

P001
NOVEL COMBINED DELIVERY OF ANTIMICROBIAL PEPTIDES AND ANTI‐INFLAMMATORY AGENTS FOR TREATMENT OF CLOSTRIDIUM DIFFICILE INFECTION AND DISEASE
D. Kennedy, H. Humphries, M. Devocelle
Royal College of Surgeons in Ireland
Clostridium difficile is the main cause of nosocomial diarrhoea in developed countries. The incidence of disease due to this bacterium has been increasing in recent years, with several outbreaks, including from strains such 027 associated with increased severity and mortality.1 The number of antibiotics for clinical treatment of Clostridium difficile infection is limited.2 Antimicrobial peptides offer promise as novel anti‐infective agents to which the development of resistance by microbes may be restricted.3 We have developed a novel approach to combined delivery of antimicrobial peptides and agents that protect host epithelia for treatment of Clostridium difficile infection and disease.
1. McFarland, L. V., Update on the changing epidemiology of Clostridium difficile‐ associated disease. Nat Clin Pract Gastroenterol Hepatol, 2008, 5(1), 40–48.
2. Bricker, E.; Garg, R.; Nelson, R.; Loza, A.; Novak, T.; Hansen, J., Antibiotic treatment for Clostridium difficile‐associated diarrhea in adults. Cochrane Database Syst Rev, 2005(1), CD004610.
3. Hancock, R. E.; Sahl, H. G., Antimicrobial and host‐defense peptides as new anti‐ infective therapeutic strategies. Nat Biotechnol, 2006, 24(12), 1551–1557.
P002
CORRELATIONS OF PROTEIN DYNAMICS WITH FUNCTION IN HIV‐1 PROTEASE CATALYSIS
V. Torbeev,1 H. Raghuraman,1 D. Hamelberg,2 M. Tonelli,3 M. Westler,3 E. Perozo1 and S. Kent1
1Institute for Biophysical Dynamics, Department of Chemistry, University of Chicago, Chicago, IL 60637; 2Department of Chemistry, Georgia State University, Atlanta, GA 30302; 3National Magnetic Resonance Facility at Madison, Madison, WI 53706
Total chemical synthesis was used to prepare a series of unique analogues of the HIV‐1 protease (HIV‐1 PR), where we systematically substituted the residues Gly51, Gly51' at the tips of the mobile flaps' (residues 37‐61 in each domain of the protein homodimer) with L‐Ala, D‐Ala in both symmetric and asymmetric fashion. Such substitutions, although in regions distant from the catalytic aspartates, led in most cases to very substantial reductions of catalytic activity. In contrast to this, a chemically synthesized 203 amino acid residue ‘covalent dimer’ protein with L‐Ala51 in one flap and D‐Ala51' in the other flap has native‐like enzyme activity. To gain insight into the molecular details of such phenomena, we have applied a variety of biophysical methods including NMR and pulse‐EPR spectroscopies, as well as X‐ray crystallography and appropriate enzymatic assays. Both, NMR relaxation experiments with 15N‐spin labeled proteins and pulse‐EPR measurements of nitroxide‐labeled analogues have revealed that [L‐Ala51,51'] HIV‐1 PR has nearly wild‐type flap mobility on sub‐ns time scale, whereas [D‐Ala51,51'] HIV‐1 PR has much more rigid flaps both on sub‐ns and μ ;s‐ms time scale. Molecular dynamics simulations further demonstrated that the asymmetric combination of D‐Ala/L‐Ala flaps, but not the symmetric D/D or L/L, uniquely stabilizes the conformational state of catalytic residues Asp25 and Asp25' with the nucleophilic water molecule being prearranged for catalysis. The results obtained also confirm that the viral HIV‐1 protease and the cell‐encoded monomeric aspartic proteases share the same intrinsically asymmetric chemical mechanism.
P003
DESIGN AND CHARACTERIZATION OF A PH SENSITIVE HOMO‐ DIMERIC ANTI‐PARALLEL COILED COIL
R. Nagarkar and J. Schneider
Department of Chemistry and Biochemistry, University of Delaware, Newark, DE 19716
A homodimeric anti‐parallel coiled coil that folds under acidic conditions has been designed. Structure‐based design was used starting from the anti‐parallel coiled coil BCR30‐65from the Bcr‐Abl oncoprotein. This coiled coil contains a naturally occurring glutamic acid (Glu52) in its hydrophobic core. By modulating the protonation state of Glu52 we could potentially regulate coiled coil formation based on electrostatic destabilization of the hydrophobic core by a buried charge. However, denaturation of this peptide revealed no significant changes in its thermal stability over a wide pH range. This pH‐independent behavior was purported to arise from a stabilizing salt bride between Arg55 and Glu52 that compensated for buried negative charge centered at Glu52. To induce pH responsive behavior, firstly, an Arg55Ala mutation to unmask the effect of Glu52 and second, Ile42 in the ‘a‐d’ layer of Glu52 was mutated to Glu, to magnify the charge repulsions inhibiting coiled coil formation. Detailed thermodynamics, AUC and orientation analyses showed that while deleting the Arg55/Glu 52 salt bridge does not significantly compromise its thermal stability, the ability of the protein to adopt an anti‐parallel coiled coil is lost; a mixture of anti‐parallel and parallel coiled coils is realized. However, Ile42Glu adopts an anti‐parallel homodimeric coiled coil in a pH‐dependant manner with only a marginal decrease in thermal stability.
P004
SPECTROSCOPIC ANALYSIS OF A β‐HAIRPIN‐FORMING MINIPROTEIN
M.P.D. Hatfield, R.F. Murphy, S. Lovas
Department of Biomedical Sciences, Creighton University, Omaha, NE
Electronic and vibrational circular dichroism are often used to determine the secondary structure of proteins, because each secondary structure has a unique spectrum. In order to determine these spectral features, polypeptides that are known to adopt the desired conformation along their entire length are studied ideally. Little is known about the vibrational circular dichroism spectroscopic features of β‐hairpins. In this study, we used a decapeptide, YYDPETGTWY (CLN025), which forms a stable β‐hairpin that is stabilized by intramolecular weakly polar interactions and hydrogen bonds. CLN025 was synthesized by microwave‐assisted solid phase peptide synthesis with N‐a‐Fmoc protected amino acids. During purification on C18 reversed phase HPLC columns it was found that CLN025 adopts two or more conformations, which was shown, so far, only for much larger polypeptides. The structure of CLN025 in aqueous and organic solvents was examined using electronic and vibrational circular dichroism spectroscopy. The presence of β‐hairpin was confirmed by ECD and a unique VCD signal was observed. Molecular Dynamics simulations were used to correlate conformational features with CD spectra for CLN025 in the same aqueous and organic solvents.
This work was supported by the NIH‐INBRE grant P20 RR016469 and the Carpenter Endowed Chair in Biochemistry, Creighton University.
P005
NATURE OF AMIDE CARBONYL–CARBONYL INTERACTIONS IN PROTEINS
A. Choudhary and R. Raines
University of Wisconsin‐Madison
Non‐covalent interactions define and modulate biomolecular structure, function, and dynamics. In many protein secondary structures, an intimate interaction exists between adjacent carbonyl groups of the main‐chain amide bonds. As this short contact contributes to the energetics of protein conformational stability as well as protein–ligand interactions, understanding its nature is crucial. The intimacy of the carbonyl groups could arise from a charge–charge or dipole–dipole interaction, or n→π* electronic delocalization. This last putative origin, which is reminiscent of the Bürgi–Dunitz trajectory, involves delocalization of the lone pairs (n) of the oxygen (Oi–1) of a peptide bond over the antibonding orbital (p*) of the carbonyl group (Ci=Oi) of the subsequent peptide bond. By installing isosteric chemical substituents in a peptidic model system, and using NMR spectroscopy, X–ray diffraction analysis, and ab initio calculations to analyze the consequences, the intimate interaction between adjacent carbonyl groups is shown to arise primarily from n→π* electronic delocalization. Additionally, computational analysis on an Ac–Ala4– NHMe peptidic model system suggests widespread n→π* electronic delocalization in the allowed regions of the Ramachandran plot. These finding have implications for organic, biological, and medicinal chemistry.
Figure .
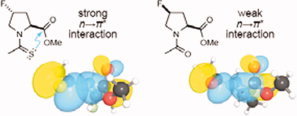
P006
HIGH‐RESOLUTION CRYSTAL DATA: REVISION OF HYDROGEN BONDING IN PROTEINS
G. Marshall, D. Kuster
Washington University
Critical analysis of high‐resolution crystallographic structures indicated that protein helices populate a single smoothly peaked distribution, rather than the weighted bimodal distribution expected from mixed populations of α‐helices and 310‐helices. The helical population was centered at (f = −62, y = −43), a coordinate intermediate between the classical parameters for the ideal α‐helix or 310‐helix. These data contradict the prevailing dogma that helices in proteins exist at or near one of the classic “ideal” helical configurations. Further, the Protein DataBank (PDB) was shown to manifest a clear bias towards supposedly ideal helices at nominal resolutions greater than 2Å, where structure models are insufficiently constrained by experimental electron density. A new model for helices in proteins that accounts for the contributions of shared (bifurcated) hydrogen bonds evolved from a straightforward interpretation of high‐resolution crystallographic data and physicochemical constraints. This new molten helical model unifies experimental observations with the concept of the protein helix as a stable secondary structure that samples conformational space from a single smooth potential well located between the classical α‐helical and 310‐helical coordinates on the energy surface. Together, these observations regarding helical models suggest simple improvements in backbone hydrogen‐bond potentials, molecular mechanics force‐field parameters (particularly for electrostatics), scoring functions for protein folding, and helical peptidomimetic design. The bias of an abundance of α‐helical structures seen in low‐resolution (>2.0) crystal structure in the PDB severely compromised this structural database and is hypothesized to arise form the use of monopole electrostatics in modeling programs. A detailed statistical analysis of the impact of crystallographic resolution on protein secondary structures was suggested as well as a thorough comparison of the impact of quantum mechanics approximations and force‐field methodology on the potential surfaces of bifurcated hydrogen bonds. We anticipate, based on our recent analyses of helical hydrogen bonding in high‐resolution crystal structures of proteins in the PDB, that a detailed re‐interpretation of all secondary structures in lower‐resolution (nominal resolution >2.0) structural models of proteins may be necessary.
P007
SHORT, HYPERSTABLE BETA SHEETS WITHOUT TURNS
B. Kier and N. Andersen
University of Washington
Beta‐hairpin peptides have become popular model systems of beta sheet structure in proteins, but their reliance on high propensity reversing turns limits their usefulness in modeling the behavior of natural beta sheets with flexible loops or large contact order between strands. Herein we report a class of homodimeric turnless beta sheets linked by a central cystine residue. These constructs range from 6 to 13 residues per strand, adopt well‐defined beta‐sheet structures, and contain no unnatural amino acids. Though some betasheet structure was observed for a number of diverse homodimeric sequences, the most stable of these (Tm > 70 C) employed a specific beta capping motif. This motif takes the form (Pr/Ac‐WċWTG) and caps both ends of the beta sheet. These constructs should provide useful minimized systems for studying beta sheet structure, and serve to illustrate both the potency of the beta capping motif and the context‐dependent superfluity of nucleating backbone turn sequences.
P008
CRITICAL ASSESSMENT OF MINI‐PROTEIN STABILITY USING MOLECULAR SIMULATIONS, CIRCULAR DICHROISM, CALORIMETRY AND NMR
J. Feng, G. Marshall
Washington University in St. Louis
Designing mini‐proteins with enhanced stability is a challenging problem. The thermal stability of a 28‐residue synthetic mini‐protein, FSD‐1, and chimeric analogs were analyzed by using molecular simulations, circular dichroism, differential scanning calorimetry and NMR. Experimental melting curves were determined by using both CD and DSC. Melting curves were also calculated from an explicit solvent replica‐exchange molecular dynamics simulation. Simulation results showed that FSD‐1 exhibited only weak cooperativity between its alpha helix and beta hairpin. The beta hairpin was relatively unstable but its formation was necessary for the formation of a hydrophobic core and folding. The experimental melting curves resembled that of a slowly unfolding helix instead of a two‐state folder. The weak stability of FSD‐1 was further supported by NMR measurements at 7°C.
P009
LONG CHAIN PEG CONSTRUCTION WITH NATIVE CHEMICAL LIGATION ON GLASS SUBSTRATES
R. Hamel, E. Wieczerzak, V. Chabot, V. Aimez, P. Charette, M. Grandbois, E. Escher
Université de Sherbrooke
We have shown that polypeptides can be rapidly immobilized on glass substrates by means of thiol catalyzed native chemical ligation (NCL) [Biopolymers/PeptideScience 90, 415; 2008]. The aim of the present contribution is to build long molecules on glass surfaces for biosensors. α‐Amino‐ω‐carboxy PEG‐amino acids of 10 kDa (PEG = polyethylene glycol) were to be attached on glass surfaces and extended to 20 kDa oligomers. For this purpose, a first Boc‐Cys(Boc)‐PEG‐thioester was synthesized and coupled to Cys modified glass surfaces by NCL. Reactions were followed by surface plasmon resonance (SPR). A second PEG unit was then attached by the same procedure after dilute aqueous HCl deprotection of the Boc protection of the first Cys‐PEG unit. In order to measure the length of the attached constructions, the surfaces were probed by atomic force microscopy (AFM). For this purpose an α‐biotinyl PEG (10kDa)‐ω‐thioester was prepared and coupled, either instead of the first PEG or as the second PEG, thus giving opportunity of molecular recognition. In conjunction with this, AFM cantilevers were coated with biotinyl BSA and then with streptavidin. The AFM was configured in force‐scope mode and the biotinyl‐PEG surface was probed. PEG length was measured by pulling perpendicularly and the pulling force was recorded. Lengths at rupture point were close to the theoretical PEG length and with a rupture force similar to the avidin‐biotin bond, confirming correct tether immobilization, length and functionality.
P010
HOW DO CYCLOTIDES EXERT THEIR MODE OF ACTION? THE IMPORTANCE OF CELL MEMBRANES ON THEIR DIVERSE ACTIVITIES
S. Henriques,1,2 Y. Huang,1 N. Daly,1 D. Craik1
1The University of Queensland, Institute of Molecular Bioscience, Brisbane, QLD 4072, Australia; 2Institute of Molecular Medicine, Medicine School, University of Lisbon, 1649‐028 Lisbon, Portugal
Cyclotides are plant‐derived peptides that incorporate a unique structural motif comprising three disulfide bonds and a circular backbone that engenders a particularly high degree of stability to enzymatic and chemical assault. Divided in two major subfamilies termed Möbius and bracelet, cyclotides are currently the largest known family of circular proteins and a range of intrinsic activities have been identified, including anti‐microbial, anti‐cancer, haemolytic, antifouling and insecticidal activities. A major question regarding the biology of cyclotides remains unanswered: How do cyclotides exert their biological effects? The plasma membrane is the first cellular barrier and the interaction of cyclotides with membranes seems to be the key‐feature for their biological activities. In the current study we have examined the membrane interaction of cyclotides from the two major subfamilies along with a natural hybrid of the two families. Fluorescence spectroscopy, surface plasmon resonance, confocal microscopy and optical density have been used to analyse the peptides with model membranes. All the cyclotides studied show a preference for membranes containing phosphatidylethanolamine phospholipids whereas negatively‐charged phospholipids did not affect membrane binding extension. The presence of cholesterol to modulate membrane fluidity did not alter membrane preference. These results clearly show that cyclotides do not prefer more rigid domains in the membrane, such as raft domains, and that the peptide‐membrane binding is mainly governed by hydrophobic rather than electrostatic interactions. Cyclotides belonging to the bracelet family are able to disintegrate the membrane bilayers, whereas Möbius peptides appear to form pores without membrane destruction. Overall, we have shown that even with similar membrane selectivity, different cyclotides have different effects on membrane stability, which explains the diversity of this family and also their diverse activities.
P011
NUCLEAR LOCALIZATION OF THE AMYLOID β‐PEPTIDE DURING OXIDATIVE STRESS IN NEURONAL CELLS
J. Bailey, D. Lahiri
Indiana University School of Medicine, Indianapolis, IN
Alzheimer's disease (AD) pathology is characterized by two distinct lesions: neurofibrillary tangles comprised of hyperphosphorylated microtubule associated tau protein, and neuritic plaque comprised principally of the amyloid β‐peptide (Aβ). Plaque formation is thought to be key in the development of AD and much effort has been spent in the study of Aβ aggregation and attempts to remove or prevent formation of Aβ plaque. The physiological role of Aβ is much less well understood. We have recently shown that Aβ binds specific G‐rich DNA sequences and suggested a possible role for Aβ as a transcription factor (Society for Neuroscience meeting, 2008). To better understand the physiological role of Aβ, we studied the uptake of fluorescine‐conjugated Aβ by cultured neuron‐like PC12 cells under oxidative stress. In response to oxidative stress, Aβ was localized to the nuclei of a portion of viable cells and nearly all dead cells, while very little nuclear localization was observed in unstressed cells. This pattern of nuclear labeling is suggests a role for Aβ in the oxidative death cascade, which is an important player in AD. These results provide clues to the physiological role of Aβ and the Aβ‐mediated toxicity seen in AD.
P012
AN ALTERNATIVE BINDING MODE OF THE NSH2 DOMAIN OF PHOSPHATASE SHP2
Y. Zhang
The Ohio State University
Protein tyrosine phosphatase SHP2 participates in signaling events through the interaction of its tandem SH2 domains with various activated receptors. The binding specificity of its N‐terminal SH2 (N‐SH2) domain was previously determined by a combinatorial library approach. It was found that the N‐SH2 domain could recognize four distinct classes of binding motifs. In this study, the co‐crystal structure of a class IV motif, VIpYFVP, bound to the N‐SH2 domain was determined, which revealed a noncanonical 2:1 (peptide: protein) binding mode. This binding mode was confirmed in solution phase by 15N‐1H HSQC, 13C‐1H HSQC, and 31P NMR experiments.
P013
APP‐DERIVED PEPTIDOMIMETIC SUBSTRATES MODULATE BACE ACTIVITY
J.P. Juliano,1 L. Pattenden,1 P. Perlmutter,2 M.I. Aguilar,1
1Department of Biochemistry & Molecular Biology, Monash University, Clayton, Melbourne, Australia; 2School of Chemistry, Monash University, Clayton, Melbourne, Australia
Beta‐site APP Cleaving enzyme (BACE) is a membrane‐associated enzyme involved in the production of Amyloid‐beta which is implicated in the formation of amyloid plaques within Alzheimer's disease brains. Performing the primary cleavage of Amyloid Precursor Protein (APP) of the amyloid‐forming pathway, BACE serves as a favourable therapeutic target. While APP is implicated as a modulator of such activity, numerous other known biological and synthetic substrates may also play a role. Mechanisms that regulate such cleavage activity by mature BACE remain unclear. In this study, we explored the role of different sequence domains within APP to modulate BACE activity. Specifically, we utilised the BACE cleavage domain of APP and other numerous substrates as peptide scaffolds to form a superset of BACE substrate analogues. These were generated via a series of truncations, extensions, termini capping, residue substitutions and side chain modifications via solid‐phase peptide synthesis. A Quenched Fluorescent Substrate BACE assay in conjunction with LC/MS analysis were used to investigate cleavage susceptibility and inhibition potency under competitive and non‐competitive conditions to define a subset of peptides that function to modulate BACE activity, identifying both inhibitory and stimulatory structural motifs. These results provide insight into the possible modulatory role of APP in BACE function in vivo and as an avenue to further explore novel BACE inhibitor design.
P014
STRUCTURE‐ACTIVITY RELATIONSHIP STUDY OF NEW FK228 ANALOGUES AS ANTITUMOR AGENTS
S. Di Maro, J. Ahn, R. Pong, J. Hsieh
University of Naples “Federico II”
Histone deacetylases (HDACs) are enzymes that repress gene transcription by removing acetyl groups from histones. Aberrant recruitment and expression of HDACs have been correlated to tumorigenesis process and provided a rationale for the use of HDAC inhibitors to treat cancers. Among many compounds under preclinical investigation, a bicyclic depsipeptide FK228 is considered as the most potent HDAC inhibitor in vitro and in vivo; however, its synthetic challenges have made in‐depth SAR studies difficult, which would facilitate the understanding of structural requirements of the natural product. Herein, we report novel FK228 analogues in which the most synthetically challenging unit, (3S,4E)‐3‐hydroxy‐7‐mercapto‐heptenoic acid was modified by simple isosteric substitutions. These modifications did not alter the backbone conformation and enabled facile production of a large number of analogues. For high synthetic efficiency, we have developed a solidphase strategy that allowed rapid synthesis with high overall yield (>75%) and purity (>80%). Over 60 compounds were constructed to explore the positions of L‐Val, Z‐Dhb and D‐Val of the original FK228 structure with a variety of side chain functional groups. To examine anticancer activity, the prepared FK228 analogues were evaluated on various cancer cells, and new potent FK228 analogues were identified (IC50 values between 20‐50 nM).
P015
TOTAL SYNTHESIS OF HISTONE H3 ACETYLATED AT LYSINE 56
J. Shimko, J. Ottesen
The Ohio State University
The eukaryotic genome is organized by the periodic wrapping of the DNA double helix around histone octamers to form nucleosomes. The DNA entry‐exit region of the nucleosome is a vital interface between the DNA double helix and the histone octamer. Dynamic acetylation of histone H3 lysine 56, buried beneath the DNA in the entry‐exit region, plays an important role in DNA repair and transcription regulation. Here, we present the synthesis of histone H3 site‐specifically acetylated at Lys 56 (H3‐K56Ac) by sequential native chemical ligation of three peptide segments. Nucleosomes are reconstituted with synthetic, chemically modified H3‐K56Ac along with DNA and recombinant histones H2A, H2B, and H4. Biochemical and biophysical study of the resultant semi‐synthetic, modified nucleosomes allows for the direct characterization of the effects of H3‐K56Ac on nucleosome structure and dynamics.
P016
SYNTHESIS AND PROPERTIES OF BCOX‐DNA
S.Luisier, C. Leumann
University of Chicago
Bc1‐ and tc2‐nucleosides (Figure 1) are nucleoside analogues with a conformational restriction. In both modified nucleosides, the orientation of the C(5')‐OH group around the C(4')‐C(5') bond is different to that found in unmodified DNA.3 Tc‐DNA exhibits a high RNA affinity over Watson‐Crick base pairing4 and has shown promising properties as antisense oligonucleotides.5
Figure .

Structure and atom numbering of bc‐, tc‐, and bcOX‐ nucleosides.
The anionic nature of the backbone of an oligonucleotide represents a major challenge for its cellular uptake. We reasoned that the introduction of a lipophilic substituent on modified nucleosides could improve the cellular uptake of a modified oligonucleotide. In addition, the modification of the bicyclic core of bc‐nucleoside could induce a correction of the misaligned torsion angle γ, resulting in a higher RNA affinity.
A new thymidine bicyclo‐nucleoside, bcox‐T, carrying a benzyl substituent was synthesized, using a β‐face selective nucleosidation. Its structure was determined by x‐ray crystallography. The modified nucleoside was incorporated into oligonucleotide by solid phase synthesis using phosphoramidite chemistry. The stability of DNA duplexes and DNA‐RNA hybrid duplexes containing modified nucleotides was determined by UV‐melting experiment, and the shape of the duplexes were analyzed by CD‐spectroscopy. The base pairing selectivity of modified oligonucleotides was determined by measuring the stability of DNA duplexes containing a mismatched base opposite to the modified nucleotide. Finally, fluorescein‐labeled oligonucleotides were transfected into HeLa cell, and the cells were analyzed under fluorescence microscopy.6
1. Tarkoy M., Leumann C., Angew. Chem. Int. Ed. Eng., 1993, 32, 1432. Tarkoy M., Bolli M., Leumann C., Helv. Chim. Acta., 1994, 77, 716.
2. Steffens R., Leumann C., Helv. Chim. Acta, 1997, 80, 2426.
3. Pallan P., Ittig D., Heroux A., Wawrzak Z., Leumann C., Egli M., Chem. Commun., 2008, 883.
4. Steffens R., Leumann C., J. Amer. Chem. Soc., 1997, 119, 11598.
5. Renneberg, D., Mouliong E., Reber U., Schumperli D., Leumann C., Nucleic Acid res., 2002, 30 (13), 2751. Ittig D., Renneberg D., Schumperli D., Leumann C., Nucleic Acids Res., 2004, 32, 346.
6. Luisier S., Leumann C., ChemBioChem., 2008, 9, 2244.
P017
VIRTUAL SCREENING TARGETING THE PHOP RESPONSE REGULATOR TO INHIBIT VIRULENCE
Y. Tang, T. Latifi, E. Groisman, G. Marshall
Washington University in St. Louis, MO
A computational approach using virtual screening combined with consensus scoring revealed drug‐like compounds that inhibit the function of the PhoP response regulator by disrupting formation to its active dimer state. PhoP, the regulatory receiver of the PhoQ/PhoP two‐component signal transduction system, senses and responds to extracellular Mg2+ levels by controlling the transcription level of key virulence genes in Salmonella typhimurium, as well as a number of other pathogenic species including Yersinia pestis, the causative pathogen for plague. The inactive monomeric state of PhoP is activated via phosphorylation by PhoQ, the sensor histidine kinase, at low extracellular Mg2+ levels. Phosphorylation leads to formation of its active dimer state, that bind to its DNA promoter. Compounds that bind to the surface of the dimerization interface can disrupt its DNA‐binding ability, and most importantly, interfere with its function as a transcription factor regulating virulence. Experimental methods including electrophoretic mobility‐shift assays and gel filtration chromatography were used to test the predictability of the computational approach and confirmed the mechanism of action for the predicted inhibitors. This study serves as a proof‐of‐principle for targeting the dimerization interface of a response regulator to inhibit its function and modulate gene regulation of the expression of virulence. With the increasing resistance of pathogenic bacteria to current antibiotics, the success of this strategy suggests targeting two‐component signal transduction systems as a promising approach for development of novel antibiotics.
P018
TOWARDS THE TOTAL SOLID‐PHASE SYNTHESIS OF THE CYCLIC LIPODEPSIPEPTIDE DAPTOMYCIN
E. Ezeigwe*, J.P. Malkinson
*Department of Pharmaceutical and Biological Chemistry, School of Pharmacy, University of London, 29‐39 Brunswick Square, London WC1N 1AX, UK ebele.ezeigwe@pharmacy.ac.uk
The cyclic lipodepsipeptides are a diverse class of natural products with significant pharmacotherapeutic potential.
Daptomycin is a semi‐synthetic 13‐membered cyclic lipodepsipeptide, with a decadepsipeptide core ring. It is produced from the fermentation products of Streptomyces roseosporus. Daptomycin is an effective bactericidal agent against multi‐drug resistant Grampositive bacteria such as MRSA.
Successful solid‐phase synthesis of various cyclic lipodepsipeptide daptomycin analogues was achieved using glutamic acid in place of (2S,3R)‐methylglutamic acid at position 12 in the cyclic core structure. The C‐terminal kynurenine residue, utilised for depsipeptide bond formation, was substituted with tryptophan, valine or isoleucine.
Macrolactamisation took place on the solid‐phase between the Glu12 α‐carboxyl and the Trp/Val/Ile α‐amino group.
Synthesis of the protected (2S,3R)‐methylglutamic acid for buildingblock was also accomplished via enzymatic resolution of racemic Na‐acetyl β‐methylaspartic acid β‐methyl ester. Through the successful synthesis of the daptomycin analogues and the non‐proteineogenic amino acid βMeGlu, we demonstrate the feasibility of the total SPPS of daptomycin.
P019
ANTIMICROBIALS DERIVED FROM HOST DEFENSE PEPTIDES USING MULTIVALENT AND COMBINATORIAL LIBRARY STRATEGIES
Z. Liu and N. Kallenbach
Department of Chemistry, New York University, 2 New York, NY 10003
As natural effectors of innate immunity, host defense peptides (HDPs) exhibit rapid killing and a broad spectrum of activity against Gram‐positive and ‐negative bacteria, fungi, and viruses. HDPs have thus inspired a variety of designs for antimicrobial therapeutics. Recently, we and others have demonstrated that simple combinations of Arginine (R) and Trptophan (W) side chains constitute the pharmacophore for one class of HDPs. Based on this result, we have employed two strategies aimed at identifying potent antimicrobial agents for the ultimate therapeutic treatment of multidrug‐resistant (MDR) bacterial infections. The first derives from the application of the principle of multivalency to create antimicrobial agents using a variety of scaffolds to link short RW peptides to afford multivalent constructs with different topology. In the second strategy, we designed and screened several combinatorial libraries based on a 1, 3, 5‐triazine scaffold to mimic the hydrophobic and charge patterns detected in the pharmocophore metioned above. A set of compounds have been identified from these strategies with potent antimicrobial activity and low hemolytic activity. These provide a set of new antimicrobial leads that open possible routes to novel antimicrobials with more drug‐like pharmaceutical properties than natural peptides.
P020
THE EFFECT OF LOCATION AND THE NUMBER OF POSITIVELY CHARGED RESIDUES ON THE POLAR FACE OF AN AMPHIPATHIC α‐HELICAL ANTIMICROBIAL PEPTIDE ON BIOPHYSICAL PROPERTIES AND BIOLOGICAL ACTIVITIES
Z. Jiang
University of Colorado, Denver, School of Medicine
Due to the growing increase in antibiotic resistance, antimicrobial peptides (AMPs) have become important candidates as potential therapeutic agents. They have two unique features: 1) a net positive charge of +2 or greater, owing to an excess of basic amino acids (Lys, Arg) over acidic amino acids (Asp, Glu); 2) an amphipathic nature, with a non‐polar face and a polar face. Their main target is the cell membrane of microorganisms. Our lead compound, a 26‐residue peptide, V13K, showed that a single valine to lysine substitution (compared to its parent peptide) in the center of the non‐polar face dramatically reduced toxicity and increased the therapeutic index. We then systematically substituted positively charged residues on the polar face to give a net positive charge from +5 to +11 as well as changing the relative location of these charged residues while maintaining the identical non‐polar face for all analogs. We evaluated these peptide analogs for their antimicrobial activity against six clinical strains of Pseudomonas aeruginosa and their hemolytic activity to human red blood cells. Increasing net positive charge and varying the location of these charged residues had a dramatic effect on antimicrobial activity and the resulting therapeutic index.
P021
DESIGN AND SYNTHESIS OF NEW POTENT MELANOCORTIN PEPTIDES WITH CANDIDACIDAL ACTIVITY
L. Auriemma,1 S. Malfi,1 S. Di Maro,1 P. Campiglia,2 E. Novellino,1 A. Catania3, P. Grieco1
1Department of Chimica Farmaceutica e Tossicologica, University of Naples Federico II; 2Department of Scienze Farmaceutiche, University of Salerno, 3Divisions of Internal Medicine and Liver Transplantation, Ospedale Maggiore di Milano IRCCS, 20122 Milano, Italy
In the last two decades the incidence of human fungal infections has increased, mainly in immunocompromised patients and additionally many microorganisms have shown resistance to common antibiotics. Therefore alternative antimicrobial agents have to be found.
Natural antimicrobial peptides are optimal candidates because they fight against resistant microorganisms and they have a broad‐spectrum activity. Recently we have reported that α‐MSH, an endogenous linear peptide, and particularly the C‐terminal tripeptide Lys‐Pro‐Val have a potent antimicrobial activity against two representative pathogens: Staphylococcus aureus and C. albicans. Indeed the candidacidal effect of α‐MSH is linked to the increase of intracellular cAMP in the C.albicans and it has been possible to hypothesize an interaction of peptide with a yeast isoform of melanocortin receptor.
Previous studies allowed to obtain [(D)Nal‐7, Phe‐12]‐ a ‐ MSH(6‐13), originated from changes into the sequence of α‐MSH(6‐13). This α‐MSH analog showed the most potent candidacidal activity and it became our lead compound. T o improve antifungal activity the residue of glycine of lead compound was replaced with natural and unnatural amino acids and new potent antifungal compounds were identified.
P022
DESIGN AND SYNTHESIS OF ANTIMICROBIAL PEPTIDE
S. Karbalaee‐Mohammad, H. Naderi‐manesh, H. Hossein nejad Ariani
Tarbiat Modares University
There is a wide range of antimicrobial peptide (AMPs) in nature. The activity of these peptide usually depends on several parameters such as: sequence, size, degree of structure formation, hydrophobicity , and Amphipathicity. In this work we plan to analysis these parameters in order to design a potent AMPcandidate. Although these parameters are infleuenced on activity but not directly correlated.AMPs due to their negative charge usually have anticancer activity. In this work we focus on the group of peptides and choose aurein 1.2. which designed on it a target peptide. The designed peptide is synthesised and analysed conformation and the MIC test will be compared with aurein 1.2.
P023
SHEPHERIN‐I AND ANALOGUES: SYNTHESES AT 60°C AND PROPERTIES
C. Remuzgo1, G.M. Machado‐Santelli2, S. Daffre3 and M.T.M. Miranda 11
Dept. of Biochemistry, Institute of Chemistry; 2Dept. of Cell and Developmental Biology, 3Dept. of Parasitology, Institute of Biomedical Sciences; University of São Paulo, 05508‐900, São Paulo, Brazil
Glycine‐rich proteins and peptides (GRP) are found in animals and plants. As little is known about their chemical synthesis, structure, structure‐activity relationship and mode of action, we studied shepherin I (ShepI; an antimicrobial GRP with 67.9% of Gly and 28.6% of His) and analogues. Stepwise solid‐phase synthesis at 60°C and NIR‐FT‐Raman spectra of the growing peptide‐resins indicated that Shep I is prone to aggregation. Among the analogues studied, the C‐terminally amidated Shep Ia, Shep I (3‐28)a and Shep I (6‐28)a showed to be the most attractive as they were as active as Shep I against C. albicans strains. Anticandidal activity of Shep Ia, Shep I (3‐28)a and Shep I (6‐28)a was inhibited in high salt concentration solutions, but significantly enhanced in 10 μM ZnCl2 solutions. At 62.5 μM (5 MIC), Shep Ia killed C. albicans MDM8 in 30 min. It caused low hemolysis in phosphate buffered saline and isotonic glucose phosphate buffer. Confocal microscopy and flow cytometry analyses revealed that Shep Ia modified with carboxyfluorescein (FAM‐Shep Ia) was rapidly internalized into C. albicans MDM8 cells. Such process, energy and temperature‐dependent, was not affected in high salt concentration. Altogether, the results suggest that (i) Shep I (3‐28)a mimics the fungicidal activity of Shep I, (ii) Shep Ia has an intracellular target and is internalized into the fungal cells via endocitosis.
Supported by FAPESP and CNPq.
P024
DESIGN, SYNTHESIS AND CONFORMATIONAL STUDIES OF NEW ANALOGUES OF TEMPORINS A AND L
S. Malfi, M.R. Saviello, L.M.H. Gaddi, M.L. Mangoni, A. Carotenuto, L. Auriemma, C. Marcozzi, I. Gomez‐Monterrey, P. Campiglia, E.Novellino, P. Grieco
University of Naples, “Federico II”
Temporins, antimicrobial peptides (AMPs) isolated from the skin of Rana temporaria, are short, linear 10‐14 residues long peptides, with a net charge positive and an amidate Cterminal. To get insight into mechanism of action and biological activity of these compounds, we have investigated two members of this family, temporin L (TL) and temporin A (TA). The first (FLPLIGRVLSGIL‐NH2) is preferentially active against Gram‐positive bacterial strains and hasn't haemolytic activity. The second (FVQWFSKFLGRIL‐NH2) has the highest activity among all temporins but it shows a haemolytic activity at the concentrations required to kill bacteria. We have investigated the preferential conformation of TL and TA in SDS and DPC solutions. On the bases of the NMR and CD results, we have designed and synthesized new TA and TL analogues called Pro3TL and Gln3TA to understand the exact mechanism of the action. In addition, we have synthesized other compounds containing D‐isomers and a Retro‐TL analogue to define the structural characteristics for the design of the feature peptides.
P025
PROBING THE EFFECT OF GOMESIN AND ITS LINEAR ANALOGUE ON GIANT UNILAMELLAR VESICLES VIA OPTICAL MICROSCOPY
T. Domingues, K. Riske, A. Miranda
Federal University of São Paulo
Gomesin (Gm) is a potent antimicrobial peptide. This molecule has two disulfide bridges (2/15 and 6/11), and adopts a β‐hairpinlike structure (pGlu‐CRRLCYKQRCVTYCRGR‐NH2). Gm and its non active linear analogue, [Ser2,6,11,15]‐Gm, were synthesized by solid‐phase methodology. We show here results obtained with optical and fluorescence microscopy of giant unilamellar vesicles (GUVs) composed of mixtures of a neutral (POPC) and a negatively charged (POPG) lipid. Two different setups were used. First we observed the effect of injecting a peptide solution with a micropipette placed at the vicinities of the GUVs. As a result of peptide‐lipid interaction, GUVs burst suddenly and stable pores were hardly ever observed. As control, in the absence of peptide, the GUVs were never spontaneously disrupted. This fact leads us to speculate that gomesin disrupt the membrane via carpeting mode. Injections of fluorescently labeled peptides show that Gm first accumulates on the vesicle surface, then domains are formed and eventually the vesicle burst. In the second setup, a GUV solution was mixed with increasing concentration of the peptides to quantify their lytic action against GUVs of different compositions. The observed effect on the lytic activity were around 90% with 3 μM gomesin and with 10 μM of [Ser2,6,11,15]‐ Gm. From our results we conclude that both peptides strongly interact with phospholipids vesicles and induce leakage of their content in a surface charge‐dependent manner. Supported by FAPESP, CNPq and FADA/UNIFESP.
P026
STRUCTURE‐ANTIBACTERIAL ACTIVITY RELATIONSHIP OF CYCLIC LIPODEPSIPEPTIDE ANTIBIOTIC FUSARICIDIN A
N. Bionda, D. Treitl, M. Stawikowski, and P. Cudic
Department of Chemistry and Biochemistry, Florida Atlantic University, 777 Glades Road, Boca Raton, FL 33431, USA
Fusaricidin A is a cyclic lipodepsipeptide originally isolated from Bacillus polymyxa KT‐8 strain, and exhibits promising activity against various kinds of fungi and Gram‐positive bacteria, including methicillin‐resistant S. aureus, In order to elucidate structural basis for its antibacterial activity we have synthesized fusaricidin A's analogues that differ in the lipid tail part, and its alanine‐scanning library using standard Fmoc solid‐phase methodology. Library screening assays revealed that lipophylic tail and amino acids involved in creation of an ester bond are crucial for peptides antibacterial activity. Complete loss of antibacterial activity was obtained for the depsipeptides without lipid tail and when peptide was cyclized via Lys side chain amino group instead of hydroxyl group found in the natural product.
P027
NOVEL PLEUROCIDIN‐AMP ANALOGS AND BIOASSAYS
K. Tamminedi, B. Chaudhary, J. Sperry, R. LaCroix, and L. Martin*
Department of Cell and Molecular Biology, University of Rhode Island, Kingston, RI 02881 USA
Existing research shows that the C‐terminal amide analog of pleurocidin is more active antibacterial agent than the native C‐terminal acid. Other evidence suggests that the amphipathic α‐helical structure is required for antibacterial activity and pleurocidin can assume an amphipathic alpha‐helical conformation that is similar to other antimicrobial peptides (AMPs) in the presence of hexafluoroisopropanol. Although the exact mechanism by which this antimicrobial peptide kills the bacterial cell is still unknown, it is believed that the cationic charge of the peptide is initially attracted to the negatively charged bacterial surface lipids. Our hypothesis is that specific alterations in the sequence of amino acids of native pleurocidin can increase the antimicrobial activity. We have tested the effect of addition of N‐terminal tryptophan to the pleurocidin C‐terminal amide, and developed high‐throughput bioassays that evaluated the effects of both AMP analogs on four diverse types of pathogenic bacteria.
P028
VIRTUAL SCREENING TARGETING THE BACTERIAL PHOP RESPONSE REGULATOR TO INHIBIT VIRULENCE
Y.T. Tang, T. Latifi, E.A. Groisman, G.R. Marshall
Washington University
A computational approach using virtual screening combined with consensus scoring revealed drug‐like compounds that inhibit the function of the PhoP response regulator by disrupting formation to its active dimer state. PhoP, the regulatory receiver of the PhoQ/PhoP two‐component signal transduction system, senses and responds to extracellular Mg2+ levels by controlling the transcription level of key virulence genes in Salmonella typhimurium, as well as a number of other pathogenic species including Yersinia pestis, the causative pathogen for plague. The monomeric state of PhoP is activated via phosphorylation by PhoQ, the sensor histidine kinase, at low extracellular Mg2+ levels. Phosphorylation leads to formation of its active PhoP dimer, that bind to its DNA promoter. Compounds that bind to the surface of the dimerization interface can disrupt its DNA‐binding ability, and most importantly, interfere with its role as a transcription factor regulating virulence. Experimental methods including electrophoretic mobility‐shift assays and gel‐filtration chromatography were used to test the predictability of the computational approach and confirm the mechanism of action for the predicted inhibitors. This study serves as a proof‐of‐principle for targeting the dimerization interface of a response regulator to inhibit its function and modulate gene regulation of the expression of virulence. With the increasing resistance of pathogenic bacteria to current antibiotics, the success of this strategy suggests targeting two‐component signal transduction as a promising approach for development of novel antibiotics.
P029
CHARACTERIZATION OF THE PEPTIDE‐MEDIATED INHIBITION OF PORPHYROMONAS GINGIVALIS/STREPTOCOCCUS GORDONII BIOFILM FORMATION
C. Daep,1 R. Lamont,2 and D. Demuth1
1University of Louisville, School of Dentistry, Louisville, KY 40202; 2University of Florida, School of Dentistry, Gainesville, FL 32610
Periodontal disease is an inflammatory disease that, while predominantly affecting the gums, has been linked to the onset of other severe systemic diseases. The disease is caused by the formation and accumulation of a bacterial biofilm in the sub‐gingival pocket and in particular by presence of Porphyromonas gingivalis. P. gingivalis is an asaccharolytic, acid‐intolerant, obligate anaerobe which interacts with specific bacterial species such as the oralis group streptococci to successfully colonize the oral cavity. The interaction between these bacteria is mediated through a species‐specific protein‐protein interaction occurring between the minor fimbrial protein Mfa1 of P. gingivalis and the SspB Adherence Region (BAR) of the antigen I/II protein (e.g., SspB) that is expressed by streptococci. Our studies have shown that this inter‐bacterial co‐aggregation can be inhibited using a synthetic peptide based on BAR and that the amino acid motif, NITVK is essential for this interaction. Current studies have expanded the interacting interface between Mfa1 and show that a eukaryotic nuclear receptor co‐activator‐like motif, VXXLL, is also involved in facilitating the Mfa1/SspB (BAR) interaction. Maintaining an amphipathic α‐helix through this region is important for the binding of Mfa1 to BAR. In addition, positively charged lysine residues, Lys1174, 1175, 1179, 1180 that flank VXXLL were shown to contribute to the interaction. Both the VXXLL and NITVK motifs are conserved among the oralis streptococci but these sequences have diverged in antigen I/II proteins expressed by streptococci that do not interact with P. gingivalis. These comparisons also identified a conserved proline residue upstream of VXXLL. A Pro1171/Gln substitution resulted in the loss of the peptide's inhibitory activity, suggesting that Pro1171 may also play an important structural role in the interaction of BAR with Mfa1. Together, these studies have defined the protein‐protein interaction that may drive the initial colonization of the oral cavity by P. gingivalis and may facilitate the development of targeted therapeutics to prevent colonization.
P030
MUTATIONAL STUDIES ON α‐CONOTOXIN VC1.1
R. Halai,1 R. Clark,1 S. Nevin,2 D. Adams2 and D. Craik1
The University of Queensland, 1Institute for Molecular Bioscience and 2Queensland Brain Institute, Brisbane, Queensland, 4072, Australia
Vc1.1 is a small, synthetic disulfide‐bonded conotoxin under development as a therapeutic for neuropathic pain. It has been shown to induce analgesia in rat models of human neuropathic pain and has been reported to target the a9a10 nAChR, with low nM potency. We conducted mutational studies on Vc1.1, replacing individual residues with either an alanine, lysine or aspartic acid, except for the highly conserved cysteines. Two key positions were identified, position 4 and position 9, which once mutated led to a significant increase in the potency of Vc1.1 at the a9a10 nAChR. Subsequently, a second generation of analogues was synthesised, with further mutations at these positions, all of which were shown to be equally, if not more potent than Vc1.1 at the a9a10 nAChR. The most potent mutants were also tested on a human a9 rat a10 hybrid clone. For potential changes in the selectivity profile of Vc1.1, these potent mutants were also tested on other nAChR subtypes, namely a3β2, a3β4 and a7. All the second generation analogues were more selective for the a9a10, with 4 out of the 7 analogues exclusively selective for a9a10 over the other nAChR subtypes screened. This study has shown that the potency of Vc1.1 can be improved, whilst maintaining its selectivity, by strategic substitutions at key positions identified through mutational studies. Overall, the mutational studies have provided a great foundation for the design and synthesis of novel conotoxins with marked improvement in potency and selectivity towards the a9a10 nAChR.
P031
NEW DEVELOPMENTS FOR THE DESIGN, SYNTHESIS AND BIOLOGICAL EVALUATION OF POTENT SARS‐COV 3CLPRO INHIBITORS
D. Sarma,1 R. Thomas,1 K. Hidaka,1 U. Bacha,2 E. Freire,2 Y. Hayashi,1,3* and Y. Kisoa*
1Department of Medicinal Chemistry, Center for Frontier Research in Medicinal Science, 21st Century COE Program, Kyoto Pharmaceutical University, Kyoto 607‐8412, Japan. 2Department of Biology, Johns Hopkins University, Baltimore, MD, USA. 3Department of Medicinal Chemistry, School of Pharmacy, Tokyo University of Pharmacy and Life Sciences, Tokyo 192‐0392, Japan
It has been reported that a novel coronavirus is the causative agent of severe acute respiratory syndrome (SARS). In 2003, the SARS epidemic resulted in over 8000 infections, about 10% of which resulted in death. Several attempts have been made to develop compounds that can inhibit SARS‐CoV. However, no effective therapy is available so far and hence it is still a matter of necessity to develop new potent compounds in case the disease re‐emerges. Herein, we disclose the design, synthesis and activity of a series of trifluoromethyl, benzothiazolyl and thiazolyl ketone‐containing peptidic compounds as SARS‐CoV 3CL protease inhibitors. Three candidates had encouraging results for the development of new anti‐SARS compounds.
Figure .

1. Sydnes, M. O.; Hayashi, Y.; Sharma, V. K.; Hamada, T.; Bacha, U.; Barrila, J.; Freire, E.; Kiso, Y. Tetrahedron 2006, 62, 8601.
2. Bacha, U.; Barrila, J.; Gabelli, B.; Kiso, Y.; Amzel, L. M.; Freire, E. Chem. Biol. Drug Des. 2008, 72, 34.
P032
THE USE OF ONE‐BEAD TWO‐COMPOUNDS PEPTIDE AND LIPOPEPTIDE LIBRARIES FOR IDENTIFICATION OF CELL SIGNALING AND PROAPOPTOTIC LIGANDS FOR CANCER CELLS
Y. Wang,1 D. Hanna,2 K. Lam,1 P. Kumar1
1Division of Hematology & Oncology, Department of Internal Medicine, UC Davis Cancer Center, University of California, Davis, Sacramento, CA; 2UC Davis Medical School, University of California, Davis, Sacramento, CA
In the last decade, we have successfully used one‐bead‐one‐compound (OBOC) [1] combinatorial library method to identify ligands that target cancer cell surface molecule [2, 3]. We were able to demonstrate that these ligands can be used as effective optical and radioimaging agents for cancer. Recently, we have developed a one‐bead two‐compounds (OBTC) combinatorial library method which can facilitate the discovery of ligands that affect cell function while interacting with specific receptors on the cell surface. In this new method, we used topographically segregated bilayer bead methods [4] to generate chemical library beads with two distinct chemical molecules displayed on the bead surface and a coding tag in the bead interior. One of these molecules could be a high affinity α4β1 integrin ligand such as LLP2A [3] that will bind strongly to both T‐ and B‐lymphoid cancer cells. The other molecule (e.g. peptide or lipopeptide), however, is totally random, encoded by the coding tag in the bead interior, and unique on each bead.
After incubation of beads displaying a random peptide or lipopeptide and LLP2A with live lymphoma cells (Molt‐4, a T lymphoma cell line) for 30 minutes, the majority of these beads were coated with the added cells. Upon adding (LAsp) 2 rhodamine 110 (D2R) 48 hours later, we detected rare beads that were covered entirely by apoptotic cells, indicating that the second ligand displayed on the bead surface has the capacity to induce apoptosis of lymphoid cancer cells. Work is currently underway to evaluate the proapoptotic mechanism of these molecules.
1. Lam, K.S., et al., A new type of synthetic peptide library for identifying ligand‐ binding activity. Nature, 1991, 354, p. 82–84.
2. Aina, O.H., et al., From combinatorial chemistry to cancer‐targeting peptides. Mol. Pharm. 2007. 4(5): p. 631–51.
3. Li, P., et al., Combinatorial chemistry identifies high‐affinity peptidomimetics against a4β1 integrin for in vivo tumor imaging. Nature Chem. Biol., 2 (7), 2006, p. 381–389.
4. Liu, R., et al., A novel peptide‐based encoding system for one‐bead one‐compound peptidomimetic and small molecule combinatorial libraries. J. Am. Chem. Soc., 2002, 124, p. 7678–7680.
P033
ON‐BEAD SCREENING OF COMBINATORIAL LIBRARIES: REDUCTION OF NONSPECIFIC BINDING BY DECREASING SURFACE LIGAND DENSITY
X. Chen and D. Pei
Department of Chemistry and Ohio State Biochemistry Program, The Ohio State University, 100 West 18th Avenue, Columbus, OH 43210
On‐bead screening of one‐bead‐one‐compound (OBOC) libraries provides a powerful method for the rapid identification of active compounds against biological targets. However, on‐bead screening is susceptible to interference from nonspecific binding, which results in biased screening and false positives. In this work, we have found that a major source of nonspecific binding is derived from the high ligand loading on the library beads, which permits a macromolecular target (e.g. protein) to simultaneously interact with multiple ligands on the bead surface. To circumvent this problem, we have synthesized a phosphotyrosyl (pY)‐containing peptide library on spatially segregated TentaGel microbeads, which feature a 10‐fold reduced peptide loading on the bead surface but a normal peptide loading in the bead interior. The library was screened against 10 Src homology 2 (SH2) domains including Csk, Fyn and SLAP, and the specific recognition motif(s) was successfully identified for each domain. In contrast, when the SH2 domains were screened against a control library that contained unaltered (high) ligand loading at the bead surface, six of them exhibited varying degrees of sequence biases. The reduction of the ligand loading on the bead surface represents a simple, effective strategy to largely eliminate the interference from nonspecific binding, while preserving sufficient amounts of materials in the bead interior for compound identification. This finding should further expand the utility of OBOC libraries in biomedical research.
P034
3‐NITRO‐TYROSINE AS AN INTERNAL QUENCHER OF AUTOFLUORESCENCE ENHANCES THE COMPATIBILITY OF FLUORESCENCE BASED SCREENING OF OBOC COMBINATORIAL LIBRARIES
J. Townsend, B. Sanii, A. Lehman, A. Do, S. Dixon, A. Parikh, K. Lam
UC Davis Medical Center
In the one‐bead‐one‐compound (OBOC) combinatorial method, compounds are constructed on bead resin via split‐mix synthesis such that multiple copies of the library compound are displayed on each bead. These libraries are rapidly screened with ELISA, fluorescent, radiometric, or whole‐cell binding assays. While fluorescence‐based probes are powerful tools in OBOC screening, their utility is greatly limited by the intrinsic fluorescence of many commonly used solid supports (e.g. TentaGel [TG] resin), residual coupling reagents, and library compounds. To overcome this problem, we topologically partitioned TG resin with a thin Fmoc‐protected outer layer and an unprotected inner core. The inner core was derivatized with 3‐nitrotyrosine, followed by random peptide library construction. Spectral scans from a confocal microscope showed a dramatic decrease in the autofluorescence of blank beads and OBOC peptide libraries across a broad range of the optical spectrum. The quenching capacity of 3‐nitro‐tyrosine was visualized in fluorescent micrographs for both bead samples. Using biotin/streptavidin as a model ligand/receptor system, we demonstrated a marked increase in visibility of multiple commercially available fluorescent probes binding to quenched beads, and increased feasibility of using a robust and efficient fluorescence‐based, bead sorting platform known as COPAS®. These data show that using 3‐nitro‐tyrosine as an internal quencher greatly enhances the compatibility of fluorescence‐based applications and OBOC combinatorial screening.
P035
IDENTIFICATION OF POTENT CYCLIC PEPTIDYL PIN1 INHIBITORS FROM A COMBINATORIAL LIBRARY
T. Liu, Y. Liu, H. Kao, D. Pei
The Ohio State University
Pin1 is a peptidyl‐prolyl isomerase which plays a significant role in cell‐cycle regulation by altering the conformation, thus the function/stability of targeted phosphoproteins. Recent studies have suggested Pin1 as a potential target for anticancer drug design. A Dphosphoserine and D‐phosphothreonine containing head‐to‐tail cyclic peptide library was built based on Pin1 structure and substrate specificity. After screening the library against Pin1, a few peptides were selected, individually synthesized and test for binding. The peptides were shown to bind to Pin1 with a Kd ranging from 20nM ‐ 1.6uM by isothermal titration calorimetry. The inhibition potency was confirmed by in vitro enzymatic assays. To test whether Pin1 inhibitor is active in the cells, we examined the effects of one of the Pin1 inhibitor on the protein levels of two known Pin1 targets, PML and SMRT. Our data show that treatment of HeLa cells with the Pin1 inhibitor leads to increases in the protein levels of PML and SMRT.
P036
SCREENING OF A ONE BEAD ONE COMPOUND C‐TERMINAL LIBRARY FOR THE SEQUENCE SPECIFICITY OF PDZ, BUZ, AND BRCT DOMAINS
R. Hard and D. Pei
The Ohio State University
Postsynaptic Density‐95/Discs Large/Zona Occluden‐1 (PDZ) domains bind to the free C‐termini of target proteins to mediate protein interactions and display either unique or overlapping specificity. The BUZ (binder of ubiquitin zinc‐finger) domain binds to the free C‐terminus of ubiquitin and therefore helps mediate ubiquitin‐based signaling events. The BRCA1 C‐terminal (BRCT) domain interacts with a variety of proteins involved in the DNA damage response and has been shown to associate with the C‐termini of many proteins in a phosphorylation dependent manner.
The one‐bead one‐compound (OBOC) combinatorial library approach allows for the determination of individual binding sequences to proteins of interest, which can be compiled and analyzed to determine the specificity of a particular protein domain.
A C‐terminal OBOC library was used to define the sequence specificity of the PDZ domains of Tiam 1 and Tiam 2, the BUZ domains of HDAC6 and Ubp‐M, and the BRCT domains of BRCA1, MDC1, MCPH1, TopBP1, and PTIP.
P037
INCORPORATION OF AZA‐AMINO ACIDS IN THE GHRP‐6 PEPTIDE FOR INDUCING SELECTIVITY TOWARDS THE CD36 VS GHS‐R1A RECEPTOR
C. Proulx, D. Boeglin, K. Zaniolo, S. Chemtob, H. Ong and W. Lubell
Département de Chimie, Université de Montréal, C.P. 6128, Succursale Centre Ville, Montréal, Québec, H3C 3J7, Canada
Aza‐amino acids are amino acids in which the α‐carbon has been replaced by a nitrogen atom. Lone pair repulsion between the adjacent nitrogens atoms in aza‐amino acid residues have suggested to induce turn conformation as observed by X‐ray diffraction, NMR spectroscopy and computational analysis. Aza‐peptide analogs of GHRP‐6 (His‐D‐Trp‐Ala‐Trp‐D‐Phe‐Lys‐NH2), a hexapeptide that exhibits dual affinity towards both the GHSR‐1a and the CD36 receptors, were synthesized using solid‐phase chemistry and split‐and‐mix protocols to make librariesi. Certain analogs exhibited selectivity to the CD36 receptor. Our presentation will describe the synthesis of GHRP‐6 aza‐peptides, binding affinity values for the GHS‐R1a and CD36 receptors, as well as in vitro antiangiogenic properties of the best analogs.iBoeglin, D.; Lubell, W.D.; J.Comb.Chem. 2005, 7, 864–878.
P038
SCREENING A CYCLIC PEPTIDE LIBRARY FOR ORGANOCATALYSTS
J. Shen and J. Pei
Department of Chemistry, The Ohio State University, 100 West 18th Avenue, Columbus, OH 43210
Cyclic peptides have the potential to mimic selection processes evolved by enzymes. Linear peptides containing N‐methylimidazole moiety have been established as catalysts for kinetic resolution of certain secondary alcohols. However, cyclic peptides N‐methylimidazole moiety have not been studied in catalytic reactions. In this work, a new type of cyclic peptides is being developed as acyl transfer catalysts. A cyclic peptide library (2.15 million peptides) containing five random positions and N‐methyl histidine was synthesized on PL‐AMS resin on which fluorescent pH sensor and corresponding linear peptide were also employed. The library was then screened against several acylation reactions. Selection of the brightest beads from the library identified the most active catalysts. Resynthesis of these cyclic peptides were then carried out. Preliminary results showed the selected cyclic peptides can catalyze acylation reactions.
P039
BIOMATERIALS BASED ON SELF‐ASSEMBLING α,β‐PEPTIDE MOTIFS
R. Nagarkar and J. Schneider
Department of Chemistry and Biochemistry, University of Delaware, Newark, DE 19716
A responsive self‐assembling peptide consisting of an amphiphilic β‐hairpin fused to a helical domain capable of forming coiled coils has been designed. This design allows the study of synergistic contributions to material formation from multiple distinct motifs within the self‐assembling peptide. Folding of each domain (helical and β‐sheet) can be triggered by pH or temperature. CD and FTIR were employed to probe changes in secondary conformation from α‐helical at low temperatures to predominately β‐sheet at high temperatures. Aggregation state of the coiled coils appended to the β‐hairpin was investigated using AUC. The fusion peptides form physically crosslinked hydrogels with fibrillar nanostructure. Fibril dimensions, as measured by TEM elucidate the underlying self‐assembly mechanism. Rheological studies revealed that upon heating the 5 wt% peptide solutions at pH 9, viscoelastic gels formed from self‐assembled β‐sheet rich fibrillar networks, which when cooled to room temperature exhibited increased mechanical rigidity concomitant with refolding of the helical domains.
P040
METAL‐TRIGGERED SELF‐ASSEMBLING COLLAGEN SCAFFOLDS
D. Przybyla and J. Chmielewski
Department of Chemistry, Purdue University
Self‐assembling systems provide the unique bottom up approach for creating macro‐sized objects from smaller fragments. These systems are of particular interest in the field of regenerative medicines, where self‐assembling systems have successfully been used as scaffolds for cellular proliferation. Specifically, collagen based scaffolds are particularly desirable because it is the major component of the extra cellular matrix. To date, several groups have developed self‐assembling collagen fibers by incorporating a variety of N‐ and C‐terminal sticky ends that grow through a linear mechanism. However, these systems all generate similarly shaped structures, and provide no control over the 3‐dimensional architecture. Consequently, we have successfully designed and synthesized three metal‐triggered self‐assembling peptides (H‐byp, H‐(byp)2, and H‐(byp)3). These collagen mimetic peptides are capable of assembling through a radial mechanism into a variety of unique structures, fibers, disks, and hollow spheres, and may provide enhanced biological properties to the current collagen based scaffolds.
P041
SELF‐ASSEMBLY OF MULTIPLE HOMOTRIMERIC COLLAGEN PEPTIDES BY ELECTROSTATIC INTERACTION
J. Lee, J. Chmielewski
Department of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, Indiana 47907, USA
Collagen fibers are important components in skin, tendon, cartilage and blood vessel. Controlling the self‐assembly of collagen upon environmental condition is an essential feature for collagen‐based biomaterial applications. Electrostatic interactions have been one of the most popular strategies to promote self‐assembly of the collagen peptides. Herein we disclose the self‐assembly of the collagen peptides using positively and negatively charged collagen peptides. Each positive and negative charge was introduced to the (POG)9 peptide by incorporation of O‐alkylated hydroxyproline with carboxylate and amine functionalities. These collagen peptides display electronic charges that exposed to environmental conditions while stabilized as triple helix at neutral pH. We successfully showed to control self‐assembly of multiple homotrimers upon pH conditions through electrostatic interactions of displayed charges on the peptides. Our designs are distinct from the previous research in that thermal unfolding of the collagen peptide is the necessary process to promote self assembly of the peptides. The self‐assembled peptides formed 200nm cone shaped structures that were confirmed by Dynamic Light Scattering and Atomic Force Microscopy. We anticipate that our design of self assembly of the collagen peptide can offer the new possibility of strategy for various collagen biomaterial applications.
P042
SELF‐ASSEMBLY OF COLLAGEN PEPTIDES INTO MICROFLORETTES VIA METAL COORDINATION
M. Pires, J. Chmielewski
Purdue University, West Lafayette, IN
The hierarchical assembly of nanoscale building blocks into micron‐scaled functional materials is a powerful strategy for the design of devices for regenerative medicine and tissue engineering. Essential features of successful tissue engineering, for instance, include the control of scaffold morphology at the nm to micro scale, along with the temporal and spatial release of specific growth factors to promote tissue growth and repair. Herein, we designed and synthesized a peptide composed of a core of (Pro‐Hyp‐Gly)9 flanked by the metal binding moieties nitrilotriacetic acid (NTA) and a di‐histidine unit. We report that this peptide rapidly and reversibly assembles in the presence of metal ions to form microflorettes of reproducible size and shape. In addition, we demonstrate that unsatisfied metal/ligands exist on the surface and within the microflorettes, and that these may be easily modified with His‐tag functionalized molecules. Two distinct His‐tagged fluorescent proteins were used to demonstrate that these proteins could be incorporated within the structures with spatial control. Critical to their use in biomedical applications is their ability to associate with mammalians cells. We show that the microflorettes are capable of avidly binding to various adherent and suspended cells. These unprecedented microscopic structures offer opportunities in many areas, including tissue engineering and regeneration.
P043
THREE‐DIMENSIONAL ENCAPSULATION AND CULTURING OF PRIMARY BOVINE CHONDROCYTES IN INJECTABLE β‐HAIRPIN PEPTIDE HYDROGELS
C. Sinthuvanich and J. Schneider
Department of Chemistry and Biochemistry, University of Delaware, Newark, DE
Articular cartilage plays an important role in bearing mechanical loads and shear forces in the joint. Due to its avascular nature, articular cartilage has a limited capacity to repair itself making its injury difficult to treat. The current therapies such as joint replacement and autologous cell implantation are often invasive leading to neighboring tissue morbidity. Moreover, these treatments suffer from unfavorable complications including implant adhesion failure, cell sedimentation and leakage. To address this, we have developed a β‐hairpin peptide hydrogel, MAX8, capable of injectable chondrocyte delivery. Hydrogelation of MAX8 can be triggered in the presence of cells allowing homogeneous cell encapsulation under physiological conditions. MAX8 also has a unique ability to shearthin upon application of shear force and recover nearly to its original rigidity after the removal of shear. For applications in cartilage repair, MAX8 can be used as a delivery vehicle and 3D scaffold to deliver cells in a minimally invasive manner. The objectives of current study are to evaluate the chondrocyte responses phenotypically and genotypically to MAX8 after syringe delivery. The viability of primary encapsulated chondrocytes is assessed. Production of cartilage‐specific ECM molecules is examined by histochemical staining and biochemical assays. Expression levels of cartilagespecific genes are also monitored by quantitative PCR. Collectively, the data suggest that MAX8 is a potential candidate as an injectable hydrogel scaffold for chondrocyte delivery and cartilage repair.
P044
RELEASE OF MODEL MACROMOLECULES FROM SELF‐ASSEMBLING PEPTIDE HYDROGELS FOR INJECTABLE DELIVERY
M. Branco1, N. Wagner1, D. Pochan3, J. Schneider2
1Department of Chemical Engineering, University of Delaware, Newark, DE 19716, USA; 2Department of Chemistry and Biochemistry, University of Delaware, Newark, DE; 3Department of Materials Science and Engineering, University of Delaware, Newark, DE
Advances in biotechnology techniques have led to the rapid development of small protein and antibody therapeutics. However, several limitations remain in the preparation and delivery of these drugs due to the susceptibility of proteins to degrade during storage and upon administration. To address this problem, hydrogels have been used as delivery devices for these protein drugs. We have designed a class of self‐assembling peptides that undergo triggered hydrogelation in response to physiological pH and salt conditions (pH 7.4, 150 mM NaCl). The peptides fold into a β‐hairpin, and subsequently, selfassemble to form a rigid hydrogel stabilized by non‐covalent cross‐links. For these peptides, it is possible to control the folding and assembly kinetics to form hydrogels with different rigidities. These changes affect the porous morphology within the hydrogel system, and subsequently influence the rate of macromolecular diffusion within the peptide fibrillar network. This study focuses on determining the mass transport properties of model proteins from self‐assembled MAX8 hydrogels. Proteins of varying molecular weights and pIs were chosen to assess the effect of fibrillar charge density and hydrogel mesh size on their release properties from these networks. Proteins are added into solution during folding and self‐assembling, yielding gels with macromolecules directly encapsulated into the network.
P045
GOLD NANOPARTICLE SELF‐ASSEMBLY PROMOTED BY A NON‐COVALENT, CHARGE‐COMPLEMENTED COILED‐COIL PEPTIDE
D. Ernenwein, P. Ghosh, V. Rotello, J. Chmielewski
Purdue University, West Lafayette, IN
Nanomaterials have been explored in bottom‐up assembly patterns to produce effective biosensing, optoelectronic and catalytic systems. Numerous attempts to assemble gold nanoparticles (GNPs) to various biomolecules via covalent linkages are present in the literature; however non‐covalent interactions have not been studied as extensively. To this end, we have designed a system using cationic GNPs and anionic coiled‐coil peptides to facilitate assembly via noncovalent interactions. An anionic variant of the leucine zipper region of the transcription factor GCN4 was designed. The hydrophobic residues of the heptad repeat remained unaltered to facilitate the natural parallel, dimeric oligomerization of the peptide, but five solvent exposed residues were modified to Glu residues to yield GCN4‐E. Circular dichroism (CD), UV‐vis spectroscopy, dynamic light scattering (DLS) and transmission electron microscopy (TEM) were used to characterize and confirm the assembly and aggregation of GNPs in the presence of GCN4‐E. To establish the importance of the hydrophobic face and the cofacial anionic face, two control peptides, GCN4‐X, scrambled version of GCN4‐E, and the native GCN4‐p1 peptide were also tested. Neither showed any evidence of facilitating GNP aggregation. The designed system effectively shows that non‐covalent interactions are responsible for the bottom‐up self assembly of GNPs. Potential future applications of this work lies in the area of self‐replicating catalytic systems.
P046
CONTROLLING THE DEGRADATION OF β‐HAIRPIN SELF ASSEMBLED HYDROGELS BY PROTEOLYSIS WITH TRYPSIN
M. Giano, D. Salick, J. Schneider
University of Delaware
Hydrogels have emerged as biomaterials to act as scaffolds for tissue regenerative therapies and drug delivery. The ability of the hydrogel scaffold to temporarily support the injured tissue, by providing mechanical properties consistent with the tissue during the wound healing process, allows for optimal tissue regeneration. Controlled degradation of the implanted material can aid the successful integration of the new tissue formed. For many applications, the rate of degradation of the hydrogel scaffold should approximate the rate of new tissue formation. If the hydrogel material is peptide‐based, proteases, which hydrolyze amide bonds in a sequence specific manner, could provide a means of degrading the hydrogel with temporal resolution. The degradation profile of β‐hairpin peptide hydrogels in vitro by trypsin, a model enzyme, was assessed by HPLC, mass spectrometry and rheology. Exogenous trypsin proteolysis studies were conducted on the peptide gels and respective fragments were separated by analytical HPLC and characterized utilizing electrospray ionization mass spectrometry. Bulk material properties were assessed with oscillatory shear rheology as a function of enzyme exposure time and were consistent with the rate of proteolysis measured by HPLC and mass spectrometry. Results demonstrated that tailoring the amino acid sequence towards the specificity of trypsin can be useful in tuning the degradation of the β‐hairpin peptide hydrogel.
P047
SELF‐ASSEMBLING PEPTIDIC NANOSTRUCTURES VIA ASYMMETRIC HYDROPHOBIC COLLAPSE
H. Cui, E.T. Pashuck, A. Cheetham, W. Tsai, S. Mui, S. Stupp
Department of Materials Science and Engineering, Department of Chemistry, and Feinberg School of Medicine, Northwestern University, 2220 Campus Drive, Evanston, Illinois 60208
The impact of nanotechnology on medicine and energy relies on the development and improvement of fabrication methods that produce highly pure nano‐objects with novel structural features in a simple manner. One underlying goal of synthetic peptide science is to devise simple protocols that can program the assembly kinetics of synthetically facile peptides into desired sizes and shapes with necessary surface chemistries that lead the nanostructures to pack into hierarchically well‐defined larger objects. We report here our recent efforts to design and assemble small molecular peptides into discrete nanostructures, such as cylinders, belts, multi‐walled tubes and ribbons, and their subsequent assembly at a larger length scale into highly ordered crystalline structures. The key feature of the designed molecular structural motif is the alternating hydrophobic and hydrophilic amino acid sequence that is covalently grafted onto hydrophobic tails, either a short alkyl tail or semiconducting oligomers. We have explored their applications in regenerative medicine, cell signaling, and photoelectric energy conversion.
P048
THE CHEMICAL SYNTHESIS AND NMR PROPERTIES OF BPTI ANALOGUES
J. Wojnar,1 M. Weiss,2 S. Kent1
1Department of Biochemistry & Molecular Biology, Institute for Biophysical Dynamics, The University of Chicago, Chicago, IL 60637; 2Department of Biochemistry, Case Western Reserve University, Cleveland, OH 44106
Chemical synthesis of proteins gives exquisite control over their covalent structure, enabling for example the incorporation of non‐coded amino acids. In this research, total syntheses of wild‐type bovine pancreatic trypsin inhibitor (BPTI) and several chemical analogues (with appropriate controls) were undertaken by native chemical ligation of two unprotected peptide segments. BPTI is the archetypal member of the Kunitz‐type serine protease inhibitors, organized around a short (∼50 amino acid) alpha/beta fold constrained by three disulfide bonds. This fold occurs in a variety of mammalian proteins, some of which are implicated in cancer metastasis and tumor growth.
A [G37A]BPTI mutant reportedly destabilized the protein by 5 kcal/mol, a surprisingly large destabilization thought to result from the disruption of an unusual NH‐aromatic‐NH network of interactions that involves the amide –NH‐ of Gly 37. We explored the effects of a [G37D‐Ala] substitution at this position of BPTI, as this offered novel opportunities to look for damping effects on aromatic ring flipping and subglobal amide proton exchange. NMR properties of the unique chemical analogues and control molecules will be reported.
P049
O‐ACYL ISOPEPTIDE METHOD: TOWARD AN EFFICIENT CHEMICAL PREPARATION OF PEPTIDES/PROTEINS USING RACEMIZATION‐FREE SEGMENT CONDENSATION METHOD
T. Yoshiya, H. Kawashima, Y. Sohma, T. Kimura and Y. Kiso*
Department of Medicinal Chemistry, Center for Frontier Research in Medicinal Science, 21st Century COE Program, Kyoto Pharmaceutical University, Japan
The convergent synthetic method has greatly facilitated the assembly of peptides/proteins. However, a fundamental drawback of convergent synthesis is that epimerization at the C‐terminal residue of an N‐segment occurs during the condensation reaction with a C‐segment, thereby limiting the N‐segment to contain either a C‐terminal Gly or Pro residue. We herein report a racemization‐free segment condensation based on the O‐acyl isopeptide method with successful synthesis of bioactive peptides. This method allows the use of an N‐segment possessing a C‐terminal Ser/Thr residue for segment condensation without any epimerization, because of a C‐terminal urethane‐protected Ser/Thr residue. Additionally, final deprotected peptides were effectively purified by HPLC, because an O‐acyl structure remarkably changes the physicochemical properties of the native peptide. Finally, an O‐to‐N intramolecular acyl migration reaction triggers the native amide bond formation under neutral conditions [1]. Using this method, the segment condensation becomes possible at positions other than those bearing a C‐terminal Gly or Pro residue at the N‐segment, and suggesting that O‐acyl isopeptide segment condensation as being advantageous for synthesizing larger peptides and proteins [2]. Considering these features, the O‐acyl isopeptide method‐based segment condensation method would contribute to the field of chemical synthesis of peptides and proteins.
[1] Sohma, Y. et al. Chem. Commun. 2004, 124–125.
[2] Yoshiya, T. et al. Tetrahedron Lett. 2006, 47, 7905–7909.
P050
PEPTIDE PURIFICATION: OPTIMIZATION OF A REVERSED‐PHASE CHROMATOGRAPHIC PURIFICATION STEP AND EFFECT OF CLEANING IN PLACE
N. Forrer 1, D. Gétaz2, M. Gençoglu, R. Rüede, and M. Morbidelli2
1Zeochem AG, 8707 Uetikon, Switzerland, 2ETH Zurich, 8093, Zurich, Switzerland
Nowadays, peptides are gaining more and more importance in the treatment of various diseases. They can be produced by solid phase synthesis or fermentation processes. Reversed‐phase chromatography is one of the most important steps in the purification of peptides. Especially after a fermentation process, the downstream process and, therefore, the chromatographic steps, are responsible for the high cost of the peptide. As a consequence, the chromatographic process has to be optimized to achieve maximum yield and purity.
Another important problem that is often seen in peptide purification is the progressive column performance dropping as a consequence of irreversibly adsorbing impurities. To restore the separation performance, so‐called cleaning in place is often performed. This step involves washing the stationary phase with a high pH solution to remove the irreversibly adsorbing impurities. An undesired consequence of cleaning in place (seen especially on silica‐based reversed‐phase materials) is the progressive destruction of the stationary phase.
In this poster a crude peptide mixture will be studied. The reversed phase chromatographic purification step will be optimized leading to the best separation between the target peptide and the impurities. Moreover, the effect of cleaning in place will be studied. The deterioration and the purification performance change of the stationary phase will be followed after different cleaning in place steps.
P051
MOLECULAR DYNAMICS SIMULATIONS OF CHEMOSELECTIVE BONDS IN BOVINE RIBONUCLEASE A
C.E. Winsor, R.J. Broadbridge and B. Howlin
University of Surrey
Chemoselective ligation can be used to join peptide fragments together to create longer peptides than can be synthesised as a single chain, through the formation of non‐amide linkages, and it is a possible tool in the production of synthetic proteins. For this to be used as a general method it is important to assess what impact these unnatural linkages may have on the structure and consequentially on the activity of biologically active molecules.
Chemoselective bonds formed from reaction of an aldehyde to hydrazine and amino‐oxy groups have been used to assemble bovine ribonuclease A, masking the aldehyde as an amino alcohol to allow sequential ligation of several peptide fragments. The structures of these non‐amide bonds have been modeled within the structure of ribonuclease and molecular dynamics simulations have been run to allow theoretical predictions of the effect of these unnatural bonds to be compared to experimental results of the catalytic activity of the enzyme.
P052
SYNTHESIS OF LIPOPEPTIDES AND THEIR SUPRAMOLECULAR FORMATIONS
A. Suzuki 1, Y. Suzuki1, and T. Inazu1,2
1Department of Applied Chemistry, School of Engineering, 2Institute of Glycoscience, Tokai University, Hiratsuka, Kanagawa 259‐1292 Japan. inz@keyaki.cc.u‐tokai.ac.jp
Recently many kinds of self‐assembly peptide have been reported. In this work synthesis of artificial lipopeptides and their supramolecular formation were studied. First we synthesized Fmoc‐Lys(Hdc)‐OH and Fmoc‐Glu(Hda)‐OH with long alkyl chains. A long alkyl chain consists of hexaethleneglycol and decanoic acid or decanamine. Next we also synthesized lipopeptides (Ac‐Tyr‐Ser‐Gln‐Glu(Hda)‐Gln‐Ser‐Ser‐Ser‐Ser‐Gln‐Glu(Hda)‐Gln‐Ser‐Gly‐NH2) using the artificial amino acid derivatives by the solid‐phase method. The synthesized lipopeptide was measured by AFM, and TEM. From these results, we found the synthesized lipopeptide was formed a cylindrical micelle. Details of the investigation will be discussed.
Figure .

P053
PYRAZINOYLATION AND BENZOYLATION OF ANOPLIN: A PLAUSIBLE APPROACH OF PREPARING NOVEL POTENT ANTIBACTERIAL DRUGS
M.A.O. Tan,1 P.M.G. Sabido1,2
1Institute of Chemistry, College of Science, University of the Philippines, Diliman, Quezon City, 1101, Philippines; 2Natural Sciences Research Institute, University of the Philippines, Diliman, Quezon City, 1101, Philippines
The balanced interplay of amphiphilic properties grants antimicrobial peptides their target specificity and selective toxicity. While recent literature reports the enhancement of peptide activity from acylation with fatty acids of different lengths, N‐terminal conjugation of peptides with aromatic carboxylic acids can produce derivatives that structurally resemble other antibacterial drugs. Using anoplin as the model peptide in this study, pyrazinoylation and benzoylation of the glycine residue through HBTU/HOBt/DIEA coupling would yield N‐peptidyl substituted pyrazinamide and hippuric acid carboxamide derivatives yield. This synthetic approach may prove useful in preparing novel antibacterial peptide conjugate drugs
P054
ACID CATALYZED MONODEHYDRO‐2,5‐DIKETOPIPERAZINE FORMATION TOWARD NATURAL PRODUCT SYNTHESIS
Y. Yamazaki, Y. Mori, A. Oda, Y. Kiso, Y. Hayashi
Tokyo University of Pharmacy and Life Sciences
There are many naturally occurring monodehydro‐2,5‐diketopiperazines (monodehydroDKPs) possessing biological activity such as phenylahistin (PLH), which is a lead compound of NPI‐2358 under Phase I/II clinical trial in the US as a vascular disrupting agent (VDA) [1]. These cyclicdipeptides monodehydroDKPs are useful building blocks and templates for combinatorial chemistry.
To develop an efficient synthetic route of monodehydroDKPs with no racemization for natural product synthesis, in our previous study, we focused our work on Gladiali et al.'s report [2] that the reaction of α‐ketoester and Boc‐NH2 in the presence of a catalytic amount of p‐TsOH resulted in the formation of a dehydroamino acid, and successfully applied this method to N‐α‐ketoacyl‐Phe‐NH2 to synthesize monodehydroDKPs in a reasonable yield with no racemization. In the present study, we have also successfully expanded this method to other amino acid residues such as Leu and Ser(Bzl) in case that β‐aliphatic‐α‐keto acid derivatives were used. Therefore, this method would be effective for the synthesis of natural products that contain β‐aliphatic‐monodehydroDKPs.
[1] Nicholson, B., Hayashi, Y., et al. (2006) Anti‐Cancer Drugs, 17, 25‐31.[2] Gladiali, S., Pinna, G. (1991) Tetrahedron: Asymmetry, 2, 623–632.
P055
SYNTHESIS OF MULTIPLE O‐TYROSINE SULFATED PEPTIDES BY FMOC/TBU SOLID PHASE PEPTIDE SYNTHESIS TO STUDY THE C5A‐RECEPTOR
A. Bunschoten, J. Ippel, J. Kemmink, J. Kruijtzer, C. de Haas, J. van Strijp, R. Liskamp
Utrecht University
Although much less known than protein phosphorylation, the significance of protein sulfation is rapidly gaining momentum and has been identified to be crucial in certain protein‐protein interactions. To broaden the research into the function of O‐sulfated tyrosine residues within proteins, a reliable synthesic approach has to be developed. The selective introduction of an O‐sulfated tyrosine in a peptide is still considered a challenge because of the acid lability of the aryl sulfate ester during the acidic deprotection steps. Within this contribution we would like to present a new selective Fmoc/tBu SPPS approach for the introduction of multiple O‐sulfated tyrosine residues.1 Following this approach we synthesized several representatives of the N‐terminal part of the C5a‐Receptor containing two O‐sulfated tyrosine residues. The binding of Chemotaxis Inhibitory Protein of Staphylococcus aureus (CHIPS) to these C5aRmimics is studied with ITC and the structures of CHIPS2 and of CHIPS in complex with our C5aR‐mimics are solved with NMR spectroscopy, both clearly showing the importance of the sulfated tyrosine residues within the C5a‐Receptor.
1. Bunschoten et al., Chem. Commun., 2009, Accepted for publication.
2. Haas et al., J. Mol. Bio., 2005, 353, 859–872.
P056
CHEMICAL SYNTHESIS AND RACEMIC CRYSTALLIZATION OF THE SNOW FLEA ANTIFREEZE PROTEIN
B. Pentelute, Z. Gates, V. Tereshko, J. Dashnau, J. Vanderkooi, A. Kossiakoff, S. Kent
Harvard Medical School
Chemical protein synthesis and X‐ray crystallography were used to investigate the L and D forms of a recently discovered snow flea antifreeze protein (sfAFP) [1‐2]. This interesting glycine rich protein has no sequence homology with any known proteins and no experimental structure has been reported. Chemically synthesized L and D‐sfAFP were found to have the expected antifreeze activity in an ice recrystallization inhibition assay. The X‐ray structure was determined by an largely unexplored approach called racemic protein crystallization. It was found that crystal formation from a racemic solution containing equal amounts of the chemically synthesized proteins D‐sfAFP and L‐sfAFP occurred much more readily than for L‐sfAFP alone. More facile crystal formation also occurred from a quasi‐racemic mixture of D‐sfAFP and L‐Se‐sfAFP, where L‐Se‐sfAFP is an analogue. The novel structure of sfAFP is made up of six antiparallel left‐handed PPII helixes wrapped to form a compact bricklike structure with a hydrophilic and hydrophobic face.
1. Pentelute, B.L., Gates, Z.P., Tereshko, V., Dashnau, J.L, Vanderkooi, J.M., Kossiakoff, A.A., Kent, S.B.H. J. Am. Chem. Soc., 130: 9695–9701 (2008).
2. Pentelute, B.L., Gates, Z.P., Dashnau, J.L, Vanderkooi, J.M., Kent, S.B.H. J. Am. Chem. Soc., 130: 9702–9707 (2008).
P057
CHEMICAL SYNTHESIS AND BIOTINYLATION OF ANTIANGIOGENIC THROMBOSPONDIN TYPE 1 REPEAT ANALOGS
T. Tiefenbrunn and P. Dawson
The Scripps Research Institute
The type 1 repeat domain from thrombospondin has potent antiangiogenic activity and a structurally interesting fold, making it an attractive target for protein engineering. Chemical synthesis is an attractive approach for studying protein domains since it enables the use of unnatural amino acids for site‐specific labeling and detailed structure‐function analysis. Here, we demonstrate the first total chemical synthesis of the thrombospondin type 1 repeat domain by native chemical ligation. In addition to the natural domain, five sites for side chain modification were evaluated and two were found to be compatible with oxidative folding. Several challenges were encountered during peptide synthesis due to the functional complexity of the domain. These challenges were overcome through the use of new solid supports, scavengers, and the testing of multiple ligation sites, as well as through utilizing a combination of Boc‐ and Fmoc‐based SPPS for individual peptide segments. Synthetic access to this domain has enabled the synthesis of a number of variants containing arginine or tryptophan analogs that modify the core cation‐p stack of the TSR2 domain. The relative stabilities of these varaints, investigated via circular dichroism thermal melting and differential scanning calorimetry, provide insights into the stability afforded by cation‐p interactions in the context of a native protein fold. Synthesized variants of TSR2 can be used to further our understanding of the biochemical interaction network of thrombospondin and provide insight into the structure and function of this important anti‐tumorogenic protein domain.
P058
SOLID PHASE CHEMICAL SYNTHESIS OF N‐LINKED GLYCOPEPTIDES
R. Chen and T. Tolbert
Department of Chemistry, Indiana University, Bloomington, Indiana 47405
Growing interest in developing and designing N‐linked glycopeptidebased vaccines and therapeutics stimulates the study of the synthesis of N‐glycopeptides. Although several strategies of in‐solution glycopeptide synthesis have been developed, repetitive purification of intermediates is still a problem that lowers yields. Solid phase synthesis, on the other hand, has the advantage of automating the repetitive coupling steps and drives the coupling reactions to completion by using large excesses of building blocks. Hence, solid phase synthesis of glycopeptides has many benefits, yet this strategy requires large amounts of N‐oligosaccharides, which are extremely hard to obtain, thereby slowing down the development of solid phase synthesis of N‐linked glycopeptide. In our lab, we have developed the capability of producing large amounts of N‐linked oligosaccharides, and this allows the exploration of high‐mannose glycopeptide synthesis on the solid phase. Monosaccharide and oligosaccharide Fmoc‐Asparagine building blocks have been synthesized and applied to glycopeptide synthesis on different resins to determine the effect of resin type on glycosylamino acid coupling. In addition, on‐resin glycosylamine coupling strategies have also been explored. These strategies have been employed for a variety of biological relevant glycopeptides, such as HIV gp120 sequences.
P059
FMOC SOLID‐PHASE SYNTHESIS OF C‐TERMINAL PEPTIDE THIOESTERS BY FORMATION OF A PYROGLUTAMYL IMIDE MOIETY
A.P. Tofteng, K. Sørensen, K. Conde‐Frieboes, T.S Hoeg‐Jensen, K. Jensen
Faculty of Life Sciences, University of Copenhagen
The extensive use of C‐terminal peptide thioesters in protein chemistry, i.e. native chemical ligation or other chemoselective reactions, has provided the impetus for the search for optimal solid‐phase strategies for their synthesis. Numerous approaches for Fmoc based SPPS have been published; methods for reliable synthesis of peptide thioesters with a C‐terminal glycine have been established. However, Fmoc based methods for the syntheses of peptide thioesters with a C‐terminal chiral amino acid are less reliable. We have very recently introduced a novel method for the synthesis of C‐terminal peptide thioesters. A C‐terminal glutamic acid residue with a highly labile side‐chain protective group is first anchored to a solid support. The desired peptide is then assembled on resin, followed by selective removal of the glutamic acid side‐chain protecting group with dilute TFA. Strong activation, e.g. PyBrOP/DIEA in NMP, of the deprotected carboxylic acid results in formation of the pyroglutamyl imide moiety on‐resin. Nucleophilic displacement by treatment with thiol results in release of the thioester from the solid support. Here we present optimized results where the overall yields have been improved and the epimerization problems have been addressed.
Figure .

P060
THIOACID CAPTURE LIGATION AT VALINE RESIDUE
F. Li and C. Liu
School of Biological Sciences, Nanyang Technological University, Singapore 637551
Thioester‐mediated peptide ligation at valine was reported recently[1]. The method utilizes an N‐terminal penicillinamine to mediate the ligation reaction, which is followed by desulfuration to give a Val residue at the ligation site. However, the slow kinetics of the ligation step severely limits the practical value of this method. Herein, we report that thioacid capture ligation can overcome this problem and be used for ligation at Phe‐Val, Leu‐Val and even Pro‐Val junctions. Comparative studies using small model peptides showed that thioacid capture ligation exhibited much faster reaction kinetics than native chemical ligation. Using thioacid capture ligation, we successfully synthesized a histone protein H2B, whereas attempts to synthesize the protein using native chemical ligation at the same junction were not successful.
[1] Christian Haase, H. R., Oliver Seitz (2008). “Native Chemical Ligation at Valine.” Angew. Chem. Int. Ed 47: 6807‐6810.
Figure .

P061
LINEAR CHEMICAL SYNTHESIS OF LYSM DOMAIN PROTEINS
K. Srensen,1 J. Stougaard,2 K. Jensen1
Centre for Carbohydrate Recognition and Signalling, 1University of Copenhagen, 2University of Aarhus; kso@life.ku.dk, kjj@life.ku.dk
The structural requirements for recognition of complex polysaccharides and the role of ligand‐receptor interactions between different cells and organisms remain unsolved for many systems. We are here focusing on the ‘communication’ between rhizobium and leguminous plants, which leads to symbiosis and nitrogen fixation. The main focus is understanding the interaction between lipochitin oligosaccharide signal molecules and the plant receptors they are predicted to interact with.The LysM domain is a common protein moiety and may have a general peptidoglycan binding function. The LysM domains are predicted to have a ‐á‐á‐ secondary structure with the two helices packing onto the same side of an anti‐parallel beta sheet. Here we present the linear solid‐phase synthesis of LysM domains from the plant receptors in order to study their structure and the interaction with the lipochitin oligosaccharide signal molecules. The segments were the LysM 2 and LysM 3 domains of receptor kinase (NFR5) originating from Lotus japonicus filicaulis. These small proteins ranged in size from 44 to 71 residues.
The synthesis was optimized, especially to overcome problems in the assembly of the C‐ and N‐terminal ends. Microwave heating using a custom‐made semi‐automated synthesizer was a key element in the improved protocol. Other aspects of the synthesis were also improved. These small proteins were subjected to various conditions for folding and disulfide bridge formation. Binding to lipochitin oligosaccharides was studied using a range of biophysical techniques.
P062
A REPETITIVE METHOD FOR THE SYNTHESIS OF HIGHLY PURE PROTECTED PEPTIDES IN GOOD YIELD
Y. Helou,1 and J. Burton2
1Department of Chemistry, Emmanuel College, Boston, MA; 2Psyche Pharmaceuticals, Inc., 676 Concord Avenue, Cambridge, MA 02138
Both the homogeneity and yield of protected peptides are impediments to the cost‐effective chemical synthesis of proteins by fragment condensation. Use of the EPG (Excluded Protecting Group) Method to overcome these issues is examined by synthesis of the 11peptide Fmoc‐Leu‐Lys(Boc)‐Cys(Acm)‐Ser(tBu)‐Tyr(tBu)‐Ala‐Gln‐Val‐Glu(OtBu)‐Phe‐Gly. The C‐terminal glycyl‐residue was esterified to allocholesterol by μwave irradiation and reacted with Fmoc‐Phe‐OPfp. The dipeptide product was deprotected and purified by gel permeation chromatography and the cycle repeated to produce the 11peptide. Homogeneity of intermediates was determined both by measuring coupling reaction kinetics and mass spectrometry. Coupling reaction half‐times were <2 minutes. As little as 0.03% uncoupled amino component could be accurately measured at each step by ES‐MS with deletion sequences comprising ∼0.26% of the 11peptide. An approximately three‐fold increase in the weight of the product is obtained over the course of synthesis. Removal of the C‐terminal allocholesterol group was readily effected by [ϕ3P]4Pd°/SiMe3H. The high yield of an essentially homogeneous protected peptide should allow chemical synthesis by fragment condensation to become cost‐competitive with biologic methods of protein production.
P063
PSK — A NOVEL AMPHIBIAN SKIN BOWMAN‐BIRK‐LIKE TRYPSIN AND TMPRSS2 INHIBITOR
H. Chen, W. Chen, S. Hawthorne, T. Chen, C. Shaw, B. Walker
Molecular Therapeutics Research, School of Pharmacy, Queen's University, McClay Research Centre, 97 Lisburn Road, Belfast BT9 7BL, Northern Ireland, UK
Here we describe a novel C‐terminally amidated heptadecapeptide, isolated from the skin secretion of the frog of the genus Rana pipiens, {SAPRGCWTK SYPPKPCK‐amide (pSK)} that contains a disulphide loop between Cys6 and Cys16. pSK shares a high degree of homology with the core inhibitory motif found in Bowman–Birk type protease inhibitors (BBI) such as sunflower seed trypsin inhibitor (aSFTI). A synthetic replicate of pSK, was found to be a potent inhibitor of trypsin with a Ki just slightly less than 96 nM, a value falling between those determined for aSFTI (2.2 nM) and the core inhibitory loop of BBI (119.0 nM). pSK also exhibited inhibitory activity (Ki = 6.85 μM) against TMPRSS2 (type II transmembrane‐bound serine protease), a trypsin‐like serine protease involved in tumor proliferation. This compares very favorably with the degree of inhibition observed with aSFTI (Ki = 6.97 μM) and BBI (Ki = 19.55 μM). pSK one of the first examples of a naturally occurring inhibitor of TMPRSS2 and could prove useful in delineating the role of this protease in tumour proliferation and invasion.
Key words: Frog, Skin, Peptide, Protease, Inhibitor, Prostate cancer, TMPRSS2, Type II transmembrane serine protease.
* Corresponding author.
E‐mail address: hchen10@qub.ac.uk (H.Chen).
P064
PEGYLATION OF INTERMEDIATE PEPTIDES: THYMOSIN α1 AND GROWTH HORMONE‐RELEASING FACTOR
A. Felix and S. Veech
Ramapo College of New Jersey, School of Theoretical and Applied Science, Mahwah, NJ 07430, U.S.A.
Recently, several biologically active peptides and proteins have been successfully pegylated to improve their metabolic stabilities and increase their biological half‐lives. These pegylated proteins have been shown to have improved biological profiles, require fewer injections than their non‐pegylated counterparts, and some are being used as therapeutics. The goal of our work was to develop general conditions for the pegylation of small proteins e.g. thymosin a1, a naturally occurring thymic peptide consisting of 28‐amino acids that possesses immunomodulatory activity.
Using a model pentapeptide, [H‐Leu‐Lys(Boc)‐Lys(Boc)‐Gly‐Thr(tBu)‐NH2], conditions were optimized for pegylation using an aminoalkylation procedure with PEG‐aldehyde followed by reduction with NaCNBH3. Purification of the crude pegylated pentapeptide was achieved by dialysis and lyophilization. Deprotection of the Boc/tBuprotecting groups resulted in the expected positive ninhydrin reaction. Thymosin a1 was then pegylated by coupling with either PEG2000‐aldehyde or PEG5000‐aldehyde by the same aminoalkylation procedure using a novel H2O‐trifluoroethanol solvent in place of methanol to maintain homogeneity. Confirmation of structure was provided by hydrolysis of the PEG‐thymosin a1 followed by thin layer chromatography, which revealed the presence of amino acids present in thymosin a1. The aminoalkylation procedure has been extended to include pegylation of GRF(1‐29)‐NH2 and GRF(1‐44)‐NH2. Biological studies of PEG‐thymosin a1 are in progress.
P065
ISODGR: A NEW SEQUENCE FOR INTEGRIN LIGANDS
E. Otto,1 D. Heckmann,1 G. Zahn,2 D. Vossmeyer,2 R. Stragies,2 H.Kessler1
1Department of Chemistry TU München, Lichtenbergstr. 4, 85747 Garching, Germany; 2Jerini AG, Invalidenstr. 130, 10115 Berlin, Germany
The peptide sequence Arg‐Gly‐Asp (RGD) is by far the most prominent ligand to promote specific cell adhesion through integrin stimulation. Recently, it was shown that the mutation of the RGD sequence in Fibronectin to the RGE sequence still exhibits binding affinity, although RGE is known not to be recognized by integrins. However, these fibrils display a slightly different phenotype than wild‐type Fibronectin. A hypothetical model for these unexpected results was proposed by Curnis et al.. The Asn‐Gly‐Arg sequence in fibronectin FN‐I5 repeat is able to undergo a rearrangement to isoAsp‐Gly‐Arg, which shows activity on αvβ3 and ‐ with less potency ‐ on α5β1. The mechanism of Asn‐deamidation is already known for a long time and has widely been considered to be a process of degradation, acting as a biochemical clock that limits protein lifetimes in vivo. Curnis et al. were the first to show that the deamidation process increases protein function instead.
These findings stimulated us to create a library of cyclic peptides containing the isoDGR sequence. A screening of this library showed various new peptides with high activity towards the integrin receptor αvβ3 or the receptor α5β1.
Further studies, including NMR‐based structure determinations and docking experiments are currently in progress in order to elucidate the relationship between the three‐dimensional arrangement of the isoDGR‐sequence and the biological activity found in cellular assays.
P066
PEPTIDE REGULATORS OF PROTEIN–PROTEIN INTERACTIONS WITH SELECTIVITY FOR SPECIFIC SUB‐CELLULAR COMPARTMENTS
N. Qvit, M. Disatnik and D. Mochly‐Rosen
Department of Chemical and Systems Biology, Stanford University School of Medicine, Stanford, CA, 94305, USA
Multiple protein‐protein interactions (PPI) govern the function of the protein kinase C (PKC) family of isozymes. We previously generated selective regulator peptides derived from the C2 domain of individual PKC isozymes, since the C2 domain is the site of many PPIs. The agonist and antagonist peptide regulators were used in vitro, in vivo and are currently being tested in humans. Selective antagonists inhibit PPI between a specific PKC and its RACK. The agonists interfere with intra‐molecular inhibitory PPI within a given PKC isozyme and cause its activation. These isozyme‐selective inhibitors and activators regulate all the cellular functions mediated by that isozyme. For example, we showed that δPKC induces apoptosis, inhibits ATP regeneration, induces endoplasmic reticulum stress and induces cell adhesion by phosphorylation of different substrates at each of those sub‐cellular compartments. The current PPI inhibitor of δPKC, δV1‐1, inhibits all the above functions. Here we sought to identify regulators of one of these δPKC‐mediated functions. Such regulators have the potential to affect only the PKC function that is associated with a particular pathology, without affecting other functions of these ubiquitous enzymes. Guided by the structure of the C2 domain, we developed a series of short peptide regulators of δPKC. One peptide, derived from a unique site in δPKC induces δPKC translocation to the mitochondria when using a myocardial infarction model, but not to other sub‐cellular components and increases the phosphorylation of only a subset of δPKC substrates. In contrast, a previous δPKC agonist, δRACK, induces translocation of δPKC to the mitochondria, plasma membrane and endoplasmic reticulum. The characterization of the new selective δPKC agonist will be provided.
P067
EXENATIDE COULD UPREGULATE SIRT1 PROTEIN IN RINM‐5F CELLS
X. Wang, J. Yu, J. Wang, S. Yang, W. Li
Jilin University
Sirt1, a class III histone protein deacetylase,is essential in aging process. It has been reported that Sirt1 is involved in glucose metabolism and can improve insulin sensitivity by repressing PTP1B [1]. Furthermore,it has been testified by Laura Bordone that Sirt1 can regulate insulin secretion by repressing UCP2 in pancreatic βcells [2]. Exenatide, which has been used to treat T2DM, is a 39 amino acid peptide. We have shown in our experiment that exenatide in RINm‐5F cells could upregulate not only the protein level of sirt1 but also the gene expression. There may be some correlationship with the previous result we have got that exenatide could downregulate p53 protein level [3].
P068
NOVEL μ AGONIST, Δ ANTAGONIST AND NK1 ANTAGONIST PEPTIDE CHIMERAS
I. Kumarasinghe, V. Hruby, S. Tumati, E. Varga, J. Lai, F. Porecca
Department of Chemistry, University of Arizona, Tucson, AZ85721; Department of Pharmacology, University Of Arizona, Tucson, AZ85721
Chronic pain such as neuropathic pain can take place due to nerve injuries and is considered to be the most difficult to treat. Opioids often are the first line of treatment. Due to their side effects including tolerance, prolonged treatment with opioids is not feasible. Development of tolerance can occur for many reasons. It has been found that the co‐administeration of opioid mu agonist and delta antagonist or Neurokinin1 (NK1) antagonist reduces tolerance. These observations prompted us to create a single peptide chimera which could act as an antagonist at the delta and NK1 receptors and an agonist at the mu receptor. We made a series containing H2N‐Dmt‐Tic as an N terminal delta antagonist pharmacophore and Trp‐CONHBzl(CF3)2 as a C terminal NK1 pharmacophore. We found that mu and delta affinities of the series increased when the length between these pharmacophore was decreased. We found an optimized ligand that has good affinity constant (Ki) for both the mu and delta receptors 32.2 nM and 0.79 nM respectively. Hybrid molecules having mu agonist, delta and NK1 antagonist activities will be therapeutically advantageous for patients who do not show any response with current analgesics. We anticipate that these new molecules will show a high antinociceptive effect, less development of tolerance and less respiratory depression in animal assay models. Supported by the grants from the US Public Health Service, National Institutes of Health.
P069
DEVELOPMENT OF COMPSTATIN‐ALBUMIN BINDING PEPTIDE CONJUGATES FOR PROLONGED PEPTIDE HALF‐LIFE
H. Qu, P. Magotti, J. Lambris
University of Pennsylvania
Complement is the first line of defense against invading pathogens and a bridge to adaptive immunity. However, inappropriate or excessive complement activation can cause host tissue damage and is implicated in many autoimmune diseases including Alzheimer's disease, rheumatoid arthritis, age‐related macular degeneration (AMD), sepsis, ischemia‐reperfusion injuries, transplantation etc. Our lab has developed a 13‐residue disulfide‐bridged peptide named Compstatin that is able to selectively bind to complement component 3 and effectively inhibit complement activation. A Compstatin analogue is currently in phase I clinically trials for the treatment of AMD. However, it has relatively short half‐life in vivo due to its small size. One of the ways to improve peptide in vivo half‐life is by conjugation to a molecule that binds to human albumin, which has a long half‐life of 19 days. Therefore, we have designed chimeric peptides with a cyclic albumin binding peptide conjugated to either N‐terminal or C‐terminal of [W(Me)4]‐Compstatin via a short PEG linker. The peptides were synthesized by a combination of solid phase and solution phase chemistry to avoid crossing of the two disulfide bonds. In vitro assays show that those chimeric peptides are able to bind to albumin and C3 separately, and simultaneously. Thus, the chimeric peptides show great promise of having longer in vivo half‐life and for the treatment of a broader range of complement associated diseases.
P070
NOVEL N‐TERMINALLY MODIFIED PYY3‐36 ANALOGS: DESIGN, STRUCTURAL ANALYSIS, AFFINITY STUDY AND IN VIVO EVALUATION
S. Pedersen, N. Vrang, K. Jensen
University of Copenhagen
The prevalence of obesity is increasing with an alarming rate world wide and there is a need for anti‐obesity drugs. PYY3‐36 have been demonstrated to possess appetite suppressing potential thus novel analogs with high Y2R selectivity and potency have potential as drugs for the treatment of obesity. It has been hypothesized that PYY3‐36 and possibly related peptide hormones bind to the membrane prior to interaction with the receptors. Based on this hypothesis, we have designed novel PYY3‐36 analogs for increased membrane binding.
The N‐terminal segments (residue 1‐12) of the PP fold family of peptides have proven very important for the Y receptor selectivity. We have taken advantage of this phenomenon in the design of two classes of novel PYY3‐36 analogs. First, the N‐terminal segment was replaced with different amphiphatic heptad‐repeat sequences which were designed to self‐aggregate into protein‐like structures. Binding experiments demonstrated high selectivity for the Y2R. Second, different N‐terminal segments derived from the PP fold family of peptides were introduced at an e amine of an additional Lys positioned before Ser‐13 of the native sequence. Our hypothesis is that this gives rise to a tighter back‐folding, which consequently makes the N‐terminal partly inaccessible for the receptors and also to some extent towards proteases. Binding experiments back up the hypothesis by displaying interesting YR affinities and DIO mice studies show prolonged efficacy for at least one analog.
P071
GUIDING VLP'S THE RIGHT WAY: COATING OF VIRUS LIKE PARTICLES WITH PEPTIDIC INTEGRIN LIGANDS
B. Laufer,1 N. F. Steinmetz,2 V. Hong,3 M. Manchester,2 H. Kessler,1 M.G. Finn3
1Institute for Advanced Study, Technische Universitt München, Department Chemie, Lichtenbergstr. 4, 85747 Garching, Germany; 2Department of Cell Biology, The Scripps Research Institute, 10550 N. Torrey Pines Road, La Jolla, CA 9203, USA; 3Department of Chemistry, The Scripps Research Institute, 10550 N. Torrey Pines Road, La Jolla, CA 9203, USA
Virus like particles are one of the most exciting kind of nanoparticles that can be used for medicinal approaches, as they might serve as polyvalent building blocks that provide a stable and well characterized surface that can be used for many different chemical reactions. So far, such particles have successfully been used for imaging purposes or to gain antibody responses. However, they still suffer from unselectivity concerning internalization if only one specific cell has to be attacked or binding to cell surfaces.
Integrins constitute a family of heterodimeric, transmembrane cell adhesion receptors for a variety of extracellular matrix proteins, e.g. vitronectin or fibronectin. The pioneering observation that integrins ‐ especially αvβ3 and α5β1 ‐ are hallmarks of metastatic cancer and are seriously involved in the process of tumor angiogenesis turned them into attractive targets for cancer therapy. For more than two decades the RGD sequence is known to be recognized by several integrins, e.g. αvβ3, αvβ5 and α5β1.
Here we report the successful surface coating of a virus like particle with a cyclic integrin binding peptide using click chemistry to gain selective binding towards cells presenting αvβ3, αvβ5 or α5β1 integrin receptors on their surface.
P072
HIGHLY ACTIVE AND SELECTIVE INTEGRIN LIGANDS FOR MAPPING INTEGRIN EXPRESSION
S. Neubauer,1 D. Heckmann,1 J. Eckardt,1 G. Zahn,2 D. Vossmeyer,2 R. Stragies,2 H.‐J. Wester,3 H. Kessler1
1Institute for Advanced Study, Technische Universitt München, Department Chemie, Lichtenbergstr. 4, 85747 Garching, Germany; 2Jerini AG, Invalidenstraße 130, 10115 Berlin, Germany; 3Nuklearmedizinische Klinik und Poliklinik, Klinikum rechts der Isar, Technische Universitt München, 81675 München, Germany
Integrins constitute a family of heterodimeric, transmembrane cell adhesion receptors for a variety of extracellular matrix proteins, e.g. vitronectin or fibronectin. The pioneering observation that integrins ‐ especially αvβ3 and α5β1 ‐ are hallmarks of metastatic cancer and seriously involved in the process of tumor angiogenesis turned them into attractive targets for cancer therapy. Recent reevaluation of the integrins αvβ3 and α5β1 pointed out the importance of the subtype α5β1 in angiogenesis in contrast to αvβ3 as assumed so far. Out of that, the inhibition of integrin function is a major challenge in medicinal chemistry.
There are different radiolabeled αvβ3 antagonists for the non‐invasive investigation of angiogenesis and metastasis in vivo. The first radiolabeled antagonist [123I]cyclo(RGDyV) was presented by R. Haubner et al.. Recently, T. D. Harris et al. published the first nonpeptidic αvβ3 antagonists for tumor imaging.
Until now, there is no radiolabeled subtype selective α5β1 ligand for imaging reported in literature. Starting from a recently published α5β1‐selective integrin ligand, we report the synthesis of the first selective α5β1 ligand for tumor imaging application and a new highly selective αvβ3 tumor imaging ligand.
P073
ENGINEERING OF BETTER HIV ENTRY INHIBITOR PEPTIDES THROUGH CHEMICAL MODIFICATION
J. Xiao 1 and T. Tolbert1,2
1 Interdisciplinary Biochemistry Graduate Program† and Department of Chemistry, 2 Indiana University, Bloomington, Indiana 47405
Entry inhibitor peptides are a new class of antiretroviral drugs for the treatment of HIV infection. T20 (also known as Fuzeon, Enfuvirtide, or DP‐178) is the only one in this class approved by FDA. Although T20 has been successfully used in the treatment of HIV infection, it still faces many challenges. For example, T20 is a synthetic peptide requires 106 steps which is costly and time‐consuming. It has low solubility at neutral pH, short half‐life in human serum and HIV strains resistant to T20 have been reported recently. Herein, we describe a successful method to express the T20 and another HIV entry inhibitor peptide C37H6 in bacteria and efforts to improve their solubility, half‐life and biological activity through chemical modification of expressed peptides.
P074
NEW INSIGHT INTO THE BINDING MODE OF CYCLIC MELANOCORTIN LIGANDS AT THE MC4 RECEPTOR
D. Brancaccio, A. Carotenuto, P. Grieco, V. Hruby, E. Novellino
University of Naples, “Federico II”
The melanocortin receptors are involved in many physiological functions, including pigmentation, sexual function, feeding behavior, and energy homeostasis, making them potential targets to treat obesity, sexual dysfunction, etc. Understanding the basis of the ligand receptor interactions is crucial for the design of potent and selective ligands for these receptors. Cyclic melanocortin MTII and SHU9119 were investigated by solution NMR spectroscopy in aqueous solution of DPC (dodecylphosphocholine), used as a membrane mimetic environment. NMR derived structures of these analogues were then docked within the hMC4 receptor, where they act as agonist and antagonist, respectively. Interesting differences in the binding mode of the two ligands were observed and will be widely discussed. The detailed structural information on ligand‐receptor complexes obtained in this study may assist future attempts to optimize the selectivity, potency, and efficacy of agonist and antagonist at MC4 receptor, which is involved in feeding.
MTII: Ac – Nle‐c[Asp – His – DPhe – Arg – Trp – Lys]
SHU9119: Ac – Nle‐c[Asp – His – DNal – Arg – Trp – Lys]
P075
PROTEINOUS MICROSPHARES CONTAINING KLVFF PEPTIDES STRONGLY INHIBIT AMYLOID‐BETA AGGREGATION AND TOXICITY
M. Richman, S. Willick, N. Skirtenko, A. Gedanken, and S. Rahimipour
Department of Chemistry, Bar‐Ilan University, Ramat‐Gan 52900, Israel
Alzheimer's disease (AD) is the most common neurodegenerative disease. More than 20 million people suffer from it worldwide. AD has been identified as a protein misfolding disease due to the accumulation of aggregated amyloid beta (Aβ) proteins in the brains of the patients. Therefore, compounds that can selectively interfere with Aβ aggregation and accumulation are desperately needed. Previous studies have demonstrtaed that the hydrophobic core of Aβ, KLVFF, is an essentional sequence for the self‐assembly of Aβ. It binds the homologous sequence in Aβ, prevents its aggregation and reduces its toxicity, in vitro. Using a multivalent variant of KLVFF, Scheper and coworkers have demonstrated that the affinity and specifity to Aβ can be significantly improved. This strategy was found to be more effective in preventing Aβ aggregation than the native KLVFF. Here we describe how proteinous microsphares containing different copies of KLVFF fragments on their surface can potently prevent Aβ aggregation and reduce its toxicity to the cells. These microsphares are prepared from ultrasonic irradiation of various proteins, such as bovine serum albumin, with the presence of different amounts of KLVFF analogs. We demonstrate that these microspheres have a narrow size distribution (1‐5 μm) and are highly stable in different conditions. Moreover, using confocal microscopy and fluorescently labeled KLVFF analog, we show that the peptide is presented mainly on the surface of the microsphares, while the internal volume can be filled with different contrasting agents.
P076
COMBINATION TREATMENT WITH HER‐2 AND VEGF PEPTIDE MIMICS INDUCES POTENT ANTI‐TUMOR AND ANTI‐ ANGIOGENIC RESPONSES IN VITRO AND IN VIVO
K. Foy,1, 2 S. Rawale,2 P.T.P. Kaumaya 1, 2, 3
1Department of Microbiology, 2Department of Obstetrics and Gynecology, and the 3Arthur G. James Comprehensive Cancer Center, The Ohio State University
HER‐2 is a member of the EGFR family and is overexpressed in 20‐30% of breast cancer patients.VEGF is a glycoprotein containing an anti‐parallel structure with inter and intra chain disulfide bonds. It belongs to the cysteine‐knot family and is a ligand for class III tyrosine kinase growth receptors. Both HER‐2 and VEGF are good targets for cancer treatments. Pertuzumab and Bevacizumab are two humanized monoclonal antibodies (mAb) that target HER‐2 and VEGF respectively. The overexpression of HER‐2 is associated with increased expression of VEGF at both the RNA and protein level in human breast cancer cells [1] and exposure of HER‐2 overexpressing cells to trastuzumab significantly decreases VEGF expression. Furthermore, a positive association between HER‐2 and VEGF expression in breast cancer patients has been identified [2]. We have created novel peptide molecules based on the known crystal structures of HER‐2 in complex with pertuzumab, and VEGF with Avastin and VEGFR2. Because of the potential synergy between HER‐2 and VEGF inhibitors, we want to test if combination therapy interacts in a synergistic or additive manner, killing tumor cells or retarding tumor development.
[1] Liang, Y., and Hyder M, Endocrinology 146:3632 (2005).
[2] Konecny, G. E., Meng, G., et al Clin Cancer Res 10:1706 (2004).
P077
SAR AND MECHANISTIC STUDIES OF TETRAPEPTIDE INHIBITORS OF Aβ42‐INDUCED NEUROTOXICITY
H. Li and G. Bitan
Department of Neurology, David Geffen School of Medicine, University of California, Los Angeles, CA 90095
Neurotoxic oligomers of amyloid β‐protein (Aβ) are believed to be the main cause of Alzheimer's disease (AD). Recently, we identified the C‐terminal tetrapeptide, Aβ(39–42), as an inhibitor of Aβ42‐induced neurotoxicity (IC50 = 16 5 M) and showed that Aβ(39–42) rescued Aβ42‐induced inhibition of synaptic activity. To characterize the binding site(s) of Aβ(39–42) on Aβ42 we used Tyr as a probe along the Aβ42 sequence and monitored changes in intrinsic fluorescence as a function of Aβ(39–42) binding. In addition, we performed structure–activity relationship (SAR) studies and identified key structural features, including chirality, side chain identity, and a free N‐terminus, that control Aβ(39–42) inhibitory activity. The results demonstrate that despite the small size of Aβ(39–42) and the hydrophobic aliphatic nature of all four side chains, the interaction of Aβ(39‐42) and its derivatives with Aβ42 is based on specific interactions. The data will serve as basis for design and testing of new peptidomimetics derivatives with improved activity and metabolic stability.
P078
INVESTIGATION OF Aβ42 C‐TERMINAL FRAGMENTS AS INHIBITORS OF Aβ42 ASSEMBLY AND NEUROTOXICITY
H. Li,1 B. H. Monien,1,4 A. Lomakin,2 E. A. Fradinger,1,5 S. M. Spring,1 B. Urbanc,3 G. B. Benedek,2 and G. Bitan1
1Department of Neurology, David Geffen School of Medicine, University of California, Los Angeles; 2Department of Physics, Massachusetts Institute of Technology; 3Physics Department, Drexel University; 4Current address: Deutsches Institut für Ernhrungsforschung Potsdam‐Rehbrücke, Germany; 5Current address: Department of Biology, Whittier College
Alzheimer's disease (AD) likely is caused by neurotoxic oligomers of amyloid β‐protein (Aβ). Disruption of Aβ oligomerization is a promising approach for developing therapeutics for AD. We have prepared a series of Aβ42 C‐terminal fragments (CTFs), evaluated their bioactivity, and identified lead inhibitors of Aβ42 assembly and neurotoxicity (Fradinger et al., Proc. Natl. Acad. Sci. USA (2008) 105: 14175–14180). To decipher the mechanisms by which CTFs affect Aβ assembly and neurotoxicity, we investigated the solubility, secondary structure, morphology, self‐assembly, and interaction with full‐length Aβ42 of CTFs and of control Aβ‐derived fragments. The results demonstrated that the biophysical properties of CTFs were roughly length‐dependent. In contrast, inhibition of Aβ42 assembly and neurotoxicity correlated poorly with CTF length and was sequence‐specific. Understanding the relationship between CTF sequence and biophysical/biological properties provides insight into their mechanism of action and will be used for design and testing of improved peptidomimetic inhibitors of Aβ42 toxicity as drug candidates for AD.
P079
SYNTHESIS AND CHARACTERIZATION OF PEPTIDE HORMONE‐BASED PRODRUGS
A. De and R. DiMarchi*
Department of Chemistry, Indiana University, Bloomington, Indiana 47405, USA*rdimarch@indiana.edu
Peptides represent a rich natural source of potential medicines with a notable pharmaceutical limitation being their relatively short duration of action. A particularly good example of this phenomenon is glucagon‐like peptide 1 (GLP). In the native form GLP demonstrates an extremely short half‐life in plasma and a relatively narrow therapeutic index with notable adverse gastrointestinal pharmacology. We explored a set of GLP‐based prodrugs as a means to extend the duration of action and broaden the therapeutic index of the native hormone. A set of N‐terminal dipeptideextended hormone analogs was synthesized and characterized by chemical and biochemical methods. GLP‐analogs with amide‐based dipeptide extensions demonstrated much reduced in vitro potency relative to the native hormone, but little propensity to generate diketopiperazines (DKP) or diketomorpholines (DMP), under physiological conditions. Analogous ester‐based prodrugs of GLP were observed to generate the parent peptide and a cyclized dipeptide when incubated at physiological pH and temperature. The relative rate of DKP and DMP formation with simultaneous formation of a highly active and otherwise chemically stable GLP was optimized for pharmacological use. The speed of cleavage was observed to be a function of the conformational propensity of the dipeptide extension to reside in a cisconfiguration coupled with the relative strength of the cyclization nucleophile. A set of GLP ester‐based prodrugs of variable half‐live has been identified and biochemically characterized.
P080
BIOACTIVITY OF INSULIN ANALOGS WITH ALTERED B‐CHAIN SECONDARY STRUCTURE
P. Mroz, V. Gelfanov and R. DiMarchi
Department of Chemistry, Indiana University, Bloomington, Indiana 47405, USA *pamroz@indiana.edu
The central β‐helix of the insulin B‐chain formed by residues 9‐19 is crucial for the structural stability of the hormone and its biological activity. Previous studies have investigated numerous amino acid changes with a particular emphasis on natural amino acids. The reported results while informative are highly restrictive in the depth and breath of secondary structural change explored given the limited structural diversity of natural amino acids. However, these analogs are easier to obtain since the vast majority can be prepared by rDNA methods where the native conformation is directed by biosynthesis of the native three disulfides. We report here the synthesis and biological analysis of insulin analogs with select helix stabilizing elements such as substitution with an Aib residue, salt bridge insertion and backbone lactam cyclization. L‐Lysine or L‐Ornithine was introduced at position B9 or B10 to facilitate salt bridge formation or lactam cyclization with the native GluB13 residue of insulin. The analogs were synthesized via a 1+2 chain combination procedure developed by Dr. Jie Han and reported elsewhere in this meeting. Sequence dependent, synthetic difficulties were encountered in the lactam cyclization step. Most of the insulin analogs demonstrated increased helical content. Significant decreases in insulin receptor binding and in vitro biochemical signaling were observed for all analogs in this series. One of the conclusions from this set of analogs is that secondary structure alteration at the specific sites studied had a negative effect on bioactivity and appears to result from more than local alterations in conformation.
P081
EXPLORING THE N‐TERMINAL HYDROPHOBIC FACES OF GLUCAGON AND GLUCAGON‐LIKE PEPTIDE‐1
B. Ward*, B. Finan, V. Gelfanov and R. DiMarchi
Department of Chemistry, Indiana University, Bloomington, Indiana 47405, USA; *bpward@indiana.edu
The aromatic face in the N‐terminal region of glucagon and glucagon‐like peptide‐1 (GLP‐1) is well recognized for its importance in achieving high affinity interactions with their respective receptors. To further examine the significance of these residues in glucagon and GLP‐1, an amino acid scan of positions 6, 10 and 13 was performed. The basis for this study is an observation relating similar aromatic faces in glucagon, GLP‐1 and calcitonin. Work by Fowler et al. 1 on human and salmon calcitonin variants stimulated our formulation of a “leucine hypothesis” in the quest to find a receptor superagonist. The structural series included all three natural aromatic amino acid residues (phenylalanine, tryptophan and tyrosine) and leucine at positions 6, 10 and 13 for both peptides. Our results illustrate that in contrast to calcitonin, glucagon and GLP‐1 do not tolerate simultaneous replacement of all aromatic character with leucine in the N‐terminal region. More specifically, residues 6 and 13 were found to be sensitive to all changes studied. Position 10 in each of the glucagon‐based peptides was observed to be more permissive to substitution where observed bioactivity changes were more subtle relative to the native hormones. These results warrant additional study to explain the clear difference in recognition of peptide ligands by the calcitonin and glucagon receptors, as well as additional investigation pertaining to the degree of structural change that can be tolerated at residue 10.
1. Fowler, S. B., Poon, S., Muff, R., Chiti, F., Dobson, C. M. and Zurdo, J. PNAS (2005) 102, 10105–10110.
P082
MOLECULAR‐BASIS FOR SPECIFICITY IN BIOLOGICAL ACTION AT THE HOMOLOGOUS GLUCAGON AND GLP‐1 RECEPTORS
J. Day, J. Patterson, V. Gelfanov and R. DiMarchi
Department of Chemistry, Indiana University Bloomington, Indiana 47405, USA
Glucagon and glucagon‐like peptide‐1 (GLP‐1) are two highly homologous hormones involved in the maintenance of glucose homeostasis and of sizable importance to the clinical management of diabetes. These two peptides are highly specific in association with their native receptors through structural elements within different regions of their sequences that confer biological specificity. We explored the structure‐activity relationship of these two hormones through the use of single residue substitutions and hybrid peptides. We have identified a set of novel 29‐residue peptides which exhibit high potency and balanced co‐agonism at the glucagon and GLP‐1 receptors. Modifications to the glucagon C‐terminal sequence resulted in a nearly complete loss of specificity, with minimal change to inherent activity. Three modifications to native glucagon and addition of a C‐terminal amide produced a peptide with a 100‐fold increase in potency at the GLP‐1 receptor and potency equivalent to glucagon. The significant increase in potency at the GLP‐1 receptor can be attributed to an increase in alpha helicity as well as enhanced positional interactions at the receptor. These high potency glucagon‐based co‐agonists represent a refined set of peptides that vary in selectivity within a dynamic range that spans a ten‐fold preference for one or the other receptor while maintaining full potency at a one of the two receptors. The analogs identified provide the basis for investigating through in vivo studies the relative efficacy and safety obtainable at each receptor for purposes of optimizing metabolism and body weight.
P083
SOLID PHASE SYNTHESIS OF LIPIDATED PEPTIDOTRIAZOLES AS BISUBSTRATE INHIBITORS OF PROTEIN GERANYLGERANYLTRANSFERASE‐I
A. Aditya, R.A. Gibbs
School of Pharmacy, Purdue University, West Lafayette, IN
Protein prenylation is an important post‐translational modification that controls the subcellular localization and membrane trafficking of more than hundred human proteins, particularly G‐proteins belonging to the Ras and Rho families. Prenylation is executed by three protein prenyltransferases ‐ Farnesyltransferase (FTase) and Geranylgeranyl‐ transferase‐I & II (GGTase) that catalyze the transfer of a C15 or C20isoprenoid to the cysteine thiol present close to the C‐terminal of the protein. FTase and GGTase‐I have been established as effective targets for development of cancer therapeutics. These bisubstrate enzymes utilize their protein substrates (or peptides containing a C‐terminal CaaX‐motif) along with the respective isoprenoid pyrophosphates for their catalytic turnover.
Our bisubstrate inhibitor design involves the conjunction of the peptide and the isoprenoid fragments by utilizing Cu‐catalyzed Hüisgen cycloaddition of isoprenoid alkynes with the N‐terminal azide of the peptide moiety. These peptidotriazoles were assembled on chlorotrityl resin by sequential on‐resin diazo transfer and click chemistry. The potency of these inhibitors was fine‐tuned by systematically modifying the diversity elements such as the “aaX” motif, the linker length between the peptide and triazole, and length of the isoprenoid chain. This approach has led to the discovery of potent and highly selective bisubstrate inhibitors of GGTase‐I with IC50 values ranging from 500 nM to 5 μM.
P084
SYNTHESIS AND BIOLOGICAL EVALUATION OF CONSTRAINED V3‐PEPTIDE ANTI HIV‐1 IMMUNOGENS CONJUGATED TO A T‐CELL EPITOPE
S. Tantry,1 A. Moseri,2 B. Arshava,1 F. Naider1 and J. Anglister2
1Department of Chemistry, College of Staten Island, CUNY, NY 10314, USA; 2Department of Structural Biology, Weizmann Institute of Science, Rehovot, Israel
Human immunodeficiency virus type 1 (HIV‐1) is responsible for a pandemic which has infected ∼1% of the world's adult population. Two envelope glycoproteins, gp41 and gp120 play critical roles in facilitating the entry of the retroviral genome into macrophages and T‐cells. The third variable loop V3 of gp120, consisting of ∼35 amino acids plays a critical role in the binding of gp120 to CXCR4 or CCR5 co‐receptors and is a dominant antigenic determinant. Based on NMR structures of bound peptide antigens we designed and synthesized, three constrained V3‐loop analogues conjugated to a T‐Cell epitope. (P1) KQIINMWQEVGKAMYA‐RPNNNTRKSIHIGPGRAFYTTGEI, a conserved V3 loop T‐cell epitope conjugate, (P2) KQIINMWQEVGKA MYA‐RPNNN‐cyclo[CRKSIHIGPGRAFYTTGEC], a T→C and I→C mutant of P1 and (P3) KQIINMWQEVGKAMYA‐RPNNNTR‐cyclo [CSIHIGPGRAFYTC]GEI, a K→C and T→C mutant of P1. Cystine replacement enabled cyclization through disulfide bonds and stabilized the conserved V3‐loop region of the gp120 in a β‐hairpin like structure. These 41‐residue peptides were synthesized using standard solid phase Fmoc‐chemistry on an Applied biosystems 433A peptide synthesizer. Cyclization of the HPLC‐purified linear peptide was performed using a glutathione (GSSG/GSH) oxidation of the linear peptide. Final peptides were recovered in >95% homogeneity with the expected molecular weights. Constrained V3 peptides analogous to P2 and P3 exhibited strong binding to 447‐52D, a broadly neutralizing human anti‐HIV‐1 antibody, and the P2 and P3 immunogens elicited stronger immune responses than P1. In addition the P2 elicited antibody response was more effective in neutralizing HIV‐1 primary isolates. These results indicate that constrained V3 peptides will be a useful component of an anti‐HIV‐1 vaccine.
P085
MEMBRANE‐BOUND PEPTIDES DESIGNED TO ELICIT ANTIBODIES AGAINST HIV‐1
T. Reichart, F. Brunel, V. Luna, S. Sligar, C. Stout, M. Zwick, P. Dawson
The Scripps Research Institute, La Jolla, CA 92037
The membrane proximal external region (MPER) of the HIV surface glycoprotein gp41 is the binding site for several broadly neutralizing antibodies. Antigens derived from this region have the potential to be an important components of a vaccine directed against HIV‐1.1 Peptide epitopes for these antibodies are highly conserved but sterically obscured from the immune system by an intimate association with the viral membrane and by the presence of a large trimeric glycoprotein spike. In order to better mimic the structural features of the MPER domain with an eye towards better understanding of epitope‐antibody interactions near membranes, peptide mimics derived from HIV gp41 have been synthesized with a variety of structural changes affecting characteristics such as helicity, orientation, and positioning of the epitope with respect to the membrane. These peptide mimics have been incorporated into membrane bilayer mimics called “Nanodiscs”. These Nanodiscs are ∼10 nm‐diameter structures composed of a phospholipid bilayer ringed by an apolipoprotein‐derived scaffold protein.2 We have examined the affinity of HIV‐1 neutralizing antibodies to these membrane‐bound peptides to better understand the binding of these neutralizing antibodies to epitopes on or near a membrane. These constructs will enable both immunization studies to give insight into the affects of membrane proximity on immune response as well as the design of simple peptides that are capable of self‐assembly into stable Nanodiscs.
1) Burton, D. R., 2002. “Antibodies, viruses, and vaccines”. Nature Reviews Immunology 2, 706–713.
2) Denisov, I. G., Grinkova, Y. V., Lazarides, A. A., & Sligar, S. G., 2004, “Directed Self‐Assembly of Monodisperse Phospholipid Bilayer Nanodiscs with Controlled Size”. J. Am. Chem. Soc., 126, 3477–3487.
P086
SOLID‐PHASE SYNTHESIS OF EUROPIUM‐LABELLED HUMAN INSL3 AS A NOVEL PROBE IN THE STUDY OF LIGAND‐ RECEPTOR INTERACTIONS
F. Shabanpoor, R. Hughes, R. Bathgate, F. Separovic, J. Wade
Howard Florey Institute, University of Melbourne
The assessment of the receptor binding affinity of analogues of INSL3 typically requires the use of 125I‐labelled INSL3 which has a number of disadvantages including its short half‐life, high cost, the need to be prepared prior to use, and radioactivity safety issues. To overcome these problems, we have developed a fluorescent‐lanthanide receptor binding assay. Initially, INSL3 was labelled post‐synthesis with a europium chelate at free amino groups. This method of labelling gave a mixture of mono‐ and multi‐labelled peptide which was difficult to purify. Consequently we have developed an efficient solid phase synthesis technique to prepare specifically mono‐labelled peptide using DTPA chelate. Eu‐DTPA‐labelled INSL3 had receptor binding affinity (9.05 ± 0.03, n=3) similar to 125I‐labelled INSL3 (9.59 ± 0.09, n=3). The receptor binding affinity (pKi) of INSL3 was determined to be 9.27 ± 0.06, n=3 using Eu‐DTPA‐INSL3 as a labelled ligand, which again is similar to that obtained when 125I‐INSL3 was used as labelled ligand (9.34 ± 0.02, n=4). The lanthanide‐based binding assay has a very low background and is highly reproducible.
P087
QUANTIFICATION OF MULTIPLE PHOSPHORYLATED TAU‐ EPITOPES FOR DIAGNOSIS OF ALZHEIMER'S DISEASE
D.Singer, I. Alafuzoff, R. Hoffmann
Leipzig University
Alzheimer's disease (AD) is characterized by two major hallmarks, the intracellular aggregation of hyperphosphorylated protein τ into paired helical filaments (PHF‐τ) and the extra cellular deposition of Aβ‐peptides. Several lines of evidence suggest that the analysis of Aβ, total‐τ and especially phospho‐τ might be favorable for early and specific AD diagnosis. To establish a sensitive, AD‐specific Immuno‐PCR (polymerase chain reaction) based Sandwich‐ELISA, the peptides τ207‐219[p212/p214] and τ226‐240[p231/p235] containing two PHF‐τ‐specific phosphoepitopes were used for immunization. The obtained monoclonal antibodies (mAb) HPT‐1 and HPT‐101/HPT‐103 were specific for the doubly phosphorylated epitopes p212/p214 and p231/p235 respectively. HPT‐104 and HPT‐110 were specific for monophosphorylated Thr231 or Ser235. The PHF‐τ‐specificity of the mAbs was confirmed by ELISA, immunoblotting and histochemical analysis of brain tissue from AD patients. Thus, PHF‐specific mAbs HTP‐1 and HPT‐101 were combined in a Sandwich‐ELISA to detect phospho‐τ in CSF at the low pg/mL range. After digoxigenin labeling of the detector mAb and assay optimization a detection limit of 2 pg/mL PHF‐τ‐in buffer and spiked in CSF from healthy individuals was achieved. In combination with mAbs specific for p231 and unphosphorylatedτ‐protein, a quantification of the fourfold phosphoepitope p212/p214/p231/p235, the single phosphorylation at Thr231 and total‐τ in post mortem CSF samples was accomplished.
P088
DEVELOPMENT OF AN EUROPIUM(III) DOTA‐BASED LUMINESCENCE ASSAY FOR DETECTION OF LIGAND‐RECEPTOR INTERACTIONS
C. De Silva, J. Vagner, R. Lynch, R. Gillies, V. Hruby
University of Arizona
Lanthanide‐based luminescent ligand binding assays are superior to traditional radiolabel assays due to improved sensitivity and affordability in high throughput screening while eliminating the use of radioactivity. Accordingly, dissociation enhanced lanthanide fluoroimmunoassay (DELFIA) technology is rapidly emerging as a bioanalytical tool using Eu(III)‐coordinated chelators such as DTPA (diethylenetriaminepentaacetic acid). However, DELFIA has not been successfully used with more stable chelators such as DOTA (1,4,7,10‐tetraaza‐1,4,7,10‐tetrakis(carboxymethyl)‐cyclododecane) due to the incomplete release of Eu(III). Here we report a modified and optimized DELFIA procedure for the use of Eu(III)‐DOTA labeled peptides to study the ligand‐receptor interactions. NDP‐a‐MSH (melanocyte stimulating hormone) ligand labeled with Eu(III)‐DOTA was synthesized and the binding affinity of this peptide to cells overexpressing human melanocortin‐4 receptors was evaluated using an acid treatment protocol. Complete release of Eu(III) from the DOTA chelate was observed using HCl acid (2M) prior to the luminescent enhancement step. Saturation binding data indicate that the Eu(III)‐DOTA linked peptide binds to these cells with an affinity similar to its DTPA analogue. Modified DELFIA procedure will be used to monitor the binding of heterobivalent peptides to the cells expressing both MSH and CCK (Cholecystokinin) receptors. Supported by grants from the U. S. Public Health Service National Cancer Institute and the Arizona Biological Research Commission.
P089
OPTIMIZATION OF DOTA‐VIP CONJUGATES FOR RADIOPEPTIDE IMAGING AND THERAPY
M. Martin, B. Schafer, and M.S. O'Dorisio
Department of Pediatrics, Division of Hematology/Oncology, Carver College of Medicine, University of Iowa, Iowa City, IA
Peptide‐based radioligands have become an important tool in the diagnosis and treatment of malignant tumors. Many types of tumors overexpress G‐protein coupled receptors for regulatory peptides making them an excellent target for cancer diagnosis and therapy. Vasoactive intestinal peptide (VIP) is a 28 amino acid neuropeptide with high affinity for VPAC1 and VPAC2 receptors. VIP has potential as a radiopeptide imaging agent because several of the most frequently occurring human tumors, including breast, ovarian, prostate, bladder, pancreatic, colonic, and esophageal carcinoma, have been found to express VIP receptors. For use as an imaging agent, a radionuclide chelator needs to be incorporated into the peptide sequence to stably complex the radiometal ion. DOTA (1,4,7,10‐tetraazacyclododecane‐1,4,7,10‐tetraacetic acid) is often used for this purpose as it is able to form stable complexes with trivalent metal ions in biological systems. However, the key residues for receptor affinity and biological activity of VIP are distributed throughout the entire 28‐amino acid peptide sequence. Because of this, DOTA will need to be conjugated to the peptide in a position where it will not interfere with either receptor binding or activity. In this study, we have conjugated DOTA to multiple positions within the VIP sequence to determine the optimal placement of the metal chelator to preserve receptor binding affinity. We have also studied the effect of DOTA conjugation on peptide stability and stimulation of cAMP generation as compared to VIP.
P090
PREPARATION OF FLUORESCENT PROBES OF TRANSCRIPTION BASED ON AMANITIN
D. Dietrich, D. Perrin
University of British Columbia
The bicyclic octapeptide α‐amanitin is a potent inhibitor of eukaryotic transcription. It binds tightly to RNA Pol II (KD∼10‐9) with high selectivity over other polymerases. This exceptional binding occurs at the bridge‐helix site of the polymerase. We have chosen to use this octapeptide scaffold to create a series of probes that could be used to aid in the elucidation of various transcriptionally related processes. Specifically, we prepared fluorescent and photo‐activatible probes that provide the ability to visualize peptide localization as well as enable control of transcription in a spatio‐temporal manner. Unfortunately, poor cellular uptake (as demonstrated by fluorescent microscopy) rendered these probes unsuitable to transcription studies. It was hypothesized that our mutation of (1S, 2R, 3R) δ.δ,d‐dihydroxyisoleucine for isoleucine directly led to this poor uptake. Following the stereoselective synthesis of this monomer, we prepared a small library of our amanitin probes varying the identity at this amino acid, and studied its effects on cellular penetration. The results of the synthesis of these probes, as well as their cellular uptake will be discussed here.
P091
SPECIFIC TARGETING OF CELLS BY HETEROMULTIVALENT LIGANDS & ITS IMPLICATIONS IN CANCER
J. Josan, L. Xu, J. Vagner, R. Gillies, V. Hruby
Bio5 Institute, University of Arizona
Current cancer therapies exploit either differential metabolism or overexpression of specific individual gene products in aberrant cells. We propose an alternative approach – to specifically target combinations of cell‐surface receptors using heteromultivalent ligands (htMVLs). For proof‐of‐concept, heterobivalent ligands (htBVLs) were constructed with binding motifs as melanocortin peptide ligand combined with either a cholecystokinin peptide ligand or the delta‐opioid specific ligand. The two ligands were tethered with linkers of varying rigidity and length and constructed from natural and/or synthetic building blocks. Using lanthanide based competitive binding assay, the htBVLs were found to bind with higher affinity (apparent cooperativity) to their cognate receptors compared to their monovalent binding, in cell lines transiently co‐expressing hMC4R/CCK‐2R and hMC4R/δOR receptors. There is up to a 80‐fold decrease in IC50 for CCK‐6 ligand in hMC4R/CCK‐2R system and up to a 50‐fold decrease for MSH‐7 ligand hMC4R/δOR system in the bivalent binding mode. Cell‐surface labeling demonstrated the high avidity and specificity of heteromultivalent targeting and validity of the receptor combination approach. This approach to non‐covalently crosslink heterologous receptors and target individual cells opens up new possibilities for specific targeting in vivo for theragnostic purposes. Supported by grants from the U.S. Public Health Service, NCI
P092
ENGINEERING HETEROCHIRAL PEPTIDES
J. Kulp, T. Clark
Naval Research Laboratory
Heterochiral peptides promise new routes to important compounds for use in sensors, smart materials, and catalysts. These peptides, which contain amino acids with periodic alternating chirality, exist in Nature: gramicidin, polytheonamide, and feglymycin. These few peptides all reside in membrane environments, so are heterochiral peptides limited to hydrophobic conditions for proper folding? When compared to peptides containing all alpha‐amino acids, heterochiral peptides differ not only in structural space—these peptides fold into single‐ and double‐stranded helices stabilized by beta‐sheet‐type hydrogen bonding—but also differ in stabilizing mechanisms. For instance, the alternating updown configuration of the amide carbonyl groups along the backbone results in a smaller, or absent, macrodipole; all L‐peptide helices have an overall dipole moment. This talk will communicate general synthetic and structural principles for heterochiral peptides by presenting the design, synthesis, and biophysical characterization of a set of hydrophobic‐ and hydrophilic‐heterochiral peptides.
Figure .

P093
SYNTHESIS AND INVESTIGATION OF NOVEL DIRECT THROMBIN INHIBITORS
E. Girnys, K. Williams, H. Mosberg
University of Michigan
The serine protease thrombin is the main clotting enzyme in the hemostatic system, in addition to being an effective platelet activator. Thrombin has two substrates that are G‐protein coupled receptors, protease‐activated receptors 1 and 4 (PAR1 and PAR4). The PARs carry their own cryptic ligand, and thrombin cleaves a fragment of the N‐terminus to reveal this ligand, as the neo‐Nterminus, which then binds intramolecularly to activate platelet aggregation. Thus, the development of a direct thrombin inhibitor offers an approach for the treatment of myocardial infarctions and other acute coronary syndromes through a two‐prong modulation of the hemostatic system: blocking thrombus formation by inhibiting both clotting and platelet aggregation. Previous studies have shown that the pentapeptide Arg‐Pro‐Pro‐Gly‐Phe, a bradykinin breakdown product, inhibits the function of thrombin by interacting with its active site in a retro‐binding fashion, as well as binding to PAR1 to prevent cleavage by thrombin. Structure‐activity studies led to the development of a lead compound, FM 19, with the sequence D‐Arg‐Octahydroindole‐2‐carboxylic acid‐Pro‐D‐Ala‐Phe(p‐Me)‐NH2. The recently determined x‐ray structure of FM 19 in the active site of thrombin has revealed potential modifications to improve binding, resulting in two analogs, BG 5 and BG 6, which each show an over 30‐fold increase in potency over FM 19. Additional modifications are planned, including changes to improve bioavalability, to develop potential orally bioavailable direct thrombin inhibitors.
P094
NMR STUDIES ON A DOUBLE TRANSMEMBRANE‐ CONTAINING FRAGMENT OF A G PROTEIN‐COUPLED RECEPTOR
L.S.Cohen, 1,2 A. Neumoin,3 B. Arshava,1 M. Hauser,4 J.M. Becker,4 O. Zerbe,3 F. Naider1,2
1Department of Chemistry, The College of Staten Island, City University of New York (CUNY), Staten Island, NY 10314; 2Department of Biochemistry, The Graduate Center, CUNY; 3Institute of Organic Chemistry, University of Zurich, Switzerland; 4Department of Microbiology, University of Tennessee, Knoxville, TN 37996
Structural studies of integral membrane proteins (IMPs) are often hampered by difficulties in crystallizing them in a lipid‐like environment. Alternatively, NMR has been applied and most detailed results have been so far obtained from the analysis of fragments containing one or two TM domains. In this study, the structure and dynamic properties of an 80‐residue fragment of Ste2p, the G protein‐coupled receptor for the ‐mating factor of S. cerevisiae, was studied in lyso‐palmitoylphosphatidylglycerol (LPPG) micelles and trifluoroethanol:water using solution NMR. The fragment, Ste2p (G31‐T110; TM1‐TM2), consisted of 19 residues from the N‐terminal domain, the first transmembrane helix (TM1), the first cytoplasmic loop, the second transmembrane helix (TM2) and 7 residues from the first extracellular loop. Multi‐dimensional NMR experiments on [15N], [15N,13C], [15N,13C,2H]‐labeled TM1‐TM2 and this fragment selectively labeled at specific amino acids or protonated at selected methyl groups resulted in >95% assignment of backbone and side chain nuclei in both media. The NMR investigation revealed the secondary structure of specific residues of TM1‐TM2 and a β‐hairpin like structure in LPPG. The LPPG structure is consistent with the results of biochemical experiments that identified the ligand binding site within this region. Furthermore, superposition of the backbone atoms of TM1‐TM2 with a rhodopsin‐templated model of Ste2p yielded an RMSD of 1.8Å justifying the fragment approach towards determination of the full length structure of GPCRs.
P095
FULL‐LENGTH INSULIN ANALOGUES AND THE EFFECT OF N‐ METHYLATION OF PEPTIDE BONDS AT POSITIONS B24, B25 OR B26
E. Antolková,1 L. Žáková,1 A. Cooper,2 T. Kraus,1 J. Jirácek1
1Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, v.v.i., Flemingovo nám. 2, 166 10 Praha 6, Czech Republic, and 2Department of Chemistry, Glasgow University, Glasgow G12 8QQ, Scotland, UK
The study of the insulin analogues may help provide a better understanding of the structure‐function relationship of insulin and its receptor. The amino acids in the C‐terminus of the B‐chain of insulin, especially PheB24, PheB25 and TyrB26, are involved in the recognition of insulin receptor. The C‐terminus of the B‐chain, and in particular residues B24 and B26‐B28, are also essential for insulin dimer formation. Dimerization of insulin has an important role in the delivery of the hormone from the pancreas to the circulation or in the administration of insulin to diabetic patients. The N‐methylation of particular peptide bonds of insulin may result in the elimination of the ability to form intermolecular or intramolecular hydrogen bonds, which are important for stability and correct folding of the insulin monomer, dimer formation, and interaction with its receptor. Here we present several novel full‐length insulin analogues with the N‐methylation of the peptide bond at position B24, B25 or B26. We determined the binding affinities of the analogues for insulin receptor in rat adipose membranes. We used the isothermal microcalorimetry method to compare the energetics of dissociation of the analogues with the wild‐type insulin (which forms dimers) and with des(B23‐B30)octapeptide insulin (which does not form dimers). This method allowed us to deduce several clear conclusions about the role of individual peptide bond amides for the dimerization and binding affinity of insulin.
P096
(αME)PRO VERSUS PRO β‐TURN FORMATION PROPENSITY
M. De Poli,1 A. Moretto,1 M. Crisma,1 C. Peggion,1 F. Formaggio,1 B. Kaptein,2 Q.B. Broxterman2 and C. Toniolo1
1ICB, Padova Unit, CNR, Department of Chemistry, University of Padova, 35131 Padova, Italy, 2DSM Pharmaceutical Products, Advanced Synthesis, Catalysis, and Development, 6160 MD Geleen, The Netherlands; matteo.depoli.1@unipd.it
Ca‐Methyl L‐proline, L‐(aMe)Pro, is one of the most conformationally constrained α‐amino acids. In particular, its ω and ϕ torsion angles are restricted to about 180 (trans) and ‐60, respectively, and only two ranges of values are expected for ψ, in dior longer peptides, namely about ‐30 (cis', helical structure) or 140 [trans', poly‐(L‐Pro)n II structure]. In this work, we examined the tendency of a number of Na‐acyl dipeptide N'‐alkylamides of the type RCO‐(αMe)Pro‐Xxx‐NHR' or RCO‐ Xxx ‐(αMe)Pro‐NHR', where Xxx is L (or D)‐Ala, Aib, or L (or D)‐(αMe)Pro, long enough to fold into intramolecularly H‐bonded β‐turns. The results are compared with those obtained for the corresponding dipeptides based on the prototypical Pro, a well known turn forming residue. For the crystal‐state 3D‐structural analysis we exploited X‐ray diffraction, while for our solution conformational study we relied heavily on the CD, FT‐IR absorption, and 1H and 13C NMR techniques.
P097
“CLICK PEPTIDE” BASED ON “O‐ACYL ISOPEPTIDE METHOD”: IN SITU PRODUCTION OF MONOMER AMYLOID β PEPTIDE FROM WATER‐SOLUBLE PRECURSOR ANALOGUES
A. Taniguchi, Y. Sohma, M. Skwarczynski, Y. Hirayama, T. Okada, K. Ikeda, H. Prakash, H. Mukai, T. Kimura, Y. Hayashi, S. Hirota, K. Matsuzaki, Y. Kiso
Kyoto Pharmaceutical University
In Alzheimer's disease (AD) research, low water‐solubility and uncontrolled aggregation of amyloid β peptide (Aβ) 1‐42 are known to cause difficulties in preparing monomer Aβ1‐42, resulting in irreproducible or discrepant outcomes in toxicological studies. To solve these problems, we disclosed water‐soluble “click peptides” [1] based on an “O‐acyl isopeptide method” [2]. The peptide had no self‐assembling nature due to an O‐acyl isopeptide structure at Gly25‐Ser26. The peptide converted to intact Aβ1‐42 under physiological conditions via an O‐to‐N intramolecular acyl migration upon being triggered by “click” (e.g. pH‐change or photo‐irradiation). The produced Aβ1‐42 underwent self‐assembling events starting from a monomeric random coil structure. These results indicated that in situ production of monomer Aβ1‐42 from click peptides can be used to establish a reliable experimental system to investigate the pathological functions of Aβ1‐42 in AD.
[1] (a) A. Taniguchi, Y. Kiso et al., J. Am. Chem. Soc. 2006, 128, 696; (b) A. Taniguchi, Y. Kiso et al., ChemBioChem 2008, 9, 3055; (c) Y. Kiso, A. Taniguchi, Y. Sohma in Wiley Encyclopedia of Chemical Biology (Ed.: T. P. Begley), Wiley, Hoboken, 2008, 1, 379; (d) A. Taniguchi, Y. Kiso et al., ChemBioChem in press, doi: 10.1002/cbic.200800765.
[2] (a) Y. Sohma, Y. Kiso et al., Chem. Commun. 2004, 124; (b) Y. Sohma, Y. Kiso et al., Biopolymers (Pept. Sci.) 2007, 88, 253.
P098
TOTAL CHEMICAL SYNTHESIS AND MEMBRANE INTERACTION STUDIES OF ISLET AMYLOID POLYPEPTIDE AND ITS PRECURSORS
P. Van de Vijver, L. Khemtémourian, D. Suylen, L. Scheer, G.L. Casarramona, H.J.D. Meeldijk, B. de Kruijff, J.W.M. Höppener, J.A. Killian, & T.M. Hackeng
Maastricht University
The human islet amyloid polypeptide, also known as hIAPP or amylin, is a 37‐residue peptide that is the major component of the amyloid deposits frequently found in patients suffering from diabetes mellitus type 2. Although it is not yet clear whether these amyloid deposits are a cause, consequence or side effect of the disease, it is hypothesized that the interaction between hIAPP and cellular membranes is a cause of IAPP cytotoxicity, leading to β‐cell death.
To provide further insights in the interaction between cell membranes and IAPP, we chemically synthesized human (h) and murine (m) ProIAPP, ProIAPP1‐48 as well as mature IAPP. mIAPP differs at only 6 residues from hIAPP but is known not to aggregate into amyloid deposits.
IAPP and its precursors were synthesized by manual Boc‐SPPS. ProIAPP and ProIAPP1‐48 were prepared by native chemical ligation of N‐terminal fragments with mercaptopropionyl‐leucine functionalized C‐termini and C‐terminal fragments starting with cysteine. Oxidative folding and purification yielded IAPP and IAPP precursors.
Amyloid fibril formation of IAPP, IAPP‐precursors and combinations thereof was studied in synthetic membranes as well as in cells, and a model was developed describing (protective) effects of IAPPprecursors on IAPP fibril formation.
P099
THE DESIGN AND SYNTHESIS OF A NOVEL MULTIFUNCTIONAL PEPTIDE TO STUDY DELTA OPIOID RECEPTORS
W. Hartsock,*1 M. Del Borgo,1 D. Moore,2 J. Aldrich1
1Department of Medicinal Chemistry and 2Microscopy and Analytical Imaging Laboratory, The University of Kansas, Lawrence, KS 66045 USA
We are interested in preparing novel probes to study receptor‐ligand interactions of delta opioid receptors (DOR). Our approach utilizes a multilabeled peptide containing a fluorescent moiety along with isolation tags and an electrophilic affinity label to aid in the elucidation of the specific DOR amino acids that bind these peptides.
The multifunctional peptide is a derivative of the high affinity, DOR selective antagonist TIPP (Tyr‐Tic‐Phe‐Phe, Tic =1,2,3,4‐tetrahydroisoquinoline‐3‐carboxylic acid). The multifunctional peptide was successfully prepared by Fmoc solid phase peptide synthesis using an aldehyde‐functionalized resin. Multiple lysine residues bearing different side chain protecting groups were employed to allow for selective deprotection and subsequent functionalization of the side chain amines with the various labels during peptide synthesis. PEG (poly(ethylene glycol))‐like linkers were included to enhance water solubility and to separate the TIPP portion of the peptide from the various labels incorporated at the C‐terminus.
The interaction of this peptide with Chinese hamster ovary (CHO) cells expressing DOR was assessed using fluorescence microscopy. These experiments provide evidence that this novel peptide binds covalently and specifically to DOR and demonstrates its utility to visualize DOR. This research was supported by NIDA grant DA010035.
P100
THE 13Cα AND 13Cβ SHIFTS REVEAL CLEAR PATTERN ALONG STRANDS IN β‐HAIRPINS
I. Shu, J. Stewart, B. Kier, M. Scian, N. Andersen
Department of Chemistry, University of Washington, Seattle WA 98195
It has long been recognized that 13Cα and 13Cβ chemical shift deviations (CSDs) from random coil values have different characteristics for residues in α‐helical versus β‐sheet regions of proteins; CSDs of 13Cα (and 13C') are positive and those of 13Cβ are negative in α‐helices, while these are reversed in β‐sheets. Although Rico and co‐workers were early advocates of using the 13Cα and 13Cβ shifts qualitatively (and even quantitatively) as a means for assessing β‐hairpin fold populations, unlike the case of Hα and HN shifts, the more detailed pattern of the 13C CSDs along the β‐strands does not appear to have been recognized in literature yet. In protein β‐sheets, many amide units have H‐bonds with two strands and the range of 13C CSDs occurring in β‐sheets overlaps with the narrower range for α‐helices. There also appears to be a discrepancy in the prediction of H‐bonding effects on 13C shifts in helices versus hairpins. To answer these questions, 13C structuring shifts have been determined for a series of designed hairpins of varying fold stability. Our study reveals that the diagnostic 13C shifts for β‐structuring are associated, almost exclusively, with the cross‐strand H‐bonded residues, and instances in which aromatic ring current effects on 13C CSDs can be as large as the secondary structure shifts. Results from a further search of 13C shifts in β‐structures of proteins will also be reported as a test of the generality of the conclusions based on the model hairpins.
P101
DETERMINATION OF UNIQUE AGRP AND MC4R INTERACTIONS THE USE OF STEREOCHEMICAL MODIFICATIONS OF LIGANDS AND RECEPTOR MUTAGENESIS
E. Haslach, Z. Xiang, and C. Haskell‐Luevano
Department of Pharmacodynamics, University of Florida, Gainesville, Florida 32601 USA
Agouti‐related protein (AGRP) is competitive antagonist at the melanocortin 4‐ receptor which is implicated to be involved in energy homeostasis. It is hypothesized that not all disulfide bridges of hAGRP(87‐132) are necessary for binding and activation. It is proposed that DArg111 interacts with mMC4R Asn115 and DPhe113 interacts with mMC4R Phe176 by the use of homology modeling. Derivatives of AGRP will be synthesized and tested at mutant receptors to determine if there are unique AGRP‐MC4R interactions. This work is supported by NIH grant DK57080.
[1] Wilczynski, A, Wang, X.S., Joseph,C.G, Xiang,, Z., Bauzo, R.M. Scott, J.W.,Sorensen, N.B., Shaw, A.M., Millard, W.J., Richards, N.G., Haskell‐ Luevano, C. J. Med. Chem. 2004, 47, 2194‐2207.
[2] Joseph, C.G, Wang, X.S., Scott, J.W., Bauzo, R.M., Xiang,Z., Richards, N.G., Haskell‐Luevano, C. J. Med. Chem. 2004, 47, 6702‐6710.
P102
LABELING THE ANGIOTENSIN II TYPE 1 RECEPTOR WITH A DOPA SUBSTITUTED ANGIOTENSIN II ANALOGUE
J. Arsenault, H. Jarine, G. Guillemette, R. Leduc, P. Lavigne, E. Escher
University of Sherbrooke
Biologically active GPCR structures are not yet accessible to analytical methods. The human angiotensin II type 1 receptor (hAT1) has been investigated with photoaffinity labeling approaches using Angiotensin II (DRVYIHPF, AngII) analogues with biologically acceptable photolabile moieties. Many ligand‐receptor contact points were found for most ligand residues; nevertheless, the central amino acid of AngII, the Tyrosine 4 residue could never be addressed due to inadequate SAR results with all attempted substitutions. To remediate this difficulty a DOPA (3'‐hydroxy Tyr) was introduced into position 4 of [Sar1] AngII, maintaining ∼1nM affinity. Once oxidized with NaIO4, the DOPA moiety may form a covalent bond to a cystein residue of the target receptor trough a Michael addition of the free thiol. Oxidized [Sar1,DOPA4] AngII could thus label a free SH‐ present in the immediate binding environment. To assure feasibility, this analogue was treated with NaIO4in presence of 1mM H‐Cys‐OH and incorporation of Cys was confirmed by MALDI‐TOF MS. In order to be able to trace a labeled receptor and receptor fragments, mono‐iodination of [Sar1,DOPA4] AngII was performed and confirmed with MS but for oxidized [Sar1, 2'‐I‐DOPA4] AngII no Cys incorporation was found. Therefore two pathways are explored, one with [Biotin0,DOPA4] AngII and streptavidine HRP tracing and another with a [Sar1,DOPA4, Tyr8] AngII, using DOPA acetonide protection and radioiodination of position 8 for radiotracing. Both analogues display affinities sufficient for receptor analysis.
P103
THE MINIMAL FUNCTIONAL UNIT OF THE MU OPIOID RECEPTOR IS MONOMERIC
J. Anand,1 A. Kusak,2 S. Pitchiaya,3 N. Walter,3 R. Sunahara,2 H. Mosberg1
Departments of Medicinal Chemistry1, Pharmacology2, and Chemistry3 University of Michigan, Ann Arbor
Despite the medical significance of the mu opioid receptor (MOR), which plays a key role in the mediation of analgesia, very little is known about the hysiologically significant state. Early dogma maintained that MOR, like other G‐protein coupled receptors (GPCRs), functions as a monomer. However, a significant body of evidence has been reported that suggests that GPCRs may act as dimers or higher order oligomers. We describe here the development of selective, fluorescent, peptidic ligands for MOR and their use to probe the function of monomeric MOR. Ligands with either Cy3 or Cy5 fluorescent tags covalently attached to a C‐terminal Cys were synthesized. The profile of these ligands has been established in C6 cells expressing either mu or delta opioid receptors, as well as CHO cells expressing kappa opioid receptors. These ligands have been used to probe the state of yellow fluorescent protein tagged MOR (YMOR) in vitro in a forced monomeric state using single molecule fluorescence and TIRF. This was achieved by reconstituting purified YMOR in HDL particles that mimic the lipid bilayer of plasma membranes. A monomeric state can be forced by limiting the diameter of the HDL particles. The MOR agonist [Lys7, Cys8] Dermorphin was used to show that the minimal functional unit of the mu opioid receptor is monomeric. Additionally, a novel method for studying ligand binding of monomeric GPCRs in vitro was developed.
P104
CELL SECRETED MATRIX METALLOPROTEINASE‐9 MEDIATED TRIGGERED RELEASE OF LIPOSOMAL CONTENTS FROM OPTIMIZED LIPOSOMAL FORMULATION INCORPORATING COLLAGEN MODEL LIPO‐PEPTIDES
J. Banerjee, A. Hanson, B. Gadam, B. Ganguly, A. Wagh, D. Srivastava, B. Law, S. Mallik
Introduction and Purpose: Matrix metalloproteinases (MMPs) are metalloenzymes responsible for degradation of extracellular matrix. Out of 26 different types of MMPs, matrix metalloproteinase‐9 (MMP‐9) is ubiquitously over expressed in almost every type of tumor cells undergoing metastasis. Our objective was to develop and optimized a novel methodology for releasing liposomal contents triggered by human recombinant and cancer cell secreted MMP‐9. For this purpose we formulated liposomes by incorporating commercially available phosphatidylcholines as the major component and a synthesized MMP‐9 cleavable lipid‐peptide conjugate of varying triple helicity as the minor component. The liposomes were encapsulated with a self quenching dye carboxyfluorescein.
Experimental Methods: The lipopeptides were synthesized by employing a microwave peptide synthesizer (Liberty from CEM Inc.). The triple helical peptides were purified and characterized by reverse phase HPLC, mass spectrometry (MALDI‐ToF) and circular dichroism (CD) spectrometry. The fluorescence emission of carboxyfluorescein from liposomes was recorded with a spectrophotometer (Spectramax, Molecular Devices). Cell based studies were done using optimized liposomal formulations with human breast adenocarcinoma MCF‐7 and human colorectal HT‐29 cultured cell lines.
Results and Discussion: The lipopeptides synthesized have the general sequence : St‐GPQGIAGQR(GPO)n GG. The peptides contain a Stearic acid (St) at the N‐terminal of Gly, a MMP‐9 sessile bond (underlined) and varying GPO units from n = 3 to n = 5. An optimized liposome formulation was obtained for POPC (70 mole %) and lipopetide (30 mole%) with 4 units of GPO (n = 4). This formulation releases about 50% of the dye at caner associated concentration of recombinant human MMP‐9. When tested against cultured cell lines we found that while metastatic MCF‐7 cells secreting active MMP‐9 was able to release the encapsulated dye, HT‐29 cells which do not secrete much of active MMP‐9 was unable to show any significant release of the dye over the same time period.
Conclusions: The triggered release methodology showed significant promise in in‐vitro release experiments in cancer cells. This methodology can be next applied on an animal model for in‐vivo studies and in future can be fine tuned to act as a drug delivery vehicle for inhibition of MMP‐9 over expressed under various pathological conditions.
P105
GASTRIN‐RELEASING PEPTIDE (GRP) DOES NOT MEDIATE GNTI‐INDUCED EXCESSIVE SCRATCHING OR THE ANTIPRURITIC ACTIVITY OF NALFURAFINE IN MICE
Saadet Inan,1,* Nae J. Dun1 and Alan Cowan1,2
Department of Pharmacology1 and Center for Substance Abuse Research2, Temple University School of Medicine, Philadelphia
We investigated if GRP plays a role in compulsive, hindleg scratching of the neck in mice by the kappa opioid antagonist, 5‐guanidinonaltrindole (GNTI), or in the antipruritic activity of the kappa agonist, nalfurafine. We showed that GNTI (0.03‐1 mg/kg, s.c. behind the neck) elicits dose‐related scratching and that nalfurafine (0.001‐0.02 mg/kg, s.c.) inhibits this stereotyped behavior. Utilizing immunohistochemistry, we located GRP positive nerve fibers in the epidermis and dermis of mouse skin and superficial layer of the dorsal horn of the spinal cord as well as GRP positive small and medium sized cells in the dorsal root ganglion. In a double staining study, GRP positive nerve fibers were near the cells expressing c‐fos in response to GNTI injection, located on the lateral aspect of the superficial layer of the dorsal horn. Cervical spinal cord GRP mRNA levels were similar in mice treated with saline‐saline, saline‐GNTI, nalfurafine‐saline or nalfurafine‐GNTI. Pretreating mice with RC‐3095 (10 mg/kg, s.c.), a GRP receptor antagonist, did not markedly suppress GNTI‐induced scratching. We conclude that GRP mediates neither GNTI‐induced scratching nor the antipruritic activity of nalfurafine in mice. (P30DA013429 and NS18710).
P106
FUNCTIONAL ASSOCIATION OF N‐TERMINAL RESIDUES OF EX‐4 WITH THE CENTRAL & C‐TERMINAL REGIONS OF THE HORMONE
J. Patterson*, V. Gelfanov and R. DiMarchi
Department of Chemistry, Indiana University, Bloomington, Indiana 47405, USA *jtpatter@indiana.edu
GLP‐1 is an incretin hormone involved in the regulation of glucose metabolism and its primary action is insulin secretion in a glucose‐dependent manner upon nutrient ingestion. Treatment of Type II diabetics with GLP‐1 receptor (GLP‐1R) agonist Exendin‐4 (Ex‐4) has proven clinical benefit in lowering blood glucose and the reduction of body weight. Ex‐4 is a paralog of GLP‐1 that has increased GLP‐1R potency and extended physiological action compared to the native hormone. Both peptides are highly homologous and are reported to adopt an alpha helical secondary structure. Unlike GLP‐1, Ex‐4 contains a C‐terminal extension, termed Cex. We have confirmed that the addition of Cex to GLP‐1 modestly improves GLP‐1R‐mediated cAMP induction in engineered reporter cells. We have prepared GLP‐1 analogs that incorporate the native Ex‐4 residue Glu.16 Our results demonstrate that this single substitution enhances GLP‐1R agonism to be near‐equivalent to Ex‐4 even in the absence of the terminal Cex extension peptide. Furthermore, GLP‐1 and Ex‐4 contain several divergent residues, one of which is a GLP‐1 Ala2 as opposed to Ex‐4 Gly2. This single change was observed to yield divergent results when studied in the context of the two different hormonal sequences. We further demonstrated that Ex‐4 tolerates Gly2 through selective amino acid differences located within the mid‐ to C‐terminal region of its sequence. These amino acids were identified by site specific mutation and their location indicates that the N‐terminal end of Ex‐4 and GLP‐1 are functionally linked to the middle and C‐terminal segments.
P107
AMINO‐LACTAM PEPTIDE SYNTHESIS USING FMOC‐ PROTECTED CYCLIC SULFAMIDATES ON SOLID‐PHASE
N. Boutard,1 A. Jamieson,1 K. Beauregard2 M. Bodas,1 H. Ong,3 C. Quiniou,2 S. Chemtob,2 W. D. Lubell1
1Chemistry Department, 3Faculty of Pharmacy, Université de Montréal, C.P. 6128, Succursale Centre‐Ville, Montreal, Quebec H3C 3J7, Canada; 2St.‐Justine Hospital Research Centre, Université de Montréal, 3175 Cte‐Ste‐Catherine, Montreal, Quebec, H3T 1C5 Canada.
Incorporation of amino lactams into biologically active peptides may restrict conformational mobility, enhance selectivity and increase potency. Fmoc‐protection has been used in a novel strategy for the solid‐phase synthesis of peptides containing <‐ and ®amino ©‐lactams. Lactams were introduced into the peptide chain by Nalkylation using five‐ and six‐membered cyclic sulfamidates and lactam annulation under microwave heating. Employing this solidphase protocol on the growth hormone secretagogue GHRP‐6 as well as on the allosteric modulator of the IL1 receptor 101.10 has furnished lactam peptides for biological studies. The binding affinity IC50 values of the GHRP‐6 lactam analogs on both GHS‐R1a and CD36 are reported as well as inhibition of thymocyte proliferation measurements for the 101.10 lactam analogs. In these cases, lactam analogs were prepared exhibiting similar and improved properties compared with the parent peptide.
P108
MULTIPLE INSERTION OF α‐AMINO γ‐LACTAM (AGL) RESIDUES IN PEPTIDE SEQUENCES BY PARALLEL SOLID‐PHASE SYNTHESIS ON SYNPHASE LANTERNS
L. Ronga and W. Lubell
Départment de Chimie, Université de Montréal, C.P. 6128, Succursale Centre‐Ville, Montreal, Quebec H3C 3J7, Canada
The presence of lactams in natural products, such as penicillin, as well as in peptide analogs with enhanced potency and greater receptor selectivity [1], underline their importance as synthetic tools in drug discovery. In particular, the insertion of lactam motif in linear peptides limits conformational freedom stabilizing β‐turn structures with potential bioactivity [2]. Here, we present the synthesis of lactam‐bridged peptide sequences performed by a solidphase approach on SynPhase lanterns [3] using cyclic sulfamidate to annulate the amino lactam residue onto the peptide chain. Parallel synthesis of α‐amino γ‐lactam (Agl) analogs of the allosteric modulator of IL‐1 receptor 101.10 (rytvela) [4] was performed by split and mix chemistry on the lanterns. In particular, the double insertion of α‐amino γ‐lactams in the same peptide sequence has been accomplished by this effective method for the solid‐supported combinatorial synthesis of lactam‐bridged peptides.
[1] Freidinger R.M. J. Med. Chem. 2003, 46 (26), 5553–5566 and references within.
[2] Perdih A.; Kikelj D. Curr. Med. Chem. 2006, 13, 1525–1556 and references within.
[3] Ede N.J. J. Immunol. Methods. 2002, 267(1):3–11.
[4] Quiniou C. et al. J Immunol. 2008, 180(10), 6977–87.
P109
SYNTHESIS OF CONFORMATIONALLY RESTRICTED α‐HELIX MIMICS AS REV MULTIMERIZATION INHIBITORS
S. Pawar,1 T. Vercruysse,2 W. Dehaen,1 D. Daelemans,2 W. De Borggraeve1
1Molecular Design and Synthesis, 2Rega Institute for Medical Research, KULeuven, 3000 Leuven, Belgium. Sonalika.pawar@chem. kuleuven.be
The replication of the type 1 human immunodeficiency virus (HIV) is regulated by its viral mRNA expression. Control of HIV RNA expression is complex and involves the interplay of viral transactivators and several cellular proteins. Rev is one of these proteins. The HIV‐1 Rev protein is an essential regulator of the HIV‐1 mRNA expression, through multimerization Rev molecules bind throughout the Rev‐responsive element RRE and promotes the export of unspliced and partially spliced mRNA. It is suggested that the multimerization regions of Rev are two separate α‐helices comprising residues 9‐24 and 34‐62.
Here we report the synthesis of an α‐helix mimic of the Rev protein domain located between amino acid 10 and 24 which is responsible for Rev multimerization.
Synthesis of several α‐helical, cyclic peptides of the Rev protein domain mimics was carried out by SPPS, using Fmoc strategy. The a helical nature of these peptides has shown by Circular Dichroism (CD) and NMR spectroscopy. The Fluorescence Resonance Energy Transfer (FRET) assay for inhibition of Rev‐multimerization, will also be discussed.
P110
THE PROTEOLYTIC STABILITY AND CYTOTOXICITY STUDIES OF L‐ASPARTIC AND L‐DIAMINOPROPIONIC ACID DERIVED OLIGOMERS
S. Ahmed and K. Kaur
Faculty of Pharmacy and Pharmaceutical Sciences, University of Alberta, Edmonton, AB, Canada, T6G 2N8
The use of peptides as drugs in pharmaceutical application is hindered by their susceptibility to enzymatic degradation (proteolysis) and so low bioavailability. Peptidomimetic oligomers, such as β‐peptides with an additional methylene group in the backbone, are gaining recognition from biological and pharmaceutical stand points as they are considerably more resilient to proteolysis and metabolism. Recently we have reported two new classes of β‐peptides, β3‐ and β2‐peptides derived from L‐aspartic acid and L‐diaminopropionic acid, respectively. Here we studied the proteolytic stability of these β‐peptides, and a mixed a/β‐peptide against three proteolytic enzymes (pronase, trypsin, and elastase) as well as human serum. The stability of these peptides was compared to an α‐peptide, to evaluate their potential as peptidomimetics. Peptides containing β‐linkages were resistant to all conditions, whereas, the mixed α/β‐peptide exhibited proteolysis in the presence of trypsin and pronase but not elastase. The rate of degradation of the mixed α/β‐peptide was slower than that would be expected for an α‐peptide, a feature important for drug design. The toxicity profile and application of these oligomers in the design of biological active peptides will be highlighted.
Figure .

P111
EFFICIENT SOLID‐PHASE SYNTHESIS OF TRIS‐BENZAMIDES FOR A RAPID PRODUCTION OF ALPHA‐HELIX MIMETICS
T. Lee, J. Ahn
University of Texas at Dallas
α‐Helix involves widely in protein‐protein interaction and modulation of this key process would be of great interest in studying many fundamental biochemical pathways. However, developing non‐peptidic small molecules for this goal is still a challenging area of research. Suitable α‐helix mimetics should be able to hold a structure to properly place functional groups as presented in a helix and also to be easily synthesized for a production of a number of compounds for efficient biochemical studies. Recently, we have reported a tris‐benzamide as a scaffold to mimic the side chains of amino acids found at the i, i+4, and i+7 positions of an α‐helix. Its simple and straightforward synthesis uses conventional and high‐yielding reactions that can be readily amenable to solid‐phase combinatorial synthesis. Thus, we report herein an efficient solid‐phase strategy to synthesize a number of tris‐benzamides. The library construction involved several repeating reactions including removal of an allyl ester, an amidebond formation via O‐N acyl migration, and an O‐alkylation on a solid support, and each step was carried out with high synthetic yield. This facile and efficient approach allowed the production of tris‐benzamides containing functional groups that represent hydrophobic (e.g., Ala, Val, Leu, Phe, Trp) and hydrophilic (e.g., Glu, Lys) amino acids, in high overall yield and purity.
P112
ISOTOPICALLY LABELLED β‐TURN PEPTIDE MIMICS AS TOOL FOR UNDERSTANDING ALLOSTERIC ANTAGONISM OF GPCR ACTIVITY
C.B. Bourguet, W. D. Lubell
Département de Chimie, Université de Montréal, Montréal, Québec H3C 3J7, Canada
Preterm labor is a significant medical and social burden, estimated at 9 billion per year in the USA (the highest per patient cost of any disorder). Furthermore, preterm labor has steadily increased over the last thirty years. A member of the G‐protein coupled receptor family, the prostaglandin F2a (FP) receptor is a valid target for the development of a method to prevent preterm labor, because it is directly involved in uterine contractions and over‐expressed at onset and during labor. Potent allosteric modulators of the FP receptor have been developed, which feature an indolizidinone dipeptide mimic as a type II' β‐turn surrogate (Fig.1).1 In addition to delaying labor and stopping uterine contractions in vivo, the mimic THG113.824 has been used in vitro as atool for studying secondary messenger signalling on FP binding to its receptor. Isotopically labelled and photo‐reactive analogs of THG113.824 are being pursued to better understand its mechanism of action. Our presentation will discuss the synthesis, biological activity and determination of the binding site of these novel allosteric modulators of FP receptor activity.
Figure .
1. Peri, K.G.; Polyak, F.; Thouin, E.; Lubell, W.D., Chemtob, S. PCT Int. Appl. serial No. 60/387, 424, June 11, 2002.
P113
DISRUPTION OF P53/MDM2/MDMX PROTEIN‐PROTEIN INTERACTIONS
M. Topper, M. Zhou, L. Anderson, M. McLaughlin
University of South Florida
The overall balance of pro and anti‐apoptotic protein interactions controls the susceptibility of a cell towards programmed cell death. Overexpression of the anti‐apoptotic proteins, MDM2 and MDMX, is associated with chemo and radiation therapy resistance. NMR studies have shown that the tumor suppressor p53 protein, forms an amphipathic α‐helix in the NH2 terminal that directly interacts (binds) with a hydrophobic groove on the globular MDM2 and MDMX domains; resulting in inhibition of apoptosis, allowing the continuation of tumor growth. Synthesis of derivatives that mimic the α‐helix portion of p53's NH2 activation site, capable of disrupting the p53/MDM2 and p53/MDMX interactions, could represent a great potential for medicinal chemistry studies. There have been documented syntheses of various potent alpha helix mimics that successfully bind to NH2 activation site of the p53 protein. However, these compounds have hydrophobic scaffolds that limit their usefulness as drugs. Presented, is the design and synthesis of a new family of α‐helical mimics based on a functionalized hybrid scaffold containing both piperazine and pyrimidine based repeat units, holding amino acid‐like side chain residues in positions that mimic the ith, ith+4, and ith+7 positions of an alpha helix. This novel scaffold has a high degree of polarity therefore increasing its degree of solubility and potential as a novel therapeutic when compared to previous α‐helix mimics.
P114
EFFECT OF HELIX‐PROMOTING STRATEGIES ON THE BIOLOGICAL ACTIVITY OF NOVEL ANALOGUES OF THE B‐CHAIN OF INSL3
F. Shabanpoor, R. Hughes, R. Bathgate, F. Separovic, J. Wade
Howard Florey Institute, University of Melbourne
Insulin‐like 3 (INSL3) is a novel circulating peptide hormone that is produced by testicular Leydig cells and ovarian thecal and luteal cells. In males, INSL3 is responsible for testicular descent during foetal life and suppresses germ cell apoptosis in adult males, whereas in females, it causes oocyte maturation. Antagonists of INSL3 thus have significant potential clinical application as contraceptives in both males and females. Previous work has shown that the INSL3 receptor binding region is largely confined to the B‐chain central _‐helix of the hormone and a conformationally constrained analogue of this has modest receptor binding and INSL3 antagonist activity. We have employed and evaluated several approaches for increasing the _‐helicity of this peptide in order to better present the key receptor binding residues and increase its affinity for the receptor. Analogues of INSL3 with higher _‐helicity generally had higher receptor binding affinity although other structural considerations limit their effectiveness.
P115
HIGH‐THROUGHPUT SEQUENCING OF PEPTOIDS AND PEPTIDE‐ PEPTOID HYBRIDS BY PARTIAL EDMAN DEGRADATION AND MASS SPECTROMETRY
A. Thakkar and D. Pei
Department of Chemistry, The Ohio State University, 100 West 18th Avenue, Columbus, Ohio 43210
A method for the rapid sequence determination of peptoids [oligo(N‐substituted glycines)] and peptide‐peptoid hybrids selected from one‐bead‐one‐compound combinatorial libraries has been developed. In this method, beads carrying unique peptoid (or peptide‐peptoid) sequences were subjected to multiple cycles of partial Edman degradation (PED) by the treatment with a 1:3 (mol/mol) mixture of phenyl isothiocyanate (PITC) and 9‐fluorenylmethyl chloroformate (Fmoc‐Cl) to generate a series of N‐terminal truncation products for each resin‐bound peptoid. After PED, the Fmoc group was removed from the N‐terminus and any reacted side chains via piperidine treatment. The resulting mixture of the full‐length peptoid and its truncation products was analyzed by matrix‐assisted laser desorption ionization (MALDI) mass spectrometry, to reveal the sequence of the full‐length peptoid. This rapid, high‐throughput, sensitive, and inexpensive sequencing method should greatly expand the utility of combinatorial peptoid libraries in biomedical and materials research.
P116
SELF–PEPTIDOMIC REPERTOIRE OF THE HUMAN PRE‐NODAL LYMPH
C. Clement, R. Maitra, R. Sahu, L. Santambrogio
Pathology, Albert Einstein College of Medicine, Bronx, NY
The reticular network within the lymph node is a conduit system forming the infrastructure for the fast delivery of soluble substances from the afferent lymph to the lumen of high endothelial venules (HEVs). Conduit associated dendritic cells (DC) are capable of taking up and processing soluble antigens by directly fishing into the nanometer size conduits transporting the afferent lymph. No data are currently available on the antigenic composition of the human lymph available to the resident dendritic cells, which constantly sample the self‐antigenic repertoire. In this study we are reporting important sequence information of peptides from the human lymph of healthy donors. Using advanced proteomics approaches, such as high resolution electrospray ionization mass spectroscopy sequencing (LTQ‐MS/MS) together with proteins MS/MS database search using Mascot and SEQUEST algorithms we were able to sequence more than three hundred peptides present in the pre‐nodal lymph. A significant majority of these peptides derived from the in vivo processing and turnover of extracellular matrix proteins such as collagens, laminins, fibronectins, mucins. Sequenced peptides were then screened for their potential affinity for HLA‐DR1 and HLA‐DR4 MHC class II proteins using the Syf‐P9 algorithm. The best hit candidates were further screened and found to have high binding affinity for either HLA‐DR1 or HLA‐DR4.
P117
β‐AMINO ACID SYNTHESIS AND THE TOTAL SOLID‐PHASE SYNTHESIS OF MIXIRINS A AND B
G. Kemp*, J. P. Malkinson
*Department of Pharmaceutical and Biological Chemistry, School of Pharmacy, University of London, 29‐39 Brunswick Square, London WC1N 1AX, UK gary.kemp@pharmacy.ac.uk
Mixirins A, B and C were recently isolated from a marine Bacillus sp. bacterium and have shown cytotoxic activity against human colon tumor cells. The mixirins were subsequently identified as a mixture of cyclic lipopeptides belonging to the iturin class of compounds. The iturins contain 7 proteinogenic α‐amino acid residues and one unusual lipophilic β‐amino acid residue of undetermined absolute stereochemistry.
The β‐amino acid moieties of mixirins A and B were synthesised enantioselectively by a novel α‐amino acid homologation procedure, yielding both enantiomers of the β‐amino acid residue. The lipophilic β‐amino acids were then suitably protected for subsequent Fmoc SPPS.
Four novel potential anticancer compounds have been synthesised by solid‐phase techniques: mixirin A and its β‐amino acid epimer, and mixirin B and its β‐amino acid epimer. Their synthesis and evaluation will be reported.
P118
DESIGN, TOTAL CHEMICAL SYNTHESIS, AND X‐RAY STRUCTURE OF A PROTEIN MOLECULE OF NOVEL TOPOLOGY BASED ON CRAMBIN
K. Mandal,1,3* B. Pentelute,2,3 D. Bang,1,3 Z. Gates,2,3 V.Y. Torbeev,1,3 and S.B.H. Kent1,2,3
1Department of Biochemistry and Molecular Biology, 2Department of Chemistry, 3Institute for Biophysical Dynamics, The University of Chicago, *Email: kmandal@uchicago.edu
Crambin possesses an important salt bridge between the Arg10 side chain and the α‐carboxylate of Asn46 that guides the formation of correct disulfide bonds and contributes to the tightly folded overall structure.1 We set out to use chemistry to replace the salt bridge by a covalent bond. This led us to ask: will the artificial covalent bond allow the molecule to fold with correct disulfide bond formation? Replacement of a salt bridge by amide bond has never been done for a protein and is only possible by total chemical synthesis. The resulting novel topology is fascinating because the polypeptide chain will have a start (N‐termianl) but no end (C‐terimanl). This novel topology is reminiscent of but not identical to the naturally occurring cyclotides and lasso proteins. Chemical synthesis of such a unique topological analogue is highly challenging. We have devised an efficient convergent synthetic route to the crambin‐derived protein of novel topology. The synthetic challenge represented by this protein molecule of novel topology has led to the development of ‘kinetically controlled ligation’.2 We have used a novel quasi‐racemic crystallization technique3 to crystallize the synthetic topologue, and determined the molecular structure by X‐ray crystallography. The X‐ray structure reveals that the novel topologue shares a fold similar to native crambin with correct' disulfide bond formation even in the presence of an artificial covalent bond replacing a salt bridge.
1. Bang, D., PhD thesis. 2005. Chapter 6.
2. Bang, D.; Pentelute, B. L.; Kent, S.B.H. Angew. Chem. Int. Ed. 2006. 45, 3985–3988.
3. Pentelute, B. L.; Gates, Z. P.; Tereshko, V.; Dashnau, J. L.; Vanderkooi, J. M.; Kossiakoff, A. A.; Kent, S. B. H., J. Am. Chem. Soc. 2008, 130, 9695–9701.
P119
SYNTHESIS OF N‐ALPHA‐FMOC‐L‐GAMMA‐CARBOXY‐ GLUTAMIC ACID
D. Smith, J. Schneider
University of Delaware
L‐γ‐Carboxyglutamic acid (Gla) can be incorporated into peptides to mimic the calcium‐ and mineral‐binding domains of naturally occurring proteins. The incorporation of this unnatural amino acid residue into peptides using solid‐phase peptide synthesis (SPPS) is quite costly. This has motivated many research groups to develop more cost‐effective syntheses of this residue which contain the correct Fmoc‐ and t‐Bu‐protecting groups necessary for SPPS. Herein, N‐a‐Fmoc‐L‐γ‐carboxyglutamic acid‐γ,γ'‐tert‐butyl ester was synthesized from an easily prepared racemic amino acid ester precursor which was resolved using papain. Our approach begins with protecting the amine of glycine methyl ester as the benzyl‐imine. Subsequent coupling of di‐tert‐butyl methylenemalonate to the α‐carbon via a Michael addition, acidic hydrolysis of the imine, followed by enzymatic resolution of the L‐isomer using papain provided the enantiomerically pure amino acid suited for N‐protection with N‐(9‐fluorenylmethoxycarbonyloxy)succinimide (Fmoc‐OSu). This economical synthesis from simple starting materials provides a fully protected unnatural amino acid residue suitable for use in Fmoc‐based solid‐phase peptide synthesis.