

Summary
As an approach to determining the aetiology of chronic lymphocytic leukaemia (CLL), we searched for a virus expressed in human CLL B‐cells by combining high‐throughput sequencing and digital subtraction. Pooled B‐cell mRNA transcriptomes from five CLL patients and five healthy donors were sequenced with 454 Life Sciences technology. Human reads were excluded by BLAST (Basic Local Alignment Search Tool) and BLAT (BLAST‐like alignment tool) searches. Remaining reads were screened with BLAST against viral databases. Purified B‐cells from two CLL patients, with and without stimulation by phorbol‐esters, were sequenced using Illumina technology to achieve depth of sequencing. Burrows‐Wheeler Aligner mapping and BLAST searches were used for the Illumina data. Pyrosequencing resulted in about 400 000 reads per sample. No viral candidate could be found. Illumina single‐end sequencing for 115 cycles yielded an average of 26 ± 2·5 million filtered reads per sample, of which 2·2 ± 0·6 million remained unmapped to human references. BLAST searches of these reads against viral and human databases assigned nine reads to an Epstein‐Barr virus origin, in one sample following phorbol‐ester stimulation. Other reads showing a putative viral origin were dismissed after further analysis. Despite an in‐depth analysis of the CLL transcriptome reaching more than 100 million sequences, we have not found evidence for a putative viral candidate in CLL.
Keywords: chronic lymphocytic leukaemia, aetiology, virus, high‐throughput sequencing, digital transcriptome subtraction
Introduction
Chronic Lymphocytic Leukaemia (CLL) is the most common form of leukaemia in Western countries and mainly affects elderly individuals. It follows an extremely variable course, with survival ranging from months to decades. Available treatments often induce disease remission, but almost all patients will relapse and there is a consensus that CLL remains incurable. Multiple instances of the disease in some families and a low incidence among individuals of Japanese origin (including those who migrated to Hawaii), suggest that genetic influences are stronger than environmental factors in its pathogenesis (Weiss, 1979). Genome‐wide association studies have detected some loci influencing CLL risk (Sellick et al, 2007; Crowther‐Swanepoel et al, 2010; Slager et al, 2011) and recent whole‐genome and whole‐exome sequencing studies addressed the repertoire of somatic mutations and other genetic lesions in the disease (Fabbri et al, 2011; Puente et al, 2011; Wang et al, 2011; Quesada et al, 2012). Among the few genes found recurrently mutated, NOTCH1 and SF3B1 emerged as predictors of poor survival (Sportoletti et al, 2010; Fabbri et al, 2011; Puente et al, 2011; Rossi et al, 2011; Wang et al, 2011; Quesada et al, 2012). Several studies have started to shed light on the nature of genetic predisposition of CLL, however none of the reported genetic aberrations (mutations, deletions or trisomies) have been shown to be a major cause of the disease and the basis of this disorder remains unknown (Sellick et al, 2007; Dighiero & Hamblin, 2008).
Since the Peyton Rous discovery in 1911 that avian sarcoma could be transmitted by non‐cellular filtrates, followed by the discovery of mammary tumour virus, murine leukaemia virus and polyoma virus between 1931 and 1958, at least six different viruses have been implicated in about 15% of human cancers (Klein, 2002). They include the DNA viruses Epstein‐Barr virus (EBV), human papilloma virus, hepatitis B virus and Human Herpes Virus 8 (HHV‐8), as well as RNA viruses, such as hepatitis C or human adult T cell leukaemia virus (HTLV). Class I or direct‐transforming RNA tumour viruses carry cellular oncogenes but are not known to play any role in tumour causation in nature. Class II or chronic RNA tumour viruses do not carry cell‐derived oncogenes but act through proviral DNA insertion into the immediate neighbourhood of a cellular oncogene. Feline, murine, and avian leukaemia viruses belong to this category. HTLV and Bovine Leukaemia Virus (BLV) expand the pre‐neoplastic cell population through transactivation induced by viral Tax protein, thereby providing the seed for secondary cellular changes.
An animal model of CLL, Enzootic Bovine Leukaemia (EBL), is induced by BLV, a retrovirus belonging to the Deltaretrovirus genus and closely related to the HTLV‐1 virus. This virus induces hyper lymphocytosis of CD5+ B‐cells in a significant number of animals and causes aggressive disease in about 10% of cases (Gillet et al, 2007). Despite this suggestive animal model, little research has been devoted to this issue by using classical polymerase chain reaction (PCR) technology and no known virus has been identified in the case of human CLL (Hermouet et al, 2003). Attempts have been made to associate EBV with CLL, but accumulated data argue against a role of EBV in the early development of the disease and would only suggest a possible involvement in the generation of secondary malignancies (Tsimberidou et al, 2006; Dolcetti & Carbone, 2010; Tarrand et al, 2010). Recent studies have also addressed the presence of Merkel cell polyomavirus (MCPyV) in CLL (Koljonen et al, 2009; Shuda et al, 2009; Pantulu et al, 2010; Tolstov et al, 2010; Teman et al, 2011), but the results regarding MCPyV DNA are contradictory, showing extremely low MCPyV copy number if detected. Given the high seroprevalence of this virus in the analysed populations and the progressive immunodeficiency associated to CLL, MCPyV detection might be more likely explained by background infection or low‐level viral reactivation in CLL‐induced immunodeficiency (Koljonen et al, 2009; Shuda et al, 2009; Pantulu et al, 2010; Tolstov et al, 2010; Teman et al, 2011).
In recent years, molecular techniques have been successfully applied in the identification of infectious agents, such as Borna virus, Kaposi sarcoma‐associated herpesvirus (HHV‐8), West Nile virus, and the Severe Acute Respiratory Syndrome (SARS) coronavirus. Such efforts fail, however, when the agents in question are truly novel or sufficiently distant in sequence from related agents to allow hybridization, or if they are poorly expressed at the RNA level. The advent of high‐throughput sequencing in combination with bioinformatics analysis could pave the way to the discovery of new infectious pathogens. This approach led to the identification of a new species of arenavirus in patients suffering from an infectious disease for which a causative pathogen could not be detected using any of the available diagnostic procedures (Palacios et al, 2008). Similarly, the novel MCPyV was characterized from Merkel cell carcinoma samples and recognized as a contributing factor in the pathogenesis (Feng et al, 2008). Thus, this methodology is suitable for applications in which the infectious pathogen is unknown. In the current study, we searched for the presence of a virus expressed at the RNA level in human CLL, by using massive sequencing technology associated with a digital transcriptome subtraction method.
Methods
Patients
Peripheral blood samples were obtained from seven CLL patients meeting the diagnostic criteria of the International Workshop on Chronic Lymphocytic Leukaemia‐sponsored Working Group (Hallek et al, 2008) and five healthy donors. Written informed consent was obtained in accordance with the ethical regulations of Uruguay and the Declaration of Helsinki. The selected CLL patients were diagnosed with a progressive phenotype (Table 1). Peripheral blood mononuclear cells (PBMCs) were isolated by centrifugation on Ficoll‐Hypaque (Pharmacia Fine Chemicals, Uppsala, Sweden) and immediately cryopreserved in liquid nitrogen.
Table 1.
Clinical and molecular characteristics of patients
| Patient | Sex | Age (years) | Binet | CD38 | LPL | MS | Outcome |
|---|---|---|---|---|---|---|---|
| 454 pool | |||||||
| CLL 046 | F | 45 | A | Neg | Pos | UM | Progressor |
| CLL 072 | M | 61 | C | Neg | Pos | UM | Indolent |
| CLL 080 | F | 63 | C | Neg | Pos | UM | Progressor |
| CLL 083 | M | 60 | B | Pos | ND | UM | Progressor |
| CLL 096 | M | 70 | C | Neg | Neg | UM | Progressor |
| Illumina | |||||||
| CLL 238 | M | 76 | C | Neg | Neg | M | Progressor |
| CLL 250 | M | 57 | B | ND | Pos | UM | Progressor |
Binet, Binet stage; LPL, lipoprotein lipase; MS, mutational status; F, female; M, male; Neg, negative; Pos, positive; ND, no data; UM, unmutatated; M, mutated.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
454 Life Sciences pyrosequencing
Total RNA from five CLL patients and five healthy donors was extracted using the RNeasy Midi Kit (Qiagen, Alameda, CA, USA). The integrity of the RNA was analysed on the Agilent 2100 Bioanalyser (Quantum Analytics, Foster City, CA, USA). Poly‐adenylated RNA was purified with Dynabeads® mRNA Purification Kit (Invitrogen, Carlsbad, CA, USA). Two double‐stranded cDNA samples were synthesized for a pool of the five CLL patients and a pool of the normal individuals with oligo(dT) primer using the SuperScript Double‐stranded cDNA Synthesis Kit (Invitrogen).
Libraries were made from both pools and each library was pyrosequenced on one full LR70 plate (two plates total) using the Standard 454 Life Sciences FLX Sequencing Chemistry (454 Life Sciences, Branford, CT, USA).
Illumina Genome Analyzer sequencing
Two additional CLL patients were sequenced individually. Purification of the B‐cell population was performed by flow cytometry (MoFlo cell sorter; Beckman Coulter Inc., Brea, CA, USA) with phycoerythrin conjugated anti‐CD19 monoclonal antibody (DAKO, Glostrup, Denmark). The purity of isolated sub‐populations was shown to be greater than 97% after flow cytometric evaluation.
For activation of isolated B‐cells, 12‐O‐tetradecanoylphorbol‐13 acetate (TPA) was dispensed from 0·3 mmol/l stock solution in dimethyl sulfoxide to a final concentration of 0·15 μmol/l. Following TPA addition, CLL B‐cells were incubated for 17 h. Maturation of TPA‐treated cells was documented by a 20% increase in mean cell volume and by characteristic changes in cell morphology.
Total RNA from activated and non‐activated B‐cells was isolated with TRIzol® (Invitrogen) and quality control was performed as previously described. Libraries were prepared using a slightly modified version of the pre‐release protocol for directional RNA‐Seq available from Illumina (Illumina Inc., San Diego, CA, USA). Normalization using duplex‐specific thermostable nuclease (DSN; Evrogen, Moscow, Russia) was then performed before the cluster generation step. The normalization is based on denaturation of DNA libraries followed by addition of DSN after partial renaturation. Highly expressed genes, such as ribosomal RNA, tRNA and house‐keeping genes, rapidly renature to form double‐stranded DNA and are degraded by the DSN enzyme, while DNA molecules derived from less abundant transcripts are preserved (Zhulidov et al, 2004; Bogdanova et al, 2009).
Single‐read sequencing was conducted for 115 cycles on the Illumina Genome Analyzer IIx, using one flowcell lane per library. Base calling and quality scores were produced using the illumina genome analyzer software v1.6.
Bioinformatics analysis
Sequences were filtered and trimmed as appropriate for the sequencing technology used in each case. After removal of ribosomal sequences, reads were mapped against the human genome and transcriptome references downloaded from UCSC (University of California, Santa Cruz) Genome Browser (Kent et al, 2002) and Ensembl (Flicek et al, 2011) databases. Remaining reads (i.e., those for which we could not determine a human origin) were screened through BLAST (Basic Local Alignment Search Tool) (Altschul et al, 1997) searches in RefSeq (Pruitt et al, 2009) viral databases to identify putative xenobiotic sequences, if present. These steps follow the main idea of the original digital subtraction proposal (Weber et al, 2002), but we introduced modifications or changed alignment tools in order to cope with the different attributes of the sequence data produced by each technology (Fig 1A–B summarizes the pipelines employed for 454 and Illumina data, respectively). A detailed description of the analysis and simulations conducted is given in online supplementary Data S1 Fig S2 and Table SI.
Figure 1.
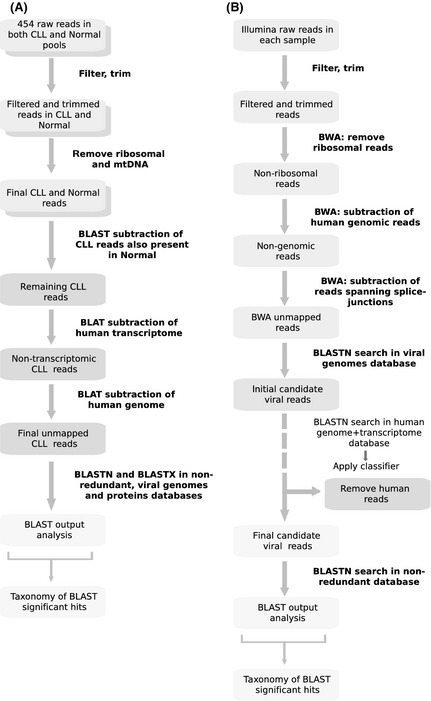
Computational subtraction approach developed to identify non‐human sequences in the chronic lymphocytic leukaemia (CLL) transcriptome. (A) Subtraction strategy for 454 data. (B) Subtraction strategy for Illumina data.
Results
Analysis of 454 data
Pyrosequencing resulted in 400 324 and 454 150 raw reads for the pooled CLL and Normal mRNA samples, respectively (Fig S1). After a trimming and filtering step including removal of ribosomal reads, 376 731 and 433 255 reads remained for the CLL and Normal samples, respectively [CLL average length: 224 ± 56 nucleotides (nts); Normal average length: 241 ± 47 nts).
To isolate reads present only in the CLL pooled sample, an initial search using BLAST allowed us to discard 259 881 CLL reads that were also present in the Normal sample. The remaining sequences were sequentially aligned with BLAT (BLAST‐like alignment tool) (Kent, 2002) to the human transcriptome and genome, returning a list of 2968 candidate reads (reads with no or poor alignments). No significant matches were found for these reads in viral databases, the few recovered alignments were short and had high E‐values (Fig S1). Though we did not necessarily expect high similarity and alignment quality to known viral agents, when carefully re‐examined, these reads also displayed alignments of similar quality against the human transcriptome or genome. Thus, we were not able to find any putative sequence of viral origin (known or novel) that was present in the CLL pooled sample but absent in the Normal pool.
Analysis of Illumina data
The 454 sequencing was limited to approximately 400 000 sequences from the CLL transcriptome, a sequencing depth that bioinformatics analysis suggested would be insufficient to detect rare viral transcripts (see details below). We therefore decided to study two additional patients using Illumina sequencing technology, which offers greatly increased depth at the expense of shorter read length. Both patients were diagnosed with a progressive malignancy, even in the IgG non‐mutated case (Table 1). Two substantial improvements in this dataset were the preparation of libraries from normalized total RNA (rather than only poly‐adenylated mRNAs) and a 90‐fold increase in the amount of reads, enabling the detection of transcripts with very low expression level. Furthermore, to exclude the possibility that the virus could be integrated in the genome and barely transcribed, we analysed the isolated cells prior to and after treatment with phorbol‐ester, a potent protein kinase C (PKC) activator (Blumberg, 1988).
Illumina sequencing yielded 36 ± 2·7 million raw reads per sample, from which 26 ± 2·5 million reads per sample remained after filtering steps (Table 2, Data S1). As previously mentioned, given this very large amount of sequence data, changes to the mapping tools and final BLAST searches of the subtraction pipeline were necessary. To begin with, we used Burrows‐Wheeler Aligner (BWA) (Li & Durbin, 2009) to discard reads of human origin by successive mapping to ribosomal RNAs, the human genome and transcriptome databases. A summary of BWA mapping results (Table 2) shows that the ribosomal content of our total RNA libraries was 18·0% ± 3·3%, a figure comparable to other ribosomal depletion protocols. Regarding non‐ribosomal reads, 67·3% ± 6·2% and 7·2% ± 0·6% aligned to genome and transcriptome databases respectively, while 2·2 ± 0·6 million reads remained unmapped.
Table 2.
Illumina sequencing, filtering and mapping statistics
| Reads | CLL250 | CLL250_act | CLL238 | CLL238_act |
|---|---|---|---|---|
| Raw counts | 34 248 055 | 38 889 936 | 33 948 020 | 38 421 323 |
| Filtering steps | ||||
| Purity filter | 28 771 020 | 32 477 113 | 29 210 502 | 31 924 855 |
| Additional filter | 22 479 555 | 27 956 701 | 26 039 540 | 27 613 647 |
| Burrows‐Wheeler Aligner mapping steps | ||||
| Ribosomal | 5 050 513 | 5 022 371 | 3 851 214 | 4 627 416 |
| Genomic | 13 790 644 | 18 914 191 | 18 738 268 | 17 791 494 |
| Transcriptomic | 1 634 399 | 2 050 829 | 1 677 217 | 2 144 262 |
| Final unmapped | 2 004 005 | 1 969 310 | 1 772 841 | 3 050 475 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
In the second phase, these unmapped reads were first subjected to a nucleotide BLAST similarity search against a database of viral genomes [Fig 1B; see Data S1 for a discussion on the BLASTN (nucleotide BLAST) protocol used]. An average of 219 499 ± 5111 reads per sample had matches in at least one viral genome (Table 3). These reads were thus considered as initial candidate viral reads. However, many candidate viral sequences also matched the human genome or transcriptome and could, by means of a linear discriminant function (see Data S1 for details), be assigned to a human origin (Table 3). Next, BLAST similarity searches were performed for both unmapped candidate viral reads and reads with a poor match in human (posterior probability lower than 0·9), against the whole non‐redundant nucleotide database (nt), followed by a careful inspection of the results.
Table 3.
BLASTN results of unmapped reads
| CLL250 | CLL250_act | CLL238 | CLL238_act | |
|---|---|---|---|---|
| Initial viral reads | 230 221 | 200 276 | 163 277 | 284 222 |
| Non‐human readsa | 689 | 551 | 705 | 790 |
| Final viral readsb | 0 | 0 | 0 | 9 |
All initial viral reads were BLASTN searched in human databases. Non‐human reads are those that were not aligned and, if aligned, those classified by the discriminant function either as ‘viral’ or ‘human’ with posteriors lower than 0·9.
After a careful inspection of BLASTN searches in the non‐redundant database, only nine reads were conclusively assigned to a viral origin.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
As expected, reads with higher similarity to mammalian sequences constituted an abundant class in the four samples (Fig 2; Table SII and Data S2). Most reads matching viral sequences mapped anti‐sense to the primer binding site (PBS) of the Mason‐Pfizer monkey virus (MPMV). At its 5′ end, this primate betaretrovirus carries two long terminal repeats separated by 63 nucleotides complementary to tRNA‐Lys, the usual primer for reverse transcription of the viral genome (Sonigo et al, 1986). Further analysis revealed that these reads would be chimeric‐like sequences, where the first stretch of the read derives from human tRNA‐Lys precursors and the following region is variable (Data S1 and Fig S3). As supported by the absence of additional MPMV reads in our transcriptomes (i.e., reads aligning to MPMV genome loci other than PBS), we definitively excluded a true viral origin for these reads (Data S1). Other reads showing a putative viral origin were also dismissed because: (i) they aligned to viral genomes with qualities not better than to human [most of them were classified as human by linear discriminant analysis (LDA), although with a posterior probability lower than 0·9]; (ii) had low complexity repeats; (iii) were unknown to be related to human disease (see Grouper iridovirus, Sindbis virus, Glypta fumiferanae and a Lausannevirus isolate in online supplementary Data S2). Finally, nine reads from one of the two phorbol‐ester activated samples (Table 3 and Data S2) showed high similarity to the Epstein‐Barr virus genome (EBV), in a region of the BcLF1 gene. In this case, alignments to the EBV genome involved the full length of the reads with non‐hits to neither human genome nor transcriptome. Thus, these nine reads were considered to have a true viral origin.
Figure 2.
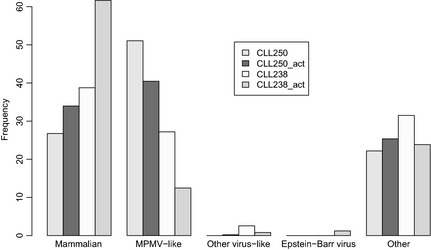
Taxonomy of BLASTN results of the final candidate viral reads in the non‐redundant database. The shown classes were defined arbitrarily (online Data S1 and Table SII). Only the ‘Epstein‐Barr virus’ class involves reads with a final viral assignment. MPMV, Mason‐Pfizer monkey virus.
Illumina versus 454 data
As expected, the use of Illumina sequencing technology drastically increased the transcriptome depth reached. Comparison of read counts from all CLL samples showed: (i) 14 980 and 20 436 genes were detectable by 454 and Illumina, respectively; (ii) no gene present in 454 data was absent from Illumina data; (iii) absent genes from 454 showed counts with medians higher than 0 in Illumina samples (Fig S4 and Data S1). For instance, 3993 genes absent from 454 data were detected by at least five reads with Illumina sequencing (3355 fulfilled the additional requirement of being present in at least two samples). In addition, we focused on two well‐known genes in CLL disease: ADAM29 and LPL. Specifically, these genes are known to be differentially expressed in mutated and unmutated CLL patients respectively, showing an opposite behaviour between them (Oppezzo et al, 2005). Raw read counts from the 454 assay (pool of unmutated patients) could only detect seven reads for LPL and one read for ADAM29. Meanwhile, Illumina technology accounted for 477 reads for LPL and only one read for ADAM29, in the case of the unmutated sample. For the mutated patient, Illumina raw read counts were one and 420 for LPL and ADAM29, respectively. Notably, these expected LPL/ADAM29 expression ratios could only be detected by Illumina sequencing, demonstrating the higher resolution of this method.
Discussion
The first draft of the human genome enabled the development of new techniques to detect the presence of foreign sequences in a human transcriptome through in silico filtering of the transcripts against the reference genome (Weber et al, 2002). Xu et al (2003) showed that this simple digital subtraction approach was successful in detecting viral sequences from an Epstein‐Barr virus infected tissue. With the advent of massively parallel sequencing technologies, a renewed interest in the discovery of novel pathogens arose and new viruses have recently been identified as causative agents of human disease (Feng et al, 2008; Palacios et al, 2008).
To date, 454 Life Sciences has been the chosen technology to discover new pathogens from transcriptome data, mainly because of the longer read length, which simplifies identification of the novel agent once the non‐human reads are detected. However, as Illumina technology has achieved increased read lengths and developed protocols for paired‐end sequencing, the advantage of 454 has become overpowered by the impressive amount of sequence data produced by Illumina Genome Analyzer and HiSeq sequencers, at least in cases where the foreign agent is expected to be weakly expressed in the sample library. In this respect, Illumina sequencing paired with a digital subtraction strategy has recently been shown to be sensitive enough to mine RNA‐Seq libraries with decreasing amounts of a spiked RNA‐virus (Moore et al, 2011). Recently, small RNA Illumina sequencing also proved suitable for discovering viruses in plants and insects, a strategy based on the small interfering RNA (siRNA) immune response that these organisms trigger against virus infection (Kreuze et al, 2009; Wu et al, 2010; Ma et al, 2011).
Presently, no causative factor has been firmly linked to the aetiology of CLL, the most common form of leukaemia among Caucasian populations. To analyse the putative involvement of a viral agent in its onset, we conducted two surveys on CLL transcriptomes acquired by massively parallel sequencing technologies. First, we applied digital subtraction of human sequences to 454 sequencing data for a polyA‐positive RNA from a pool of five CLL patients. To improve sensitivity, we then prepared paired samples of B‐cells with and without prior treatment with a potent cell activator for two additional patients. Normalized libraries for total RNA were sequenced with Illumina technology, increasing the data yield over 454 sequencing. As we have shown, this approach allowed an important increase in transcript detection, very pronounced for genes expressed at low levels.
Nine reads of viral origin (EBV) were detected in one activated sample. EBV prevalence in humans is approximately 90% and the evidence accumulated so far strongly support its involvement in the pathogenesis of a wide spectrum of human malignancies, particularly in the case of immuno‐compromised patients (Thompson & Kurzrock, 2004). As for the role of EBV in CLL development, EBV infection has been demonstrated to only play a role in the case of secondary malignancies, generally involving a new clone (Tsimberidou et al, 2006; Dolcetti & Carbone, 2010; Tarrand et al, 2010). Thus, given available data, the assignment of nine reads to an EBV origin were considered neither unexpected nor relevant to this work.
Retroviruses were first discovered as tumour‐inducing agents in animals. The discovery of HTLV‐1 and its role in adult T cell leukaemia confirmed that retroviruses can also be oncogenic in humans, initiating extensive research on retroviral aetiology of human chronic diseases and cancer. A caveat of our study is the fact that a retroviral candidate of CLL would not be detectable using this strategy if it shows high similarity to human endogenous retroviral (HERV) sequences, allowing reads to be mapped to the human genome assembly and not further analysed (Voisset et al, 2008). Also, a HERV transcribed and related to CLL would not be detected by our pipeline. However, it is worth mentioning that the bovine analogous model of CLL is produced by a deltaretrovirus (BLV). As there are no HERVs related to deltaretroviruses, we did not expect to specifically face these confounding factors. Furthermore, with the single exceptions of HIV and HTLV, the role of retroviruses in human diseases remains highly controversial (Voisset et al, 2008; Weiss, 2010). For instance, the last of the RNA ‘rumour’ viruses, the Xenotropic murine leukaemia virus‐related virus, seems to have been erroneously related to disease as consequence of a contaminant artefact (Robinson et al, 2011; Simmons et al, 2011).
In conclusion, despite having analysed CLL transcriptomes from seven different patients by massively parallel sequencing technology and provided the depth needed to detect a putative viral agent, we failed to identify any candidate viral sequence. Although this search was conducted with two powerful, up‐to‐date technologies, our data do not definitively exclude a viral origin for human CLL because the existence of a novel virus sufficiently distant in sequence from any related agents would be difficult to detect. Given that our study included a limited number of patients, we cannot exclude that an infectious agent could be found in a small percentage of patients, as we cannot exclude the possibility that the virus could operate as a hit and run agent. Also, an infectious agent might operate indirectly through attacking microenvironment cells.
Further studies at the genomic level might be required to definitively exclude the presence of an integrated retrovirus. A truly interesting and novel concept would be a population genomics study, designed to explore the potential role of polymorphic HERVs as aetiological factors of CLL.
Authorship contributions
SB, PM and HP performed the experiments; NR and HN conducted the bioinformatics analysis; AK, HP and AP contributed to the analysis; SB, PO, CR, GD and OP designed the study; GD and OP coordinated the study. All authors were involved in the analysis and the interpretation of the results. All authors read, gave comments and approved the final version of the manuscript.
Conflict of interests
The authors reported no potential conflicts of interest.
Supporting information
Data S1. Methods and results.
Data S2. Similarity search results of the BLAST search of final viral candidate reads (Illumina reads) against NCBI's non‐redundant database.
Fig S1. Computational subtraction approach developed to identify non‐human sequences in the CLL transcriptome obtained with the 454 technology.
Fig S2. Simulated reads from HIV and EBV genomes were pooled and searched using different BLASTN protocols in the viral genome database.
Fig S3. Compositional analysis of 1203 reads that aligned anti‐sense to the primer binding site PBS of the MPMV genome (sample CLL250).
Fig S4. Median values of Illumina raw read counts for genes with 0‐5 read counts when previously analysed by 454.
Table SI. Performance of different BLASTN protocols when searching simulated HIV reads.
Table SII. Distribution of BLASTN hits in the non‐redundant database.
Acknowledgements
This work was supported by a grant from Agencia Nacional de Investigación e Innovación, Fondo Clemente Estable (FCE 2009_2611), and Centre Nationale de la Recherche Scientifique (CNRS – LIA Laboratoire Franco‐Uruguayen sur la Pathogénèse virale des leucémies).
NR and SB contributed equally to this manuscript.
References
- Altschul, S.F. , Madden, T.L. , Schaffer, A.A. , Zhang, J. , Zhang, Z. , Miller, W. & Lipman, D.J. (1997) Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Research, 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumberg, P.M. (1988) Protein kinase C as the receptor for the phorbol ester tumor promoters: sixth Rhoads memorial award lecture. Cancer Research, 48, 1–8. [PubMed] [Google Scholar]
- Bogdanova, E.A. , Shagina, I.A. , Mudrik, E. , Ivanov, I. , Amon, P. , Vagner, L.L. , Lukyanov, S.A. & Shagin, D.A. (2009) DSN depletion is a simple method to remove selected transcripts from cDNA populations. Molecular Biotechnology, 41, 247–253. [DOI] [PubMed] [Google Scholar]
- Crowther‐Swanepoel, D. , Broderick, P. , Di Bernardo, M.C. , Dobbins, S.E. , Torres, M. , Mansouri, M. , Ruiz‐Ponte, C. , Enjuanes, A. , Rosenquist, R. , Carracedo, A. , Jurlander, J. , Campo, E. , Juliusson, G. , Montserrat, E. , Smedby, K.E. , Dyer, M.J. , Matutes, E. , Dearden, C. , Sunter, N.J. , Hall, A.G. , Mainou‐Fowler, T. , Jackson, G.H. , Summerfield, G. , Harris, R.J. , Pettitt, A.R. , Allsup, D.J. , Bailey, J.R. , Pratt, G. , Pepper, C. , Fegan, C. , Parker, A. , Oscier, D. , Allan, J.M. , Catovsky, D. & Houlston, R.S. (2010) Common variants at 2q37.3, 8q24,21, 15q21,3 and 16q24.1 influence chronic lymphocytic leukemia risk. Nature Genetics, 42, 132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dighiero, G. & Hamblin, T.J. (2008) Chronic lymphocytic leukaemia. Lancet, 371, 1017–1029. [DOI] [PubMed] [Google Scholar]
- Dolcetti, R. & Carbone, A. (2010) Epstein‐Barr virus infection and chronic lymphocytic leukemia: a possible progression factor? Infectious Agent and Cancer, 5, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri, G. , Rasi, S. , Rossi, D. , Trifonov, V. , Khiabanian, H. , Ma, J. , Grunn, A. , Fangazio, M. , Capello, D. , Monti, S. , Cresta, S. , Gargiulo, E. , Forconi, F. , Guarini, A. , Arcaini, L. , Paulli, M. , Laurenti, L. , Larocca, L.M. , Marasca, R. , Gattei, V. , Oscier, D. , Bertoni, F. , Mullighan, C.G. , Foá, R. , Pasqualucci, L. , Rabadan, R. , Dalla‐Favera, R. & Gaidano, G. (2011) Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. The Journal of Experimental Medicine, 208, 1389–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, H. , Shuda, M. , Chang, Y. & Moore, P.S. (2008) Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science, 319, 1096–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek, P. , Amode, M.R. , Barrell, D. , Beal, K. , Brent, S. , Chen, Y. , Clapham, P. , Coates, G. , Fairley, S. , Fitzgerald, S. , Gordon, L. , Hendrix, M. , Hourlier, T. , Johnson, N. , Kähäri, A. , Keefe, D. , Keenan, S. , Kinsella, R. , Kokocinski, F. , Kulesha, E. , Larsson, P. , Longden, I. , McLaren, W. , Overduin, B. , Pritchard, B. , Riat, H.S. , Rios, D. , Ritchie, G.R. , Ruffier, M. , Schuster, M. , Sobral, D. , Spudich, G. , Tang, Y.A. , Trevanion, S. , Vandrovcova, J. , Vilella, A.J. , White, S. , Wilder, S.P. , Zadissa, A. , Zamora, J. , Aken, B.L. , Birney, E. , Cunningham, F. , Dunham, I. , Durbin, R. , Fernández‐ Suarez, X.M. , Herrero, J. , Hubbard, T.J. , Parker, A. , Proctor, G. , Vogel, J. & Searle, S.M. (2011) Ensembl 2011. Nucleic Acids Research, 39, D800–D806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet, N. , Florins, A. , Boxus, M. , Burteau, C. , Nigro, A. , Vandermeers, F. , Balon, H. , Bouzar, A.B. , Defoiche, J. , Burny, A. , Reichert, M. , Kettmann, R. & Willems, R. (2007) Mechanisms of leukemogenesis induced by bovine leukemia virus: prospects for novel anti‐retroviral therapies in human. Retrovirology, 4, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallek, M. , Cheson, B.D. , Catovsky, D. , Caligaris‐Cappio, F. , Dighiero, G. , Döhner, H. , Hillmen, P. , Keating, M.J. , Montserrat, E. , Rai, K.R. , Kipps, T.J. & International Workshop on Chronic Lymphocytic Leukemia. (2008) Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute–Working Group 1996 guidelines. Blood, 111, 5446–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermouet, S. , Sutton, C.A. , Rose, T.M. , Greenblatt, R.J. , Corre, I. , Garand, R. , Neves, A.M. , Bataille, R. & Casey, J.W. (2003) Qualitative and quantitative analysis of human herpesviruses in chronic and acute B cell lymphocytic leukemia and in multiple myeloma. Leukemia, 17, 185–195. [DOI] [PubMed] [Google Scholar]
- Kent, W.J. (2002) BLAT‐the BLAST‐like alignment tool. Genome Research, 12, 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, W.J. , Sugnet, C.W. , Furey, T.S. , Roskin, K.M. , Pringle, T.H. , Zahler, A.M. & Haussler, D. (2002) The human genome browser at UCSC. Genome Research, 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, G. (2002) Perspectives in studies of human tumor viruses. Frontiers in Bioscience, 7, d268–d274. [DOI] [PubMed] [Google Scholar]
- Koljonen, V. , Kukko, H. , Pukkala, E. , Sankila, R. , Böhling, T. , Tukiainen, E. , Sihto, H. & Joensuu, H. (2009) Chronic lymphocytic leukaemia patients have a high risk of Merkel‐cell polyomavirus DNA‐positive Merkel‐cell carcinoma. British Journal of Cancer, 101, 1444–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuze, J.F. , Perez, A. , Untiveros, M. , Quispe, D. , Fuentes, S. , Barker, I. & Simon, R. (2009) Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: a generic method for diagnosis, discovery and sequencing of viruses. Virology, 388, 1–7. [DOI] [PubMed] [Google Scholar]
- Li, H. & Durbin, R. (2009) Fast and accurate short read alignment with Burrows‐Wheeler Transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, M. , Huang, Y. , Gong, Z. , Zhuang, L. , Li, C. , Yang, H. , Tong, Y. , Liu, W. & Cao, W. (2011) Discovery of DNA viruses in wild‐caught mosquitoes using small RNA high trhoughput sequencing. PLoS One, 6, e24758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, R.A. , Warren, R.L. , Freeman, D. , Gustavsen, J.D. , Chénard, C. , Friedman, J.M. , Suttle, C.A. , Zhao, Y. & Holt, R.A. (2011) The sensitivity of massively parallel sequencing for detecting candidate infectious agents associated with human tissue. PLoS One, 6, e19838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppezzo, P. , Vasconcelos, Y. , Settegrana, C. , Jeannel, D. , Vuillier, F. , Legarff‐Tavernier, M. , Kimura, E.Y. , Bechet, S. , Dumas, G. , Brissard, M. , Merle‐Beral, H. , Yamamoto, M. , Dighiero, G. , Davi, F. & French Cooperative Group on CLL. (2005) The LPL/ADAM29 expression ratio is a novel prognosis indicator in chronic lymphocytic leukemia. Blood, 106, 650–657. [DOI] [PubMed] [Google Scholar]
- Palacios, G. , Druce, J. , Du, L. , Tran, T. , Birch, C. , Briese, T. , Conlan, S. , Quan, P.L. , Hui, J. , Marshall, J. , Simons, J.F. , Egholm, M. , Paddock, C.D. , Shieh, W.J. , Goldsmith, C.S. , Zaki, S.R. , Catton, M. & Lipkin, W.I. (2008) A new arenavirus in a cluster of fatal transplant‐associated diseases. The New England Journal of Medicine, 358, 991–998. [DOI] [PubMed] [Google Scholar]
- Pantulu, N.D. , Pallasch, C.P. , Kurz, A.K. , Kassem, A. , Frenzel, L. , Sodenkamp, S. , Kvasnicka, H.M. , Wendtner, C.M. & Zur Hausen, A. (2010) Detection of a novel truncating Merkel cell polyomavirus large T antigen deletion in chronic lymphocytic leukemia cells. Blood, 116, 5280–5284. [DOI] [PubMed] [Google Scholar]
- Pruitt, K.D. , Tatusova, T. , Klimke, W. & Maglott, D.R. (2009) NCBI Reference Sequences: current status, policy and new initiatives. Nucleic Acids Research, 37, D32–D36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente, X.S. , Pinyol, M. , Quesada, V. , Conde, L. , Ordóñez, G.A. , Villamor, N. , Escaramis, G. , Jares, P. , Beà, S. , González‐Díaz, M. , Bassaganyas, L. , Baumann, T. , Juan, M. , López‐Guerra, M. , Colomer, D. , Tubío, J.M. , López, C. , Navarro, A. , Tornador, C. , Aymerich, M. , Rozman, M. , Hernández, J.M. , Puente, D.A. , Freije, J.M. , Velasco, G. , Gutiérrez‐Fernández, A. , Costa, D. , Carrió, A. , Guijarro, S. , Enjuanes, A. , Hernández, L. , Yagüe, J. , Nicolás, P. , Romeo‐ Casabona, C.M. , Himmelbauer, H. , Castillo, E. , Dohm, J.C. , de Sanjosé, S. , Piris, M.A. , de Alava, E. , San Miguel, J. , Royo, R. , Gelpí, J.L. , Torrents, D. , Orozco, M. , Pisano, D.G. , Valencia, A. , Guigó, R. , Bayés, M. , Heath, S. , Gut, M. , Klatt, P. , Marshall, J. , Raine, K. , Stebbings, L.A. , Futreal, P.A. , Stratton, M.R. , Campbell, P.J. , Gut, I. , López‐Guillermo, A. , Estivill, X. , Montserrat, E. , López‐Otín, C. & Campo, E. (2011) Whole‐genome sequencing identifies recurrent mutationa in chronic lymphocytic leukaemia. Nature, 475, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesada, V. , Conde, L. , Villamor, N. , Ordóñez, G.R. , Jares, P. , Bassaganyas, L. , Ramsay, A.J. , Beà, S. , Pinyol, M. , Martínez‐Trillos, A. , López‐Guerra, M. , Colomer, D. , Navarro, A. , Baumann, T. , Aymerich, M. , Rozman, M. , Delgado, J. , Giné, E. , Hernández, J.M. , González‐Díaz, M. , Puente, D.A. , Velasco, G. , Freije, J.M. , Tubío, J.M. , Royo, R. , Gelpí, J.L. , Orozco, M. , Pisano, D.G. , Zamora, J. , Vázquez, M. , Valencia, A. , Himmelbauer, H. , Bayés, M. , Heath, S. , Gut, M. , Gut, I. , Estivill, X. , López‐Guillermo, A. , Puente, X.S. , Campo, E. & López‐Otín, C. (2012) Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nature Genetics, 44, 47–52. [DOI] [PubMed] [Google Scholar]
- Robinson, M.J. , Tuke, P.W. , Erlwein, O. , Tettmar, K.I. , Kaye, S. , Naresh, K.N. , Patel, A. , Walker, M.M. , Kimura, T. , Gopalakrishnan, G. , Tedder, R.S. & McClure, M.O. (2011) No evidence of XMRV or MuLV sequences in prostate cancer, diffuse large B‐cell lymphoma, or the UK blood donor population. Advances in Virology, 2011, doi: 10.1155/2012/782353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi, D. , Bruscaggin, A. , Spina, V. , Rasi, S. , Khiabanian, H. , Messina, M. , Fangazio, M. , Vaisitti, T. , Monti, S. , Chiaretti, S. , Guarini, A. , Del Giudice, I. , Cerri, M. , Cresta, S. , Deambrogi, C. , Gargiulo, E. , Gattei, V. , Forconi, F. , Bertoni, F. , Deaglio, S. , Rabadan, R. , Pasqualucci, L. , Foà, R. , Dalla‐Favera, R. & Gaidano, G. (2011) Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine‐refractoriness. Blood, 118, 6904–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellick, G.S. , Goldin, L.R. , Wild, R.W. , Slager, S.L. , Ressenti, L. , Strom, S.S. , Dyer, M.J. , Mauro, F.R. , Marti, G.E. , Fuller, S. , Lyttelton, M. , Kipps, T.J. , Keating, M.J. , Call, T.G. , Catovsky, D. , Caporaso, N. & Houlston, R.S. (2007) A high‐density SNP genome‐wide linkage search of 206 families identifies susceptibility loci for chronic lymphocytic leukemia. Blood, 110, 3326–3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuda, M. , Arora, R. , Kwun, H.J. , Feng, H. , Sarid, R. , Fernández‐ Figueras, M.T. , Tolstov, Y. , Gjoerup, O. , Mansukhani, M.M. , Swerdlow, S.H. , Chaudhary, P.M. , Kirkwood, J.M. , Nalesnik, M.A. , Kant, J.A. , Weiss, L.M. , Moore, P.S. & Chang, Y. (2009) Human Merkel cell polyomavirus infection I. MCV T antigen expression in Merkel cell carcinoma, lymphoid tissues and lymphoid tumors. International Journal of Cancer, 125, 1243–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons, G. , Glynn, S.A. , Komaroff, A.L. , Mikovits, J.A. , Tobler, L.H. , Hackett, J., Jr , Tang, N. , Switzer, W.M. , Heneine, W. , Hewlett, I.K. , Zhao, J. , Lo, S.C. , Alter, H.J. , Linnen, J.M. , Gao, K. , Coffin, J.M. , Kearney, M.F. , Ruscetti, F.W. , Pfost, M.A. , Bethel, J. , Kleinman, S. , Holmberg, J.A. , Busch, M.P. & Blood XMRV Scientific Research Working Group (SRWG). (2011) Failure to confirm XMRV/MLVs in the blood of patients with chronic fatigue syndrome: a multi‐laboratory study. Science, 334, 814–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slager, S.L. , Rabe, K.G. , Achenbach, S.J. , Vachon, C.M. , Goldin, L.R. , Strom, S.S. , Lanasa, M.C. , Spector, L.G. , Rassenti, L.Z. , Leis, J.F. , Camp, N.J. , Glenn, M. , Kay, N.E. , Cunningham, J.M. , Hanson, C.A. , Marti, G.E. , Weinberg, J.B. , Morrison, V.A. , Link, B.K. , Call, T.G. , Caporaso, N.E. & Cerhan, J.R. (2011) Genome‐wide association study identifies a novel susceptibility locus at 6p21.3 among familial CLL. Blood, 117, 1911–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonigo, P. , Barker, C. , Hunter, E. & Waint‐Hobson, S. (1986) Nucleotide sequence of a Mason‐Pfizer monkey virus: an immunosuppresive D‐type retrovirus. Cell, 45, 375–385. [DOI] [PubMed] [Google Scholar]
- Sportoletti, P. , Baldoni, S. , Cavalli, L. , Del Papa, B. , Bonifacio, E. , Ciurnelli, R. , Bell, A.S. , Di Tommaso, A. , Rosati, E. , Crescenzi, B. , Mecucci, C. , Screpanti, I. , Marconi, P. , Martelli, M.F. , Di Ianni, M. & Falzetti, F. (2010) NOTCH1 PEST domain mutation is an adverse prognostic factor in B‐CLL. British Journal of Haematology, 151, 402–412. [DOI] [PubMed] [Google Scholar]
- Tarrand, J.J. , Keating, M.J. , Tsimberidou, A.M. , O'Brien, S. , LaSala, R.P. , Han, X.Y. & Bueso‐ Ramos, C.E. (2010) Epstein‐Barr virus latent membrane protein 1 mRNA is expressed in a significant proportion of patients with chronic lymphocytic leukemia. Cancer, 116, 880–887. [DOI] [PubMed] [Google Scholar]
- Teman, C.J. , Tripp, S.R. , Perkins, S.L. & Duncavage, E.J. (2011) Merkel cell polyomavirus (MCPyV) in chronic lymphocytic leukemia/small lymphocytic lymphoma. Leukemia Research, 35, 689–692. [DOI] [PubMed] [Google Scholar]
- Thompson, M.P. & Kurzrock, R. (2004) Epstein‐Barr virus and cancer. Clinical Cancer Research, 10, 803–821. [DOI] [PubMed] [Google Scholar]
- Tolstov, Y.L. , Arora, R. , Scudiere, S.C. , Busam, K. , Chaudhary, P.M. , Chang, Y. & Moore, P.S. (2010) Lack of evidence for direct involvement of Merkel cell polyomavirus (MCV) in chronic lymphocytic leukemia (CLL). Blood, 115, 4973–4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsimberidou, A.M. , Keating, M.J. , Bueso‐ Ramos, C.E. & Kurzrock, R. (2006) Epstein‐Barr virus in patients with chronic lymphocytic leukemia: a pilot study. Leukemia & Lymphoma, 47, 827–836. [DOI] [PubMed] [Google Scholar]
- Voisset, C. , Weiss, R.A. & Griffiths, D.J. (2008) Human RNA “rumor” viruses: the search for novel human retroviruses in chronic disease. Microbiology and Molecular Biology Reviews, 72, 157–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Lawrence, M.S. , Wan, Y. , Stojanov, P. , Sougnez, C. , Stevenson, K. , Werner, L. , Sivachenko, A. , DeLuca, D.S. , Zhang, L. , Zhang, W. , Vartanov, A.R. , Fernandes, S.M. , Goldstein, N.R. , Folco, E.G. , Cibulskis, K. , Tesar, B. , Sievers, Q.L. , Shefler, E. , Gabriel, S. , Hacohen, N. , Reed, R. , Meyerson, M. , Golub, T.R. , Lander, E.S. , Neuberg, D. , Brown, J.R. , Getz, G. & Wu, C.J. (2011) SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. The New England Journal of Medicine, 365, 2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, G. , Shendure, J. , Tanenbaum, D.M. , Church, G.M. & Meyerson, M. (2002) Identification of foreign gene sequences by transcript filtering against the human genome. Nature Genetics, 30, 141–142. [DOI] [PubMed] [Google Scholar]
- Weiss, N.S. (1979) Geographical variation in the incidence of the leukemias and the lymphomas. National Cancer Institute Monograph, 53, 139–142. [PubMed] [Google Scholar]
- Weiss, R.A. (2010) A cautionary tale of virus and disease. BMC Biology, 8, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Q. , Luo, Y. , Lu, R. , Lau, N. , Lai, E.C. , Li, W.X. & Ding, S.W. (2010) Virus discovery by deep sequencing and assembly of virus‐derived small silencing RNAs. Proceedings of the National Academy of Sciences of the United States of America, 107, 1606–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Y. , Stange‐Thomann, N. , Weber, G. , Bo, R. , Dodge, S. , David, R.G. , Foley, K. , Beheshti, J. , Harris, N.L. , Birren, B. , Lander, E.S. & Meyerson, M. (2003) Pathogen discovery from human tissue by sequence‐based computational subtraction. Genomics, 81, 329–335. [DOI] [PubMed] [Google Scholar]
- Zhulidov, P.A. , Bogdanova, E.A. , Shcheglov, A.S. , Vagner, L.L. , Khaspekov, G.L. , Kozhemyako, V.B. , Matz, M.V. , Meleshkevitch, E. , Moroz, L.L. , Lukyanov, S.A. & Shagin, D.A. (2004) Simple cDNA normalization using kamchatka crab duplex‐specific nuclease. Nucleic Acids Research, 34, e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Methods and results.
Data S2. Similarity search results of the BLAST search of final viral candidate reads (Illumina reads) against NCBI's non‐redundant database.
Fig S1. Computational subtraction approach developed to identify non‐human sequences in the CLL transcriptome obtained with the 454 technology.
Fig S2. Simulated reads from HIV and EBV genomes were pooled and searched using different BLASTN protocols in the viral genome database.
Fig S3. Compositional analysis of 1203 reads that aligned anti‐sense to the primer binding site PBS of the MPMV genome (sample CLL250).
Fig S4. Median values of Illumina raw read counts for genes with 0‐5 read counts when previously analysed by 454.
Table SI. Performance of different BLASTN protocols when searching simulated HIV reads.
Table SII. Distribution of BLASTN hits in the non‐redundant database.