

Abstract
Background
Inflammatory bowel disease (IBD) is an umbrella term used to describe a group of chronic, progressive inflammatory disorders of the digestive tract. Crohn's disease and ulcerative colitis are the two main types. Fatigue is a common, debilitating and burdensome symptom experienced by individuals with IBD. The subjective, complex nature of fatigue can often hamper its management. The efficacy and safety of pharmacological or non‐pharmacological treatments for fatigue in IBD is not yet established through systematic review of studies.
Objectives
To assess the efficacy and safety of pharmacological and non‐pharmacological interventions for managing fatigue in IBD compared to no treatment, placebo or active comparator.
Search methods
A systematic search of the databases Embase, MEDLINE, Cochrane Library, CINAHL, PsycINFO was undertaken from inception to July 2018. A top‐up search was run in October 2019. We also searched the Cochrane IBD Group Specialized Register, the Cochrane Central Register of Controlled Trials, ongoing trials and research registers, conference abstracts and reference lists for potentially eligible studies.
Selection criteria
Randomised controlled trials of pharmacological and non‐pharmacological interventions in children or adults with IBD, where fatigue was assessed as a primary or secondary outcome using a generic or disease‐specific fatigue measure, a subscale of a larger quality of life scale or as a single‐item measure, were included.
Data collection and analysis
Two authors independently screened search results and four authors extracted and assessed bias independently using the Cochrane 'Risk of bias' tool. The primary outcome was fatigue and the secondary outcomes included quality of life, adverse events (AEs), serious AEs and withdrawal due to AEs. Standard methodological procedures were used.
Main results
We included 14 studies (3741 participants): nine trials of pharmacological interventions and five trials of non‐pharmacological interventions. Thirty ongoing studies were identified, and five studies are awaiting classification. Data on fatigue were available from nine trials (1344 participants). In only four trials was managing fatigue the primary intention of the intervention (electroacupuncture, physical activity advice, cognitive behavioural therapy and solution‐focused therapy).
Electroacupuncture Fatigue was measured with Functional Assessment of Chronic Illness Therapy ‐ Fatigue (FACIT‐F) (scores range from 0 to 52). The FACIT‐F score at week eight was 8.00 points higher (better) in participants receiving electroacupuncture compared with no treatment (mean difference (MD) 8.00, 95% CI 6.45 to 9.55; 1 RCT; 27 participants; low‐certainty evidence). Results at week 16 could not be calculated. FACIT‐F scores were also higher with electroacupuncture compared to sham electroacupuncture at week eight (MD 5.10, 95% CI 3.49 to 6.71; 1 RCT; 30 participants; low‐certainty evidence) but not at week 16 (MD 2.60, 95% CI 0.74 to 4.46; 1 RCT; 30 participants; low‐certainty evidence). No adverse events were reported, except for one adverse event in the sham electroacupuncture group.
Cognitive behavioural therapy (CBT) and solution‐focused therapy Compared with a fatigue information leaflet, the effects of CBT on fatigue are very uncertain (Inflammatory Bowel Disease‐Fatigue (IBD‐F) section I: MD ‐2.16, 95% CI ‐6.13 to 1.81; IBD‐F section II: MD ‐21.62, 95% CI ‐45.02 to 1.78; 1 RCT, 18 participants, very low‐certainty evidence). The efficacy of solution‐focused therapy on fatigue is also very uncertain, because standard summary data were not reported (1 RCT, 98 participants).
Physical activity advice One 2 x 2 factorial trial (45 participants) found physical activity advice may reduce fatigue but the evidence is very uncertain. At week 12, compared to a control group receiving no physical activity advice plus omega 3 capsules, FACIT‐F scores were higher (better) in the physical activity advice plus omega 3 group (FACIT‐F MD 6.40, 95% CI ‐1.80 to 14.60, very low‐certainty evidence) and the physical activity advice plus placebo group (FACIT‐F MD 9.00, 95% CI 1.64 to 16.36, very low‐certainty evidence). Adverse events were predominantly gastrointestinal and similar across physical activity groups, although more adverse events were reported in the no physical activity advice plus omega 3 group.
Pharmacological interventions Compared with placebo, adalimumab 40 mg, administered every other week ('eow') (only for those known to respond to adalimumab induction therapy), may reduce fatigue in patients with moderately‐to‐severely active Crohn's disease, but the evidence is very uncertain (FACIT‐F MD 4.30, 95% CI 1.75 to 6.85; very low‐certainty evidence). The adalimumab 40 mg eow group was less likely to experience serious adverse events (OR 0.56, 95% CI 0.33 to 0.96; 521 participants; moderate‐certainty evidence) and withdrawal due to adverse events (OR 0.48, 95%CI 0.26 to 0.87; 521 participants; moderate‐certainty evidence).
Ferric maltol may result in a slight increase in fatigue, with better SF‐36 vitality scores reported in the placebo group compared to the treatment group following 12 weeks of treatment (MD ‐9.31, 95% CI ‐17.15 to ‐1.47; 118 participants; low‐certainty evidence). There may be little or no difference in adverse events (OR 0.55, 95% CI 0.26 to 1.18; 120 participants; low‐certainty evidence)
Authors' conclusions
The effects of interventions for the management of fatigue in IBD are uncertain. No firm conclusions regarding the efficacy and safety of interventions can be drawn. Further high‐quality studies, with a larger number of participants, are required to assess the potential benefits and harms of therapies. Future studies should assess interventions specifically designed for fatigue management, targeted at selected IBD populations, and measure fatigue as the primary outcome.
Plain language summary
Treatments for extreme tiredness and lack of energy (fatigue) in inflammatory bowel disease
Review question
What are the effects of drug and non‐drug treatments on fatigue in individuals with inflammatory bowel disease (IBD) compared to no treatment, placebo (e.g. a sugar pill) or active comparator (e.g. a known effective treatment)?
Background
IBD is a life‐long illness that causes inflammation and ulceration in the gut. Crohn's disease and ulcerative colitis are the two main types of IBD. People living with IBD often experience fatigue, which can be burdensome and negatively impact on their quality of life. Different treatments, such as medications and exercise, may improve fatigue. However, it is unclear what the effects of such treatments on fatigue in IBD are. This review presents the available evidence of the effectiveness of treatments on fatigue in IBD.
Search date
Extensive searches were undertaken from inception up to July 2018. A top‐up search was run in October 2019.
Study characteristics
Fourteen studies (3741 participants with IBD) met the inclusion criteria. Nine different drug trials, four non‐drug trials and one multimodular trial were included in the review. Thirty ongoing studies were also identified and five studies are awaiting classification. In only four trials was managing fatigue the aim of the intervention. In the remaining trials the interventions were aimed at managing other symptoms, including fatigue. Data on fatigue were not available for the fourteen trials, therefore, the findings of this review are based on 1344 participants in nine trials. Most studies were small in size and had low or very low quality of evidence.
Key results and quality of evidence
The evidence suggests electroacupuncture may result in a large reduction in fatigue compared to control and sham electroacupuncture, however, the overall certainty of the evidence is low due to sparse data. No adverse events were reported, except for one adverse event in the sham acupuncture group.
We are very uncertain about the effect of cognitive behavioural therapy and solution‐focused therapy on fatigue, as the quality of the evidence is very low.
One small study found that physical activity advice plus omega 3 and physical activity advice plus placebo may reduce fatigue compared to no physical activity advice plus omega 3. Adverse events were similar across physical activity groups, although more adverse events were reported in the no physical activity advice plus omega 3 group. Adverse events were mainly mild gastrointestinal events like diarrhoea and bloating
Compared with placebo, the drug alimumab 40 mg, administered every other week, may reduce fatigue in patients with moderately‐to‐severely active Crohn's disease, who are already known to respond to adalimumab treatment, but the evidence is very uncertain. People taking adalimumab 40 mg weekly were less like to experience serious adverse events or withdraw from the trial due to adverse events, compared to people taking placebo.
The evidence suggests ferric maltol results in a slight increase in fatigue in participants with Crohn's disease and ulcerative colitis, in remission or with mild‐to‐moderate disease activity. Following 12 weeks of ferric maltol treatment, less fatigue was reported in the placebo group compared to the treatment group, however, the quality of evidence is low.
Conclusion
The effects of interventions for the management of fatigue on IBD are uncertain, with limited evidence available. No firm conclusions regarding the benefits and harms (e.g. side effects) can be drawn, Further high‐quality studies, with a larger number of participants, are needed to determine the potential effect of treatments on fatigue in IBD. Future studies should assess fatigue as a primary outcome, be specifically designed for fatigue management and targeted at specific IBD populations.
Summary of findings
Summary of findings for the main comparison. Electroacupuncture compared to no treatment for participants with quiescent IBD.
| Electroacupuncture compared to control for participants with quiescent IBD | ||||||
| Patient or population: participants with quiescent IBD Setting: outpatients from a single centre in Spain Intervention: electroacupuncture Comparison: no treatment | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with control | Risk with electroacupuncture | |||||
| Fatigue assessed with: FACIT‐Fatigue follow‐up: 8 weeks | The mean fatigue score was 25.2 | MD 8 higher (6.45 higher to 9.55 higher) | ‐ | 27 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | FACIT‐F scores ranged from 0 to 52, with higher scores indicating less fatigue. The difference in fatigue levels at the week‐16 follow‐up could not be calculated. |
| Quality of life assessed with: IBDQ‐9 follow‐up: 8 weeks | The mean quality of life score was 57.0 | MD 4.5 higher (3.37 higher to 5.63 higher) | ‐ | 27 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | IBDQ‐9 scores ranged from 9 to 63, with higher scores indicating better quality of life. |
| Adverse events follow‐up: 16 weeks | See comment | ‐ | 34 (1 RCT) | ⊕⊕⊝⊝ LOW 2 | Safety evaluations occurred throughout the treatment period; no events were reported in either group. | |
| Serious adverse events follow‐up: 16 weeks | See comment | ‐ | 34 (1 RCT) | ⊕⊕⊝⊝ LOW 2 | Safety evaluations occurred throughout the treatment period; no events were reported in either group. | |
| Withdrawal due to adverse events follow‐up: 16 weeks | See comment | ‐ | 34 (1 RCT) | ⊕⊕⊝⊝ LOW 2 | Safety evaluations occurred throughout the treatment period; no events were reported in either group. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded two levels due to very serious imprecision as the number of participants was small and the confidence interval was wide.
2 Downgraded two levels due to very serious imprecision as the number of participants was small and an unvalidated outcome measure was used.
Summary of findings 2. Electroacupuncture compared to sham electroacupuncture for participants with quiescent IBD.
| Electroacupuncture compared to sham electroacupuncture for participants with quiescent IBD | ||||||
| Patient or population: participants with quiescent IBD Setting: outpatients from a single centre in Spain Intervention: electroacupuncture Comparison: sham electroacupuncture | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with sham electroacupuncture | Risk with electroacupuncture | |||||
| Fatigue assessed with: FACIT‐F follow‐up: 16 weeks | The mean fatigue score was 28.8 | MD 2.6 higher (0.74 higher to 4.46 higher) | ‐ | 30 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | FACIT‐F scores ranged from 0 to 52, with higher scores indicating less fatigue. |
| Quality of life assessed with: IBDQ‐9 follow‐up: 16 weeks | The mean quality of life score was 58.6 | MD 2.2 higher (0.98 higher to 3.42 higher) | ‐ | 30 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | IBDQ‐9 scores ranged from 9 to 63, with higher scores indicating better quality of life. |
| Adverse events follow‐up: 16 weeks | See comment | OR 0.32 (0.01 to 8.27) | 36 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | One adverse event was reported in the sham acupuncture group. | |
| Serious adverse events follow‐up: 16 weeks | See comment | ‐ | 36 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | Safety evaluations occurred throughout the treatment period; no events were reported in either group. | |
| Withdrawal due to adverse events follow‐up: 16 weeks | See comment | OR 0.32 (0.01 to 8.27) | 36 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | There was one withdrawal due to adverse events in the sham acupuncture group. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded two levels for very serious imprecision; small number of participants from a single study and the confidence interval was wide.
Summary of findings 3. CBT with therapist support compared to fatigue information leaflet only for participants with IBD.
| CBT with therapist support compared to fatigue information leaflet only for participants with IBD | ||||||
| Patient or population: participants with IBD Setting: outpatients from a single centre in the United Kingdom Intervention: CBT with therapist support Comparison: fatigue information leaflet only | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with fatigue information leaflet only | Risk with CBT with therapist support | |||||
| Fatigue assessed with: IBD‐F Section I follow‐up: 3 months | The mean fatigue score was 9.45 | MD 2.16 lower (6.13 lower to 1.81 higher) | ‐ | 18 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | IBD‐F Section I scores ranged from 0 to 20, with higher scores indicating greater levels of fatigue. |
| Fatigue assessed with: IBD‐F Section II Scale from: 0 to 120 follow‐up: 3 months | The mean fatigue score was 47.33 | MD 21.62 lower (45.02 lower to 1.78 higher) | ‐ | 16 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | IBD‐F Section II scores ranged from 0 to 120, with higher scores indicating greater impact of fatigue. |
| Quality of life assessed with: UK‐IBDQ follow‐up: 3 months | The mean quality of life score was 95.7 | MD 0.19 higher (9.32 lower to 9.7 higher) | ‐ | 19 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | UK‐IBDQ scores ranged from 32 to 224, with higher scores indicating better quality of life. |
| Adverse events | ‐ | ‐ | ‐ | ‐ | This outcome was not measured. | |
| Serious adverse events | ‐ | ‐ | ‐ | ‐ | This outcome was not measured. | |
| Withdrawal due to adverse events | ‐ | ‐ | ‐ | ‐ | This outcome was not measured. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded two levels for very serious imprecision; small number of participants from a single study and the confidence interval was wide.
2 Downgraded one level due to risk of bias in blinding of participants and personnel and blinding in outcome assessment.
Summary of findings 4. Physical activity advice plus omega 3 compared to no physical activity advice plus omega 3 for participants with quiescent IBD.
| Physical activity advice plus omega 3 compared to no physical activity advice plus omega 3 for participants with quiescent IBD | ||||||
| Patient or population: participants with quiescent IBD Setting: outpatients from a single centre in the United Kingdom Intervention: physical activity advice plus omega 3 Comparison: no physical activity advice plus omega 3 | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with no physical activity advice plus omega 3 | Risk with physical activity advice plus omega 3 | |||||
| Fatigue assessed with: FACIT‐F follow‐up: 12 weeks | The mean fatigue score was 32.1 | MD 6.4 higher (1.8 lower to 14.6 higher) | ‐ | 25 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | FACIT‐F scores ranged from 0 to 52, with higher scores indicating less fatigue. |
| Fatigue assessed with: MFI follow‐up: 12 weeks | The mean fatigue score was 14.1 | MD 0.5 lower (3.88 lower to 2.88 higher) | ‐ | 25 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | MFI scores ranged from 0 to 84, with higher scores indicating greater fatigue. |
| Fatigue assessed with: IBD‐F Section I follow‐up: 12 weeks | The mean fatigue score was 9.6 | MD 3.1 lower (6.67 lower to 0.47 higher) | ‐ | 25 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | IBD‐F Section I scores ranged from 0 to 20, with higher scores indicating greater levels of fatigue. |
| Fatigue assessed with: IBD‐F Section II follow‐up: 12 weeks | The mean fatigue score was 34.8 | MD 13.1 lower (29.37 lower to 3.17 higher) | ‐ | 25 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | IBD‐F Section II scores ranged from 0 to 120, with higher scores indicating greater impact of fatigue. |
| Quality of life assessed with: IBDQ follow‐up: 12 weeks |
The mean QoL score was 167 | MD 4.00 higher (18.46 lower to 26.46 higher) | ‐ | 25 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | IBDQ scores ranged from 32 to 224, with higher scores indicating better quality of life. |
| Adverse events assessed with: Medication diary follow‐up: 12 weeks | There were five reported adverse events in the physical activity advice plus omega 3 group and 14 adverse events in the control group, including epigastric pain, bloating, and nausea and vomiting. | ‐ | 25 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | Adverse events were recorded in the medication diary and assessed by the researcher during the 6 follow‐up contact time points. | |
| Serious adverse events | ‐ | ‐ | ‐ | ‐ | This outcome was not reported. | |
| Withdrawal due to adverse events | ‐ | ‐ | ‐ | ‐ | This outcome was not reported. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded two levels due to very serious imprecision as the number of participants was small, confidence interval was wide and pre‐protocol analyses used.
2 Downgraded one level as high risk of bias for blinding of participants and personnel and blinding of outcome assessment.
Summary of findings 5. Physical activity advice plus placebo compared to no physical activity advice plus placebo for participants with quiescent IBD.
| Physical activity advice plus placebo compared to no physical activity advice plus placebo for participants with quiescent IBD | ||||||
| Patient or population: participants with quiescent IBD Setting: outpatients from a single centre in the United Kingdom Intervention: physical activity advice plus placebo Comparison: no physical activity advice plus placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with no physical activity advice plus placebo | Risk with physical activity advice plus placebo | |||||
| Fatigue assessed with: FACIT‐F follow‐up: 12 weeks | The mean fatigue was 38.4 | MD 2.7 higher (2.48 lower to 7.88 higher) | ‐ | 27 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | FACIT‐F scores ranged from 0 to 52, with higher scores indicating less fatigue. |
| Fatigue assessed with: MFI follow‐up: 12 weeks | The mean fatigue was 15.3 | MD 2.6 lower (4.7 lower to 0.5 lower) | ‐ | 27 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | MFI scores ranged from 0 to 84, with higher scores indicating greater fatigue. |
| Fatigue assessed with: IBD‐F Section I follow‐up: 12 weeks | The mean fatigue was 8.5 | MD 1.7 lower (4.04 lower to 0.64 higher) | ‐ | 27 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | IBD‐F Section I scores ranged from 0 to 20, with higher scores indicating greater levels of fatigue. |
| Fatigue assessed with: IBD‐F Section II follow‐up: 12 weeks | The mean fatigue was 27.9 | MD 8.5 lower (21.57 lower to 4.57 higher) | ‐ | 27 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | IBD‐F Section II scores ranged from 0 to 120, with higher scores indicating greater impact of fatigue. |
| Adverse events: assessed with: Medication diary follow‐up: 12 weeks | There were four reported adverse events in the physical activity advice plus placebo group and five reported adverse events in the no physical activity advice plus placebo group, including epigastric pain, diarrhoea, bloating, nausea and vomiting, headache, and molluscum | ‐ | 27 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | Adverse events were recorded in the medication diary and assessed by the researcher during the 6 follow‐up contact time points. | |
| Serious adverse events ‐ not reported | ‐ | ‐ | ‐ | ‐ | This outcome was not reported. | |
| Withdrawal due to adverse events ‐ not reported | ‐ | ‐ | ‐ | ‐ | This outcome was not reported. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded two levels due to very serious imprecision as the number of participants was small, the confidence interval was wide and per protocol analyses used.
2 Downgraded one level due to high risk of bias for blinding of participants and personnel and blinding of outcome assessments.
Summary of findings 6. Physical activity advice plus placebo compared to no physical activity advice plus omega 3 for participants with quiescent IBD.
| Physical activity advice plus placebo compared to no physical activity advice plus omega 3 for participants with quiescent IBD | ||||||
| Patient or population: participants with quiescent IBD Setting: outpatients from a single centre in the United Kingdom Intervention: physical activity advice plus placebo Comparison: no physical activity advice plus omega 3 | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with no physical activity advice plus omega 3 | Risk with physical activity advice plus placebo | |||||
| Fatigue assessed with: FACIT‐F follow‐up: 12 weeks | The mean fatigue score was 32.1 | MD 9 higher (1.64 higher to 16.36 higher) | ‐ | 29 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | FACIT‐F scores ranged from 0 to 52, with higher scores indicating less fatigue. |
| Fatigue assessed with: MFI follow‐up: 12 weeks | The mean fatigue score was 14.1 | MD 1.4 lower (4.39 lower to 1.59 higher) | ‐ | 29 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | MFI scores ranged from 0 to 84, with higher scores indicating greater fatigue. |
| Fatigue assessed with: IBDF Section 1 follow‐up: 12 weeks | The mean fatigue score was 9.6 | MD 2.8 lower (5.93 lower to 0.33 higher) | ‐ | 29 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | IBD‐F Section I scores ranged from 0 to 20, with higher scores indicating greater levels of fatigue. |
| Fatigue assessed with: IBDF Section 2 follow‐up: 12 weeks | The mean fatigue score was 34.8 | MD 15.4 lower (30.51 lower to 0.29 lower) | ‐ | 29 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | IBD‐F Section II scores ranged from 0 to 120, with higher scores indicating greater impact of fatigue. |
| Adverse events: assessed with: Medication diary follow‐up: 12 weeks | There were four reported adverse events in the physical activity advice plus placebo group and fourteen reported adverse events in the no physical activity advice plus omega 3 group, including epigastric pain, diarrhoea, bloating, nausea and vomiting, IBD flare, joint pain, and ankle injury. | ‐ | 29 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | Adverse events were recorded in the medication diary and assessed by the researcher during the 6 follow‐up contact time points. | |
| Serious adverse events ‐ not reported | ‐ | ‐ | ‐ | ‐ | This outcome was not reported. | |
| Withdrawal due to adverse events ‐ not reported | ‐ | ‐ | ‐ | ‐ | This outcome was not reported. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded two levels due to very serious imprecision as the number of participants was small, confidence interval was wide and per protocol analyses used.
2 Downgraded one level due to high risk of bias for blinding of participants and personnel and blinding of outcome assessments.
Background
Description of the condition
Inflammatory bowel disease (IBD) represents a group of chronic, progressive, complex inflammatory disorders of the digestive tract, and approximately five million people have a diagnosis of IBD worldwide (Wilson 2012). Crohn's disease (CD) and ulcerative colitis (UC) are the two most common forms of IBD. Both diseases are characterised by periods of relapse and remission, and they have overlapping and distinct pathological and clinical features (Bernstein 2010). Individuals with CD or UC experience a wide range of symptoms including diarrhoea, abdominal pain, fatigue, weight loss and rectal bleeding (Cronin 2005).
Fatigue has been identified as one of the most burdensome symptoms experienced by individuals with IBD (Farrell 2013; Farrell 2016). This symptom is particularly problematic during active disease with prevalence rates as high as 86% reported (Van Langenberg 2010). However, patients also continue to experience fatigue during remission (41% to 48%) (Van Langenberg 2010). These high rates of fatigue are comparable with rates experienced by oncology patients (Stone 2008). Furthermore, a study by Jelsness‐Jørgensen 2011a found that chronic fatigue, defined as substantial fatigue with duration of more than six months, was significantly more common in patients with UC and CD than healthy controls. In addition, patients with IBD experiencing chronic fatigue have significantly higher levels of disease‐related worries and concerns (Jelsness‐Jørgensen 2012). Reduced energy level is a leading and consistent concern among individuals with IBD (Casati 2000; Casellas 2001; De Rooy 2001; Drossman 1989; Jelsness‐Jørgensen 2011b). In addition, IBD‐related fatigue negatively impacts on health‐related quality of life and activities of daily living (Czuber‐Dochan 2013a; Czuber‐Dochan 2013b; Graff 2011; Jelsness‐Jørgensen 2011c; Minderhoud 2003; Opheim 2014). Despite the high prevalence of chronic fatigue in IBD, this subjective complaint remains largely ignored in the IBD literature, particularly regarding the investigation of underlying mechanisms and treatment strategies for fatigue.
Fatigue has been difficult to delineate due to the subjective nature of the symptom. In chronic diseases, fatigue has been defined as a ‘persistent, overwhelming sense of tiredness, weakness or exhaustion resulting in a decreased capacity for physical and mental work’ (Dittner 2004; Lai 2003). Although some studies have continued to measure fatigue in IBD from a unidimensional perspective, for example, in terms of prevalence (Minderhoud 2003), or severity (Opheim 2014), it is now generally accepted that fatigue is a multidimensional phenomenon, characterised by diminished perceived physical energy, mental capacity and psychological status (Van Langenberg 2010). These physical, cognitive and affective dimensions of fatigue form the components of generic fatigue measures such as the Multidimensional Fatigue Inventory. More recent studies have assessed fatigue using multiple dimensions, such as severity/intensity, frequency, duration, distress and impact (Bager 2012; Czuber‐Dochan 2013a). Furthermore, the characteristics of fatigue are captured to varying degrees by the diverse range of symptom and quality of life measures available, including both generic and disease‐specific indices (Czuber‐Dochan 2014c; Hjollund 2007). It is known that IBD‐related fatigue is associated with a number of physical, psychological and situational factors, with increased disease activity, depression, anxiety and stress found to be consistently associated with greater levels of fatigue (Czuber‐Dochan 2013a). As a result, the nonspecific, subjective, complex nature of fatigue can often hamper the management of this burdensome symptom. Although healthcare professionals perceive fatigue as an important and problematic symptom in patients with IBD, the management of fatigue remains poorly understood (Czuber‐Dochan 2014a). Healthcare professionals have identified the need for more information and education to facilitate the management of fatigue in clinical practice (Czuber‐Dochan 2014b). However, the effectiveness of interventions for fatigue in IBD has not been systematically reviewed.
Description of the intervention
Given the multidimensional nature of fatigue involving biological, psychosocial, and behavioural processes (Opheim 2014), pharmacological and non‐pharmacological interventions either alone or in combination may help to improve or alleviate fatigue. Pharmacological interventions involve the administration of drugs through any route. Non‐pharmacological interventions may include any type of physical, psychological, psychosocial, behavioural or educational interventions. Interventions have been developed to address the problem of IBD‐related fatigue either directly or indirectly. For example, this could be directly in terms of an intervention specifically aimed at improving or alleviating fatigue (Vogelaar 2014), or indirectly in terms of an intervention aimed at the overall management of IBD which assesses fatigue as a secondary outcome (Garcia‐Vega 2004). However, there is uncertainty regarding the effectiveness of these interventions in alleviating fatigue, particularly in the long term.
How the intervention might work
Interventions may address the physical, psychological or situational factors contributing to fatigue. It is important that these contributory factors are clearly understood in order to target interventions effectively. For example, where fatigue is related to a physical problem such as anaemia, iron supplements or intravenous iron therapy may be beneficial. Alternatively, if the physical issue is inflammation due to a disease flare, a pharmacological intervention such as biological therapy may be valuable. If altered mood is a factor contributing to fatigue, psychosocial behavioural interventions may be valuable. Often fatigue is influenced by a number of factors, therefore a multicomponent intervention may be an effective approach.
Why it is important to do this review
The incidence of IBD has been increasing over time (Molodecky 2012). Fatigue has been identified as the most burdensome symptom experienced by individuals with IBD that impacts negatively on all aspects of daily life (Farrell 2013; Farrell 2016; Wilson 2012). Due to the increasing prevalence, debilitating character and unknown aetiology, interventions for IBD‐related fatigue have received increased attention. Recently, in other chronic conditions which are associated with fatigue, there has been an increase in the number of Cochrane reviews on interventions for fatigue. For example, for cancer‐related fatigue, there are reviews assessing the effect of pharmaceutical interventions (Minton 2010), blood transfusions (Preston 2012), exercise (Cramp 2012), education (Bennett 2009), and psychosocial interventions (Goedendorp 2009). However, unlike cancer and other chronic conditions, such as multiple sclerosis (Heine 2015), peripheral neuropathy (White 2014) and rheumatoid arthritis (Cramp 2013), no systematic review has been undertaken to assess the effects of interventions for fatigue in IBD. It is therefore proposed to systematically review and synthesise existing evidence on the effects of interventions for the management of fatigue in individuals with IBD.
Objectives
The aim of this review is to assess the efficacy and safety of pharmacological and non‐pharmacological interventions on fatigue in IBD compared to no treatment, placebo or active comparator.
Methods
Criteria for considering studies for this review
Types of studies
All types of randomised controlled trials (RCTs), including cluster and cross‐over trials, were considered for inclusion.
Types of participants
Children, adolescents and adults of all ages with a clinical diagnosis of Crohn's disease, ulcerative colitis (with or without a total colectomy), or any other form of IBD (e.g. indeterminate colitis or IBD unclassified) were considered for inclusion. Participants were included regardless of whether disease status was active or in remission.
Types of interventions
Any pharmacological and non‐pharmacological interventions designed to help alleviate fatigue in individuals with IBD were included. To be eligible for inclusion, an intervention must have a focus on fatigue explicitly stated in its aims, content, or as a primary or secondary outcome measure.
The following comparisons were considered:
Pharmacological versus non‐pharmacological;
Pharmacological versus pharmacological (different drugs or same drugs with different doses and time intervals);
Pharmacological versus usual or standard care;
Non‐pharmacological versus usual or standard care;
Non‐pharmacological versus non‐pharmacological (different non‐pharmacological interventions or same non‐pharmacological intervention with different formats); or
Any of the above versus placebo.
Interventions may be delivered in any form, for example, but not limited to face to face, telephone, the internet, or technology in the case of non‐pharmacological interventions. Interventions may be delivered individually or be group‐focused and occur in different settings such as a clinic or home environment.
Types of outcome measures
Primary outcomes
The primary outcome for this review was fatigue. Therefore, eligible studies for inclusion must have fatigue or loss of energy measured as a primary or secondary outcome. Measures of fatigue are self‐reported as it is a subjective phenomenon and these instruments may be generic or disease‐specific. Examples of generic self‐reported measures include but are not limited to: the Fatigue Severity Scale (FSS) (Krupp 1989), Chalder Fatigue Scale (CFQ) (Chalder 1993), Fatigue Impact Scale (FIS) (Fisk 1994; Fisk 2002), Visual Analogue Scale of Fatigue (VAS‐F) (Lee 1991), Piper Fatigue Scale (PFS) (Piper 1998), Functional Assessment of Chronic Illness Therapy – Fatigue (FACIT‐F) (Yellen 1997), Multidimensional Fatigue Inventory (MFI) (Smets 1996), and the Multidimensional Assessment of Fatigue (MAF) (Tack 1991). An example of a disease‐specific measure includes the Inflammatory Bowel Disease‐Fatigue scale (IBD‐F) (Czuber‐Dochan 2014c).
In addition, studies that reported data on fatigue, loss of energy, vigour and vitality which was assessed as a single question or as a subscale of a questionnaire (e.g. the vitality subscale of the Short Form‐36 (SF‐36) or Inflammatory Bowel Disease Questionnaire (IBDQ) ( Irvine 1999; Ware 1992) were included. Multidimensional characteristics of fatigue symptoms may be measured. For example, these characteristics may include intensity, severity, frequency, duration, distress or dimensions including physical fatigue, mental fatigue or general fatigue.
Secondary outcomes
Secondary outcomes included:
Any measure of quality of life (e.g. validated generic or disease‐specific quality of life measures, such as the SF‐36 or IBDQ); and
Adverse events.
Adverse events included:
The proportion of participants who experience any adverse event (i.e. an unfavourable outcome occurring during, but not necessarily caused by, the intervention);
Serious adverse events (i.e. an adverse event that results in death, requires hospitalisation or a life‐threatening event, resulting in a persistent or significant disability); and
Withdrawal due to adverse events.
Search methods for identification of studies
Electronic searches
The databases Embase, MEDLINE, CINAHL, and PsycINFO were searched from inception to July 2018 and these searches were updated in October 2019. We also searched the Cochrane IBD Group Specialized Register and the Cochrane Central Register of Controlled trials (CENTRAL) for applicable RCTs. The search strategies were modified for each database. Search limits included humans and publication in English language only. The search strategies used the relevant database filters or the recommended Cochrane search string for the identification of RCTs (Lefebvre 2011). The search strategies for each database are reported in Appendix 1, Appendix 2, Appendix 3, Appendix 4, and Appendix 5. One author (DF) liaised with the Cochrane IBD Group Trials Search Coordinator for the identification of potentially eligible studies.
Searching other resources
To identify other relevant published, unpublished and ongoing trials we:
Examined the reference lists of included studies and review articles for additional citations;
Searched ongoing trials and research registers including the Current Controlled Trials register (www.controlled‐trials.com), ClinicalTrials.gov (www.clinicaltrials.gov) and the WHO International Clinical Trials Registry Platform (ICTRP) (www.who.int/ictrp/en/), using the search terms 'fatigue' and 'inflammatory bowel disease' or 'ulcerative colitis' or 'Crohn’s disease';
Contacted trial authors to identify further published and unpublished trials and asked if they were willing to disclose their unpublished data;
Searched published abstracts from conference proceedings, including the European Crohn’s and Colitis Organisation Congress, Digestive Disease Week and Advances in Inflammatory Bowel Diseases; and
Searched relevant journals (Journal of Crohn's and Colitis; Inflammatory Bowel Diseases; Gastroenterology; Gastrointestinal Endoscopy).
Data collection and analysis
Selection of studies
Initially, two review authors (DF and MA) independently screened and examined the eligibility of the titles and abstracts identified by the search based on the predetermined inclusion criteria described above. Full‐text papers were retrieved for all studies appearing to meet the inclusion criteria and were read independently by two review authors (DF and MA). Trials with a heterogeneous sample of disorders were included, only if relevant data from participants with IBD could be extracted. All trial authors were contacted regarding information that was unclear or missing in order to reach a decision about inclusion. In case of disagreement about the selection of a study, arbitration was sought from a third author with content expertise (CN) and a decision made by consensus.
Data extraction and management
For each included study, two review authors independently extracted and documented the relevant data using standardised data extraction forms. The lead author (DF) extracted data for all included studies (Appendix 6). Second independent extraction of data from included studies was shared between three review authors (WCD or LPJJ or MA). All trial authors were contacted to provide additional (unpublished) relevant information. Any disagreements regarding inclusion or exclusion were resolved through discussion and by consultation with another author (CN) as necessary.
Assessment of risk of bias in included studies
For each study, the reviewers who extracted the data also independently assessed methodological quality using the Cochrane 'Risk of bias' tool (Higgins 2011a). We assessed trials for random sequence generation, allocation concealment, blinding (participants, personnel and outcome assessors), incomplete outcome data, outcome misclassification, selective outcome reporting and other potential sources of bias. We then made a judgement on each of these criteria relating to the risk of bias, of 'low risk of bias', 'high risk of bias' or 'unclear risk of bias'. Judgement justification was provided in the Characteristics of included studies section of the review. When agreement was not achieved by the two paired review authors (DF and WCD or LPJJ or MA) through discussion, another review author (CN) provided consensus assessment.
Measures of treatment effect
We used the Cochrane Collaboration’s Review Manager Software, RevMan 5, for all analyses. Outcomes were recorded both at the end of the intervention period and at the end of the follow‐up for the purpose of comparison between the intervention and control groups. These were the only time points at which outcomes were recorded in studies with multiple time points. We calculated the mean difference (MD) and the corresponding 95% confidence interval (CI) for continuous outcomes. We calculated the risk ratio (RR) and 95% CI for dichotomous outcomes.
Unit of analysis issues
The level at which randomisation occurred was accounted for in the data analysis. We planned that the unit of analysis would be individuals for participants individually randomised to one of two groups. Where groups of individuals were randomised together to the same intervention (i.e. cluster‐randomised trials), we planned to contact the trial authors for further information if these group data were not reported. Where individuals underwent more than one intervention during the period of the study (i.e. cross‐over trial), we planned to only include the first part of the study (i.e. before the cross‐over) to avoid potential carry‐over effects. For multi‐arm pharmacological trials with a single placebo group and two treatment dose groups, we planned to split the placebo group in half to avoid a unit of analysis error (Higgins 2011b). We did not find any available cluster‐randomised or cross‐over studies. However, we did find study designs where multiple treatment attempts were used, therefore, we selected the dose group that reflected clinical practice as the comparison for all continuous variables, as a mean and standard deviation for each placebo comparator was not possible.
Dealing with missing data
Data were analysed on an intention‐to‐treat basis. Where data were missing, we contacted the trial authors and requested the missing data. The trial authors who replied to our additional information request are detailed in the 'Acknowledgements' section. If this information was unattainable, we planned to undertake an available case analysis by analysing only the available data (i.e. ignoring the missing data). If change scores were not available and the mean change could be calculated, we planned to impute standard deviations from baseline data using methods recommended in Chapter 16 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b). If the number of patients randomised to each group and the number of dropouts were known, we planned to calculate a worst case intention‐to‐treat (ITT) analysis, where by all dropouts were assumed to be treatment failures.
Assessment of heterogeneity
The studies were assessed for clinical homogeneity with regards to participants, interventions and outcomes. We planned to assess statistical heterogeneity in terms of the difference in the effects of interventions, by firstly visually inspecting the forest plots and secondly using statistical tests of variation (Chi² and I² statistics). We planned to investigate heterogeneity by visually inspecting the forest plots to identify outliers. If outliers were identified, we would conduct sensitivity analysis to explore potential explanations for the heterogeneity. For the Chi² test, a P value of less than 0.1 would be considered statistically significant. We planned to use the I² statistic to quantify heterogeneity (I² 0% ‐ 40%: low heterogeneity; I² 30% – 60%: moderate heterogeneity; 50% ‐ 90%: substantial heterogeneity; 75 – 100%: considerable heterogeneity) (Higgins 2011b). However, the size of the I² would be interpreted in light of the size and direction of effects, as well as the strength of evidence for heterogeneity (e.g. P value from Chi² test). If heterogeneity was suspected, the possibility of utilising a random‐effects model of meta‐analysis would be considered as recommended in Chapter 9 of the Cochrane Handbook for Systematic Reviews of Interventions (Deek 2011). We pooled statistically homogeneous studies (I² < 50%) using a fixed‐effect model.
Assessment of reporting biases
All studies were assessed for reporting bias. We assessed selective reporting by comparing outcomes that were prespecified in study protocols to those reported in study manuscripts. If protocols were not available for the included studies, we assessed reporting bias by comparing the outcomes specified in the methods section of the manuscript to those reported in the results section. If there were more than 10 included studies in a pooled analysis, we planned to investigate publication bias by constructing funnel plots (Sterne 2011).
Data synthesis
We combined data from individual studies when the interventions, participant groups and outcomes were sufficiently similar, which was determined by consensus. When pooling studies was not possible, we narratively summarised the results of individual studies. For continuous outcomes, we calculated the pooled mean difference (MD) and corresponding 95% CI. For continuous outcomes that utilised difference scales to measure the same underlying construct, we planned to calculate the standardised mean difference (SMD) and corresponding 95% CI. For dichotomous outcomes, we planned to calculate the pooled risk ratio (RR) and 95% CI.
Subgroup analysis and investigation of heterogeneity
If sufficient power and data were available, we planned to perform subgroup analyses to investigate possible reasons for variations in fatigue results across trials for the following subsets: disease type (Crohn’s disease, ulcerative colitis); disease activity (active disease, inactive disease), sex (male, female); age groups (child, adolescents, adults, elderly (aged 65 years and old)), comorbidities, and intervention type (pharmacological, non‐pharmacological).
Sensitivity analysis
We planned to perform sensitivity analysis, where appropriate, to explore the effects of risk of bias on fatigue. For example, studies identified as having high risk of bias would be excluded from the pooled analysis to see if the effect estimate changed in a substantive way. However, due to insufficient data, sensitivity analysis was deemed not appropriate in this review.
Summary of findings and assessment of the certainty of the evidence
We used the GRADE criteria (risk of bias, inconsistency, imprecision, indirectness and publication bias) to assess the overall quality of evidence for the prespecified primary and secondary outcomes (Schünemann 2011a; Schünemann 2011b). Using this approach, outcome data were rated high, moderate, low or very low certainty. All decisions to downgrade the quality of the evidence were explained using footnotes.
Using the GRADEpro software, a 'Summary of findings' table was created for the following outcomes:
Fatigue;
Quality of life;
Adverse events:
Serious adverse events; and
Withdrawal due to adverse events.
Comparisons where the primary intention of the intervention was management of fatigue were prioritised for presentation in summary of findings tables. Other comparisons, where the management of fatigue was not the primary intention of the intervention with fatigue being assessed as a secondary outcome, were presented as additional tables.
Results
Description of studies
See Characteristics of included studies and Characteristics of excluded studies.
Results of the search
The electronic search identified a total of 3183 citations (Figure 1) of which 3055 were excluded based on titles and abstracts alone. The search of conference abstracts, relevant journals, reference lists, ongoing trials and research registers, and contact with trial authors yielded 18 further potentially eligible citations. The full texts of 91 reports were examined in detail. A total of 34 citations were excluded, mostly as fatigue outcome data were not assessed or reported (n = 24), not an RCT design (n = 7), or data from IBD participants was not presented separately (n = 3). A total of fourteen trials were identified as meeting the inclusion criteria of this review (Artom 2018; Colombel 2007; Colombel 2017; Feagan 2013; García‐Vega 2004; Gasche 2015; Hetzel 2013a; Horta 2017; McNelly 2016; Raftery 2013; Sandborn 2013; Therkelsen 2016a; Therkelsen 2016b; Vogelaar 2014). Furthermore, 27 ongoing trials were identified as eligible and will be included in future updates of this review (ACTN12617000586314P; EudraCT Number: 2008‐004277‐17; EudraCT Number: 2011‐002122‐43; EudraCT Number: 2012‐005644‐26; NCT02193750; NCT02208310; NCT02517151; NCT02704624; NCT02707068; NCT02772965; NCT02849717; NCT02861053; NCT02891226; NCT02963246; NCT03104413; NCT03105102; NCT03105128; NCT03107793; NCT03162575; NCT03266484; NCT03345823; NCT03345836; NCT03345849; NCT03398135; NCT03398148; NCT03456752; NCT03466411) (Characteristics of ongoing studies).
1.

Study flow diagram
The electronic search was re‐run in October 2019, which identified an additional 337 citations (Figure 1). Five potentially relevant studies (Ghosh 2019; Louis 2019; O' Connor 2019; Sands 2018; Tew 2019) and three ongoing trials (ACTRN12619000150145; ISRCTN11470370; NCT03574948) were identified. Additional information is needed from the study authors and these studies will be assessed for inclusion at the next update (Characteristics of studies awaiting classification and Characteristics of ongoing studies)
Included studies
Fourteen trials published in peer‐reviewed journals (28 citations) were included in this review. Multiple records for ten of the trials were identified: four trials were reported in conference proceeding abstracts (and published in journals) (Artom 2018; Horta 2017; Hetzel 2013a; Raftery 2013) and six trials were reported in journal articles and conference proceeding abstracts (Colombel 2007; Colombel 2017; Feagan 2013; McNelly 2016; Sandborn 2013; Vogelaar 2014). The primary reference for four trials did not report fatigue (Colombel 2007; Colombel 2017; Feagan 2013; Sandborn 2013), however secondary publications reported the outcome (Loftus 2008; Panaccione 2018; Rubin 2009; Rubin 2018). Therefore, for the purpose of this review, the trials were included as eligible studies and the primary publication (Colombel 2007; Colombel 2017; Feagan 2013; Sandborn 2013) was used as the study identifier. In one trial (Colombel 2007), results for all randomised participants were presented in a conference proceedings abstract (Rubin 2009) and results from a subset of participants (participants who responded [achieved clinically meaningful change in disease activity by week 4] to open‐label adalimumab induction therapy) were presented in a full‐text paper (Loftus 2008). For the purpose of data synthesis, the results from all randomised participants and randomised responders were presented separately in this review, as the sample sizes and fatigue data differed between the datasets. Another secondary publication (Rubin 2018) presented fatigue subcomponent data from both the GEMINI I and II trials in a conference proceedings abstract, therefore, these two trials were presented as eligible studies in this review (Feagan 2013; Sandborn 2013). A total of five trials were reported in insufficient detail to allow for inclusion in the analysis (Colombel 2017; Feagan 2013; Hetzel 2013a; Raftery 2013; Sandborn 2013), however we are currently awaiting additional information from one trial (Colombel 2017).
Type of studies
All studies used standard therapy as a comparator. In three studies, a three‐arm design was used, with two studies including two intervention arms and a control group (Colombel 2007; García‐Vega 2004) and one study including an intervention and sham arm and a control group (Horta 2017). In one study, a four‐arm design was used (McNelly 2016).
Six studies were double‐blinded (Colombel 2007; Feagan 2013; Gasche 2015; Hetzel 2013a; Raftery 2013; Sandborn 2013), with the remaining studies single‐blinded (Horta 2017; Therkelsen 2016a; Therkelsen 2016b) or unblinded (Artom 2018; Colombel 2017; García‐Vega 2004; McNelly 2016; Vogelaar 2014). Four studies used open allocation (García‐Vega 2004; Therkelsen 2016a; Therkelsen 2016b; Vogelaar 2014), three used an interactive voice response system (Colombel 2007; Colombel 2017; Gasche 2015) and two studies used sealed opaque envelopes (Artom 2018; Horta 2017). One study used a combination of open allocation and sequentially named drug containers of identical appearance (McNelly 2016). Four studies (Feagan 2013; Hetzel 2013a; Raftery 2013; Sandborn 2013) did not present information on group allocation.
The follow‐up time ranged from 21 days (Therkelsen 2016a; Therkelsen 2016b) to 56 weeks (Colombel 2007). Only two studies had a follow‐up period longer than 12 months (Colombel 2007; García‐Vega 2004).
Populations
All studies were conducted on adults. Sample size ranged from 27 participants (Raftery 2013) to 1115 participants (Sandborn 2013). Most sample sizes were small (< 100 participants) (Artom 2018; García‐Vega 2004; Horta 2017; McNelly 2016; Raftery 2013; Therkelsen 2016a; Therkelsen 2016b; Vogelaar 2014). Settings varied from single (Artom 2018; García‐Vega 2004; Horta 2017; McNelly 2016; Therkelsen 2016a; Therkelsen 2016b) to multicentred studies (Colombel 2007; Colombel 2017; Feagan 2013; Gasche 2015; Raftery 2013; Sandborn 2013; Vogelaar 2014). It is unclear if one study (Hetzel 2013a) was a single or multicentred study. The multicentred studies ranged from two centres (Raftery 2013) to 285 centres (Sandborn 2013). Most studies recruited participants from hospital‐based settings, specifically inflammatory bowel disease clinics (Artom 2018; García‐Vega 2004; Gasche 2015; Horta 2017; McNelly 2016; Therkelsen 2016a; Therkelsen 2016b), with the specific location of recruitment unclear for some studies, although it appeared to be hospital‐based (Colombel 2007; Colombel 2017; Feagan 2013; Hetzel 2013a; Raftery 2013; Sandborn 2013; Vogelaar 2014). Most studies were conducted in European countries (Artom 2018; García‐Vega 2004; Gasche 2015; Horta 2017McNelly 2016; Raftery 2013; Therkelsen 2016a; Therkelsen 2016b; Vogelaar 2014), with four trials being conducted worldwide (Colombel 2007; Colombel 2017; Feagan 2013; Sandborn 2013).
Some studies sampled those with a diagnosis of Crohn's disease (Colombel 2007; Colombel 2017; García‐Vega 2004; Raftery 2013; Sandborn 2013; Therkelsen 2016b), with two studies focused specifically on ulcerative colitis only (Feagan 2013; Therkelsen 2016a). The remaining studies recruited individuals with Crohn's disease and ulcerative colitis (Gasche 2015; McNelly 2016) or inflammatory bowel disease (Crohn's disease, ulcerative colitis and unclassified) (Artom 2018; Hetzel 2013a; Horta 2017; Vogelaar 2014). In all studies, results were presented as an overall group rather than by disease type.
Disease status or activity was defined as an inclusion criterion in all studies, except one (Hetzel 2013a). Some studies recruited participants in remission only (Artom 2018; García‐Vega 2004; Horta 2017; McNelly 2016; Raftery 2013; Vogelaar 2014). Other studies recruited individuals with mixed disease activity, including remission or mild‐ to‐moderate disease (Gasche 2015), mild‐to‐moderately active disease (Therkelsen 2016a; Therkelsen 2016b) and moderate‐to‐severely active disease (Colombel 2007; Colombel 2017; Sandborn 2013). For Crohn's disease, the Crohn's Disease Activity Index (CDAI) was most commonly used, with remission defined as a CDAI score < 150 (Raftery 2013; Vogelaar 2014), mild‐to‐moderately active disease defined as a CDAI score < 220 (Gasche 2015) and moderate‐to‐severely active disease defined as a CDAI score 220 to 450 (Colombel 2007; Colombel 2017; Sandborn 2013). The Harvey Bradshaw Index (HBI) (García‐Vega 2004; McNelly 2016) and the short CDAI (Therkelsen 2016b) were also used to characterise disease activity for Crohn's disease populations. For ulcerative colitis, the Simple Clinical Colitis Activity Index (SSCAI) (Gasche 2015; McNelly 2016), the Clinical Activity Index (CAI) (Therkelsen 2016a; Vogelaar 2014) and the Mayo score (Feagan 2013) were used. Although there was consistency in defining disease activity in Crohn's disease, disparity existed in the cut‐off points for the ulcerative colitis disease activity indices. For example, remission was defined as SSCAI score < 4 (Gasche 2015) and also as a score < 3 (McNelly 2016). Remission was also defined in a study as CAI score < 10 (Vogelaar 2014), however, a CAI score ≥ 3 indicated a mild‐to‐moderate active disease (Therkelsen 2016a). Some studies (Colombel 2017; McNelly 2016; Sandborn 2013; Vogelaar 2014) also used c‐reactive protein (CRP) levels as a measure of disease activity, however, the studies differed in terms of the cut‐off used to define remission [ranging from <5 mg/dL (McNelly 2016) to < 10mg/dL (Vogelaar 2014)] and moderately‐to‐severely active disease [ranging from > 2.87 mg/L (Sandborn 2013) to ≥ 5 mg/L (Colombel 2017). Endoscopy disease activity scores (Colombel 2017; Feagan 2013; Sandborn 2013) and faecal calprotectin (Colombel 2017) were also used to evaluate disease activity of eligible participants.
Studies used different inclusion criteria, such as a history of TNF‐antagonist treatment (Colombel 2007; Feagan 2013; Sandborn 2013) or drug treatments (sulfasalazine or 5ASA) (García‐Vega 2004), iron deficiency anaemia (Gasche 2015; Hetzel 2013a) and fatigue severity (Artom 2018; Horta 2017; McNelly 2016; Vogelaar 2014).
The loss to follow‐up ranged from 0% (García‐Vega 2004) to 34% (Therkelsen 2016b), although most studies had less than 25% loss to follow‐up (Colombel 2007; Colombel 2017; Feagan 2013; Gasche 2015; Horta 2017; McNelly 2016; Sandborn 2013; Therkelsen 2016a; Vogelaar 2014). Information regarding loss to follow‐up was not available for two studies (Hetzel 2013a; Raftery 2013).
Interventions
Most studies measured fatigue as a secondary outcome, with only four trials specifically designed for managing fatigue (Artom 2018; Horta 2017; McNelly 2016; Vogelaar 2014). Mostly trials were pharmacological (Colombel 2007; Colombel 2017; Feagan 2013; Gasche 2015; Hetzel 2013a; Raftery 2013; Sandborn 2013; Therkelsen 2016a; Therkelsen 2016b), with four studies being non‐pharmacological (Artom 2018; Horta 2017; García‐Vega 2004; Vogelaar 2014). One study used a multi‐interventional approach of both pharmacological and non‐pharmacological interventions (McNelly 2016). The pharmacological interventions included adalimumab (Colombel 2007), vedolizumab (Feagan 2013; Sandborn 2013), iron therapies (Gasche 2015; Hetzel 2013a), and supplements, such as agaricus blazei murill‐based mushroom extract (Therkelsen 2016a; Therkelsen 2016b), omega 3 (McNelly 2016) and vitamin D3 (Raftery 2013). One trial used a combination customised pharmacological therapy, including prednisone, adalimumab and azathioprine (Colombel 2017). The non‐pharmacological interventions included cognitive behavioural therapy with therapist support (Artom 2018), electroacupuncture (Horta 2017), stress management (García‐Vega 2004), solution‐focused therapy (Vogelaar 2014) and physical activity advice (McNelly 2016).
Most of the pharmacological interventions were oral therapies administered daily (Colombel 2017; McNelly 2016; Raftery 2013) or twice daily (Gasche 2015; Therkelsen 2016a; Therkelsen 2016b), although five studies involved intravenous (Feagan 2013; Hetzel 2013a; Sandborn 2013) or subcutaneous injection treatments (Colombel 2007; Colombel 2017), administered either weekly (Colombel 2007; Colombel 2017), every other week (Colombel 2007; Colombel 2017; Sandborn 2013), as two stat doses (three to eight days apart) (Hetzel 2013a) or every four or eight weeks (Feagan 2013; Sandborn 2013). The duration of administration of regularly administered pharmacological interventions ranged from 21 days (Therkelsen 2016a; Therkelsen 2016b) to 58 weeks (Feagan 2013; Sandborn 2013). The duration of the pharmacological interventions for three studies was 12 weeks (Gasche 2015; McNelly 2016; Raftery 2013).
The non‐pharmacological interventions ranged from face‐to‐face or telephone/Skype individuals sessions (Artom 2018; Horta 2017; McNelly 2016; García‐Vega 2004), group sessions (Vogelaar 2014) to self‐directed sessions using a written guide and audiotape (García‐Vega 2004). The intensity of the interventions varied from a one‐off 15‐minute consultation on physical activity (McNelly 2016) to nine electroacupuncture sessions over an eight week period (Horta 2017). These non‐pharmacological interventions were delivered in the hospital (Horta 2017; McNelly 2016; Vogelaar 2014) or in the participants' homes (Artom 2018; García‐Vega 2004).
Outcomes
Fatigue
Fatigue was assessed in studies either using a generic or disease‐specific fatigue scale, as a subscale of a broader questionnaire or as a single‐item question. Predominantly, fatigue was assessed using a single scale (Artom 2018; Colombel 2007; Colombel 2017; Feagan 2013; Horta 2017; Gasche 2015; Hetzel 2013a; Raftery 2013; Sandborn 2013), the remaining four studies used two (Therkelsen 2016a; Therkelsen 2016b; Vogelaar 2014) or three different fatigue scales (McNelly 2016). Fatigue was most commonly assessed using the generic fatigue scale, the Functional Assessment of Chronic Illness Therapy ‐ Fatigue (FACIT‐F) scale, in five studies (Colombel 2007; Colombel 2017; Horta 2017; Hetzel 2013a; McNelly 2016). Other generic scales of fatigue, included the Multidimensional Fatigue Inventory (MFI) (McNelly 2016; Raftery 2013), the Fatigue Questionnaire (Therkelsen 2016a; Therkelsen 2016b), the Checklist of Individual Strength (Vogelaar 2014) and the Fatigue Severity Scale (Vogelaar 2014). Six studies presented fatigue data from a single‐item question (García‐Vega 2004), or as a subscale or subcomponent of quality of life questionnaires (Feagan 2013; Gasche 2015; Sandborn 2013; Therkelsen 2016a; Therkelsen 2016b). Only two studies assessed fatigue using a disease‐specific fatigue measure, namely the IBD‐Fatigue scale (IBD‐F) (Artom 2018; McNelly 2016). Summary data for fatigue were not available in five studies (Colombel 2017; Feagan 2013; Hetzel 2013a; Raftery 2013; Sandborn 2013), therefore, although eligible studies, the findings could not be included in the analysis of this review.
Quality of life
The secondary outcome of this review, quality of life, was assessed in twelve of the fourteen studies (Artom 2018; Colombel 2007; Colombel 2017; Feagan 2013; Gasche 2015; Horta 2017; McNelly 2016; Raftery 2013; Sandborn 2013; Therkelsen 2016a; Therkelsen 2016b; Vogelaar 2014). Four studies measured the outcome using both a generic and disease‐specific quality of life questionnaire (Colombel 2007; Colombel 2017; Gasche 2015; Vogelaar 2014). Six studies (Artom 2018; Feagan 2013; Horta 2017; McNelly 2016; Raftery 2013; Sandborn 2013) used a disease‐specific measure of quality of life and two studies used a generic measure (Therkelsen 2016a; Therkelsen 2016b). The most commonly used generic quality of life questionnaire was the Short Form‐36 (SF‐36) (Colombel 2007; Gasche 2015; Therkelsen 2016a; Therkelsen 2016b; Vogelaar 2014), however, the EuroQual (EQ‐5D) was also used (Vogelaar 2014). The most common disease‐specific quality of life questionnaire used was the Inflammatory Bowel Disease Questionnaire (IBDQ) (Colombel 2007; Feagan 2013; Gasche 2015; McNelly 2016; Raftery 2013; Sandborn 2013; Vogelaar 2014), the UK IBDQ (Artom 2018) and the IBDQ‐9 (Horta 2017).
Of the twelve studies, five studies presented total quality of life scores (Artom 2018; Colombel 2007; Gasche 2015; Horta 2017; McNelly 2016). Two studies reported not analysing total quality of life scores (Therkelsen 2016a; Therkelsen 2016b), whereas other studies did not publish summary data for quality of life (Colombel 2017; Feagan 2013; Raftery 2013; Sandborn 2013; Vogelaar 2014).
Adverse events
Adverse (AEs) were assessed in ten of the fourteen eligible studies (Colombel 2007; Colombel 2017; Feagan 2013; Gasche 2015; Hetzel 2013a; Horta 2017; McNelly 2016; Sandborn 2013; Therkelsen 2016a; Therkelsen 2016b). Three non‐pharmacological trials did not assess adverse events (Artom 2018; García‐Vega 2004; Vogelaar 2014), whereas most of the pharmacological and multimodular trials reported adverse events. It is unclear if one pharmacological study assessed adverse events as it was not presented in the report and the information remains unavailable (Raftery 2013). The assessment of adverse events varied considerably across the studies, with methods including the MedDRA system organ class and preferred terms (Colombel 2007; Colombel 2017; Feagan 2013; Gasche 2015; Sandborn 2013), medication diaries (McNelly 2016) and interviews (Horta 2017; Therkelsen 2016a; Therkelsen 2016b) being employed. Specific predefined adverse events were reported in one trial (Hetzel 2013a).
Other outcomes
Due to the diverse nature of the eligible trials included in this review, other outcomes were also assessed in the trials. For example, the iron therapy interventions assessed Hgb concentration and transferrin saturation (Gasche 2015; Hetzel 2013a), along with serum ferritin concentration (Gasche 2015), whereas the physical activity advice intervention assessed physical activity and body composition (McNelly 2016). Disease activity was assessed in a number of studies using established measures (Artom 2018; Colombel 2007; Colombel 2017; Gasche 2015; McNelly 2016; Raftery 2013; Therkelsen 2016a; Therkelsen 2016b), and faecal calprotectin levels (Colombel 2017; Therkelsen 2016a; Therkelsen 2016b; Vogelaar 2014). Many of the pharmacological trials assessed disease activity response and remission as primary outcomes of interest (Colombel 2007; Colombel 2017; Feagan 2013; Sandborn 2013). Some of the studies assessed a number of blood tests, such as inflammatory levels (CRP) (Colombel 2007; Colombel 2017; McNelly 2016; Raftery 2013; Sandborn 2013; Vogelaar 2014), full blood count (Raftery 2013), ethylenediamine tetraacetic acid (EDTA) (Therkelsen 2016a; Therkelsen 2016b), and serum 25(OH)D (Raftery 2013). Psychological outcomes, such as depression (Artom 2018; Colombel 2017; Horta 2017; Vogelaar 2014) and anxiety (Artom 2018; Horta 2017; Vogelaar 2014), and self‐reported outcomes including sleepiness (Artom 2018; Horta 2017), sleep quality (Vogelaar 2014) and illness perception (Artom 2018) were other outcomes assessed. Of these outcomes, only those related to fatigue, quality of life and adverse events were extracted for this review.
Excluded studies
Of the 91 reports reviewed, 34 citations were excluded. The majority (n = 24) were excluded as fatigue was not assessed as a primary or secondary outcome in the trial. These trials assessed quality of life using generic or disease‐specific measures, so potentially they were eligible to be included. However, as the subscale data on fatigue, loss of energy, vitality or vigour were not reported, these were excluded (Boye 2011; Colombel 2010; Cosnes 2013; Dewint 2014; Feagan 2003; Leiper 2001; Lichtenstein 2002; Loftus 2017; Maragkoudaki 2016; Mikocka‐Walus 2017; Paramsothy 2017; Pena Rossi 2009; Reusch 2016; Sands 2008; Sands 2013; Schmidt 2016; Schreiber 2007; Smith 2011; Smith 2013; Steinhart 2002; Targan 2007; Valentine 2009; Van Assche 2012; Vermeire 2017). Seven studies were excluded as a randomised controlled trial design was not employed (Loftus 2009; Minderhoud 2007; NCT01991314; NCT02148718; NCT02162862; Persoons 2007; Szigethy 2016). For example, trials were single‐group assignment open trials (NCT01991314; NCT02148718; Szigethy 2016) or non‐randomised trials (Minderhoud 2007; NCT02162862). Three studies were excluded as data from IBD‐specific participants were not presented separately (Ford 2016; Hetzel 2013b; Scholten 2018).
Details of the 34 excluded studies are presented in the Characteristics of excluded studies table.
Studies awaiting classification
Five studies are awaiting classification following the top‐up search conducted in October 2019 (Ghosh 2019; Louis 2019; O' Connor 2019; Sands 2018; Tew 2019). Two studies are presented in full text and three are published as conference proceedings. Additional information is needed from the study authors and these studies will be assessed for inclusion at the next update of the review.
Risk of bias in included studies
Two of the fourteen included trials (Colombel 2007; Gasche 2015) were judged as adequately meeting all criteria (Figure 2; Figure 3).
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
3.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Details on the risk of bias for each individual trial are located within the 'Characteristics of included studies' table.
Allocation
All studies were randomised, however, for two studies, the method of sequence generation could not be ascertained as sufficient description of the process was not provided to confirm true randomisation (Hetzel 2013a; Raftery 2013). Studies used sequence generation methods such as random number tables (García‐Vega 2004), interactive voice response system (Colombel 2007; Colombel 2017; Gasche 2015), computer‐generated simple randomisation (Artom 2018; Feagan 2013; Horta 2017; Sandborn 2013), computer‐generated block randomisation (McNelly 2016; Vogelaar 2014), or manual block randomisation (Therkelsen 2016a; Therkelsen 2016b).
Studies also applied measures to conceal allocation using an interactive voice response system (Colombel 2007; Colombel 2017; Gasche 2015), sequential named drug containers of identical appearance provided by a pharmacy (capsule assignment) (McNelly 2016) or sealed opaque envelopes (Artom 2018; Horta 2017). Other studies employed open allocation (high risk of bias) (García‐Vega 2004; McNelly 2016) or physical activity advice intervention (Therkelsen 2016a; Therkelsen 2016b). Concealment of allocation was unclear for the remaining five studies (Feagan 2013; Hetzel 2013a; Raftery 2013; Sandborn 2013; Vogelaar 2014) due to insufficient information available. These studies were described as randomised, however convenience allocation may have been employed.
Blinding
With regards to performance bias, studies varied from double‐blinded (Colombel 2007; Feagan 2013; Gasche 2015; Sandborn 2013), single‐blinded (Horta 2017; Therkelsen 2016a; Therkelsen 2016b) to part‐blinded (McNelly 2016) and unblinded (Artom 2018; Colombel 2017; García‐Vega 2004; Vogelaar 2014). It remained unclear from two studies (Hetzel 2013a; Raftery 2013) if blinding occurred due to limited information provided, although one study stated that the trial was double‐blinded, however, further information was not provided (Raftery 2013). Most drug trials were double‐blinded (Colombel 2007; Feagan 2013; Gasche 2015; Sandborn 2013), resulting in a low risk of bias classification. Participants, sponsors, clinical researchers and clinical staff were blinded to the treatment allocation in these studies, whereas only the participants were blinded (single‐blinded) in three studies (Horta 2017; Therkelsen 2016a; Therkelsen 2016b). Personnel administering the intervention were not blinded resulting in an unclear risk of bias classification (Therkelsen 2016a; Therkelsen 2016b). However, in another study, the participants and evaluators, but not the therapist, were blinded to group assignment, therefore, deemed at low risk of bias (Horta 2017). A high risk of bias was observed in one study which blinded participants and researchers to the capsule (supplement) type, however, were unable to blind personnel to the consultation type due to the nature of the physical activity advice intervention (McNelly 2016). Three studies were unblinded due to the inherent psychosocial nature of the interventions (Artom 2018; García‐Vega 2004; Vogelaar 2014). Although one study (García‐Vega 2004) tried to blind outcome assessment undertaken by a gastroenterologist, all studies were judged to have a high risk of bias.
The primary outcome of interest, fatigue, is a subjective outcome self‐reported in all studies, therefore, the unblinded (Artom 2018; Colombel 2017; García‐Vega 2004; Vogelaar 2014) and part‐blinded (McNelly 2016) studies were classified as at high risk of detection bias. Participants were blinded in the remaining double‐blinded (Colombel 2007; Feagan 2013; Gasche 2015; Sandborn 2013) and single‐blinded (Horta 2017; Therkelsen 2016a; Therkelsen 2016b) studies, therefore, these studies were judged to have a low risk of detection bias. An unclear risk of bias was recorded for two studies (Hetzel 2013a; Raftery 2013), as no explicit statement about blinding status of participants, healthcare providers, data collectors and outcome adjudicators was presented.
Incomplete outcome data
There was a relatively low dropout rate for the psychosocial intervention trials (Artom 2018; García‐Vega 2004; Horta 2017; McNelly 2016; Vogelaar 2014) and loss to follow‐up was balanced across groups with reasons comparable to the pharmacological trials (Colombel 2007; Colombel 2017; Feagan 2013; Gasche 2015; McNelly 2016; Sandborn 2013; Therkelsen 2016a; Therkelsen 2016b), yielding a low risk of bias classification. One study had no loss to follow‐up (García‐Vega 2004). The remaining two trials provided insufficient information on missing outcome data (Hetzel 2013a; Raftery 2013).
Selective reporting
Most studies were judged as having a low risk of bias in terms of selective outcome reporting as all information on outcomes was presented and outcomes assessed were reported or provided upon request (Colombel 2007; García‐Vega 2004; Gasche 2015McNelly 2016; Therkelsen 2016a; Therkelsen 2016b). However, selective outcome reporting was an issue in the remaining studies, where the results were presented for subgroups only or certain outcomes were not reported. For example, two trial authors provided information on the outcomes upon request (Artom 2018; Horta 2017). However, the findings are only published as conference abstracts at present, therefore, we rated these studies as having unclear risk of bias for selective reporting. One study presented data comparing the intervention group with the overall placebo population control group rather than an IBD placebo control group (Hetzel 2013a). Also, one study did not report the fatigue and quality of life scores for the intervention and control groups, but rather, data for a subgroup based on the 25‐hydroxyvitamin D (25OHD) levels were presented (Raftery 2013). Another study presented only percentages for fatigue scores, with no effect sizes or P values reported (Vogelaar 2014). Additional information requested was not provided by trial authors, therefore, these five trials were all classified as having high risk of bias (Feagan 2013; Hetzel 2013a; Raftery 2013; Sandborn 2013; Vogelaar 2014). Fatigue and quality of life data were not published for one trial (Colombel 2017), however, we are awaiting outcome data from the trial author, so this trial, therefore, was judged as having unclear risk of bias for selective outcome reporting.
Other potential sources of bias
Selection bias
Ten studies reported similar baseline characteristics between treatment groups (Artom 2018; Colombel 2007; Colombel 2017; Feagan 2013; Gasche 2015; McNelly 2016; Sandborn 2013; Therkelsen 2016a; Therkelsen 2016b; Vogelaar 2014) minimising the potential for selection bias. Unbalanced groups at baseline were noted in one study only (García‐Vega 2004), so this study was rated as having a high risk of selection bias. It remained unclear from two studies if baseline characteristics of groups differed due to limited or no information reported and so selection bias was rated as being unclear for these studies (Hetzel 2013a; Raftery 2013).
Use of validated assessment instruments
Ten studies used a validated fatigue assessment instrument to measure fatigue (Artom 2018; Colombel 2007; Colombel 2017; Feagan 2013; Gasche 2015; Hetzel 2013a; McNelly 2016; Raftery 2013; Sandborn 2013; Vogelaar 2014), however, in three trials, fatigue was assessed as a subscale or subcomponent of a broader quality of life scale only (Feagan 2013; Gasche 2015; Sandborn 2013). One study assessed the average frequency and severity of 'tiredness' (fatigue) as a single item from a 10‐item disease symptom diary (García‐Vega 2004). The validity of single‐item or subscale measures of fatigue are unknown, hence, the reliability of the study findings need to be interpreted with caution.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6
The effects of 'pharmacological versus placebo'; 'pharmacological versus pharmacological', 'non‐pharmacological versus usual or standard care', 'non‐pharmacological versus non‐pharmacological' and 'pharmacological versus non‐pharmacological' were the comparisons included. No trial made the remaining comparisons considered eligible for this review. See Table 3; Table 2; Table 4; Table 6; Table 5; Table 1; Table 7; Table 8; Table 9; Table 10; Table 11; Table 12; Table 13. The summary of effects of the interventions on the predefined outcomes (fatigue, quality of life and adverse events) of the included trials are presented below.
1. Adalimumab 40 mg every other week compared to placebo for fatigue in inflammatory bowel disease.
| Adalimumab 40 mg every other week compared to placebo for fatigue in inflammatory bowel disease | ||||||
| Patient or population: participants with moderately‐to‐severely active disease Setting: 92 centres in the United States, Europe, Canada, Australia and South Africa Intervention: adalimumab 40 mg every other week Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with adalimumab 40 mg every other week | |||||
| Fatigue assessed with: FACIT‐Fatigue follow‐up: 56 weeks | The mean fatigue score was 32.5 | MD 4.3 higher (1.75 higher to 6.85 higher) | ‐ | 337 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | FACIT‐F scores ranged from 0 to 52, with higher scores indicating less fatigue. |
| Fatigue assessed with: SF‐36 Vitality follow‐up: 56 weeks | The mean fatigue score was 54.8 | MD 2.8 higher (5.1 lower to 10.7 higher) | ‐ | 143 (1 RCT) | ⊕⊕⊝⊝ LOW 2 | SF‐36 vitality subscale scores ranged from 0 to 100, with higher scores indicating greater vitality (less fatigue). |
| Quality of life assessed with: IBDQ follow‐up: 56 weeks | The mean quality of life score was 172.9 | MD 10.4 higher (0.53 lower to 21.33 higher) | ‐ | 145 (1 RCT) | ⊕⊕⊝⊝ LOW 2 | IBDQ scores ranged from 32 to 224, with higher scores indicating better quality of life. |
| Adverse events assessed with: MedDRA follow‐up: 56 weeks | 847 per 1,000 | 888 per 1,000 (826 to 930) | OR 1.44 (0.86 to 2.41) | 521 (1 RCT) | ⊕⊕⊕⊝ MODERATE 3 | |
| Serious adverse events assessed with: MedDRA follow‐up: 56 weeks | 153 per 1,000 | 92 per 1,000 (56 to 148) | OR 0.56 (0.33 to 0.96) | 521 (1 RCT) | ⊕⊕⊕⊝ MODERATE 3 | |
| Withdrawal due to adverse events assessed with: MedDRA follow‐up: 56 weeks | 134 per 1,000 | 69 per 1,000 (39 to 119) | OR 0.48 (0.26 to 0.87) | 521 (1 RCT) | ⊕⊕⊕⊝ MODERATE 3 | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded two levels due to very serious imprecision as the confidence interval was wide and the fatigue data represented the last‐observation‐carried‐forward when a participant had missing value, dropped out or switched to open‐label therapy.
2 Downgraded two levels due to serious imprecision as the number of participants was small and confidence interval was wide.
3 Downgraded one level due to serious imprecision as the number of participants was small.
2. Adalimumab 40 mg weekly compared to placebo for fatigue in inflammatory bowel disease.
| Adalimumab 40 mg weekly compared to placebo for fatigue in inflammatory bowel disease | ||||||
| Patient or population: fatigue in inflammatory bowel disease Setting: 92 centres in the United States, Europe, Canada, Australia and South Africa Intervention: adalimumab 40 mg weekly Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with adalimumab 40 mg weekly | |||||
| Fatigue assessed with: FACIT‐Fatigue follow‐up: 56 weeks | The mean fatigue score was 32.5 | MD 2.5 higher (0.26 lower to 5.26 higher) | ‐ | 327 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | FACIT‐F scores ranged from 0 to 52 with higher scores indicating less fatigue. |
| Fatigue assessed with: SF‐36 vitality follow‐up: 56 weeks | The mean fatigue score was 54.8 | MD 3.2 higher (4.72 lower to 11.13 higher) | ‐ | 145 (1 RCT) | ⊕⊕⊝⊝ LOW 2 | SF‐36 vitality subscale scores ranged from 0 to 100, with higher scores indicating greater vitality (less fatigue). |
| Quality of life assessed with: IBDQ follow‐up: 56 weeks | The mean quality of life score was 172.9 | MD 9.4 higher (1.63 lower to 20.43 higher) | ‐ | 147 (1 RCT) | ⊕⊕⊝⊝ LOW2 | IBDQ scores ranged from 32 to 224, with higher scores indicating better quality of life. |
| Adverse events assessed with: MedDRA follow‐up: 56 weeks | 847 per 1,000 | 856 per 1,000 (785 to 906) | OR 1.08 (0.66 to 1.75) | 518 (1 RCT) | ⊕⊕⊕⊝ MODERATE 3 | |
| Serious adverse events assessed with: MedDRA follow‐up: 56 weeks | 153 per 1,000 | 81 per 1,000 (48 to 135) | OR 0.49 (0.28 to 0.86) | 518 (1 RCT) | ⊕⊕⊕⊝ MODERATE 3 | |
| Withdrawal due to adverse events assessed with: MedDRA follow‐up: 56 weeks | 134 per 1,000 | 47 per 1,000 (24 to 88) | OR 0.33 (0.15 to 0.70) | 518 (1 RCT) | ⊕⊕⊕⊝ MODERATE 3 | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded two levels due to very serious imprecision as the confidence interval was wide and the fatigue data represented the last‐observation‐carried‐forward when a participant had missing value, dropped out or switched to open‐label therapy.
2 Downgraded two levels due to serious imprecision as the number of participants was small and confidence interval was wide.
3 Downgraded one level due to serious imprecision as the number of participants was small.
3. Adalimumab maintenance compared to placebo for fatigue in inflammatory bowel disease.
| Adalimumab maintenance compared to placebo for fatigue in inflammatory bowel disease | ||||||
| Patient or population: participants with moderately‐to‐severely active Crohn's disease Setting: 92 centres in the United States, Europe, Canada, Australia and South Africa Intervention: adalimumab maintenance Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with adalimumab maintenance | |||||
| Fatigue assessed with: SF‐36 Vitality follow‐up: 56 weeks | The mean fatigue score was 54.8 | MD 3 higher (4.25 lower to 10.25 higher) | ‐ | 246 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | SF‐36 vitality subscale scores ranged from 0 to 100, with higher scores indicating better vitality (less fatigue). |
| Quality of life assessed with: SF‐36 Physical Component Summary follow‐up: 56 weeks | The mean quality of life score was 47.7 | MD 1.3 higher (1.3 lower to 3.99 higher) | ‐ | 240 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | SF‐36 physical component summary scores ranged from 0 to 100, with higher scores indicating better physical quality of life. |
| Quality of life assessed with: SF‐36 Mental Component Summary follow‐up: 56 weeks | The mean quality of life score was 48.1 | MD 2.2 higher (1.28 lower to 5.68 higher) | ‐ | 240 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | The SF‐36 mental component summary scores ranged from 0 to 100, with higher scores indicating better mental quality of life. |
| Quality of life assessed with: IBDQ follow‐up: 56 weeks | The mean quality of life score was 172.9 | MD 9.9 higher (0.4 lower to 20.2 higher) | ‐ | 250 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | IBDQ scores ranged from 32 to 224, with higher scores indicating better quality of life. |
| Adverse events assessed with: MedDRA follow‐up: 56 weeks | 847 per 1,000 | 873 per 1,000 (817 to 913) | OR 1.24 (0.81 to 1.89) | 521 (1 RCT) | ⊕⊕⊕⊝ MODERATE 2 | |
| Serious adverse events assessed with: MedDra follow‐up: 56 weeks | 153 per 1,000 | 88 per 1,000 (56 to 131) | OR 0.53 (0.33 to 0.83) | 521 (1 RCT) | ⊕⊕⊕⊝ MODERATE 2 | |
| Withdrawal due to adverse events assessed with: MedDRA follow‐up: 56 weeks | 134 per 1,000 | 58 per 1,000 (36 to 93) | OR 0.40 (0.24 to 0.66) | 521 (1 RCT) | ⊕⊕⊕⊝ MODERATE 2 | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded two levels due to serious imprecision as the number of participants was small and confidence interval was wide.
2 Downgraded one level due to serious imprecision as the number of events was small.
4. AndoSan compared to placebo for participants with mild‐to‐moderately active Crohn's disease.
| AndoSan compared to placebo for participants with mild‐to‐moderately active Crohn's disease | ||||||
| Patient or population: participants with mild‐to‐moderately active Crohn's disease Setting: outpatients from a single centre in Oslo, Norway Intervention: AndoSan Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with AndoSan | |||||
| Fatigue assessed with: Total Fatigue score follow‐up: 21 days | The mean fatigue score was 16.13 | MD 1.63 lower (3.51 lower to 0.26 higher) | ‐ | 100 (2 RCTs) | ⊕⊕⊝⊝ LOW 1 2 | Total fatigue scores ranged from 0 to 33, with higher scores indicating greater fatigue. |
| Fatigue assessed with: SF‐36 Vitality follow‐up: 21 days | The mean fatigue score was 40.61 | MD 3.68 higher (1.64 lower to 9 higher) | ‐ | 100 (2 RCTs) | ⊕⊕⊝⊝ LOW 1 2 | SF‐36 vitality subscale scores ranged from 0 to 100, with higher scores indicating greater vitality (less fatigue). |
| Quality of life ‐ not measured | ‐ | ‐ | ‐ | ‐ | This outcome was not measured. | |
| Adverse events ‐ not reported | ‐ | ‐ | ‐ | ‐ | This outcome was not reported. | |
| Serious adverse events ‐ not reported | ‐ | ‐ | ‐ | ‐ | This outcome was not reported. | |
| Withdrawal due to adverse events ‐ not reported | ‐ | ‐ | ‐ | ‐ | This outcome was not reported. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded one level due to serious imprecision as the number of participants was small and confidence interval was wide.
2 Downgraded by one level due to high risk of bias for allocation bias (open allocation).
5. Ferric maltol compared to placebo for participants in remission or mild‐to‐moderately active Crohn's disease.
| Ferric maltol compared to placebo for participants in remission or mild‐to‐moderately active Crohn's disease | ||||||
| Patient or population: participants in remission or mild‐to‐moderately active Crohn's disease Setting: outpatients from 4 centres in Austria, Germany, Hungary and the UK Intervention: ferric maltol Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with ferric maltol | |||||
| Fatigue assessed with: SF‐36 vitality follow‐up: 12 weeks | The mean fatigue score was 53.23 | MD 9.31 lower (17.15 lower to 1.47 lower) | ‐ | 118 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | SF‐36 vitality subscale scores ranged from 0 to 100, with higher scores indicating greater vitality (less fatigue). |
| Quality of life assessed with: IBDQ follow‐up: 12 weeks | The mean quality of life score was 176 | MD 3.7 higher (7.89 lower to 15.29 higher) | ‐ | 120 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | IBDQ scores ranged from 32 to 224, with higher scores indicating better quality of life. |
| Adverse events assessed with: MedDRA follow‐up: 12 weeks | 717 per 1,000 | 582 per 1,000 (397 to 749) | OR 0.55 (0.26 to 1.18) | 120 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | |
| Serious adverse events assessed with: MedDRA follow‐up: mean 12 weeks | 33 per 1,000 | 33 per 1,000 (5 to 202) | OR 1.00 (0.14 to 7.34) | 124 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | |
| Withdrawal due to adverse events assessed with: MedDRA follow‐up: 12 weeks | 83 per 1,000 | 133 per 1,000 (45 to 334) | OR 1.69 (0.52 to 5.51) | 120 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded two levels due to serious imprecision as the number of participants was small and confidence interval was wide.
6. Guided stress management compared to conventional medical treatment for participants with Crohn's disease in remission.
| Guided stress management compared to conventional medical treatment for participants with Crohn's disease in remission | ||||||
| Patient or population: participants with Crohn's disease in remission Setting: outpatients from a single centre in Spain Intervention: guided stress management Comparison: conventional medical treatment | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with conventional medical treatment | Risk with guided stress management | |||||
| Fatigue assessed with: Average frequency of tiredness follow‐up: 12 months | The mean fatigue score was 51.4 | MD 0 (30.77 lower to 30.77 higher) | ‐ | 30 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | The average frequency of tiredness was calculated as the number of symptomatic days divided by total reported days x 100. Higher mean scores indicated higher average frequency of tiredness. |
| Fatigue assessed with: Severity of tiredness follow‐up: 12 months | The mean fatigue score was 1 | MD 0.3 higher (0.39 lower to 0.99 higher) | ‐ | 30 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | The severity of tiredness scores ranged from 0 to 3, with higher scores indicating greater severity of tiredness. |
| Quality of life ‐ not measured | ‐ | ‐ | ‐ | ‐ | This outcome was not measured. | |
| Adverse events ‐ not measured | ‐ | ‐ | ‐ | ‐ | This outcome was not measured. | |
| Serious adverse events ‐ not measured | ‐ | ‐ | ‐ | ‐ | This outcome was not measured. | |
| Withdrawal due to adverse events ‐ not measured | ‐ | ‐ | ‐ | ‐ | This outcome was not measured. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded by two levels due to serious imprecision as the number of participants was small and the confidence interval was wide.
2 Downgraded by two levels due to high risk of bias for most criteria.
7. Self‐directed stress management compared to conventional medical treatment for participants with Crohn's disease in remission.
| Self‐directed stress management compared to conventional medical treatment for participants with Crohn's disease in remission | ||||||
| Patient or population: participants with Crohn's disease in remission Setting: outpatients from a single centre in Spain Intervention: self‐directed stress management Comparison: conventional medical treatment | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with conventional medical treatment | Risk with self‐directed stress management | |||||
| Fatigue assessed with: Average frequency symptom (tiredness) follow‐up: 12 months | The mean fatigue score was 51.4 | MD 29.6 lower (58.68 lower to 0.52 lower) | ‐ | 30 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | The average frequency symptom (tiredness) was calculated as the number of symptomatic days divided by the total reported days x 100. Higher scores indicated greater average frequency of tiredness. |
| Fatigue assessed with: Severity of tiredness follow‐up: 12 months | The mean fatigue score was 1 | MD 0.3 lower (0.98 lower to 0.38 higher) | ‐ | 30 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | Severity of tiredness scores ranged from 0 to 3, with higher scores indicating greater severity of tiredness. |
| Quality of life ‐ not measured | ‐ | ‐ | ‐ | ‐ | This outcome was not measured. | |
| Adverse events ‐ not measured | ‐ | ‐ | ‐ | ‐ | This outcome was not measured. | |
| Serious adverse events ‐ not measured | ‐ | ‐ | ‐ | ‐ | This outcome was not measured. | |
| Withdrawal due to adverse events ‐ not measured | ‐ | ‐ | ‐ | ‐ | This outcome was not measured. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded two levels due to serious imprecision as the number of participants was small and confidence interval was wide.
2 Downgraded by two levels due to high risk of bias for most criteria.
Pharmacological interventions versus placebo
Eight trials made this comparison (Colombel 2007; Feagan 2013; Gasche 2015; Hetzel 2013a; Raftery 2013; Sandborn 2013; Therkelsen 2016a; Therkelsen 2016b), however fatigue data were only available for four of the trials (Colombel 2007; Gasche 2015;Therkelsen 2016a; Therkelsen 2016b). In four trials, the mean difference in the primary and secondary outcomes could not be calculated, as data were not available. These comparisons included ferumoxytol versus placebo (Hetzel 2013a); vedolizumab maintenance versus placebo (Feagan 2013; Sandborn 2013); and vitamin D3 versus placebo (Raftery 2013).
a. Adalimumab versus placebo
The effect of adalimumab therapy was assessed in one trial (Colombel 2007). Adalimumab was administered either 40 mg every other week (eow) or 40 mg weekly. In this review, dichotomous data were presented for both dosing regimens individually and also combined (adalimumab maintenance therapy). Continuous data were presented for the adalimumab maintenance therapy group and the adalimumab 40 mg eow group only, to avoid a unit of analysis error with multi‐arm trials. As it was not possible to calculate a mean and standard deviation for each placebo comparator, the adalimumab 40 mg eow was selected as the dose group that reflects clinical practice.
Primary outcome ‐ Fatigue
Fatigue, as measured using the FACIT‐F and SF‐36 vitality subscale, was assessed in one trial (Colombel 2007). The mean difference between adalimumab maintenance therapy and the placebo groups could not be calculated for FACIT‐F, as summary data were not published or available upon request. However, fatigue data, as measured using the FACIT‐F, were available for randomised responders (participants who had previously responded [achieved clinically meaningful change in disease activity by week 4] to open‐label adalimumab induction therapy) only. Although fatigue was also assessed for all randomised participants, data were not available upon request. Furthermore, the FACIT‐F data represented the last‐observation‐carried‐forward when a participant had a missing value, dropped out, or switched to open‐label therapy. A difference was found in fatigue levels between adalimumab 40 mg eow, compared to the placebo at the 56‐week follow‐up, as measured by the FACIT‐F. The mean fatigue score after 56 weeks of treatment was 36.8 in the in the adalimumab 40 mg eow group compared to 32.5 in the placebo group (MD 4.30, 95% CI 1.75 to 6.85, very low‐certainty evidence) (Analysis 2.1).
2.1. Analysis.

Comparison 2 Adalimumab 40 mg eow versus placebo, Outcome 1 Fatigue: FACIT‐ F.
Fatigue, as measured using the SF‐36 vitality subscale, revealed no difference between adalimumab maintenance therapy and the adalimumab 40 mg eow group, compared to the placebo group, for all randomised participants or for randomised previous responders (low‐certainty evidence) (Analysis 1.1; Analysis 2.2).
1.1. Analysis.

Comparison 1 Adalimumab maintenance versus placebo, Outcome 1 Fatigue: SF‐36 Vitality.
2.2. Analysis.

Comparison 2 Adalimumab 40 mg eow versus placebo, Outcome 2 Fatigue: SF‐36 Vitality.
Secondary outcomes ‐ Quality of life and adverse events
Quality of life data, as measured using the SF‐36 and IBDQ, were reported in one trial (Colombel 2007). There was no difference in quality of life scores, as measured by the SF‐36 between the adalimumab maintenance therapy group and adalimumab 40 mg eow group, compared to the placebo group, for all randomised participants or randomised previous responders (low‐certainty evidence) (Analysis 1.2; Analysis 1.3; Analysis 2.3; Analysis 2.4). However, when quality of life was measured using the disease‐specific IBDQ, the mean quality of life score after 56 weeks of treatment was 187.1 in the adalimumab maintenance therapy group compared to 173.3 in the placebo group for randomised responders (MD 13.80, 95% CI 1.99 to 25.61, low‐certainty evidence) (Analysis 1.4).
1.2. Analysis.
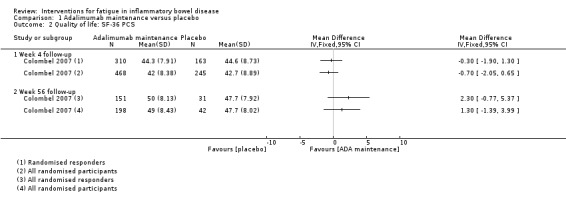
Comparison 1 Adalimumab maintenance versus placebo, Outcome 2 Quality of life: SF‐36 PCS.
1.3. Analysis.

Comparison 1 Adalimumab maintenance versus placebo, Outcome 3 Quality of life: SF‐36 MCS.
2.3. Analysis.

Comparison 2 Adalimumab 40 mg eow versus placebo, Outcome 3 Quality of life: SF‐36 PCS.
2.4. Analysis.
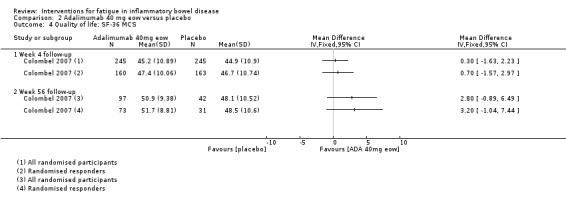
Comparison 2 Adalimumab 40 mg eow versus placebo, Outcome 4 Quality of life: SF‐36 MCS.
1.4. Analysis.

Comparison 1 Adalimumab maintenance versus placebo, Outcome 4 Quality of life: IBDQ.
The treatment‐emergent adverse events during double‐blind treatment for the intention‐to‐treat subjects revealed that there was no difference in adverse events for adalimumab maintenance therapy, adalimumab 40 mg eow and adalimumab 40 mg weekly compared to placebo (Analysis 1.5; Analysis 2.6; Analysis 3.6). Serious adverse events and withdrawal due to adverse events were reported in 9% (24/260) and 7% (18/260) of participants in the adalimumab 40 mg eow group compared to 15% (20/130) and 13% (17/130) in the placebo group, respectively (OR 0.56, 95% CI 0.30 to 1.06 and OR 0.49, 95% CI 0.25 to 1.00, moderate‐certainty evidence) (Analysis 2.7; Analysis 2.8). However, the adalimumab maintenance and adalimumab 40 mg weekly groups were less likely to experience serious adverse events and withdrawal due to adverse events compared to placebo. Serious adverse events were reported in 8.7% (45/517) and 8.2% (21/257) of participants in the treatment groups compared to 15.3% (40/261) and 15.4% (20/130) of participants in the placebo groups, respectively (OR 0.53, 95% CI 0.33 to 0.83 and OR 0.49, 95% CI 0.25 to 0.94, moderate‐certainty evidence) (Analysis 1.6; Analysis 3.7). Withdrawal due to adverse events was reported in 5.8% (30/517) and 4.7% (12/257) of participants in the treatment groups compared to 13.4% (35/261) and 13.1% (17/130) of participants in the placebo groups for the adalimumab maintenance therapy and the adalimumab 40 mg weekly groups, respectively (OR 0.40, 95% CI 0.24 to 0.66 and OR 0.33 95% CI 0.15 to 0.70, moderate‐certainty evidence) (Analysis 1.7; Analysis 3.8).
1.5. Analysis.

Comparison 1 Adalimumab maintenance versus placebo, Outcome 5 Adverse events.
2.6. Analysis.

Comparison 2 Adalimumab 40 mg eow versus placebo, Outcome 6 Adverse events.
3.6. Analysis.

Comparison 3 Adalimumab 40 mg weekly versus placebo, Outcome 6 Adverse events.
2.7. Analysis.

Comparison 2 Adalimumab 40 mg eow versus placebo, Outcome 7 Serious AEs.
2.8. Analysis.

Comparison 2 Adalimumab 40 mg eow versus placebo, Outcome 8 Withdrawal due to AEs.
1.6. Analysis.

Comparison 1 Adalimumab maintenance versus placebo, Outcome 6 Serious AEs.
3.7. Analysis.

Comparison 3 Adalimumab 40 mg weekly versus placebo, Outcome 7 Serious AEs.
1.7. Analysis.

Comparison 1 Adalimumab maintenance versus placebo, Outcome 7 Withdrawal due to AEs.
3.8. Analysis.

Comparison 3 Adalimumab 40 mg weekly versus placebo, Outcome 8 Withdrawal due to AEs.
b. AndoSanTM versus placebo
Primary outcome ‐ Fatigue
Fatigue data, as measured using the Fatigue Questionnaire and SF‐36 vitality subscale, were available in two trials (Therkelsen 2016a; Therkelsen 2016b). Data from these trials were pooled as they were assessed as clinically homogenous with regards to participants, interventions, and outcomes, with I² = 0% for total fatigue score and I² = 61% for SF‐36 vitality. At 21 days post‐intervention, there was no difference in fatigue, as measured by the Fatigue Questionnaire, between the AndoSanTM group compared to the placebo group for those with ulcerative colitis (MD ‐1.80, 95% CI ‐4.39 to 0.79, low‐certainty evidence) (Therkelsen 2016a) or Crohn's disease (MD ‐1.36,95% CI ‐4.07 to 1.35, low‐certainty evidence) (Therkelsen 2016b) (Analysis 4.1). Similar results were found when fatigue was measured using the SF‐36 vitality subscale (Therkelsen 2016a; Therkelsen 2016b). At 21 days post‐intervention, there was no difference in fatigue scores between AndoSanTM and the placebo group for those with ulcerative colitis (MD 8.60, 95% CI ‐1.09 to 18.29, low‐certainty evidence) (Therkelsen 2016a) and those with Crohn's disease (MD ‐3.72, 95% CI ‐15.31 to 7.87, low‐certainty evidence) (Therkelsen 2016b) (Analysis 4.2).
4.1. Analysis.

Comparison 4 AndoSan versus placebo, Outcome 1 Fatigue: Total Fatigue Score.
4.2. Analysis.

Comparison 4 AndoSan versus placebo, Outcome 2 Fatigue: SF‐36 Vitality subscale.
Secondary outcomes ‐ Quality of life and adverse events
Quality of life was assessed using the SF‐36, however, total physical component summary scores and mental component summary scores were not calculated. Rather, findings from the eight health dimensions of the scale were presented (author information).
Adverse events were assessed by the trial author via interviews with participants at all visits. Although participants tolerated AndoSanTM well, the trial authors did not record AEs (author information).
c. Ferric maltol versus placebo
Primary outcome ‐ Fatigue
Fatigue data, as measured using the SF‐36 vitality subscale, were available in one trial (Gasche 2015). At 12 weeks post‐intervention, there was a difference in the mean vitality scores between the ferric maltol and placebo groups. The mean vitality score was 43.92 in the treatment group compared to 53.23 in the placebo group, with higher scores indicating better vitality (less fatigue) (MD ‐9.31, 95% CI ‐17.15 to ‐1.47, low‐certainty evidence) (Analysis 5.1).
5.1. Analysis.

Comparison 5 Ferric maltol versus placebo, Outcome 1 Fatigue: SF‐36 Vitality subscale.
Secondary outcomes ‐ Quality of life and adverse events
Quality of life data, as measured using the IBDQ, were available in one trial (Gasche 2015). At 12 weeks post‐intervention, there was no difference in quality of life scores between the ferric maltol group and the placebo group (MD 3.70, 95% CI ‐7.89 to 15.29, low‐certainty evidence) (Analysis 5.2).
5.2. Analysis.

Comparison 5 Ferric maltol versus placebo, Outcome 2 Quality of life: IBDQ.
Adverse events were reported in 51% (35/60) of participants in the ferric maltol group compared to 71% (43/60) of participants in the placebo group (OR 0.55, 95% CI 0.26 to 1.18, low‐certainty evidence) (Analysis 5.3). Serious adverse events were reported in 8% (8/60) of participants in the ferric maltol group compared to 13% (6/60) of participants in the placebo group (OR 1.00,95% CI 0.14 to 7.34, low‐certainty evidence) (Analysis 5.4). No difference in withdrawal due to adverse events was found between the groups (OR 1.69, 95% CI 0.52 to 5.51, low‐certainty evidence) (Analysis 5.5).
5.3. Analysis.

Comparison 5 Ferric maltol versus placebo, Outcome 3 Adverse events.
5.4. Analysis.

Comparison 5 Ferric maltol versus placebo, Outcome 4 Serious AEs.
5.5. Analysis.

Comparison 5 Ferric maltol versus placebo, Outcome 5 Withdrawal due to AEs.
Pharmacological interventions versus pharmacological interventions
One trial made this comparison, however, the mean difference in the primary and secondary outcomes could not be calculated as data were not available (Colombel 2017).
Non‐pharmacological interventions versus usual or standard care
Three trials made this comparison (García‐Vega 2004; Horta 2017; Vogelaar 2014).
a. Electroacupuncture versus no treatment
Primary outcome ‐ Fatigue
Fatigue data, as measured using the FACIT‐F, were available in one trial (Horta 2017). There was a difference in fatigue levels found between the electroacupuncture group, compared to the control group. The mean fatigue score after eight weeks of treatment was 33.2 in the electroacupuncture group compared to 25.2 in the control group, with higher scores indicating better vitality (less fatigue) (MD 8.00, 95% CI 6.45 to 9.55, low‐certainty evidence) (Analysis 6.1). The difference in fatigue levels at the week 16 follow‐up could not be calculated. The summary data for the control group were not available at follow‐up, as the group was offered open‐label electroacupuncture treatment.
6.1. Analysis.

Comparison 6 Electroacupuncture versus no treatment, Outcome 1 Fatigue: FACIT‐F.
Secondary outcomes ‐ Quality of life and adverse events
Quality of life data, as measured using the IBDQ‐9, were available in one trial (Horta 2017). At the week eight follow‐up, there was a difference in IBDQ‐9 scores between the electroacupuncture group compared to the control group. The mean quality of life score after eight weeks of treatment was 61.5 in the treatment group, compared to 57.0 in the control group (MD 4.50, 95% CI 3.37 to 5.63, low‐certainty evidence) (Analysis 6.2). The IBDQ‐9 summary data were not available at the week 16 follow‐up to calculate the mean difference, as the group was offered open‐label electroacupuncture treatment. No adverse events, serious adverse events and withdrawal due to adverse events were reported by participants in the electroacupuncture or control groups.
6.2. Analysis.

Comparison 6 Electroacupuncture versus no treatment, Outcome 2 Quality of life: IBDQ‐9.
b. Electroacupuncture versus sham electroacupuncture
Primary outcome ‐ Fatigue
Fatigue data, as measured using the FACIT‐F, were available in one trial (Horta 2017). At the week eight and 16 follow‐up, there was a difference in fatigue scores between the electroacupuncture group and the sham electroacupuncture group. The mean vitality score after eight weeks of treatment was 33.2 in the electroacupuncture group compared to 28.1 in the sham electroacupuncture group, with higher scores indicating better vitality (less fatigue) (MD 5.10, 95% CI 3.49 to 6.71, low‐certainty evidence) (Analysis 7.1). At 16‐week follow‐up, the mean vitality score was 31.4 in the electroacupuncture group compared to 28.8 in the sham electroacupuncture group (MD 2.60, 95% CI 0.74 to 4.46, low‐certainty evidence) (Analysis 7.1).
7.1. Analysis.

Comparison 7 Electroacupuncture versus sham electroacupuncture, Outcome 1 Fatigue: FACIT‐F.
Secondary outcomes ‐ Quality of life and adverse events
Quality of life data, as measured using the IBDQ‐9, were available in one trial (Horta 2017). A mean difference was found in the quality of life scores following treatment at week eight and at the week 16 follow‐up. The mean quality of life score after eight weeks of treatment was 61.5 in the electroacupuncture group compared to 57.8 in the sham electroacupuncture group (MD 3.70, 95% CI 2.66 to 4.74, low‐certainty evidence) (Analysis 7.2). At 12‐week follow‐up, the mean quality of life score was 60.8 in the electroacupuncture group compared to 58.6 in the sham electroacupuncture group (MD 2.20, 95% CI 0.98 to 3.42, low‐certainty evidence) (Analysis 7.2). One adverse event was reported in the sham electroacupuncture group (Analysis 7.3). No serious adverse events were reported by participants in either group (Analysis 7.4). There was one withdrawal due to adverse events in the sham electroacupuncture group (Analysis 7.5).
7.2. Analysis.

Comparison 7 Electroacupuncture versus sham electroacupuncture, Outcome 2 Quality of life: IBDQ‐9.
7.3. Analysis.
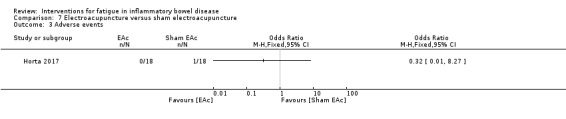
Comparison 7 Electroacupuncture versus sham electroacupuncture, Outcome 3 Adverse events.
7.4. Analysis.

Comparison 7 Electroacupuncture versus sham electroacupuncture, Outcome 4 Serious AEs.
7.5. Analysis.

Comparison 7 Electroacupuncture versus sham electroacupuncture, Outcome 5 Withdrawal due to AEs.
c. Guided stress management programme versus conventional medical treatment
Primary outcome ‐ Fatigue
Fatigue data, measured as the average frequency of tiredness and the severity of tiredness, were available in one trial (García‐Vega 2004). At post‐intervention and 12‐month follow‐up, there were no differences in the average frequency of tiredness in the guided stress management programme compared to the conventional medical treatment group (MD ‐22.10, 95% CI ‐55.47 to 11.27 and MD 0.00, 95% CI ‐30.77 to 30.77, respectively, very low‐certainty evidence) (Analysis 8.1). Likewise, the severity of tiredness was not different in the guided stress management group, compared to the conventional medical treatment group post‐intervention or at 12‐month follow‐up (MD ‐0.40, 95% CI ‐1.18 to 0.38 and MD 0.30, 95% CI ‐0.39 to 0.99, very low‐certainty evidence) (Analysis 8.2), respectively.
8.1. Analysis.

Comparison 8 Guided stress management versus conventional medical treatment, Outcome 1 Fatigue: Average frequency symptom (tiredness).
8.2. Analysis.

Comparison 8 Guided stress management versus conventional medical treatment, Outcome 2 Fatigue: Average severity of tiredness.
Secondary outcomes ‐ Quality of life and adverse events
Quality of life and adverse events were not assessed in this trial (García‐Vega 2004). The author stated that no adverse events were reported (author information).
d. Self‐directed stress management programme versus conventional medical treatment
Primary outcome ‐ Fatigue
Fatigue data, measured as the average frequency of tiredness and the severity of tiredness, were available in one trial (García‐Vega 2004). At post‐intervention, there was no difference in the average frequency of tiredness between the self‐directed stress management group and the conventional medical treatment group (MD ‐17.20, 95% CI ‐50.91 to 16.51, very low‐certainty evidence), However, at 12 months follow‐up, a difference in the average frequency of tiredness was found. The average frequency of tiredness scores was 21.8 in the self‐directed stress management group compared to 51.4 in the conventional medical treatment group (MD ‐29.60, 95% CI ‐58.68 to ‐0.52, very low‐certainty evidence) (Analysis 9.1). There was no difference in the severity of tiredness post‐intervention (MD ‐0.20, 95% CI ‐1.01 to 0.61, very low‐certainty evidence), or at 12‐month follow‐up (MD ‐0.30, 95% CI ‐0.98 to 0.38, very low‐certainty evidence) (Analysis 9.2).
9.1. Analysis.
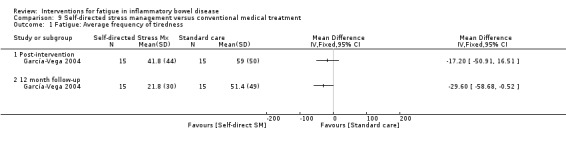
Comparison 9 Self‐directed stress management versus conventional medical treatment, Outcome 1 Fatigue: Average frequency of tiredness.
9.2. Analysis.

Comparison 9 Self‐directed stress management versus conventional medical treatment, Outcome 2 Fatigue: Average severity of tiredness.
Secondary outcomes ‐ Quality of life and adverse events
Quality of life and adverse events were not assessed in this trial (García‐Vega 2004). The author stated that no adverse events were reported (author information).
e. Solution‐focused therapy versus care‐as‐usual
Primary outcome ‐ Fatigue
Fatigue data, as measured using the CIS‐F and FSS‐9, were available in one trial (Vogelaar 2014). The mean difference in fatigue between the solution‐focused therapy and care‐as‐usual groups could not be calculated because standard summary data were not reported. However, when data from the CIS‐F were presented as a dichotomous variable, the solution‐focused therapy group showed a greater reduction in fatigue across the first six months compared with the CAU group (P < 0.001). No significant differences between the two groups were found at nine months.
Secondary outcomes ‐ Quality of life and adverse events
Quality of life was assessed in one trial (Vogelaar 2014). However, the mean difference in quality of life scores between the solution‐focused therapy and care‐as‐usual groups could not be calculated because summary data were not reported. Adverse events were not assessed in this trial (Vogelaar 2014).
Non‐pharmacological interventions versus non‐pharmacological interventions
One trial made this comparison (Artom 2018).
a. Cognitive behavioural therapy with therapist support versus fatigue information leaflet
Primary outcome ‐ Fatigue
Fatigue data, as measured using the IBD‐F, were available in one trial (Artom 2018). At month three follow‐up, there was no difference in IBD‐F section I scores and IBD‐F section II scores between the cognitive behavioural therapy with therapist support group and the fatigue information leaflet group. The mean fatigue level score was 7.29 in the treatment group compared to 9.45 in the control group (MD ‐2.16, 95% CI ‐6.13 to 1.81, very low‐certainty evidence) (Analysis 10.1). The mean impact of the fatigue score was 25.71 in the cognitive behavioural therapy with therapist support group compared to 47.33 in the fatigue information leaflet group (MD ‐21.62, 95% CI ‐45.02 to 1.78, very low‐certainty evidence) (Analysis 10.2).
10.1. Analysis.

Comparison 10 CBT with therapist support versus fatigue information leaflet only, Outcome 1 Fatigue: IBD‐F Section I.
10.2. Analysis.

Comparison 10 CBT with therapist support versus fatigue information leaflet only, Outcome 2 Fatigue: IBD‐F Section II.
Secondary outcomes ‐ Quality of life and adverse events
Quality of life data, as measured using the UK‐IBDQ, were available in one trial (Artom 2018). No difference in quality of life scores were found between the cognitive behaviour therapy with therapist support group, compared to the fatigue information leaflet group, at the month three follow‐up (MD 0.19, 95% CI ‐9.32 to 9.70, very low‐certainty evidence) (Analysis 10.3). Adverse events were not assessed in this trial (Artom 2018).
10.3. Analysis.
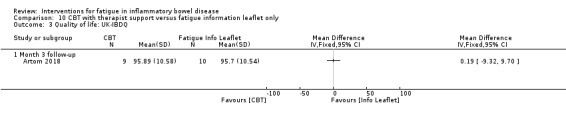
Comparison 10 CBT with therapist support versus fatigue information leaflet only, Outcome 3 Quality of life: UK‐IBDQ.
Multimodular intervention
One trial used multimodular comparisons (McNelly 2016). This 2 x 2 factorial RCT examined six comparisons, namely:
1. No physical activity advice plus omega 3 versus no physical activity advice plus placebo;
2. Physical activity advice plus omega 3 versus no physical activity advice plus omega 3;
3. Physical activity advice plus placebo versus no physical activity advice plus placebo;
4. Physical activity advice plus omega 3 versus physical activity advice plus placebo;
5. Physical activity advice plus omega 3 versus no physical activity advice plus placebo;
6. Physical activity advice plus placebo versus no physical activity advice plus omega.
In order to avoid a unit of analysis error, it was not possible to calculate mean and standard deviations for each placebo comparator, therefore the following three key comparisons were selected:
a. Physical activity advice plus omega 3 versus no physical activity advice plus omega 3
Primary outcome ‐ Fatigue
Fatigue data, as measured using the FACIT‐F, MFI and IBD‐F, were available in one trial (McNelly 2016). At the 12‐week follow‐up, there was no difference in fatigue levels between the physical activity advice plus omega 3 group, compared to the no physical activity advice plus omega 3 group, as measured using the FACIT‐F (MD 6.40, 95% CI ‐1.80 to 14.60, very low‐certainty evidence) (Analysis 11.1) or the MFI (MD ‐ 0.50, 95% CI ‐3.88 to 2.88, very low‐certainty evidence) (Analysis 11.2). Likewise, there was no difference between the groups when fatigue was measured using the disease‐specific IBD‐F scale section I (MD ‐ 3.10, 95% CI ‐6.67 to 0.47, very low‐certainty evidence) (Analysis 11.3) and section II (MD ‐13.10, 95% CI ‐29.37 to 3.17, very low‐certainty evidence) (Analysis 11.4).
11.1. Analysis.

Comparison 11 Physical activity advice plus omega 3 versus no physical activity advice plus omega 3, Outcome 1 Fatigue: FACIT‐F.
11.2. Analysis.

Comparison 11 Physical activity advice plus omega 3 versus no physical activity advice plus omega 3, Outcome 2 Fatigue: MFI.
11.3. Analysis.

Comparison 11 Physical activity advice plus omega 3 versus no physical activity advice plus omega 3, Outcome 3 Fatigue: IBD‐F Section I.
11.4. Analysis.

Comparison 11 Physical activity advice plus omega 3 versus no physical activity advice plus omega 3, Outcome 4 Fatigue: IBD‐F Section II.
Secondary outcomes ‐ Quality of life and adverse events
At the 12‐week follow‐up, there was no difference in quality of life scores between the physical activity advice plus omega 3 group, and the no physical activity advice plus omega 3 group (MD 4.00, 95% CI ‐18.46 to 26.46, very low‐certainty evidence) (Analysis 11.5).
11.5. Analysis.

Comparison 11 Physical activity advice plus omega 3 versus no physical activity advice plus omega 3, Outcome 5 Quality of life: IBDQ.
At the 12‐week follow‐up, there were five reported adverse events in the physical activity advice plus omega 3 group, namely, epigastric pain (n = 1), bloating (n = 1), headache (n = 1), leg pain (n = 1) and feeling unwell (n = 1). There were fourteen reported adverse events in the no physical activity advice plus omega 3 group, namely, epigastric pain (n = 1), diarrhoea (n = 1), bloating (n = 2), nausea and vomiting (n = 3), IBD flare (n = 1), pins and needles (n = 1), joint pain (n = 1), ankle injury (n = 1), rash (n = 1) and feeling unwell (n = 2). Serious adverse events and withdrawal due to adverse events were not reported.
b. Physical activity advice plus placebo versus no physical activity advice plus placebo
Primary outcome ‐ Fatigue
Fatigue data, as measured using the FACIT‐F, MFI and IBD‐F, were available in one trial (McNelly 2016). At the 12‐week follow‐up, there was no difference in fatigue levels between the physical activity advice plus placebo group, and the no physical activity advice plus placebo group, as measured using the FACIT‐F (MD 2.70, 95% CI ‐2.48 to 7.88, very low‐certainty evidence) (Analysis 12.1), however a difference was identified when measured using the MFI. The mean fatigue score was 12.7 in the treatment group compared to 15.3 in the control group, with higher scores indicating greater fatigue (MD ‐ 2.60, 95% CI ‐4.70 to ‐0.50, very low‐certainty evidence) (Analysis 12.2). There was no difference between the groups when fatigue was measured using the disease‐specific IBD‐F scale section I (MD ‐ 1.70, 95% CI ‐4.04 to 0.64, very low‐certainty evidence) (Analysis 12.3) and section II (MD ‐8.50, 95% CI ‐21.57 to 4.57, very low‐certainty evidence) (Analysis 12.4).
12.1. Analysis.

Comparison 12 Physical activity advice plus placebo versus no physical activity advice plus placebo, Outcome 1 Fatigue: FACIT‐F.
12.2. Analysis.

Comparison 12 Physical activity advice plus placebo versus no physical activity advice plus placebo, Outcome 2 Fatigue: MFI.
12.3. Analysis.

Comparison 12 Physical activity advice plus placebo versus no physical activity advice plus placebo, Outcome 3 Fatigue: IBD‐F Section I.
12.4. Analysis.

Comparison 12 Physical activity advice plus placebo versus no physical activity advice plus placebo, Outcome 4 Fatigue: IBD‐F Section II.
Secondary outcomes ‐ Quality of life and adverse events
At the 12‐week follow‐up, there was no difference in quality of life scores between the physical activity advice plus placebo group and the no physical activity advice plus placebo group (MD 9.00, 95% CI ‐15.72 to 33.72, very low‐certainty evidence) (Analysis 12.5).
12.5. Analysis.

Comparison 12 Physical activity advice plus placebo versus no physical activity advice plus placebo, Outcome 5 Quality of life: IBDQ.
At the 12‐week follow‐up, there were four reported adverse events in the physical activity advice plus placebo group, namely, epigastric pain (n = 1), diarrhoea (n = 1), bloating (n = 1), and nausea and vomiting (n = 1). There were five reported adverse events in the no physical activity advice plus placebo group, namely epigastric pain (n = 2), diarrhoea (n = 1), headache (n = 1), and molluscum (n = 1). Serious adverse events and withdrawal due to adverse events were not reported.
c. Physical activity advice plus placebo versus no physical activity advice plus omega 3
Primary outcome ‐ Fatigue
At the 12‐week follow‐up, a difference in fatigue levels was found between the physical activity advice plus placebo group, and the no physical activity advice plus omega 3 group, as measured using the FACIT‐F. The mean vitality scores after 12 weeks of treatment was 41.1 in the physical activity advice plus placebo group compared to 32.1 in the no physical activity advice plus omega 3, with higher scores indicating greater vitality (less fatigue) (MD 9.00, 95% CI 1.64 to 16.36, very low‐certainty evidence) (Analysis 13.1). No difference was identified when fatigue was measured using the MFI (MD ‐1.40, 95% CI ‐4.39 to 1.59, low‐certainty evidence) (Analysis 13.2). There was no difference between the groups when fatigue was measured using the disease‐specific IBD‐F scale section I, however, the mean level of fatigue scores after 12 weeks of treatment was 6.8 for the physical activity advice plus placebo group compared to 9.6 for the no physical activity advice plus omega 3 group (MD ‐2.80, 95% CI ‐5.93 to 0.33, very low‐certainty evidence) (Analysis 13.3). A difference was evident between the groups when measured by section II of the IBD‐F. The mean impact of the fatigue scores was 19.4 for the physical activity advice plus placebo group compared to 34.8 in the no physical activity advice plus omega 3 group (MD ‐15.40, 95% CI ‐30.51 to ‐0.29, very low‐certainty evidence) (Analysis 13.4).
13.1. Analysis.
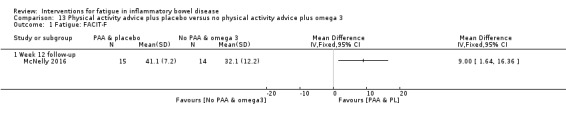
Comparison 13 Physical activity advice plus placebo versus no physical activity advice plus omega 3, Outcome 1 Fatigue: FACIT‐F.
13.2. Analysis.

Comparison 13 Physical activity advice plus placebo versus no physical activity advice plus omega 3, Outcome 2 Fatigue: MFI.
13.3. Analysis.

Comparison 13 Physical activity advice plus placebo versus no physical activity advice plus omega 3, Outcome 3 Fatigue: IBD‐F Section I.
13.4. Analysis.

Comparison 13 Physical activity advice plus placebo versus no physical activity advice plus omega 3, Outcome 4 Fatigue: IBD‐F Section II.
Secondary outcomes ‐ Quality of life and adverse events
At the 12‐week follow‐up, there was no difference in quality of life scores between the physical activity advice plus placebo group, and the no physical activity advice plus omega 3 group (MD 4.00, 95% CI ‐18.20, to 26.20, very low‐certainty evidence) (Analysis 13.5).
13.5. Analysis.

Comparison 13 Physical activity advice plus placebo versus no physical activity advice plus omega 3, Outcome 5 Quality of life: IBDQ.
At the 12‐week follow‐up, there were four reported adverse events in the physical activity advice plus placebo group, namely, epigastric pain (n = 1), diarrhoea (n = 1), bloating (n = 1), and nausea and vomiting (n = 1). There was fourteen reported adverse events in the no physical activity advice plus omega 3 group, namely, epigastric pain (n = 1), diarrhoea (n = 1), bloating (n = 2), nausea and vomiting (n = 3), IBD flare (n = 1), pins and needles (n = 1), joint pain (n = 1), ankle injury (n = 1), rash (n = 1) and feeling unwell (n = 2). Serious adverse events and withdrawal due to adverse events were not reported.
Discussion
Summary of main results
This systematic review included fourteen randomised controlled trials (3741 participants) that evaluated the efficacy and safety of different pharmacological and non‐pharmacological interventions on fatigue in IBD. The fourteen trials in the review included nine pharmacological, four non‐pharmacological and one multimodular trial. Only four trials assessed fatigue as a primary outcome (Artom 2018; Horta 2017; McNelly 2016; Vogelaar 2014). Outcome data on fatigue were not available for analysis from all fourteen trials, therefore, the findings of this review are based on 1344 participants in nine trials. Only two of the studies were pooled, due to the diversity and limited number of studies for each intervention.
Given the high prevalence and burden of fatigue in IBD, there is very limited evidence on interventions that help fatigue. In the Colombel 2007 trial, at week 56 the mean fatigue scores were lower in the adalimumab 40 mg administered every other week group (36.8) than in the placebo group (32.5) in participants with Crohn's disease who were already known to respond to adalimumab treatment, with higher scores indicating less fatigue. The mean difference between the groups was 4.30 points and this is likely to be a clinically meaningful improvement in fatigue levels (Cella 2005; Cella 2002). However, the GRADE analysis indicated that the overall certainty of the evidence for this outcome was very low due to due to serious inconsistency and very serious imprecision as the confidence interval was wide and the fatigue data measured using the FACIT‐F represented the last‐observation‐carried‐forward when a participant had missing values, dropped out or switched to open‐label therapy. No difference in fatigue was evident for adalimumab maintenance therapy or adalimumab 40 mg administered every other week in all randomised participants. Results must be interpreted with caution as the fatigue findings differ when measured using the FACIT‐F and the SF‐36 vitality subscale, and the indication for the intervention was disease activity, rather than the primary outcome of this review. Further research is needed before any firm conclusions can be drawn about potential fatigue benefits with adalimumab 40 mg administered every other week in patients with Crohn's disease.
The evidence suggests that ferric maltol results in a slight increase in fatigue in patients with Crohn's disease or ulcerative colitis, in remission or with mild‐to‐moderate disease activity (Gasche 2015). Mean fatigue scores were lower in the ferric maltol group (43.92) compared to placebo (53.23), with higher scores indicating better vitality (less fatigue). The mean difference between ferric maltol and placebo groups was 9.31 points and this is likely to be a minimally important differences in fatigue in people with Crohn's disease (Coteur 2009). However, these results should be interpreted with caution as the GRADE analysis indicated that the overall certainty of the evidence was low due to sparse data and a wide confidence interval, therefore, further research is needed.
Results from one trial on self‐directed stress management intervention suggest that the average frequency of tiredness is lower compared to the standard care group, with a mean difference of 29.6 points found at 12 months (García‐Vega 2004). However, there was no difference in the severity of tiredness between the self‐directed stress management group and those receiving standard care. The assessment measure used has not been validated and it is unclear whether observed improvements in the frequency of tiredness are clinically meaningful. Thus, we are very uncertain about the effects of self‐directed stress management on fatigue.
There is evidence to suggest that electroacupuncture may result in a large reduction in fatigue and increase in quality of life compared to no treatment after eight weeks of treatment and also compared to sham electroacupuncture post‐treatment and eight weeks following treatment (Horta 2017). The mean difference between electroacupuncture and control groups post‐treatment was 8 points and this is likely to be a clinically meaningful improvement in fatigue levels (Cella 2005; Cella 2002). The scores were not available for the control group to calculate the mean difference between the groups at week 16, as participants reporting no improvement in fatigue (FACIT‐F score < 40) at week eight were offered open electroacupuncture in this cross‐over design trial. Mean fatigue scores were also higher in the electroacupuncture group compared to the sham electroacupuncture group both at week eight and week 16 follow‐up, with higher scores indicating less fatigue. A mean difference of 5.10 points and 2.60 points was found at week eight and week 16, respectively. This suggests a likely clinically meaningful improvement in fatigue post‐treatment, however, the difference may not be clinically important eight weeks following treatment (Cella 2002; Cella 2005). The GRADE analysis indicated that the overall certainty of the evidence supporting these results were low due to sparse data and wide confidence intervals. Further research is needed before any strong conclusions can be drawn on the efficacy of electroacupuncture on fatigue.
We found that physical activity advice may also reduce fatigue but the evidence is very uncertain. One 2 x 2 factorial RCT (McNelly 2016) examined six comparisons, therefore, in order to avoid a unit of analysis error, only three key comparisons were selected. The mean differences in fatigue scores between the physical activity advice plus omega 3 and no physical activity advice plus omega 3 groups using the different fatigue scales (FACIT‐F 6.40; MFI ‐0.50; IBDF Section I ‐3.10; IBDF Section II ‐13.10) were not statistically significant, however, may be clinically meaningful for the FACIT‐F scale (Cella 2002; Cella 2005). It is unclear whether the observed improvements in fatigue for the IBDF scale are important, due to the lack of clearly defined minimally important thresholds. In contrast, the mean difference scores were lower for the physical activity advice plus placebo and no physical activity advice plus placebo comparison across the FACIT‐F (MD 2.7), MFI (‐2.6), IBDF Section I (‐1.70) and IBDF Section II (‐8.50) scales. Although, a statistically significant difference was found, as measured by the MFI scale, the mean difference may not be clinically important (Goligher 2008; Pouchot 2008). For the third comparison, it was found that the mean fatigue scores were higher in the physical activity advice plus placebo group (41.1) than the no physical activity advice plus omega 3 group (32.1) using the FACIT‐F scale, with higher scores indicating less fatigue. The mean fatigue scores were lower in the physical activity advice plus placebo group (MFI 12.7; IBDF Section I 6.8; IBDF Section II 19.4) than the no physical activity advice group plus omega 3 group (MFI 14.1; IBDF Section I 9.6; IBDF Section II 34.8), with lower scores indicating less fatigue. The mean difference for the FACIT‐F scale (MD 9.00) is likely to be clinically meaningful (Cella 2002; Cella 2005), however not important based on the MFI mean difference (MD ‐ 1.40) (Goligher 2008; Pouchot 2008) and unclear for the IBDF scale (IBDF Section I MD ‐ 2.80; IBDF Section II MD ‐ 15.40). Results must be interpreted with caution as the fatigue findings differ when measured using different fatigue scales and the overall GRADE analysis indicated that the certainty of the evidence supporting these results is low due to high risk of bias, sparse data and wide confidence intervals.
Partial data were available from some of the fatigue outcomes assessed in the solution‐focused therapy (SFT) trial (Vogelaar 2014). There is evidence to suggest that SFT shows a greater reduction in fatigue at six months follow‐up compared with care‐as‐usual, however, data were reported in a non‐standard way and the study rated as high risk of bias for selective reporting, therefore, we are very uncertain about the efficacy and safety of solution‐focused therapy on fatigue,
No difference in fatigue levels were found for any of the other comparisons, including adalimumab 40 mg weekly (Colombel 2007), AndoSanTM (Therkelsen 2016a; Therkelsen 2016b), guided stress management (García‐Vega 2004), physical activity advice and/or omega 3 (McNelly 2016) and cognitive behavioural therapy with therapist support (Artom 2018).
Fatigue data were not available for the trials investigating the effect of customised combination therapy (tight control management) (Colombel 2017), vedolizumab maintenance therapy (Feagan 2013; Sandborn 2013), vitamin D3 (Raftery 2013) or ferumoxytol (Hetzel 2013a). This was primarily due to fatigue not being a primary outcome in these trials.
Secondary outcomes relevant to our review, namely quality of life and adverse events, were both assessed in nine trials (Colombel 2007; Colombel 2017; Feagan 2013; Gasche 2015; Horta 2017; McNelly 2016; Sandborn 2013Therkelsen 2016a; Therkelsen 2016b). One trial (Hetzel 2013a) assessed adverse events only; three trials assessed quality of life only (Artom 2018; Raftery 2013; Vogelaar 2014) and one trial (García‐Vega 2004) assessed neither secondary outcomes relevant to our review. There was no difference in physical or mental quality of life, as measured using a generic quality of life scale, for adalimumab maintenance therapy or adalimumab 40 mg administered every other week compared to placebo (Colombel 2007). In contrast, when quality of life was measured using a disease‐specific measure, results suggest a difference in scores for the adalimumab maintenance therapy and adalimumab 40 mg administered every other week compared to placebo, for randomised previous responders only. Mean fatigue scores were higher in the adalimumab maintenance group (187.1) and the adalimumab 40 mg administered every other week group (187.9) than in the placebo group (173.3) at 56 weeks in participants with Crohn's disease who were already known to respond to adalimumab treatment, with higher scores indicating better quality of life. However, the mean differences between the groups were 13.80 and 14.60 points, respectively, and this is likely to not be a clinically meaningful improvement in quality of life (Cella 2005; Cella 2002). The evidence suggests that electroacupuncture may result in a large increase in quality of life, compared to control and sham electroacupuncture. The mean quality of life scores were higher in the electroacupuncture group (61.5) than the control group (57) and sham electroacupuncture (57.8) at week eight. The mean quality of life scores were also higher in the electroacupuncture group (60.8) compared to the sham electroacupuncture group (58.6) at week 16. However, it is unclear whether the observed improvements in quality of life are clinically important, due to the lack of clearly defined minimally important difference thresholds for the IBDQ‐9 scale. The GRADE analysis indicated that the overall certainty of the evidence supporting these results on quality of life was low due to sparse data and wide confidence intervals. Further research is needed before any strong conclusions can be drawn on the efficacy and safety of adalimumab maintenance therapy, adalimumab 40 mg administered every other week and electroacupuncture on quality of life.
No differences in quality of life were found for ferric maltol (Gasche 2015), cognitive behavioural therapy with therapist support (Artom 2018), and the physical activity advice and omega 3 comparisons (McNelly 2016). Although assessed, quality of life data were not available for the trials investigating the effect of customised combination therapy (tight control management) (Colombel 2017), vedolizumab maintenance therapy (Feagan 2013; Sandborn 2013), vitamin D3 (Raftery 2013), solution‐focused therapy (Vogelaar 2014), and guided and self‐directed stress management (García‐Vega 2004). Total physical and mental component quality of life scores were not reported in two trials (Therkelsen 2016a; Therkelsen 2016b).
In terms of safety, there was no difference in adverse events reported in the three adalimumab treatment groups compared to placebo, however, the adalimumab maintenance and adalimumab 40 mg weekly groups were significantly less likely to experience serious adverse events and withdrawal due to adverse events, compared to control, for all randomised participants (Colombel 2007). The multimodular trial (McNelly 2016) reported a range of adverse events, including epigastric pain, diarrhoea, bloating, nausea and vomiting, IBD flare, headache, pins and needles, joint pain, ankle injury, leg pain, rash, molluscum and feeling unwell. Adverse events varied across groups, with the no physical activity advice plus omega 3 group reporting more adverse events (n = 14), compared to the other three groups (n = 4 or n = 5). These adverse events were considered to be mild in nature. This study did not report serious adverse events or withdrawal due to adverse events. There was no difference in adverse events reported for any of the other trials. Additional information from the trial authors revealed that there were no adverse events reported by participants involved in the guided and self‐directed stress management (García‐Vega 2004) and the AndoSanTM trials (Therkelsen 2016a; Therkelsen 2016b). Adverse events were not reported in the solution‐focused therapy trial (Vogelaar 2014), cognitive behavioural therapy with therapist support trial (Artom 2018) or for the randomised responders in the adalimumab trial (Colombel 2007).
The evidence presented in this review is limited due the diversity of interventions, missing data, low or very low evidence of certainty due to single studies, small sample sizes, and unclear or high risk of bias in trials (see 'Characteristics of included studies'). The results reported in this review must therefore be interpreted with caution. Most studies included in this review were published in the last five years, with a large number of ongoing trials (n = 30) also identified as potentially eligible in future updates, indicating the growing interest in IBD fatigue.
Overall completeness and applicability of evidence
This review included fourteen studies (3741 participants), however, as outcome data on fatigue were not available for all included trials, the findings are based on 1344 participants in nine trials, so completeness of evidence is a concern. Due to insufficient and non‐standard reporting, fatigue data were available for analysis from only eight of the 14 eligible trials. Data on the two secondary outcomes were presented in five of the trials. Although some additional information was obtained from the trial authors, insufficient data was a difficulty in this review. For example, data on the primary outcome relevant to this review, although assessed, were not available for analysis in four trials (Feagan 2013; Hetzel 2013a; Raftery 2013; Sandborn 2013). Furthermore, one trial presented fatigue data as estimates and effect sizes for the intervention and control groups at each time point, rather than standard reporting of mean and standard deviation scores (Vogelaar 2014). Pharmacological companies have ownership of data for one of the large pharmacological trials and a data request has been submitted and is awaited (Colombel 2017). Three of the nine eligible trials did not present baseline participant characteristics for the groups (Hetzel 2013a; Raftery 2013; Vogelaar 2014). However, most studies did present sufficient baseline information on age, gender, disease type, disease activity or disease duration.
The studies tended to include participants with Crohn's disease only, or both ulcerative colitis or Crohn's disease, whereas the focus of trials on only individuals with ulcerative colitis remains limited (Feagan 2013; Therkelsen 2016a). Crohn's disease is a more complex disease and it has been previously suggested that this disease type may be more responsive to therapy, as Crohn's disease is associated with higher levels of fatigue and greater impairment in quality of life. Studies with mixed populations did not report subgroup analysis according to disease type. As most interventions were not specifically targeted at IBD fatigue, there was no subgroup analysis on those with other pre‐existing fatigue related comorbidities, for example, depression and anxiety. Furthermore, subgroup analysis was not possible in this review due to the small sample sizes and diversity across trials.
As most participants were not recruited because of fatigue, it seems likely in some studies that participants experienced low levels of fatigue at baseline and perhaps may have had limited scope for improvement in fatigue levels. For example, in the two trials investigating the effect of AndoSanTM on fatigue in participants with ulcerative colitis (Therkelsen 2016a) and Crohn's disease (Therkelsen 2016b), the baseline total fatigue scores were relatively low.
There is some evidence of the effect of pharmacological interventions on IBD fatigue, however, there were fewer studies examining the efficacy of non‐pharmacological interventions. The cost implications and potential side effects of pharmacological therapies suggest there may be difficulty using such type of treatment specifically for managing IBD fatigue. In contrast, four of the five non‐pharmacological interventions were specifically designed and developed for managing IBD fatigue (Artom 2018; Horta 2017; McNelly 2016; Vogelaar 2014). More research is needed, in particular, studies specifically developed and targeted at managing IBD fatigue in well‐defined patient groups. For example, a specific type of therapy may work best for participants with Crohn's disease, rather than those with ulcerative colitis, in improving fatigue.
The duration of interventions ranged from 21 days to 56 weeks. Given the multidimensional nature of fatigue and the complexities of IBD, the potential benefit of longer intervention periods should also be explored. More research is needed to be able to draw any firm conclusions about the efficacy and safety of pharmacological and non‐pharmacological interventions on fatigue in individuals with IBD.
Quality of the evidence
The quality of evidence presented in this review is limited. Two studies were assessed as high quality on all criteria (Figure 2; Figure 3). Our assessment of risk of bias was considerably compromised by insufficient publication of information. Most studies used adequate procedures for random sequence generation. Methods of allocation concealment was rated low risk in only five trials. Most pharmacological trials were either double‐blinded (Colombel 2007; Feagan 2013; Gasche 2015; Raftery 2013; Sandborn 2013) or single‐blinded (Therkelsen 2016a; Therkelsen 2016b). In contrast, most non‐pharmacological interventions were unblinded, due to the inherent nature of the type of intervention (Artom 2018; García‐Vega 2004; McNelly 2016; Vogelaar 2014). Therefore, the trend of positive effect found in these studies may have been as a result of a placebo response. Standard care or care‐as‐usual were frequently used as the control, however, limited information on the care received or the visits schedule was typically provided. There is little evidence if and to what extent conditions between the intervention and control groups were kept similar in non‐pharmacological trials, in order to reduce the risk of performance bias.
The primary outcome of this review was a subjective, self‐reported outcome, therefore, detection bias was rated high for all unblinded trials and low for double‐ and single‐ (participant) blinded trials. There were no concerns over attrition bias in most studies, however, the small sample size in many trials was problematic. Selection bias was not an issue in terms of comparable baseline characteristics of groups. This may be as a result of the random sampling strategies used in trials. There were concerns over the quality of reporting in six trials, where data were presented insufficiently (Colombel 2017; Feagan 2013; Hetzel 2013a; Raftery 2013; Sandborn 2013) and reported in a non‐standard way (Vogelaar 2014). Overall, our assessment, based on GRADE analyses, suggests that the certainty of the evidence supporting the outcomes of this review ranges was low or very low. As a result of this uncertainty, no firm conclusions can be drawn regarding the efficacy and safety of pharmacological and non‐pharmacological interventions on fatigue in IBD.
Potential biases in the review process
We performed a comprehensive literature search to minimise bias related to study selection. We are confident that our search strategy has identified all eligible pharmacological and non‐pharmacological randomised controlled trials assessing fatigue or loss of energy as a primary or secondary outcome in IBD published up until October 2019. Our search was limited to English, therefore, there may be a language bias. Two authors reviewed the studies for inclusion and exclusion, extracted data independently, and reviewed study quality. Additional information was retrieved from authors for eight of the 14 eligible trials (Artom 2018; Colombel 2007; García‐Vega 2004; Gasche 2015; Horta 2017; McNelly 2016; Therkelsen 2016a; Therkelsen 2016b), with one request for additional information still outstanding (Colombel 2017) and five studies identified from the top‐up search run in October 2019 awaiting assessment needing additional information from the trial authors. Some studies where insufficient data were reported were included in the review demonstrating presence of reporting bias (Feagan 2013; Hetzel 2013a; Raftery 2013; Sandborn 2013; Vogelaar 2014). However, as data on the primary outcome remain unavailable, these studies did not contribute to the overall findings of the review.
Limitations of this systematic review include the diversity of interventions, the limited number of studies with a small number of participants and sparse data. Only one pooled analysis was undertaken. The small sample sizes in the trials may have been insufficient to detect a small effect. All studies that assessed fatigue or loss of energy as a primary or secondary outcome using a generic or disease‐specific fatigue scale, a subscale of a broader questionnaire or as a single question were deemed eligible for inclusion. All generic and disease‐specific measures used in the trials demonstrated adequate psychometric properties, however, caution needs to be exercised when interpreting findings measured using non‐validated subscales or single‐item measures. For example, one trial assessed the average frequency and severity of tiredness using a single item on a Crohn's disease symptom diary (García‐Vega 2004). In future updates of this review, further consideration needs to be given to the inclusion of studies that use subscales or single‐item measures of fatigue. As there are a lack of clearly defined minimal clinically important differences (MCID) for some fatigue measures established in the IBD literature, this limits the interpretation of results and conclusions drawn. Three review authors (WCD, MA, CN) were involved in two eligible trials included in this review. These authors were not involved in extracting data or conducting 'Risk of bias' assessments for these trials (Artom 2018; McNelly 2016).
Agreements and disagreements with other studies or reviews
A review has appraised the management of fatigue in IBD (Czuber‐Dochan 2013a). Similar to the present review, a paucity of evidence was found. It supported the findings regarding the potential benefit of adalimumab, stress management and solution‐focused therapy for fatigue in IBD. Furthermore, the evidence from this review is broadly in agreement with some other Cochrane reviews on fatigue management in other chronic illnesses. For example, Cochrane reviews suggest that exercise therapy may be an effective and safe treatment for reducing fatigue in adults with chronic fatigue syndrome (Larun 2017), multiple sclerosis (Heine 2015), rheumatoid arthritis (Cramp 2013) and people with breast and prostate cancer both during and after cancer treatment (Cramp 2012), which supports the findings of this review in relation to the potential benefit of physical activity advice (McNelly 2016). A Cochrane review evaluating the effect of biologic therapies on fatigue in adults with rheumatoid arthritis found a small to moderate improvement (Almeida 2016), which is in agreement with the findings of this review on adalimumab treatment (Colombel 2007). Other Cochrane reviews have found no specific drugs can be recommended as an effective treatment of fatigue in palliative care patients with advanced chronic diseases (Mücke 2015) and multiple sclerosis (Pucci 2007; Tejani 2012).
Cochrane reviews have also demonstrated the benefit of psychosocial interventions in managing fatigue in rheumatoid arthritis (Cramp 2013) and people with cancer receiving active treatment (Goedendorp 2009). In particular, cognitive behavioural therapy (Price 2008) and educational interventions (Bennett 2016) have shown an effect on fatigue in adults with chronic fatigue syndrome and cancer, respectively. There is no evidence in this review suggesting that cognitive behavioural therapy may be beneficial in managing fatigue, however this trial (Artom 2018) was a pilot with a limited sample size. Furthermore, there is no evidence available from our review regarding the effectiveness of educational interventions on fatigue in IBD, which may be a useful strategy that could be incorporated into routine care of individuals with IBD, if proven effective. Similar to our review, Cochrane reviews investigating the effectiveness and safety of pharmacological and non‐pharmacological interventions on fatigue in the area of primary brain tumours (Day 2016), amyotrophic lateral sclerosis (Gibbons 2018) and Parkinson's disease (Elbers 2015) have found insufficient and low‐quality evidence, resulting in no firm conclusion being reached. This suggests that further research also needs to be conducted in the area of fatigue management across other chronic illnesses. Due to the complex nature of fatigue, it appears likely that the optimal management of fatigue would require a multidimensional approach.
Authors' conclusions
Implications for practice.
The data were not sufficient or of high enough quality to identify specific interventions that might be associated with a positive effect on fatigue management in individuals with IBD. Limited available data, the heterogeneity of studies and fatigue not being the primary endpoint of most studies are limitations of the evidence base. Furthermore, the optional duration and suitability of interventions remains unclear. Biologic therapy (adalimumab 40 mg administered every other week only for those known to respond to adalimumab induction therapy) may reduce fatigue and iron therapy (ferric maltol) may result in a slight increase in fatigue, however the evidence is very uncertain. Non‐pharmacological treatments, such as electroacupuncture and physical activity may reduce fatigue in individuals with IBD. The small sample sizes and high risk of bias in these trials means the benefit of these treatments is uncertain and suggests that further research is needed.
Implications for research.
Research aimed at managing fatigue as a major IBD burden is lacking and hopefully, this review will act as a catalyst to initiate these studies. To date, there is a lack of high‐quality research evaluating the effect of interventions on fatigue in IBD, despite the high prevalence and burdensome nature of this symptom. The evidence in this review applies to a broad range of therapies (pharmacological and non‐pharmacological). Further investigation is warranted. Reporting in some of the trials was insufficient to assess the efficacy and safety of some therapies. This review focused on presenting clinically important findings, however, at times, it was difficult to determine if the observed improvements in fatigue on some scales (IBDF, total fatigue score, average frequency of tiredness, average severity of tiredness) were clinically meaningful. There is need for clearly defined minimal clinically important difference thresholds for fatigue measures in the IBD population.
There is a need for considerably more robust, well‐designed randomised controlled trials to identify effective treatments for fatigue in IBD. Further research is required to test interventions specifically designed to manage fatigue in patients with IBD. In order to identify the types and specific elements of interventions that are effective in improving fatigue, there should be more research targeted at selected IBD populations. This will identify subgroups of patients most likely to benefit from certain interventions. Alternative non‐pharmacological treatments found to be effective in other chronic illnesses, such as educational interventions, warrant investigation in the IBD population. Future research needs to use validated generic and/or disease‐specific measures of fatigue and measure the long‐term effects of interventions. Trials should recruit a sufficient sample size, as predetermined by a power calculation, in order to be adequately powered to detect differences between intervention and control groups. Methodological standards of clinical trials, particularly for non‐pharmacological interventions, need to be improved.
Acknowledgements
We wish to express our sincere thanks to the Cochrane IBD/FBD Group, especially the Managing Editor, John K. MacDonald, and the Information Specialists, Claire Parker and Tran Nguyen, who provided valuable support throughout the review process.
We would like to thank all the investigators who provided additional information about their trials including Micol Artom, Diana Horta, Elena García‐Vega, Angela McNelly, Stig Palm Therkelsen and Christoph Gasche. We also received help from several investigators of studies which were eventually excluded from this review. We would also like to express our thanks to the team at AbbVie Inc, in particular, Kristin Costello, Cynthia Holas, Katie Saveraid, Gregory Petinatos and Christa Rambert for their assistance with data sharing and to the Research and Legal teams at University College Cork, including David O' Connell, Lucy Wallis and Catherine‐Ellen O’Keeffe, for their advice and guidance on data sharing.
We would also like to thank the Health Research Board, Ireland, for funding this review through a Cochrane Fellowship awarded to Dr. Dawn Farrell and acknowledge University College Cork for hosting this grant.
Funding for the IBD/FBD Review Group (September 1, 2010 ‐ August 31, 2015) has been provided by the Canadian Institutes of Health Research (CIHR) Knowledge Translation Branch (CON ‐ 105529) and the CIHR Institutes of Nutrition, Metabolism and Diabetes (INMD); and Infection and Immunity (III) and the Ontario Ministry of Health and Long Term Care (HLTC3968FL‐2010‐2235).
Miss Ila Stewart has provided support for the IBD/FBD Review Group through the Olive Stewart Fund.
Appendices
Appendix 1. Embase search strategy
1. random$.mp.
2. factorial$.mp.
3. (crossover$ or cross over$ or cross‐over$).mp.
4. placebo$.mp.
5. single blind.mp.
6. double blind.mp.
7. triple blind.mp.
8. (singl$ adj blind$).mp.
9. (double$ adj blind$).mp.
10. (tripl$ adj blind$).mp.
11. assign$.mp.
12. allocat$.mp.
13. crossover procedure/
14. double blind procedure/
15. single blind procedure/
16. triple blind procedure/
17. randomized controlled trial/
18. or/1‐17
19. exp Crohn disease/ or crohn*.mp.
20. (colitis and ulcerat*).mp. or exp ulcerative colitis/
21. (inflammatory bowel disease* or IBD).mp.
22. or/19‐21
23. exp fatigue/
24. exp chronic fatigue syndrome/
25. (physical fatigue OR mental fatigue OR muscle fatigue).mp.
26. (energy OR tired* OR sleep* OR drows* OR letharg* OR lassitude OR weari*).mp.
27. (exhaust* OR listless* OR apath* OR malaise).mp.
28. ((asthenia OR asthenic) adj3 syndrome).tw.
29. (((lack or loss or lost) adj3 energy) or vigo* or vitality).tw.
30. or/23‐29
31. 18 and 22 and 30
Limit to Human
Appendix 2. MEDLINE search strategy
1. random$.mp.
2. factorial$.mp.
3. (crossover$ or cross over$ or cross‐over$).mp.
4. placebo$.mp.
5. single blind.mp.
6. double blind.mp.
7. triple blind.mp.
8. (singl$ adj blind$).mp.
9. (double$ adj blind$).mp.
10. (tripl$ adj blind$).mp.
11. assign$.mp.
12. allocat$.mp.
13. randomized controlled trial/
14. or/1‐13
15. exp Crohn disease/ or crohn*.mp.
16. (colitis and ulcerat*).mp. or exp ulcerative colitis/
17. (inflammatory bowel disease* or IBD).mp
18. or/15‐17
19. exp fatigue/
20. exp chronic fatigue syndrome/
21. (physical fatigue OR mental fatigue OR muscle fatigue).mp.
22. (energy OR tired* OR sleep* OR drows* OR letharg* OR lassitude OR weari*).mp.
23. (exhaust* OR listless* OR apath* OR malaise).mp.
24. ((asthenia OR asthenic) adj3 syndrome).tw.
25. (lack OR loss OR lost) adj3 (energy OR vigo* OR vitality).tw.
26. or/19‐25
27. 14 and 18 and 26
Appendix 3. CINAHL search strategy
1. (TI fatigue or AB fatigue) OR (TI energy or AB energy) OR (TI sleep* or AB sleep*) OR (TI drows* or AB drows*) OR (TI lethargy* or AB lethargy*) OR (TI lassitude or AB lassitude) OR (TI weari* or AB weari*) OR (TI exhaust* or AB exhaust*) OR (TI listless* or AB listless*) OR (TI apath* or AB apath*) OR (TI malaise or AB malaise)
2. (TI inflammatory bowel or AB inflammatory bowel) OR (TI IBD or AB IBD) OR (TI Crohn* or AB Crohn*) OR (TI CD* or AB CD*) OR (TI colitis* or AB colitis*) OR (TI UC or AB UC) OR (TI proctitis or AB proctitis) OR (TI ileitis or AB ileitis)
3. 1 AND 2
Appendix 4. PsycINFO search strategy
TI(fatigue OR energy OR sleep* OR drows* OR lethargy* OR lassitude OR weari* OR exhaust* OR listless* OR apath* OR malaise) AND TI(inflammatory bowel OR IBS OR Crohn* OR CD OR colitis OR UC OR proctitis OR ileitis)
Appendix 5. CENTRAL search strategy
#1 MeSH descriptor: [Inflammatory Bowel Diseases] explode all trees
#2 crohn*
#3 ulcerative colitis
#4 colitis
#5 proctitis
#6 ileitis
#7 #1 or #2 or #3 or #4 or #5 or #6
#8 MeSH descriptor: [Fatigue] explode all trees
#9 "chronic fatigue" or "physical fatigue" or "mental fatigue" or "muscle fatigue"
#10 energy or tired* or sleep* or drows* or letharg* or lassitude or weari* or exhaust* or listless* or apath* or malaise
#11 #8 or #9 or #10
#12 #7 and #11
Appendix 6. Data extraction
Study
Study aim
Participant characteristics
Number of participants
Setting of study
Country of origin
Demographic characteristics such as age and gender
Disease characteristics such as disease type, disease status and disease duration
Inclusion and exclusion criteria for participation in the study
Intervention characteristics
For each arm:·
Assignment to groups·
The aim, type, mode and content of the intervention·
Time points of delivery ·
Duration of the intervention, number and duration of sessions/dose
Providers of the intervention
Comparison intervention/s
Setting of the intervention
Participant adherence
Outcomes
Time, frequency and duration at which outcomes are measured
Instruments used for key primary and secondary outcomes
Outcome scoring methods
Adverse events
Others
Funding
Declaration of interest
Sample size and evidence of power calculation
Follow‐up – withdrawal/dropout
Data and analyses
Comparison 1. Adalimumab maintenance versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Fatigue: SF‐36 Vitality | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 Week 4 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Week 56 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Quality of life: SF‐36 PCS | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 Week 4 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Week 56 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Quality of life: SF‐36 MCS | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.1 Week 4 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Week 56 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Quality of life: IBDQ | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4.1 Week 4 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Week 56 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Adverse events | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6 Serious AEs | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7 Withdrawal due to AEs | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 2. Adalimumab 40 mg eow versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Fatigue: FACIT‐ F | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 Week 4 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Week 56 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Fatigue: SF‐36 Vitality | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 Week 4 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Week 56 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Quality of life: SF‐36 PCS | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.1 Week 4 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Week 56 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Quality of life: SF‐36 MCS | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4.1 Week 4 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Week 56 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Quality of life: IBDQ | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 5.1 Week 4 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Week 56 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Adverse events | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7 Serious AEs | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8 Withdrawal due to AEs | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
2.5. Analysis.

Comparison 2 Adalimumab 40 mg eow versus placebo, Outcome 5 Quality of life: IBDQ.
Comparison 3. Adalimumab 40 mg weekly versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Fatigue: FACIT‐Fatigue | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 Week 4 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Week 56 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Fatigue: SF‐36 vitality | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 Week 4 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Week 56 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Quality of life: SF‐36 PCS | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.1 Week 4 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Week 56 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Quality of life: SF‐36 MCS | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4.1 Week 4 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Week 56 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Quality of life: IBDQ | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 5.1 Week 4 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Week 56 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Adverse events | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7 Serious AEs | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8 Withdrawal due to AEs | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
3.1. Analysis.

Comparison 3 Adalimumab 40 mg weekly versus placebo, Outcome 1 Fatigue: FACIT‐Fatigue.
3.2. Analysis.

Comparison 3 Adalimumab 40 mg weekly versus placebo, Outcome 2 Fatigue: SF‐36 vitality.
3.3. Analysis.

Comparison 3 Adalimumab 40 mg weekly versus placebo, Outcome 3 Quality of life: SF‐36 PCS.
3.4. Analysis.

Comparison 3 Adalimumab 40 mg weekly versus placebo, Outcome 4 Quality of life: SF‐36 MCS.
3.5. Analysis.

Comparison 3 Adalimumab 40 mg weekly versus placebo, Outcome 5 Quality of life: IBDQ.
Comparison 4. AndoSan versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Fatigue: Total Fatigue Score | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.1 Day 14 follow‐up | 2 | 100 | Mean Difference (IV, Fixed, 95% CI) | ‐1.63 [‐3.51, 0.26] |
| 1.2 Day 21 follow‐up | 2 | 100 | Mean Difference (IV, Fixed, 95% CI) | ‐1.59 [‐3.46, 0.28] |
| 2 Fatigue: SF‐36 Vitality subscale | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1 Day 14 follow‐up | 2 | 100 | Mean Difference (IV, Fixed, 95% CI) | 3.84 [‐3.78, 11.47] |
| 2.2 Day 21 follow‐up | 2 | 100 | Mean Difference (IV, Fixed, 95% CI) | 3.53 [‐3.91, 10.96] |
Comparison 5. Ferric maltol versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Fatigue: SF‐36 Vitality subscale | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 Week 12 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Quality of life: IBDQ | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 Week 12 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Adverse events | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4 Serious AEs | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 5 Withdrawal due to AEs | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 6. Electroacupuncture versus no treatment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Fatigue: FACIT‐F | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 Week 8 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Quality of life: IBDQ‐9 | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 Week 8 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Adverse events | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4 Serious AEs | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 5 Withdrawal due to AEs | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
6.3. Analysis.

Comparison 6 Electroacupuncture versus no treatment, Outcome 3 Adverse events.
6.4. Analysis.

Comparison 6 Electroacupuncture versus no treatment, Outcome 4 Serious AEs.
6.5. Analysis.

Comparison 6 Electroacupuncture versus no treatment, Outcome 5 Withdrawal due to AEs.
Comparison 7. Electroacupuncture versus sham electroacupuncture.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Fatigue: FACIT‐F | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 Week 8 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Week 16 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Quality of life: IBDQ‐9 | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 Week 8 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Week 16 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Adverse events | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4 Serious AEs | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 5 Withdrawal due to AEs | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 8. Guided stress management versus conventional medical treatment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Fatigue: Average frequency symptom (tiredness) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 Post‐intervention | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 12 month follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Fatigue: Average severity of tiredness | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 Post‐intervention | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 12 month follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 9. Self‐directed stress management versus conventional medical treatment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Fatigue: Average frequency of tiredness | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 Post‐intervention | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 12 month follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Fatigue: Average severity of tiredness | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 Post‐intervention | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 12 month follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 10. CBT with therapist support versus fatigue information leaflet only.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Fatigue: IBD‐F Section I | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 Month 3 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Fatigue: IBD‐F Section II | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 Month 3 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Quality of life: UK‐IBDQ | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.1 Month 3 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 11. Physical activity advice plus omega 3 versus no physical activity advice plus omega 3.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Fatigue: FACIT‐F | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 Week 12 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Fatigue: MFI | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 Week 12 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Fatigue: IBD‐F Section I | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.1 Week 12 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Fatigue: IBD‐F Section II | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4.1 Week 12 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Quality of life: IBDQ | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 5.1 Week 12 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 12. Physical activity advice plus placebo versus no physical activity advice plus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Fatigue: FACIT‐F | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 Week 12 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Fatigue: MFI | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 Week 12 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Fatigue: IBD‐F Section I | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.1 Week 12 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Fatigue: IBD‐F Section II | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4.1 Week 12 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Quality of life: IBDQ | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 5.1 Week 12 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 13. Physical activity advice plus placebo versus no physical activity advice plus omega 3.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Fatigue: FACIT‐F | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 Week 12 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Fatigue: MFI | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 Week 12 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Fatigue: IBD‐F Section I | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.1 Week 12 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Fatigue: IBD‐F Section II | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4.1 Week 12 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Quality of life: IBDQ | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 5.1 Week 12 follow‐up | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Artom 2018.
| Methods | Pilot RCT with nested qualitative study | |
| Participants | Adults (aged 18 years old and over) with a diagnosis of IBD (record of diagnostic endoscopy in patient clinical notes) and reported currently experiencing fatigue Exclusion criteria: Currently experiencing bowel symptoms they would associate with a relapse of their disease; received CBT for fatigue in the last year; enrolled in a trial involving a non‐licensed pharmacological intervention; pregnant women or planning a pregnancy or were unable to give informed consent A total of 31 participants were randomised to the CBT group (n = 15) or information leaflet group (n = 16). Age [Mean(SD) years]: CBT group 37.00 (8.71); Information leaflet 39.13 (10.49) Gender: CBT group (33.3% Male, 67.7% female); Information leaflet (37.5% male; 62.5% female) Disease duration [Mean(SD) years]:CBT group 14.26 (18.19); information leaflet 18.08 (20.18) Loss to follow‐up: n = 9; CBT group (n = 5); information leaflet (n = 4) |
|
| Interventions | CBT manual about the management of fatigue with the support of a qualified CBT therapist Mode of delivery: Telephone/Skype sessions with a therapist Duration: 60‐minute session and seven 30‐minute sessions over an eight‐week period Setting of trial: Single centre in the UK Setting of intervention: Participants home Comparision treatment: The Crohn’s and Colitis UK (IBD charity) "Fatigue in IBD" Information Sheet without therapist assistance Co‐interventions: none |
|
| Outcomes | Fatigue was assessed using the Inflammatory Bowel Disease‐Fatigue (IBD‐F) scale. Quality of life was assessed using the UK IBDQ and adverse events were not assessed. Other outcomes: Disease activity, illness perceptions, daytime sleepiness, anxiety and depression Assessment time points: Baseline and month 3 |
|
| Notes | Information provided by trial author. Full‐text paper submitted for publication to Pilot and Feasibility Studies Data analysed with per protocol analyses No power calculation conducted, however a pilot study |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised at the individual level using a random number generator with a 1:1 ratio in the Statistical Package for the Social Sciences (SPSS) Version 22. |
| Allocation concealment (selection bias) | Low risk | Sealed opaque envelopes (author information) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants and therapists not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Self‐reported measure and participants were not blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Loss to follow‐up relatively similar across the groups |
| Selective reporting (reporting bias) | Unclear risk | All information provided by trial author on outcomes were transparent, however, findings published as a conference abstract at present |
| Other bias | Low risk | Baseline characteristics were similar between treatment groups. No apparent sources of bias |
Colombel 2007.
| Methods | Phase III, RCT, double‐blinded | |
| Participants | Crohn's disease, moderately to severely active disease (CDAI 220‐450). History of having received TNF‐anatagonist therapy but discontinued at least 12 weeks before study entry; other stable dosages of concurrent treatment Exclusion criteria: Ulcerative colitis; symptomatic obstructive disease; bowel resection within the past 6 months; an ostomy; extensive small bowel resection or short bowel syndrome; currently receiving total parenteral nutrition; history of cancer, listeria, human immunodeficiency virus, central nervous system demyelinating disease, or untreated tuberculosis; had received investigational chemical agents within 30 days or investigational biologic therapy within 3 months before screening; had received antibiotic treatment for non‐CD‐related infections within 3 weeks before screening; were pregnant or breastfeeding; had a history of significant drug or alcohol abuse within the past years; had poorly controlled medical conditions; had received treatment with adalimumab or participated in an adalimumab clinical study; had received enema therapy within 2 weeks before screening; had received cyclosporine, mycophenolate mofetil, or tacrolimus within 8 weeks of screening; had a positive clostridium difficile stool assay; or had clinically significant deviations in prespecified laboratory parameters A total of 854 participants enrolled in the study and received open‐label adalimumab 80 mg subcutaneously followed by a 40 mg dose at week 2. At week 4, participants were assessed for response and randomised (n = 778) to one of the three treatment arms (adalimumab 40 mg every other week (eow) (n = 260), adalimumab 40 mg weekly (n = 257), or placebo weekly (n = 261)). Participants who experienced a decrease in CDAI scores of 70 or more points from baseline were considered responders (n = 499). A total of 170 participants were placebo randomised responders; 172 participants were adalimumab 40 mg every other week (eow) randomised responders; 157 participants were adalimumab 40 mg weekly randomised responders Participant characteristics for all randomised participants (Rubin 2009): Age [Mean(SD) years): placebo: 36.9 (11.43); adalimumab maintenance therapy (40 mg eow): 36.8 (11.48); adalimumab maintenance therapy (40 mg weekly): 37.8 (12.09) Gender: placebo (99 males, 162 females); adalimumab maintenance therapy (40 mg eow) (97 males; 163 females); adalimumab maintenance therapy (40 mg weekly) (100 males, 157 females) Disease status [Mean(SD) CDAI score]: placebo: 315.8 (65.72); adalimumab maintenance therapy (40 mg eow): 309.6 (60.70); adalimumab maintenance therapy (40 mg weekly): 308.2 (55.26) (author information) Loss to follow‐up: placebo (n = 114; 43.7%); adalimumab maintenance therapy (40 mg eow) (n = 94; 36.2%); adalimumab maintenance therapy (40 mg weekly) (n = 65; 25.3%) Participant characteristics for randomised responders (Loftus 2008): Age [Mean(SD) years]: placebo: 36.9 (11.9); adalimumab maintenance therapy (40 mg eow): 36.4 (11.1); adalimumab maintenance therapy (40 mg weekly): 36.9 (11.8) Gender: placebo (65 males, 105 females); adalimumab maintenance therapy (40 mg eow) (61 males; 111 females); adalimumab maintenance therapy (40 mg weekly) (62 males, 95 females) Disease status [Mean(SD) CDAI score]: placebo: 321.1 (67.1); adalimumab maintenance therapy (40 mg eow): 315.7 (61.5); adalimumab maintenance therapy (40 mg weekly): 312.6 (58.3) Loss to follow‐up: placebo (n = 60; 35.3%); adalimumab maintenance therapy (40 mg eow) (n = 57; 33.1%); adalimumab maintenance therapy (40 mg weekly) (n = 26; 16.6%) |
|
| Interventions | Adalimumab maintenance therapy Dosing regimen: a) 40 mg every other week; b) 40 mg weekly Mode of delivery: Subcutaneously administered Duration: Four weeks open‐label adalimumab followed by up to 52 weeks of randomised study drug. Setting of trial: 92 centres in the United States, Europe, Canada, Australia, and South Africa Comparison treatment: placebo weekly (open‐label 80 mg adalimumab at baseline (week 0) and 40 mg adalimumab at week 2, plus placebo sc injections following randomisation (week 4) Co‐interventions: Not reported |
|
| Outcomes | Fatigue was assessed using the FACIT‐F and the SF‐36 vitality subscale. Quality of life was assessed using the IBDQ and the SF‐36. Adverse events were assessed for each Medical Dictionary for Drug Regulatory Affairs (MedDRA) system organ class and preferred term. Other outcomes assessed: Disease activity (CDAI), CRP concentration, number of cutaneous fistulas draining, concomitant medication Assessment time points: Baseline, week 4, week 12, week 26, week 56 (AE also assessed at week 2, week 6, week 8, week 16, week 20, week 32, week 40, week 48, week 60) |
|
| Notes | 3‐arm trial (2 experimental, 1 control) The secondary publication (Rubin 2009) presented data from all randomised participants regardless of week 4 response status to pre‐trial dose of adalimumab (n = 778). The secondary publication (Loftus 2008) presented data from a subgroup of participants who responded to pre‐trial dose of adalimumab (n = 499). The study was funded and supported by Abbot Laboratories. Additional study information was supplied by AbbVie (formally Abbot Laboratories). Data analysed with intention‐to‐treat analyses, however, FACIT‐F data represented the last‐observation‐carried‐forward when a participant had a missing value, dropped out, or switched to open‐label therapy. Adalimumab maintenance group represented data from both the 40 mg every other week and 40 mg weekly groups combined. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | At week 4, all subjects were randomised to either adalimumab 40 mg eow, adalimumab 40 mg weekly or placebo, stratified by responder status and previous anti‐TNF use. The subject's number and treatment regimen of each stratum was assigned by a central randomisation schedule, utilising an interactive voice response system. The randomisation schedule was prepared by the Statistics Department of Abbott (author information). |
| Allocation concealment (selection bias) | Low risk | "All patients were randomised centrally using an interactive voice response system". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Patients, study coordinators, and study investigators were blinded to treatment assignment throughout the blinded portion of the study". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The investigators and participants were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Loss to follow‐up relatively similar across the groups and reasons comparable |
| Selective reporting (reporting bias) | Low risk | All information on outcomes were transparent and outcomes assessed were reported/provided upon request. |
| Other bias | Low risk | Baseline characteristics were similar between treatment groups. No apparent sources of bias |
Colombel 2017.
| Methods | Phase III, RCT, open‐label | |
| Participants | Ileal, colonic (including rectal), or ileocolonic Crohn's disease (not more than 6 years prior to baseline). Moderately‐to‐severely active disease (CDAI 220 ‐ 450 if not receiving prednisone; CDAI 220 ‐ 450 if receiving prednisone ≤ 20 mg; CDAI 150 ‐ 450 if receiving prednisone > 20 mg greater than or equal to 7 days before baseline); active endoscopic disease (CDEIS > 6; sum of CDEIS subscores of > 6 in one or more segments with ulcers); CRP of 5 mg/L or more, fecal calprotectin of 250 μg/g or more, or both Exclusion criteria: Previous or current use of biologic or immunomodulators; more than two previous courses of corticosteroids, or current use of corticosteroids for more than 3 months before screening; fibrotic stricture/draining perianal fistulas/non‐perianal fistula; poorly controlled medical conditions; positive C. difficile stool assay at screening A total of 255 participants enrolled in the study and received the prednisone induction therapy. A total of 244 participants were randomly allocated to the tight control (n = 122) or clinical management (n = 122) groups. Age [Mean(SD) years]: clinical management group 31.1 (11.4); tight control group 32.1 (12.0) Gender: clinical management group (53 males, 69 females); tight control group (50 males, 72 females) Disease status [Mean(SD) CDAI score]: clinical management group 267.7 (58.4); tight control group 273.3 (59.5) Disease duration [Mean (SD) years]: clinical management group 0.9 (1.7); tight control group 1.0 (2.3) Loss to follow‐up: clinical management group (n = 29); tight control management group (n = 32) |
|
| Interventions | Tight Control Management ‐ customised therapy (after prednisone induction therapy, management escalated in a stepwise manner, from no treatment, to adalimumab induction followed by adalimumab every other week, adalimumab every week, and lastly to both weekly adalimumab and daily azathioprine) based on disease activity (CDAI, high sensitivity C‐reactive protein, fecal calprotectin, and corticosteroid use) Dosing regimen: (i) Prednisone 40 mg/day for 2 weeks (max dose), followed by a taper with a schedule that was set by each investigator at his or her discretion for 6 weeks, however prednisone treatment could continue on the basis of the rapidity and tolerance of the taper (ii) Adalimumab 160 mg induction dose at week 0, followed by 80 mg at week 3 and 40 mg eow as a maintenance dose (participants who met any of the failure criteria 1 week before group allocation). Increase to 40 mg weekly if response inadequate and de‐escalated to 40 mg eow in participants who did not meet the treatment failure criteria (iii) Azathioprine 2.5 mg/kg/day (normal thiopurine methyltransferase [TPMT])/1.25 mg/kg/day oral (intermediate TMPT). Dose‐adjusted according to abnormalities of white blood cell (WBC) count, platelet count, liver function tests (LFTs; i.e. alanine transaminase [ALT], aspartate transaminase [AST], phosphatase), lipase, blood urea nitrogen (BUN), and serum creatinine. Therapy was escalated based on meeting treatment failure criteria, which differed between groups (tight control group before and after random assignment: faecal calprotectin ≥ 250 µg/g, CRP ≥ 5 mg/L, CDAI ≥ 150, or prednisone use in the previous week; clinical management group before random assignment: CDAI decrease of 200; clinical management group after random assignment: CDAI decrease of < 100 points compared with baseline or CDAI ≥ 200, or prednisone use in the previous week). Participants who did not meet the treatment failure criteria stayed on their previously assigned treatment option. Mode of delivery: (i) sc (ii) oral (iii) oral Duration: 56 weeks (8 weeks of prednisone induction treatment and 48 weeks of intervention/active control treatment) Setting of trial: 22 countries at 74 hospitals and outpatient centres Comparision treatment: Clinical Management ‐ customised therapy (as per intervention group) based on disease activity (CDAI and corticosteroid use). Therapy was escalated according to prespecified failure criteria using less stringent criteria: CDAI decrease ≥ 70 (CR‐70) compared to baseline at visit 1 or CDAI < 200 at 1 week prior to visit 1; CDAI decrease of ≥ 100 (CR‐100) compared to baseline or CDAI < 200, and absence of prednisone during the preceding week at visit 3, 4, and 5 Co‐intervention: Not reported |
|
| Outcomes | Fatigue was assessed using the FACIT‐F. Quality of life was assessed using the IBDQ and the SF‐36. Adverse events were assessed for each Medical Dictionary for Drug Regulatory Affairs (MedDRA) system organ class and preferred term. Other outcomes assessed: Mucosal healing and no deep ulcerations; deep remission; biologic remission; endoscopic response; endoscopic disease activity; clinical disease activity; Crohn's disease flare; clinical remission; steroid‐free remission; hospitalisation; length of stay in hospital; CD‐related surgeries; Crohn's disease behaviour; High Sensitivity CRP; fecal calprotectin; total dose of prednisone; work productivity; depression; Assessment time points: Baseline, week 12, week 24, week 48 |
|
| Notes | The study was funded and supported by AbbVie. Awaiting additional study information from AbbVie via the Vivli Platform |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "At week 9 (earlier if active disease present), patients were randomly assigned to the tight control or clinical management group in a 1:1 ratio, stratified by smoking status (yes or no), weight (< 70 kg or ≥ 70 kg) and disease duration (≤ 2 years or > 2 years). The patient number and group of each stratum were assigned by a central randomisation schedule generated by AbbVie (Chicago, IL, USA) using WebRando software for randomisation and ClinPhone, an interactive voice and web response system for patient allocation. The subject randomisation schedule was generated by a designated person in the AbbVie statistics department who was not involved in the rest of the study". |
| Allocation concealment (selection bias) | Low risk | "All patients were randomised centrally using an interactive voice and web response system (ClinPhone)". |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Treatments were open‐label ‐ unblinded. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Subjective outcome and participants were not blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Loss to follow‐up relatively similar across the groups and reasons comparable |
| Selective reporting (reporting bias) | Unclear risk | Fatigue and quality of life outcomes assessed but not reported. Awaiting outcome data from trial authors |
| Other bias | Low risk | Baseline characteristics were similar between treatment groups. No apparent sources of bias |
Feagan 2013.
| Methods | Phase III, RCT, double‐blinded | |
| Participants | Ulcerative colitis, moderately‐to‐severely active disease (Mayo Clinic score of 6 to 12, with a sigmoidoscopy subscore of at least 2, and disease that extended ≥ 15 cm or more from the anal verge). No response to or unacceptable side effects from ≥ 1 of the following: glucocorticoids, immunosuppressive agents, or TNF antagonists Exclusion criteria: previously treatment with vedolizumab, natalizumab, efalizumab, or rituximab; received TNF antagonists within 60 days before enrollment or cyclosporine, thalidomide, or investigational drugs within 30 days before enrollment; participants with toxic megacolon, abdominal abscess, symptomatic colonic stricture, stoma, a history of colectomy, an increased risk of infectious complications, clinically meaningful laboratory abnormalities, pregnancy or lactation, an unstable or uncontrolled medical disorder, an anticipated requirement for major surgery, colonic dysplasia or adenomas, and malignant neoplasms A total of 895 participants enrolled in the study and were randomised in a 3:2 ratio to received DB placebo (n = 149), DB vedolizumab (n = 225) or open‐label vedolizumab (n= 521) 300 mg at week 0 and week 2. At week 6, participants who demonstrated clinical response (reduction in Mayo Clinic score of ≥ 3 and a decrease of ≥ 30% from baseline, with an accompanying decrease in the rectal bleeding subscore of ≥ 1 or an absolute rectal bleeding subscore of 0 or 1) were then randomly assigned in a 1:1:1 ratio to receive DB vedolizumab every 8 weeks (n = 122), vedolizumab every 4 weeks (n = 125), or placebo (n = 126), for up to 52 weeks. Participants who did not have a clinical response at week 6 to vedolizumab induction therapy received vedolizumab at a dose of 300 mg every 4 weeks, for up to 52 weeks (n = 330). Age [Mean(SD) years): placebo 41.2 (12.5); vedolizumab DB induction therapy 40.1(13.1); vedolizumab open‐label induction therapy 40.1(13.3) Gender: placebo (92 males, 57 females); vedolizumab DB induction therapy (132 males; 93 females); vedolizumab open‐label induction therapy (301 males; 220 females) Disease duration [Mean(SD) years]: placebo 7.1 (7.2); vedolizumab DB induction therapy 6.1 (5.1); vedolizumab open‐label induction therapy 7.2 (6.6) Disease status [Mean(SD) Mayo Clinical score]: placebo 8.6 (1.7); vedolizumab DB induction therapy 8.5 (1.8); vedolizumab open‐label induction therapy 8.6 (1.8) Loss to follow‐up for maintenance phase: placebo (n = 78); vedolizumab maintenance therapy (300 mg every 8 weeks) (n = 45); vedolizumab maintenance therapy (300 mg every 4 weeks) (n = 41) |
|
| Interventions | Vedolizumab maintenance therapy Dosing regimen: a) 300 mg every 8 weeks; b) 300 mg every 4 weeks Mode of delivery: Intravenous infusion Duration: Six weeks DB or open‐label vedolizumab, followed by up to 52 weeks of randomised vedolizumab. Setting of trial: 211 medical centres (15 centres discontinued enrollment) in 34 countries worldwide Comparision treatment: placebo (double‐blind placebo intravenous infusions at week 0 and week 2, followed by every 4 weeks from week 6) Co‐interventions: Not reported |
|
| Outcomes | Fatigue was assessed using the IBDQ subcomponent 'fatigue' and 'energy level' (Rubin 2018). Quality of life was assessed using the IBDQ. Adverse events were assessed according to the Medical Dictionary for Regulatory Activities, version 15. Other outcomes assessed: Clinical remission, CRP, clinical response, glucocorticoid‐free clinical remission; durable clinical remission Assessment time points: Baseline, week 2, week 4, week 6 in the trial of induction therapy and every 4 weeks thereafter during the trial of maintenance therapy until week 52. IBDQ was assessed at week 6, week 30 and week 52. |
|
| Notes | 3‐arm trial (2 experimental, 1 control) The study was supported by Millennium Pharmaceuticals. Additional study information request from Takeda Oncology (formally Millennium Pharmaceuticals) via the Vivli Platform, was declined as fatigue was considered a tertiary endpoint in the study, thus, not meeting the eligibility criteria for this review (primary or secondary outcome). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomization was performed centrally with the use of computer‐generated randomization schedules". |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Participant, Care Provider and Investigator were blinded to treatment assignment throughout the maintenance phase of the study". |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The investigators and participants were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Loss to follow‐up relatively similar across the groups and reasons comparable |
| Selective reporting (reporting bias) | High risk | Fatigue and quality of life outcomes assessed but not reported in full‐text publication or available upon request |
| Other bias | Low risk | "No clinically important differences in baseline characteristics". No apparent sources of bias evident |
García‐Vega 2004.
| Methods | RCT, not blinded | |
| Participants | Crohn’s disease, in remission (HBI < 5). All receiving sulfasalazine or 5 ASA Exclusion criteria: dietary restrictions A total of 45 participants were randomised to one of three treatment groups, two intervention groups (guided stress management n = 15 and self‐directed stress management n = 15) and a control group (conventional medical treatment n = 15) Age [Mean(SD) years]: guided stress management group 28.7 (6.4); self‐directed stress management group 31.0 (5.7); control group 35.3 (9.1) Gender: guided stress management group (5 males, 10 females); self‐directed stress management group (5 males, 10 females); control group (6 males, 9 females) Disease duration [Mean(SD) years]: guided stress management group 5.6 (6.0); self‐directed stress management group 5.7 (4.8); control group 8.2 (5.7) Loss to follow‐up: No loss to follow‐up (author information) |
|
| Interventions | Stress management programme: a) guided stress management (relaxation practice, problem‐solving, coping in everyday life) b) self‐directed stress management (personal planning skills, autogenic training) Mode of delivery: a) face to face; delivered by psychotherapist, b) introduction by psychotherapist, written guide and audiotape Duration: a) 2 sessions, followed by weekly sessions over 6 weeks, daily practice, b) 2 sessions, daily practice for 8 weeks Setting of trial: Single centre in Spain, gastroenterology department Setting of intervention: Participants home Comparision treatment: none (standard medical therapy ‐ salicylates [author information]) Co‐interventions: 5 ‐ ASA compounds |
|
| Outcomes | Fatigue was assessed using a self‐reported daily diary (recorded between evening dinner and retiring). Tiredness (a subjective feeling of tiredness) was one of the 10 Crohn's disease symptoms assessed for presence or absence and rated for severity (according to the scale: 1 = mild, 2 = moderate and 3 = severe). Quality of life and adverse events were not assessed, however, the author reported that there were no adverse events. Other outcomes: "Symptom reduction score" for individual symptoms assessed in those afflicted only Assessment time points: Baseline, post‐intervention, month 6, month 12 |
|
| Notes | 3‐arm trial (2 experimental, 1 control) No evidence of power calculation statistical analysis Non‐validated fatigue measure Additional information was supplied by the author. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number table was used (author information). |
| Allocation concealment (selection bias) | High risk | Allocation was performed by therapist (author information) ‐ open allocation. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Due to the psychosocial nature of this intervention, participants and personnel could not be blinded to the intervention receiving/delivering. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Subjective outcome and participants were not blinded. A gastroenterologist performed the blind evaluation (authors information). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants included in the analysis (author information) |
| Selective reporting (reporting bias) | Low risk | All information on outcomes were transparent and outcomes assessed were reported. |
| Other bias | High risk | Some diversity in baseline characteristics of groups Tiredness was assessed using a single item of a Crohn's disease symptom outcome assessment. The validity of the outcome measure not reported |
Gasche 2015.
| Methods | Phase III, RCT, double‐blinded | |
| Participants | Crohn's disease or ulcerative colitis, in remission or mild‐to‐moderate disease activity (CDAI < 220; SCCAI < 4). All required to have a mild‐to‐moderate iron‐deficiency anaemia (IDA) (Hb concentration ≥ 9.5 g/dL and < 12.0 g/dL for females and ≥ 9.5 g/dL and < 13.0 g/dL for males and serum ferritin levels < 30 ug/L) and previously failed on treatment with oral ferrous products (OFP). Participants receiving protocol‐allowed immunosuppressive and immunomodulatory agents at screening were required to have been on a stable dose for ≥ 4 weeks before randomisation. Exclusion criteria: patients with anaemia unrelated to iron deficiency or who had received depot iron preparations, erythropoietin, or blood transfusions within 12 weeks of screening; oral iron treatment within 4 weeks of randomisation; treatment with immunosuppressants known to induce anaemia; folate deficiency; uncorrected vitamin B12 deficiency; serum creatinine > 2.0 mg/dL (176 ųmol/L); abnormal liver function tests and pregnancy A total of 128 participants were randomised to the ferric maltol group (n = 64) or placebo group (n = 64) Age [Mean(SD) years]: ferric maltol group 40.1 (13.5); placebo group 38.5 (12.3) Gender: ferric maltol group (24 males, 40 females); placebo group (21 males, 43 females) Disease type: ferric maltol group (35 CD, 29 UC); placebo group (35 CD, 29 UC) Disease duration [Mean(SD) years]: ferric maltol group UC 9.0 (8.28)/CD 11.25 (9.3); placebo group UC 10.99 (11.43)/CD 11.01 (8.09) (author information) Loss to follow‐up: n = 20 in total. Ferric maltol group (n = 9); due to adverse event (n = 5), participant withdrawal (n = 3), physician decision (n = 1); Placebo group (n = 11); due to adverse event (n = 4), protocol violation (n = 1), participant withdrawal (n = 5), physician decision (n = 1) |
|
| Interventions | Ferric maltol Dosing regimen: 231.5 mg of ferric maltol (equivalent to 30 mg of elemental iron) twice daily Mode of delivery: Oral Duration: 12 weeks Setting of trial: 4 centres in Austria, Germany, Hungary and the UK Setting of intervention: IBD clinic (author information) Comparision treatment: Placebo ‐ the capsules had the same excipients as ferric maltol without the active ingredient (author information) Co‐interventions: Study participants did not receive any additional co‐interventions during the trial, apart from the ones they were already on (author information). |
|
| Outcomes | Fatigue was assessed using the SF‐36 vitality subscale. Quality of life was assessed using the SF‐36 and the IBDQ. Adverse events were recorded according to MedDRA preferred terms. Treatment‐emergent adverse events assessed were the following: abdominal pain, diarrhoea, constipation, nasopharyngitis, flatulence, abdominal discomfort, rectal haemorrhage, arthralgia, abdominal distension, gastroesophageal reflux disease, fatigue, worsening of CD, headache, worsening of UC, vomiting, upper respiratory tract infection, oropharyngeal pain, Hb decreased, seasonal allergy, pruritis, nausea Other outcomes assessed: change in Hb concentration; serum ferritin concentration; percentage transferrin saturation (TSAT); clinical symptoms (SCCAI; CDAI) Assessment time points: Randomisation (baseline) and week 12 |
|
| Notes | Funders were Iron Therapeutics (UK) Ltd, now Shield TX (UK) Ltd (author information). Additional information was supplied by the author. Data analysed with intention‐to‐treat analyses |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation codes were used to randomise participants. Generation of randomisation codes and drug packaging were performed by independent providers (ALMAC, AXIO and Piramal, respectively). The subject and kit randomisation lists were generated using Statistical Analysis System (SAS) v9.2 (author information). |
| Allocation concealment (selection bias) | Low risk | "Randomisation to either ferric maltol or placebo was conducted through an interactive voice response system (IXRS system ‐ author information) according to a centralised randomisation list". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Patients and all sponsors, clinical research and clinical staff were blinded to the randomisation code until all randomized trial processes were complete". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The investigators and participants were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Loss to follow‐up similar in the intervention (n = 9) and control groups (n = 11) and reasons comparable |
| Selective reporting (reporting bias) | Low risk | All information on outcomes were transparent and outcomes assessed were reported. |
| Other bias | Low risk | Baseline demographics and disease characteristics were generally comparable between the intervention and control groups. |
Hetzel 2013a.
| Methods | Two studies, Phase III, RCTs | |
| Participants | Patients with inflammatory bowel disease and iron deficiency (baseline haemogloblin (Hgb) < 10g/dL and > 7g/dL and transferrin saturation (TSAT) < 20%) Exclusion criteria: Not reported A total of 116 participants were included in the analysis of Study 1. Participants were randomised to the ferumoxytol group (n = 79; Study 1 n = 47), iron sucrose group (n = 25) and placebo group (n = 12). Loss to follow‐up: Not reported |
|
| Interventions | Ferumoxytol Dosing regimen: 2 injections each 510 mg, 3‐8 days apart Mode of delivery: Intravenous Duration: 5 weeks Setting: Not reported Comparision treatment: Study 1 ‐ placebo (comparator) Co‐interventions: Not reported |
|
| Outcomes | Fatigue was assessed using the Functional Assessment of Chronic Illness Therapy‐Fatigue (FACIT‐F) Quality of life was not assessed Adverse events (AEs) were assessed as follows: all AEs; related AEs; serious AEs; related serious AEs, AEs of special interest – protocol defined (included protocol defined signs and symptoms of hypotension and hypersensitivity); cardiovascular AE composite endpoint (included myocardial infarctions, heart failure, moderate to severe hypertension, and hospitalisation due to any cardiovascular cause); AEs resulting in study discontinuation; death Other outcomes assessed: Change in Hgb, transferrin saturation Assessment time points: Baseline, week 2, week 3, week 4, week 5 |
|
| Notes | No additional information supplied by the author Study 2 (active comparator ‐ iron sucrose) excluded as fatigue not assessed |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information on the methods used to generate a sequence provided |
| Allocation concealment (selection bias) | Unclear risk | No information on how allocation to groups occurred |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information on whether the study personnel, participant, clinical staff were blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Subjective outcome but unclear if blinding occurred |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The number of participants randomised to each group and the number included in the analysis for study one was unclear. No information on the number of withdrawals and exclusion following randomisation and the reasons for this |
| Selective reporting (reporting bias) | High risk | Presented data on change from baseline in FACIT‐F score for the intervention group and the overall placebo population, but not the IBD placebo group |
| Other bias | Unclear risk | No information presented on the baseline characteristics of the interventions and placebo groups in study one or no information of the disease activity status of the participants |
Horta 2017.
| Methods | RCT, single‐blinded, cross‐over design | |
| Participants | Diagnosis of IBD (CD or UC) and clinically quiescent disease (HBI < 5 for CD or Mayo score ≤ 2 for UC; absence of biochemical markers of disease activity) for at least six months, experiencing persistent fatigue (defined as 2 consecutive FACIT‐F scores (4 weeks apart) lower than 40 points) Exclusion criteria: previous or active neoplasia; pregnancy or lactation; anaemia, defined as haemoglobin lower than 12 g/dL in women and < 14g/dL in men; previous treatment with acupuncture; contraindications for acupuncture; participation in other therapeutic trials; concomitant chronic diseases not related to IBD that might contribute to the presence or the severity of fatigue. Participants who presented clinical relapse during the study period were withdrawn from the study. A total of 52 participants were randomised to the electroacupuncture (EAc: n = 18), sham acupuncture (ShaEAc: n = 18) or control group (WL: n = 16). Age [Mean(SD) years): EAc: 44.6 (12.1); ShaEAc: 38.6 (8.3); control: 42.6 (10.7) Gender: EAc (10 males, 8 females); ShaEAc (13 males; 5 females); control (11 males, 15 females) Disease type: EAc (17 CD, 1 UC); ShaEAc (17 CD; 1 UC); control (12 CD, 4 UC) Disease duration [Mean(SD) years]: EAc: 10.6 (4.8); ShaEAc: 11.4 (5.4); control: 10.5 (4.7) Loss to follow‐up: EAc (n = 3; 17%); ShaEAc (n = 3; 17%); control (n = 4; 25%) |
|
| Interventions | Participants in EAC and sham EAC groups performed a total of 9 acupuncture sessions during eight weeks (2 sessions/first week and one session per week during 7 weeks). (a) Electroacupuncture; (b) sham EAc Mode of delivery: (a) 20 acupoints were selected, needles were inserted to a depth of 20 mm and connected to a 6‐channel electroacupuncture device (ITO® ES‐160, Japan). A pulsed electrical stimulation (asymmetric balanced, rectangular shape, 8 to 100 Hz frequency) was used and needles were left in place connected to the electroacupuncture device for 20 minutes. (b) 8 'non‐acupuncture' points were selected, needles were inserted to a depth of 20 mm and connected to a 6‐channel electroacupuncture device (ITO® ES‐160, Japan), but electric stimulation was not applied and needles were left in place for 20 minutes. The sessions for both groups was delivered by 3 senior acupuncturists with at least five years of experience. Duration: 9 sessions during eight weeks (2 sessions during the first week and one session per week during 7 weeks) Setting of trial: Single centre in Spain, outpatient clinic of a Digestive Diseases department Setting of intervention: IBD unit in separate rooms Comparision treatment: none (waiting‐list group was offered an open EAc treatment at the end of follow‐up) Co‐interventions: Usual pharmacological treatments were maintained and drug dosage unchanged during the study. |
|
| Outcomes | Fatigue was assessed using the FACIT‐FS. Quality of life was assessed using the IBDQ‐9. Adverse events, including acupuncture abnormalities such as bleeding, haematoma, pain in the acupuncture sites, increased blood pressure, fainting during acupuncture and other adverse reactions were evaluated (during the treatment period). Other outcomes assessed: depression, anxiety and sleepiness Assessment time points: baseline, week 4, week 8 and week 16 |
|
| Notes | Additional information was supplied by author. Full‐text paper submitted for publication 3‐arm trial (1 experimental, 2 control) Cross‐over of participants who had no improvement in fatigue (FACIT‐FS score < 40) after 8 weeks post‐treatment occurred (participants from EAc group to ShEAc group and vice versa) and also the control group started EAc. However, due to loss to follow‐up and dropouts, data analysis was not performed. Data analysed with per protocol analyses. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Simple randomisation was performed by generating a random number table using the SPSS 20.0 software. The first number in the random number table was used as a starting point to create random assignment cards, which were then sealed in opaque envelopes. The envelopes were numbered and kept secure by one of the authors (AF) (author information). |
| Allocation concealment (selection bias) | Low risk | The acupuncturists were given randomly generated treatment allocations within sealed opaque envelopes (author information). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Participants and evaluators were blinded to group assignment. Personnel administering the intervention were not blinded (author information). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Participants were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Loss to follow‐up relatively similar across the groups (author information) |
| Selective reporting (reporting bias) | Unclear risk | All information provided by trial author on outcomes was transparent, however, findings published only as a conference abstract at present |
| Other bias | Low risk | Baseline demographics were generally comparable between groups (author information). |
McNelly 2016.
| Methods | RCT, 2 x 2 factorial design, part‐blinded | |
| Participants | Diagnosed with Crohn's disease or ulcerative colitis in remission (CRP < 5 mg/dL and HBI < 5/SCCI < 3), who are self‐reporting fatigue, willing to increase current activity levels and able to take medication with ingredients derived from animal or fish sources. Exclusion criteria: At least 60 minutes of moderate‐to‐vigorous exercise performed per week; concurrent course of anticoagulant medications; concurrent participation in another randomised control trial; concurrent pregnancy; consumption of oily fish at least twice per week or eight times per month; Omega‐3 fatty acid supplements taken in the 12 weeks before screening; fatigue‐related comorbidities. A total of 74 participants were randomised (n = 52 completed the trial according to protocol) to one of four treatment groups (No physical activity advice (PAA) and omega 3 n = 14; PAA and omega 3 n = 11; No PAA and placebo n = 12; PAA and placebo n = 15). Age [Median(IQR) years] : No PAA with omega 3: 45 (36, 51); PAA with omega 3: 31 (29, 55); No PAA and placebo: 31 (27, 51); PAA and placebo: 35 (28, 43) Gender: No PAA and omega 3 (7 males, 7 females); PAA and omega 3 (6 males and 5 females); No PAA and placebo (4 males, 8 females); PAA and placebo (8 males and 7 females) Disease type: No PAA and omega (7 CD, 7 UC); PAA and omega 3 (6 CD and 5 UC); No PAA and placebo (6 CD, 6 UC); PAA and placebo (6 CD, 8 UC, 1 unclassified) Loss to follow‐up: n = 8; No PAA and omega 3 n = 0; PAA and omega 3 n = 4; no PAA and placebo n = 2; PAA and placebo n = 2 (author information). Reasons included clinically active disease (n = 3), pregnancy (n = 2), anaemia (n = 1); musculoskeletal injury (n = 1) and lack of time (n = 1). |
|
| Interventions | a) Physical activity advice ‐ received advice about initiating and maintaining motivation (using techniques of imagery, goal‐setting [for each week and the whole programme]) and discussed overcoming barriers to exercise (physical limitations and fears of worsening symptoms of IBD) b) Active supplementation ‐ two capsules containing a total of 2970 mg of omega‐3 fatty acids (EPA, 2250 mg; DHA, 150 mg) Mode of delivery a) face‐to‐face individual consultation with a personal trainer and one researcher b) orally with food Duration: a) 15 minutes b) daily for 12 weeks Setting of trial: Single‐centre London tertiary referral hospital (author information) Setting of delivery: IBD outpatient clinic. The exercise programme was undertaken either at home or at a gym. Comparision treatment: a) Placebo (capsules containing capric and caprylic acid) b) No physical activity advice (15 minute face‐to‐face individual consultation with the researcher discussing current dietary habits and general health) Co‐interventions: None |
|
| Outcomes | Fatigue was assessed using the FACIT‐F; MFI and IBD‐F. Quality of life was assessed using the IBDQ. Adverse events were recorded in the medication diary and assessed by the researcher during the 6 follow‐up contact time points. Other outcomes assessed: Depression; physical activity (diary and accelerometer), disease activity (HBI; SCCAI); body composition; Hgb; CRP Assessment time points: Baseline and week 12. All groups were contacted by the researcher on six occasions (a week following their second study visit, and then every two weeks) using email, text or telephone to discuss progress. |
|
| Notes | 4‐arm trial Additional information was supplied by the author. The study was supported by the Big Lottery Fund [grant number GFTTAFR] and Crohn's and Colitis UK was the fund holder. No power calculation undertaken Data analysed with per protocol analyses |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Block randomisation using computer‐generated random numbers |
| Allocation concealment (selection bias) | Unclear risk | For the capsule assignment, sequentially named drug containers of identical appearance were provided by UCHL pharmacy. The first author performed the randomisation and enrollment of the participants for the physical activity advice intervention (open allocation) (author information). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants and researchers were blinded to capsule type, but could not be blinded to the consultation type due to the nature of the intervention. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Self‐reported outcomes and participants not blinded to the physical activity advice group |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Loss to follow‐up relatively similar across the four groups and reasons comparable |
| Selective reporting (reporting bias) | Low risk | All information on outcomes were transparent and outcomes assessed were reported. |
| Other bias | Low risk | Baseline characteristics were similar between treatment groups. No apparent sources of bias |
Raftery 2013.
| Methods | RCT, double‐blinded | |
| Participants | Crohn’s disease, in remission and on stable drug therapies for at least 1 month at study entry Exclusion criteria: Symptomatic CD (CDAI > 150); pregnancy; previous extensive small bowel research; presence of an ileostomy or colostomy; known hypersensitivity to vitamin D; hypercalcaemia; those currently using supplemental vitamin D > 800 IU/D; diagnosis of any of the following: active tuberculosis, sarcoidosis, hyperparathyroidism, renal failure, pseudohyperparathyroidism, malignancy, active lymphoma, short bowel syndrome; antibiotic use in the 4 weeks prior to enrollment; current use of bisphosphonates; renal impairment, diabetes mellitus; participants participating in a concurrent RCT; alcohol dependency A total of 27 participants were randomised to the intervention (vitamin D3, n = not reported) or control group (placebo, n = not reported) Loss to follow‐up: Not reported |
|
| Interventions | Vitamin D3 Dosing regimen: 2000 IU per day Mode of delivery: Oral Duration: 3 months Setting: 2 centres in the Republic of Ireland Comparision treatment: Placebo (soya bean oil) Co‐interventions: None |
|
| Outcomes | Fatigue was assessed using the MFI. Quality of life was assessed using the IBDQ. Adverse events were not assessed. Other outcomes assessed: Change in hand grip strength; disease activity (CDAI), FBC, CRP, serum 25OHD Assessment time points: Baseline and month 3 |
|
| Notes | No additional information supplied by the author | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind but no further information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Self‐reported outcomes but details of blinding not explicit |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The number of participants included in the analysis was not reported. |
| Selective reporting (reporting bias) | High risk | Reported only the difference in muscle strength scores for the intervention and control groups. Changes in fatigue and quality of life scores from baseline to month 3 presented for those who achieved 25OHD levels ≥ 75nmol/L compared to those with 25OHD levels < 75nmol/L only. |
| Other bias | Unclear risk | Baseline group characteristics not reported |
Sandborn 2013.
| Methods | Phase III, RCT, double‐blinded | |
| Participants | Crohn's disease, moderately‐to‐severely active disease (CDAI 220‐450) and one of the following: a serum CRP > 2.87 mg/L, colonoscopic findings showing ≥ 3 large ulcers or ≥ 10 aphthous ulcers, or fecal calprotectin > 250 μg/g of stool plus evidence of ulcers on CT or MRI, small‐bowel radiography, or capsule endoscopy. No response to or unacceptable side effects from ≥ 1 of the following: glucocorticoids, immunosuppressive agents, or TNF antagonists Exclusion criteria: previous treatment with vedolizumab, natalizumab, efalizumab, or rituximab; treatment with adalimumab within 30 days before enrollment and treatment with infliximab or certolizumab pegol within 60 days before enrollment; participants with a stoma, ≥ 3 small‐bowel resections, short‐bowel syndrome, extensive colonic resection, intestinal stricture, abdominal abscess, active or latent tuberculosis, or cancer A total of 1115 participants enrolled in the study and were randomised in a 3:2 ratio to receive DB placebo (n = 148), DB vedolizumab (n = 220) or open‐label vedolizumab (n= 748) 300 mg at week 0 and week 2. At week 6, participants who demonstrated clinical response (≥ 70‐point decrease in the CDAI score) were then randomly assigned, in a 1:1:1 ratio to continue in a blinded fashion to receive vedolizumab every 8 weeks (n = 154), vedolizumab every 4 weeks (n = 154), or placebo (n = 153), for up to 52 weeks. Participants who did not have a clinical response at week 6 to vedolizumab induction therapy received vedolizumab at a dose of 300 mg every 4 weeks, for up to 52 weeks (n = 412). Age [Mean(SD) years): placebo 38.6 (13.2); vedolizumab DB induction therapy 36.3 (11.6); vedolizumab open‐label induction therapy: 35.6 (12.0) Gender: placebo (69 males, 79 females); vedolizumab DB induction therapy (105 males; 115 females); vedolizumab open‐label induction therapy (346 males, 401 females) Disease duration [Mean(SD) years]: placebo 8.2 (7.8); vedolizumab DB induction therapy 9.2 (8.2);vedolizumab open‐label induction therapy 9.2 (7.6) Disease status [Mean(SD) CDAI score]: placebo: 325 (78); vedolizumab DB induction therapy 327 (71) ; vedolizumab open‐label induction therapy 322 (67) Loss to follow‐up in maintenance phase: placebo (n = 89); vedolizumab maintenance therapy (300 mg every 8 weeks) (n = 81); vedolizumab maintenance therapy (300 mg every 4 weeks) (n = 72) |
|
| Interventions | Vedolizumab maintenance therapy Dosing regimen: a) 300 mg every 8 weeks; b) 300 mg every 4 weeks Mode of delivery: Intravenous infusion Duration: Six weeks DB or open‐label vedolizumab, followed by up to 52 weeks of randomised vedolizumab Setting of trial: 285 medical centres (15 centres discontinued enrollment) in 39 countries worldwide Comparision treatment: placebo (double‐blind placebo intravenous infusions at week 0 and week 2, followed by every 4 weeks from week 6) Co‐interventions: Not reported |
|
| Outcomes | Fatigue was assessed using the IBDQ subcomponent 'fatigue' and 'energy level' (Rubin, 2008). Quality of life was assessed using the IBDQ. Adverse events were assessed according to the Medical Dictionary for Regulatory Activities, version 15. Other outcomes assessed: Clinical remission, CRP, clinical response, glucocorticoid ‐free clinical remission; durable clinical remission Assessment time points: Baseline, week 2, week 4, week 6 in the trial of induction therapy and every 4 weeks thereafter during the trial of maintenance therapy until week 52. IBDQ was assessed at week 6, week 30 and week 52. |
|
| Notes | 3‐arm trial (2 experimental, 1 control) The study was supported by Millennium Pharmaceuticals, a wholly owned subsidiary of Takeda Pharmaceuticals. Additional study information request from Takeda Oncology (formally Millennium Pharmaceuticals) via the Vivli Platform, was declined as fatigue was considered a tertiary endpoint in the study, thus deeming it not to meet the eligibility criteria for this review (primary or secondary outcome). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomization was computer‐generated and was performed at a central location". |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participant, Care Provider and Investigator were blinded to treatment assignment throughout the maintenance phase of the study. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The investigators and participants were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Loss to follow‐up relatively similar across the groups and reasons comparable |
| Selective reporting (reporting bias) | Unclear risk | Fatigue and quality of life outcomes assessed but not reported in full‐text publication or available upon request |
| Other bias | Low risk | "Baseline disease characteristics were similar across groups". No apparent sources of bias evident |
Therkelsen 2016a.
| Methods | RCT, single‐blinded | |
| Participants | Ulcerative colitis, mild‐to‐moderate disease activity (CAI ≥ 3) Exclusion criteria: Pregnancy, biological treatment with antibodies to TNFα, daily use of more than 5 mg of prednisolone, change of medication and/or consumption of mushroom products from two weeks before till end of the study A total of 62 participants were randomised to the intervention (AndoSan™, n = 31) or control group (placebo, n = 31). Age [Mean(SD) years]: AndoSan™ group 41.6 (12.8); placebo group 38.5 (8.9) (author information) Gender: AndoSan™ group (13 males, 11 females); placebo group (12 males, 14 females) Disease duration [Mean(SD) years]: AndoSan™ group: 8.7 (7.3); placebo group: 7.2 (6.6) (author information) Loss to follow‐up: AndoSan™ group (n = 7): due to 3 with change of medication, 2 with missing laboratory data and 2 with missing attendance. Placebo group (n = 5): due to 2 with change of medication, 2 with missing laboratory data, and 1 with missing attendance |
|
| Interventions | Agaricus blazei Murill‐ based mushroom extract (AndoSan™) Dosing regimen: 30 mL twice daily (60 mL/day) Mode of delivery: Oral Duration: 21 days Setting: Single centre in Oslo, Norway Comparison treatment: Placebo (colour‐like drink with ionised water containing 0.5 mL per litre of caramel colour [E150c] with salt) Co‐interventions: None (author information) |
|
| Outcomes | Fatigue was assessed using an 11‐item total fatigue score (Fatigue Questionnaire) and the vitality subscale of the SF‐36 (IQOLA SF‐36 Norwegian Version 1.2). Quality of life was assessed using the SF‐36 (IQOLA SF‐36 Norwegian Version 1.2). Adverse events were assessed during an interview between the first author and participant at all visits. Other outcomes assessed: EDTA blood test; faecal calprotectin; CAI Assessment time point: Baseline, day 14 and day 21 |
|
| Notes | Sample size was calculated for prospective differences of 20% between the experimental and placebo group and assumed standard deviation of 20% for the different parameters with a significant level of 5% and a power of 90% (ß = 0.10), demanded about 25 participants per randomised arm (calculated in cooperation with Oslo Center for Biostatistics and Epidemiology, Oslo University Hospital). Two of the authors (GH and EJ) had patent/patent applications and financial interests relating to material (AndoSan™) pertinent to this article: i) WO2005065063 A2, Appl. No.:10/ 585600, NO‐ and PCT‐filed Jan 2004 and Jan 2005 respectively, by Inventor Hetland Geir, and ii) NO20090003383, Appl. No.: NO20090003383 20091119, by Inventors Hetland Geir and Johnson Egil and filed by Applicant Immunopharma AS in Nov 2009 and financial interest of Geir Hetland as shareholder in Immunopharma AS of Norway, commercialising AndoSan™. Grants received from the University of Oslo (author information) Additional information was supplied by the author. SF‐36 PCS and MCS not calculated (author information) Data analysed with per protocol analyses |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "50 patients were divided into 13 groups and manually randomised with the overall allocation ratio of one to one. Block randomisation was done after the phone interview, with uneven and even numbers given for AndoSan™ or placebo respectively. The patients, one by one, were placed in one pile, and the group affiliations were placed in another pile, both anonymized". |
| Allocation concealment (selection bias) | High risk | "The first author performed the randomisation, enrolled the participants, and assigned participants to interventions" ‐ open allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Single‐blinded trial (participants). Personnel administering the intervention were not blinded (author information). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Participants were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 7 lost to follow‐up from intervention group, 5 from placebo group. Missing data balanced across groups with similar reasons. The included 50 symptomatic participants had no missing data. |
| Selective reporting (reporting bias) | Low risk | All information on outcomes were transparent and outcomes assessed were reported. |
| Other bias | Unclear risk | Baseline characteristics similar between treatment groups. Mixed models corrected for baseline values were used for measuring P values between the study groups. Two authors (GH and EJ) had patent/patent applications and financial interest relating to material (AndoSan™) pertinent to these articles. Therkelsen 2016a and Therkelsen 2016b published as two distinct trials, however, the trials have the same ethical approval number. No mention of subjects being stratified according to disease type |
Therkelsen 2016b.
| Methods | RCT, single‐blinded | |
| Participants | Crohn's disease, mild‐to‐moderate disease activity (SCDAI ≥ 3), aged 18 years old and older Exclusion criteria: Pregnancy, biological treatment with antibodies to TNFα, daily use of more than 5 mg of prednisolone, change of medication and/or consumption of mushroom products from two weeks before till end of the study A total of 76 participants were randomised to the intervention (AndoSan™: n = 37) or control group (placebo: n = 39) Age [Mean(SD) years]: AndoSan™ group: 44.92 (13.96) years; placebo group: 43.28 (13.74) years (author information) Gender: AndoSan™ group (11 males, 14 females); placebo group (10 males, 15 females) Disease duration [Mean(SD) years]: AndoSan™ group: 9.7 (0.5 ‐ 46) years; placebo group: 8.0 (0.5 ‐ 42) years Loss to follow‐up: AndoSan™ group (n = 12): due to 11 with missing report for symptoms and 1 with missing laboratory data. Placebo group (n = 14): due to 9 with missing report for symptoms, 4 missing laboratory data, and 1 with missing attendance |
|
| Interventions | Agaricus blazei Murill‐ based mushroom extract (AndoSan™) Dosing regimen: 30 mL twice daily (60 mL/day) Mode of delivery: Oral Duration: 21 days Setting: Single centre in Oslo, Norway Comparison treatment: Placebo (colour‐like drink with ionised water containing 0.5 mL per litre of caramel colour [E150c] with salt) Co‐interventions: None (author information) |
|
| Outcomes | Fatigue was assessed using an 11‐item total fatigue score (Fatigue Questionnaire) and the vitality subscale of the SF‐36 (IQOLA SF‐36 Norwegian Version 1.2). Quality of life was assessed using the SF‐36 (IQOLA SF‐36 Norwegian Version 1.2). Adverse events were assessed during an interview between the first author and participant at all visits, however, such events were not registered (author information). Other outcomes assessed: EDTA blood test; faecal calprotectin; SCDAI Assessment time point: Baseline, day 14 and day 21 |
|
| Notes | Sample size was calculated for prospective differences of 20% between the experimental and placebo group and assumed standard deviation of 20% for the different parameters with a significant level of 5% and a power of 90% (ß = 0.10), demanded about 25 participants per randomised arm (calculated in cooperation with Oslo Center for Biostatistics and Epidemiology, Oslo University Hospital). Two of the authors (GH and EJ) had patent/patent applications and financial interests relating to material (AndoSan™) pertinent to this article: i) WO2005065063 A2, Appl. No.:10/ 585600, NO‐ and PCT‐filed Jan 2004 and Jan 2005 respectively, by Inventor Hetland Geir, and ii) NO20090003383, Appl. No.: NO20090003383 20091119, by Inventors Hetland Geir and Johnson Egil and filed by Applicant Immunopharma AS in Nov 2009 and financial interest of Geir Hetland as shareholder in Immunopharma AS of Norway, commercialising AndoSan™. Grants received from the University of Oslo (author information) Additional information was supplied by the author. SF‐36 PCS and MCS not calculated (author information). Data analysed with per protocol analyses |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Block randomization was done after the phone interview, with uneven and even numbers given for AndoSan™ or placebo respectively. The patients, one by one, were placed in one pile, and the group affiliations were placed in another pile. The randomization was performed by combining one selection from each pile, both anonymized." |
| Allocation concealment (selection bias) | High risk | "The first author performed the randomization, enrolled the participants, and assigned participants to interventions" ‐ open allocation. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Single‐blinded trial (participants). The first author (SPT) was responsible for the inclusion and randomisation of participants, the implementation of the practical aspects of and in meeting with the participants and also in the analysis of the results. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Participants were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 12 lost to follow‐up from intervention group, 14 from placebo group. Missing data balanced across groups with similar reasons. The included 50 symptomatic participants had no missing data. |
| Selective reporting (reporting bias) | Low risk | All information on outcomes transparent and outcomes assessed were reported. |
| Other bias | Unclear risk | Baseline characteristics similar between treatment groups. Mixed models corrected for baseline values were used for measuring P values between the study groups. Two authors (GH and EJ) had patent/patent applications and financial interest relating to material (AndoSan™) pertinent to these articles. Therkelsen 2016a and Therkelsen 2016b published as two distinct trials, however, the trials have the same ethical approval number. No mention of subjects being stratified according to disease type |
Vogelaar 2014.
| Methods | RCT, not blinded | |
| Participants | Inflammatory bowel disease, in remission (CDAI < 150 or CAI < 10) and CRP < 10) experiencing fatigue (CIS‐fatigue score of ≥ 35). Exclusion criteria: Pregnant or breastfeeding women; history of lymphoproliferative disease or cancer, other than skin basocellular carcinoma; other gastrointestinal disease than IBD; listeriosis; HIV infection; immunodeficiency syndrome; CNS demyelinating disease; chronic hepatitis B or C virus infection or untreated tuberculosis; poorly controlled medical conditions, including anaemia, low iron levels, diabetes mellitus, kidney disease, liver disease and unstable Ischaemic heart disease; a known pre‐existing condition that could interfere with the participant’s participation such as psychiatric conditions or CNS trauma or active seizure disorders; surgery in the past 12 weeks prior to the screening visit; history of clinically significant drug or alcohol abuse in the last 2 years A total of 98 participants were randomised to the intervention group (SFT: n =48 [1 participant declined further participation after randomisation]) or control group (CAU n = 49) Baseline characteristics for each treatment group were not presented. Loss to follow‐up: One participant declined further participation after randomisation. |
|
| Interventions | Solution‐focused therapy (psychotherapy/coping styles for fatigue) Mode of delivery: 6 face‐to‐face group sessions during 3 months and a final booster session at 6 months. Each group consisted of 7 participants. In the fifth session, a partner, family member or close relative participated. Duration: 7 sessions, 1.5 hours per session Setting: 2 centres in the Netherlands Comparision treatment: none (care‐as‐usual) Co‐interventions: Not reported |
|
| Outcomes | Fatigue was assessed using the CIS and the FSS‐9. Quality of life was assessed using the IBDQ, SF‐36 and EQ‐5D. Adverse events were not reported. Other outcomes assessed: faecal calprotectin; depression and anxiety; sleep quality; disease activity; medication use; side effect of medication and laboratory parameters (CRP, leucocytes and haemoglobin) Assessment time point: Baseline, month 3, month 6, month 9 |
|
| Notes | Baseline characteristics were reported to be similar between the two groups, however, the data were not presented. No additional information supplied by the author |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were randomised to the treatment or control arm in blocks of 14 subjects using randomisation lists drawn from a computer generated series of random numbers". |
| Allocation concealment (selection bias) | Unclear risk | Randomisation was conducted by the second author. The randomisation lists were anonymised for the randomisation process. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants were informed about the study and the design of the treatment. Due to the psychosocial nature of this intervention, participants and personnel could not be blinded to the intervention receiving/delivering. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Self‐reported outcomes and participants were informed about the study and the design of the treatment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Only one participant was excluded from the analysis after randomisation due to participant declining further participation. |
| Selective reporting (reporting bias) | High risk | Non‐standard reporting of CIS, FSS‐9, IBDQ, SF‐36 and EQ‐5D scores. Only fixed mixed models and estimates and effect sizes presented |
| Other bias | Low risk | Baseline characteristics similar between the treatment groups, however, the data were not presented. No apparent sources of bias |
AE: Adverse events
ALT: Alanine transaminase
ASA: aminosalicylate
AST: Aspartate transaminase
BUN: Blood urea nitrogen
CAI: Clinical Activity Index
CAU: Care‐as‐usual
CBT: Cognitive behavioural therapy
CD; Crohn's disease
CDAI: Crohn's Disease Activity Index
CDEIS: Crohn's Disease Endoscopic Index of Severity
CIS: Checklist of Individual Strength
CNS: Central nervous system
CRP: C‐reactive protein
CT: Computed tomography
DB: Double‐blind
DHA: Docosahexaenoic acid
EAc: Electroacupuncture
EDTA: Ethylenediaminetetraacetic acid
eow: every other week
EPA: Eicosapentanoic acid
EQ‐5D: EuroQol 5 dimensions
FACIT‐F: Functional Assessment of Chronic Illness Therapy‐Fatigue
FACIT‐FS: Functional Assessment of Chronic Illness Therapy‐Fatigue Scale
FBC: Full blood count
FSS‐9: 9‐item Fatigue Severity Scale
g/dL: grams per decilitre
Hb: Haemogloblin
HBI: Harvey Bradshaw Index
Hgb: Haemoglobin
HIV: Human immunodeficiency viruses
IBD: Inflammatory bowel disease
IBD‐F: Inflammatory Bowel Disease Fatigue scale
IBDQ: Inflammatory Bowel Disease Questionnaire
IBDQ‐9: 9‐item Inflammatory Bowel Disease Questionnaire
IDA: Iron deficiency anaemia
IL: Illinois
IQOLA: International Quality of Life Assessment
IU/D: International unit of Vitamin D
IXRS: Interactive voice response system
kg: Kilogram
LFT: Liver Function Test
MCS: Mental component summary
MedDRA: Medical Dictionary for Regulatory Activities
MFI: Multidimensional Fatigue Inventory
mg: milligrams
mg/kg/day: milligrams per kilograms per day
ml/L: millilitres per Liters
MRI: Magnetic Resonance Imaging
OFP: Oral ferrous products
PAA: Physical Activity Advice
PCS: Physical component summary
RCT: Randomised controlled trial
SAS: Statistical Analysis System
sc: Subcutaneous
SCCAI : Simple Clinical Colitis Activity Index
SCCI: Simple Clinical Colitis Index
SCDAI: Short Crohn's Disease Activity Index
SD: Standard deviation
SF‐36: 36‐Item Short Form
SFT: Solution‐focused therapy
ShaEAc: Sham electroacupuncture
SPSS: Statistical Package for the Social Sciences
SSCAI: Simple Clinical Colitis Activity Index
TNF: Tumor necrosis factor
TPMT: Thioprurine methyltransferase
TSAT: Transferrin saturation
UC: Ulcerative colitis
UCHL: University College London Hospitals
UK: United Kingdom
UK IBDQ: British version of the Inflammatory Bowel Disease Questionnaire
USA: United States of America
WBC: White blood cells
WL: Waiting list
μg/g: microgram per gram
ųmol/L: micromole/litre
25OHD: 25‐hydroxycholecalciferol or 25‐hydroxyvitamin D
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Boye 2011 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the IBDQ not reported |
| Colombel 2010 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the IBDQ not reported |
| Cosnes 2013 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the IBDQ not reported |
| Dewint 2014 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the IBDQ not reported |
| Feagan 2003 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the IBDQ and SF‐36 not reported |
| Ford 2016 | Participants: Data from patients with IDA and underlying gastrointestinal disorders ‐ IBD‐specific data not presented |
| Hetzel 2013b | Participants: Data from patients with IDA and underlying gastrointestinal disorders ‐ IBD‐specific data not presented |
| Leiper 2001 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the inflammatory bowel disease quality of life index not reported |
| Lichtenstein 2002 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the IBDQ not reported |
| Loftus 2009 | Type of study: Not a RCT. Only presented data from baseline to week 4 prior to commencement of RCT |
| Loftus 2017 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the IBDQ or SF‐36 not reported |
| Maragkoudaki 2016 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the IMPACT‐III scale not reported |
| Mikocka‐Walus 2017 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the WHOQoL not reported |
| Minderhoud 2007 | Type of study: Not a RCT |
| NCT01991314 | Type of study: Not a RCT |
| NCT02148718 | Type of study: Not a RCT |
| NCT02162862 | Type of study: Not a RCT |
| Paramsothy 2017 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the IBDQ not reported |
| Pena Rossi 2009 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the IBDQ not reported |
| Persoons 2007 | Type of study: Not a RCT |
| Reusch 2016 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the Short Form‐12 Health Survey not reported |
| Sands 2008 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the IBDQ and SF‐36 not reported |
| Sands 2013 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the IBDQ and SF‐36 not reported |
| Schmidt 2016 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the IBDQ and SF‐36 not reported |
| Scholten 2018 | Participants: Data from patients with IBS and IBD not analysed separately |
| Schreiber 2007 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the IBDQ not reported |
| Smith 2011 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the IBDQ and SF‐36 not reported |
| Smith 2013 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the IMPACT‐III survey not reported |
| Steinhart 2002 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the IBDQ not reported |
| Szigethy 2016 | Type of study: Not a RCT |
| Targan 2007 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the IBDQ and SF‐36 not reported |
| Valentine 2009 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the IBDQ and SF‐36 not reported |
| Van Assche 2012 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the IBDQ not reported |
| Vermeire 2017 | Outcome: Data on fatigue, loss of energy, vigour and vitality as a subscale of the IBDQ or SF‐36 not reported |
IBD: Inflammatory bowel disease IBDQ: Inflammatory Bowel Disease Questionnaire IBS: Irritable bowel syndrome IDA: Iron deficiency anemia RCT: Randomised controlled trial SF‐36: 36‐Item Short Form WHOQoL: World Health Organisation Quality of Life
Characteristics of studies awaiting assessment [ordered by study ID]
Ghosh 2019.
| Methods | A phase 3, double‐blind RCT |
| Participants | Adults (aged 16 years and older) with a diagnosis of active ulcerative colitis (adapted Mayo score of 5 to 9 points and endoscopic subscore of 2 to 3) who have demonstrated an inadequate response to, loss of response to, or intolerance to at least one of the following treatments including: oral aminosalicylates, corticosteroids, immunosuppressants, and/or biologic therapies in the opinion of the investigator |
| Interventions | Experimental: Extended‐release updacitinib 7.5, 15, 30, 45 mg orally, once daily for 8 weeks Comparator: Placebo, orally, once daily for 8 weeks |
| Outcomes | Patient‐reported outcomes utilising the newly developed 17‐item Ulcerative Colitis Symptom Questionnaire (UC‐SQ). Overall scores of UC‐SQ range from 17 to 85; higher scores indicate greater symptom burden. Participants at selected study sites completed the UC‐SQ at baseline (BL) and week 2, 4, and 8. |
| Notes | Data from the Phase 2b study U‐ACHIEVE (NCT02819635) 110 participants completed the UC‐SQ questionnaire. Conference proceeding only ‐ additional information on item scores for fatigue to be requested |
Louis 2019.
| Methods | A phase II, multicentre, randomised, double‐blind, multiple dose, placebo‐controlled, parallel‐group study |
| Participants | Adults (aged 18 ‐ 75 years) with a diagnosis of moderate‐to‐severe active Crohn' disease (CDAI ≥ 220 and ≤ 450) with mucosal ulcers and CDEIS >= 7 (or >= 4 in participants with isolated ileitis on ileocolonoscopy). Patients who are naive or experienced to 1 or more TNF antagonists (infliximab, adalimumab, or certolizumab pegol) at a dose approved for CD were included. |
| Interventions | Experimental: Risankizumab (200 mg or 600 mg) or placebo IV Q4W for 12 weeks as induction therapy. In the extended induction/washout phase (weeks 14‐26, period 2), those not in deep remission at week 12 received open‐label RZB 600 mg IV Q4W for 12 weeks and those in deep remission at week 12 entered a washout phase until week 26. Patients in clinical remission at week 26 entered the maintenance phase and received open‐label RZB 180 mg SQ Q8W for 26 weeks (weeks 26‐52, period 3); those not in clinical remission discontinued. In period 2 and 3, only participants who received open‐label RZB treatment were analysed. |
| Outcomes | Percentages of participants with IBDQ response (increase in IBDQ total score >= 16); IBDQ remission (IBDQ total score >= 170); and mean change from baseline (BL) in IBDQ total, domain, and selected individual item scores were calculated at weeks 12, 26, and 52. |
| Notes | Data from phase 2 trial of risankizumab in Crohn's disease (NCT02031276) 121 participants were analysed in period 1. Recent conference proceeding ‐ additional information on individual item scores for fatigue to be requested |
O' Connor 2019.
| Methods | A single‐centred randomised, placebo‐controlled, open trial |
| Participants | 40 adults (aged 18 years and older) with IBD (20 adults with Crohn's disease and 20 adults with ulcerative colitis) in clinical and biochemical remission with no other obvious identifiable explanation for their fatigue. Remission for CD will be defined as HBI < 5 and CRP < 5. For UC, it will be defined as partial Mayo score ≤ 2 and CRP < 5. For both groups a faecal calprotectin (FC) of > 250 ug/g will be regarded as indicating active disease and will exclude participants from the study. Participants will also be excluded if they are anaemic (Hb < 14g/dL for males, 11.5g/dL for females). In the CD arm, participants will be assessed for Vitamin B12 and Vitamin D status and excluded if B12 is less than 300 pmol/L or Vitamin D less than 30 ng/mL. |
| Interventions | Experimental: Structured psychoeducational intervention centred around a self‐management booklet and group work (3 small group sessions over 6 months) Comparator: Standard care |
| Outcomes | Primary outcome: Fatigue severity and impact (CCUK fatigue score at 20 weeks) Secondary outcomes: Anxiety/depression (HADS at 20 weeks) Somatisation (PHQ15 at 20 weeks) IBS symptoms (Rome III at 20 weeks) Quality of life (SIBDQ and SF‐36 at 20 weeks) Disease activity (HBI/Mayo score; CRP and FC) Activity diaries (for four weeks) Fatigue scores and disease activity (as measured by HBI/Mayo score, CRP, FC), quality of life (as measured by SF‐36 & SIBDQ), anxiety and depression (HAD) or somatisation (PHQ15) |
| Notes | Estimated enrollment: 40 participants Small pilot RCT recently published |
Sands 2018.
| Methods | A phase 3, randomised, double‐blind, placebo‐controlled, parallel‐group multicentre study |
| Participants | Adults (aged 18 years and older) with moderately‐to‐severely active CD (CDAI 220‐450) who had inadequate response or intolerance to TNF antagonists (UNITI‐1, N = 741) or to conventional therapy (UNITI‐2, N = 627) were randomised 1:1:1 to receive Ustekinumab (UST) ˜6 mg/kg, UST 130 mg or placebo (PBO) intravenously (IV) at week 0 and the IBDQ score was assessed at baseline and week 8. |
| Interventions | Experimental: Ustekinumab ˜6 mg/kg or 130 mg intravenously at week 0 Active comparator: Placebo intravenously at week 0 |
| Outcomes | Specific items of quality of life (IBDQ assessed at baseline and week 8) |
| Notes | Post hoc analysis of the UNITI‐ I & 2 phase 3 trials. Participants included those who had an inadequate response or intolerance to TNF antagonists (UNITI‐1, N = 741) or to conventional therapy (UNITI‐2, N = 627). Recent conference proceeding ‐ additional information on individual item scores for fatigue to be requested |
Tew 2019.
| Methods | A multicentred RCT |
| Participants | Adults (aged between 16 and 65 years) with a clinical diagnosis of Crohn's disease (for at least 4 weeks before the screening visit) with mildly active (150 to 219 on Crohn’s Disease Activity Index [CDAI]) or inactive (< 150 on CDAI) disease. Faecal calprotectin < 250 mcg/g recorded no greater than 4 weeks before the screening visit. Stable medications for at least 4 weeks before the screening visit. Able to provide written informed consent and complete the study questionnaires. Able to travel to the research centre for assessment visits and exercise sessions. 53 participants were assessed for eligibility and 36 (68%) were randomised. |
| Interventions | Experimental: High‐intensity training group ‐ participants will be invited to complete three exercise sessions each week for 12 consecutive weeks. All exercise will be performed on a stationary upright cycle ergometer. Each session will begin with a 5‐minute warm‐up of easy cycling. The main body of each session will involve ten, 1‐minute bouts of hard cycling, interspersed with 1‐minute bouts of easy cycling. The session will end with a 3‐minute cool‐down of easy cycling. The resistance level on the cycle ergometers will be progressed after 4 and 8 weeks of training. Experimental: Moderate‐intensity training group ‐ participants will be invited to complete three exercise sessions each week for 12 consecutive weeks. All exercise will be performed on a stationary upright cycle ergometer. Each session will begin with a 5‐minute warm‐up of easy cycling. The main body of each session will involve 30 minutes of cycling at a moderate intensity. The session will end with a 3‐minute cool‐down of easy cycling. The resistance level on the cycle ergometers will be progressed after 4 and 8 weeks of training. Comparator: Participants continue as normal, and do not undertake any additional activity. |
| Outcomes | Primary outcomes: Recruitment rates and intervention adherence rates (calculated when intervention delivery period is complete). Missing data rates and retention rates are calculated when follow‐up is complete. Secondary outcomes: Blood markers of inflammation (e.g. IL‐6, CRP) are measured at baseline and 13 week in all participants, and week 7 in exercise group participants. Body mass, cardiorespiratory fitness (ventilatory threshold and peak oxygen uptake), disease symptoms (CDAI), bowel inflammation (faecal calprotectin), resting blood pressure, resting heart rate and waist circumference is determined at baseline and 13 weeks. Health status (EuroQol EQ‐5D‐5L), anxiety and depression (Hospital Anxiety and Depression Scale), fatigue (Inflammatory Bowel Disease Fatigue Scale), quality of life (Inflammatory Bowel Disease Quality of Life Questionnaire) and physical activity (International Physical Activity Questionannaire) is measured using the questionnaire at baseline, 13 and 26 weeks. |
| Notes | Findings from pilot study recently published |
BL: Baseline CCUK: Crohn's & Colitis UK CD: Crohn's disease CDAI: Crohn's Disease Activity Index CDEIS: Crohn's Disease Endoscopic Index of Severity CRP: C‐reactive protein CT: Computed tomography FACIT‐F: Functional Assessment of Chronic Illness Therapy‐Fatigue FC: Faecal calprotectin HADS: Hospital Anxiety and Depression Scale Hb: Haemogloblin HBI: Harvey Bradshaw Index IBD: Inflammatory bowel disease IBDQ: Inflammatory Bowel Disease Questionnaire IBS: Irritable bowel syndrome IL: Interleukin IV: intravenous MD: Doctor of Medicine PHQ15: Patient Health Questionnaire 15 PI: Principal Investigator Q4W: Every 4 weeks
R CT: Randomised controlled trials RZB: Risankizumab SF‐36: 36‐Item Short Form SIBDQ: Short Inflammatory Bowel Disease Questionnaire SQ: Subcutaneous TNF: Tumor necrosis factor UC: Ulcerative colitis UC‐SQ: Ulcerative Colitis Symptom Questionnaire UST: Ustekinumab
Characteristics of ongoing studies [ordered by study ID]
ACTN12617000586314P.
| Trial name or title | A prospective randomised multicentre study to evaluate the effect of intravenous iron infusion compared to oral iron supplementation on the quality of life in inflammatory bowel disease with non‐anaemia hypoferritinanaemia |
| Methods | A phase 4 multicentre RCT |
| Participants | 58 adults (aged 17 ‐ 80 years) with a diagnosis of IBD for at least 3 months, disease in remission for at least 3 months, ferritin equal or less than 30 ug/L and haemoglobin equal or greater than 13 0g/L in males and equal or greater than 120 g/L in females |
| Interventions | Experiment: one off visit for administration of intravenous Ferric Carboxymaltose (Ferrinject) 1000 mg Comparator: oral ferrous sulphate 1 x 325 mg tablet once daily for 6 weeks |
| Outcomes | Quality of life (Shortened IBD Questionnaire and SF‐36 at week 6) Fatigue (IBD‐F at week 6) Disease Activity (HBI and SCCAI at 6 weeks) |
| Starting date | May 2017 |
| Contact information | Principal Investigator: Dr Stephen Inns Hutt Hospital, High Street, Lower Hutt 5010, New Zealand +6445666999 stephen.inns@huttvalleydhb.org.nz Contact person: Dr Sylvia Wu +6445666999 sylvia.wu@huttvalleydhb.org.nz |
| Notes | Anticipated date of last participant enrollment: May 2018 No published data identified in latest search Unable to contact trial authors to determine the status of this trial |
ACTRN12619000150145.
| Trial name or title | Influence of extra virgin olive oil intake on disease activity and gut microbiota profile of community dwelling adults with ulcerative colitis in comparison to healthy subjects |
| Methods | RCT, parallel, open allocation |
| Participants | Individuals with ulcerative colitis (aged >= 18 years) for > 3 months duration of any extent. Non‐IBD subjects (aged >= 18 years) with stable medication and willing to be randomised into one of the two study arms and participate in the intervention prescribed |
| Interventions | Experimental: total replacement of all free dietary fats with extra virgin olive oil (EVOO) Active comparator: usual care, no dietary replacement |
| Outcomes | Primary outcomes: Ulcerative colitis disease activity (partial Mayo scoring Index); gut microbiota variety and richness (faecal samples) Secondary outcomes: Diet quality (3‐day weighted food record submitted and Food Frequency Questionnaire); malnutrition risk (Mini‐Nutritional Assessment); Food Frequency Questionnaire; medication intake; fatty acid profile; polyphenol content; diet Inflammatory Index (3‐day food diary as determined by FoodWorks); plasma oleic acid levels; faecal short chain fatty acid content; plasma hydroxityrosol sulfate (gas chromatography–mass spectrometry); self‐reported symptoms (weekly status check); bone density in the lumbar spine and hip (dual‐energy X‐ray absorptiometry); serum levels of Tumour Necrosis Factor Alpha (enzyme‐linked immunosorbent assays ELISA); blood C‐Reactive Protein; Erythrocyte Sedimentation Rate (sedimentation rate test); faecal IgA (ELISA); blood lipopolysaccharides (LPS); serum levels of folate; serum levels of calcium; Serum levels of vitamin D; Quality of life (IBDQ and SF‐36); Depressive Symptoms (patient health questionnaire ‐ 9); Depression and Anxiety (HADS); Fatigue (IBD‐F); 7‐day physical activity (Axivity MEMS 3‐axis accelerometer); adverse events ( weekly status check); plasma tyrosol (gas chromatography–mass spectrometry); IgA coating bacteria in stool analysis (16S rRNA sequencing); faecal levels of Interleukin (IL) 1, IL6, IL10, IL12 (ELISA); faecal LPS (Pro Q Emerald 300 Gel Staining kit); food‐related quality of life (FR‐QoL‐29) |
| Starting date | April 2019 |
| Contact information | Principal investigator: Prof Maria A. Fiatarone Singh, MD, FRACP University of Sydney Cumberland Campus Building K, Room K221 75 East Street, Lidcombe, 2141 NSW Australia +61 2 9351 9755 maria.fiataronesingh@sydney.edu.au Contact Person: Mr Kenneth Daniel University of Sydney Cumberland Campus Building K, Room K220 75 East Street, Lidcombe, 2141 NSW Australia +61 2 9351‐9138 kdan2775@uni.sydney.edu.au |
| Notes | Anticipated date of data collection: April 2020 Trial forms part of a PhD programme No published data identified in latest search Estimated enrollment: 50 participants |
EudraCT Number: 2008‐004277‐17.
| Trial name or title | The effectiveness and tolerability of GlobiFer (haem iron) tablets compared to ferrous sulphate tablets in inflammatory bowel disease: a randomised‐controlled trial |
| Methods | A phase 2, multicentre RCT |
| Participants | Adolescents and adults (> 12years) with established inactive IBD (defined by CDAI < 150 with normal CRP or SCCAI < 4 for patients with Crohn's colitis or colitis and terminal ileal disease and ulcerative colitis respectively) and iron deficiency anaemia (haemoglobin levels at least 1 g/dL below the sex‐specific lowest value [13 g/dL for men and 12 g/dL for women] and either mean cell volume ≤ 80 fl or ferritin ≤100 g/L or transferrin saturation < 20%) |
| Interventions | Experimental: Oral GlobiFer Forte (iron‐integrated haemoglobin powder equal to 18 mg Fe++) Active comparator: Oral ferrous sulphate 65 mg iron ++ per tablet |
| Outcomes | Primary outcome: 1 g/dL increase in haemoglobin over baseline at 12 weeks Secondary outcomes: Clinical assessment at each visit: FBC, serum ferritin, transferrin saturation, CRP, ESR, disease activity (CDAI or SCCAI); quality of life (IBDQ); fatigue (10‐point visual analogue scale); analysis of faecal microbiota at baseline, week 12, week 24; safety of treatment ‐ symptoms and side effects (weekly diary card); adverse and serious adverse events |
| Starting date | Starting date not reported |
| Contact information | Globifer International bvba, Belgium |
| Notes | No published data identified in latest search The study was initiated but never completed as there were not enough participants. Findings from the data retrieved were not published, however, a small internal report was completed. 21 participants were enrolled (11 with GlobiFer Forte, 10 with ferrous sulphate) and the conclusion was that GlobiFer Forte seemed to be better tolerated than ferrous sulphate. But it was a small number of participants and a non‐homogeneous data situation so difficult to make a strong conclusion (personal correspondence with Globifer International bvba). |
EudraCT Number: 2011‐002122‐43.
| Trial name or title | Iron therapy in IBD patients with normal levels of haemoglobin and chronic fatigue |
| Methods | A multicentre, randomised, placebo‐controlled, double‐blinded trial |
| Participants | Adults (aged > 18 years) with IBD and chronic fatigue (MFI‐20 > 13). At least 12‐month history of IBD. Inactive Crohn's disease or ulcerative colitis (HBI: < 5, Mayo clinical score: ≤ 2) for at least 6 months. Concurrent stable doses of 5‐aminosalicylate, azathioprine, methotrexate or anti‐tumour necrosis factor alpha. Iron deficiency (ferritin < 100 ng/mL or ferritin 100‐300 ng/mL when transferring saturation < 20%) |
| Interventions | Experimental: Ferric carboxymaltose 50 mg/mL Comparator: Placebo |
| Outcomes | Primary outcome: chronic fatigue remission at week 24 (MFI‐20 < 13) Secondary outcomes: clinical response at week 24, defined as decrease in MFI‐20 of at least 4 points, but with a score > 13 |
| Starting date | Not reported |
| Contact information | Instituto Clinico Humanitas, via Manzoni 56, Rozzano, Italy, 20089 Telephone number: 02 82244033 Fax number: 02 82247208 Email: francesco.minuti@humanitas.it |
| Notes | No published data identified in latest search Study is still ongoing ‐ personal correspondence with Francesco Minuti (July 2018) Estimated enrollment: 60 participants |
EudraCT Number: 2012‐005644‐26.
| Trial name or title | Prospective Open label study of Parenteral vs Enteral iron in Young IBD patients and Effect on physical fitness ‐ POPEYE study |
| Methods | A randomised, placebo‐controlled, multicentre trial |
| Participants | 150 children (aged 8 ‐ 18 years) with a diagnosis of CD, UC or IBDU |
| Interventions | Experiment: Ferric Carboxymaltose 50 mg/mL intravenous Comparator: Ferrous Fumarate 100 mg oral |
| Outcomes | Primary outcome: Exercise capacity (6‐minute walk test at week 4, month 3 and month 6) Secondary outcomes: Haemoglobin levels (Hbg at week 4, month 3 and month 6) Quality of life (IMPACT‐III at week 4, month 3 and month 6) Fatigue (PedsQL Multidimensional Fatigue Scale at week 4, month 3 and month 6) Disease activity (PCDAI and PUCAI at week 4, month 3 and month 6) Iron stores (Ht, cell indices, thrombocytes, ferritin, transferrin, serum iron level, transferrin saturation, reticulocytes/retHb , sTfR (soluble transferrin receptor), soluble transferrin receptors to log ferritin (sTfR‐F ratio), transferrin/log ferritin ratio, hepcidin at week 4, month 3 and month 6) Side effects of IV iron therapy on liver functioning (AST, ALT, AF, total protein, albumin at week 4, month 3 and month 6) Side effects on electrolyte homeostasis (phosphate at week 4, month 3 and month 6) Daily Activity and sleep (IV accelerometers at week 4, month 3 and month 6) |
| Starting date | February 2018 |
| Contact information | Dr. Els Van de Vijver Wilrijkstraat 10 Edegem 2650 Belgium 003238215524 els.vandevijver@uza.be |
| Notes | No published data identified in latest search Estimated enrollment: 107 participants |
ISRCTN11470370.
| Trial name or title | Effects of a 6‐month practical resistance training programme on muscle function and bone mineral density in adults with inactive or mildly active Crohn’s disease: study protocol for a randomised controlled trial |
| Methods | A mulitcentre RCT |
| Participants | Adults (aged 16 years and older) diagnosed with Crohn's disease with inactive (CDAI < 150) or mild disease (CDAI 150 ‐ 219) and faecal calprotectin < 250 mcg/g |
| Interventions | Experimental: A six‐month resistance training programme involving a combination of supervised and unsupervised exercise sessions. Comparator: Usual care |
| Outcomes | Primary outcomes: Bone mineral density (dual energy X‐ray absorptiometry); maximum voluntary isometric and isokinetic strength (isokinetic dynamometry); handgrip strength (handgrip dynamometer); lower limb muscle endurance (30‐second chair sit‐to‐stand test); upper limb muscle endurance (30‐s arm bicep curl test) Secondary outcomes: Quality of life (Inflammatory Bowel Disease Quality of Life Questionnaire); health status (EuroQol 5‐dimensions, 5‐level questionnaire); fatigue (IBD‐F); body mass (balance beam scales); stature (stadiometer); disease activity (CDAI); bowel inflammation (faecal calprotectin); blood markers of inflammation; physical activity (Scottish Physical Activity Questionnaire); feasibility and acceptability outcomes |
| Starting date | October 2016 |
| Contact information | Dr Garry Tew Associate Professor of Exercise and Health Sciences Department of Sport Exercise and Rehabilitation Northumbria University Northumberland Building Northumberland Road Newcastle upon Tyne NE1 8ST United Kingdom garry.tew@northumbria.ac.uk |
| Notes | Estimated enrollment: 50 participants No published data identified in latest search Overall trial end date: October 2019 |
NCT02193750.
| Trial name or title | Assessing the tolerability of oligosaccharide supplementation in patients with Crohn's disease: a randomized, controlled trial |
| Methods | RCT |
| Participants | Adults (aged >/= 19 years) with a diagnosis of Crohns disease for >/= 6 months, currently in remission (HBI score </= 4 points and CRP < 5 mg/L) |
| Interventions | Experimental: Moderate Oligosaccharide Group ‐ 1 placebo muesli bar and 1 serving intervention muesli per day (3.25 g total fructans/GOS) Experimental: High Oligosaccharide Group ‐ 1 intervention muesli bar and 1 serving intervention muesli per day (5.43 total fructans/GOS) Comparator: Placebo Group ‐ 1 placebo muesli bar and 1 serving placebo muesli per day (0.55 g total fructans/GOS) |
| Outcomes | Primary Outcome: Overall GI symptoms (VAS in diet diaries) at baseline and study completion Secondary Outcomes: Tolerability (individual gastrointestinal symptoms (abdominal bloating, abdominal pain, gut rumbling, flatulence) quantified by the VAS), fatigue (Fatigue Impact Scale), quality of life (Physical Component Summary and Mental Component Summary), mood (STPI), disease activity, adherence assessment at baseline and study completion |
| Starting date | August 2015 |
| Contact information | Cherry E. Galorport Telephone: 604‐806‐9440 Email: cgalorport@gmail.com PI: Brian Bressler, MD, Division of Gastroenterology, Department of Medicine St. Paul's Hospital, Vancouver, BC Cananda PI: Peter Gibson, MD, Department of Gastroenterology Alfred Hospital, Melbourne, Australia |
| Notes | Estimated study completion date: December 2017 Estimated Enrollment: 48 participants No published data identified in latest search Unable to contact trial authors to determine the status of this trial |
NCT02208310.
| Trial name or title | A randomised controlled trial of high‐dose vitamin D in Crohn's disease |
| Methods | A multicentre RCT |
| Participants | Adults (aged 18‐75 years) with a diagnosis of Crohn's disease and vitamin D deficiency or insufficiency (serum 25‐hydroxyvitamin D < 30 ng/mL) |
| Interventions | Experimental: High dose vitamin D ‐ cholecalciferol 10,000 IU daily for 30 days. At that point, if their vitamin D levels remain below 50 ng/mL, the 30‐day course will be repeated. For participants who enrol in the summer, levels will be rechecked in March and if < 50 ng/mL, a 30‐day course will be administered. Active comparator: Low dose vitamin D ‐ cholecalciferol 400 IU once daily for 30 days. To maintain the blind, a random few will be given another round at the 30‐day mark. For participants who enrol in the summer, a random few will again receive 400 IU cholecalciferol in March. |
| Outcomes | Primary outcomes: Primary composite outcome at year 1 ‐ Crohn's disease(CD)‐related hospitalisations, CD‐related surgeries, CD‐related ER visits, steroid prescriptions; hypercalcaemia at year 1 (calcium > 10.8 mg/dL); incidence of nephrolithiasis associated with hypercalcaemia at year 1 (documented by imaging) Secondary outcomes: Crohn's related hospitalisation at year 1; steroid prescriptions at year 1; Crohn's disease‐related surgeries at year 1; change in modified HBI at year 1; change in CRP at year 1; change in fecal calprotectin at year 1 ; percent with escalation of therapy at year 1; quality of life measure changes at year 1 (IBDQ and CD‐PRO); change in fatigue measurements at year 1 (FACIT‐F); Crohn's related ED visits at year 1 |
| Starting date | April 2015 |
| Contact information | Principal Investigator: Peter Higgins MD, PhD, MSc, University of Michigan Contact name: Kelli Porzondek Telephone number: 007346470507 Email: kporzond@med.umich.edu |
| Notes | No published data identified in latest search. The clinical trial registers webpage (https://clinicaltrials.gov/ct2/show/NCT02208310) stated that the study has been terminated, unable to enroll at rate anticipated, insufficient low vitamin D in clinical remission at 5 sites. Actual enrollment: 11 participants |
NCT02517151.
| Trial name or title | Effects of iron therapy in patients with chronic fatigue and IBD |
| Methods | A phase 2, randomised, placebo‐controlled trial |
| Participants | Adults (aged 18 ‐ 75 years) with a diagnosis of Crohn's disease or ulcerative colitis for a least 6 months prior to day 1 by endoscopy and/or imaging. At least 6 months of clinical remission (HBI ≤ 5; Mayo clinical score ≤ 2). Therapy with mesalamine, immunosuppressors or anti‐tumour necrosis factor alpha at stable doses for at least 3 months prior to enrollment; steroids are not permitted from 6 months prior to baseline. Chronic fatigue symptoms (MFI‐20 > 13). Iron deficiency (ferritin < 100 microg/L or < 300 microg/L in case of transferrin‐iron saturation percentage < 20% |
| Interventions | Experimental: Ferric carbxymaltose 200 mg in normal saline 100 mL administered IV at day 0 and then every 4 weeks up to week 24 Comparator: Placebo ‐ normal saline 100 mL administered IV at day 0 and then every 4 weeks up to week 24 |
| Outcomes | Primary outcome: chronic fatigue remission at week 24 (MFI‐20 < 13) Secondary outcomes: chronic fatigue reduction at week 24 (MFI‐20 reduction of at least 4 points. Absolute MFI‐20 > 13) Chronic fatigue remission at week 12 (MFI‐20 < 13) Anxiety evaluation (State‐Trait Anxiety Inventory (STAI) at week 4, week 12, week 24) Depression evaluation (BDI‐II at week 4, week 12, week 24) Quality of life (IBDQ at week 4, week 12, week 24) |
| Starting date | October 2014 |
| Contact information | Silvio Danese, MD, PhD IBD Center, Rozzano, MI, Italy, 20089 Telephone number: 0039028224 ext 5555 Email: IBDclinicaltrials@humanitas.it or sdanese@hotmail.com |
| Notes | No published data identified in latest search Study is still ongoing (personal correspondence with Dr. Danese July 2018) Estimated enrollment: 36 participants |
NCT02704624.
| Trial name or title | The impact of serum vitamin D and calcium levels on the body composition, bone mineral density, muscle strength, exercise tolerance, fatigue and inflammatory activity in patients with Crohn's disease: a randomized controlled trial |
| Methods | A single‐centred randomised, placebo‐controlled, double‐blinded trial |
| Participants | 110 adults (aged 18 ‐ 50 years) with a clinical diagnosis of Crohn's disease, with remission of Crohn's activity and reduced blood levels of vitamin D |
| Interventions | Experimental: Tablets with 50000 IU cholecalciferol (vitamin D3) will be administered, weekly, for six months and nutritional instructions from an experienced nutritionist regarding the consumption of vitamin D and calcium rich foods. Placebo Comparator: The participants selected for the placebo group will receive inert content tablets without therapeutic effect, weekly, for six months. |
| Outcomes | Primary outcome: Grip strength after 6 months of supplementation with vitamin D Secondary outcomes: Mineral bone density (DXA at 6 months) Fecal calprotectin levels (at 6 months) Inflammatory biomarkers (TNF‐α; IL6; IL17; CRP at 6 months) Exercise capacity (shuttle walk test at 6 months) Lean body mass (DXA at 6 months) Fatigue (Chalder Fatigue Questionnaire at 6 months) |
| Starting date | December 2016 |
| Contact information | Lorena NO Pinto, Lecturer Universidade Federal de Juiz de Fora, Juiz de Fora, Minas Gerais, Brazil Telephone Number: +55‐32‐98490‐8718 Email: lorenanagme@gmail.com Carla Malaguti, Professor Universidade Federal de Juiz de Fora, Juiz de Fora, Minas Gerais, Brazil Telephone Number: +55‐32‐99199‐3329 Email: carlamalaguti@gmail.com Principal Investigator: Júlio MF Chebli, Professor Federal University of Juiz de Fora |
| Notes | No published data identified in latest search Estimated study completion date: February 2020 Estimated enrollment: 110 participants |
NCT02707068.
| Trial name or title | Quality Of LIfe Tool for IBD (QOLITI): pilot testing of a self‐administered intervention to target psychological distress in inflammatory bowel disease |
| Methods | A single‐centred randomised, placebo‐controlled, open trial |
| Participants | 62 adults (aged 18 years and older) with a clinical diagnosis of inflammatory bowel disease |
| Interventions | Experimental: QOLITI Intervention group receives the QOLITI ('Quality Of LIfe Tool for IBD') manual immediately to work with over the course of several weeks along with 3 x 30 minutes of telephone support by a trained healthcare professional. Telephone calls will occur at two, four and six weeks post‐randomisation. Comparator: Waiting‐List Control group (WLC) Waiting‐list control group waits until after the study finishes to receive the same manual, but without telephone support sessions. |
| Outcomes | Primary outcomes: Depression (Patient Health Questionnaire ‐ 9 at 10 weeks post‐intervention) Anxiety (Generalised Anxiety Disorder ‐ 7‐item scale at 10 weeks post‐intervention) Generic quality of life (EQ‐5D‐5L at 10 weeks post‐intervention) Specific quality of life (IBDQ at 10 weeks post‐intervention) Secondary outcomes: Semi‐structured qualitative Interviews: Participants will be invited to discuss their experiences after the end of the actual study. These interviews are no obligatory part of the QOLITI study. Fatigue (Chalder Fatigue Scale at 10 weeks post‐intervention) Illness perception (Illness Preception Questionnaire at 10 weeks post‐intervention) Disease activity (Patient‐Modified Simple Clinical Colitis Activity Index or Crohns Disease Activity Index for research surveys) |
| Starting date | January 2016 |
| Contact information | Lyndsay D Hughes, PhD Kings College London Email: Lyndsay.hughes@kcl.ac.uk |
| Notes | The study has been completed however no fatigue data published yet (personal correspondence July 2018). Actual enrollment: 62 participants |
NCT02772965.
| Trial name or title | A randomized, double‐blind, placebo‐controlled, multi‐center pragmatic clinical trial to evaluate the effectiveness of low dose oral methotrexate In patients with pediatric Crohn's disease initiating anti‐TNF therapy |
| Methods | A randomised, double‐blind, placebo‐controlled, multicentre trial |
| Participants | 425 paediatric Crohn's disease (PCD) patients, < 21 years of age, ≥ 20 kg, initiating anti‐TNF therapy with infliximab or adalimumab (including biosimilars) |
| Interventions | Experiment: Methotrexate (10, 12.5, or 15 mg), once weekly. Weight‐based dosing. Ondansetron (4 mg), twice weekly, 1 hour prior to methotrexate dose and the morning after methotrexate dose. Folic Acid (1 mg) daily Comparator: Placebo (sugar pill) for methotrexate, once weekly. Placebo for ondansetron, twice weekly, 1 hour prior to methotrexate placebo dose and the morning after methotrexate placebo dose. Folic Acid (1 mg) daily |
| Outcomes | Primary outcomes Time to treatment failure (Short Pediatric Crohn's Disease Activity Index (SPCDAI) through week 104); hospitalisation for active irritable bowel disease or abdominal surgery after week 25; use of oral prednisone or prednisolone, enteral release budesonide, or intravenous (IV) methylprednisolone for over 10 weeks cumulatively, beyond week 16 (not inclusive of steroids used as premed for anti‐TNF administration or steroids used for conditions other than CD); or discontinuation of anti‐TNF or study drug for lack of effectiveness or toxicity) Secondary outcomes: Pain (patient‐reported outcome measurement and Information system pain interference T score at week 52 and 104) Fatigue (patient‐reported outcome measurement and information system fatigue T score at week 52 and 104) Positive anti‐TNF antibody status (between week 91 and week 104) |
| Starting date | October 2016 |
| Contact information | Michael D Kappelman, MD, University of North Carolina, Chapel Hill |
| Notes | Estimated study completion date: December 2020 Estimated enrollment: 425 participants |
NCT02849717.
| Trial name or title | Pre‐habilitation exercise intervention for patients scheduled for colorectal surgical resection |
| Methods | RCT |
| Participants | Adults (18 years and older) with a primary diagnosis of colon/rectal cancer or IBD or diverticular disease who are scheduled for elective (non‐emergent) surgery. Patients with approval from their treating physician, study physician, or physician's designee to participate in maximal physiological fitness testing and a low‐to‐moderate home‐based walking and progressive resistance exercise programme and those who are able to read English are eligible to participate. |
| Interventions | Experiment: A standardised, daily, home‐based, progressive exercise programme (walking and resistance exercise treatment) Comparator: Standard care |
| Outcomes | Primary outcome: Fatigue (FACIT‐F fatigue subscale) at baseline and 12 weeks Secondary Outcomes: aerobic capacity (VO2 maximum testing), skeletal muscle mass (bio‐electrical impedance assessment of muscle mass and CT assessment of muscle mass), pro‐inflammatory cytokines (IL‐6, IL‐8,IL‐10, IL‐1B,and IFN‐y), TNFr1 cytokine receptor, postoperative complications (Clavien‐Dindo classification of surgical complications), quality of life (Profile of Mood States and Spielberg State/Trait Anxiety Inventory) at baseline and 12 weeks |
| Starting date | Pilot trial start date: March 2016 |
| Contact information | PI Fergal Fleming, MD from the University of Rochester fergal.fleming@urmc.rochester.edu) |
| Notes | This study will be eligible to be included in future updates of the review if sufficient numbers of IBD participants are included to conduct a subgroup analysis (personal correspondence with the PI Fergal Fleming, MD from the University of Rochester ‐ fergal.fleming@urmc.rochester.edu). Data collection ongoing (personal correspondence July 2018) |
NCT02861053.
| Trial name or title | Inflammatory bowel disease: could a regular physical activity reduce patients fatigue? |
| Methods | A single‐centre RCT |
| Participants | Adults (18 ‐ 45 years old) with IBD in remission (CRP < 5) with fatigue (FACIT score < 30). Patients with haemoglobin dosage > 10 g/dL, BMI > 18 and < 30kg/m2, women with no risk of pregnancy (menopausal women or with contraceptive drugs), affiliated to the social security and living not far from the centre where the rehabilitation programme will be performed, are eligible to be included. |
| Interventions | Experimental: Regular and moderate physical activity will be done 3 times per week more than usual Sham Comparator: No regular and moderate physical activity more than usual |
| Outcomes | Primary Outcome: Fatigue (FACIT) at baseline and 3 months Secondary Outcome: Quality of life (IBDQ and SF‐36) at baseline and 3 months |
| Starting date | September 2016 |
| Contact information | David Debeaumont, MD Rouen University Hospital Rouen, France Email: david.debeaumont@chu‐rouen.fr Julien Blot ‐ julien.blot@chu‐rouen.fr |
| Notes | Estimated study completion date: April 2018 Estimated enrollment: 10 participants No published data identified in latest search Unable to contact trial authors to determine the status of this trial |
NCT02891226.
| Trial name or title | A phase 2, multicentre, randomized, parallel‐arm, placebo‐controlled study of LY3074828 in subjects with active Crohn's disease (SERENITY) |
| Methods | A Phase 2, multicentre, randomised, parallel‐arm, placebo‐controlled trial |
| Participants | Adults (aged 18 ‐75 years) with active Crohn's disease (SES‐CD and participant‐reported stool frequency and abdominal pain) and inadequate response or failure to tolerate at least one of the following: aminosalicylates; budesonide; systemic corticosteroids; immunosuppressants (e.g. azathioprine, 6‐mercaptopurine, or methotrexate); or prior exposure to biologics for the treatment of CD |
| Interventions | Experimental: Mirikizumab dose level 1 Period 1 (Weeks 0 ‐12) Mirikizumab dose level 1 Period 2 (Weeks 12 ‐ 52) Mirikizumab dose level 1 or dose level 4 or dose level 3 Period 3 (Weeks 52 ‐ 104) Mirikizumab dose level 4 Experimental: Mirikizumab dose level 2 Period 1 (Weeks 0 ‐12) Mirikizumab dose level 2 Period 2 (Weeks 12 ‐ 52) Mirikizumab dose level 2 or dose level 4 or dose level 3 Period 3 (Weeks 52 ‐ 104) Mirikizumab dose level 4 Experimental: Mirikizumab dose level 3 Period 1 (Weeks 0 ‐12) Mirikizumab dose level 3 Period 2 (Weeks 12 ‐ 52) Mirikizumab dose level 3 or dose level 4 Period 3 (Weeks 52 ‐ 104) Mirikizumab dose level 4 Comparator: Placebo Period 1 (Weeks 0 ‐12) Placebo Period 2 (Weeks 12 ‐ 52) Mirikizumab dose level 3 Period 3 (Weeks 52 ‐ 104) Mirikizumab dose level 4 |
| Outcomes | Primary outcome: Endoscopic disease activity (Simple Endoscopic Activity Score‐Crohn's Disease (SES‐CD)) at baseline and week 12 Secondary outcomes: Discontinuation rate and pharmacokinetics (area under the curve Mirikizumab) at baseline through week 104. Endoscopic remission, patient‐reported outcome remission, patient global rating (severity and change (PGR‐S and PGR‐C) Crohn's disease score), quality of life (IBDQ and SF‐36), fatigue (FACIT‐F) at baseline and week 12 |
| Starting date | December 2016 |
| Contact information | Eli Lilly and Company Telephone: 1‐877‐CTLILLY (1‐877‐285‐4559) or 1‐317‐615‐4559 |
| Notes | Estimated study completion date: April 2021 Estimated enrollment: 180 participants |
NCT02963246.
| Trial name or title | Effects of a mindfulness therapy intervention for individuals with inflammatory bowel disease: a randomized controlled trial |
| Methods | RCT single‐blinded |
| Participants | Adults (18 ‐55 years) with a diagnosis of inflammatory bowel disease in remission (non‐active disease symptoms in the last 3 months). Participants with access to the internet and basic informatics knowledge are eligible to be included. |
| Interventions | Experimental: Mindfulness‐Based Cognitive Therapy (psychological programme designed to help manage depressive and stress symptoms) for 12 months Comparator: Treatment‐as‐usual control |
| Outcomes | Primary Outcome: Quality of life (IBDQ‐32) at baseline and 12 months Secondary Outcome: Inflammation stress markers (CRP and FC) at baseline and 12 months |
| Starting date | February 2017 |
| Contact information | Dr. Jose Miguel Soria Lopez (PI) Professor, Cardenal Herrera University Contact: Dr. Xavier Cortés or Dr Juan F Lisón Telephone: 686774074 or 606503108 Email: xavier.cortes@uchceu.es orjuanfran@uchceu.es |
| Notes | Eligible only if analysis of the subcomponents of IBDQ is undertaken Estimated study completion date: March 2018 Actual enrollment: 60 participants No published data identified in latest search Unable to contact trial authors to determine the status of this trial |
NCT03104413.
| Trial name or title | A multicenter, randomized, double‐blind, placebo‐controlled induction study to assess the efficacy and safety of risankizumab in subjects with moderately to severely active Crohn's disease who failed prior biologic treatment |
| Methods | A multicentre, randomised, placebo‐controlled, double‐blinded trial |
| Participants | Patients (aged 16 ‐ 80 years ‐ Where locally permissible, subjects 16 to < 18 years of age who meet the definition of Tanner stage 5 for development at the baseline visit) with a diagnosis of Crohn's disease for at least 3 months prior to baseline. A confirmed diagnosis of moderate‐to‐severe CD as assessed by stool frequency (SF), abdominal pain (AP) score, and Simple Endoscopic Score for Crohn's Disease (SES‐CD). Demonstrated intolerance or inadequate response to biologic therapy for CD. If female, subject must meet the contraception recommendations. |
| Interventions | Placebo Comparator: Placebo (induction period 1) ‐ Subjects randomised to receive placebo for risankizumab in induction period 1. Experimental: Risankizumab dose 1 and dose 2 in induction period 1 Experimental: Risankizumab dose 1 (induction period 2): Participants who received placebo in period 1 and participants with inadequate response at week 12 in period 1 randomised to receive risankizumab dose 1 administered by intravenous (IV) infusion in period 2. Experimental: Risankizumab dose 2 (induction period 2) Participants with inadequate response at week 12 in period 1 randomised to receive risankizumab dose 2 administered by subcutaneous (SC) injection in period 2. Experimental: Risankizumab dose 3 (induction period 2) Participants with inadequate response at week 12 in period 1 randomised to receive risankizumab dose 3 administered by subcutaneous (SC) injection in period 2. |
| Outcomes | Primary outcomes: Clinical remission (average daily SF and average daily AP at week 12) Endoscopic response (SES‐CD at week 12) Secondary outcomes: Clinical response (average daily SF and average daily AP at week 4 and week 12) Clinical remission (CDAI at week 4 and week 12) Clinical response and endoscopic response (at week 12) Endoscopic healing (SES‐CD at week 12) Severity of Crohn's disease symptoms (Crohn's Symptom Severity at week 12) Resolution of extra‐intestinal manifestation (at week 12) Hospitalisation (through to week 12) Draining fistula (at week 12) Fatigue (FACIT‐F at week 12) Overall health status (SF‐36 at week 12) Crohn's disease‐related surgeries (through to week 12) |
| Starting date | June 2017 |
| Contact information | AbbVie Call Center Telephone: 8472838955 Email: abbvieclinicaltrials@abbvie.com |
| Notes | Estimated study completion date: June 2019 Estimated enrollment: 579 participants |
NCT03105102.
| Trial name or title | A multicenter, randomized, double‐blind, placebo controlled 52‐week maintenance and an open‐label extension study of the efficacy and safety of risankizumab in subjects with Crohn's disease who responded to induction treatment in M16‐006 or M15‐991 |
| Methods | The study consists of 3 substudies, as follows:
|
| Participants | Adults (16 ‐ 80 years) with Crohn's disease who have completed study M16‐006 or study M15‐991 and have achieved clinical response |
| Interventions | Experimental: Double‐blind risankizumab dose 1 (substudy 1) ‐ Participants randomised to receive double‐blind risankizumab dose 1 for 52 weeks Experimental: Double‐blind risankizumab dose 2 (substudy 1) ‐ Participants randomised to receive double‐blind risankizumab dose 2 for 52 weeks. Placebo Comparator: Double‐blind placebo for risankizumab (substudy 1) ‐ Participants randomised to receive double‐blind placebo for risankizumab for 52 weeks Experimental: Maintenance risankizumab dose 1 (substudy 2) ‐ Participants randomised to receive 1 dose of double‐blind risankizumab dose 1 followed by open‐label risankizumab for 52 weeks Experimental: Maintenance risankizumab dose 2 (substudy 2) ‐ Participants randomised to receive 1 dose of double‐blind risankizumab dose 2 followed by open‐label risankizumab for 52 weeks Experimental: Open‐label risankizumab (substudy 3) ‐ Participants who completed substudy 1 or substudy 2 will receive open‐label risankizumab beginning at Week 56 |
| Outcomes | Primary outcomes: Clinical remission (average daily SF and average daily AP at week 52) Endoscopic response (SES‐CD at week 52) Secondary outcomes: Clinical remission (CDAI at week 52) Discontinued corticosteroid use and achieved clinical remission (average daily SF and average daily AP score at Week 52) Discontinued corticosteroid use (at week 52) Sustained clinical remission (average daily SF and average daily AP score at both week 0 and week 52) Enhanced clinical response (decrease in average daily SF and/or decrease in average daily AP score, and/or clinical remission per average daily SF and average daily AP score at week 52) Clinical remission (average daily stool frequency and average daily abdominal pain score) and endoscopic response (SES‐CD at week 52) Endoscopic healing (SES‐CD at week 52) Severity of Crohn's disease symptoms (Crohn's Symptom Severity at week 52) Resolution of extra‐intestinal manifestation (at week 52) Deep remission, both clinical remission (per average daily SF and average daily AP score) and endoscopic healing (SES‐CD at week 52) Hospitalisation (through to week 52) Draining fistula (at week 52) Fatigue (FACIT‐F at week 52) Overall health status (SF‐36 at week 12) Crohn's disease‐related surgeries (through to week 12) |
| Starting date | September 2017 |
| Contact information | AbbVie Call Center Telephone: 8472838955 Email: abbvieclinicaltrials@abbvie.com |
| Notes | Estimated completion date: September 2022 Estimated enrollment: 912 participants |
NCT03105128.
| Trial name or title | A multicenter, randomized, double‐blind, placebo controlled induction study of the efficacy and safety of risankizumab in subjects with moderately to severely active Crohn's disease |
| Methods | A multicentre, randomised, double‐blind, placebo‐controlled induction trial |
| Participants | Adults (aged 16 ‐ 80 years ‐ where locally permissible, subjects 16 to < 18 years of age who meet the definition of Tanner stage 5 for development at the baseline visit) with a diagnosis of Crohn's disease for at least 3 months prior to baseline. Confirmed diagnosis of moderate‐to‐severe Crohn's disease assessed by stool frequency, abdominal pain score and simple endoscopic score for Crohn's disease (SES‐CD). Demonstrated intolerance or inadequate response to conventional or to biologic therapy for Crohn's disease. If female, subject must meet the contraception recommendations. |
| Interventions | Placebo Comparator: Placebo (period 1)‐ Participants randomised to receive placebo for risankizumab administered by intravenous (IV) infusion Experimental: Risankizumab dose 1 (period 1) ‐ Participants randomised to receive risankizumab dose 1 administered by intravenous (IV) infusion Experimental: risankizumab dose 2 (period 1) ‐ Participants randomised to receive risankizumab dose 2 administered by intravenous (IV) infusion Experimental: risankizumab dose 1 (period 2) ‐ Participants who received placebo in period 1 and participants with inadequate response at week 12 in period 1 randomised to receive risankizumab dose 1 administered by intravenous (IV) infusion in period 2 Experimental: Risankizumab dose 2 (period 2) ‐ Participants with inadequate response at week 12 in period 1 randomised to receive risankizumab dose 2 administered by subcutaneous (SC) injection in period 2 Experimental: Risankizumab dose 3 (period 2) ‐ Participants with inadequate response at week 12 in period 1 randomised to receive risankizumab dose 3 administered by subcutaneous (SC) injection in period 2 |
| Outcomes | Primary outcomes: Clinical remission (average daily SF and average daily AP at week 12) Endoscopic response (SES‐CD at week 12) Secondary outcomes: Clinical response (average daily SF and average daily AP score at week 4 and week 12) Clinical remission (CDAI at week 12) Clinical remission (average daily SF and average daily AP score at week 4) Clinical response and endoscopic response (at week 12) Endoscopic healing (SES‐CD at week 12) Severity of Crohn's disease symptoms (Crohn's Symptom Severity at week 12) Resolution of extra‐intestinal manifestation (at week 12) Hospitalisation (through to week 12) Draining fistula (at week 12) Fatigue (FACIT‐F at week 12) Overall health status (SF‐36 at week 12) Crohn's disease‐related surgeries (through to week 12) |
| Starting date | May 2017 |
| Contact information | AbbVie Call Center Telephone: 8472838955 Email: abbvieclinicaltrials@abbvie.com |
| Notes | Estimated completion date: February 2020 Estimate enrollment: 940 participants |
NCT03107793.
| Trial name or title | Study of treat to target versus routine care maintenance strategies in Crohn's disease patients treated with ustekinumab |
| Methods | RCT |
| Participants | Adults (aged 18 years and older) with active moderate‐to‐severe Crohn's disease (CDAI score >/= 220 and </= 450 and endoscopic evidence of active Crohn's disease (SES‐CD score >/= 3, excluding the contribution of the narrowing component score). Patient who has had an inadequate response with, lost response to, was intolerant to, or had medical contraindications to either conventional therapy, or one previous biologic therapy approved for the treatment of Crohn's disease and are eligible according to tuberculosis (TB) infection screening criteria can participate. |
| Interventions | Experimental: All participants At week (wk) 0, all eligible participants will initiate intravenous (IV) induction treatment with ustekinumab (UST) on a weight‐tiered basis at a dose of approximately 6 milligram per kilogram (mg/kg). At week 8, all participants will receive a 90 milligram (mg) subcutaneous (SC) injection of ustekinumab. At week 16, participants who do not achieve a Crohn's Disease Activity Index (CDAI) improvement of greater than or equal to (>=) 70 points versus week 0 (CDAI 70) will leave the study. Remaining participants will be randomised in a 1:1 ratio to either one of two arms for open‐label maintenance treatment up to week 48: the treat‐to‐target arm or the routine care arm. Experimental: Routine care arm In the routine care arm, assessment visits will be scheduled according to the timing of maintenance treatment injections, which will be in compliance with the EU Summary of Product Characteristics for ustekinumab for the treatment of Crohn's disease, in which dosing every 12 weeks is recommended. At week 16, (that is, 8 weeks after the first SC dose) participants continuing in the study will have demonstrated a CDAI‐70 response. Nonetheless, participants who have not shown adequate response based on the investigator's judgement may receive a second SC dose at week 16. During the routine care maintenance treatment period, in case of clinical worsening reported by the participant, consistent with disease flare in the investigator's judgement, clinical assessments of disease flare will be performed at the investigator's discretion. Experimental: Treat‐to‐Target (T2T) arm UST maintenance treatment assignment will be based on centrally‐read colonoscopy (at wk 16). Participants with < 25% improvement in SES‐CD score at wk16 will be assigned to Q8 (8‐weekly) treatment and will receive UST 90 mg SC at wk 16. In contrast, participants with >= 25% improvement in SES‐CD score at wk 16 will be assigned to Q12 treatment and will receive the next UST dose (90 mg SC) at wk 20. At assessment visits (from wk 24 for participants assigned to the Q8 regimen or from wk 20 for the Q12 group) UST maintenance treatment will be directed by T2T assessments. Participants meeting the target will continue on the same UST dosing frequency. The dosing frequency will be optimised for all participants failing to meet the target at the assessment visit. Those previously on Q12 regimens will be adjusted to Q8 dosing; those previously on Q8 regimens will be adjusted to Q4 dosing. Participants subsequently failing to meet the target will not be able to adjust further and will leave the study. |
| Outcomes | Primary Outcome: Endoscopic response (reduction from baseline in simple endoscopic score for Crohn's disease (SES‐CD) of >/= 50%) at week 48 Secondary Outcomes: Endoscopic remission (SES‐CD score </= 2), mucosal healing (complete absence of mucosal ulcerations in any ileocolonic segment), clinical remission (CDAI score < 150 points), clinical response (>/= 100‐point reduction from the baseline CDAI score, or a CDAI score < 150), corticosteroid‐free clinical remission (CDAI score < 150), corticosteroid‐free endoscopic response (a reduction from baseline in SES‐CD score of >= 50%), inflammation (serum CRP and fecal calprotectin), quality of life (IBDQ), work productivity (Work Productivity and Activity Impairment Questionnaire score), health‐related quality of life (European Quality Of Life 5 Dimensions 5 Level), fatigue (FACIT‐F), depression and anxiety (Hospital Anxiety and Depression Scale), time lost from work (number of days lost from work due to Crohn's disease), adverse events at baseline and week 48 |
| Starting date | April 2017 |
| Contact information | Janssen Cilag Ltd. 844‐434‐4210 JNJ.CT@sylogent.com |
| Notes | Estimated study completion date: April 2019 Estimated enrollment: 650 participants |
NCT03162575.
| Trial name or title | The possible beneficial effects of mindfulness‐based cognitive therapy (MBCT) in fatigued adult patients with Inflammatory Bowel Disease (IBD) |
| Methods | RCT |
| Participants | Adults (18 ‐ 75 years) with a diagnosis of Crohn's disease and ulcerative colitis in remission with severe fatigue (subscale 'subjective fatigue' of the CIS (8 items) ≥ 35). Participants with no expectation of a surgery in the upcoming 3 months, able to attend 8 weekly group sessions of 2.5 hours in the hospital, and able to read, write and speak Dutch are eligible to participate. |
| Interventions | Experimental: The intervention consists of 8 weekly sessions of Mindfulness‐Based Cognitive Therapy. Each session will be administered in a group and will last 2.5 hours. Comparator: The waiting‐list control group will receive no intervention for three months and afterwards will receive Mindfulness‐Based Cognitive Therapy. |
| Outcomes | Primary Outcome: Fatigue (Checklist Individual Strength (CIS‐20) at baseline, 3, 6 and 12 months
Secondary Outcomes: Fatigue interference (Fatigue Symptom Inventory), anxiety (Generalised Anxiety Disorder Assessment (GAD 7)), depression (Beck Depression Inventory‐II (BDI‐II)), IBD‐specific quality of life (IBDQ), sleep quality (Pittsburgh Sleep Quality Index), labour participation (assessed with several questions) at baseline, 3, 6 and 12 months. Satisfaction with treatment (assessed with several questions) at 3 months |
| Starting date | January 2017 |
| Contact information | Dr. Annika Tovote University Medical Center Groningen, Groningen, Netherlands Telephone: 0031(0)503632955 Email: k.a.tovote@umcg.nl |
| Notes | Estimated study completion date: February 2019 Estimated enrollment: 128 participants |
NCT03266484.
| Trial name or title | Effect of dietary therapy with a probiotic mixture on the gut microbiome and fatigue symptoms in patients with quiescent inflammatory bowel disease ‐ a clinical trial |
| Methods | A randomised, double‐blind, placebo‐controlled trial |
| Participants | 100 adults (aged 18 ‐ 75 years) with a confirmed diagnosis of CD, UC or IBD‐unspecified, quiescent disease and persistent ongoing fatigue symptoms |
| Interventions | Experiment: Probiotic mixture taken twice a day for 12 weeks. Comparator: Placebo |
| Outcomes | Primary outcomes: Gut microbiome (16sRNA sequencing at week 12) Serum inflammatory cytokines levels (Biosciences Cytometric Bead Array kits at week 12) Metabolomic profiles (quantitative and semi‐quantitative gas chromatography‐mass spectrometry and liquid chromatography‐mass spectrometry methods at week 12) Fatigue (FACIT‐F at week 4 and week 12) |
| Starting date | November 2017 |
| Contact information | Principal Investigator: Ashwin Ananthakrishnan Assistant Professor of Medicine, Massachusetts General Hospital Contact: Nynke Borren, MD 617‐726‐1997 nborrent@mgh.harvard.edu Contact Christine Wong 617‐724‐7559 cywong4@mgh.harvard.edu |
| Notes | Estimated study completion date: December 2019 Estimated enrollment: 100 participants |
NCT03345823.
| Trial name or title | A multicenter, randomized, double‐blind, placebo‐controlled maintenance and long‐term extension study of the efficacy and safety of upadacitinib (ABT‐494) in subjects with Crohn's disease who completed the studies M14‐431 or M14‐433 |
| Methods | A multicentre, randomised, double‐blind, placebo‐controlled maintenance and long‐term extension study |
| Participants | 738 participants who receive double‐blind treatment in Study M14‐431 or Study M14‐433 and achieve clinical response, and complete study procedures in the parent study. |
| Interventions | Experimental: Group A ‐ Arm B: This is a maintenance group with 52 weeks which includes participants who achieved clinical response to upadacitinib dose A in studies M14‐431 and M14‐433 and will receive dose C. Experimental: Group B ‐ Arm C: This is a long‐term extension group with 240 weeks which includes participants who complete group A. Experimental: Group B ‐ Arm A: This is a long‐term extension group with 240 weeks which includes participants who complete group A and will receive dose B. Experimental: Group B ‐ Arm B: This is a long‐term extension group with 240 weeks which includes participants who complete group A and will receive dose C. Experimental: Group A ‐ Arm A: This is a maintenance group with 52 weeks which includes participants who achieved clinical response to upadacitinib dose A in studies M14‐431 and M14‐433 and will receive dose B. |
| Outcomes | Primary outcomes: Clinical remission (average daily stool frequency and average daily abdominal pain score at week 52) Endoscopic response (SES‐CD at week 52) Secondary outcomes: Endoscopic remission (SES‐CD at week 52) Fatigue (FACIT‐F at week 52) Enhanced Clinical Response (decrease in average daily SF and/or decrease in average daily AP score at week 52) Quality of life (SF‐36 at week 52) Hospitalisations due to CD (at week 52) Discontinuation of corticosteroid use for CD and achieve clinical remission (at week 52) Clinical remission (average daily stool frequency and average daily abdominal pain (AP) score at week 52) Clinical remission (CDAI < 150 at week 52) Endoscopic remission (SES‐CD at week 52) Draining fistulas (at week 52) Reduction in draining fistulas (>= 50% at week 12) Crohn's Symptoms Severity Questionnaire (CSS at week 52) Discontinuation of corticosteroid use for CD (at week 52) |
| Starting date | March 2018 |
| Contact information | AbbVie |
| Notes | Estimated study completion date: March 2023 Estimated enrollment: 738 participants |
NCT03345836.
| Trial name or title | A study of the efficacy and safety of upadacitinib (ABT‐494) in subjects with moderately to severely active Crohn's disease who have inadequately responded to or are intolerant to biologic therapy |
| Methods | A multicentre, randomised, double‐blind, placebo‐controlled induction study |
| Participants | 855 adults (aged 18 ‐ 75 years) with a confirmed diagnosis of Crohn's disease for at least 3 months prior to baseline, moderate‐to‐severe disease activity with evidence of mucosal inflammation and previous inadequate response or intolerance to any biologic therapy |
| Interventions | Experimental:upadacitinib dose A (oral; once daily) for 12 weeks or open‐label upadacitinib dose A (oral; once daily) for 12 weeks. Comparator: matching placebo for upadacitinib |
| Outcomes | Primary outcomes: Clinical remission (average daily stool frequency and average daily abdominal pain score at week 12) Endoscopic response (SES‐CD at week 12) Secondary outcomes: Fatigue (FACIT‐F at week 12) Enhanced Clinical Response (decrease in average daily SF and/or decrease in average daily AP score at week 2) Quality of life (SF‐36 at week 12) Hospitalisations due to CD (at week 12) Clinical remission (average daily stool frequency and average daily abdominal pain (AP) score at week 12) Clinical remission (CDAI < 150 at week 12). Endoscopic remission (SES‐CD at week 12) Reduction in draining fistulas (>= 50% at week 12) Crohn's Symptoms Severity Questionnaire (CSS at week 12) Discontinuation of corticosteroid use for CD (at week 12) |
| Starting date | November 2017 |
| Contact information | AbbVie Call centre Telephone: 847.283.8955 Email: abbvieclinicaltrials@abbvie.com |
| Notes | Estimated study completion date: March 2020 Estimated enrollment: 645 participants |
NCT03345849.
| Trial name or title | A multicenter, randomized, double‐blind, placebo‐controlled induction study of the efficacy and safety of upadacitinib (ABT‐494) in subjects with moderately to severely active Crohn's disease who have inadequately responded to or are intolerant to conventional therapies but have not failed biologic therapy |
| Methods | A multicentre, randomised, double‐blind, placebo‐controlled induction study |
| Participants | 300 adults (aged 18 ‐ 75 years) with a confirmed diagnosis of Crohn's disease for at least 3 months prior to baseline, moderate‐to‐severe disease activity with evidence of mucosal inflammation and previous inadequate response or intolerance to conventional therapies |
| Interventions | Experimental: upadacitinib dose A (oral; once daily) for 12 weeks Comparator: matching placebo for upadacitinib |
| Outcomes | Primary outcomes: Endoscopic response (SES‐CD at week 12) Clinical remission (average daily stool frequency and average daily abdominal pain score at week 12) Secondary outcomes: Crohn's Symptoms Severity Questionnaire (CSS at week 12) Enhanced Clinical Response (decrease in average daily SF and/or decrease in average daily AP score at week 2) Discontinuation of corticosteroid use for CD (at week 12) Clinical remission (average daily stool frequency and average daily abdominal pain (AP) score at week 12) Fatigue (FACIT‐F at week 12) Quality of life (SF‐36 at week 12) Endoscopic remission (SES‐CD at week 12) Reduction in draining fistulas (>= 50% at week 12) Clinical remission (CDAI < 150 at week 12). Hospitalisations due to CD (at week 12) |
| Starting date | December 2017 |
| Contact information | AbbVie Call centre Telephone: 847.283.8955 Email: abbvieclinicaltrials@abbvie.com |
| Notes | Estimated study completion date: March 2020 Estimated enrollment: 300 participants |
NCT03398135.
| Trial name or title | A multicenter, randomized, double‐blind, placebo controlled 52‐week maintenance and an open‐label extension study of the efficacy and safety of risankizumab in subjects with ulcerative colitis who responded to induction treatment in M16‐067 or M16‐065 |
| Methods | A multicentre, randomised, double‐blind, placebo‐controlled 52‐week maintenance and an open‐label extension study |
| Participants | 760 adults (aged 16 ‐ 80 years) who have completed Study M16‐065 or Study M16‐067 and have achieved clinical response |
| Interventions | Experimental: Substudy 2: Open‐label risankizumab dose 1: Participants randomised to receive risankizumab dose 1 administered by subcutaneous (SC) injection Experimental: Substudy 1: Double‐blind risankizumab dose 2: Participants randomised to receive risankizumab dose 2 administered by subcutaneous (SC) injection Experimental: Substudy 2: Open‐label risankizumab dose 2: Participants randomised to receive risankizumab dose 2 administered by subcutaneous (SC) injection Experimental: Substudy 3: Open‐label extension risankizumab: Participants who completed substudy 1 or 2 receive open‐label risankizumab in substudy 3 Placebo Comparator: Substudy 1: Double‐blind placebo: Participants randomised to receive placebo for risankizumab administered by subcutaneous (SC) injection Experimental: Substudy 1: Double‐blind risankizumab dose 1: Participants randomised to receive risankizumab dose 1 administered by subcutaneous (SC) injection |
| Outcomes | Primary outcome: Clinical remission (adapted Mayo Score at week 52) Secondary outcomes: Clinical remission (adapted Mayo Score at week 52 in subjects with a clinical remission at week 0) Discontinuation of corticosteroid use at week 52 in subjects who were taking steroids at baseline (of induction) (at week 52) Discontinued corticosteroid use, remained corticosteroid free for 90 days, and achieved clinical remission per adapted Mayo Score at week 52 in subjects taking steroids at baseline (of induction) (at week 52) Endoscopic remission (endoscopy subscore at week 52) Ulcerative colitis symptoms (UC‐SQ at week 52) Ulcerative Colitis (UC)‐related surgeries (through week 52) Hospitalisation (through week 52) Health status (SF‐36 at week 52) Quality of life (IBDQ at week 52) Fatigue (FACIT‐Fatigue at week 52) Endoscopic improvement (endoscopy subscore at week 52) Clinical remission (full Mayo Score at week 52) Endoscopic improvement (endoscopy subscore at week 52) Clinical response (adapted Mayo score at week 52) Histologic remission (Geboes Score at week 52). Mucosal healing (endoscopic and histologic remission at week 52) |
| Starting date | August 2018 |
| Contact information | AbbVie Call centre Telephone: 847.283.8955 Email: abbvieclinicaltrials@abbvie.com |
| Notes | Estimated study completion date: December 2023 Estimated enrollment: 760 participants |
NCT03398148.
| Trial name or title | A multicenter, randomized, double‐blind, placebo controlled induction study to evaluate the efficacy and safety of risankizumab in subjects with moderately to severely active ulcerative colitis who have failed prior biologic therapy |
| Methods | A multicentre, randomised, double‐blind, placebo‐controlled induction study |
| Participants | 720 participants (aged 16 to <= 80 years) with a confirmed diagnosis of ulcerative colitis (UC) for at least 3 months prior to baseline, active UC and intolerance or inadequate response to one or more biologic therapies |
| Interventions | Experimental: Substudy 1, induction 2: Double‐blind risankizumab dose 2: Participants who received risankizumab with inadequate response in induction 1 randomised to receive risankizumab dose 2 administered by subcutaneous (SC) injection in induction 2 Experimental: Substudy 2, induction 1: Open‐label risankizumab dose 2: Participants randomised to receive risankizumab dose 2 administered by intravenous (IV) infusion Experimental: Substudy 2, induction 2: Double‐blind risankizumab dose 1(a): Participants who received placebo with inadequate response in induction 1 randomised to receive risankizumab dose 1 administered by intravenous (IV) infusion in induction 2 Experimental: Substudy 2, induction 2: Double‐blind risankizumab dose 3: Participants who received risankizumab with inadequate response in induction 1 randomised to receive risankizumab dose 3 administered by subcutaneous (SC) injection in induction 2 Experimental: Substudy 1, induction 1: Double‐blind risankizumab dose 1: Participants randomised to receive risankizumab dose 1 administered by intravenous (IV) infusion Experimental: Substudy 1, induction 1: Double‐blind risankizumab dose 2: Participants randomised to receive risankizumab dose 2 administered by intravenous (IV) infusion Experimental: Substudy 1, induction 2: Double‐blind risankizumab dose 1(a): Participants who received placebo with inadequate response in induction 1 receive risankizumab dose 1 administered by intravenous (IV) infusion in induction 2 Experimental: Substudy 1, induction 2: Double‐blind risankizumab dose 1(b): Participants who received risankizumab with inadequate response in induction 1 randomised to receive risankizumab dose 1 administered by intravenous (IV) infusion in induction 2 Experimental: Substudy 2, induction 1: Double‐blind risankizumab dose 1: Participants randomised to receive risankizumab dose 1 administered by intravenous (IV) infusion Experimental: Substudy 1, induction 1: Double‐blind risankizumab dose 3: Participants randomised to receive risankizumab dose 3 administered by intravenous (IV) infusion Placebo Comparator: Substudy 1, induction 1: Double‐blind placebo: Participants randomised to receive placebo for risankizumab administered by intravenous (IV) infusion Experimental: Substudy 2, induction 2: Double‐blind risankizumab dose 2: Participants who received risankizumab with inadequate response in induction 1 randomised to receive risankizumab dose 2 administered by subcutaneous (SC) injection in induction 2 Experimental: Substudy 1, induction 2: Double‐blind risankizumab dose 3: Participants who received risankizumab with inadequate response in induction 1 randomised to receive risankizumab dose 3 administered by subcutaneous (SC) injection in induction 2 Placebo Comparator: Substudy 2, induction 1: Double‐blind placebo: Participants randomised to receive placebo for risankizumab administered by intravenous (IV) infusion Experimental: Substudy 2, induction 2: Double‐blind risankizumab dose 1(b): Participants who received risankizumab with inadequate response in induction 1 randomised to receive risankizumab dose 1 administered by intravenous (IV) infusion in induction 2 Experimental: Substudy 1, induction 1: Open‐label risankizumab dose 3: Participants receive risankizumab dose 3 administered by intravenous (IV) infusion |
| Outcomes | Primary outcomes: Clinical remission (Mayo score at week 12) Secondary outcomes: Clinical remission (full Mayo score at week 12) Severity of Crohn's symptoms (Ulcerative Colitis Symptom Questionnaire (UC‐SQ) at week 12) Quality of life (IBDQ at week 12) Fatigue (FACIT‐Fatigue at week 12) Clinical response (adapted Mayo score at week 12) Endoscopic remission (endoscopy subscore at week 12) Clinical response (partial adapted Mayo score at week 4) Mucosal healing (endoscopic and histologic remission at week 12) Health status (SF‐36 at week 12) Hospitalisation (through week 12) Ulcerative Colitis (UC)‐related surgeries (through week 12) Endoscopic improvement (endoscopy subscore at week 12) |
| Starting date | March 2018 |
| Contact information | AbbVie Call centre Telephone: 847.283.8955 Email: abbvieclinicaltrials@abbvie.com |
| Notes | Estimated study completion date: December 2021 Estimated enrollment: 720 participants |
NCT03456752.
| Trial name or title | The impact of perioperative dexamethasone on postoperative outcome in inflammatory bowel diseases. |
| Methods | A randomised, double‐blind, placebo‐controlled study |
| Participants | 302 adults (aged 18 ‐ 75 years) undergoing elective open and laparoscopic small and large bowel operations for IBD, including CD and UC |
| Interventions | Experiment: Dexamethasone 8 mg intravenously prior to anaesthesia induction Comparator: Placebo of normal saline 8 mg intravenously prior to anaesthesia induction |
| Outcomes | Primary outcome: Prolonged ileus (defined as two or more of the following five criteria are met on day 4 postoperatively: nausea or vomiting; inability to tolerate an oral diet over last 24 h; absence of flatus over last 24 h; abdominal distension; and radiologic confirmation) Secondary outcomes: Postoperative nausea and vomiting, and additional antiemetics given within 24 hr after surgery Postoperative pain on postoperative day (POD) 1, 3, and 5 (VAS for pain) Postoperative fatigue on POD 1, 3, and 5 (FACIT‐F) GI‐2 recovery (time to upper (first tolerance of solid food) and lower (first bowel movement) GI recovery) Blood WBC levels (preoperative and on POD 1, 3 and 5) Blood neutrophil percentage (preoperative and on POD 1, 3 and 5) Serum C‐reactive protein (CRP) level (preoperative and on POD 1, 3 and 5) Serum Interleukin‐6 (IL‐6) level (preoperative and on POD 1, 3 and 5) Serum procalcitonin (PCT) (preoperative and on POD 1, 3 and 5) Body composition (bioelectrical impedance analysis (BIA) preoperative and on POD 1) Postoperative length of stay in days Postoperative morbidity (comprehensive complication index (CCI)) Postoperative surgical site infections (SSIs) (superficial SSIs and deep SSIs) Overall cost of treatment (in Chinese Yuan) |
| Starting date | June 2018 |
| Contact information | Jianfeng Gong, MD, Department of General Surgery, Jinling Hospital, Medical School of Nanjing University Nanjing, Jiangsu, China 210000 +86‐25‐80860036 gongjianfeng@hotmail.com |
| Notes | Estimated study completion date: October 2019 Estimated enrollment: 302 participants |
NCT03466411.
| Trial name or title | A phase 2/3, randomized, double‐blind, placebo‐ and active‐controlled, parallel‐group, multicenter protocol to evaluate the efficacy and safety of guselkumab in participants with moderately to severely active Crohn's disease |
| Methods | A phase 2/3, randomised, double‐blind, placebo‐ and active‐controlled, multicentre trial |
| Participants | 2000 adults (aged 18 years and older) with a diagnosis of CD or fistulising Crohn's disease of at least 3 months duration with colitis, ileitis, or ileocolitiis. Moderate‐to‐severe CD with intolerance or inadequate response to conventional or to biologic therapy for CD |
| Interventions | Experiment: Phase 2 (GALAXI 1): Group 1 (Guselkumab) participants will receive guselkumab (dose 1) by intravenous (IV) infusion, followed by guselkumab (dose 2) by subcutaneous (SC) injection. Participants who are eligible and willing to continue guselkumab may enter the Long‐Term Extension (LTE) phase and continue to receive guselkumab. Experiment: Phase 2 (GALAXI 1): Group 2 (Guselkumab) participants will receive guselkumab (dose 3) by intravenous (IV) infusion, followed by guselkumab (dose 2) by subcutaneous (SC) injection. Participants who are eligible and willing to continue guselkumab may enter the LTE phase and continue to receive guselkumab. Experiment: Phase 2 (GALAXI 1): Group 3 (Guselkumab) participants will receive guselkumab (dose 4) by intravenous (IV) infusion, followed by guselkumab (dose 5) by subcutaneous (SC) injection. Participants who are eligible and willing to continue guselkumab may enter the LTE phase and continue to receive guselkumab. Active Comparator: Phase 2 (GALAXI 1): Group 4 (Ustekinumab) participants will receive Ustekinumab by intravenous (IV) infusion, followed by subcutaneous (SC) injection. Participants who are eligible and willing to continue Ustekinumab may enter the LTE and continue to receive Ustekinumab. Experimental: Phase 2 (GALAXI 1): Group 5 (Placebo/Ustekinumab) participants will receive placebo administered by intravenous (IV) infusion. At week 12, non‐responders will receive active treatment (Ustekinumab) administered by intravenous (IV) infusion followed by subcutaneous (SC) injection. Participants who are eligible and willing to continue placebo/Ustekinumab may enter the LTE and continue to receive placebo/Ustekinumab. Experimental: Phase 3 (GALAXI 2 and 3): Group 1 and Group 2 (Guselkumab) participants will receive guselkumab by intravenous (IV) infusion, followed by guselkumab by subcutaneous (SC) injection. Participants who are eligible and willing to continue guselkumab may enter the LTE phase and continue to receive guselkumab. Active Comparator: Phase 3 (GALAXI 2 and 3): Group 3 (Ustekinumab) participants will receive Ustekinumab by intravenous (IV) infusion, followed by subcutaneous (SC) injection. Participants who are eligible and willing to continue Ustekinumab may enter the LTE phase and continue to receive Ustekinumab. Experimental: Phase 3 (GALAXI 2 and 3): Group 4 (Placebo/Ustekinumab) participants will receive placebo administered by intravenous (IV) infusion. At week 12, non‐responders will receive active treatment (Ustekinumab) administered by intravenous (IV) infusion followed by subcutaneous (SC) injection. Participants who are eligible and willing to continue placebo/Ustekinumab may enter the LTE and continue to receive placebo/Ustekinumab. |
| Outcomes | Primary outcomes: Phase 2: Change from baseline in the Crohn's Disease Activity Index (CDAI) Score at week 12 Phase 3: Clinical remission (CDAI at week 12) Secondary outcomes: Phase 2: Clinical remission (CDAI at week 12) Phase 2: Clinical response (CDAI at week 12) Phase 2 and Phase 3: Patient‐Reported Outcome (PRO)‐2 remission (average daily stool frequency and average daily abdominal pain score at week 12) Phase 2: Clinical‐biomarker response (CDAI and CRP/fecal calprotectin at week 12) Phase 2 and Phase 3: Endoscopic response (SES‐CD at week 12) Phase 3: Clinical remission (CDAI at week 48) Phase 3: Durable clinical remission (CDAI at week 48) Phase 3: Corticosteroid‐free clinical remission (CDAI at week 48) Phase 3: PRO‐2 remission (average daily stool frequency and average daily abdominal painscore.at week 48) Phase 3: Fatigue (Patient‐Reported Outcomes Measurement Information System (PROMIS). Fatigue Short Form Response at week 12) Phase 3: Endoscopic response (SES‐CD at week 48) |
| Starting date | April 2018 |
| Contact information | Janssen Research & Development, LLC Clinical Trial |
| Notes | Estimated study completion date: November 2024 Estimated enrollment: 2000 participants |
NCT03574948.
| Trial name or title | Multicentric, double‐blind, placebo controlled clinical trial with 5‐hydroxytryptophan (5‐HTP) in patients with inflammatory bowel disease in clinical and biologic remission: effect on fatigue scores |
| Methods | A phase 2, multicentred, double‐blind, randomised placebo‐controlled trial, cross‐over assignment |
| Participants | Adults (aged 18 ‐ 60 years) with a documented diagnosis of Crohn's disease or ulcerative colitis in clinical remission over 3 months (physical global assessment; SCCAI ≤ 2 for ulcerative colitis or Harvey Bradshaw index ≤ 4 for Crohn's disease) reporting fatigue (score 5 or more on a 1 ‐ 10 visual analogue scale). Treated with biologicals and/or immunosuppressives since at least 6 months with stable dose over last 3 months and in biologic remission at day 0 (CRP < 10 mg/L and faecal calprotectin value < 250 mg/kg) |
| Interventions | Experimental: 5‐HTP ‐ 8 weeks active substance 5‐HTP (2 x 100 mg per day) Placebo comparator: 8 weeks placebo oral capsule (2 x 1 capsule per day) |
| Outcomes | Primary outcome: Fatigue (visual analog scale) Secondary outcome: Fatigue (FACIT‐F); Depression (short Depression Anxiety and Stress Scale); Physical activity (Adapted International Physical Activity Questionnaire); serum 5‐HydroxyTryptophane (5‐HTP); serum 5‐hydroxyindoleacetic acid; serum serotonin levels; serum melatonin levels; faecal microbiome/metabolome |
| Starting date | December 2018 |
| Contact information | Dr Martine De Vos 0032 9 332 23 71 martine.devos@uzgent.be Dr Triana Lobaton Ortega 0032 9 332 23 89 Triana.LobatonOrtega@UZGENT.be |
| Notes | Estimated study completion date:December 2020 Estimated enrollment: 180 participants |
5‐HTP: 5‐hydroxytryptophan AF: ALT: alanine transaminase AP: Abdominal pain AST: Aspartate transaminase BDI‐II: Beck Depression Inventory BIA: Bioelectrical impedance analysis BMI: Body Mass Index CCI: Comprehensive complication index CD: Crohn's disease CDAI: Crohn's Disease Activity Index CD‐PRO: Crohn's disease ‐ Patient Reported Outcomes CIS‐20: Checklist Individual Strength ‐ 20 CRP: C‐reactive protein CSS:Crohn's Symptoms Severity CT: Computerised tomography DXA: Dual‐energy X‐ray absorptiometry ED: Emergency department ER: Emergency room ESR: Erythrocyte sedimentation rate EVOO: extra virgin olive oil FACIT‐F: Functional Assessment of Chronic Illness Therapy ‐ Fatigue FBC: Full blood count FC: Faecal calprotectin fl: femtolitre FR‐QoL‐29: Food‐related quality of life GAD: Generalised Anxiety Disorder GI: Gastrointestinal GOS: Galacto‐oligosaccharides HAD: Hospital Anxiety and Depression Hbg: Hemoglobin HBI: Harvey Bradshaw Index Ht: hematocrit IBD: Inflammatory bowel disease IBD‐F: Inflammatory Bowel Disease ‐ Fatigue IBDQ: Inflammatory Bowel Disease Questionnaire IBDU: Inflammatory bowel disease unclassified IFN‐y: interferon gamma IgA: Immunoglobulin A IL: Interleukin IU: International units IV: Intravenous LPS: Lipopolysaccharidde LTE: Long‐term extension mg: milligram mg/kg: milligram per kilogram MFI: Multidimensional Fatigue Inventory PCD: Paediatric Crohn's disease PCDAI: Paediatric Crohn's Disease Activity Index PCT: Procalcitonin PedsQL: Pediatric Quality of Life Inventory PGR‐C: Patient Global rsting ‐ Change
PGR‐S: Patient Global Rating ‐ Severity PHQ15: Patient Health Questionnaire 15 POD: Postoperative day PROMIS: Patient‐Reported Outcomes Measurement Information System PUCAI: Pediatric Ulcerative Colitis Activity Index Q4: 4 weekly
Q8: 8 weekly
Q12: 12 weekly QOLITI: Quality of Life Tool for IBD RCT: Randomised controlled trial retHb: reticulocyte hemoglobin rRNA: Ribosomal ribonucleic acid SC: Subcutaneous SCCAI: Simple Clinical Colitis Activity Index SES‐CD: Simple Endoscopic Score for Crohn's Disease SF: Stool Frequency SF‐36:36‐Item Short Form SIBDQ: Short Inflammatory Bowel Disease Questionnaire SPCDAI:Short Pediatric Crohn's Disease Activity Index SSIs:surgical site infections STAI: State‐Trait Anxiety Inventory sTfR:soluble transferrin receptors
sTfR(‐F): soluble transferrin receptors to log ferritin STPI: State‐Trait Personality Inventory TB: Tuberculosis TNFα: Tumor necrosis factor alpha TNFr1: Tumor necrosis factor receptor 1 T2T:Treat‐to‐Target UC: Ulcerative colitis UC‐SQ: Ulcerative Colitis Symptom Questionnaire UST: Ustekinumab VAS: Visual Analogue Scale VO2: maximal oxygen uptake WBC: White blood cells wk: week WLC: Waiting‐list control
Differences between protocol and review
The order of existing authors was changed to reflect their relative contribution to the review.
The term 'managing' was included in the aim of the review to provide further clarity regarding the focus of the review.
The same two authors (DF, MA) completed the initial and full screening of search hits to identify eligible trials for inclusion in the review.
Under 'Assessment of risk of bias in included studies', the term 'upgrade' was removed as not relevant to this review of RCTs (quality of evidence downgraded only).
Contributions of authors
Dawn Farrell: conceived and designed the review, developed the protocol, coordinated the review, participated in devising the search strategy, undertook searches and retrieved papers for the review, screened the literature, extracted the data, appraised the quality of papers, wrote to authors of papers for additional information, analysed and interpreted data, managed and entered data into RevMan, wrote the review, sourced funding for the review, and is responsible for the full review and update.
Micol Artom: content expert, screened the literature, extracted the data, appraised the quality of papers, analysed and interpreted data.
Wladyslawa Czuber‐Dochan: content expert, extracted the data, appraised the quality of papers, analysed and interpreted data.
Lars P Jelsness‐Jørgensen: content expert, extracted the data, appraised the quality of papers, analysed and interpreted data.
Christine Norton: content and methodological expert, critically evaluated the protocol, provided general advice on the review and interpretation of results, edited the discussion and conclusions.
Eileen Savage: methodological expert, critically evaluated protocol, screened the literature, provided general advice on the review and interpretation of results.
Sources of support
Internal sources
-
University College Cork, Ireland.
Provided infrastructure support to the primary author to carry out the Cochrane review
External sources
-
Health Research Board, Ireland.
Provided a Cochrane Fellowship to the Principal Investigator
Declarations of interest
Dawn Farrell: This paper presents independent research funded by the Health Research Board of Ireland under the Cochrane Fellowship programme (Reference Number CFT‐2014‐887). Infrastructure support was provided from the host institution, University College Cork, to conduct this review.
Micol Artom: This paper presents independent research funded by the UK National Institute for Health Research (NIHR) under its Programme Grants for Applied Research Programme (Reference Number RP‐PG‐0216‐20001). The views expressed are those of the author and not necessarily those of the NHS, the NIHR or the Department of Health.
Wladyslawa Czuber‐Dochan: Serves as a member of Scientific Committee for Crohn's and Colitis UK, and Nurse‐European Crohn's and Colitis Organisation. She has also received speaker fees from Pfizer. This paper presents independent research funded by the UK National Institute for Health Research (NIHR) under its Programme Grants for Applied Research Programme (Reference Number RP‐PG‐0216‐20001). The views expressed are those of the author and not necessarily those of the NHS, the NIHR or the Department of Health.
Lars‐Petter Jelsness‐Jørgensen has received unrestricted research grants from Ferring Pharmaceuticals and Tillots Pharma. and has also acted as consultant and speaker for Abbvie. All of these activities are outside the submitted work.
Christine Norton: Tillotts, Takeda, AbbVie, Ferring (lecture fees) (outside submitted work). This paper presents independent research funded by the UK National Institute for Health Research (NIHR) under its Programme Grants for Applied Research Programme (Reference Number RP‐PG‐0216‐20001). The views expressed are those of the author and not necessarily those of the NHS, the NIHR or the Department of Health.
Eileen Savage: None known.
New
References
References to studies included in this review
Artom 2018 {published and unpublished data}
- Artom M, Czuber‐Dochan W, Sturt J, Norton C. Cognitive behavioural therapy for the management of inflammatory bowel disease‐fatigue with a nested qualitative element: study protocol for a randomised controlled trial. Trials 2017;18:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artom M, Czuber‐Dochan W, Sturt J, Proudfoot H, Norton C. Cognitive behavioural therapy for the management of inflammatory bowel disease‐fatigue: a feasibility randomised controlled trial. Pilot Feasibility Studies 2019;5(145). [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISRCTN17917944. Management of inflammatory bowel disease‐fatigue: a pilot cognitive‐behavioural therapy interventional study. http://www.isrctn.com/ISRCTN17917944 (first received 2 September 2016).
Colombel 2007 {published data only}
- Colombel JF, Sandborn WJ, Rutgeerts P, Enns R, Hanauer SB, Panaccione R, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn's disease: the CHARM trial. Gastroenterology 2007;132(1):52‐65. [DOI] [PubMed] [Google Scholar]
- Loftus EV, Feagan BG, Colombel J‐F, Rubin DT, Wu EQ, Yu AP, et al. Effects of adalimumab maintenance therapy on health‐related quality of life of patients with Crohn’s disease: patient‐reported outcomes of the CHARM Trial. American Journal of Gastroenterology 2008;103(7):3132‐41. [DOI] [PubMed] [Google Scholar]
- Rubin D, Rutgeerts P, Colombel J, Wu E, Yu A, Chao J, et al. Adalimumab maintenance therapy and health related quality of life in TNT‐antagonist‐naïve patients with Crohn’s disease. Journal of the Canadian Association of Gastroenterology 2009;23(1):S118. [Google Scholar]
Colombel 2017 {published data only}
- Colombel JF, Panaccione R, Bossuyt P, Lukas M, Baert F, Vaňásek T, et al. Effect of tight control management on Crohn's disease (CALM): a multicentre, randomised, controlled phase 3 trial. Lancet 2017;390(10114):2779‐89. [DOI] [PubMed] [Google Scholar]
- NCT01235689. An open‐label, multicenter, efficacy and safety study to evaluate two treatment algorithms in subjects with moderate to severe Crohn's Disease. clinicaltrials.gov/ct2/show/NCT01235689 (first received 5 November 2010).
- Panaccione R, Colombel JF, Bossuyt P, Baert F, Vanasek T, Danalioglu A, et al. DOP071 Tight control with adalimumab‐based treatment is associated with improved quality of life outcomes in patients with moderate to severely active Crohn’s disease: data from CALM. Journal of Crohn's and Colitis 2018;12:S078‐9. [Google Scholar]
Feagan 2013 {published data only}
- Feagan BG, Rutgeerts P, Sands BE, Hanauer S, Colombel JF, Sandborn WJ, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. New England Journal of Medicine 2013;369(8):699‐710. [DOI] [PubMed] [Google Scholar]
- NCT00783718. A phase 3, randomized, placebo‐controlled, blinded, multicenter study of the induction and maintenance of clinical response and remission by MLN0002 in patients with moderate to severe ulcerative colitis. clinicaltrials.gov/ct2/show/NCT00783718 (first received 2 November 2008).
- Rubin DT, Tudor D, Khalid JM, Patel H. P570 Improvements in subcomponents of the inflammatory bowel disease questionnaire in patients treated with vedolizumab: results from GEMINI trial data. Journal of Crohn's and Colitis 2018;12(Supplement 1):S394‐5. [Google Scholar]
García‐Vega 2004 {published data only}
- García‐Vega E, Fernandez‐Rodriguez C. A stress management programme for Crohn’s disease. Behaviour Research and Therapy 2004;42(4):367‐83. [DOI] [PubMed] [Google Scholar]
Gasche 2015 {published data only}
- Gasche C, Ahmad T, Tulassay Z, Baumgart DC, Bokemeyer B, Büning C, et al. Ferric Maltol is effective in correcting iron deficiency anaemia in patients with inflammatory bowel disease: results from a Phase‐3 clinical trial program. Inflammatory Bowel Diseases 2015;21(3):579‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hetzel 2013a {published data only}
- Hetzel D, Barish C, Dahl N, Bernard K, Lau G, Strauss W. Efficacy and safety of intravenous Ferunoxytol for the treatment of iron deficiency anaemia in patients with inflammatory bowel disease. Inflammatory Bowel Diseases 2013;19(Supplement 1):S81‐2. [Google Scholar]
- Hetzel D, Ford D, Dahl N, Strauss W. Intravenous (IV) ferumoxytol (FER) for the treatment of iron deficiency anaemia (IDA) in patients with inflammatory bowel disease (IBD): efficacy, safety and health‐related quality of life (HRQOL). Journal of Crohn's and Colitis 2014;8:S248‐9. [Google Scholar]
Horta 2017 {published and unpublished data}
- Horta D, Sanchez‐Lloansi M, Lira A, Villoria A, Teggiachi M, Garcia D, et al. A prospective randomised study: electroacupuncture vs. sham procedure for the treatment of fatigue in patients with quiescent inflammatory bowel disease. United European Gastroenterology Journal 2017;5(Supplement 1):A293‐4. [Google Scholar]
McNelly 2016 {published data only}
- McNelly AS, Nathan I, Monti M, Grimble GK, Norton C, Bredin F, et al. Inflammatory bowel disease and fatigue: the effect of physical activity and/or omega‐3 supplementation. Journal of Crohn's and Colitis 2016;10:S370‐1. [Google Scholar]
- McNelly AS, Nathan I, Monti M, Grimble GK, Norton C, Bredin F, et al. The effect of increasing physical activity and/or omega‐3 supplementation on fatigue in inflammatory bowel disease. Gastrointestinal Nursing 2016;14(8):39‐50. [Google Scholar]
Raftery 2013 {published data only}
- NCT01792388. Vitamin D and its effects on inflammation and intestinal permeability in Crohn's disease in remission. clinicaltrials.gov/ct2/show/NCT01792388 (first received 15 February 2013).
- Raftery TC, Healy M, Cox G, McNamara D, O’ Sullivan M. Vitamin D supplementation improves muscles strength, fatigue and quality of life in patients with Crohn’s disease in remission: results of a randomised double‐blind placebo‐controlled study. Gastroenterology 2013;144(5):S227. [Google Scholar]
Sandborn 2013 {published data only}
- NCT00783692. A phase 3, randomized, placebo‐controlled, blinded, multicenter study of the induction and maintenance of clinical response and remission by vedolizumab (MLN0002) in patients with moderate to severe Crohn's Disease. clinicaltrials.gov/ct2/show/study/NCT00783692 (first received 2 November 2008).
- Rubin DT, Tudor D, Khalid JM, Patel H. P570 Improvements in subcomponents of the inflammatory bowel disease questionnaire in patients treated with vedolizumab: results from GEMINI trial data. Journal of Crohn's and Colitis 2018;12(Supplement 1):S394‐5. [Google Scholar]
- Sandborn WJ, Feagan BG, Rutgeerts P, Hanauer S, Colombel JF, Sands BE, et al. Vedolizumab as induction and maintenance therapy for Crohn's disease. New England Journal of Medicine 2013;369(8):711‐21. [DOI] [PubMed] [Google Scholar]
Therkelsen 2016a {published data only}
- Therkelsen SP, Hetland G, Lyberg T, Lygren I, Johnson E. Effect of a medicinal agaricus blazei murill‐based mushroom extract, AndoSan™, on symptoms, fatigue and quality of life in patients with ulcerative colitis in a randomized single‐blinded placebo controlled study. PlOS One 2016;11(3):e0150191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Therkelsen 2016b {published data only}
- Therkelsen SP, Hetland G, Lyberg T, Lygren I, Johnson E. Effect of the medicinal agaricus blazei murill‐based mushroom extract, AndoSanTM, on symptoms, fatigue and quality of life in patients with Crohn’s Disease in a randomized single‐blinded placebo controlled study. PLOS One 2016;11(7):e0159288. [DOI] [PMC free article] [PubMed] [Google Scholar]
Vogelaar 2014 {published data only}
- Vogelaar L, Van't Spijker A, Timman R, Tilburg A, Bac D, Kuipers EJ, et al. Fatigue in IBD patients decreases with psychotherapy: results of a randomized controlled trial. Journal of Crohn's and Colitis 2013;7:S211‐12. [Google Scholar]
- Vogelaar L, Van’t Spijker A, Timman R, Tilburg AJP, Bac D, Vogelaar T, et al. Fatigue management in patients with IBD: a randomised controlled trial. Gut 2014;63:911‐8. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Boye 2011 {published data only}
- Boye B, Lundin KE, Jantschek G, Leganger S, Mokleby K, Tangen T, et al. INSPIRE study: does stress management improve the course of inflammatory bowel disease and disease specific quality of life in distressed patients with ulcerative colitis or Crohn's disease? A randomised controlled trial. Inflammatory Bowel diseases 2011;17(9):1863‐73. [DOI] [PubMed] [Google Scholar]
Colombel 2010 {published data only}
- Colombel JF, Sandborn WJ, Reinisch W, Mantzaris GJ, Kornbluth A, Rachmilewitz D, et al. Infliximab, azathioprine, or combination therapy for Crohn's disease. New England Journal of Medicine 2010;362(15):1383‐95. [DOI] [PubMed] [Google Scholar]
Cosnes 2013 {published data only}
- Cosnes J, Bourrier A, Laharie D, Nahon S, Bouhnik Y, Carbonnel F, et al. Early administration of azathioprine vs conventional management of Crohn's disease: a randomised controlled trial. Gastroenterology 2013;145(4):758‐65. [DOI] [PubMed] [Google Scholar]
Dewint 2014 {published data only}
- Dewint P, Hansen BE, Verhey E, Oldenburg B, Hommes DW, Pierik M, et al. Adalimumab combined with ciprofloxacin is superior to adalimumab monotherapy in perianal fistula closure in Crohn's disease: a randomised, double‐blind, placebo controlled trial (ADAFI). Gut 2014;63(2):292‐9. [DOI] [PubMed] [Google Scholar]
Feagan 2003 {published data only}
- Feagan BG, Yan S, Bala M, Bao W, Lichtenstein GR. The effects of infliximab maintenance therapy on health‐related quality of life. American Journal of Gastroenterology 2003;98(10):2232‐8. [DOI] [PubMed] [Google Scholar]
Ford 2016 {published data only}
- Ford DC, Dahl NV, Strauss WE, Barish CF, Hetzel DJ, Bernard K, et al. Ferumoxytol versus placebo in iron deficiency anaemia: efficacy, safety, and quality of life in patients with gastrointestinal disorders. Clinical and Experimental Gastroenterology 2016;9:151‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hetzel 2013b {published data only}
- Hetzel D, Strauss W, Bernard K. IV iron treatment of iron deficiency anaemia with ferumoxytol in patients with gastrointestinal disorders unable to take oral iron: a randomised controlled trial versus iron sucrose. Journal of Crohn's and Colitis 2013;7:S204. [Google Scholar]
Leiper 2001 {published data only}
- Leiper K, Woolner J, Mullan MMC, Parker T, Vliet M, Fear S, et al. A randomised controlled trail of high versus low long chain triglyceride whole protein feed in active Crohn's disease. Gut 2001;49(6):790‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lichtenstein 2002 {published data only}
- Lichtenstein GR, Bala M, Chenglong H, DeWoody K, Schaible T. Infliximab improves quality of life in patients with Crohn’s disease. Inflammatory Bowel Diseases 2002;8(4):237‐43. [DOI] [PubMed] [Google Scholar]
Loftus 2009 {published data only}
- Loftus E, Colombel J, Pollack P, Majethia S, Chen N. Adalimumab treatment associated with rapid and significant improvement in health‐related quality of life in patients with Crohn’s disease. Journal of the Canadian Association of Gastroenterology 2009;23(1). [Google Scholar]
Loftus 2017 {published data only}
- Loftus EV, Colombel JF, Feagan BG, Vermeire S, Sandborn WJ, Sands BE, et al. Long‐term efficacy of vedolizumab for ulcerative colitis. Journal of Crohn's and Colitis 2017;11(4):400‐11. [DOI] [PubMed] [Google Scholar]
Maragkoudaki 2016 {published data only}
- Maragkoudaki M, Chouliaras G, Papadopoulou A, Christaki M. Feasibility and effectiveness of a psycho‐educational program in adolescents with Inflammatory Bowel Disease (IBD). Journal of Pediatric Gastroenterology and Nutrition 2016;61:363. [Google Scholar]
Mikocka‐Walus 2017 {published data only}
- Mikocka‐Walus A, Hughes PA, Bampton P, Gordon A, Campaniello MA, Mavrangelos C, et al. Fluoxetine for maintenance of remission and to improve quality of life in patients with Crohn’s disease: a pilot randomized placebo‐controlled trial. Journal of Crohn's and Colitis 2017;11(4):509‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Minderhoud 2007 {published data only}
- Minderhoud IM, Samsom M, Oldenburg B. Crohn’s disease, fatigue, and infliximab: Is there a role for cytokines in the pathogenesis of fatigue?. World of Journal of Gastroenterology 2007;13(14):2089‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT01991314 {published data only}
- NCT01991314. Treatment of iron deficiency anaemia in adults and adolescents with inflammatory bowel disease using ferrous sulphate: tolerance and effects on haemoglobin, mood, quality of life and fatigue. clinicaltrials.gov/ct2/show/NCT01991314 (first received 25 November 2013).
NCT02148718 {published data only}
- NCT02148718. Rapidity of onset of response to adalimumab in luminal Crohn's disease (RAPIDA Study). clinicaltrials.gov/ct2/show/NCT02148718 (first received 28 May 2014).
NCT02162862 {published data only}
- NCT02162862. Treating disrupted sleep in individuals with inflammatory bowel disease: a novel adjunctive therapy for chronic inflammatory illness. clinicaltrials.gov/ct2/show/NCT02162862 (first received 13 June 2014).
Paramsothy 2017 {published data only}
- Paramsothy S, Kamm MA, Kaakoush NO, Walsh AJ, Bogaerde J, Samuel D, et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: a randomised placebo‐controlled trial. Lancet 2017;389(10075):1218‐28. [DOI] [PubMed] [Google Scholar]
Pena Rossi 2009 {published data only}
- Pena Rossi C, Hanauer SB, Tomasevic R, Hunter JO, Shafran I, Graffner H. Interferon beta‐1a for the maintenance of remission in patients with Crohn's disease: results of a phase II dose‐finding study. BMC Gastroenterology 2009;9:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Persoons 2007 {published data only}
- Persoons P, Demyttenaere K, Vandenberghe J, Oudenhove L, Rugeerts P. The evolution of fatigue in patients with Crohn's disease after treatment with infliximab: a prospective study. Gastroenterology 2004;126(4):A476. [Google Scholar]
Reusch 2016 {published data only}
- Reusch A, Weiland R, Gerlich C, Dreger K, Derra C, Mainos D, et al. Self‐management education for rehabilitation inpatients suffering from inflammatory bowel disease: a cluster randomized controlled trial. Health Education Research 2016;31(6):782‐91. [DOI] [PubMed] [Google Scholar]
Sands 2008 {published data only}
- Sands BE, Sandborn WJ, Feagan B, Löfberg R, Hibi T, Wang T, et al. A randomised, double‐blind, sham‐controlled study of granulocyte/monocyte apheresis for active ulcerative colitis. Gastreoenterology 2008;135(2):400‐9. [DOI] [PubMed] [Google Scholar]
Sands 2013 {published data only}
- Sands BE, Katz S, Wolf DC, Feagan BG, Wang T, Gustofson L‐M, et al. A randomised, double‐blind, sham‐controlled study of granulocyte apheresis for moderate to severe Crohn's disease. Gut 2013;62(9):1288‐94. [DOI] [PubMed] [Google Scholar]
Schmidt 2016 {published data only}
- Schmidt C, Ahmad T, Tulassay Z, Baumgart DC, Bokemeyer B, Howaldt S, et al. Ferric maltol therapy for iron deficiency anaemia in patients with inflammatory bowel disease: long‐term extension data from a phase 3 study. Alimentary Pharmacology and Therapeutics 2016;44:259‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Scholten 2018 {published data only}
- Scholten AM, Vermeulen E, Dhonukshe‐Rutten RA, Verhagen T, Visscher A, Olivier A, et al. Surplus vitamin B12 use does not reduce fatigue in patients with irritable bowel syndrome or inflammatory bowel disease: a randomized double‐blind placebo‐controlled trial. Clinical Nutrition European Society for Clinical Nutrition and Metabolism 2018;23:48‐53. [DOI] [PubMed] [Google Scholar]
Schreiber 2007 {published data only}
- Schreiber S, Keshavarzian A, Isaacs KL, Schollenberger J, Guzman JP, Orlandi C, et al. A randomised, placebo‐controlled, phase II study of tetomilast in active ulcerative colitis. Gastroenterology 2007;132(1):76‐86. [DOI] [PubMed] [Google Scholar]
Smith 2011 {published data only}
- Smith JP, Bingaman SI, Ruggiero F, Mauger DT, Mukherjee A, McGovern CO, et al. Therapy with the opioid antagonist naltrexone promotes mucosal healing in active Crohn's disease: a randomised placebo‐controlled trial. Digestive Diseases and Sciences 2011;56(7):2088‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
Smith 2013 {published data only}
- Smith JP, Field D, Bingaman S, Evans R, Mauger D. Safety and tolerability of low dose naltrexone therapy in children with moderate to severe Crohn's disease: a pilot study. Journal of Clinical Gastroenterology 2013;47(4):339‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Steinhart 2002 {published data only}
- Steinhart AH, Feagan BG, Wong CJ, Vandervoort M, Mikolainis S, Croitoru K, et al. Combined budesonide and antibiotic therapy for active Crohn's disease: a randomised controlled trial. Gastroenterology 2002;123(1):33‐40. [DOI] [PubMed] [Google Scholar]
Szigethy 2016 {published data only}
- Szigethy E, McAuliff K, Strassburger M, Hashash J, Vachon A, Rode NM, et al. Brief behavioral therapy and bupropion for sleep and fatigue in adolescents and young adults with inflammatory bowel disease. Journal of the American Academy of Child and Adolescent Psychiatry 2016;55(10):S210. [Google Scholar]
Targan 2007 {published data only}
- Targan SR, Feagan BG, Fedorak RN, Lashner BA, Panaccione R, Present DH, et al. Natalizumab for the treatment of active Crohn's disease: results of the ENCORE trial. Gastroenterology 2007;132(5):1672‐83. [DOI] [PubMed] [Google Scholar]
Valentine 2009 {published data only}
- Valentine JF, Fedorak RN, Feagan B, Fredlund P, Schmitt R, Ni P, et al. Steriod‐sparing properties of sargramostim in patients with corticosteroid‐dependent Crohn's disease: a randomised, double‐blind, placebo‐controlled, phase 2 study. Gut 2009;58(10):1354‐62. [DOI] [PubMed] [Google Scholar]
Van Assche 2012 {published data only}
- Assche G, Vermeire S, Ballet V, Gabriels F, Noman M, D'Haens G, et al. Switch to adalimumab in patients with Crohn's disease controlled by maintenance infliximab: prospective randomised SWITCH trial. Gut 2012;61(2):229‐34. [DOI] [PubMed] [Google Scholar]
Vermeire 2017 {published data only}
- Vermeire S, Loftus EV, Colombel JF, Feagan BG, Sandborn WJ, Sands BE. Long‐term efficacy of vedolizumab for Crohn’s Disease. Journal of Crohn's and Colitis 2017;11(4):412‐24. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Ghosh 2019 {published data only}
- Ghosh S, Aberra F, Cross R, Zhou W, Chen N, Lee WJ, et al. Effect of upadacitinib on patient‐reported symptoms by the new ulcerative colitis symptoms questionnaire (UC‐SQ) in patients with moderate to severe ulcerative colitis: data from the Phase 2b study U‐ACHIEVE. Journal of Crohn's and Colitis 2019;13:S247‐S248. [Google Scholar]
- NCT02819635. A multicenter, randomized, double‐blind, placebo‐controlled study to evaluate the safety and efficacy of upadacitinib (ABT‐494) for induction and maintenance therapy in subjects with moderately to severely active ulcerative colitis. clinicaltrials.gov/ct2/show/NCT02819635 (first received 30 June 2016).
Louis 2019 {published data only}
- Louis E, Sandborn WJ, D'Haens G, Baert F, Kalabic J, Wallace K, et al. Effect of risankizumab on improved and sustained quality of life in patients with moderate to severe Crohn's disease: phase 2 trial results. Journal of Crohn's and Colitis 2019;13:S291‐S292. [Google Scholar]
- NCT02031276. A phase II, multicenter, randomized, double‐blind, multiple dose, placebo‐controlled, parallel‐group study to evaluate the efficacy, pharmacokinetics, and safety of BI 655066/ABBV‐066 (risankizumab), and IL‐23 p19 antagonist monoclonal antibody in patients with ,moderately to severely active Crohn's disease who are naïve to, or were previously treated with, anti‐TNF therapy. clinicaltrials.gov/ct2/show/NCT02031276 (first received 9 January 2014).
O' Connor 2019 {published data only}
- NCT02709434. The efficacy of a structured psychoeducational inflammatory bowel disease group on the control of fatigue in an adult outpatient setting for patients with objectively quiescent disease. ClinicalTrials.gov/show/NCT02709434 (first received 16 March 2016).
- O'Connor A, Ratnakumaran R, Warren L, Pullen D, Errington A, Gracie DJ, et al. Randomized controlled trial: a pilot study of a psychoeducational intervention for fatigue in patients with quiescent inflammatory bowel disease. Therapeutic Advances in Chronic Disease 2019;10:1‐12. [DOI: 10.1177/2040622319838439] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sands 2018 {published data only}
- Sands BE, Pires A, Gasink C, Laliman V, Han C, Feagan BG. Post hoc analysis of the impact of ustekinumab treatment on specific items of the Inflammatory Bowel Disease Questionnaire in the Uniti‐1 & 2 programs. Gastroenterology 2018;154:S‐811. [Google Scholar]
Tew 2019 {published data only}
- ISRCTN13021107. A randomised controlled trial investigating the feasibility and acceptability of high‐intensity interval training and moderate‐intensity continuous training in adults with inactive or mildly active Crohn’s disease. www.isrctn.com/ISRCTN13021107 (first received 2 December 2015).
- Tew GA, Leighton D, Carpenter R, Anderson S, Langmead L, Ramage J, et al. High‐intensity interval training and moderate‐intensity continuous training in adults with Crohn's disease: a pilot randomised controlled trial. BMC Gastroenterology 2019;19(1):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to ongoing studies
ACTN12617000586314P {published data only}
- ACTRN12617000586314P. A prospective randomized multicentre study to evaluate the effect of intravenous iron infusion compared to oral iron supplementation on the quality of life in inflammatory bowel disease with non‐anaemia hypoferritinanaemia. https://www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=372758 (first received 17 April 2017).
ACTRN12619000150145 {published data only}
- ACTRN12619000150145. Influence of extra virgin olive oil intake on disease activity and gut microbiota profile of community dwelling adults with ulcerative colitis in comparison to healthy subjects. https://www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=375208 (first received 24 January 2019).
EudraCT Number: 2008‐004277‐17 {published data only}
- EudraCT Number: 2008‐004277‐17. The effectiveness and tolerability of GlobiFer (haem iron) tablets compared to ferrous sulphate tablets in inflammatory bowel disease: a randomised‐controlled trial. https://www.clinicaltrialsregister.eu/ctr‐search/trial/2008‐004277‐17/GB (first received 15 March 2010).
EudraCT Number: 2011‐002122‐43 {published data only}
- EudraCT Number: 2011‐002122‐43. Iron therapy in IBD patients with normal levels of haemoglobin and chronic fatigue. https://www.clinicaltrialsregister.eu/ctr‐search/search?query=2011‐002122‐43 (first received).
EudraCT Number: 2012‐005644‐26 {published data only}
- EUCTR2012‐005644‐26‐BE. Administration of intravenous ferric carboxymaltose to children with IBD. https://www.clinicaltrialsregister.eu/ctr‐search/search?query=EUCTR2012‐005644‐26‐BE (first received ).
ISRCTN11470370 {published data only}
- ISRCTN11470370. Effects of a 6‐month practical resistance training programme on muscle function and bone mineral density in adults with inactive or mildly active Crohn’s disease: Study protocol for a randomised controlled trial. http://www.isrctn.com/ISRCTN11470370 (first received 11 December 2017).
NCT02193750 {published data only}
- NCT02193750. Assessing the tolerability of oligosaccharide supplementation in patients with Crohn's disease: A randomized, controlled trial. https://clinicaltrials.gov/ct2/show/NCT02193750 (first received 18 July 2014).
NCT02208310 {published data only}
- NCT02208310. A randomised controlled trial of high‐dose vitamin D in Crohn's disease. https://clinicaltrials.gov/ct2/show/NCT02208310 (first received 5 August 2014).
NCT02517151 {published data only}
- NCT02517151. Effects of iron therapy in patients with chronic fatigue and IBD. https://clinicaltrials.gov/ct2/show/NCT02517151 (first received 6 August 2015).
NCT02704624 {published data only}
- NCT02704624. The impact of serum vitamin D and calcium levels on the body composition, bone mineral density, muscle strength, exercise tolerance, fatigue and inflammatory activity in patients with Crohn's disease: a randomized controlled trial. https://ClinicalTrials.gov/show/NCT02704624 (first received 10 March 2016).
NCT02707068 {published data only}
- NCT02707068. Quality Of LIfe Tool for IBD (QOLITI): pilot testing of a self‐administered intervention to target psychological distress in inflammatory bowel disease. https://clinicaltrials.gov/show/NCT02707068 (first received 14 March 2016).
NCT02772965 {published data only}
- NCT02772965. A randomized, double‐blind, placebo‐controlled, multi‐center pragmatic clinical trial to evaluate the effectiveness of low dose oral methotrexate in patients with pediatric Crohn's disease initiating anti‐TNF therapy. https://clinicaltrials.gov/ct2/show/NCT02772965 (first received 16 May 2016).
NCT02849717 {published data only}
- NCT02849717. Pre‐habilitation exercise intervention for patients scheduled for colorectal surgical resection. clinicaltrials.gov/ct2/show/NCT02849717 (first received 29 July 2016).
NCT02861053 {published data only}
- NCT02861053. Inflammatory Bowel Disease: Could a regular physical activity reduce patients fatigue?. https://clinicaltrials.gov/ct2/show/NCT02861053 (first received 10 August 2016).
NCT02891226 {published data only}
- NCT02891226. A phase 2, multicenter, randomized, parallel‐arm, placebo‐controlled study of LY3074828 in subjects with active Crohn's Disease (SERENITY). https://clinicaltrials.gov/ct2/show/NCT02891226 (first received 7 September 2016).
NCT02963246 {published data only}
- NCT02963246. Effects of a mindfulness therapy intervention for individuals with inflammatory bowel disease: a randomized controlled trial. https://clinicaltrials.gov/ct2/show/NCT02963246 (first received 15 November 2016).
NCT03104413 {published data only}
- NCT03104413. A multicenter, randomized, double‐blind, placebo‐controlled induction study to assess the efficacy and safety of Risankizumab in subjects with moderately to severely active Crohn's disease who failed prior biologic treatment. https://clinicaltrials.gov/show/NCT03104413 (first received 7 April 2017).
NCT03105102 {published data only}
- NCT03105102. A multicenter, randomized, double‐blind, placebo controlled 52‐week maintenance and an open‐label extension study of the efficacy and safety of Risankizumab in subjects with Crohn's disease who responded to induction treatment in M16‐006 or M15‐991; or completed M15‐989. ClinicalTrials.gov/show/NCT03105102 (first received 7 April 2017).
NCT03105128 {published data only}
- NCT03105128. A multicenter, randomized, double‐blind, placebo controlled induction study of the efficacy and safety of Risankizumab in subjects with moderately to severely active Crohn's disease. ClinicalTrials.gov/show/NCT03105128 (first received 7 April 2017).
NCT03107793 {published data only}
- NCT03107793. Study of treat to target versus routine care maintenance strategies in Crohn's disease patients treated with Ustekinumab. clinicaltrials.gov/ct2/show/NCT03107793 (first received 11 April 2017).
NCT03162575 {published data only}
- NCT03162575. The possible beneficial effects of mindfulness‐based cognitive therapy (MBCT) in fatigued adult patients with Inflammatory Bowel Disease (IBD). clinicaltrials.gov/ct2/show/NCT03162575 (first received 22 May 2017).
NCT03266484 {published data only}
- NCT03266484. Effect of dietary therapy with a probiotic mixture on the gut microbiome and fatigue symptoms in patients with quiescent inflammatory bowel disease ‐ A clinical trial. clinicaltrials.gov/ct2/show/NCT03266484 (first received 30 August 2017).
NCT03345823 {published data only}
- NCT03345823. A multicenter, randomized, double‐blind, placebo‐controlled maintenance and long‐term extension study of the efficacy and safety of Upadacitinib (ABT‐494) in subjects with Crohn's disease who completed the studies M14‐431 or M14‐433. clinicaltrials.gov/ct2/show/NCT03345823 (first received 17 November 2017).
NCT03345836 {published data only}
- NCT03345836. A multicenter, randomized, double‐blind, placebo‐controlled induction study of the efficacy and safety of Upadacitinib (ABT‐494) in subjects with moderately to severely active Crohn's disease who have inadequately responded to or are intolerant to biologic therapy. clinicaltrials.gov/ct2/show/NCT03345836 (first received 17 November 2017).
NCT03345849 {published data only}
- NCT03345849. A multicenter, randomized, double‐blind, placebo‐controlled induction study of the efficacy and safety of Upadacitinib (ABT‐494) in subjects with moderately to severely active Crohn's disease who have inadequately responded to or are Intolerant to conventional and/or biologic therapies. clinicaltrials.gov/ct2/show/NCT03345849 (first received 17 November 2017).
NCT03398135 {published data only}
- NCT03398135. A multicenter, randomized, double‐blind, placebo controlled 52‐week maintenance and an open‐label extension study of the efficacy and safety of Risankizumab in subjects with ulcerative colitis who responded to induction treatment in M16‐067 or M16‐065. clinicaltrials.gov/ct2/show/NCT03398135 (first received 12 January 2018).
NCT03398148 {published data only}
- NCT03398148. A multicenter, randomized, double‐blind, placebo controlled induction study to evaluate the efficacy and safety of Risankizumab in subjects with moderately to severely active ulcerative colitis who have failed prior biologic therapy. clinicaltrials.gov/ct2/show/NCT03398148 (first received 12 January 2018).
NCT03456752 {published data only}
- NCT03456752. The Impact of perioperative dexamethasone on postoperative outcome in inflammatory bowel diseases. clinicaltrials.gov/ct2/show/NCT03456752 (first received 7 March 2018).
NCT03466411 {published data only}
- NCT03466411. A phase 2/3, randomized, double‐blind, placebo‐ and active‐controlled, parallel‐group, multicenter protocol to evaluate the efficacy and safety of Guselkumab in participants with moderately to severely active Crohn's disease. clinicaltrials.gov/ct2/show/NCT03466411 (first received 15 March 2018).
NCT03574948 {published data only}
- NCT03574948. Multicentric, double‐blind, placebo controlled clinical trial with 5‐hydroxytryptophan (5‐HTP) in patients with inflammatory bowel disease in clinical and biologic remission: effect on fatigue scores. clinicaltrials.gov/ct2/show/NCT03574948 (first received 2 July 2018).
Additional references
Almeida 2016
- Almeida C, Choy EHS, Hewlett S, Kirwan JR, Cramp F, Chalder T, et al. Biologic interventions for fatigue in rheumatoid arthritis. Cochrane Database of Systematic Reviews 2016, Issue 6. [DOI: 10.1002/14651858.CD008334] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bager 2012
- Bager P, Befrits R, Wikman O, Lindgren S, Moum B, Hjortswang H, et al. Fatigue in out‐patients with inflammatory bowel disease is common and multifactorial. Alimentary Pharmacology and Therapeutics 2012;35(1):133‐41. [DOI] [PubMed] [Google Scholar]
Bennett 2009
- Bennett S, Purcell A, Meredith P, Beller E, Haines T, Fleming J. Educational interventions for the management of cancer‐related fatigue in adults. Cochrane Database of Systematic Reviews 2009, Issue 4. [DOI: 10.1002/14651858.CD008144] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bennett 2016
- Bennett S, Pigott A, Beller EM, Haines T, Meredith P, Delaney C. Educational interventions for the management of cancer‐related fatigue in adults. Cochrane Database of Systematic Reviews 2016, Issue 11. [DOI: 10.1002/14651858.CD008144.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bernstein 2010
- Bernstein C, Fried M, Krabshuis JH, Cohen H, Eliakim R, Fedail S, et al. World Gastroenterology Organization Practice Guidelines for the diagnosis and management of IBD in 2010. Inflammatory Bowel Diseases 2010;16(1):112‐24. [DOI] [PubMed] [Google Scholar]
Casati 2000
- Casati J, Toner BB, Rooy EC, Drossman DA, Maunder RG. Concerns of patients with inflammatory bowel disease: a review of emerging themes. Digestive Diseases and Sciences 2000;45(1):26‐31. [DOI] [PubMed] [Google Scholar]
Casellas 2001
- Casellas F, López‐Vivancos J, Badia X, Vilaseca J, Malagelada JR. Influence of inflammatory bowel disease on different dimensions of quality of life. European Journal of Gastroenterology and Hepatology 2001;13(5):567‐72. [DOI] [PubMed] [Google Scholar]
Cella 2002
- Cella D, Eton DT, Lai JS, Peterman AH, Merkel DE. Combining anchor and distribution‐based methods to derive minimal clinically important differences on the Functional Assessment of Cancer Therapy (FACT) anemia and fatigue scales. Journal of Pain and Symptom Management 2002;24(6):547‐61. [DOI] [PubMed] [Google Scholar]
Cella 2005
- Cella D, Yount S, Sorensen M, Chartash E, Sengupta N, Grober J. Validation of the Functional Assessment of Chronic Illness Therapy Fatigue Scale relative to other instrumentation in patients with rheumatoid arthritis. Journal of Rheumatology 2005;32(5):811‐9. [PubMed] [Google Scholar]
Chalder 1993
- Chalder T, Berelowitz G, Pawlikowska T, Watts L, Wessely S, Wright D, et al. Development of a fatigue scale. Journal of Psychosomatic Research 1993;37(2):147‐53. [DOI] [PubMed] [Google Scholar]
Coteur 2009
- Coteur G, Feagan B, Keininger DL, Kosinski M. Evaluation of the meaningfulness of health‐related quality of life improvements as assessed by the SF‐36 and the EQ‐5D VAS in patients with active Crohn’s disease. Alimentary Pharmacology & Therapeutics 2009;29(9):1032‐41. [DOI] [PubMed] [Google Scholar]
Cramp 2012
- Cramp F, Byron‐Daniel J. Exercise for the management of cancer‐related fatigue in adults. Cochrane Database of Systematic Reviews 2012, Issue 11. [DOI: 10.1002/14651858.CD006145.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cramp 2013
- Cramp F, Hewlett S, Almeida C, Kirwan JR, Choy EHS, Chalder T, et al. Non‐pharmacological interventions for fatigue in rheumatoid arthritis. Cochrane Database of Systematic Reviews 2013, Issue 8. [DOI: 10.1002/14651858.CD008322.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cronin 2005
- Cronin CC, Shanahan F. Understanding symptoms and signs in inflammatory bowel disease. In: Targan SR, Shanahan F, Karp LC editor(s). Inflammatory Bowel Disease: From Bench to Bedside. Springer, 2005:253‐67. [Google Scholar]
Czuber‐Dochan 2014a
- Czuber‐Dochan W, Norton C, Bredin F, Darvell M, Nathan I, Terry H. Healthcare professionals' perceptions of fatigue experienced by people with IBD. Journal of Crohn's and Colitis 2014;8(8):835‐44. [DOI] [PubMed] [Google Scholar]
Czuber‐Dochan 2014b
- Czuber‐Dochan W, Norton C, Bredin F, Forbes A, Nathan I, Berliner S, et al. Assessing fatigue in patients with inflammatory bowel disease. Gastrointestinal Nursing 2014;12(8):13‐21. [Google Scholar]
Czuber‐Dochan 2014c
- Czuber‐Dochan W, Norton C, Bassett P, Berliner S, Bredin F, Darvell M, et al. Development and psychometric testing of inflammatory bowel disease fatigue (IBD‐F) patient self‐assessment scale. Journal of Crohn's and Colitis 2014;8(11):1398‐406. [DOI] [PubMed] [Google Scholar]
Czuber‐Dochan 2013a
- Czuber‐Dochan W, Ream E, Norton C. Review article: description and management of fatigue in inflammatory bowel disease. Alimentary Pharmacology and Therapeutics 2013;37(5):505‐16. [DOI] [PubMed] [Google Scholar]
Czuber‐Dochan 2013b
- Czuber‐Dochan W, Dibley LB, Terry H, Ream E, Norton C. The experience of fatigue in people with inflammatory bowel disease: an exploratory study. Journal of Advanced Nursing 2013;69(9):1987‐99. [DOI] [PubMed] [Google Scholar]
Day 2016
- Day J, Yust‐Katz S, Cachia D, Wefel J, Katz LH, Tremont I, et al. Interventions for the management of fatigue in adults with a primary brain tumour. Cochrane Database of Systematic Reviews 2016, Issue 4. [DOI: 10.1002/14651858.CD011376.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
De Rooy 2001
- Rooy EC, Toner BB, Maunder RG, Greenberg GR, Baron D, Steinhart AH, et al. Concerns of patients with inflammatory bowel disease: results from a clinical population. American Journal of Gastroenterology 2001;96(6):1816‐21. [DOI] [PubMed] [Google Scholar]
Deek 2011
- Deeks JJ, Higgins JPT, Altman DG (editors). Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane–handbook.org..
Dittner 2004
- Dittner AJ, Wessely SC, Brown RG. The assessment of fatigue: a practical guide for clinicians and researchers. Journal of Psychosomatic Research 2004;56(2):157‐70. [DOI] [PubMed] [Google Scholar]
Drossman 1989
- Drossman DA, Patrick DL, Mitchell CM, Zagami EA, Appelbaum MI. Health‐related quality of life in inflammatory bowel disease. Digestive Diseases and Sciences 1989;34(9):1379‐86. [DOI] [PubMed] [Google Scholar]
Elbers 2015
- Elbers RG, Verhoef J, Wegen EEH, Berendse HW, Kwakkel G. Interventions for fatigue in Parkinson's disease. Cochrane Database of Systematic Reviews 2015, Issue 10. [DOI: 10.1002/14651858.CD010925.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Farrell 2013
- Farrell D. Symptom Burden in Individuals with Inflammatory Bowel Disease: a Mixed Methods Study [PhD thesis]. University College Cork, Ireland, 2013. [Google Scholar]
Farrell 2016
- Farrell D, Savage E, McCarthy G. Self‐reported symptom burden in individuals with inflammatory bowel disease. Journal of Crohn’s and Colitis 2016;10(3):315‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fisk 1994
- Fisk JD, Ritvo PG, Ross L, Haase DA, Marrie TJ, Schlech WF. Measuring the functional impact of fatigue: initial validation of the fatigue impact scale. Clinical Infectious Diseases 1994;18(Supplement 1):S79‐83. [DOI] [PubMed] [Google Scholar]
Fisk 2002
- Fisk JD, Doble SE. Construction and validation of a fatigue impact scale for daily administration (D‐FIS). Quality of Life Research 2002;11(3):263‐72. [DOI] [PubMed] [Google Scholar]
Garcia‐Vega 2004
- Garcia‐Vega E, Fernandez‐Rodriguez C. A stress management programme for Crohn’s disease. Behaviour Research and Therapy 2004;42(4):367‐83. [DOI] [PubMed] [Google Scholar]
Gibbons 2018
- Gibbons C, Pagnini F, Friede T, Young CA. Treatment of fatigue in amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database of Systematic Reviews 2018, Issue 1. [DOI: 10.1002/14651858.CD011005.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Goedendorp 2009
- Goedendorp MM, Gielissen MFM, Verhagen CAHHVM, Bleijenberg G. Psychosocial interventions for reducing fatigue during cancer treatment in adults. Cochrane Database of Systematic Reviews 2009, Issue 1. [DOI: 10.1002/14651858.CD006953.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Goligher 2008
- Goligher EC, Pouchot J, Brant R, Kherani RB, Avina‐Zubieta JA, Lacaille D, et al. Minimal clinically important difference for 7 measures of fatigue in patients with systemic lupus erythematosus. Journal of Rheumatology 2008;35(4):635‐42. [PubMed] [Google Scholar]
Graff 2011
- Graff LA, Vincent N, Walker JR, Clara I, Carr R, Ediger J, et al. A population‐based study of fatigue and sleep difficulties in inflammatory bowel disease. Inflammatory Bowel Diseases 2011;17(9):1882‐9. [DOI] [PubMed] [Google Scholar]
Heine 2015
- Heine M, Port I, Rietberg MB, Wegen EEH, Kwakkel G. Exercise therapy for fatigue in multiple sclerosis. Cochrane Database of Systematic Reviews 2015, Issue 9. [DOI: 10.1002/14651858.CD009956.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JPT, Deeks JJ, Altman DG (editors). Chapter 16: Special topics in statistics. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Hjollund 2007
- Hjollund NH, Andersen JH, Bech P. Assessment of fatigue in chronic disease: a bibliographic study of fatigue measurement scales. Health Quality Life Outcomes 2007;5(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Irvine 1999
- Irvine EJ. Development and subsequent refinement of the inflammatory bowel disease questionnaire: a quality‐of‐life instrument for adult patients with inflammatory bowel disease. Journal of Pediatric Gastroenterology and Nutrition 1999;28(4):S23‐7. [DOI] [PubMed] [Google Scholar]
Jelsness‐Jørgensen 2011b
- Jelsness‐Jørgensen L‐P, Moum B, Bernklev T. Worries and concerns among inflammatory bowel disease patients followed prospectively over one year. Gastroenterology Research and Practice 2011;2011:492034. [DOI: 10.1155/2011/492034] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jelsness‐Jørgensen 2012
- Jelsness‐Jørgensen L‐P, Bernklev T, Henriksen M, Torp R, Moum B. Chronic fatigue is associated with increased disease‐related worries and concerns in inflammatory bowel disease. World Journal of Gastroenterology 2012;18(5):445‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jelsness‐Jørgensen 2011a
- Jelsness‐Jørgensen L‐P, Bernklev T, Henriksen M, Torp R, Moum BA. Chronic fatigue is more prevalent in patients with inflammatory bowel disease than in healthy controls. Inflammatory Bowel Diseases 2011;17(7):1564‐72. [DOI] [PubMed] [Google Scholar]
Jelsness‐Jørgensen 2011c
- Jelsness‐Jørgensen L‐P, Bernklev T, Henriksen M, Torp R, Moum BA. Chronic fatigue is associated with impaired health‐related quality of life in inflammatory bowel disease. Alimentary Pharmacology and Therapeutics 2011;33(1):106‐14. [DOI] [PubMed] [Google Scholar]
Krupp 1989
- Krupp LB, LaRocca NG, Muir‐Nash J, Steinberg AD. The fatigue severity scale: application to patients with multiple sclerosis and systemic lupus erythematosus. Archives of Neurology 1989;46(10):1121‐3. [DOI] [PubMed] [Google Scholar]
Lai 2003
- Lai J‐S, Cella D, Chang C‐H, Bode RK, Heinemann AW. Item banking to improve, shorten and computerize self‐reported fatigue: an illustration of steps to create a core item bank from the FACIT‐Fatigue Scale. Quality of Life Research 2003;12(5):485‐501. [DOI] [PubMed] [Google Scholar]
Larun 2017
- Larun L, Brurberg K, Odgaard‐Jensen J, Price JR. Exercise therapy for chronic fatigue syndrome. Cochrane Database of Systematic Reviews 2017, Issue 4. [DOI: 10.1002/14651858.CD003200.pub7] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lee 1991
- Lee KA, Hicks G, Nino‐Murcia G. Validity and reliability of a scale to assess fatigue. Psychiatry Research 1991;36(3):291‐8. [DOI] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JPT, Green S. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Minderhoud 2003
- Minderhoud IM, Oldenburg B, Dam PS, Berge Henegouwen GP. High prevalence of fatigue in quiescent inflammatory bowel disease is not related to adrenocortical insufficiency. American Journal of Gastroenterology 2003;98(5):1088‐93. [DOI] [PubMed] [Google Scholar]
Minton 2010
- Minton O, Richardson A, Sharpe M, Hotopf M, Stone P. Drug therapy for the management of cancer‐related fatigue. Cochrane Database of Systematic Reviews 2010, Issue 7. [DOI: 10.1002/14651858.CD006704.pub3] [DOI] [PubMed] [Google Scholar]
Molodecky 2012
- Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012;142(1):46‐54.e42. [DOI] [PubMed] [Google Scholar]
Mücke 2015
- Mücke M, Mochamat, Cuhls H, Peuckmann‐Post V, Minton O, Stone P, et al. Pharmacological treatments for fatigue associated with palliative care. Cochrane Database of Systematic Reviews 2015, Issue 5. [DOI: 10.1002/14651858.CD006788.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Opheim 2014
- Opheim R, Fagermoen MS, Bernklev T, Jelsness‐Jorgensen L‐P, Moum B. Fatigue interference with daily living among patients with inflammatory bowel disease. Quality of Life Research 2014;23(2):707‐17. [DOI] [PubMed] [Google Scholar]
Piper 1998
- Piper BF, Dibble SL, Dodd MJ, Weiss MC, Slaughter RE, Paul SM. The revised Piper Fatigue Scale: psychometric evaluation in women with breast cancer. Oncology Nursing Forum 1998;25(4):677‐84. [PubMed] [Google Scholar]
Pouchot 2008
- Pouchot J, Kherani RB, Brant R, Lacaille D, Lehman AJ, Ensworth S, et al. Determination of the minimal clinically important difference for seven fatigue measures in rheumatoid arthritis. Journal of Clinical Epidemiology 2008;61:705‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Preston 2012
- Preston NJ, Hurlow A, Brine J, Bennett MI. Blood transfusions for anaemia in patients with advanced cancer. Cochrane Database of Systematic Reviews 2012, Issue 2. [DOI: 10.1002/14651858.CD009007.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Price 2008
- Price JR, Mitchell E, Tidy E, Hunot V. Cognitive behaviour therapy for chronic fatigue syndrome in adults. Cochrane Database of Systematic Reviews 2008, Issue 3. [DOI: 10.1002/14651858.CD001027.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pucci 2007
- Pucci E, Brañas Tato P, D'Amico R, Giuliani G, Solari A, Taus C. Amantadine for fatigue in multiple sclerosis. Cochrane Database of Systematic Reviews 2007, Issue 1. [DOI: 10.1002/14651858.CD002818.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2011a
- Schünemann HJ, Oxman AD, Higgins JPT, Vist GE, Glasziou P, Guyatt GH. Chapter 11: Presenting results and ‘Summary of findings' tables. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Schünemann 2011b
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Smets 1996
- Smets EM, Garssen B, Cull A, Haes JC. Application of the multidimensional fatigue inventory (MFI‐20) in cancer patients receiving radiotherapy. British Journal of Cancer 1996;73(2):241‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JAC, Egger M, Moher D (editors). Chapter 10: Addressing reporting biases. In: Higgins JPT, Green S. Cochrane Handbook for Systematic Reviews of Intervention. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Stone 2008
- Stone P, Minton O. Cancer‐related fatigue. European Journal of Cancer 2008;44(8):1097‐104. [DOI] [PubMed] [Google Scholar]
Tack 1991
- Tack BB. Dimensions and Correlates of Fatigue in Older Adults with Rheumatoid Arthritis [PhD thesis]. San Francisco: University of California, 1991. [Google Scholar]
Tejani 2012
- Tejani AM, Wasdell M, Spiwak R, Rowell G, Nathwani S. Carnitine for fatigue in multiple sclerosis. Cochrane Database of Systematic Reviews 2012, Issue 5. [DOI: 10.1002/14651858.CD007280.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Van Langenberg 2010
- Langenberg DR, Gibson PR. Systematic review: fatigue in inflammatory bowel disease. Alimentary Pharmacology and Therapeutics 2010;32(2):131‐43. [DOI] [PubMed] [Google Scholar]
Ware 1992
- Ware JE Jr, Sherbourne CD. The MOS 36‐item short‐form health survey (SF‐36). I. Conceptual framework and item selection. Medical Care 1992;30(6):473‐83. [PubMed] [Google Scholar]
White 2014
- White CM, Doorn PA, Garssen MPJ, Stockley RC. Interventions for fatigue in peripheral neuropathy. Cochrane Database of Systematic Reviews 2014, Issue 12. [DOI: 10.1002/14651858.CD008146.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wilson 2012
- Wilson B, Lönnfors S, Vermeire S. The true impact of IBD: a European Crohn's and Ulcerative Colitis patient life. IMPACT Survey 2010‐2011. 21st Century Challenges Autoimmune Diseases. European Federation of Crohn’s and Ulcerative Colitis Associations (EFCCA), Brussels, Belgium; European Crohn’s and Colitis Organization (ECCO); 2012; Vienna, Austria. 2012.
Yellen 1997
- Yellen SB, Cella DF, Webster K, Blendowski C, Kaplan E. Measuring fatigue and other anaemia‐related symptoms with the Functional Assessment of Cancer Therapy (FACT) measurement system. Journal of Pain and Symptom Management 1997;13(2):63‐74. [DOI] [PubMed] [Google Scholar]