

Abstract
Infectious diseases comprise some of the leading causes of death and disability worldwide. Interactions between pathogen and host proteins underlie the process of infection. Improved understanding of pathogen‐host molecular interactions will increase our knowledge of the mechanisms involved in infection, and allow novel therapeutic solutions to be devised. Complete genome sequences for a number of pathogenic microorganisms, as well as the human host, has led to the revelation of their protein‐protein interaction (PPI) networks. In this post‐genomic era, pathogen‐host interactions (PHIs) operating during infection can also be mapped. Detailed systematic analyses of PPI and PHI data together are required for a complete understanding of pathogenesis of infections. Here we review the striking results recently obtained during the construction and investigation of these networks. Emphasis is placed on studies producing large‐scale interaction data by high‐throughput experimental techniques.
Keywords: Infection mechanism, Infectious disease, Pathogen‐host interaction, Protein‐protein interaction
Pathogen‐host interactions (PHIs): Infectious microorganisms (pathogens) communicate with human cells through interactions with human proteins on the cell's surface as well as intracellular proteins. These interactions allow the microorganisms to enter the host cell and to manipulate cellular mechanisms in order to use the host cell's capabilities to their own advantage, resulting in infection in the host organism. This article reviews these networks, which may enable us to better elaborate the mechanisms of infection and to identify improved strategies to prevent or cure infectious diseases.
Abbreviations:
EBV, Epstein‐Barr virus; HCV, hepatitis C virus; HIV, human immunodeficiency virus; KSHV, Kaposi sarcoma‐associated herpesvirus; PHI, pathogen‐host interaction; PPI, protein‐protein interaction; SARS‐CoV, severe acute respiratory syndrome coronavirus; VZV, varicella‐zoster virus
1 Introduction
Despite immense technological advances in medicine, pathogenic organisms remain the source of much human morbidity and mortality. HIV/AIDS, acute lower respiratory tract infections, hemorrhagic fever, diarrheal diseases, tuberculosis and malaria are particularly notorious for high mortality rates [1, 2, 3]. The continuous emergence of new diseases and drug‐resistant pathogens has heightened the global burden of infectious diseases in the 21st century [1, 4]. To tackle such biological threats, an improved understanding of pathogenic microorganisms and their interactions with host organisms is needed since pathogen‐host molecular interactions have crucial roles in initiating, sustaining, or preventing infection. Pathogenic microorganisms communicate with human cells through interactions with human proteins both on the surface of the cell and within the interior of the cell. These interactions allow the microorganisms to enter the host cell and manipulate cellular mechanisms in order to use the host cell's capabilities to their own advantage, resulting in infection in the host organism. Detailed knowledge of pathogen‐host protein interactions may enable us to comprehend the mechanisms of infection and to identify better strategies to prevent or cure infection [5, 6]. However, the identification of new drug and vaccine targets for infectious diseases is only possible when the molecular machinery within individual pathogenic and host organisms is understood. For instance, anti‐infection therapeutics should target essential genes in the pathogens which have no homology with human genes [7].
The very first genome sequencing was published in 1977 with the DNA sequence for the genome of a virus, bacteriophage phiX174 [8]. Following the sequencing of the bacterial pathogen Haemophilus influenzae in 1995 [9] and the human genome in 2000 [10], sequence data for prokaryotic and eukaryotic genomes have appeared at an accelerated rate. Today, genomic data for most of the pathogen and host organisms are available [11]. These data are used to study individual genes and corresponding proteins as well as to identify intra‐ and interspecies connections between proteins. In the light of these advances, the initial steps towards complete understanding of infection mechanisms through protein interactions have been recently published. In this review, the efforts to systematic determination and analysis of protein interaction networks underlying infection pathogenesis are summarized (mainly in a chronological order) to present the current picture of the research on infectious diseases.
2 PPI networks related to human pathogens
From a classical perspective, a protein is a functional unit that specifies a small, but discrete, part of the cellular physiology of an organism. In the post‐genomic era, a protein is seen to function as an element within network of its interaction, and its role should be evaluated within this network together with its interacting partners [12]. Advances in genomics and proteomics have been followed by the first large‐scale efforts to identify functional networks of interacting proteins using the two‐hybrid method [13, 14, 15], pull‐down assays [16, 17], and protein chips [18]. To increase our understanding of the mechanisms of infection, protein‐protein interaction (PPI) networks of pathogenic organisms should be determined in order to capture their functional and structural organizations. Pathogenic PPI maps reveal biological pathways and processes, allowing prediction of protein functions and discovery of new drug and vaccine targets.
The first genome‐wide protein interaction networks were determined for viruses [19, 20, 21]. The first large‐scale bacterial networks [22, 23, 24] followed successes in eukaryote mapping [15, 25, 26, 27]. Today, the genome‐wide PPI maps for a number of pathogens and hosts are available in public databases: BIND [28], BioGrid [29], DIP [30], HPRD [31], IntAct [32], MINT [33], MIPS [34], Reactome [35] and STRING [36].
2.1 PPI networks of viral pathogens
Primarily due to their small genome size, whole genome PPI maps were first constructed for viruses. The first interaction map of whole proteome was determined for Escherichia coli bacteriophage T7, mapping 25 interactions among viral proteins [19]. Subsequently, genome‐wide analyses of important human pathogens, hepatitis C virus [20, 37], vaccinia virus [21], herpesviruses [38, 39], and SARS coronavirus [40, 41] were performed through intraviral PPI maps.
Hepatitis C virus (HCV), a flaviviridae family member causing severe liver disease, is a positive‐sense single‐stranded RNA virus. It encodes only a single polyprotein which is co‐ or post‐translationally processed into at least 10 viral proteins [42]. A controlled two‐hybrid strategy based on a random genomic HCV library screen was used by Flajolet et al. [20], resulting in the identification of known and novel PPIs. Interactions among structural and non‐structural proteins were revealed in the study, leading to the conclusion that almost all of the viral proteins encoded by the genome function in the HCV life‐cycle, as in the cases of other members of the flaviviridae [43]. The roles of these functional interactions were discussed within the framework of the constructed genome‐wide interaction map. Interacting domains of the viral polyprotein were also identified to shed light on the development of anti‐viral agents [20]. Another genome‐wide PPI map of HCV was then generated for the viral non‐structural proteins [37].
Vaccinia virus, well‐known as a smallpox vaccine and also the source of potential recombinant vaccines against cancer and infectious diseases, is a member of poxviridae family. It is a large, double‐stranded DNA virus. Poxviruses replicate themselves in the cytoplasm of the host cells without depending on the host's transcriptional machinery. The large genome of vaccinia virus can potentially express more than 200 proteins [44, 45]. McCraith et al. [21] performed a comprehensive two‐hybrid analysis of full‐length vaccinia virus proteins and detected 37 PPIs (including 28 novel interactions) among both characterized and uncharacterized proteins. Many of the PPIs mapped involved one partner which was known to function in a specific process, coupled with another of unknown function, allowing functions to be assigned to previously unannotated proteins within DNA replication, transcription, virion structure, or host evasion.
Another double‐stranded DNA virus family is herpesviridae whose members encode 70‐170 proteins. Herpesviruses cause human diseases such as Kaposi sarcoma, B‐cell lymphomas, chickenpox, shingles, and nasopharyngeal carcinoma [46, 47, 48]. The genome‐wide intraviral protein interaction maps for three members of this family, Kaposi sarcoma‐associated herpesvirus (KSHV), varicella‐zoster virus (VZV), and Epstein‐Barr virus (EBV) were generated by two‐hybrid and analyzed comprehensively to reveal viral network properties [38, 39]. In the work of Uetz et al. [38], 123 PPIs for KSHV and 173 PPIs for VZV were identified, the largest dataset published to date, allowing the construction of the first viral networks. Topological network analyses of these interactome maps indicated that the viral networks appear as a single, highly coupled module (Fig. 1) with relatively many hubs and few peripheral nodes [38] in contrast to scale‐free cellular networks with well‐separated functional modules [49, 50]. Just after this study was published, Calderwood et al. [39] reported the detection of 43 PPIs among EBV proteins. The construction of a PPI map for EBV by merging these interactions with already published ones resulted in a network of 52 proteins with 60 interactions. This large‐scale network allowed the prediction of functions of uncharacterized proteins, further defining viral mechanisms. In these consecutive studies [38, 39], core proteins common to all herpesviruses and noncore ones specific to each strain were investigated thoroughly.
Figure 1.
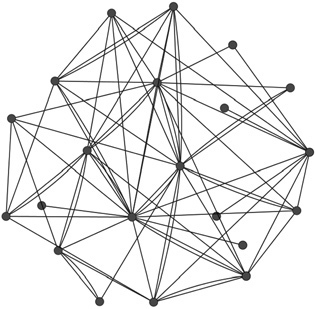
PPI map of KSHV obtained from IntAct. The figure was drawn in Cytoscape ver. 2.8.2.
The severe acute respiratory syndrome coronavirus (SARS‐CoV) is a positive‐sense single‐stranded RNA virus belonging to the family of the largest RNA viruses known, coronaviridae. Its genome encodes 14 open reading frames expressing up to 30 structural and non‐structural proteins that have roles in viral replication, assembly, and other functions for viral amplification in host cells [40, 41, 51]. For a genome‐wide analysis of PPIs of SARS‐CoV, interactions between all SARS‐CoV proteins were determined [40, 41] by two‐hybrid producing 65 and 40 interactions, respectively. Intraviral PPIs were analyzed to elucidate the functions of the proteins as well as to identify the essential proteins in viral replication. von Brunn et al. [40] compared the intraviral network topology of SARS‐CoV with a previously defined viral network [38] and cellular networks [52, 53, 54], concluding SARS‐CoV network contained similarities to the KSHV network [38]. Insights gained into molecular mechanisms and topological network properties provided by the genome‐wide analyses of intraviral PPI maps (Table 1) may be used as a basis for further characterization of the functions and mechanisms of viral proteins, especially for other members of the same virus families.
Table 1.
Examples of PPI networks of pathogens detailed in the literaturea)
| Pathogen name | Pathogen type | Number of PPIs | References | ||||
|---|---|---|---|---|---|---|---|
| E. coli bacteriophage T7 | DNA virus | 25 | [19] | ||||
| HCV | RNA virus | NA | [20] | ||||
| Vaccinia virus | DNA virus | 37 | [21] | ||||
| H. pylori | Gram‐ bacteria | 1280 | [22] | ||||
| E. coli | Gram‐ bacteria | 716 | [23] | ||||
| P. falciparum | Protozoa | 2823 | [72] | ||||
| KSHV | DNA virus | 123 | [38] | ||||
| VZV | DNA virus | 173 | [38] | ||||
| E. coli | Gram− bacteria | 11 511 | [24] | ||||
| EBV | DNA virus | 43 | [39] | ||||
| SARS‐CoV | RNA virus | 65 | [40] | ||||
| C. jejuni | Gram− bacteria | 11 687 | [55] | ||||
| T. pallidum | Gram− bacteria | 3649 | [56] | SARS‐CoV | RNA virus | 40 | [41] |
| M. pneumoniae | Bacteria without cell wall | 178 | [59] | ||||
| M. tuberculosis | Bacteria without cell wall | 8042 | [57] | ||||
| B. subtilis | Gram+ bacteria | 793 | [58] |
Abbrevations: NA, not available
2.2 PPI networks of bacterial pathogens
Having successfully built genome‐wide PPI maps for viruses, similar two‐hybrid methodology was applied to construct PPI networks for the larger, more complex genomes of pathogenic bacteria. The first prokaryotic PPI map was built for Helicobacter pylori [22]. Other large‐scale prokaryotic networks eventually emerged for Campylobacter jejuni [55], Treponema pallidum [56] Mycobacterium tuberculosis [57], and Bacillus subtilis [58]. Genome‐scale analysis of interacting proteins that assemble into protein complexes were performed for E. coli [23, 24] and Mycoplasma pneumoniae [59].
The first large‐scale intrabacterial PPI map was constructed for the human gastric pathogen, and gram‐negative bacterium H. pylori, identifying 1280 interactions between 46.6% of all 261 bacterial proteins using the two‐hybrid method [22]. The comparison of these H. pylori PPIs with previously described interactions between orthologous E. coli proteins resulted in prediction of protein functions within biological pathways such as chemotaxis and urease activity, essential for H. pylori pathogenicity. In this study, the interacting domains of H. pylori proteins were also identified and used in protein function predictions. Interacting domains may serve in mapping new functional domains, providing crucial information for antibacterial drug design studies.
Gram‐negative bacterium E. coli, the main cause of urinary tract infections and a model bacterial system, is one of the best characterized and early studied organisms [60, 61, 62]. However, any large‐scale analysis of protein complexes in E. coli was not performed until the studies of Butland et al. [23] and Arifuzzaman et al. [24]. First, 716 binary interactions involving 83 essential and 152 non‐essential proteins, were identified by pull‐down assay using tandem affinity purification‐mass spectrometry, targeting 1000 ORFs (about one‐quarter of the E. coli genome) [23]. A small number (15%) of these interactions were already available in DIP, BIND, and STRING. Ten newly described E. coli PPIs were found as orthologous to the interactions reported for H. pylori [22]. The novel interactions were analyzed for functional annotations of uncharacterized proteins, allocating them within ribosome function, RNA processing, RNA binding, and so on. The graph theoretical analysis of the PPI map of E. coli revealed scale‐free behavior and a high correlation between connectivity and the degree of conservation. The genome‐wide PPI map of E. coli K‐12 strain with 11 511 interactions among 2667 proteins was then constructed by a similar method [24]. The comprehensive analysis of this large‐scale network also validated the scale‐free nature and the connectivity‐conservation correlation found previously [23]. Arifuzzaman et al. [24] identified 107 functional units which have roles in metabolic pathways, transcriptional and translational machinery, recombination and flagella assembly. Analysis of PPIs based on this functional unit categorization provided further functional annotations.
The gram‐negative, food‐borne pathogen C. jejuni is the major cause of gastroenteritis. The proteome‐level analysis revealed 11687 interactions involving 80% of 1654 C. jejuni proteins [55], the most comprehensive bacterial PPI map determined by two‐hybrid. A scale‐free network was obtained, removing low confidence‐scored interactions. This PPI map of C. jejuni was used to identify evolutionarily conserved subnetworks through comparison with protein networks of H. pylori [22], E. coli [23] and Saccharomyces cerevisiae in DIP. Further analyses of the identified conserved sub‐networks allowed the prediction of new C. jejuni interactions using orthologous interactions. This comparative analysis also enabled the identification of essential C jejuni genes based on their orthology to essential genes in other organisms. This comprehensive interactome data were next used to predict protein roles and to map functional pathways such as chemotaxis.
The causative agent of syphilis, T. pallidum, has one of the smallest genomes known in extracellular bacteria, encoding 1039 proteins [63]. The global PPI network of T. pallidum, involving 3649 interactions connecting 726 bacterial proteins, was identified by two‐hybrid [56]. The high‐confidence subset connects 576 proteins by 991 interactions. In that study, an integrated network of DNA‐metabolism related processes was constructed and 18 proteins were functionally annotated within this network. Additionally, various orthologous interactions were predicted for completely sequenced genomes, allowing the description of phylogenetically conserved interaction patterns.
Atypical pneumonia causing human pathogen, M. pneumoniae also has one of the smallest genomes in self‐replicating organisms with 689 protein‐encoding genes, making it a good model organism to study proteome organization in prokaryotes [64]. A proteome‐wide analysis was performed by tandem affinity purification‐mass spectrometry, identifying 62 homo‐multimeric and 116 hetero‐multimeric protein complexes [59]. About a third of the found hetero‐multimeric complexes were observed to interact with proteins forming 35 larger, multiprotein complexes implying higher level of proteome organization and protein multifunctionality, allowing functional annotations of assemblies as well as prediction of biological roles of individual proteins within the complexes.
M. tuberculosis causes millions of deaths each year with tuberculosis infection [65]. After computational efforts to construct large‐scale PPI maps of M. tuberculosis [66, 67], its genome‐wide network was identified experimentally by two‐hybrid [57]. This global network is composed of 8 042 interactions among 2907 proteins which represent 74.1% of the whole proteome. The topological properties of the undirected network of these interactions were calculated and compared with those of the previously defined prokaryotic PPI networks [22, 23, 24, 55, 56]. Similar scale‐free behavior following a power‐law distribution was observed. In fact, the networks obtained by pull‐down assay [23, 24] differ in values of clustering coefficient from the networks obtained by two‐hybrid analysis [22, 55, 56]. Moreover, Wang et al. [57] performed a cross‐species network comparison analysis of M. tuberculosis interactions with the available large‐scale PPI data [22, 23, 24, 55, 56] and identified conserved sub‐networks. Additionally, the highly connected critical proteins and mechanisms of the protein secretion pathways which have roles in its pathogenesis were revealed.
A large‐scale PPI network was recently constructed for the gram‐positive bacterium B. subtilis (which is rarely pathogenic) by two‐hybrid [58]. This network of 793 interactions involves 287 bacterial proteins. Due to its role as a model organism, many studies were performed to characterize the biological functions of its PPIs in cellular processes [68, 69, 70]. However, many processes remained uncharacterized. Hence Marchadier et al. [58] performed a comprehensive analysis with the integration of transcriptomic data focusing on cell division, cell responses to stresses, the bacterial cytoskeleton, DNA replication and chromosome maintenance. These sequential efforts on construction of large‐scale PPI networks for prokaryotes (Table 1) constitute the first comprehensive description of the intraspecies mechanisms of the bacterial pathogens.
2.3 PPI networks of protozoan pathogens
The protozoan pathogen Plasmodium falciparum causes malaria which results in deaths of nearly a million of people each year [71]. A comprehensive protein interaction map of this pathogen was generated by two‐hybrid, identifying a highly interconnected, scale‐free network of 2823 interactions within 1267 proteins (∼25% of the predicted P. falciparum proteins) [72]. In this network, 33% of the interactions are between two uncharacterized proteins whereas 49% of the interactions include one such protein. Bioinformatic analysis of this network yielded functional annotations of the proteins within the processes; chromatin modification, transcription, messenger RNA stability, ubiquitination, and invasion of host cells. More detailed studies of PPIs within P. falciparum are required in order to unravel its pathogenesis mechanisms thoroughly.
2.4 The impacts of pathogenic PPI maps
Despite the increasing rate in the identification of genome‐wide PPI networks, they remain unconstructed for most pathogens. In the light of accelerating advances in genomics, proteomics, and interactomics, large‐scale maps for many more organisms are expected to be built in the near future. Increasing numbers of PPI networks will allow the comparison of networks across diverse organisms, resulting in generalized conclusions about pathogenic molecular mechanisms. The first examples of such comparative studies have been highlighted in the sections 2.1 and 2.2 above. Integration of several high‐throughput interaction datasets to generate more detailed networks is also possible, as indicated by recent examples for the E. coli system [73, 74]. The frequency of such integrated networks is expected to increase, owing to the large number of diverse data sets. These will be invaluable in defining whole proteomic maps of the pathogens.
One of the most striking results of bioinformatic analyses on the constructed PPI maps is the identification of essential proteins functioning within pathogens. These proteins should be examined thoroughly to test their potential as novel therapeutic targets. The exploration of genome‐wide PPI maps of the pathogens permits the assignment of unannotated proteins to biological pathways with function prediction. The proteins annotated to the host invasion processes may provide a launching point for pathogen‐host interaction studies.
3 Pathogen‐human interaction networks
Biochemical interactions of pathogens with their hosts are necessary to invade the host organism. These connections between pathogens and hosts include interactions between proteins, nucleotide sequences, and small ligands [75, 76]. However, the protein interactions of pathogen‐host systems have been identified as the most important, and therefore the most studied, type of pathogen‐host interactions (PHIs) [76, 77]. Since these interspecies crosstalks determine the pathogenesis, focusing on the whole PHI system, instead of investigating a pathogen or host individually, may allow us to capture critical mechanisms (i.e. strategies used by pathogens and host immune responses) during infection that cannot be provided by traditional methods.
Due to a lack of sufficient experimental PHI data until recent years, many computational PHI prediction methods have been developed [78, 79, 80, 81, 82, 83, 84]. These studies focused mainly on interactions of P. falciparum and human immunodeficiency virus (HIV), as these are some of the most threatening pathogens to humans. Very recently experiments have been carried out to determine the first large‐scale molecular interactions between human and viruses [39, 85, 86] and bacteria [87, 88]. As a result of an increase in data available for pathogen‐host systems, PHI‐specific databases have been introduced such as PHI‐base [89], VirusMINT [90], VirhostNet [91], PATRIC [92], and PHISTO [93]. Although these advances in data archiving are promising, most data relevant to PHI are still buried in the biomedical literature. Some rare efforts have been performed to obtain hidden PHIs from the literature by text mining [94, 95, 96].
3.1 Virus‐human interaction networks
As in the case of intraspecies pathogen PPIs, large‐scale PHI data were generated for viral systems before bacterial systems (Table 2). The first examples are for commonly observed human pathogens, EBV [39], HCV [85] and influenza A virus (H1N1 and H3N2) [86] and then recently for HIV [97].
Table 2.
Examples of PHI networks detailed in the literatur
| Pathogen name | Pathogen type | Number of PHIs | Number of interacting pathogen proteins | Number of interacting human proteins | References |
|---|---|---|---|---|---|
| EBV | DNA virus | 173 | 40 | 112 | [39] |
| HCV | RNA virus | 481 | 11 | 421 | [85] |
| Influenza A virus (H1N1 A/PR/8/34) | RNA virus | 135 | 10 | 87 | [86] |
| Influenza A virus (H3N2 A/Udorn/72) | RNA virus | 81 | 10 | 66 | [86] |
| B. anthracis | Gram+ bacteria | 3073 | 943 | 1748 | [87] |
| Y. pestis | Gram+ bacteria | 4059 | 1218 | 2108 | [87] |
| F. tularensis | Gram‐ bacteria | 1383 | 349 | 999 | [87] |
| Y. pestis | Gram+ bacteria | 204 | 66 | 109 | [88] |
| HIV | RNA virus | 497 | 16 | 435 | [97] |
In Calderwood et al. [39], protein interactions between herpesvirus, EBV and human were mapped by two‐hybrid in conjunction with EBV intraviral PPI mapping, providing 173 PHIs between 40 EBV proteins and 112 human proteins. A systematic analysis of these interactome maps of PPIs and PHIs enabled hypotheses of the roles of EBV proteins in pathogenesis to be generated. Furthermore, intraspecies protein interaction data for human were integrated from databases (BIND, DIP, HPRD, MIPS) and from the literature [52, 53] to analyze the organization of the human proteins targeted by EBV within human molecular machinery. It was found that EBV proteins tend to target human proteins which are highly connected (hubs) and central to many paths (bottlenecks) in the human PPI network. On the other hand, the degree distribution of the EBV‐human protein interaction network could not be fitted to any model because of its incompleteness (Fig. 2). Attempts to analyze incomplete maps of PPIs and PHIs are still able to supply a partial understanding of mechanisms underlying infection. A similar thorough analysis was earlier performed with herpesviral protein networks of KSHV and VZV and their interaction with the human proteome [38]. In that study, protein interactions between herpesviruses and human were predicted using the interacting orthologs of both proteins in other organisms [54]. Combined virus‐human networks were constructed by starting with the viral networks, adding their human protein targets, and then adding the cellular interactions among the targeted human proteins. The topological analyses of the combined herpesviruses‐human networks revealed distinct properties from both viral and human interactomes providing insights into the impact of the two organisms on each other [38].
Figure 2.
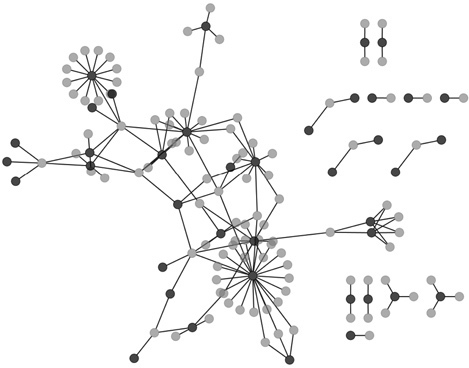
PHI map of EBV‐Human obtained from PHISTO. Light grey nodes are human proteins whereas dark grey nodes are EBV proteins. The figure was drawn in Cytoscape ver. 2.8.2.
A proteome‐wide PHI map for the flavivirus HCV was mapped by two‐hybrid and then by literature mining of previously found interactions between HCV and human [85]. A map of 481 interactions between 11 HCV proteins and 421 human proteins was generated (314 PHIs by two‐hybrid). 65% of this PHI network included novel interactions. The integrated human network of 44 223 PPIs among 9520 proteins [98] was used to evaluate the interplay between HCV and human. Very similar behavior to EBV [39] was observed for HCV in terms of attacking hub and bottleneck proteins in the human network. To assess the human pathways targeted by HCV, KEGG functional annotation pathways [99] were used. Four pathways were detected to be enriched in HCV‐targeted human proteins. Three of them were associated already with HCV clinical syndromes as insulin, TGF‐β and Jak/STAT pathways. The last enriched pathway, focal adhesion, is a novel observation as a human pathway affected during HCV infection [85].
Influenza A is a member of negative‐sense single‐stranded viruses of orthomyxoviridae family. It is the sources of all flu pandemics infecting multiple species. For H1N1 A/PR/8/34 strain of influenza virus, 31 intraviral PPIs among 10 viral proteins and 135 PHIs between 10 viral and 87 human proteins, most of which are expressed in primary human bronchial cells, were detected by two‐hybrid [86]. Some of the PHIs constructed had been published previously [100]. The topology of the constructed intraviral network revealed a highly interconnected nature, as observed previously for other viral networks [38, 101]. In the case of the influenza A‐human interaction network, important properties about connectivity of proteins were observed. First, viral proteins interact with significant number of human proteins, reflecting the multifunctionality of the small number proteins encoded in RNA viruses. Second, each of 24 human proteins connects with two or more viral proteins forming virus‐human multiprotein complexes. Additionally, it was observed that viral proteins generally target human proteins which are highly connected within their own network, as it was the case in herpesviruses‐human system [39]. In Shapira et al. [86] another PHI network was identified for strain of influenza virus, H3N2 A/Udorn/72 by the same experimental approach. This PHI network consists of 81 interactions between 10 viral and 66 human proteins, reflecting a similar nature to the network for H1N1 strain‐human system. This confirms the conserved functions of influenza virus proteins through strains. Besides direct physical interactions between viral and human proteins, host responses in bronchial cells to influenza infection was identified by expression profiling, generating a regulatory map of interactions between influenza proteins and their human targets. Comprehensive analysis of the physical and regulatory maps of the PHI system elucidated human mechanisms involved in infection. For example, NF‐κB, mitogen‐activated protein kinase, apoptosis, and Wnt signaling pathways are regulated through transcriptional and/or physical interactions during influenza A infection.
One of the most dangerous human pathogens, HIV, belongs to positive‐sense single‐stranded RNA virus family retroviridae. Acquired immunodeficiency syndrome‐causing HIV has been extensively studied since its first observation near the end of the 20th century [102, 103, 104, 105]. Similar to other RNA viruses, HIV has a small genome and depends largely on human cellular machinery to be replicated. Identifying the physical contacts between HIV and human proteins during HIV replication is critically important for a full understanding of HIV infection. Being one of the most studied pathogens, there are many PHI data for HIV‐1 in VirusMINT and PHISTO. The current PHI data have been produced mainly by small‐scale experiments [106, 107, 108]. Very recently, a global PHI network was generated for HIV‐human protein complexes by affinity tagging and purification mass spectrometry, producing 497 PHIs between 16 HIV‐1 proteins and 435 human proteins [97]. It was observed that HIV‐targeted human proteins are highly conserved across primates. The novel interactions identified in that study requires further work to detail their biological significance in terms of HIV infection. Besides whole proteins, domains of the interacting proteins were investigated and the enriched domain types in targeted human proteins were indicated for facilitat‐ing future structural modeling studies regarding HIV‐human system. The first large‐scale interaction networks between viruses and humans [39, 85, 86, 97] provide crucial clues about the viral infections, verifying the critical importance of PHI analyses in infection researches.
3.2 Bacteria‐human interaction networks
Until very recent years, the PHI data were scarce for bacterial systems because of lack of any large‐scale experiments. The first extensive bacterial PHI networks were identified for important human pathogens, Bacillus anthracis, Francisella tularensis, and Yersinia pestis [87], then another high‐throughput experimental study generating PHI data of Y. pestis was reported [88].
Gram‐positive bacteria B. anthracis and Y. pestis and gram‐negative bacterium F. tularensis are respiratory pathogens causing anthrax, bubonic plague, and acute pneumonic disease, respectively. Using a two‐hybrid assay, large‐scale interaction data were generated between these bacteria and human producing 3073 PHIs between 943 B. anthracis proteins and 1748 human proteins, 4059 PHIs between 1218 Y. pestis proteins and 2108 human proteins, and 1383 PHIs between 349 F. tularensis proteins and 999 human proteins [87]. The first conclusion of computational analyses of these comprehensive bacteria‐human networks, in combination with the integrated human PPI network from databases BIND, DIP, HPRD, IntAct, MINT, MIPS, and Reactome, was that bacterial proteins tend to target hubs and bottlenecks in the human network. Secondly, the roles of human proteins targeted by these bacteria were investigated using their gene ontology annotations [109]. The tendency of all three pathogens to target human proteins involved in immune responses was observed as previously reported [110, 111, 112]. Besides being effectors of immune signaling, the bacteria‐targeted human proteins also have crucial roles in apoptosis [87]. Thirdly, the conserved protein interaction modules of the three PHI networks were computed [113, 114] for a more systematic comparative analysis. Conserved modules revealed common attacks by the bacterial pathogens to same human pathways.
Subsequently, another PHI map was generated for plague causing Y. pestis by a different two‐hybrid strategy by choosing only potential virulence factors as bait proteins [88]. 204 PHIs were yielded between 66 Y. pestis proteins and 109 human proteins and then 23 previously reported PHIs were integrated to construct a comprehensive network between Y. pestis and human. A graph theoretic analysis confirms that Y. pestis preferentially targets hub and bottleneck proteins in the human intra‐network as concluded previously for viruses [39, 86] and bacteria [87]. Signaling pathways, crucial for human immune system, were found to be enriched in human proteins targeted by Y. pestis. These pathways include mitogen‐activated protein kinase signaling and Toll‐like receptor signaling and also pathways functioning in focal adhesion, regulation of cytoskeleton, and leukocyte transendoepithelial migration. Finally, Y. pestis‐targeted human proteins were compared with those targeted by viruses whose PHI networks were identified previously. 16 of 109 Y. pestis‐targeted human proteins are included in PHI networks of EBV [39] and HCV [85] indicating the common infection strategies of both viruses and bacteria. The recent detected first large‐scale PHI networks of bacteria‐human systems [87, 88] contribute largely to the understanding of bacterial infection mechanisms with immune evasion.
3.3 Analyses of comprehensive PHI data
As PHI data available for various pathogens increase, a need to analyze comprehensive PHI data for all pathogen types together arises in order to draw a generalized picture. Although infection mechanisms of individual pathogens have been studied through intraspecies pathogenic PPI maps and interspecies PHI maps, a general overview of infection mechanisms was missing until analyses of PHI data from different infection agents were attempted [6, 93].
In the absence of large‐scale PHI networks for bacterial, protozoan and fungal systems, Dyer et al. [6] performed the first global analysis of 10 477 protein interactions between 190 pathogen strains of viruses, bacteria, protozoa, and human through properties of targeted 1233 human proteins. Diversity of the available PHI data was not rich, 98.3% of 10 477 PHIs belonged to the virus‐human systems with 77.9% of the interaction data drawn from HIV – human interaction systems. The importance of the pathogen‐targeted proteins was evaluated within the intraspecies human PPI network of 75 457 interactions. These PHI and PPI data were integrated from public databases; MINT, IntAct, DIP, HPRD, Reactome, BIND and MIPS. Firstly, targeting hub and bottleneck proteins was concluded to be global behavior for all pathogens, as reported for individual pathogen strains previously [39, 86, 87, 88]. Gene ontology [109] functions enriched in the targeted human proteins by different pathogens revealed common infection mechanisms. Attack of human transcription factors and key proteins that control the cell cycle and regulate apoptosis and transport of genetic material across the nuclear membrane were found to be among the common viral strategies. Despite its scarcity (174 interactions in the datset), bacterial PHI data allowed identification of specific human proteins that function in the host immune response (via Toll‐like receptors and I‐κB kinase/NF‐κB signaling cascade) as a target of bacterial infection strategy [6].
Recently we performed another study with comprehensive PHI data to explore common and special infection strategies for viruses and bacteria [93]. A significant amount of bacterial PHIs, constituting 36.5% of all data, was avaiable thanks to Dyer et al. [87]. We analyzed 23435 interactions between 3419 proteins of viral, bacterial, protozoan and fungal pathogens (totally 257 strains) and 5210 proteins of human obtained from PHISTO (www.phisto.org). To generate the intraspecies human protein network, 194006 PPIs were integrated from BioGrid, DIP, IntAct, MINT and Reactome. The significant amount of bacterial and viral PHI data allowed us to focus on comparisons between their specific infection mechanisms. Firstly, attacking hub and bottleneck proteins in the human PPI network was verified as a common infection strategy of both bacteria and viruses. Furthermore, viruses were observed to target human proteins of much higher connectivity and centrality values in comparison to bacteria. Secondly, gene ontology enrichment analysis of the targeted human proteins verified the special mechanisms of bacteria and viruses use to manipulate of human immune defense mechanisms and cellular processes, respectively (as reported in Dyer et al. [6] but relying on lower amounts of PHI data). A first attempt at the investigation of the human proteins targeted by both bacteria and viruses revealed that attacking human metabolic processes is a common strategy used by both pathogens during infections [93]. Global analysis of PHI data provides insights into the strategies adapted by bacteria and viruses to subvert human cellular processes and immune system for the infection. However, large‐scale PHI networks for pathogens other than bacteria and viruses are still undetermined, leaving their pathogenesis mechanisms to be relatively uncharacterized.
3.4 The impact of PHI Networks
Research on infectious diseases through PHIs has accelerated within the post‐genomic era (Fig. 3). However, large‐scale PHI networks have been infrequently studied. Efforts to identify and analyze large‐scale PHIs for diverse pathogen types would be expected to parallel the acceleration of biotechnology and bioinformatics research. Increasing amounts of data available will allow more complete data sets to be compiled, resulting in characterization of topological properties of PHI networks. The first attempt to fit the degree distribution of EBV‐human interaction network failed due to scarcity of data [39]. On the other hand, bioinformatic analyses of the pathogen‐targeted human proteins succeeded in unraveling some infection strategies such as targeting human hubs and bottlenecks, subverting cellular processes for the usage of pathogens' own advantages and evasion of immune defenses [6, 39, 85, 87, 88, 93]. The huge amount of data expected to be generated for PHI systems will enable us to capture all details of infection processes. potentially leading to the development of new and more efficient therapeutics.
Figure 3.
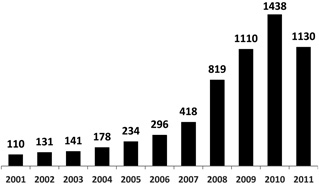
The number of scientific publications including PHI‐related terms in PubMed in the post genomic era. The searched PHI‐related terms: “pathogen host interactions”, “host pathogen interactions”, “pathogen host interaction”, “host pathogen interaction”, “pathogen‐host interactions”, “pathogen‐host interaction”, “host‐pathogen interactions”, “host‐pathogen interaction”.
Conventional treatments for infectious diseases often aim to kill pathogens by targeting their essential proteins. This approach unfortunately forces the pathogens to evolve for survival and consequently selects resistant strains (especially in the case of RNA viruses with a high mutation rate). To fight drug‐resistant pathogens, novel alternative therapeutics are emerging which target host proteins required by pathogens to replicate and persist within the host organism. If these host factors are indispensable for pathogens, but not essential for host cells, their silencing may inactivate pathogenic activity, allowing them to serve as therapeutic targets [4, 115]. In the light of PHI studies, some human factors required by viral and bacterial pathogens have been determined for HIV [115, 116, 117, 118, 119], HCV [120], West Nile virus [121], Influenza virus [122, 123], and M. tuberculosis [124] in recent years.
Despite the efforts reviewed here, the use of systems biology approaches to investigate PHI is still considered relatively undeveloped. The availability of new PHI network data, together with further topological and functional analyses of pathogen‐host systems, are expected to shed more light on infection mechanisms and novel therapeutic targets for infectious diseases in the near future.
Biographical Information

Kutlu Ö. Ülgen holds a PhD in Chemical Engineering from the University of Manchester (UMIST, UK) where she studied antibiotic production by Streptomyces coelicolor using different bioreactor operation strategies. Since 1992, she has been a faculty member in the Department of Chemical Engineering at Bogazici University. She is currently working on systems biology, computational reconstruction and analysis of signaling networks and rational design by metabolic engineering approaches.
Acknowledgements
We particularly thank Dr. Tunahan Çakir for critical reading of the manuscript and for his contributions to Figure 3. The financial support was provided by the Research Funds of Bogaziçi University, through project 5554D. The doctoral scholarship for Saliha Durmuş Tekir is sponsored by TÜBITAK, is gratefully acknowledged.
The authors declare no conflict of interest.
REFERENCES
- 1. Fauci, A. S. , Infectious diseases: for considerations the 21st century. Clin. Infect. Dis. 2001, 32, 675–685. [DOI] [PubMed] [Google Scholar]
- 2. Forst, C. V. , Host‐pathogen systems biology. Drug Discov. Today 2006, 11, 220–227. [DOI] [PubMed] [Google Scholar]
- 3. Taylor, D. J. , Leach, R. W. , Bruenn, J. , Filoviruses are ancient and integrated into mammalian genomes. BMC Evol. Biol. 2010, 10, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bünnige, M. , New Strategies to fight infectious diseases ‐ arms race on a microscale. Infecti. Res. 2011, 1–6. [Google Scholar]
- 5. Münter, S. , Way, M. , Frischknecht, F. , Signaling during pathogen infection. Science's STKE 2006, 335, re5. [DOI] [PubMed] [Google Scholar]
- 6. Dyer, M. D. , Murali, T. M. , Sobral, B. W. , The landscape of human proteins interacting with viruses and other pathogens. PLoS Pathog. 2008, 4, 2, e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barker, J. J. , Antibacterial drug discovery and structure‐based design. Drug Discov. Today 2006, 11, 391–404. [DOI] [PubMed] [Google Scholar]
- 8. Sanger, F. , Air, G. M. , Barrell, B. G. , Brown, N. L. et al., Nucleotide sequence of bacteriophage ϕX174 DNA. Nature 1977, 265, 687–695. [DOI] [PubMed] [Google Scholar]
- 9. Fleischmann, R. D. , Adams, M. D. , White, O. , Clayton, R. A. et al., Whole‐genome random sequencing and assembly of Haemophilus influenzae Rd. Science 1995, 269, 496–512. [DOI] [PubMed] [Google Scholar]
- 10. Venter, J. C. , Adams, M. D. , Myers, E. W. , Li, P. W. et al., The sequence of the human genome. Science 2001, 291, 1304–1351. [DOI] [PubMed] [Google Scholar]
- 11. Benson, D. A. , Karsch‐Mizrachi, I. , Clark, K. , Lipman, D. J. et al., GenBank. Nucleic Acids Res. 2012, 40, D48–D53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eisenberg, D. , Marcotte, E. M. , Xenarios, I. , Yeates, T. O. , Protein function in the post‐genomic era. Nature 2000, 405, 823–826. [DOI] [PubMed] [Google Scholar]
- 13. Fields, S. , Song, O. , A novel genetic system to detect protein‐protein interactions. Nature 1989, 340, 245–246. [DOI] [PubMed] [Google Scholar]
- 14. Finley, R. L. , Brent, R. , Interaction mating reveals binary and ternary connections between Drosophila cell cycle regulators. Proc. Natl. Acad. Sci. USA 1994, 91, 12980–12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fromont‐Racine, M. , Rain, J. , Legrain, P. , Toward a functional analysis of the yeast genome through exhaustive two‐hybrid screens. Nature Genet. 1997, 16, 277–282. [DOI] [PubMed] [Google Scholar]
- 16. Gavin, A.‐C. , Bosche, M. , Krause, R. , Grandi, P. et al., Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 2002, 415, 141–147. [DOI] [PubMed] [Google Scholar]
- 17. Ho, Y. , Gruhler, A. , Heilbut, A. , Bader, G. , Moore, L. , Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 2002, 415, 2–5. [DOI] [PubMed] [Google Scholar]
- 18. Zhu, H. , Bilgin, M. , Bangham, R. , Hall, D. et al., Global analysis of protein activities using proteome chips. Science 2001, 293, 2101–2105. [DOI] [PubMed] [Google Scholar]
- 19. Bartel, P. , Roecklein, J. , SenGupta, D. , A protein linkage map of Escherichia coli bacteriophage T7. Nature Genet. 1996, 12, 72–77. [DOI] [PubMed] [Google Scholar]
- 20. Flajolet, M. , Rotondo, G. , Daviet, L. , Bergametti, F. et al., A genomic approach of the hepatitis C virus generates a protein interaction map. Gene 2000, 242, 369–379. [DOI] [PubMed] [Google Scholar]
- 21. McCraith, S. , Holtzman, T. , Moss, B. , Fields, S. , Genome‐wide analysis of vaccinia virus protein‐protein interactions. Proc. Natl. Acad. Sci. USA 2000, 97, 4879–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rain, J. C. , Selig, L. , De Reus, H. , Battaglia, V. et al., The protein‐protein interaction map of Helicobacter pylori. Nature 2001, 409, 6817, 211–215. [DOI] [PubMed] [Google Scholar]
- 23. Butland, G. , Peregrin‐Alvarez, J. M. , Li, J. , Yang, W.et al. , Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature 2005, 433, 531–537. [DOI] [PubMed] [Google Scholar]
- 24. Arifuzzaman, M. , Maeda, M. , Itoh, A. , Nishikata, K. et al., Large‐scale identification of protein‐protein interaction of Escherichia coli K‐12. Genome Res. 2006, 16, 686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ito, T. , Tashiro, K. , Muta, S. , Ozawa, R. et al., Toward a protein‐protein interaction map of the budding yeast: A comprehensive system to examine two‐hybrid interactions in all possible combinations between the yeast proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Uetz, P. , Giot, L. , Cagney, G. , Mansfield, T. A. et al., A comprehensive analysis of protein‐protein interactions in Saccharomyces cerevisiae. Nature 2000, 403, 623–627. [DOI] [PubMed] [Google Scholar]
- 27. Walhout, J. M. A. , Sordella, R. , Lu, X. , Hartley, J. L. et al., Protein interaction mapping in C. elegans using proteins involved in vulval development. Science 2000, 87, 116–122. [DOI] [PubMed] [Google Scholar]
- 28. Alfarano, C. , Andrade, C. E. , Anthony, K. , Bahroos, N. et al., The biomolecular interaction network database and related tools: 2005 update. Nucleic Acids Res. 2005, 33, D418–D424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stark, C. , Breitkreutz, B. J. , Chatr‐aryamontr, A. , Boucher, L. et al., The BioGRID interaction database: 2011 update. Nucleic Acids Res. 2011, 39, D698–D704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Salwinski, L. , Miller, C. S. , Smith, A. J. , Pettit, F. K. et al., The database of interacting proteins: 2004 update. Nucleic Acids Res. 2004, 32, D449–D451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Prasad, T. S. K. , Goel, R. , Kandasamy, K. , Keerthikumar, S. , Human protein reference database. Nucleic Acids Res. 2009, 37, D767–D772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kerrien, S. , Aranda, B. , Breuza, L. , Bridge, A. et al., The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2012, 40, D841–D846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Licata, L. , Briganti, L. , Peluso, D. , Perfetto, L. et al., MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012, 40, D857–D861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Güldener, U. , Münsterkötter, M. , Oesterheld, M. , Pagel, P. et al., MPact: the MIPS protein interaction resource on yeast. Nucleic Acids Res. 2006, 34, D436–D441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Croft, D. , O'Kelly, G. , Wu, G. , Haw, R. , Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 2011, 39, D691–D697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Szklarczyk, D. , Franceschini, A. , Kuhn, M. , Simonovic, M. et al., The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dimitrova, M. , Imbert, I. , Kieny, M. P. , Schuster, C. , Protein‐protein interactions between Hepatitis C virus nonstructural proteins. J. Virol. 2003, 77, 5401–5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Uetz, P. , Dong, Y. A. , Zeretzke, C. , Atzler, C. et al., Herpesviral protein networks and their interaction with the human proteome. Science 2006, 311, 239–242. [DOI] [PubMed] [Google Scholar]
- 39. Calderwood, M. A. , Venkatesan, K. , Xing, L. , Chase, M. R. et al., Epstein‐Barr virus and virus human protein interaction maps. Proc. Natl. Acad. Sci. USA 2007, 104, 7606–7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. von Brunn, A. , Teepe, C. , Simpson, J. C. , Pepperkok, R. et al., Analysis of intraviral protein‐protein interactions of the SARS coronavirus ORFeome. PLoS One 2007, 2, e459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pan, J. , Peng, X. , Gao, Y. , Li, Z. et al., Genome‐wide analysis of protein‐protein interactions and involvement of viral proteins in SARS‐CoV replication. PLoS One 2008, 3, e3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Houghton, M. , Selby, M. , Weiner, A. , Choo, Q. L. , Hepatitis C virus: structure, protein products and processing of the polyprotein precursor. Curr. Stud. Hematol. Blood Transfus. 1994, 61, 1–11. [DOI] [PubMed] [Google Scholar]
- 43. Knipe, D. M., Howley, P. M. (Ed.), Flaviviridiae:The viruses and their replication, Lippincott–Raven Publishers, Philadelphia, 2007.
- 44. Goebel, S. J. , Johnson, G. P. , Perkus, M. E. , Davis, S. W. et al., The complete DNA sequence of vaccinia virus. Virology 1990, 179, 517–563. [DOI] [PubMed] [Google Scholar]
- 45. Moss, B. , Genetically engineered poxviruses for recombinant gene expression, vaccination, and safety. Proc. Natl. Acad. Sci. USA 1996, 93, 11341–11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Davison, A. J. , Scott, J. E. , The complete DNA sequence of varicella‐zoster virus. J. Gen. Virol. 1986, 67, 1759–1816. [DOI] [PubMed] [Google Scholar]
- 47. Chang, Y. , Cesarman, E. , Pessin, M. S. , Lee, F. et al., Identification of herpesvirus‐like DNA sequences in AIDS‐associated Kaposi's sarcoma. Science 1994, 266, 1865–1869. [DOI] [PubMed] [Google Scholar]
- 48. Dolan, A. , Addison, C. , Gatherer, D. , Davison, A. J. , McGeoch, D. J. , The genome of Epstein‐Barr virus type 2 strain AG876. Virology 2006, 350, 164–170. [DOI] [PubMed] [Google Scholar]
- 49. Barabasi, A. L. , Albert, R. , Emergence of scaling in random networks. Science 1999, 286, 509–512. [DOI] [PubMed] [Google Scholar]
- 50. Maslov, S. , Sneppen, K. , Specificity and stability in topology of protein networks. Science 2002, 296, 910–913. [DOI] [PubMed] [Google Scholar]
- 51. Snijder, E. J. , Bredenbeek, P. J. , Dobbe, J. C. , Thiel, V. et al., Unique and conserved features of genome and proteome of SARS‐coronavirus, an early split‐off from the coronavirus group 2 lineage. J. Mol. Biol. 2003, 331, 991–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rual, J.‐F. , Venkatesan, K. , Hao, T. , Hirozane, T. et al., Towards a proteome‐scale map of the human protein‐protein interaction network. Nature 2005, 437, 1173–1178. [DOI] [PubMed] [Google Scholar]
- 53. Stelzl, U. , Worm, U. , Lalowski, M. , Haenig, C. et al., A human protein‐protein interaction network: a resource for annotating the proteome. Cell 2005, 122, 957–968. [DOI] [PubMed] [Google Scholar]
- 54. Lehner, B. , Fraser, A. G. , A first‐draft human protein‐interaction map. Genome Biol. 2004, 5, R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Parrish, J. R. , Yu, J. , Liu, G. , Hines, J. A. et al., A proteome‐wide protein interaction map for Campylobacter jejuni. Genome Biol. 2007, 8, R130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Titz, B. , Rajagopala, S. V. , Goll, J. , Hauser, R. et al., The binary protein interactome of Treponema pallidum ‐ the syphilis spirochete. PLoS One 2008, 3, e2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wang, Y. , Cui, T. , Zhang, C. , Yang, M. , A global protein‐protein interaction network in the human pathogen Mycobacterium tuberculosis H37Rv. Proteome Res. 2010, 9, 6665–6677. [DOI] [PubMed] [Google Scholar]
- 58. Marchadier, E. , Carballido‐Lopez, R. , Brinster, S. , Fabret, C. et al., An expanded protein‐protein interaction network in Bacillus subtilis reveals a group of hubs: Exploration by an integrative approach. Proteomics 2011, 11, 2981–2991. [DOI] [PubMed] [Google Scholar]
- 59. Kühner, S. , van Noort, V. , Betts, M. J. , Leo‐Macias, A. et al., Proteome organization in a genome‐reduced bacterium. Science 2009, 326, 1235–1240. [DOI] [PubMed] [Google Scholar]
- 60. Buck, D. , Spencer, M. E. , Guest, J. R. , Primary structure of the succinyl‐CoA synthetase of Escherichia coli. Biochemistry 1985, 24, 6245–6252. [DOI] [PubMed] [Google Scholar]
- 61. Datsenko, K. A. , Wanner, B. L. , One‐step inactivation of chromosomal genes in Escherichia coli K‐12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gully, D. , New partners of acyl carrier protein detected in Escherichia coli by tandem affinity purification. FEBS Lett. 2003, 548, 90–96. [DOI] [PubMed] [Google Scholar]
- 63. Fraser, C. M. , Complete genome eequence of Treponema pallidum, the syphilis spirochete. Science 1998, 281, 375–388. [DOI] [PubMed] [Google Scholar]
- 64. Himmelreich, R. , Hilbert, H. , Plagens, H. , Pirkl, E. et al., Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res. 1996, 24, 4420–4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. World Health Organization. Global Tuberculosis Control 2009: Epidemiology, strategy, financing. Nonserial Publication. WHO, 2009.
- 66. Raman, K. , Chandra, N. , Mycobacterium tuberculosis interactome analysis unravels potential pathways to drug resistance. BMC Microbiol. 2008, 8, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cui, T. , Zhang, L. , Wang, X. , He, Z.‐G. , Uncovering new signaling proteins and potential drug targets through the interactome analysis of Mycobacterium tuberculosis. BMC Genom. 2009, 10, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Noirot‐Gros, M.‐F. , Dervyn, E. , Wu, L. J. , Mervelet, P. et al., An expanded view of bacterial DNA replication. Proc. Natl. Acad. Sci. USA 2002, 99, 8342–8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Duigou, S. , Ehrlich, S. D. , Noirot, P. , Noirot‐Gros, M.‐F. , DNA polymerase I acts in translesion synthesis mediated by the Y‐polymerases in Bacillus subtilis. Mol. Microbiol. 2005, 57, 678–90. [DOI] [PubMed] [Google Scholar]
- 70. Soufo, C. D. , Soufo, H. J. D. , Noirot‐Gros, M.‐F. , Steindorf, A. et al., Cell‐cycle‐dependent spatial sequestration of the DnaA replication initiator protein in Bacillus subtilis. Dev. Cell 2008, 15, 935–941. [DOI] [PubMed] [Google Scholar]
- 71. Guiguemde, W. A. , Shelat, A. A. , Bouck, D. , Duffy, S. et al., Chemical genetics of Plasmodium falciparum. Nature 2010, 465, 311–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. LaCount, D. J. , Vignali, M. , Chettier, R. , Phansalkar, A. et al., A protein interaction network of the malaria parasite Plasmodium falciparum. Nature 2005, 438, 103–107. [DOI] [PubMed] [Google Scholar]
- 73. Yellaboina, S. , Goyal, K. , Inferring genome‐wide functional linkages in E. coli by combining improved genome context methods: comparison with high‐throughput experimental data. Genome Res. 2007, 17, 527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hu, P. , Janga, S. C. , Babu, M. , Diaz‐Mejia, J. J. et al., Global functional atlas of Escherichia coli encompassing previously uncharacterized proteins. PLoS Biol. 2009, 7, e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lengeling, A. , Pfeffer, K. , Balling, R. , The battle of two genomes: genetics of bacterial host/pathogen interactions in mice. Mamm. Genome 2001, 12, 261–271. [DOI] [PubMed] [Google Scholar]
- 76. Stebbins, C. E. , Structural microbiology at the pathogen‐host interface. Cell. Microbiol. 2005, 79, 9, 1227–1236. [DOI] [PubMed] [Google Scholar]
- 77. Korkin, D., Thieu, T., Joshi, S., Mining host‐pathogen interactions, in: Yang, N.‐S. (Ed.), Systems and Computational Biology – Molecular and Cellular Experimental Systems, InTech, Rijeka, 2006, pp. 163–184.
- 78. Davis, F. P. , Barkan, D. T. , Eswar, N. , McKerrow, J. H. , Sali, A. , Host pathogen protein interactions predicted by comparative modeling. Prot. Sci. 2007, 16, 2585–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Dyer, M. D. , Murali, T. M. , Sobral, B. W. , Computational prediction of host‐pathogen protein‐protein interactions. Bioinformatics 2007, 23, i159–i166. [DOI] [PubMed] [Google Scholar]
- 80. Krishnadev, O. , Srinivasan, N. , A data integration approach to predict host‐pathogen protein‐protein interactions: application to recognize protein interactions between human and a malarial parasite. In Silico Biol. 2008, 8, 235–250. [PubMed] [Google Scholar]
- 81. Lee, S.‐A. , Chan, C. , Tsai, C.‐H. , Lai, J.‐M. et al., Ortholog‐based protein‐protein interaction prediction and its application to inter‐species interactions. BMC Bioinformatics 2008, 9, S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tastan, O. , Qi, Y. , Carbonell, J. G. , Klein‐Seetharaman, J. , Prediction of interactions between HIV‐1 and human proteins by information integration. Pacific Symposium on Biocomputing 2009, 527, 516–527. [PMC free article] [PubMed] [Google Scholar]
- 83. Evans, P. , Dampier, W. , Ungar, L. , Tozeren, A. , Prediction of HIV‐1 virus‐host protein interactions using virus and host sequence motifs. BMC Med. Genom. 2009, 2, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Doolittle, J. M. , Gomez, S. M. , Structural similarity‐based predictions of protein interactions between HIV‐1 and Homo sapiens. Virol. J. 2010, 7, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. de Chassey, B. , Navratil, V. , Tafforeau, L. , Hiet, M. S. et al., Hepatitis C virus infection protein network. Mol. Sys. Biol. 2008, 4, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Shapira, S. , Gat‐Viks, I. , Shum, B. , Dricot, A. , A physical and regulatory map of host‐influenza interactions reveals pathways in H1N1 infection. Cell 2009, 139, 1255–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Dyer, M. D. , Neff, C. , Dufford, M. , Rivera, C. G. et al., The human‐bacterial pathogen protein interaction networks of Bacillus anthracis, Francisella tularensis, and Yersinia pestis. PLoS One 2010, 5, e12089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Yang, H. , Ke, Y. , Wang, J. , Tan, Y. et al., Insight into bacterial virulence mechanisms against host immune response via the Yersinia pestis‐human protein‐protein interaction network. Infect. Immun. 2011, 79, 4413–4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Winnenburg, R. , Urban, M. , Beacham Baldwin, T. K. et al., PHI‐base update: additions to the pathogen host interaction database. Nucleic Acids Res. 2008, 36, D572–D576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chatr‐aryamontri, A. , Ceol, A. , Peluso, D. , Nardozza, A. et al., VirusMINT: a viral protein interaction database. Nucleic Acids Res. 2009, 37, D669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Navratil, V. , de Chassey, B. , Meyniel, L. , Delmotte, S. et al., VirHostNet: a knowledge base for the management and the analysis of proteome‐wide virus‐host interaction networks. Nucleic Acids Res. 2009, 37, D661–D668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Gillespie, J. J. , Wattam, A. R. , Cammer, S. A. , Gabbard, J. L. et al., PATRIC: the comprehensive bacterial bioinformatics resource with a focus on human pathogenic species. Infect. Immun. 2011, 79, 4286–4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Durmuş Tekir, S. , Çakir, T. , Ülgen, K. Ö. , Infection strategies of bacterial and viral pathogens through pathogen‐human protein‐protein interactions. Front. Microbiol. 2012, 3, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yin, L. , Xu, G. , Torii, M. , Niu, Z. , Maisog, J. , Document classification for mining host pathogen protein‐protein interactions. Artif. Intell. Med. 2010, 49, 155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Anthony, S., Sintchenko, V., Coiera, E., Text mining for discovery of host–pathogen‐interactions, in: Sintchenko, V. (Ed.), Infectious Disease Informatics, Springer Science+Business Media, New York 2010, pp. 149‐165.
- 96. Thieu, T. , Joshi, S. , Warren, S. , Korkin, D. , Literature mining of host‐pathogen interactions: comparing feature‐based supervised learning and language‐based approaches. Bioinformatics 2012, 28, 867–875. [DOI] [PubMed] [Google Scholar]
- 97. Jager, S. , Cimermancic, P. , Gulbahce, N. , Johnson, J. R. et al., Global landscape of HIV‐human protein complexes. Nature 2012, 481, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Gandhi, T. , Zhong, J. , Mathivanan, S. , Karthick, L. , Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nature Genet. 2006, 38, 285–293. [DOI] [PubMed] [Google Scholar]
- 99. Aoki‐Kinoshita, K. F., Kanehisa, M., Gene annotation and pathway mapping in KEGG, in: Nicholas, H. G. (Ed.), Comparative Genomics, Methods in Molecular Biology, Humana Press, New Jersey, 2007, pp. 71–91. [DOI] [PubMed]
- 100. Hale, B. G. , Randall, R. E. , Ortín, J. , Jackson, D. , The multifunctional NS1 protein of influenza A viruses. J. Gen. Biol. 2008, 89, 2359–2376. [DOI] [PubMed] [Google Scholar]
- 101. Bailer, S. M. , Haas, J. , Connecting viral with cellular interactomes. Curr. Opin. Microbiol. 2009, 12, 453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Brennan, R. O. , Durack, D. T. , Gay compromise syndrome. Lancet 1981, 2, 1338–1339. [DOI] [PubMed] [Google Scholar]
- 103. Weiss, R. A. , How does HIV cause AIDS? Science 1993, 260, 1273–1279. [DOI] [PubMed] [Google Scholar]
- 104. Stumptner‐Cuvelette, P. , Morchoisne, S. , Dugast, M. , Gall, S. L. et al., HIV‐1 Nef impairs MHC class II antigen presentation and surface expression. Proc. Natl. Acad. Sci. USA 2001, 98, 12144–12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Klase, Z. , Winograd, R. , Davis, J. , Carpio1, L. et al., HIV‐1 TAR miRNA protects against apoptosis by altering cellular gene expression. Retrovirology 2009, 6, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Nisole, S. , Krust, B. , Hovanessian, A. G. , Anchorage of HIV on permissive cells leads to coaggregation of viral particles with surface nucleolin at membrane raft microdomains. Exp. Cell. Res. 2002, 276, 155–173. [DOI] [PubMed] [Google Scholar]
- 107. Yedavalli, V. S. R. K. , Shih, H.‐M. , Chiang, Y.‐P. , Lu, C.‐Y. et al., Human immunodeficiency virus type 1 Vpr interacts with antiapoptotic mitochondrial protein HAX‐1. J. Virol. 2005, 79, 13735–13746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. He, B. , Qiao, X. , Klasse, P. J. , Chiu, A. et al., HIV‐1 envelope triggers polyclonal Ig class switch recombination through a CD40‐independent mechanism involving BAFF and C‐Type lectin receptors. J. Immunol. 2006, 176, 3931–3941. [DOI] [PubMed] [Google Scholar]
- 109. Ashburner, M. , Ball, C. , Blake, J. , Botstein, D. , Gene Ontology: tool for the unification of biology. Nature 2000, 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lai, X. , Golovliov, I. , Francisella tularensis induces cytopathogenicity and apoptosis in murine macrophages via a mechanism that requires intracellular bacterial multiplication. Infect. Immun. 2001, 69, 4691–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Park, J. M. , Greten, F. R. , Li, Z.‐W. , Karin, M. , Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science 2002, 297, 2048–51. [DOI] [PubMed] [Google Scholar]
- 112. Zhang, Y. , Ting, A. T. , Marcu, K. B. , James, B. , Bliska, J. B. , Inhibition of MAPK and NF‐KB pathways is necessary for rapid apoptosis in macrophages infected with Yersinia. J. Immunol. 2005, 174, 7939–7949. [DOI] [PubMed] [Google Scholar]
- 113. Flannick, J. , Novak, A. , Srinivasan, B. S. , Mcadams, H. H. , Batzoglou, S. , Græmlin: General and robust alignment of multiple large interaction networks. Genome Res. 2006, 16, 1169–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Sharan, R. , Suthram, S. , Kelley, R. M. , Kuhn, T. et al., Conserved patterns of protein interaction in multiple species. Proc. Natl. Acad. Sci. USA 2005, 102, 1974–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Murali, T. M. , Dyer, M. D. , Badger, D. , Tyler, B. M. , Katze, M. G. , Network‐based prediction and analysis of HIV dependency factors. PLoS Comput. Biol. 2011, 7, e1002164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Brass, A. L. , Dykxhoorn, D. M. , Benita, Y. , Yan, N. et al., Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [DOI] [PubMed] [Google Scholar]
- 117. König, R. , Zhou, Y. , Elleder, D. , Diamond, T. , Global analysis of host‐pathogen interactions that regulate early‐stage HIV‐1 replication. Cell 2008, 135, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Zhou, H. , Xu, M. , Huang, Q. , Gates, A. T. et al., Genome‐scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 2008, 4, 495–504. [DOI] [PubMed] [Google Scholar]
- 119. Bushman, F. D. , Malani, N. , Fernandes, J. , D'Orso, I. et al., Host cell factors in HIV replication: meta‐analysis of genome‐wide studies. PLoS Pathog. 2009, 5, e1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Li, Q. , Brass, A. L. , Ng, A. , Hu, Z. et al., A genome‐wide genetic screen for host factors required for hepatitis C virus propagation. Proc. Natl. Acad. Sci. USA 2009, 106, 16410–16415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Krishnan, M. , Ng, A. , Sukumaran, B. , Gilfoy, F. , RNA interference screen for human genes associated with West Nile virus infection. Nature 2008, 455, 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Karlas, A. , Machuy, N. , Shin, Y. , Pleissner, K. P. , Genome‐wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 2010, 463, 818–22. [DOI] [PubMed] [Google Scholar]
- 123. König, R. , Stertz, S. , Zhou, Y. , Inoue, A. et al., Human host factors required for influenza virus replication. Nature 2010, 463, 813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kumar, D. , Nath, L. , Kamal, A. , Varshney, A. et al., Genome‐wide analysis of the host intracellular network that regulates survival of Mycobacterium tuberculosis. Cell 2010, 140, 731–743. [DOI] [PubMed] [Google Scholar]