

Abstract
Breast cancer susceptibility gene 1 (BRCA1) is a tumor suppressor gene, which is frequently mutated in breast and ovarian cancers. BRCA1 plays a key role in the homologous recombination directed DNA repair, allowing its deficiency to act as a therapeutic target of DNA damaging agents. In this study, we found that inhibition of the class I histone deacetylases (HDAC) exhibited synthetic lethality with BRCA1 deficiency in breast cancer cells. Transcriptome profiling and validation study showed that HDAC inhibition enhanced the expression of thioredoxin interaction protein (TXNIP), causing reactive oxygen species (ROS)-mediated DNA damage. This effect induced preferential apoptosis in BRCA1−/− breast cancer cells where DNA repair system is compromised. Two animal experiments and gene expression-associated patients’ survival analysis further confirmed in vivo synthetic lethality between BRCA1 and HDAC. Finally, the combination of inhibitors of HDAC and bromodomain and extra-terminal motif (BET), another BRCA1 synthetic lethality target that also works through oxidative stress-mediated DNA damage, showed a strong anticancer effect in BRCA1−/− breast cancer cells. Together, this study provides a new therapeutic strategy for BRCA1-deficient breast cancer by targeting two epigenetic machineries, HDAC and BET.
KEY WORDS: BRCA1, Histone deacetylase, HDAC, Synthetic lethality, Reactive oxygen species, DNA damage, Breast cancer
Graphical abstract
Histone deacetylase (HDAC) inhibition upregulates the expression of pro-oxidant protein, thioredoxin interaction protein (TXNIP), leading to reactive oxygen species (ROS) accumulation and high DNA double-strand breaks in cells. Breast cancer susceptibility gene 1 (BRCA1)-deficient breast cancer cells were highly sensitive to HDAC inhibition due to the lack of homologous recombination DNA repair system and hence underwent synthetic lethality.
1. Introduction
Breast cancer is the most common type of cancer in women all over the world. Based on the latest estimation, 268,600 women will be diagnosed with breast cancer and 41,760 breast cancer death will occur in 2019 among women in the United States1. Although the death rate has been partially declined during the past two decades, breast cancer still accounts for the second leading cause of cancer death among women. According to the classification by molecular subtypes, breast cancer can be classified by the expression status of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2)2. Breast cancer with none of these receptors are classified as triple negative breast cancer (TNBC), which lacks efficient treatment strategies3. Recently, the tumor suppressor breast cancer susceptible gene 1 (BRCA1) was found to be frequently mutated among the TNBC patients4. BRCA1 is responsible for the repair of DNA double-strand breaks and thus is important for maintaining genomic integrity5. In addition, BRCA1 also takes part in transcription regulation and cell cycle control6. With its high mutational frequency in the triple negative breast cancer and its functional participation in a wide-spectrum of cellular pathways, BRCA1 is recognized as an emerging target for the treatment of TNBC7.
BRCA1 forms several distinct complexes with associated proteins and participates in the repair of DNA double-strand breaks by homologous recombination. Depending on the complex proteins, BRCA1 complexes have been divided into at least four complexes, including BRCA1-A, -B, -C and -D. Each complex has unique associated proteins that function to recruit BRCA1 to DNA damage foci and catalyze DNA damage repair8. The dysfunction in BRCA1 could lead to the failure of DNA damage repair, causing the cells sensitive to DNA damaging agents. Based on this feature, poly (ADP-ribose) polymerase 1 (PARP1) inhibitors have been developed as synthetic lethality drugs for BRCA1 deficient breast cancer cells7. PARP1 is an enzyme participating in the repair of DNA single-strand breaks. The single-strand breaks (nicks), if not repaired, will form double-strand breaks when DNA replication occurs at the site of the nicks, which then require BRCA1-mediated DNA repair system. By this mechanism, the combination of PARP inhibition and BRCA1 deficiency induces a synthetic lethality in the cells where multiple DNA double strand-breaks are formed9. This finding prompted the development of several PARP inhibitors for the treatment of BRCA1-deficient breast and ovarian cancers, such as olaparib, rucaparib and niraparib, which have successfully reached to clinic recently10.
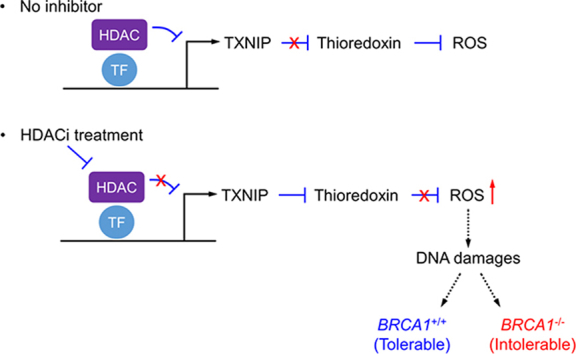
To further exploit the synthetic lethality approach to target BRCA1 mutant breast cancer, we previously screened an epigenetics compound library in BRCA1-isogenic breast cancer pairs. From the study we found several synthetic lethality targets, including bromodomain and extra-terminal domain (BET)11. We have elaborated the mechanism of the synthetic lethality between BET and BRCA1, i.e., BET inhibition abrogated MYC-dependent transcription repression of a redox regulator, thioredoxin-interacting protein (TXNIP) by switching MYC to MondoA:MLX complex on TXNIP promoter. This led to the elevated cellular reactive oxygen species (ROS) and DNA damages that are detrimental to BRCA1-deficient breast cancer cells11. In addition to BET, we also identified histone deacetylase (HDAC) as potential synthetic lethality targets for BRCA1. HDACs are a class of enzymes that remove acetyl groups from lysine residues on histones. By modifying neutral histone tails to positively charged ones, HDACs allow the DNA to tightly bind to histones and functions as a transcription corepressor12. HDAC family of proteins are involved in a variety of biological processes and recognized as a target of anticancer drugs.
In this study, we report that the inhibition of class I HDAC increases the transcription of TXNIP, an inhibitor of major antioxidant protein thioredoxin13. High expression of TXNIP induces cellular oxidative stress by elevating ROS and preferentially increases DNA damage and apoptosis in BRCA1-deficient breast cancer cells. We further show that the combination of inhibitors of BET and HDAC significantly enhances synthetic lethality effect in BRCA1-deficient breast cancer cells. Our study provides a novel, druggable synthetic lethality target for BRCA1 that can be used as a single or in combination with other synthetic lethality drugs.
2. Material and methods
2.1. Cell culture
The establishment and culture condition for T47D and HCC1937 BRCA1 isogenic cell lines were described previously14. BRCA1 wild type (MDA-MB-468, MCF-7) and mutant (MDA-MB-436, SUM-149) breast cancer cell lines were cultured in DMEM (Gibco, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco) and maintained in a CO2 incubator adjusted at 5%.
2.2. Reagents and antibodies
Entinostat (DC6909) was bought from DC chemicals (Shanghai, China). Vorinostat (S1047) and Mocetinostat (S1122) were from Selleck chemicals (Houston, TX, USA). The siRNAs and primers were purchased from Integrated DNA Technologies (Coralville, IA, USA) and the sequence information is shown in Supporting Information Tables S1 and S2. Primary antibodies against BRCA1 (sc-642), heme oxygenase 1 (HO-1) (sc-136960), GAPDH (sc-365062), PARP1 (sc-7150), acetyl-histone H4 (sc-8662) and histone H4 (sc-25260) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Primary antibodies against phospho-histone H2A.X (γ-H2AX) (Ser139) (#9718S), TXNIP (#14715S) and cleaved caspase-3 (#9661S) were purchased from Cell Signaling Technology (Danvers, MA, USA). Primary antibody against MLXIP was from Proteintech (Rosemont, IL, USA). Secondary antibodies conjugated with horseradish peroxidase (HRP) were all purchased from Santa Cruz Biotechnology. The dilution factors of different antibodies can be found in Supporting Information Table S3.
2.3. siRNA transfection
TXNIP siRNA was synthesized by Integrated DNA Technologies, Inc. In brief, 2 nmol/L of TXNIP siRNA was dissolved and incubated in Opti-MEM (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with Lipofectamine RNAi max reagent (Thermo Fisher Scientific) for 20 min. After that, the siRNA solution was added to the cells directly. Twenty-four hours later, drugs were added to the cells for evaluation. The sequence information of TXNIP siRNA can be found in Table S1.
2.4. Cell viability and clonogenic assays
For cell viability assay, cells were seeded in a 96-well plate at a density of 5 × 103 cells/well. After cells were allowed to adhere for 24 h, drugs were added and the incubation was continued for 72 h. The cell viability was evaluated by measuring the Alamarblue fluorescence at Ex560/Em590 with a SpectraMax M5 fluorescence microplate reader (Molecular Devices, Sunnyvale, CA, USA). GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA, USA) was used for the curve fitting of cell growth inhibition and analysis. For clonogenic assay, cells (2 × 103 cells/well) were plated in a 6-well plate and drugs were added 24 h post cell plating. Cells were incubated with or without drugs for 15 days until visible colonies were formed. The crystal violet regent was added to the cells for staining cell colonies.
2.5. Measurement of apoptosis
Cellular apoptosis was evaluated with three different assays, including sub-G1 cell population, annexin V staining and Hoechst33342 (HO33342) staining. For sub-G1 cell population, cells were first fixed with 75% of ice-cold ethanol for 24 h. Cells were then gently washed with PBS for three times and stained with 50 μg/mL propidium iodide (PI), 100 μg/mL RNase A and 0.2% Triton X-100 for 30 min. Accuri C6 flow cytometer (BD Biosciences, San Jose, CA, USA) was used for the cell cycle and sub-G1 population analysis. For annexin V staining, we used a staining kit from BioLegend (San Diego, CA, USA) and followed the manufacturer's instructions. Briefly, cells were co-stained with annexin V and propidium iodide regents for 30 min. Then the cells were washed by PBS and collected for test on flow cytometry. The data was analyzed with FlowJo software (FlowJo LLC, Ashland, OR, USA). For HO33342 nuclear staining, cells treated with drugs for 72 h were incubated with HO33342 at a final concentration of 0.1 μg/mL. The live-cell nuclear images were captured with Zeiss AxioObserver Z1 fluorescence microscope (Carl Zeiss, Thornwood, NY, USA).
2.6. RNA sequencing
Total RNA from cells treated with or without drugs for 24 h was isolated with RNeasy Plus Mini Kit (Qiagen, Hilden, Germany). The RNA integrity number (RIN) values were assessed with RNA 6000 Nano Kit on 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA) and were used for RNA quality evaluation. mRNA was extracted from the total RNA using NEBNext Poly(A) mRNA Magnetic Isolation Module (E7490S, New England Biolabs, Ipswich, MA, USA). cDNA library was prepared from mRNA by NEBNext Ultra Directional RNA Library Prep Kit for Illumina (E7420S, New England Biolabs). The library quality was evaluated using High Sensitivity DNA Analysis Kit on 2100 Bioanalyzer. RNA sequencing was performed with Illunima Hiseq 2500 (Illumina, San Diego, CA, USA). The raw data was analyzed with TopHat and Cuffdiff software15, 16. The gene expression profiles were uploaded to Reactome pathway database (https://reactome.org/) for signal pathway analysis.
2.7. Quantitative reverse transcription PCR
Total RNA was isolated from cells with RNeasy Mini Kit (Qiagen). cDNA was prepared with High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). The iTaq Universal SYBR Green Supermix (Bio-Rad, Hercules, CA, USA) was used for amplification of transcripts in an ABI-7500 Real-Time PCR System (Thermo Fisher Scientific). GAPDH transcript was used as an internal reference. Comparative CT values were determined to evaluate relative mRNA expression changes.
2.8. Western blotting
Total protein was extracted from cells with a radioimmunoprecipitation assay (RIPA) buffer. The quantification of protein was conducted with BCA Protein Assay Kit (Thermo Fisher Scientific). Equal amount of protein was loaded and separated via SDS-PAGE gels, and transferred to PVDF membranes. Membranes were blocked with 5% non-fat dried milk (Santa Cruz Biotechnology) and incubated with primary antibodies diluted in a blocking solution at 4 °C overnight. After washing, membranes were incubated with secondary antibodies for additional 2 h at room temperature. Blots were then incubated with Clarity™ Western ECL Substrate (Bio-Rad) and specific protein bands were analyzed under a ChemiDoc MP imaging system (Bio-Rad).
2.9. Chromatin immunoprecipitation (ChIP)
ChIP assay was performed with Imprint Chromatin Immunoprecipitation Kit (Sigma–Aldrich, St. Louis, MO, USA) according to the manufacturer's instructions. In brief, HCC1937 BRCA1−/− cells were treated with DMSO or 5 μmol/L of entinostat for 6 h. Then, 1% formaldehyde was added to the cells to crosslink DNA and proteins. The cell nuclear fraction was prepared with Nuclei Preparation Buffer. After that, shearing buffer was added to the samples for sonication by Bioruptor Sonication System (Diagenode, Denville, NJ, USA). The samples were incubated with rabbit anti-IgG or anti-acetyl-histone H4 antibody which were trapped on protein A-coated assay strips. The precipitated DNA was used for RT-qPCR with the primer pair that targets to the promoter region of TXNIP. The primer sequences are listed in Table S2.
2.10. Animal experiments
All animal experiments were carried out in accordance with the approved animal protocol by Animal Research Ethics Committee of the University of Macau, China. For the HCC1937 BRCA1 isogenic model, 4–6 weeks old NOD-SCID mice (5 mice/group) were used for generation of the xenograft models. Approximately 5 × 106 of BRCA1 wildtype or deficient cells were inoculated bilaterally (right and left flanks) in each mouse. Mice were randomized when the mean volume of tumors reached 100 mm3. Vehicle (5% DMSO, 5% tween-80 and 5% polyethylene glycol-400 in sterile saline) or entinostat (10 and 20 mg/kg) was administered to the mice via intraperitoneal injection for 14 days (once daily). After stopping drug administration, mice and tumor growth were continuously monitored for additional 25 days. For T47D BRCA1 isogenic model, 4–6 weeks old BALB/c nude mice (4 mice/group) were used for establishing a xenograft model. The drug administration was done for first 14 days and the tumor growth was monitored until 113 days. The tumor size was measured with a caliper and the tumor volume was calculated based on modified ellipsoidal formula: long axis × short axis2 × π/6. The mice body weights were measured periodically to assess toxicity. At the end of the experiment, tumors were extracted from mice for wet weight measurement. The tumor tissues were then snap frozen with liquid nitrogen for following western blot analysis.
2.11. Measurement of intracellular thioredoxin activity
Cells were treated with or without entinostat for 6 h. After washing with PBS, cells were harvested with a cell scraper. The cell suspension was sonicated using a probe sonicator at 50% magnitude for three times on ice. Thioredoxin activity was evaluated from the sonicated cell lysate with a Fluorescent Thioredoxin Activity Assay Kit (Caymen Chemicals, Ann Arbor, MI, USA). The assay was conducted according to the manufacturer's instructions.
2.12. Comet assay
Cellular DNA damage was detected at a single cell level using the comet assay (single cell gel electrophoresis assay17. Briefly, cells were treated with drugs for 48 h. Cells were then harvested, suspended in 1% low melting-point agarose gel (Affymetrix, Santa Clara, CA, USA) and spread on a glass slide. The slide was immersed in cell lysis buffer (2.5 mol/L NaCl, 100 mmol/L EDTA and 10 mmol/L Tris base) at 4 °C overnight prior to electrophoresis at 300 mA for 30 min. Propidium iodide (2.5 μg/mL) was added to the slide for DNA staining. The slide was then rinsed with PBS three times to remove residual dye. Single cell comet images were obtained under a Zeiss AxioObserver Z1 fluorescence microscope (Carl Zeiss). Image J software (NIH, Bethesda, MA, USA) was used for quantitative analysis of DNA damage.
2.13. Detection of ROS
Cellular ROS was detected with CellROX Green cell-permeable fluorescence indicator (Thermo Fisher Scientific). Cells were treated with drugs for indicated time points and CellROX Green solution (2.5 mmol/L) was added to the cells to make the final concentration of the indicator as 5 μmol/L. After incubation for 30 min, cells were rinsed with PBS three times. Cells were filtered through a mesh filter to secure single-cell suspension prior to flow cytometry analysis. Accuri C6 flow cytometry was used for ROS detection and analysis.
2.14. Drug combination analysis
The BRCA1 deficient breast cancer cell lines (HCC1937, SUM-149, and MDA-MB-436) were treated with drug alone or combination of OTX-015 and entinostat at various concentrations for 72 h. The cell growth was measured with Alamarblue and drug effects were input into CompuSyn software (ComboSyn, Paramus, NJ, USA) for drug combination analysis. The software generated combination index (CI) value for each cell line. CI value < 1 indicates synergistic effect, CI value = 1 indicates additive effect, and CI value > 1 indicates antagonistic effect.
2.15. Statistical analysis
Student's t-test was used for analyzing statistical difference between two groups. The P values less than 0.05 were considered significant. Statistical test for patient's survival was conducted with log-rank test. All the statistical evaluation was performed via GraphPad Prism 6.0 software.
3. Results
3.1. HDAC inhibition is synthetic lethal with BRCA1 deficiency in breast cancer cells
In our previous epigenetics drug library screening, HDAC inhibitors (HDACi) were identified as synthetic lethality hits in BRCA1 deficient breast cancer cells11. Here we used two BRCA1 isogenic breast cancer cell pairs, including HCC1937 BRCA1 isogenic (HCC1937-BRCA1+/+ and HCC1937-BRCA1−/−) and T47D BRCA1 isogenic (T47D-shCTRL and T47D-shBRCA1) cell lines, to validate the synthetic lethality effects of the HDACi. Three HDACi, including entinostat (class I HDACi), vorinostat (pan-HDACi), and mocetinostat (class I HDACi) showed strong synthetic lethality effects in both colony formation (Fig. 1A) and cell viability assays (Fig. 1B–D). Entinostat and mocetinostat are class-I HDACi while vorinostat is a pan-HDACi, suggesting that class I HDACs, in particular HDAC1 and 3, could be targets of the BRCA1 synthetic lethality. Among the three HDACi, entinostat showed the strongest selectivity for BRCA1-deficient breast cancer cells (Fig. 1B). Hence, we mainly used entinostat for the follow-up studies. Next we analyzed cell cycle and annexin V staining in cells treated with entinostat to assess apoptosis induction. Entinostat treatment significantly increased sub-G1 population in HCC1937-BRCA1−/−, but not in HCC1937-BRCA1+/+ cells, indicating a selective induction of apoptosis in BRCA1-deficient cells (Fig. 1E). Further, annexin V staining cleared showed that entinostat selectively induced apoptosis in HCC1937-BRCA1−/− cells (Fig. 1F). The selective apoptosis induction was further verified with nuclear morphology of the cells treated with entinostat (Fig. 1G) and Western blots of caspase-3 and PARP-1 cleavage (Fig. 1H).
Figure 1.

Synthetic lethality between HDAC inhibition and BRCA1 deficiency. (A) The effect of HDAC inhibitors on colony formation of T47D BRCA1 wildtype (shCTRL) and BRCA1 knock-down (shBRCA1) cell lines was tested. Cells were treated with the compounds for 15 days and cell colonies were stained with crystal violet reagent. (B)–(D) The effect of HDAC inhibitors on the cell viability of HCC1937 BRCA1 isogenic cell lines was tested. Cells were treated with the compounds for 3 days and cell viability was determined by alamarblue assay. (E) and (F) Flow cytometry was performed to analyze sub-G1 cell population (E) and annexin V-FITC staining (F) of HCC1937 BRCA1 isogenic cell lines after treated with 5 μmol/L entinostat for 72 h. (G) The effect of HDAC inhibitors on the nuclear morphology of HCC1937 BRCA1 isogenic cell lines was tested. Cells were treated with 5 μmol/L entinostat, vorinostat or mocetinostat for 72 h. Cell nuclei were stained with hoechst33342 reagent and observed under a fluorescent microscope. Scale bar = 100 μm. (H) Western blot analysis for PARP1 and cleaved caspase-3 in HCC1937 BRCA1 isogenic cells treated with 5 μmol/L entinostat for 72 h. Data are mean ± SD, **P < 0.01 between two groups.
3.2. Class I HDAC inhibition and BRCA1 deficiency is synthetic lethal in breast cancer in vivo
To assess the synthetic lethality effect in vivo, we conducted mouse tumor xenograft experiments with HCC1937 and T47D BRCA1 isogenic breast cancer cells. All mice were implanted bilaterally with BRCA1+/+ and BRCA1−/− breast cancer cells and were given entinostat (10 mg/kg) intraperitoneally for 2 weeks. The tumor growth was observed until 39 days for HCC1937 BRCA1-isogenic tumors and 113 days for T47D BRCA1-isogenic tumors since the start of the treatment. Entinostat treatment showed a marginal antitumor activity on HCC1937-BRCA1+/+ tumor, with the inhibition rate of 43%. Whereas it strongly inhibited the growth of HCC1937-BRCA1−/− tumors, with the inhibition rate of 74% (Fig. 2A and B). The tumor wet weight measurement also showed a selective antitumor effect of entinostat against HCC1937-BRCA1−/− tumors (Fig. 2C). Entinostat at 20 mg/kg showed a similar inhibition profile with 10 mg/kg dosage (data not shown). Similar results were observed in T47D BRCA1-isogenic tumor xenografts. Entinostat treatment almost completely stopped the growth of T47D-shBRCA1 tumors (inhibition rate = 84%), while it had marginal antitumor effect on T47D-shCTRL tumors (inhibition rate = 43%) (Fig. 2D–F). No apparent toxicity was observed under these treatment conditions (Supporting Information Figs. S1A and B). These data demonstrated that synthetic lethality between HDAC and BRCA1 occurs in vivo.
Figure 2.

In vivo synthetic lethality between HDAC inhibition and BRCA1 deficiency. (A)–(C) The effect of entinostat on the tumor growth of HCC1937 BRCA1 isogenic cancer was tested. SCID/NOD mice bearing HCC1937 BRCA1+/+ (A) or BRCA1−/− (B) tumor were given vehicle or entinostat (10 mg/kg) for 14 days and tumor volume was measured until the end of the experiment. Tumor wet weight was measured at the end of the xenograft experiments (C) (n = 5). (D)–(F) The effect of entinostat on the tumor growth of HCC1937 BRCA1 isogenic cancer was tested. Nude mice bearing T47D shCTRL (D) or shBRCA1 (E) tumor were given vehicle or entinostat (10 mg/kg) for 14 days and tumor volume was measured until the end of the experiment. Tumor wet weight was measured at the end of the xenograft experiments (F) (n = 4). (G)–(N) The effect of BRCA1 status and HDAC1/3 expression on breast cancer patient survival was analyzed using the clinical data from the METABRIC. The patients were sub-grouped according to the BRCA1 mutation status and the expression levels of HDAC1 and 3, and the overall survival from each subgroup was analyzed. Data are mean ± SD, *P < 0.05, **P < 0.01 between two groups.
We further analyzed the potential of the synthetic lethality between BRCA1 and class I HDAC using the clinical data of breast cancer patients. We downloaded 240 breast cancer patients' data with BRCA1 mutation and gene expression status of HDAC1 and 3 from METABRIC project (http://molonc.bccrc.ca/aparicio-lab/research/metabric/)18. Based on BRCA1 mutation status (+/+ or −/−) and HDAC1/3 expression status (high or low), the patients were divided and grouped into 8 subgroups, including 1) BRCA1−/−, HDAC1low/high, 2) BRCA1−/−, HDAC3low/high, 3) BRCA1−/−, HDAC1low, HDAC3low/high, 4) BRCA1−/−, HDAC1high, HDAC3low/high, 5) BRCA1+/+, HDAC1low/high, 6) BRCA1+/+, HDAC3low/high, 7) BRCA1+/+, HDAC1low, HDAC3low/high, 8) BRCA1+/+, HDAC1high, HDAC3low/high. Either the HDAC1low/high (Fig. 2G) or the HDAC3low/high (Fig. 2H) did not show significant difference in patient's survival in the BRCA1−/− patient cohort. Similar results were observed with the BRCA1+/+ patient cohort (Fig. 2K,L). Interestingly, the BRCA1−/− patient cohort with the HDAC1low, HDAC3low showed significant longer overall survival compared to those with the HDAC1low, HDAC3high (Fig. 2I) or the HDAC1high, HDAC3low/high (Fig. 2J). There was no significant difference in overall survival between the HDAC1/3 expression status in the BRCA1+/+ patient cohorts (Fig. 2M,N). These data suggested that down-regulation of both HDAC1 and 3 might provide survival benefit to patients with BRCA1 deficient breast cancer. Our results also suggested that entinostat as a broad inhibitor of class I HDACs, may serve as a possible synthetic lethality drug for patients with BRCA1 deficient breast cancer.
3.3. Inhibition of class I HDAC provokes cellular oxidative stress and DNA damage responses
To investigate the mechanism underlying the synthetic lethality, we performed transcriptome profiling of HCC1937 BRCA1 isogenic cells treated with control and entinostat. The differentially expressed genes were obtained and subjected to the Reactome signal pathway analysis19. A number of genes involved in apoptosis induction and extracellular matrix organization were up-regulated by entinostat treatment (Fig. 3A). Interestingly, genes involved in formation of the cornified envelope, keratinization and DNA damage stress pathways were also significantly upregulated. Whereas, a large part of down-regulated genes were cell cycle and mitosis related genes (Fig. 3B). HDACs, particularly class I HDACs, are highly linked to cell cycle regulation since they form a multimeric corepressor complex with E2F/RB1/RBP1/Sin3 to regulate cell cycle progression20, 21. The class I HDACs also regulate p21CIP1/WAF1 expression to regulate G1 cell cycle transition22. Furthermore, it is also well known that class I HDACs directly regulate P53 stability and transcriptional activity by regulating P53 acetylation23, which is critical for apoptosis induction24, 25. On the other hand, the two signal pathways that were up-regulated: formation of the cornified envelope and keratinization, are highly related to cellular oxidative stress26, 27. In addition, DNA damage stress genes that were up-regulated also related phenotype to cellular oxidative stress. The relationship between HDAC and cellular oxidative stress has been previously reported28, 29. The study conducted by Feingold et al.28 and Petruccelli et al.29 demonstrated that HDAC inhibition induces cellular pro-oxidant protein, TXNIP and heavily increases ROS and DNA damage. We thus hypothesized that class I HDAC inhibition could induce cellular oxidative stress, resulting in heavy DNA damages that are detrimental to BRCA1 deficient breast cancer cells. We then analyzed the protein levels that are involved in cellular oxidative response and DNA damage, such as TXNIP, HO-1 and γ-H2AX, in cells treated with entinostat. Histone acetylation status was analyzed in parallel as a positive control. Entinostat treatment significantly increased TXNIP level in a biphasic manner: increased in earlier time points (peak at 8 h) and returned to background level at 24 h, in HCC1937-BRCA1−/− cells (Fig. 3C). Oxidative stress marker HO-1 and DNA damage marker γ-H2AX levels were also significantly increased by the entinostat treatment (Fig. 3C and Fig. S1C). Similar biphasic induction of TXNIP and significant increase in HO-1 and γ-H2AX were observed in T47D-shBRCA1 cells treated with entinostat (Fig. 3D and Fig. S1D). These data suggested that entinostat indeed increased the pro-oxidant protein TXNIP and provoked cellular oxidative stress and DNA damage responses in BRCA1 deficient breast cancer cells. The RT-qPCR analysis of TXNIP mRNA level also showed a biphasic increase in TXNIP expression by entinostat (Fig. 3E). We next performed ChIP experiments to test whether the increase in TXNIP mRNA by entinostat was due to the de-repression of its transcription resulting from the inhibition of HDAC activity on the promoter of TXNIP. Treatment of the HCC1937 BRCA1−/− cells with entinostat for 6 h significantly increased the histone H4 acetylation on the TXNIP promoter (Fig. 3F). This result suggested that the increase in TXNIP expression by entinostat was from the direct inhibitory effect on HDAC activity on its promoter. We next examined whether the entinostat effect on cellular oxidative stress and DNA damage response was selective to BRCA1 deficient cells. Basal levels of TXNIP and HO-1 were higher in HCC1937-BRCA1−/− than in HCC1937-BRCA1+/+ cells (Fig. 3G), which was in agreement with previous reports that BRCA1 plays a role in antioxidant signaling. Upon entinostat treatment, HO-1 level was significantly increased in HCC1937-BRCA1−/−, but not in HCC1937-BRCA1+/+ cells. Likewise, entinostat-induced γ-H2AX was significantly higher in HCC1937-BRCA1−/− than in HCC1937-BRCA1+/+ cells. Whereas the acetylation of histones was similarly increased in both cell types by entinostat, demonstrating that entinostat-induced oxidative stress and DNA damage response was much stronger in BRCA1-deficient breast cancer cells (Fig. 3G). Similar results were observed in vorinostat (Fig. 3H) and mocetinostat treatment (Fig. 3I), suggesting that class I HDAC inhibition provoked cellular oxidative stress and DNA damage responses more selectively in BRCA1 deficient cells.
Figure 3.

Transcriptome profiling to explore the mechanism of the synthetic lethality. (A) and (B) REACTOME signal pathway analysis for the RNA sequencing results is shown. HCC1937 BRCA1 isogenic cells were treated with 5 μmol/L entinostat for 24 h and total RNA was extracted for RNA sequencing. The gene expression profiles were analyzed with REACTOME signal pathway database. The pathway genes that were significantly up- (A) and down-regulated (B) by entinostat are shown. (C) and (D) The effect of entinostat on cellular oxidative stress and DNA damage was examined. HCC1937 BRCA1−/− (C) or T47D shBRCA1 (D) cells were treated with 5 μmol/L entinostat for indicated time points and Western blots for proteins involved in oxidative stress and DNA damage responses were analyzed. GAPDH was used as an internal control. (E) RT-qPCR analysis for the TXNIP mRNA level was examined in HCC1937 BRCA1 isogenic cells treated with entinostat. (F) Chromatin immunoprecipitation (ChIP) of TXNIP promoter using anti-acetyl histone H4 antibody is shown. HCC1937 BRCA1−/− cells were treated with 5 μmol/L entinostat for 6 h and processed for ChIP using a rabbit IgG (control) or anti-acetyl histone H4 antibody. The ChIP DNA was subjected to PCR amplification with a primer pair specific for TXNIP promoter. (G) and (I) The effect of HDAC inhibitors, entinostat (class I-HDACi, G), vorinostat (pan-HDACi, H), and mocetinostat (class I-HDACi, I) on histone acetylation, cellular oxidative stress and DNA damage responses was examined in HCC1937 BRCA1 isogenic cell lines. Data are mean ± SD, *P < 0.05, **P < 0.01 between two groups.
3.4. Entinostat induces ROS and DNA damage, which is the major cause for synthetic lethality between HDAC inhibition and BRCA1 deficiency
Our expression profile showed that entinostat treated breast cancer cells provoked strong oxidative stress and DNA damage responses. We then analyzed cellular ROS and DNA damages in cell treated with entinostat. Entinostat increased cellular ROS level in both HCC1937-BRCA1+/+ and HCC1937-BRCA1−/− cells (Fig. 4A). The level of ROS induction was much higher in HCC1937-BRCA1−/− cells, which is consistent with the TXNIP and HO-1 levels in these cells (Fig. 3E). In comet assay, entinostat showed a marginal effect in DNA damage in HCC1937-BRCA1+/+, while it induced severe DNA damages in HCC1937-BRCA1−/− cells (Fig. 4B and C). Co-treatment with an antioxidant N-acetylcysteine (NAC) significantly reversed the entinostat-induced DNA damage in HCC1937-BRCA1−/− cells (Fig. 4B and C), suggesting that DNA damages induced by entinostat was largely due to the ROS. The NAC rescue effect was also observed in the synthetic lethality where entinostat inhibition on HCC1937-BRCA1−/− cell viability was significantly reversed by NAC co-treatment (Fig. 4D and E). These results suggested that entinostat induced cellular ROS and this effect facilitated DNA damages in HCC1937-BRCA1−/− cells where DNA double-strand break repair function is impaired.
Figure 4.

Induction of reactive oxygen species (ROS) and DNA damage in BRCA1−/− cells by entinostat. (A) The effect of entinostat on cellular ROS in HCC1937 BRCA1 isogenic cells was tested. HCC1937 BRCA1 isogenic cells were treated with 5 μmol/L entinostat for 6 h and intracellular ROS was detected under a flow cytometry with CellROX green fluorescence dye. (B) The effect of entinostat on DNA damage in HCC1937 BRCA1 isogenic cells was tested with the comet assay. HCC1937 BRCA1 isogenic cells were treated with entinostat alone (5 μmol/L) or in combination with N-acetylcysteine (NAC, 5 mmol/L) for 24 h and the DNA damage levels were assessed by comet assay. Scale bar = 100 μm. (C) The DNA tails (50 tails/group) were measured by Image J software and the data were plotted with Graphpad prism 6.0. (D) and (E) The effect of entinostat on cell viability in HCC1937 BRCA1 isogenic cells and its reversal by NAC was tested. Representative cell images (D) and alamarblue data (E) are shown. Scale bar = 400 μm. (F) The effect of entinostat on thioredoxin (TXN) activity was examined. HCC1937 BRCA1 isogenic cells were treated with 5 μmol/L entinostat for 6 h and intracellular TXN activity was measured with a Fluorescent Thioredoxin Activity Assay Kit. (G) The effect of TXNIP silencing on entinostat-induced ROS in HCC1937 BRCA1−/− cells was tested. HCC1937 BRCA1−/− cells were transfected with 2 nmol/L TXNIP siRNA for 24 h prior to dosing with 5 μmol/L entinostat. After 6 h incubation, intracellular ROS was measured with CellROX green kit. (H) The effect of TXNIP silencing on entinostat-induced synthetic lethality in HCC1937 BRCA1−/− cells was tested. HCC1937 BRCA1−/− cells were transfected with 2 nmol/L TXNIP siRNA for 24 h prior to dosing with 5 μmol/L entinostat. After additional 72 h incubation, the cell viability was assessed with the alamarblue assay. *P < 0.05; **P < 0.01 between two indicated groups. (I) The reversal effect of TXNIP silencing on entinostat-induced oxidative stress and DNA damage responses was examined in HCC1937 BRCA1−/− cells. Data are mean ± SD, *P < 0.05, **P < 0.01 between two groups.
Since we observed that a pro-oxidant TXNIP was significantly increased in HCC1937-BRCA1−/− cells upon treatment with entinostat, we next examined the role of TXNIP in the synthetic lethality effect. TXNIP works by inhibiting an antioxidant protein thioredoxin through disulfide exchange30. We first measured cellular thioredoxin activity after treated with entinostat. Entinostat marginally, but significantly inhibited thioredoxin activity in cells with stronger effect in HCC1937-BRCA1−/− cells (Fig. 4F). Next, we silenced TXNIP in cells treated with or without entinostat and measured intracellular ROS level. Entinostat significantly increased ROS in HCC1937-BRCA1−/− cells but this effect was partially reversed by TXNIP siRNA (Fig. 4G, upper panel). The reversal effect was well correlated with TXNIP protein level (Fig. 4G, lower panel). We further observed that entinostat-induced inhibition of HCC1937-BRCA1–/–cell viability was significantly reversed by the silencing of TXNIP (Fig. 4H). As evidenced by Western blots, treatment with TXNIP siRNA strongly reversed entinostat-induced TXNIP and the expression of HO-1 and γ-H2AX in HCC1937-BRCA1−/− cells (Fig. 4I). However, TXNIP siRNA did not affect histone acetylation by entinostat. These data strongly suggested that entinostat-induced increase in TXNIP level played a key role in the synthetic lethality.
3.5. The combined inhibition of class I HDAC and BET enhanced the synthetic lethality effect in BRCA1 deficient breast cancer cells
We previously reported that BET inhibition reduced TXNIP level by inhibiting MYC and activating MondoA:MLX transcription factor11. This led to an increase in intracellular ROS and heavy DNA double strand breaks in BRCA1 deficient breast cancer cells. Our present study revealed that class I HDAC inhibition de-repressed TXNIP transcription, thereby increasing ROS. Therefore, we hypothesized that the combination of HDAC inhibitor and BET inhibitor could lead greater oxidative stress and DNA damage in BRCA1 deficient breast cancer cells compared to the single agent treatment. Indeed, co-treatment of cells with entinostat and a BET inhibitor OTX-015 significantly enhanced cellular ROS level in HCC1937-BRCA1−/− cells compared to a single agent treatment (Fig. 5A). Consistently, same result was observed in comet assay where co-treatment with HDAC and BET inhibitors greatly increased cellular DNA damage in HCC1937-BRCA1−/− cells (Fig. 5B and C). As a result, the two drug combination strongly induced synthetic lethality in HCC1937-BRCA1−/− cells by inducing apoptosis (Fig. 5D and E). Lastly, we examined the effect of the two drug combination in a panel of breast cancer cell lines with different BRCA1 status. Either single agents or combination of the two drugs showed selective inhibition on BRCA1 deficient breast cancer cells with the drug combination having stronger effect (Fig. 5F and Support Information Fig. S2). To determine whether the combination effect of HDAC and BET inhibitors is synergistic or not, we analyzed drug CI of entinostat and OTX-015 in three BRCA1 deficient breast cancer cell lines (HCC1937, SUM-149 and MDA-MB-436). The calculated CI values indicated that the combination of entinostat and OTX-015 had synergetic anti-cancer effect on the three BRCA1 deficient breast cancer cell lines (Fig. 5G–I). Altogether, our results suggested that combined inhibition of class I-HDAC and BET boosted up cellular oxidative stress and resulting DNA double-strand breaks and exerted strong synthetic lethality in BRCA1 deficient breast cancer cells.
Figure 5.

Enhanced synthetic lethality effect by the combination of bromodomain and extraterminal motif (BET) and HDAC inhibition in BRCA1 deficient breast cancer cells. (A)–(D) The effect of single or combination of the inhibitors of BET (OTX-015) and HDAC (entinostat) on ROS induction (A), DNA damage (B (Scale bar = 100 μm) and C), and cell viability (D) (Scale bar = 400 μm) in HCC1937 BRCA1 isogenic cells is shown. (E) Enhanced apoptosis induction in HCC1937 BRCA1−/− cells by the combination of BET and HDAC inhibitors is shown. Western blots of cleaved caspase-3 and PARP1 were used as apoptosis indicators. (F) The synthetic lethality effect of the combination of BET and HDAC inhibitors in a panel of breast cancer cells with different BRCA1 status was tested. The cells were treated with single or combination of 0.25 × concentration of IC50 of OTX-015 and entinostat for 72 h and alamarblue assay was performed to assess cell viability. The combination of other concentrations (4×, 2×, 1×, 0.5× IC50) is presented in Supporting Information Fig. S2. (G)–(I) BRCA1 deficient breast cancer cells, including HCC1937 (G), SUM-149 (H) and MDA-MB-436 (I) were treated with the drug alone or combination of entinostat and OTX-015 at various concentrations for 72 h and the combination indices (CI) were determined with CompuSyn software. Data are mean ± SD, **P < 0.01 between two groups.
4. Discussion
In the present study, we identified that HDAC inhibition is synthetic lethal with BRCA1 deficiency in breast cancer cells. The synthetic lethality effect has been thoroughly validated in two different BRCA1 isogenic breast cancer pairs in vitro and in vivo mouse models. Although we failed to define precise isoform(s) of HDAC that mediated the synthetic lethality, testing with the 3 class-specific HDAC inhibitors and clinical data analyses of breast cancer patients suggested that class I HDACs, in particular HDAC1 and 3, are together involved in the synthetic lethality with BRCA1. HDACs are a family of deacetylase enzymes that remove acetyl groups from an N-acetyl lysine amino acid on a histone or other non-histone protein substrates. Class I HDACs, such as HDAC1, 2, and 3 are found primarily in the nucleus, suggesting their primary role in gene transcription regulation31. Class I HDACs are known to repress the transcription and the function of a number of tumor suppressors, including P21CIP1/WAF132, E-cadherin33, P5334, BRCA135 and retinoic acid receptor-beta (RARβ2)36. Overexpression of HDACs and aberrant recruitment of HDACs to the promoter of the tumor suppressors have been observed in a variety of tumors, making it a promising drug target for cancer treatment.
A number of recent studies commonly addressed that HDAC inhibition elevated cellular ROS37, 38, 39, 40, which is likely to be one of primary mechanisms of cancer cell death. Pre-exposure of cancer cells to antioxidants was able to protect the cells from HDACi-mediated cell death41. However, the mechanism of such oxidative stress induction by HDAC inhibition remains to be fully elucidated. Accumulating evidence has suggested that HDAC directly represses cellular pro-oxidant proteins, such as TXNIP and NADPH oxidases13, 42. TXNIP has been identified from yeast-two-hybrid screen as a thioredoxin-interacting protein43. Thioredoxin is a multifunctional antioxidant protein involved in reduction of other protein disulfide44 and H2O245, 46, in DNA synthesis as a hydrogen donor47 and in redox regulation of transcription factors48, which plays an important role in cellular redox regulation. TXNIP binds to thioredoxin and inhibits its disulfide reductase activity49, thus promoting cellular oxidative stress and apoptosis. TXNIP expression is negatively correlated with tumor progression and its high expression is significantly associated with better treatment outcomes, hence being considered as a tumor suppressor50, 51. In our study, we observed that TXNIP expression was sharply increased in breast cancer cells treated with HDACi. At later time points, however TXNIP level went down to a basal level. The early increase of the TXNIP level was likely due to the HDAC inhibition-induced de-repression of TXNIP transcription as local histones around the TXNIP promoter were highly acetylated upon entinostat treatment. The level of the transcription factor MLXIP (MondoA) remained unchanged at early treatment period, while it was reduced at later treatment period, suggesting that the reduced TXNIP level at later time point was likely due to the reduction of its transcription activator level. The transient increase of TXNIP was sufficient to induce cellular oxidative stress and DNA damage responses as evidenced by prolonged expression of HO-1 and γ-H2AX under entinostat treatment. Silencing of TXNIP markedly reversed the expression of HO-1 and γ-H2AX, as well as the entinostat-induced ROS generation, DNA damage and apoptosis induction in BRCA1 deficient breast cancer cells. These data demonstrate that TXNIP is a key factor mediating the entinostat-induced synthetic lethality.
BRCA1-deficient cells are highly sensitive to DNA damaging agents. With its major role in homology-directed repair, cells with BRCA1 mutations are unable to properly repair DNA double-strand breaks, leading to increase in sensitivity to DNA damaging agents52. With this in mind, we combined an HDACi with a BETi that we previously identified as a synthetic lethality drug for BRCA1 deficient breast cancer cells. The combination of the two ROS-inducing agents significantly promoted cellular oxidative stress and DNA damages, and thereby induced apoptosis and synergetic anticancer effect in BRCA1 deficient breast cancer cells, while it showed negligible effect in BRCA1 wildtype cells. These data suggest that the combination of HDACi and BETi could provide stronger synthetic lethality effect in BRCA1 deficient breast cancer and serve as a treatment option to avoid drug resistance associated with HDACi or BETi monotherapy.
HDAC inhibition can exert pleiotropic effect in several biological pathways as HDAC is involved in general epigenetic gene expression regulation. Hence, it cannot be ruled out that mechanisms other than TXNIP-ROS pathway may be also involved in the HDAC inhibition-induced synthetic lethality in BRCA1 deficient breast cancer cells. For example, HDAC inhibition indirectly down-regulates the expression of proteins involved in DNA damage repair, such as ATM53 and RAD5254, whose loss of expression was reported to sensitize BRCA1 deficient cancer cells55, 56. Further study is necessary to elucidate the involvement of these DNA damage repair pathways in the synthetic lethality between HDAC and BRCA1.
In summary, we show that HDAC inhibition is synthetic lethal with BRCA1 deficiency in breast cancer cells via elevating oxidative stress and DNA damage-induced apoptosis. Clinical investigation of HDACi alone or in combination with BETi is warranted for patients with BRCA1 deficient breast cancer.
Acknowledgments
We thank to the members of the Genomics and Bioinformatics Core of FHS and FHS Animal Facility at the University of Macau for experimental and technical supports. Transcriptome profiling work was performed in part at the High Performance Computing Cluster (HPCC) which is supported by Information and Communication Technology Office (ICTO) of the University of Macau, China. This work was supported by the Multi-Year Research Grant of the University of Macau, China (MYRG2017-00176-FHS to Joong Sup Shim).
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2019.08.008.
Author contributions
Baoyuan Zhang, Chu-Xia Deng and Joong Sup Shim conceptualized and designed the study. Baoyuan Zhang, Junfang Lyu, Eun Ju Yang, Yifan Liu, Changjie Wu, Qiang Chen and Xiaoling Xu performed the experiments. Baoyuan Zhang, Eun Ju Yang, Lakhansing Pardeshi and Kaeling Tan analyzed the data. Baoyuan Zhang and Joong Sup Shim wrote and revised the manuscript. Joong Sup Shim obtained the funding and supervised the whole study. All authors read and approved the final manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Gatza M.L., Lucas J.E., Barry W.T., Kim J.W., Wang Q.L., Crawford M.D. A pathway-based classification of human breast cancer. Proc Natl Acad Sci U S A. 2010;107:6994–6999. doi: 10.1073/pnas.0912708107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Denkert C., Liedtke C., Tutt A., von Minckwitz G. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet. 2017;389:2430–2442. doi: 10.1016/S0140-6736(16)32454-0. [DOI] [PubMed] [Google Scholar]
- 4.Young S.R., Pilarski R.T., Donenberg T., Shapiro C., Hammond L.S., Miller J. The prevalence of BRCA1 mutations among young women with triple-negative breast cancer. BMC Cancer. 2009;9:86. doi: 10.1186/1471-2407-9-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deng C.X., Wang R.H. Roles of BRCA1 in DNA damage repair: a link between development and cancer. Hum Mol Genet. 2003;12:R113–R123. doi: 10.1093/hmg/ddg082. [DOI] [PubMed] [Google Scholar]
- 6.Deng C.X. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006;34:1416–1426. doi: 10.1093/nar/gkl010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farmer H., McCabe N., Lord C.J., Tutt A.N.J., Johnson D.A., Richardson T.B. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 8.Savage K.I., Harkin D.P. BRCA1, a ‘complex’ protein involved in the maintenance of genomic stability. FEBS J. 2015;282:630–646. doi: 10.1111/febs.13150. [DOI] [PubMed] [Google Scholar]
- 9.Lord C.J., Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355:1152–1158. doi: 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malyuchenko N.V., Kotova E.Y., Kulaeva O.I., Kirpichnikov M.P., Studitskiy V.M. PARP1 inhibitors: antitumor drug design. Acta Naturae. 2015;7:27–37. [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang B., Lyu J., Liu Y., Wu C., Yang E.J., Pardeshi L. BRCA1 deficiency sensitizes breast cancer cells to bromodomain and extra-terminal domain (BET) inhibition. Oncogene. 2018;37:6341–6356. doi: 10.1038/s41388-018-0408-8. [DOI] [PubMed] [Google Scholar]
- 12.Li Y.X., Seto E. HDACs and HDAC inhibitors in cancer development and therapy. CSH Perspect Med. 2016;6:a026831. doi: 10.1101/cshperspect.a026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou J., Bi C., Cheong L.L., Mahara S., Liu S.C., Tay K.G. The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood. 2011;118:2830–2839. doi: 10.1182/blood-2010-07-294827. [DOI] [PubMed] [Google Scholar]
- 14.Vazquez-Ortiz G., Chisholm C., Xu X., Lahusen T.J., Li C., Sakamuru S. Drug repurposing screen identifies lestaurtinib amplifies the ability of the poly (ADP-ribose) polymerase 1 inhibitor AG14361 to kill breast cancer associated gene-1 mutant and wild type breast cancer cells. Breast Cancer Res. 2014;16:R67. doi: 10.1186/bcr3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olive P.L., Banath J.P. The comet assay: a method to measure DNA damage in individual cells. Nat Protoc. 2006;1:23–29. doi: 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]
- 18.Curtis C., Shah S.P., Chin S.F., Turashvili G., Rueda O.M., Dunning M.J. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joshi-Tope G., Vastrik I., Gopinath G.R., Matthews L., Schmidt E., Gillespie M. The Genome Knowledgebase: a resource for biologists and bioinformaticists. Cold Spring Harb Symp Quant Biol. 2003;68:237–243. doi: 10.1101/sqb.2003.68.237. [DOI] [PubMed] [Google Scholar]
- 20.Brehm A., Miska E.A., McCance D.J., Reid J.L., Bannister A.J., Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- 21.Lai A., Kennedy B.K., Barbie D.A., Bertos N.R., Yang X.J., Theberge M.C. RBP1 recruits the mSIN3-histone deacetylase complex to the pocket of retinoblastoma tumor suppressor family proteins found in limited discrete regions of the nucleus at growth arrest. Mol Cell Biol. 2001;21:2918–2932. doi: 10.1128/MCB.21.8.2918-2932.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zupkovitz G., Grausenburger R., Brunmeir R., Senese S., Tischler J., Jurkin J. The cyclin-dependent kinase inhibitor p21 is a crucial target for histone deacetylase 1 as a regulator of cellular proliferation. Mol Cell Biol. 2010;30:1171–1181. doi: 10.1128/MCB.01500-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ito A., Kawaguchi Y., Lai C.H., Kovacs J.J., Higashimoto Y., Appella E. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002;21:6236–6245. doi: 10.1093/emboj/cdf616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sykes S.M., Mellert H.S., Holbert M.A., Li K., Marmorstein R., Lane W.S. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24:841–851. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamaguchi H., Woods N.T., Piluso L.G., Lee H.H., Chen J.D., Bhalla K.N. p53 acetylation is crucial for its transcription-independent proapoptotic functions. J Biol Chem. 2009;284:11171–11183. doi: 10.1074/jbc.M809268200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schafer M., Werner S. The cornified envelope: a first line of defense against reactive oxygen species. J Invest Dermatol. 2011;131:1409–1411. doi: 10.1038/jid.2011.119. [DOI] [PubMed] [Google Scholar]
- 27.Ndiaye M.A., Nihal M., Wood G.S., Ahmad N. Skin, reactive oxygen species, and circadian clocks. Antioxid Redox Signal. 2014;20:2982–2996. doi: 10.1089/ars.2013.5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feingold P.L., Surman D.R., Brown K., Xu Y., McDuffie L.A., Shukla V. Induction of thioredoxin-interacting protein by histone deacetylase inhibitor, entinostat, is associated with DNA damage and apoptosis in esophageal adenocarcinoma. Mol Cancer Ther. 2018;17:2013–2023. doi: 10.1158/1535-7163.MCT-17-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petruccelli L.A., Dupere-Richer D., Pettersson F., Retrouvey H., Skoulikas S., Miller W.H. Vorinostat induces reactive oxygen species and DNA damage in acute myeloid leukemia cells. PLoS One. 2011;6:e20987. doi: 10.1371/journal.pone.0020987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishiyama A., Matsui M., Iwata S., Hirota K., Masutani H., Nakamura H. Identification of thioredoxin-binding protein-2/vitamin D-3 up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J Biol Chem. 1999;274:21645–21650. doi: 10.1074/jbc.274.31.21645. [DOI] [PubMed] [Google Scholar]
- 31.Yang X.J., Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–218. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gui C.Y., Ngo L., Xu W.S., Richon V.M., Marks P.A. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc Natl Acad Sci U S A. 2004;101:1241–1246. doi: 10.1073/pnas.0307708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peinado H., Ballestar E., Esteller M., Cano A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol Cell Biol. 2004;24:306–319. doi: 10.1128/MCB.24.1.306-319.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Juan L.J., Shia W.J., Chen M.H., Yang W.M., Seto E., Lin Y.S. Histone deacetylases specifically down-regulate p53-dependent gene activation. J Biol Chem. 2000;275:20436–20443. doi: 10.1074/jbc.M000202200. [DOI] [PubMed] [Google Scholar]
- 35.Di L.J., Fernandez A.G., De Siervi A., Longo D.L., Gardner K. Transcriptional regulation of BRCA1 expression by a metabolic switch. Nat Struct Mol Biol. 2010;17:1406–1413. doi: 10.1038/nsmb.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cras A., Darsin-Bettinger D., Balitrand N., Cassinat B., Soulie A., Toubert M.E. Epigenetic patterns of the retinoic acid receptor beta2 promoter in retinoic acid-resistant thyroid cancer cells. Oncogene. 2007;26:4018–4024. doi: 10.1038/sj.onc.1210178. [DOI] [PubMed] [Google Scholar]
- 37.Ruefli A.A., Ausserlechner M.J., Bernhard D., Sutton V.R., Tainton K.M., Kofler R. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc Natl Acad Sci U S A. 2001;98:10833–10838. doi: 10.1073/pnas.191208598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee J.H., Choy M.L., Ngo L., Foster S.S., Marks P.A. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc Natl Acad Sci U S A. 2010;107:14639–14644. doi: 10.1073/pnas.1008522107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosato R.R., Almenara J.A., Maggio S.C., Coe S., Atadja P., Dent P. Role of histone deacetylase inhibitor-induced reactive oxygen species and DNA damage in LAQ-824/fludarabine antileukemic interactions. Mol Cancer Ther. 2008;7:3285–3297. doi: 10.1158/1535-7163.MCT-08-0385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dai Y., Rahmani M., Dent P., Grant S. Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-kappa B activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage, XIAP downregulation, and c-Jun N-terminal kinase 1 activation. Mol Cell Biol. 2005;25:5429–5444. doi: 10.1128/MCB.25.13.5429-5444.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Newbold A., Falkenberg K.J., Prince H.M., Johnstone R.W. How do tumor cells respond to HDAC inhibition?. FEBS J. 2016;283:4032–4046. doi: 10.1111/febs.13746. [DOI] [PubMed] [Google Scholar]
- 42.Chen F., Li X., Aquadro E., Haigh S., Zhou J., Stepp D.W. Inhibition of histone deacetylase reduces transcription of NADPH oxidases and ROS production and ameliorates pulmonary arterial hypertension. Free Radic Biol Med. 2016;99:167–178. doi: 10.1016/j.freeradbiomed.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vignols F., Brehelin C., Surdin-Kerjan Y., Thomas D., Meyer Y. A yeast two-hybrid knockout strain to explore thioredoxin-interacting proteins in vivo. Proc Natl Acad Sci U S A. 2005;102:16729–16734. doi: 10.1073/pnas.0506880102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holmgren A. Enzymatic reduction oxidation of protein disulfides by thioredoxin. Methods Enzymol. 1984;107:295–300. doi: 10.1016/0076-6879(84)07019-1. [DOI] [PubMed] [Google Scholar]
- 45.Kang S.W., Chae H.Z., Seo M.S., Kim K.H., Baines I.C., Rhee S.G. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-alpha. J Biol Chem. 1998;273:6297–6302. doi: 10.1074/jbc.273.11.6297. [DOI] [PubMed] [Google Scholar]
- 46.Zhang P., Liu B., Kang S.W., Seo M.S., Rhee S.G., Obeid L.M. Thioredoxin peroxidase is a novel inhibitor of apoptosis with a mechanism distinct from that of BCL-2. J Biol Chem. 1997;272:30615–30618. doi: 10.1074/jbc.272.49.30615. [DOI] [PubMed] [Google Scholar]
- 47.Holmgren A. Thioredoxin and glutaredoxin systems. J Biol Chem. 1989;264:13963–13966. [PubMed] [Google Scholar]
- 48.Schenk H., Klein M., Erdbrugger W., Droge W., Schulzeosthoff K. Distinct effects of thioredoxin and antioxidants on the activation of transcription factors NF-Kappa B and AP-1. Proc Natl Acad Sci U S A. 1994;91:1672–1676. doi: 10.1073/pnas.91.5.1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chutkow W.A., Patwari P., Yoshioka J., Lee R.T. Thioredoxin-interacting protein (Txnip) is a critical regulator of hepatic glucose production. J Biol Chem. 2008;283:2397–2406. doi: 10.1074/jbc.M708169200. [DOI] [PubMed] [Google Scholar]
- 50.Woolston C.M., Madhusudan S., Soomro I.N., Lobo D.N., Reece-Smith A.M., Parsons S.L. Thioredoxin interacting protein and its association with clinical outcome in gastro-oesophageal adenocarcinoma. Redox Biol. 2013;1:285–291. doi: 10.1016/j.redox.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cadenas C., Franckenstein D., Schmidt M., Gehrmann M., Hermes M., Geppert B. Role of thioredoxin reductase 1 and thioredoxin interacting protein in prognosis of breast cancer. Breast Cancer Res. 2010;12:R44. doi: 10.1186/bcr2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moynahan M.E., Cui T.Y., Jasin M. Homology-directed DNA repair, mitomycin-C resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res. 2001;61:4842–4850. [PubMed] [Google Scholar]
- 53.Thurn K.T., Thomas S., Raha P., Qureshi I., Munster P.N. Histone deacetylase regulation of ATM-mediated DNA damage signaling. Mol Cancer Ther. 2013;12:2078–2087. doi: 10.1158/1535-7163.MCT-12-1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen C.C., Huang J.S., Wang T.H., Kuo C.H., Wang C.J., Wang S.H. Dihydrocoumarin, an HDAC inhibitor, increases DNA damage sensitivity by inhibiting Rad52. Int J Mol Sci. 2017;18:E2655. doi: 10.3390/ijms18122655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen C.C., Kass E.M., Yen W.F., Ludwig T., Moynahan M.E., Chaudhuri J. ATM loss leads to synthetic lethality in BRCA1 BRCT mutant mice associated with exacerbated defects in homology-directed repair. Proc Natl Acad Sci U S A. 2017;114:7665–7670. doi: 10.1073/pnas.1706392114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lok B.H., Carley A.C., Tchang B., Powell S.N. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene. 2013;32:3552–3558. doi: 10.1038/onc.2012.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.