.8 Hz, 1H), 7.23 (dd, J = 4.8, 4.0 Hz, 1H), 6.75 (s, 1H), 3.80–3.66 (m, 4H), 3.09–2.95 (m, 4H). HR-MS (EI) m/z Calcd. C12H13N3O2S2 (M+) 295.0449, Found 295.0452.
4.2.32. N-(5-Morpholinothiazol-2-yl)furan-3-carboxamide (34)
34 was synthesized by the general procedure of 15 given above, 4-fluoro-2-hydroxybenzoic acid was replaced by 3-furoic acid, to afford 34 as a yellow solid. Yield: 61%, m.p. 232–235 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.08 (s, 1H), 8.50 (d, J = 0.5 Hz, 1H), 7.81 (t, J = 1.7 Hz, 1H), 7.06 (dd, J = 1.8, 0.6 Hz, 1H), 6.74 (s, 1H), 3.80–3.65 (m, 4H), 3.09–2.93 (m, 4H). HR-MS (EI) m/z Calcd. C12H13N3O3S (M+) 279.0678, Found 279.0677.
4.2.33. N-(5-Morpholinothiazol-2-yl)thiophene-3-carboxamide (35)
35 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by 3-thiophenecarbonyl chloride, to afford 35 as a yellow solid. Yield: 63%, m.p. 218–221 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.14 (s, 1H), 8.53 (d, J = 1.5 Hz, 1H), 7.70 (dd, J = 5.0, 1.0 Hz, 1H), 7.66 (dd, J = 5.0, 2.9 Hz, 1H), 6.75 (s, 1H), 3.83–3.62 (m, 4H), 3.14–2.91 (m, 4H). HR-MS (EI) m/z Calcd. C12H13N3O2S2 (M+) 295.0449, Found 295.0450.
4.2.34. 5-Chloro-N-(5-morpholinothiazol-2-yl)thiophene-2-carboxamide (36)
36 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by 5-chloro-2-thiophenecarbonyl chloride, to afford 36 as a yellow solid. Yield: 57%, m.p. 222–225 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.54 (s, 1H), 7.99 (s, 1H), 7.26 (d, J = 3.9 Hz, 1H), 6.75 (s, 1H), 3.81–3.65 (m, 4H), 3.11–2.93 (m, 4H). EI-MS m/z 329.0 (M+); 329 (100%); HR-MS (EI) m/z Calcd. C12H1235ClN3O2S2 (M+) 329.0059, Found 329.0061, Calcd. C12H12ClN3O2S2 (M+) 331.0030, Found 331.0031.
4.2.35. 5-Methyl-N-(5-morpholinothiazol-2-yl)thiophene-2-carboxamide (37)
37 was synthesized by the general procedure of 15 given above, 4-fluoro-2-hydroxybenzoic acid was replaced by 5-methyl-2-thiophenecarboxylic acid, to afford 37 as a yellow solid. Yield: 89%, m.p. 195–197 °C; 1H NMR (400 MHz, acetone-d6) δ 10.89 (s, 1H), 7.93 (d, J = 3.5 Hz, 1H), 6.92 (d, J = 2.8 Hz, 1H), 6.66 (s, 1H), 3.87–3.68 (m, 4H), 3.16–2.98 (m, 4H), 2.55 (s, 3H). HR-MS (EI) m/z Calcd. C13H15N3O2S2 (M+) 309.0606, Found 309.0607.
4.2.36. 3-Methyl-N-(5-morpholinothiazol-2-yl)thiophene-2-carboxamide (38)
38 was synthesized by the general procedure of 15 given above, 4-fluoro-2-hydroxybenzoic acid was replaced by 3-methyl-2-thiophenecarboxylic acid, to afford 38 as a yellow solid. Yield: 81%, m.p. 160–162 °C; 1H NMR (400 MHz, acetone-d6) δ 10.78 (s, 1H), 7.62 (d, J = 5.0 Hz, 1H), 7.04 (d, J = 5.0 Hz, 1H), 6.62 (s, 1H), 3.82–3.74 (m, 4H), 3.11–3.02 (m, 4H), 2.58 (s, 3H). HR-MS (EI) m/z Calcd. C13H15N3O2S2 (M+) 309.0606, Found 309.0605.
4.2.37. N-(5-Morpholinothiazol-2-yl)cyclopropanecarboxamide (39)
39 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by cyclopropanecarboxyl chloride, to afford 39 as a white solid. Yield: 61%, m.p. 196–198 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.01 (s, 1H), 6.65 (s, 1H), 3.76–3.64 (m, 4H), 3.02–2.89 (m, 4H), 1.93–1.82 (m, 1H), 0.91–0.78 (m, 4H). HR-MS (EI) m/z Calcd. C11H15N3O2S (M+) 253.0885, Found 253.0886.
4.2.38. N-(5-Morpholinothiazol-2-yl)cyclobutanecarboxamide (40)
40 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by cyclobutanecarbonyl chloride, to afford 40 as a yellow solid. Yield: 46%, m.p. 179–184 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.60 (s, 1H), 6.65 (s, 1H), 3.78–3.64 (m, 4H), 3.32–3.23 (m, 1H), 3.04–2.91 (m, 4H), 2.27–2.14 (m, 2H), 2.14–2.03 (m, 2H), 2.00–1.87 (m, 1H), 1.86–1.73 (m, 1H). HR-MS (EI) m/z Calcd. C12H17N3O2S (M+) 267.1041, Found 267.1036.
4.2.39. N-(5-Morpholinothiazol-2-yl)cyclopentanecarboxamide (41)
41 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by cyclopentanecarbonyl chloride, to afford 41 as a white solid. Yield: 64%, m.p. 175–177 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.71 (s, 1H), 6.66 (s, 1H), 3.81–3.61 (m, 4H), 3.10–2.90 (m, 4H), 2.89–2.78 (m, 1H), 1.91–1.76 (m, 2H), 1.75–1.60 (m, 4H), 1.60–1.44 (m, 2H). HR-MS (EI) m/z Calcd. C13H19N3O2S (M+) 281.1198, Found 281.1197.
4.2.40. N-(5-Morpholinothiazol-2-yl)cyclohexanecarboxamide (42)
42 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by cyclohexylcarbonyl chloride, to afford 42 as a white solid. Yield: 68%, m.p. 180–182 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.63 (s, 1H), 6.65 (s, 1H), 3.76–3.65 (m, 4H), 3.01–2.90 (m, 4H), 2.47–2.36 (m, 1H), 1.74 (t, J = 12.6 Hz, 4H), 1.63 (d, J = 8.4 Hz, 1H), 1.44–1.31 (m, 2H), 1.29–1.14 (m, 3H). HR-MS (EI) m/z Calcd. C14H21N3O2S (M+) 295.1354, Found 295.1353.
4.2.41. N-(5-Morpholinothiazol-2-yl)cycloheptanecarboxamide (43)
43 was synthesized by the general procedure of 15 given above, 4-fluoro-2-hydroxybenzoic acid was replaced by cycloheptanecarboxylic acid, to afford 43 as a white solid. Yield: 70%, m.p. 169–171 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.61 (s, 1H), 6.65 (s, 1H), 3.81–3.59 (m, 4H), 3.05–2.87 (m, 4H), 2.66–2.55 (m 1H), 1.88–1.74 (m, 2H), 1.73–1.63 (m, 2H), 1.63–1.31 (m, 8H). HR-MS (EI) m/z Calcd. C15H23N3O2S (M+) 309.1511, Found 309.1509.
4.2.42. (3r,5r,7r)-N-(5-Morpholinothiazol-2-yl)adamantane-1-carboxamide (44)
44 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by 1-adamantanecarbonyl chloride, to afford 44 as a white solid. Yield: 80%, m.p. 190–194 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.31 (s, 1H), 6.68 (s, 1H), 3.79–3.60 (m, 4H), 3.04–2.85 (m, 4H), 1.99 (s, 3H), 1.89 (d, J = 2.1 Hz, 6H), 1.68 (s, 6H). EI-MS m/z 347.2 (M+); 347 (100%); HR-MS (EI) m/z Calcd. C18H25N3O2S (M+) 347.1667, Found 347.1668.
4.2.43. N-(5-Morpholinothiazol-2-yl)acetamide (45)
45 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by acetyl chloride, to afford 45 as a white solid. Yield: 39%, m.p. 253–256 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.73 (s, 1H), 6.65 (s, 1H), 3.79–3.61 (m, 4H), 3.08–2.87 (m, 4H), 2.07 (s, 3H). EI-MS m/z 227.1 (M+); 185 (100%); HR-MS (EI) m/z Calcd. C9H13N3O2S (M+) 227.0728, Found 227.0730.
4.2.44. N-(5-Morpholinothiazol-2-yl)propionamide (46)
46 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by propionyl chloride, to afford 46 as a white solid. Yield: 65%, m.p. 198–200 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.68 (s, 1H), 6.65 (s, 1H), 3.78–3.63 (m, 4H), 3.05–2.88 (m, 4H), 2.37 (q, J = 7.5 Hz, 2H), 1.06 (t, J = 7.5 Hz, 3H). EI-MS m/z 241.1 (M+); 185 (100%); HR-MS (EI) m/z Calcd. C10H15N3O2S (M+) 241.0885, Found 241.0884.
4.2.45. N-(5-Morpholinothiazol-2-yl)butyramide (47)
47 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by butyryl chloride, to afford 47 as a white solid. Yield: 48%, m.p. 140–142 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.69 (s, 1H), 6.65 (s, 1H), 3.76–3.65 (m, 4H), 3.02–2.92 (m, 4H), 2.33 (t, J = 7.3 Hz, 2H), 1.65–1.52 (m, 2H), 0.88 (t, J = 7.4 Hz, 3H). EI-MS m/z 255.1 (M+); 185 (100%); HR-MS (EI) m/z Calcd. C11H17N3O2S (M+) 255.1041, Found 255.1040.
4.2.46. N-(5-Morpholinothiazol-2-yl)hexanamide (48)
48 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by hexanoyl chloride, to afford 48 as a white solid. Yield: 43%, m.p. 94–96 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.69 (s, 1H), 6.65 (s, 1H), 3.75–3.68 (m, 4H), 3.01–2.92 (m, 4H), 2.35 (t, J = 7.4 Hz, 2H), 1.61–1.52 (m, 2H), 1.31–1.20 (m, 4H), 0.86 (t, J = 7.0 Hz, 3H). EI-MS m/z 283.1 (M+); 185 (100%); HR-MS (EI) m/z Calcd. C13H21N3O2S (M+) 283.1354, Found 283.1353.
4.2.47. 3-Methyl-N-(5-morpholinothiazol-2-yl)butanamide (49)
49 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by isovaleryl chloride, to afford 49 as a white solid. Yield: 70%, m.p. 153–155 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.70 (s, 1H), 6.66 (s, 1H), 3.78–3.65 (m, 4H), 3.02–2.90 (m, 4H), 2.24 (d, J = 7.2 Hz, 2H), 2.12–1.97 (m, 1H), 0.90 (d, J = 6.7 Hz, 6H). HR-MS (EI) m/z Calcd. C12H19N3O2S (M+) 269.1198, Found 269.1199.
4.2.48. 4,4,4-Trifluoro-N-(5-morpholinothiazol-2-yl)butanamide (50)
To a solution of 4,4,4-trifluorobutyric acid (500 mg, 3.52 mmol) in DCM (1 mL) was added two drops of DMF and oxalyl chloride (357 μL, 4.22 mmol) dropwise at 0 °C. After stirring at 0 °C for 20 min, the mixture was warmed to room temperature for 2 h. Then, the solvent was evaporated under reduced pressure to afford crude 102, which was used directly. To a solution of 63 (200 mg, 1.08 mmol) and triethylamine (0.45 mL, 3.23 mmol) in anhydrous THF (5 mL) was added a solution of 102 obtained above in anhydrous THF (5 mL), and the mixture was stirred at room temperature for 3 h until complete, as indicated by TLC. The solvent was evaporated to dryness under reduced pressure, and the residue was purified on a silica gel column using mixtures of EtOAc/petroleum ether (1/1, v/v) as eluent, obtaining the target compound 50 as a white solid. Yield: 71%, m.p. 199–201 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.89 (s, 1H), 6.67 (s, 1H), 3.75–3.66 (m, 4H), 3.03–2.93 (m, 4H), 2.71–2.55 (m, 4H). HR-MS (EI) m/z Calcd. C11H14F3N3O2S (M+) 309.0759, Found 309.0758.
4.2.49. N-(5-Morpholinothiazol-2-yl)-2-phenylacetamide (51)
51 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by phenylacetyl chloride, to afford 51 as a yellow solid. Yield: 57%, m.p. 175–179 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.01 (s, 1H), 7.39–7.28 (m, 4H), 7.28–7.21 (m, 1H), 6.67 (s, 1H), 3.78–3.61 (m, 6H), 3.03–2.88 (m, 4H). HR-MS (EI) m/z Calcd. C15H17N3O2S (M+) 303.1041, Found 303.1042.
4.2.50. N-(5-Morpholinothiazol-2-yl)-3-phenylpropanamide (52)
52 was synthesized by the general procedure of 24 given above, benzoyl chloride was replaced by hydrocinnamoyl chloride, to afford 52 as a white solid. Yield: 72%, m.p. 188–190 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.74 (s, 1H), 7.36–7.25 (m, 2H), 7.25–7.13 (m, 3H), 6.65 (s, 1H), 3.77–3.63 (m, 4H), 3.05–2.92 (m, 4H), 2.89 (t, J = 7.6 Hz, 2H), 2.68 (t, J = 7.7 Hz, 2H). HR-MS (EI) m/z Calcd. C16H19N3O2S (M+) 317.1198, Found 317.1196.
4.2.51. N-(5-Morpholinothiazol-2-yl)-2-oxocyclohexanecarboxamide (53)
53 was synthesized by the general procedure of 50 given above, 4,4,4-trifluorobutyric acid was replaced by 2-oxocyclohexane-1-carboxylic acid, to afford 53 as a yellow solid. Yield: 21%, m.p. 175–178 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.63 (s, 1H), 6.67 (s, 1H), 3.78–3.66 (m, 4H), 3.61 (dd, J = 12.2, 5.6 Hz, 1H), 3.07–2.89 (m, 4H), 2.45–2.22 (m, 2H), 2.16–1.75 (m, 4H), 1.72–1.54 (m, 2H). HR-MS (EI) m/z Calcd. C14H19N3O3S (M+) 309.1147, Found 309.1145.
4.2.52. N-(5-Morpholinothiazol-2-yl)-3-oxobutanamide (54)
54 was synthesized by the general procedure of 50 given above, and 4,4,4-trifluorobutyric acid was replaced by 3-oxobutanoic acid, to afford 54 as a yellow solid. Yield: 25%, m.p. 172–176 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.80 (s, 1H), 6.68 (s, 1H), 3.76–3.68 (m, 4H), 3.63 (s, 2H), 3.03–2.94 (m, 4H), 2.18 (s, 3H). HR-MS (EI) m/z Calcd. C11H15N3O3S (M+) 269.0834, Found 269.0833.
4.2.53. 2-(2-Hydroxybenzamido)thiazole-5-carboxylic acid (61)
A mixture of 6 (143 mg, 0.45 mmol) and LiOH (43 mg, 1.80 mmol) in THF (10 mL) and water (5 mL) was stirred at room temperature overnight. The pH was adjusted to about 5–6 with 1 mol/L HCl, and the mixture was poured into water and extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4, filtered, and condensed to give 61 as a white solid. Yield: 92%. 1H NMR (400 MHz, DMSO-d6) δ 13.20 (s, 1H), 12.19 (s, 2H), 8.15 (s, 1H), 7.97 (d, J = 4.8 Hz, 1H), 7.49 (s, 1H), 7.14–6.94 (m, 2H).
4.2.54. 5-(Piperidin-1-yl)thiazol-2-amine (62)
To a solution of 2-amino-5-bromothiazole hydrobromide (600 mg, 2.31 mmol) in DMF (10 mL) was added potassium carbonate (478 mg, 3.46 mmol) and piperidine (422 μL, 4.61 mmol), and the mixture was heated to 60 °C for 3 h. The mixture was poured into water (40 mL) and extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4, filtered, and condensed. The residue was washed with ether to afford 62 as a brown solid. Yield: 97%. 1H NMR (400 MHz, DMSO-d6) δ 6.40 (s, 2H), 6.23 (s, 1H), 2.82–2.73 (m, 4H), 1.62–1.53 (m, 4H), 1.48–1.39 (m, 2H).
4.2.55. 5-Morpholinothiazol-2-amine (63)
63 was synthesized by the general procedure of 62 given above, piperidine was replaced by morpholine, to afford 63 as a brown solid. Yield: 68%. 1H NMR (400 MHz, DMSO-d6) δ 6.48 (s, 2H), 6.28 (s, 1H), 3.70–3.61 (m, 4H), 2.84–2.75 (m, 4H).
4.2.56. 5-(1H-Pyrrol-1-yl)thiazol-2-amine (64)
64 was synthesized by the general procedure of 62 given above, piperidine was replaced by pyrrole, to afford 64 as a brown solid. Yield: 59%. 1H NMR (400 MHz, DMSO-d6) δ 11.07 (s, 1H), 7.02 (s, 1H), 6.88 (s, 2H), 6.71 (s, 1H), 6.04–5.95 (m, 2H).
4.2.57. tert-Butyl 4-(2-aminothiazol-5-yl)piperazine-1-carboxylate (65)
65 was synthesized by the general procedure of 62 given above, piperidine was replaced by tert-butyl piperazine-1-carboxylate, to afford 65 as a white solid. Yield: 70%. 1H NMR (400 MHz, DMSO-d6) δ 6.51 (s, 2H), 6.32 (s, 1H), 3.46–3.35 (m, 4H), 2.82–2.69 (m, 4H), 1.40 (s, 9H).
4.2.58. tert-Butyl 4-(2-(2-acetoxybenzamido)thiazol-5-yl)piperazine-1-carboxylate (66)
66 was synthesized by the general procedure of 3 given above, 2-amino-5-nitrothiazole was replaced by 65, and benzoyl chloride was replaced by 2-acetoxybenzoyl chloride, to afford 66 as a white solid. Yield: 74%. 1H NMR (400 MHz, DMSO-d6) δ 12.24 (s, 1H), 7.74 (d, J = 7.6 Hz, 1H), 7.59 (t, J = 7.8 Hz, 1H), 7.38 (t, J = 7.5 Hz, 1H), 7.25 (d, J = 8.0 Hz, 1H), 6.76 (s, 1H), 3.50–3.44 (m, 4H), 3.04–2.98 (m, 4H), 2.22 (s, 3H), 1.42 (s, 9H).
4.3. Biological studies
4.3.1. Inhibition of p70S6K phosphorylation
The inhibitory activities of derivatives against the phosphorylation of p70S6K were tested by α-screen assay kit (PerkinElmer, USA). SH-SY5Y cells were seeded in 96-well plates. After grew to about 80% density, the cells were treated with varied compounds for 30 min, and then washed once with PBS. The lysis buffer from the assay kit was then added to the cells and incubated for 15–20 min at room temperature. According to the instructions of the kit, the phosphorylation level of p70S6K was detected in the cell lysates.
4.3.2. In vitro blood−brain barrier permeation assay
Brain penetration of compounds was evaluated by PAMPA–BBB assay. The PBL was obtained from Avanti Polar Lipids. The donor microplate (PVDF membrane, pore size 0.45 mm) and the acceptor microplate were both from Millipore (MA, USA). The acceptor 96-well microplate was filled with 300 μL of PBS/EtOH (7:3), and the filter membrane was impregnated with 4 μL of PBL in dodecane (20 mg/mL). Compounds were dissolved in DMSO at 5 mg/mL and diluted 50-fold in PBS/EtOH (7:3) to get the concentration of 100 μg/mL, and then 200 μL was added to the donor wells. The acceptor filter plate was carefully placed on the donor plate to form a sandwich, which was left undisturbed for 10 h at 25 °C. After incubation, the donor plate was carefully removed and the absorbance of compounds in the acceptor wells was determined using a UV spectrometer (250–500 nm). The absorbance of 200 μL compounds at 100 μg/mL in 300 μL of PBS/EtOH (7:3) was also tested as the theoretical equilibrium absorbance. Pe was calculated using the following Eq.(1).
| (1) |
where Vd is the volume in the donor well, Va is the volume in the acceptor well, A is the filter area, t is the permeation time, [drug]acceptor is the absorbance of the compound in the acceptor well, and [drug]equilibrium is the theoretical equilibrium absorbance.
4.3.3. Cell viability assay
Viability of SH-SY5Y cells and BV2 cells were respectively detected by MTT assay (Sigma, Saint Louis, MO, USA) and CCK-8 assay (YEASEN, Shanghai, China). Cells were seeded overnight at a density of 105 cells/well 96-well plates in 100 μL medium. The cells were co-incubated with different concentrations of NTZ and its derivatives for 24 h. For SH-SY5Y cells, the medium was removed and added 0.5 mg/μL MTT. For BV2 cells, the cells were added 10 μL CCK-8 directly. After incubation at 37 °C for 4 h, 100 μL of DMSO was added to SH-SY5Y cells each well, and the mixture was shaken at a low speed for 15 min to fully dissolve the formazan crystals, followed by the measurement of the absorbance at 490 nm using an MI3X spectrophotometer (Molecular Devices, Sunnyvale, CA, USA). For CCK-8 assay, the absorbance of mixture was directly measured at 450 nm.
4.3.4. Western blot assay
BV2 cells or SH-SY5Y cells were seeded in 12-well plates and treated with different compounds for 24 h, and then removed the medium and washed twice with PBS. The cells were lysed with RIPA buffer (Thermo, Waltham, MA, USA) containing a protease and phosphatase inhibitor cocktail (Thermo). BCA protein assay kits (Thermo) were used to detect the protein concentration of the samples. Samples were mixed with 2 × SDS-PAGE sample buffer (25% SDS, 62.5 mmol/L Tris-HCl, pH 6.8, 25% glycerol, 0.5 mol/L DTT and 0.1% Bromophenol Blue) and then boiled for 15 min at 99 °C. The samples were separated using SDS-PAGE and transferred to polyvinylidene difluoride membrane filters (GE, Boston, MA, USA). After incubation with the corresponding antibodies overnight, the blots were visualized using a Dura detection system (Thermo). Antibodies against P-AKT, AKT, P-mTOR, mTOR, P-ULK1, ULK1, P-p70S6K, p70S6K, p62, P199-Tau, P231-Tau, P396-Tau and GAPDH were obtained from Cell Signaling Technology (Boston, MA, USA).
4.3.5. Aβ clearance assay
BV2 cells were seeded in 12-well plates and cultured with different compounds for 24 h, followed by addition of 2 μg/mL soluble Aβ40/42, and incubated for 3 h. The culture was abandoned and BV2 cells were homogenized with 5 mol/L guanidine hydrogen chloride with a complete protease inhibitor cocktail (Thermo) for 30 min, and boiled for 15 min at 99 °C. Aβ40/42 ELISA kits were used to identify the intracellular Aβ peptide and protein concentration was determined using BCA protein assay kits, all Aβ concentration values were normalized to the total protein concentration.
4.3.6. Autophagic flux assay
Activations of NTZ and its derivatives on autophagy were investigated by using mTagRFP-mWassbi-LC3 translocation assay. BV2 cells were transfected with mRFP-GFP-LC3 plasmid via Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. The cells were treated with different compounds (20 μmol/L) with or without CQ for 24 h and then fixed with 4% paraformaldehyde. The images were acquired using a Leica confocal microscopy.
4.3.7. Animal experiment
All animal experiments and care were performed according to the Institutional Ethical Guidelines of Shanghai Institute of Materia Medica, Chinese Academy of Sciences. APP/PS1 transgenic mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Before the experiment, the genotypes of the mice were determined by tail biopsies with Tg-negative mice as negative control to confirm APP/PS1 DNA sequences. The mice were fed in standard conditions of 12 h of light/day at a temperature of 22 °C for 9 months. The test APP/PS1 transgenic mice were randomly divided into four groups with 10 mice for each group, and 10 non-transgenic normal mice were used as the negative control group. Compound 22 and NTZ were dissolved in a solution of 26% PEG400, 15% HS15 and 59% saline. The four groups of APP/PS1 transgenic mice were orally administered NTZ (30 mg/kg/day), compound 22 (10 and 30 mg/kg/day) or vehicle, respectively. After 100 days of administration, MWM assay and nesting test were used to determine the learning ability of the mice.
4.3.8. MWM assay
The mice were trained to find the invisible submerged platform in a circular pool (120 cm in diameter, 50 cm deep) filled with milk-tinted water using a variety of visual cues located on the pool walls; training was performed for 3 trials per day for 7 consecutive days. For each trial, mice were given 90 s to find the invisible platform. Each mouse was allowed to stay on the platform for 15 s, if the mouse found the platform within the given time. If the mouse failed to find the platform within the given time, it was manually placed to the platform and kept there for 15 s. On the 8th day, a probe trial was performed by removing the platform, and the animals were allowed to swim for 90 s to search for it. All data were collected for the animal performance analysis. For data analysis, the pool was divided into four equal quadrants formed by imaging lines, which intersected the center of the pool at right angles, and the quadrants were termed north, south, east and west.
4.3.9. Nesting test
Animal procedures were approved by the Institutional Animal Care and Use Committee at Shanghai Institute of Materia Medica. The mice were housed in separate cages before the experiment. A piece of cotton (5 cm × 5 cm in size) was placed into cage at 1 h before the dark cycle, and then the nesting score was determined after 12 h. The nesting score included 0–5 points, with 0 indicating that the cotton was not touched observably, 1 indicating that the cotton was moved but only bit a little, 2 indicating that the bit area was less than 30% and only on the corner or on one side of the cotton, 3 indicating that most of cotton was shredded but scattered in the corner of the cage without building to a nest, 4 indicating that the cotton was completely shredded and an identifiable but flat nest was built, 5 indicating that the cotton was fully shredded and built to a nest with a burrow.
4.3.10. Detecting Aβ level and phosphorylation of p70S6K, tau of APP/PS1 mice
The brain tissues were homogenized with RIPA buffer containing a protease inhibitor cocktail and phosphatase inhibitor cocktails and the homogenates were kept on ice for 30 min, followed by centrifugation at 20,000×g for 30 min at 4 °C. Protein concentration was determined using a BCA protein assay kit (Beyotime, Shanghai, China). Proteins were mixed with 4 × loading buffer (Thermo Scientific, Waltham, MA, USA) and boiled for 15 min at 95 °C. The Aβ level was detected by ELISA assay and the following steps referred to the cellular ELISA assay. The phosphorylation of p70S6K and tau were detected by Western blot assays and the following steps referred to the cellular assays.
Acknowledgments
Financial support for this research provided by the National Sciences and Technology Major Project of China, (2018ZX09711002-003-010), the National Natural Science Foundation of China, (81872747, 21672064), the 111 Project (B07023, China), the Chinese Postdoctoral Science Foundation (2018M641946), the Shanghai Sailing Program (19YF1412600, China), the Shanghai Morning Light Program (18CG33, China), the National Special Fund for State Key Laboratory of Bioreactor Engineering (2060204, China), and Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX18_1600, China) are gratefully acknowledged.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Author contributions
Jian Li and Xu Shen conceived and designed the study. Xiaokang Li, Xiaoxia Qiu and Jian Lu analyzed the data and wrote the manuscript. Xiaoxia Qiu, Xiaokang Li and Yixiang Xu synthesized the target compounds. Jian Lu and Lei Fan performed most of the biological experiments. Xiaokang Li, Jian Lu and Yixiang Xu revised the manuscript extensively. Jiaying Wang, Baoli Li, and Wenwen Liu assisted with carrying out experiments. Yixiang Xu, Fei Mao, and Jin Zhu assisted with analyzing experimental data. All the authors approved the final version of the manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2019.07.006.
Contributor Information
Xu Shen, Email: xshen@njucm.edu.cn.
Jian Li, Email: jianli@ecust.edu.cn.
Appendix A. Supporting information
The following are the Supplementary data to this article:
References
- 1.Blennow K., de Leon M.J., Zetterberg H. Alzheimer's disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 2.Cummings J.L., Morstorf T., Zhong K. Alzheimer's disease drug-development pipeline: few candidates, frequent failures. Alzheimer's Res Ther. 2014;6:37. doi: 10.1186/alzrt269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Selkoe D.J. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 4.Goedert M., Spillantini M.G. A century of Alzheimer's disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- 5.Tong H.J., Lou K.Y., Wang W. Near-infrared fluorescent probes for imaging of amyloid plaques in Alzheimer's disease. Acta Pharm Sin B. 2015;5:25–33. doi: 10.1016/j.apsb.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 7.Ballatore C., Lee V.M.Y., Trojanowski J.Q. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 8.Gómez-Isla T., Hollister R., West H., Mui S., Growdon J.H., Petersen R.C. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 9.Cai X.D., Golde T.E., Younkin S.G. Release of excess amyloid beta protein from a mutant amyloid beta protein precursor. Science. 1993;259:514–516. doi: 10.1126/science.8424174. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki N., Cheung T.T., Cai X.D., Odaka A., Otvos L., Jr., Eckman C. An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta-APP717) mutants. Science. 1994;264:1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 11.Hardy J., Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 12.Karran E., Mercken M., De Strooper B. The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10:698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- 13.Scott J.D., Li S.W., Brunskill A.P.J., Chen X., Cox K., Cumming J.N. Discovery of the 3-imino-1,2,4-thiadiazinane 1,1-dioxide derivative verubecestat (MK-8931) −Aβ-site amyloid precursor protein cleaving enzyme 1 inhibitor for the treatment of Alzheimer's disease. J Med Chem. 2016;59:10435–10450. doi: 10.1021/acs.jmedchem.6b00307. [DOI] [PubMed] [Google Scholar]
- 14.Kennedy M.E., Stamford A.W., Chen X., Cox K., Cumming J.N., Dockendorf M.F. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS β-amyloid in animal models and in Alzheimer's disease patients. Sci Transl Med. 2016;8:363ra150. doi: 10.1126/scitranslmed.aad9704. [DOI] [PubMed] [Google Scholar]
- 15.Henley D.B., May P.C., Dean R.A., Siemers E.R. Development of semagacestat (LY450139), a functional γ-secretase inhibitor, for the treatment of Alzheimer's disease. Expert Opin Pharmacother. 2009;10:1657–1664. doi: 10.1517/14656560903044982. [DOI] [PubMed] [Google Scholar]
- 16.Doody R.S., Raman R., Farlow M., Iwatsubo T., Vellas B., Joffe S. A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N Engl J Med. 2013;369:341–350. doi: 10.1056/NEJMoa1210951. [DOI] [PubMed] [Google Scholar]
- 17.Doody R.S., Thomas R.G., Farlow M., Iwatsubo T., Vellas B., Joffe S. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer's disease. N Engl J Med. 2014;370:311–321. doi: 10.1056/NEJMoa1312889. [DOI] [PubMed] [Google Scholar]
- 18.Siemers E.R., Sundell K.L., Carlson C., Case M., Sethuraman G., Liu-Seifert H. Phase 3 solanezumab trials: secondary outcomes in mild Alzheimer's disease patients. Alzheimers Dement. 2016;12:110–120. doi: 10.1016/j.jalz.2015.06.1893. [DOI] [PubMed] [Google Scholar]
- 19.Conde C., Cáceres A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci. 2009;10:319–332. doi: 10.1038/nrn2631. [DOI] [PubMed] [Google Scholar]
- 20.Alonso A.D.C., Zaidi T., Novak M., Grundke-Iqbal I., Iqbal K. Hyperphosphorylation induces self-assembly of τ into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A. 2001;98:6923–6928. doi: 10.1073/pnas.121119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iqbal K., Grundke-Iqbal I. Discoveries of tau, abnormally hyperphosphorylated tau and others of neurofibrillary degeneration: a personal historical perspective. J Alzheimer's Dis. 2006;9:219–242. doi: 10.3233/jad-2006-9s325. [DOI] [PubMed] [Google Scholar]
- 22.Hung S.Y., Fu W.M. Drug candidates in clinical trials for Alzheimer's disease. J Biomed Sci. 2017;24:47. doi: 10.1186/s12929-017-0355-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 24.Mizushima N., Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 25.Harris H., Rubinsztein D.C. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol. 2012;8:108–117. doi: 10.1038/nrneurol.2011.200. [DOI] [PubMed] [Google Scholar]
- 26.Wong E., Cuervo A.M. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. 2010;13:805–811. doi: 10.1038/nn.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Q., Liu Y., Sun M. Autophagy and Alzheimer's disease. Cell Mol Neurobiol. 2017;37:377–388. doi: 10.1007/s10571-016-0386-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nixon R.A., Yang D.S. Autophagy failure in Alzheimer's disease–locating the primary defect. Neurobiol Dis. 2011;43:38–45. doi: 10.1016/j.nbd.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kizilarslanoğlu M.C., Ülger Z. Role of autophagy in the pathogenesis of Alzheimer disease. Turk J Med Sci. 2015;45:998–1003. doi: 10.3906/sag-1407-75. [DOI] [PubMed] [Google Scholar]
- 30.Miners J.S., Baig S., Palmer J., Palmer L.E., Kehoe P.G., Love S. Aβ-degrading enzymes in Alzheimer's disease. Brain Pathol. 2008;18:240–252. doi: 10.1111/j.1750-3639.2008.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee M.J., Lee J.H., Rubinsztein D.C. Tau degradation: the ubiquitin–proteasome system versus the autophagy-lysosome system. Prog Neurobiol. 2013;105:49–59. doi: 10.1016/j.pneurobio.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 32.Berger Z., Ravikumar B., Menzies F.M., Oroz L.G., Underwood B.R., Pangalos M.N. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006;15:433–442. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]
- 33.Moresco E.M., Li X., Beutler B. Going forward with genetics: recent technological advances and forward genetics in mice. Am J Pathol. 2013;182:1462–1473. doi: 10.1016/j.ajpath.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fan L., Qiu X.X., Zhu Z.Y., Lv J.L., Lu J., Mao F. Nitazoxanide, an anti-parasitic drug, efficiently ameliorates learning and memory impairments in AD model mice. Acta Pharmacol Sin. 2019;40:1279–1291. doi: 10.1038/s41401-019-0220-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lafay-Chebassier C., Paccalin M., Page G., Barc-Pain S., Perault-Pochat M.C., Gil R. mTOR/p70S6k signalling alteration by Aβ exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer's disease. J Neurochem. 2005;94:215–225. doi: 10.1111/j.1471-4159.2005.03187.x. [DOI] [PubMed] [Google Scholar]
- 36.Di L., Kerns E.H., Fan K., McConnell O.J., Carter G.T. High throughput artificial membrane permeability assay for blood−brain barrier. Eur J Med Chem. 2003;38:223–232. doi: 10.1016/s0223-5234(03)00012-6. [DOI] [PubMed] [Google Scholar]
- 37.Li X., Wang H., Lu Z., Zheng X., Ni W., Zhu J. Development of multifunctional pyrimidinylthiourea derivatives as potential anti-Alzheimer agents. J Med Chem. 2016;59:8326–8344. doi: 10.1021/acs.jmedchem.6b00636. [DOI] [PubMed] [Google Scholar]
- 38.Xu Y., Zhang J., Wang H., Mao F., Bao K., Liu W. Rational design of novel selective dual-target inhibitors of acetylcholinesterase and monoamine oxidase B as potential anti-Alzheimer's disease agents. ACS Chem Neurosci. 2019;10:482–496. doi: 10.1021/acschemneuro.8b00357. [DOI] [PubMed] [Google Scholar]
- 39.Ni W., Wang H., Li X., Zheng X., Wang M., Zhang J. Novel tadalafil derivatives ameliorates scopolamine-induced cognitive impairment in mice via inhibition of acetylcholinesterase (AChE) and phosphodiesterase 5 (PDE5) ACS Chem Neurosci. 2018;9:1625–1636. doi: 10.1021/acschemneuro.8b00014. [DOI] [PubMed] [Google Scholar]
- 40.Mao F., Wang H., Ni W., Zheng X., Wang M., Bao K. Design, synthesis, and biological evaluation of orally available first-generation dual-target selective inhibitors of acetylcholinesterase (AChE) and phosphodiesterase 5 (PDE5) for the treatment of Alzheimer's disease. ACS Chem Neurosci. 2018;9:328–345. doi: 10.1021/acschemneuro.7b00345. [DOI] [PubMed] [Google Scholar]
- 41.Bjørkøy G., Lamark T., Brech A., Outzen H., Perander M., Øvervatn A. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Larsen K.B., Lamark T., Øvervatn A., Harneshaug I., Johansen T., Bjørkøy G. A reporter cell system to monitor autophagy based on p62/SQSTM1. Autophagy. 2010;6:784–793. doi: 10.4161/auto.6.6.12510. [DOI] [PubMed] [Google Scholar]
- 43.Li X., Wang H., Xu Y., Liu W., Gong Q., Wang W. Novel vilazodone-tacrine hybrids as potential multitarget-directed ligands for the treatment of Alzheimer's disease accompanied with depression: design, synthesis, and biological evaluation. ACS Chem Neurosci. 2017;8:2708–2721. doi: 10.1021/acschemneuro.7b00259. [DOI] [PubMed] [Google Scholar]
- 44.Bhat R., Crowe E.P., Bitto A., Moh M., Katsetos C.D., Garcia F.U. Astrocyte senescence as a component of Alzheimer's disease. PLoS One. 2012;7 doi: 10.1371/journal.pone.0045069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reiserer R.S., Harrison F.E., Syverud D.C., McDonald M.P. Impaired spatial learning in the APPSwe+PSEN1ΔE9 bigenic mouse model of Alzheimer's disease. Genes Brain Behav. 2007;6:54–65. doi: 10.1111/j.1601-183X.2006.00221.x. [DOI] [PubMed] [Google Scholar]
- 46.Deacon R.M.J. Assessing nest building in mice. Nat Protoc. 2006;1:1117–1119. doi: 10.1038/nprot.2006.170. [DOI] [PubMed] [Google Scholar]
- 47.Chong C.R., Sullivan D.J., Jr. New uses for old drugs. Nature. 2007;448:645–646. doi: 10.1038/448645a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.