

Abstract
For many viruses, RNA is the holder of genetic information and serves as the template for both replication and translation. While host and viral proteins play important roles in viral decision‐making, the extent to which viral RNA (vRNA) actively participates in translation and replication might be surprising. Here, the focus is on flaviviruses, which include common human scourges such as dengue, West Nile, and Zika viruses, from an RNA‐centric viewpoint. In reviewing more recent findings, an attempt is made to fill knowledge gaps and revisit some canonical views of vRNA structures involved in replication. In particular, alternative views are offered on the nature of the flaviviral promoter and genome cyclization, and the feasibility of refining in vitro‐derived models with modern RNA probing and sequencing methods is pointed out. By tracing vRNA structures from translation through encapsidation, a dynamic molecule closely involved in the self‐regulation of viral replication is revealed.
Keywords: Dengue virus (DENV), flaviviruses, host–virus interactions, viral RNA genome, untranslated region (UTR), nonstructural protein 5 (NS5), RNA‐dependent RNA polymerase (RdRp)
Flaviviral RNA is replete with unique secondary and tertiary structures that participate in the dynamic regulation of viral replication through cyclization, polymerase binding, switching between translation and replication, and host interaction and adaptation. Here, an attempt is made to summarize and also re‐examine the current understanding of how viral RNA (vRNA) functions at the molecular level.
1. Introduction
Flaviviruses include many of the most prevalent viral scourges known to humanity, such as dengue virus (DENV), West Nile virus (WNV), Zika virus (ZIKV), yellow fever virus (YFV), Japanese encephalitis virus (JEV), St. Louis encephalitis (SLEV), and Murray Valley encephalitis (MVE): a total of 53 species are listed by the International Committee on Taxonomy of Viruses as of 2017.1 These viruses contain an approximately 11 000 nt positive‐strand RNA genome (Baltimore Group IV) comprising a 5′‐untranslated region (UTR) of approximately 100 nucleotides (nt), a single open reading frame, and a 3′‐UTR of roughly 500 nt. The genome is initially translated into a single polyprotein with ≈3400 amino acids, which is subsequently proteolytically processed into ten viral proteins.2 DENV alone accounts for nearly 400 million infections3 and 500 000 hospitalizations per year worldwide.4 It is expected that factors such as global warming and urbanization will promote flaviviral infections in a manner outpacing population growth, largely due to enhanced opportunities for the proliferation of the most common flavivirus vectors, mosquitos, and ticks.5, 6 Much work is still needed to gain a comprehensive view of the flavivirus life cycle in order to develop effective treatments against these infections. In this review, we focus primarily on the flavivirus genus, although we do not hesitate to fill knowledge gaps with work involving the broader Flaviviridae family, which includes the hepatitis C virus (HCV) and the bovine diarrhea virus (BVDV), as well as work from other genera such as Picornaviridae.
The current model of the flaviviral RNA life cycle is illustrated in Figure 1. The encapsidated RNA genome exists solely as a positive single strand.7 Upon internalization, fusion of the virus membrane with the host endosomal membrane allows the release of genomic RNA into the cytoplasm. This RNA serves as the template for translation by the host ribosome at the rough endoplasmic reticulum (ER) to produce the viral nonstructural protein 5 (NS5) possessing RNA‐dependent RNA polymerase (RdRp) activity. The combination of viral nonstructural (NS) proteins that interact to amplify viral RNA (vRNA) is known as the “replicase” complex, which is localized in membranous vesicles derived from the ER.8 The negative strand is then synthesized by NS5 and is found only in association with the positive strand. This double‐stranded form of vRNA (dsRNA) serves as a template for the production of an excess of positive‐strand RNA, which may be utilized for further translation, generation of another round of negative strands, or packaged into the maturing virion. vRNA is thus found in dsRNA and (+) single‐stranded RNA (ssRNA) forms, as well as a more heterogeneous form associated with (+) ssRNA strands emerging from dsRNA.
Figure 1.
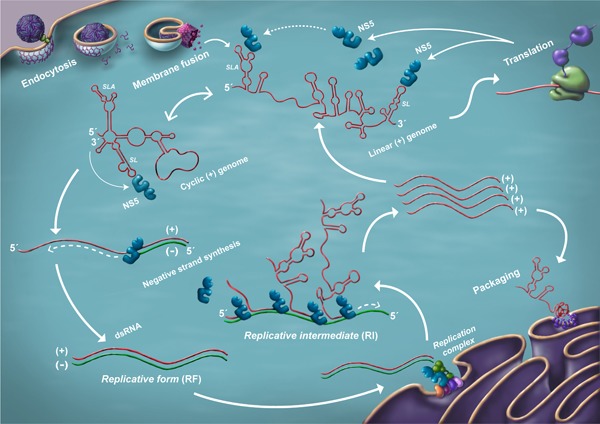
A model of the flaviviral RNA life cycle. Following RNA release into the cytoplasm, as well as translation and viral polyprotein processing, the vRNA cycle begins. The process is thought to occur primarily within or near the ER. Upon accumulation of sufficient replicase complex components, the (+) strand genome switches to a predominantly cyclized form, emphasizing (–) strand generation. The RF and RI are shown. Note that the (–) strand is only seen in a duplex with the (+) strand.
2. Translation of Flaviviral RNA Is Inefficient
Immediately following release from the viral nucleocapsid, translation is the only order of business for flaviviral RNA, as the absence of packaged viral polymerase means that immediate replication is not an option. vRNA can be translated in a canonical cap‐dependent manner, in which the 43S ribosomal component scans for a start codon.9 Though the initiation context is weak due to the lack of a Kozak consensus sequence upstream of the start codon, a downstream mini‐hairpin RNA structure termed “cHP” assists in start codon recognition.9 While not universal, the cHP can be observed in both mosquito‐borne and tick‐borne flaviviruses. Presumably, the structure serves primarily to stall the ribosome over the start codon, as the cHP's sequence is variable but its stability and location with respect to the start codon clearly correlate with initiation efficiency. It should also be noted that, while lacking a poly(A) tail, the flaviviral 3′‐UTR has been shown to interact with poly(A)‐binding protein (PABP), another hallmark of cap‐dependent translation.10
Translation of dengue vRNA has also been observed in the presence of inhibitors of canonical translation.11 However, internal ribosome entry site (IRES)‐mediated ribosome binding can be ruled out. This is because no such structures are evident in flaviviral RNA. Also, the addition of a stable 5′‐terminal structure inhibited noncanonical translation, suggesting that ribosome binding does not bypass recognition of the vRNA terminus.11 Nevertheless, we note a similarity between the hepacivirus IRES and the flavivirus 3′‐UTR: they are of similar length (350 nt and 450 nt, respectively) and contain specific structures that block degradation by the host exonuclease XRN1.12, 13 In addition, numerous studies have shown that a variety of alterations to flaviviral 3′‐UTRs affect translational output14, 15 and that these UTRs bind to host factors involved in translation (e.g., PABP10 and elongation factor 1‐α16). Conversely, the HCV 3′‐UTR and flaviviral 5′‐UTR are short relative to their opposing UTRs, and both contain poly(U) sequences of unknown function. One may wonder whether a distant evolutionary event flipped the UTRs that flanked the coding region of the hepacivirus and the flavivirus ancestor. The flaviviral 3′‐UTR, however, does not appear to directly interact with ribosome, given an absence of RNA reads in the 3′‐UTR in a ribosomal profiling study.17 This work also showed that the viral translation process is quite inefficient in comparison to that of host messenger RNA (mRNA): the authors speculate that such “lazy” translation may reduce the chances of alterations in ER homeostasis that would trigger host defense mechanisms.17
3. Numerous Genomic RNA Structures Are Involved in Replication
Following translation, the first act of the viral NS5 polymerase is to distinguish the viral (+) stranded RNA genome from host cellular RNAs and generate (–) strand vRNA, which is further used as the template for the production of (+) vRNA. It is thought that the process of vRNA replication occurs in remodeled, ER‐derived vesicles.17 Here, we highlight (+) strand RNA structures that are relevant to this process.
3.1. Is Cyclization of vRNA Firmly Established?
One observation important in generating the current picture of early steps in flaviviral replication is that of “cyclization” of (+) vRNA through the base pairing of 5′‐ and 3′‐termini (Figure 2). Electron microscopy revealed such behavior in alphavirus in the 1970s.18 In a mid‐1980s flurry of virus sequence analysis studies, Hahn et al.19 showed that a number of flaviviruses contained conserved 5′‐ and 3′‐complementary sequences (CS) that could potentially facilitate cyclization. Padmanabhan's group then showed that mutations in these CS regions impaired (–) strand synthesis, while restoring complementarity with a second set of mutations rescued replication.20 Gamarnik and colleagues later used atomic force microscopy to visualize cyclization of an approximation of DENV RNA.21 This work, however, was not without its critics; Lott and Doran22 decried an experimental setup that seemed to be designed to produce the expected cyclization result and pointed out the difficulty of distinguishing cyclization (in cis interactions) from concatemerization (in trans). Be that as it may, a long, diverse compendium of experiments apparently support flaviviral genome cyclization.23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 We note that “cyclization” of the replicating form of vRNA does not necessarily mean that the translated form of vRNA is “linear.” Indeed, it is established that translating host poly(A)‐tailed mRNAs take a circular form, with PABP and eIF4s bridging the two termini.35 It is even possible that vRNA can assume more than one cyclized form. For simplicity, we will continue to use the term “cyclization” as opposed to “5′–3′ complementarity,” but the above caveats should not be ignored.
Figure 2.
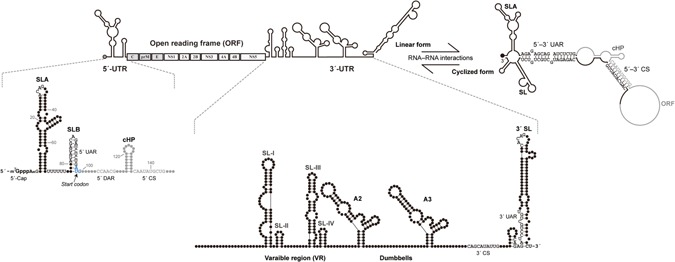
vRNA structures. The complete flaviviral genome contains a single open reading frame (gray) representing ten viral proteins. The flanking 5′‐ and 3′‐UTRs contain RNA elements of relevance to this review, which are annotated. An equilibrium between “linear” and “cyclized” forms is depicted. Upon cyclization, the 5′‐SLB structure is eliminated and the terminal 3′ structure is no longer associated with SL's stem.
3.2. Structural RNA Is Enriched in the Viral UTRs
Several groups have contributed to the identification of conserved RNA sequences and structures that play roles in flaviviral replication and translation in addition to the aforementioned 5′–3′ CS.19, 36 Of particular relevance is the conserved 3′‐terminal stem‐loop structure, designated SL (Figure 2), and two long, conserved, repeat sequences termed CS2 and RCS2 that reside in dumbbell structures (A2 and A3 in Figure 2) in many flaviviruses.37 Later work predicted a 5′‐terminal “SLA” (stem‐loop A) structure, followed by the SLB structure that contains the start codon.21 It was observed that a sequence within SLB, termed the 5′‐UAR (upstream AUG region), was complementary to one in the 5′ region of SL's stem (Figure 2). Currently, at least two other sets of sequences are suspected of engaging in 5′–3′ interactions: the “5′–3′‐DAR” (downstream AUG region)29 and multiple short sequences within the capsid coding region that are complementary with sequences within the mid‐3′‐UTR.38
3.3. Where Is the Viral Promoter?
Since the generation of the negative strand begins at the (+) 3′‐terminus of the template, it would be natural to expect an interaction between viral RdRp and RNA elements in the (+) 3′‐UTR. In 2006, Filomatori et al.21 combined a number of RNA fragments from DENV UTRs with the purified recombinant RdRp subunit of NS5 in the context of an electrophoretic mobility shift assay (EMSA). Surprisingly, RdRp was found to interact with a 160 nt (+) 5′‐terminal input, but not with a 3′ input. In vitro polymerase assays yielded parallel results; a combination of RdRp and the (+) 3′‐UTR failed to generate (–) complementary products, but a (+) 5′ template did. If both the 5′ and 3′ inputs were present with RdRp, both (–) CS were generated as previously noted in a similar experiment.20 These observations gave rise to the notion that the 5′‐terminus acts as the promoter for (–) vRNA synthesis and cyclization places 5′‐bound NS5 in proximity to the (+) 3′‐terminus. Additional EMSA and RNA footprinting analyses apparently narrowed the site of interaction down to the 5′ SLA structure.39
In our recent work using the in vivo, albeit heterologous, yeast three‐hybrid (Y3H) assay to scan for DENV NS5 and vRNA interactions over the entire viral genome, the results were in opposition to the SLA‐promoter viewpoint, revealing that NS5 interacted two orders of magnitude more strongly with the 3′ SL than with 5′ SLA in the Y3H context.40 It is interesting that both DENV SLA and SL have the same sizes of apical loops, both of which contain ACAG sequences in their 5′ portions. In fact, the consensus CACAG sequence in the SL top loop (hereafter designated SL‐TL) has been recognized as particularly conserved among flaviviruses for more than 30 years:41 a study from 2005 shows that a variety of point mutations in SL‐TL completely disable replication, but not translation.42 In contrast to previous studies conducted by Gamarnik and colleagues, our EMSA confirmed the affinity of RdRp with 3′ SL and a sensitive in vitro alpha binding assay provided an apparent K d of ≈3 nm for the interaction between RdRp and SL‐TL.40 It should be noted that two recent studies in which affinities of DENV RdRp with a 5′‐UTR fragment containing SLA were measured in solution yielded K d values ranging from 53 nm to 142 nm.43, 44
In a model endorsing a 5′ promoter, cyclization is required to bring the 3′‐terminus in contact with the viral polymerase. However, a balance between cyclized and linear forms of flaviviral genomes has been hypothesized to play roles not relating to a promoter function: to regulate replication versus translation, to insure generation of full‐length RNA strands, to control the ratio of positive to negative strands, and to control the timing of encapsidation.19, 26, 32, 37 One study demonstrated that encapsidated DENV RNA exists in cyclized form, while virion‐extracted and refolded vRNA is linear.45 HCV is also assumed to cyclize,46, 47 and the promoter for negative‐strand synthesis is thought to reside at the 3′‐UTR.48 The idea of a 3′ promoter abolishes the necessity for genomic cyclization in order for NS5 to access the 3′‐terminus.
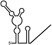
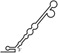
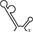
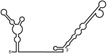
An observation supported by both bioinformatics23 and selective 2′ hydroxyl acylation analyzed by primer extension (SHAPE)49 suggests that the act of cyclization in flaviviruses alters the 3′‐terminal SL structure. Since a critical sequence for cyclization, the 3′‐UAR, overlaps with the basal 5′ region of the SL stem, a base‐pairing interaction with the 5′‐UAR in SLB results in SL stem shortening, leaving the viral 3′‐terminal region free from the stem. Clues to a role played by structural rearrangement of the 3′‐end of vRNA are found in earlier in vitro RdRp assays, where minor alterations to the 3′‐terminus produced large changes in assay output.38, 48 Also, Gamarnik's group performed a DENV RdRp assay with RNA templates that simply joined SLA to two variants of SL: in one, the 3′‐terminus was predicted to be engaged in a stem, but in the other, the terminus was expected to be unpaired.39 Though 5′–3′ interactions would not be expected with these constructs, only the construct with the unpaired terminus generated the full‐length product. Therefore, cyclization of the UTRs per se does not enable replication; rather, a free 3′‐terminus is critical. A summary of RdRp activity assays against various RNA inputs is provided in Table 1; note the obvious dependence of polymerase activity on the structure of the 3′‐terminus.
Table 1.
Summary of RdRp affinity and polymerase activity assays with various vRNA fragments
| RNAs | Poly(N) | SLA | SLA tail | SL | cySL | SLA–SL | SLA–cySL | cyUTRs | 5′ + 3′‐UTRs |
|---|---|---|---|---|---|---|---|---|---|
| 2D structures |

|
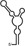
|
|
|
|
|
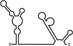
|
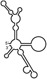
|
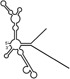
|
| Polymerase activity | Yes44 | No39 | Yes39 | No20, 21 | n.d. | No39 | Yes39 | Yes20, 21 | Yes (both 5′ and 3′ transcripts were observed)20, 21 |
| RdRp‐binding activity | No39 | Yes43 No39 | Yes39 | Yes40 No21 | Yes20, 40 | n.d. | n.d. | Yes40 | n.d. |
| Free 3′‐tail following the long stem | Yes | No | Yes | No | Yes | No | Yes | Yes | Yes |
n.d., not determined.
“cy” refers to structures as they would exist in cyclized form.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
3.4. Why Are There Two NS5‐Binding Sites?
While the ACAG motif in SL‐TL (top loop) exhibits strong conservation in all mosquito‐borne flaviviruses, the ACAG sequence within SLA is only conserved in DENV and ZIKV. In cases where the TL of 5′ SLA (SLA‐TL) has been mutated, however, it has been shown to be required for optimal replication.21, 50 SLA‐TL was also shown to be involved in 5′ cap methylation of (+) vRNA in WNV.51 Intriguingly, footprinting analysis revealed that the UAR cyclization sequence residing in the upstream SLB structure (Figure 2) became highly susceptible to RNase A digestion, thus existing in a single‐stranded form in the presence of RdRp.39 In contrast, SLA‐TL was protected from RNase PhyM in the presence of RdRp. These results suggested that RdRp binding to the SLA‐TL stimulated a conformational change of the downstream sequence.
The strong conservation of a 3′ terminal uracil (U) and penultimate cytosine (C) on both the positive and the negative flaviviral strands has been noticed in several works.52, 53 Interestingly, the conserved 3′ UUCU terminus is located 46 nt from the 5′‐end of SL‐TL, while the middle of the UUUUUU tract that straddles SLA and SLB is located 46 nt downstream from the same position in SLA‐TL, offering additional similarities between SLA and SL. It seems reasonable that NS5 could stimulate cyclization by disrupting SLB upon binding to SLA‐TL, exposing the 5′‐UAR. At the other end of the genome, cyclization triggers exposure of the 3′‐terminus, providing access to RdRp and initiates (–) vRNA synthesis.40 We propose that the dual NS5‐binding sites, SLA and SL, may act as sensors of NS5 concentration and that cyclization occurs only when sufficient quantities of NS5 have been translated. While an RdRp–SLA interaction may be important for replication, we favor the admittedly noncanonical view that the default “promoter” should be the 3′ SL structure.
3.5. Duplicated RNA Elements Play Roles in Host Switching
Repeat vRNA structures sharing strong sequence identity or multiple motifs seem to be a flavivirus theme. In addition to the aforementioned similarities between SLA and SL, the 3′‐UTR of mosquito‐borne flaviviruses contains near‐identical A2 and A3 dumbbells (Figure 2), as well as structurally similar SLI and SLII elements. These pairings are not commonly seen in flaviviruses with no known vector, making it clear that such duplication almost certainly relates to host‐switching.54, 55 With regard to the dumbbell pair, deletion of the DENV A2 sequence significantly reduced replication in mosquito cells, while the opposite effect was observed upon A3 deletion.54 When the experiment was performed in human cells, the trends held but were far less dramatic. This work also showed an accumulation of mutations within the A3 region upon multiple passages in mosquito C6/36 cells. These results were considered to suggest that one structure would be free to mutate to adapt to the host environment, while the other essential structure would remain constant. Relevance to cyclization was further implied as a mutation that eliminated a pseudoknot‐forming sequence within the A3 dumbbell, which interacts with the 3′ CS involved in 5′–3′ pairing, significantly increased viral replication in mosquito cells. Here, we would suggest that research might be furthered by a decreased focus on cyclization and increased focus on the possible protein factors that interact with the dumbbell structures. Sequestration of any host factor that has a strong influence on the immune response could be seen as the primary event and an alteration in cyclization being considered a secondary effect. It is possible that one duplicated structure binds mosquito factors, while the other binds the orthologous human factors. In such a case, repeated passaging in a single cell type would be expected to generate an abundance of mutations in one duplicated structure versus the other. Above, we suggest that the SLA/SL pair could act as sensors as NS5 concentration. It remains possible that other structural pairs could act in a similar fashion.
3.6. Extensive RNA Structure Is Evident within Coding Regions
The combination of modern deep sequencing and RNA probing tools (e.g., SHAPE and dimethyl sulfate [DMS] probing) has greatly increased the opportunities to study the vRNA structure. Not only can the entire genome be investigated, but work can be conducted in an in vivo context. Following examination of the HIV‐1,56 and HCV genomes using such an approach,57, 58 a comprehensive survey of the DENV‐2 genome was completed in 2018. As with the HIV and HCV works, a high degree of secondary and tertiary structures was uncovered even within coding regions.45 Twenty‐four RNA “elements” with high levels of predicted base pairing were revealed, 22 of them being found within coding regions. Several, though not all, cases in which synonymous mutations were introduced into DENV‐2 showed significant alterations in viral titers and/or vRNA levels in infected cells. One alteration, within the envelope coding region, had particularly potent effects. At the bioinformatic level, DENV‐2 sequence comparisons apparently revealed significant decreases in mutation rates in regions with predicted base pairing.
4. There Are Distinct Configurations of Genome‐Length vRNA
In 1967, Stollar et al.59 first investigated DENV RNA using the methods developed by Baltimore and other virology pioneers. General replication patterns noted in poliovirus60, 61 apparently held up in flavivirus: 1) generation of a dsRNA viral form shortly after infection that was termed the “replicative form” (RF), 2) high levels of (+) strand vRNA vs (–) strand vRNA, and 3) appearance of a heterogeneous, partially double‐stranded form that became known as the “replicative intermediate” (RI) (Figure 1). Later experiments involving JEV,62 SLEV,63 Kunjin,64 and WNV7 did not violate these patterns. Over the course of this work, it was found that radiolabeled nucleotide was rapidly incorporated into isolated dsRNA during replication, suggesting that the newest strands actually displaced somewhat older strands, in contrast to a model where dsRNA was only locally unwound. BVDV (Flaviviridae) was also shown to follow the above sequence of events, and RNase A resistance experiments suggested more than one nascent strand per RI.65 Another feature of flaviviral RNA was that, unlike alphaviruses, no distinct subgenomic RNAs capable of serving as viral mRNA could be detected in cells infected with flaviviruses—an observation that was important in necessitating the family division of Flaviviridae from Togaviridae.66
To date, no experiments have detected flaviviral (–) ssRNA. That is, the (–) vRNA exists only as a duplex with (+) vRNA. Using RNase digestion of WNV RNA, followed by 2D polyacrylamide gel electrophoresis analysis (“T1 fingerprinting”), Wengler et al.7 were only able to discern negative‐stranded vRNA within centrifuged dsRNA fractions, not in ssRNA fractions. However, this absence of viral (–) ssRNA is certainly not a general feature of Group IV viruses. It is, for example, established that poliovirus negative‐strand RNA can be found in a single‐stranded form, positive strands being found in vesicular structures not occupied by the negative strand.67 Some evidence even suggests that HCV may have a (–) ssRNA species.68 Conversely, it is thought that negative‐strand RNA viruses do not generate immunofluorescence‐detectable levels of dsRNA at all.69
Regarding subgenomic flaviviral RNAs (sfRNAs), though no evidence of coding‐capable sfRNAs was found, it will be of later interest to note that the early literature was not without reports of low‐molecular‐weight vRNA species with sedimentation coefficients that placed them under 100 kDa.7, 62 In the case of WNV, for example, two distinct species of (+) ssRNA were identified at 42 kDa and 65 kDa (or approximately 130 nt and 200 nt, respectively, assuming equal ratios of nucleotides), and hybridization experiments indicated positive polarity. A short vRNA that hybridized with probes representing the MVE 3′‐UTR was also found in the brains of infected mice.70 A recent wave of sfRNA research has focused on RNA products created by the degradation of the viral genome via the 5′–3′ exonuclease XRN1 until a vRNA structure halts further degradation.13 Such single‐stranded products are likely the same as those seen 40 years ago.71 At this point, we count five forms of genome‐scale flaviviral RNA: cyclized and linear (+) vRNA, dsRNA (the RF), heterogeneous (RI), and sfRNA.
5. Mechanisms Underlying the Generation of (+) Strand vRNA Remain to be Delineated
Very little research has been devoted to the means by which the viral polymerase recognizes and initiates (+) strand synthesis. Given that replication occurs within virus‐induced membranous vesicles derived from ER, dsRNA is hidden from host mechanisms that normally detect such species, meaning that NS5 may not have to discriminate between a large variety of RNAs in order to initiate vRNA synthesis. The only discrimination required would be between the two dsRNA termini, as every relevant study shows a large (10–100×) excess of positive strand to negative in Flaviviridae,65, 72, 73, 74, 75, 76 suggesting preferential recognition of one dsRNA termini versus the other. In addition to NS5 and the NS3 helicase, host factors apparently contribute to the process of (+) strand synthesis, as in vitro conversion of RF into RI was optimal in the presence of uninfected cell lysate in addition to recombinant NS5 and NS3.77 In our view, simple diffusion of NS5 to dsRNA termini is not appealing, particularly given the possibility that Flaviviridae polymerase is membrane‐anchored during the initiation of positive‐strand synthesis,75 as shown previously with poliovirus.78 Also, at least one study failed to reveal any NS5–dsRNA affinity.79 Therefore, we speculate that termini–termini interactions between the (+) strand portion of the dsRNA could be maintained, generating an RNA duplex with frayed ends. Obviously, more work is required to fully understand the molecular mechanisms that viruses utilize to exclusively produce (+) vRNA.
6. Active Switching from Translation to Replication Makes Sense
Having generated quantities of flaviviral protein and RNA, the virus is faced with the luxury of favoring translation over replication, or vice versa. It seems rational to expect that initial replication would not occur at the same time and location as translation, as the vRNA would be translated in the 5′ to 3′ direction, while negative‐strand growth would proceed in the opposite direction. Nevertheless, it could be questioned whether, probabilistically, such a ribosome–polymerase collision would be a practical concern. A number of lines of evidence suggested that viruses indeed have switching mechanisms that would minimize this conflict. Gamarnik's80 work with poliovirus replicons showed a near‐absence of vRNA synthesis in vitro shortly after the addition of the HeLa lysate; however, RNA synthesis could be induced via the simple addition of cycloheximide, an inhibitor of translation. 3CD, the poliovirus polymerase, which was previously shown to interact with a 5′ cloverleaf structure, was required for this switch to replication.81 Such a result makes sense as replication only proceeds when its components reach a critical concentration. Removal of the cloverleaf RNA structure dramatically reduced translation and, perhaps more interestingly, replication, even in the presence of adequate levels of 3CD. Moving to the flavivirus, Lo et al.,28 using WNV, and the laboratories of Gamarnik21 and Padmanabhan,82 using DENV, later showed via a replicon luciferase signal that an initial increase in translated products was followed by a decline. A new wave of translation was observed again ≈24 h after the introduction of the replicon. The downward‐sloping portion of this curve, as opposed to a continual increase in the translated products over time, would suggest a period where initial translation products are lost while replication is emphasized, followed by a new wave of translation when (+) vRNA levels reach suitable levels, i.e., an active switch from translation to replication. One HCV study also suggested that a translation/replication switch accompanied an increase in the core protein inhibiting translation.83 In addition, the combined presence of DDX6, an RNA helicase known to interact with the DENV 3′‐UTR,84 and mir‐122 at the 5′‐terminus of the HCV (+) strand, favors replication vs translation.85
Some evidence points to the cyclized form of vRNA favoring replication over translation. A variety of mutations within the DENV 3′‐UTR were shown to moderately decrease translation, as indicated via replicon luciferase signal, while alterations of the 5′ CS that weakened its interaction with the 3′ CS actually increased translation moderately.14 To complicate the picture, a double mutation that simply exchanged both CS sequences, retaining complementarity, reduced the translation signal by nearly 90%, suggesting a role for the CS sequences that extends beyond mere complementarity. Consistently, mutations or the addition of oligos that would weaken cyclization have the effect of lowering levels of replication.15, 23, 24, 25, 26, 27, 28, 29, 32, 33, 34, 86, 87
7. Do Short Derivatives of vRNA Have Functions?
Several forms of short, noncoding flaviviral RNA have been identified: defective interfering particles (DIPs), microRNA (miRNA)‐like sequences, and sfRNA. We must thus expand our collection of flaviviral RNA forms accordingly. Such RNA species likely correspond to the short RNAs found in early pulse/chase studies.7 DIPs are so named because they are incapable (defective) of generating progeny in the absence of viable virus and their presence has the effect of hindering viral propagation. Though the notion of DIPs has existed for more than 50 years, their in vivo relevance is questionable, as they are typically procured upon multiple passages of infected cells. In flavivirus, one study of dengue DIPs utilized sequencing to elucidate their nature;88 not surprisingly, the particles, ranging from 290 to 1030 nt, always contained the complete 5′‐UTR and most or all of the 3′‐UTR. We note that the 5′–3′ joining points of the sequences are significantly enriched for ACA and ACAG sequences, hinting at a role of NS5 in the initial formation of these DIPs.40
The most credible report of an miRNA‐like product generated from the flaviviral genome would concern WNV.89 In this case, the terminal stem‐loop structure (SL) is apparently processed into a 21 nt product that binds to GATA4 mRNA in a mosquito cell line and, in a noncanonical fashion, upregulates GATA4 expression, which in turn enhances viral replication. Unlike the topic of flaviviral miRNAs, that of sfRNAs has received a great deal of high‐impact attention over the last five years. sfRNAs are created by the degradation of the viral genome via the 5′–3′ exonuclease XRN1. This process is apparently halted in the 5′ region of the 3′‐UTR via a unique vRNA structure that blocks further degradation.13 While both the aforementioned RNAs do contain the terminal SL structure with which NS5 binds, no direct connections between these RNAs and NS5 interactions have been drawn to date. Several works have actually linked the presence of sfRNA to pathogenicity.13, 90 At this point, the mechanism underlying this pathogenicity remains unknown.
8. What Is Known about vRNA Packaging?
While the assembly of the virion may be described according to localization within the cell, factors whose depletion or enrichment may alter the process, and the means by which vRNA is selectively packaged within capsids, we shall concern ourselves with the latter. Given the broad range of RNAs within the host, as well as documented cases of packaging signals within vRNA (e.g., coronavirus91), it would be rational to engage in a search for repeated or conserved elements within flaviviral RNA that might specifically interact with the capsid protein. Such elements, however, have not been discovered. Amino acid sequence conservation across flaviviral capsids has been described as “low,”92 though general affinity for RNA is not in dispute.
The requirement for specific interactions between capsid and vRNA assumes an environment replete with host factors that would compete for vRNA or capsid. A more exclusive environment, however, might abrogate this need. Indeed, as mentioned above, flaviviruses are thought to carve out their own vesicle‐enclosed replication environments. In this case, we might thus look for evidence of coupling between replication and encapsidation. Such linkage has been suggested, and experiments show that only replicating vRNA (versus vRNA whose polymerase has been disabled) is packaged.93 Given the difficulty in identifying a packaging signal within flaviviral RNA, we would favor a model in which replicated (+) vRNA is directly presented to the awaiting capsid and structural proteins.
9. vRNA Interactions with Host Proteins May Confound Models Derived from In Vitro Studies
We have now examined flaviviral RNA from translation to packaging. It must be noted that most studies on vRNA structures utilize in vitro protocols. However, RNA structures derived from in vivo probing do not necessarily resemble those from in vitro and in silico studies.94 The helicase activity of ribosomes causes transient alterations in the structure,95 and a multitude of RNA‐binding proteins (RBPs) may stabilize structures that would not be preferred in a setting with minimalized components.96 Conversely, some thermostable structures have been found to exist largely in denatured states within cells.97 To complicate matters further, in vivo structures of cellular RNAs have been shown to vary over time, following virus infection.95 The potential for in‐depth analysis of in vivo vRNA structure is seen in the work of Li et al.,98 examining Zika vRNA with two different probing methods. The results showed a “moderate” correlation between in vivo and in vitro structures, in which predicted long‐range 5′–3′ interactions in vRNA were “generally consistent” with the probing results. However, an important exception was also noted: the absence of interactions between the 5′ and 3′ CS elements.
While the in vivo study of vRNA structures is in its infancy, a number of studies have attempted to globally characterize proteins that bind vRNA. The image of naked vRNA diffusing through a cell whose volume is 30% occupied by macromolecules must therefore be dispelled here.99, 100 As suggested by the aforementioned case of XRN1, the ability of the virus to utilize, evade, and sequester host factors via vRNA structures and sequences may be critical for viral propagation. While a relative paucity of vRNA‐interacting factors might be expected in the protected environment of the viral double‐membrane structures where dsRNA appears to reside, a number of experiments have demonstrated roles for host protein–vRNA interactions in translation and replication.10, 101 Flaviviruses may also sequester factors that ordinarily heighten translation of RNAs involved in an antiviral response; as one example, 3′‐UTR interactions with G3BP1, G3BP2, and CAPRIN1 may serve to dampen the interferon response.102 vRNA‐binding proteins that either increase or decrease vRNA levels in cells have been identified via protein–RNA crosslinking, followed by vRNA‐specific pulldown, identification of relevant proteins via mass spectrometry, and knockdown of proteins of interest.103 In one case, knockdown of host‐factor YBX1 resulted in an increase in vRNA inside cells despite a decrease in released viral particles, suggesting deficiencies in viral packaging. We would also point out the presence of numerous RNA helicases capable of remodeling vRNA in the handful of studies in which mass spectrometry has been utilized to examine flaviviral RNA‐interacting proteins.103, 104, 105
10. Conclusions and Outlook
We hope this review enhances a general image of a subtle, sensitive, and highly regulated flaviviral RNA. As seen in recent RNA probing studies, structural elements extend into coding regions representing 95% of the viral genome, possibly illustrating the degree to which vRNA structure and function remains to be explored. Facilitated by next‐generation sequencing (NGS), mass spectrometry, and a toolkit of probing methods, research on flaviviral RNA structure and function is entering a new era. In addition to generating entirely novel pictures of vRNA structure, such technology provides opportunities to re‐examine early studies with great precision, confirming, contradicting, and adding nuance to canonical viewpoints. It is now possible to examine the complete genome for short‐ and long‐range interactions under any variety of conditions and time points without bias. Such examinations need not be limited to purely single‐stranded forms of the genome; new insights may arise by isolating the heterogeneous (RI) form of flaviviral RNA. One may find that in vitro and in silico methods that attempted to resolve RNA structure may have in vivo alternatives that account for a vRNA that could be largely occupied by host factors. We look forward to the delineation of critical interactions involving these structures, given their obvious relevance to antiviral drug development.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
The authors thank Dr. David Michael Payne for his useful comments on the manuscript. This work was supported by Grants‐in‐Aids for Research from Mahidol University, Thailand, and by grants (RSA6080066 and IRG5780009) from the Thailand Research Fund (TRF) to S.C. K.H. is supported by Rachadaphiseksompot Fund for Postdoctoral Fellowship, Chulalongkorn University, Thailand.
Contributor Information
Trairak Pisitkun, Email: pisitkut@nhlbi.nih.gov.
Sarin Chimnaronk, Email: sarin.chi@mahidol.ac.th.
References
References
- 1. Adams M. J., Lefkowitz E. J., King A. M. Q., Harrach B., Harrison R. L., Knowles N. J., Kropinski A. M., Krupovic M., Kuhn J. H., Mushegian A. R., Nibert M., Sabanadzovic S., Sanfaçon H., Siddell S. G., Simmonds P., Varsani A., Zerbini F. M., Gorbalenya A. E., Davison A. J., Arch. Virol. 2017, 162, 2505. [DOI] [PubMed] [Google Scholar]
- 2. Nowak T., Färber P. M., Wengler G., Wengler G., Virology 1989, 169, 365. [DOI] [PubMed] [Google Scholar]
- 3. Bhatt S., Gething P. W., Brady O. J., Messina J. P., Farlow A. W., Moyes C. L., Drake J. M., Brownstein J. S., Hoen A. G., Sankoh O., Myers M. F., George D. B., Jaenisch T., Wint G. R. W., Simmons C. P., Scott T. W., Farrar J. J., Hay S. I., Nature 2013, 496, 504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Streit J. A., Yang M., Cavanaugh J. E., Polgreen P. M., Emerg. Infect. Dis. 2011, 17, 914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Slenning B. D., Vet. Pathol. 2010, 47, 28. [DOI] [PubMed] [Google Scholar]
- 6. Gubler D. J., Nat. Med. 2004, 10, 129. [DOI] [PubMed] [Google Scholar]
- 7. Wengler G., Wengler G., Gross H. J., Virology 1978, 89, 423. [DOI] [PubMed] [Google Scholar]
- 8. Welsch S., Miller S., Romero‐Brey I., Merz A., Bleck C. K. E., Walther P., Fuller S. D., Antony C., Krijnse‐Locker J., Bartenschlager R., Cell Host Microbe 2009, 5, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clyde K., Harris E., J. Virol. 2006, 80, 2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Polacek C., Friebe P., Harris E., J. Gen. Virol. 2009, 90, 687. [DOI] [PubMed] [Google Scholar]
- 11. Edgil D., Polacek C., Harris E., J. Virol. 2006, 80, 2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moon S. L., Blackinton J. G., Anderson J. R., Dozier M. K., Dodd B. J. T., Keene J. D., Wilusz C. J., Bradrick S. S., Wilusz J., PLoS Pathog. 2015, 11, e1004708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pijlman G. P., Funk A., Kondratieva N., Leung J., Torres S., van der Aa L., Liu W. J., Palmenberg A. C., Shi P. Y., Hall R. A., Khromykh A. A., Cell Host Microbe 2008, 4, 579. [DOI] [PubMed] [Google Scholar]
- 14. Chiu W. W., Kinney R. M., Dreher T. W., J. Virol. 2005, 79, 8303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Holden K. L., Stein D. A., Pierson T. C., Ahmed A. A., Clyde K., Iversen P. L., Harris E., Virology 2006, 344, 439. [DOI] [PubMed] [Google Scholar]
- 16. Blackwell J. L., Brinton M. A., J. Virol. 1997, 71, 6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reid D. W., Campos R. K., Child J. R., Zheng T., Chan K. W. K., Bradrick S. S., Vasudevan S. G., Garcia‐Blanco M. A., Nicchitta C. V., J. Virol. 2018, 92, e01766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hsu M., Kung H. J., Davidson N., Cold Spring Harb Symp. Quant. Biol. 1974, 38, 943. [DOI] [PubMed] [Google Scholar]
- 19. Hahn C. S., Hahn Y. S., Rice C. M., Lee E., Dalgarno L., Strauss E. G., Strauss J. H., J. Mol. Biol. 1987, 198, 33. [DOI] [PubMed] [Google Scholar]
- 20. You S., Padmanabhan R., J. Biol. Chem. 1999, 274, 33714. [DOI] [PubMed] [Google Scholar]
- 21. Filomatori C. V., Lodeiro M. F., Alvarez D. E., Samsa M. M., Pietrasanta L., Gamarnik A. V., Genes Dev. 2006, 20, 2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lott W. B., Doran M. R., Trends Biochem. Sci. 2013, 38, 350. [DOI] [PubMed] [Google Scholar]
- 23. Alvarez D. E., Filomatori C. V., Gamarnik A. V., Virology 2008, 375, 223. [DOI] [PubMed] [Google Scholar]
- 24. Alvarez D. E., Lodeiro M. F., Luduena S. J., Pietrasanta L. I., Gamarnik A. V., J. Virol. 2005, 79, 6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Corver J., Lenches E., Smith K., Robison R. A., Sando T., Strauss E. G., Strauss J. H., J. Virol. 2003, 77, 2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Khromykh A. A., Meka H., Guyatt K. J., Westaway E. G., J. Virol. 2001, 75, 6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kofler R. M., Hoenninger V. M., Thurner C., Mandl C. W., J. Virol. 2006, 80, 4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lo M. K., Tilgner M., Bernard K. A., Shi P. Y., J. Virol. 2003, 77, 10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Friebe P., Harris E., J. Virol. 2010, 84, 6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Friebe P., Peña J., Pohl M. O. F., Harris E., Virology 2012, 422, 346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Friebe P., Shi P. Y., Harris E., J. Virol. 2011, 85, 1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Villordo S. M., Alvarez D. E., Gamarnik A. V., RNA 2010, 16, 2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang B., Dong H., Stein D. A., Iversen P. L., Shi P. Y., Virology 2008, 373, 1. [DOI] [PubMed] [Google Scholar]
- 34. Liu Z.‐Y., Li X.‐F., Jiang T, Deng Y.‐Q., Ye Q., Zhao H., Yu J.‐Y., Qin C.‐F., eLife 2016, 5, e17636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wells S. E., Hillner P. E., Vale R. D., Sachs A. B., Mol. Cell 1998, 2, 135. [DOI] [PubMed] [Google Scholar]
- 36. Grange T., Bouloy M., Girard M., FEBS Lett. 1985, 188, 159. [DOI] [PubMed] [Google Scholar]
- 37. Proutski V., Gould E. A., Holmes E. C., Nucleic Acids Res. 1997, 25, 1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. de Borba L., Villordo S. M., Iglesias N. G., Filomatori C. V., Gebhard L. G., Gamarnik A. V., J. Virol. 2015, 89, 3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Filomatori C. V., Iglesias N. G., Villordo S. M., Alvarez D. E., Gamarnik A. V., J. Biol. Chem. 2011, 286, 6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hodge K., Tunghirun C., Kamkaew M., Limjindaporn T., Yenchitsomanus P., Chimnaronk S., J. Biol. Chem. 2016, 291, 17437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wengler G., Castle E., J. Gen. Virol. 1986, 67, 1183. [DOI] [PubMed] [Google Scholar]
- 42. Tilgner M., Deas T. S., Shi P. Y., Virology 2005, 331, 375. [DOI] [PubMed] [Google Scholar]
- 43. Bujalowski P. J., Bujalowski W., Choi K. H., J. Virol. 2017, 91, e00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kamkaew M., Chimnaronk S., Protein Expression Purif. 2015, 112, 43. [DOI] [PubMed] [Google Scholar]
- 45. Dethoff E. A., Boerneke M. A., Gokhale N. S., Muhire B. M., Martin D. P., Sacco M. T., McFadden M. J., Weinstein J. B., Messer W. B., Horner S. M., Weeks K. M., Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 11513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thurner C., Witwer C., Hofacker I. L., Stadler P. F., J. Gen. Virol. 2004, 85, 1113. [DOI] [PubMed] [Google Scholar]
- 47. Yi M., Lemon S. M., J. Virol. 2003, 77, 3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Oh J. W., Sheu G. T., Lai M. M. C., J. Biol. Chem. 2000, 275, 17710. [DOI] [PubMed] [Google Scholar]
- 49. Sztuba‐Solinska J., Teramoto T., Rausch J. W., Shapiro B. A., Padmanabhan R., Le Grice S. F. J., Nucleic Acids Res. 2013, 41, 5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lodeiro M. F., Filomatori C. V., Gamarnik A. V., J. Virol. 2009, 83, 993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang B., Dong H., Zhou Y., Shi P. Y., J. Virol. 2008, 82, 7047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Khromykh A. A., Kondratieva N., Sgro J. Y., Palmenberg A., Westaway E. G., J. Virol. 2003, 77, 10623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wengler G., Wengler G., Virology 1981, 113, 544. [DOI] [PubMed] [Google Scholar]
- 54. de Borba L., Villordo S. M., Marsico F. L., Carballeda J. M., Filomatori C. V., Gebhard L. G., Pallarés H. M., Lequime S., Lambrechts L., Sánchez vargas I., Blair C. D., Gamarnik A. V., mBio 2019, 10, e02506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Villordo S. M., Filomatori C. V., Sánchez‐Vargas I., Blair C. D., Gamarnik A. V., PLoS Pathog. 2015, 11, e1004604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Siegfried N. A., Busan S., Rice G. M., Nelson J. A. E., Weeks K. M., Nat. Methods 2014, 11, 959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mauger D. M., Golden M., Yamane D., Williford S., Lemon S. M., Martin D. P., Weeks K. M., Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pirakitikulr N., Kohlway A., Lindenbach B. D., Pyle A. M., Mol. Cell 2016, 62, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stollar V., Schlesinger R. W., Stevens T. M., Virology 1967, 33, 650. [DOI] [PubMed] [Google Scholar]
- 60. Baltimore D., Becker Y., Darnell J. E., Science 1964, 143, 1034. [DOI] [PubMed] [Google Scholar]
- 61. Baltimore D., Proc. Natl. Acad. Sci. U. S. A. 1964, 51, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zebovitz E., Leong J. K., Doughty S. C., Infect. Immun. 1974, 10, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Trent D. W., Swensen C. C., Qureshi A. A., J. Virol. 1969, 3, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Boulton R. W., Westaway E. G., Arch. Virol. 1977, 55, 201. [DOI] [PubMed] [Google Scholar]
- 65. Gong Y., Trowbridge R., Macnaughton T. B., Westaway E. G., Shannon A. D., Gowans E. J., J. Gen. Virol. 1996, 77, 2729. [DOI] [PubMed] [Google Scholar]
- 66. Westaway E.G., Brinton M. A., Gaidamovich Y., Horzinek M. C., Igarashi A., Kääriäinen L., Lvov O. K., Porterfield J. S., Russell P. K., Trent D. W., Intervirology 1985, 24, 183. [DOI] [PubMed] [Google Scholar]
- 67. Bolten R., Egger D., Gosert R., Schaub G., Landmann L., Bienz K., J. Virol. 1998, 72, 8578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shindo M., Di Bisceglie A. M., Akatsuka T., Fong T.‐L., Scaglione L., Donets M., Hoofnagle J. H., Feinstone S. M., Proc Natl Acad Sci U S A 1994, 91, 8719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Weber F., Wagner V., Rasmussen S. B., Hartmann R., Paludan S. R., J. Virol. 2006, 80, 5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Urosevic N., van Maanen M., Mansfield J. P., Mackenzie J. S., Shellam G. R., J. Gen. Virol. 1997, 78, 23. [DOI] [PubMed] [Google Scholar]
- 71. Chapman E. G., Costantino D. A., Rabe J. L., Moon S. L., Wilusz J., Nix J. C., Kieft J. S., Science 2014, 344, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Uchil P. D., Satchidanandam V., Virology 2003, 307, 358. [DOI] [PubMed] [Google Scholar]
- 73. Chen C. J., Kuo M. D., Chien L. J., Hsu S. L., Wang Y. M., Lin J. H., J. Virol. 1997, 71, 3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Fong T. L., Shindo M., Feinstone S. M., Hoofnagle J. H., Di Bisceglie A. M., J. Clin. Invest. 1991, 88, 1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tomassini J. E., Boots E., Gan L., Graham P., Munshi V., Wolanski B., Fay J. F., Getty K., LaFemina R., Virology 2003, 313, 274. [DOI] [PubMed] [Google Scholar]
- 76. Diamond M. S., Zachariah M., Harris E., Virology 2002, 304, 211. [DOI] [PubMed] [Google Scholar]
- 77. Raviprakash K., Porter K. R., Hayes C. G., Sinha M., Am. J. Trop. Med. Hyg. 1998, 58, 90. [DOI] [PubMed] [Google Scholar]
- 78. Egger D., Pasamontes L., Bolten R., Boyko V., Bienz K., J. Virol. 1996, 70, 8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Szymanski M. R., Jezewska M. J., Bujalowski P. J., Bussetta C., Ye M., Choi K. H., Bujalowski W., J. Biol. Chem. 2011, 286, 33095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gamarnik A. V., Andino R., Genes Dev. 1998, 12, 2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Andino R., Rieckhof G. E., Baltimore D., Cell 1990, 63, 369. [DOI] [PubMed] [Google Scholar]
- 82. Manzano M., Reichert E. D., Polo S., Falgout B., Kasprzak W., Shapiro B. A., Padmanabhan R., J. Biol. Chem. 2011, 286, 22521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zhang J., Yamada O., Yoshida H., Iwai T., Araki H., Virology 2002, 293, 141. [DOI] [PubMed] [Google Scholar]
- 84. Ward A. M., Bidet K., Yinglin A., Ler S. G., Hogue K., Blackstock W., Gunaratne J., Garcia‐Blanco M. A., RNA Biol. 2011, 8, 1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Biegel J. M., Henderson E., Cox E. M., Bonenfant G., Netzband R., Kahn S., Eager R., Pager C. T., Virology 2017, 507, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Deas T. S., Binduga‐Gajewska I., Tilgner M., Ren P., Stein D. A., Moulton H. M., Iversen P. L., Kauffman E. B., Kramer L. D., Shi P. Y., J. Virol. 2005, 79, 4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bredenbeek P. J., Kooi E. A., Lindenbach B., Huijkman N., Rice C. M., Spaan W. J., J. Gen. Virol. 2003, 84, 1261. [DOI] [PubMed] [Google Scholar]
- 88. Li D., Lott W. B., Lowry K., Jones A., Thu H. M., Aaskov J., PLoS One 2011, 6, e19447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hussain M., Torres S., Schnettler E., Funk A., Grundhoff A., Pijlman G. P., Khromykh A. A., Asgari S., Nucleic Acids Res. 2012, 40, 2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chapman E. G., Moon S. L., Wilusz J., Kieft J. S., eLife 2014, 3, e01892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Narayanan K., Chen C. J., Maeda J., Makino S., J. Virol. 2003, 77, 2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Byk L. A., Gamarnik A. V., Annu. Rev. Virol. 2016, 3, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Khromykh A. A., Varnavski A. N., Sedlak P. L., Westaway E. G., J. Virol. 2001, 75, 4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kwok C. K., Tang Y., Assmann S. M., Bevilacqua P. C., Trends Biochem. Sci. 2015, 40, 221. [DOI] [PubMed] [Google Scholar]
- 95. Mizrahi O., Nachshon A., Shitrit A., Gelbart I. A., Dobesova M., Brenner S., Kahana C., Stern‐Ginossar N., Mol. Cell 2018, 72, 862. [DOI] [PubMed] [Google Scholar]
- 96. Tyrrell J., McGinnis J. L., Weeks K. M., Pielak G. J., Biochemistry 2013, 52, 8777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Rouskin S., Zubradt M., Washietl S., Kellis M., Weissman J. S., Nature 2014, 505, 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Li P., Wei Y., Mei M., Tang L., Sun L., Huang W., Zhou J., Zou C., Zhang S., Qin C. F., Jiang T., Dai J., Tan X., Zhang Q. C., Cell Host Microbe 2018, 24, 875 e5 [DOI] [PubMed] [Google Scholar]
- 99. Zimmerman S. B., Trach S. O., J. Mol. Biol. 1991, 222, 599. [DOI] [PubMed] [Google Scholar]
- 100. Milo R., Bioessays 2013, 35, 1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Nagy P. D., Pogany J., Nat. Rev. Microbiol. 2011, 10, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Bidet K. , Dadlani D., Garcia‐Blanco M. A., PLoS Pathog. 2014, 10, e1004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Phillips S. L., Soderblom E. J., Bradrick S. S., Garcia‐Blanco M. A., mBio 2016, 7, e01865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Manokaran G., Finol E., Wang C., Gunaratne J., Bahl J., Ong E. Z., Tan H. C., Sessions O. M., Ward A. M., Gubler D. J., Harris E., Garcia‐Blanco M. A., Ooi E. E., Science 2015, 350, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Viktorovskaya O. V., Greco T. M., Cristea I. M., Thompson S. R., PLoS Neglected Trop. Dis. 2016, 10, e0004921. [DOI] [PMC free article] [PubMed] [Google Scholar]