

Abstract
Lung diseases belong to the major causes of death worldwide. Recent innovative methodological developments now allow more and more for the use of primary human tissue and cells to model such diseases. In this regard, the review covers bronchial air‐liquid interface cultures, precision cut lung slices as well as ex vivo cultures of explanted peripheral lung tissue and de‐/re‐cellularization models. Diseases such as asthma or infections are discussed and an outlook on further areas for development is given. Overall, the progress in ex vivo modeling by using primary human material could make translational research activities more efficient by simultaneously fostering the mechanistic understanding of human lung diseases while reducing animal usage in biomedical research.
Keywords: 3D models, alveolar, bronchial, human lung tissue, infections
Illustration of the different requirements and achievements (first and second level) for different types of 3D human pulmonary models (third level), which then can be applied in different methodological modalities and read‐out systems (fourth level) to give insight in toxicological, functional, or disease related aspects (fifth level).

1. Introduction
The human lung comprises various essential body functions, among which vital gas exchange is most central. The trachea conducts inhaled air into the lungs through dichotomic branching into tubular bronchi. They divide further into smaller bronchioles, finally becoming microscopic clusters of air sacs, called alveoli. Ciliated epithelia and mucosal glands build the inner lining of the conducting airways. These structures maintain epithelial moisture and ensure the mucociliary clearance of the airways by secreting mucus and maintaining an epithelial cell lining fluid.1 Approximately 480 million alveoli form the respiratory surface of an adult human lung,2 which is mainly composed of flat alveolar type‐I cells (AEC‐I). Together with endothelial capillaries located at the basolateral site of AEC‐I, these cells form the approximately 2 µm–600 nm (sometimes only 200 nm) thin barrier allowing for gas diffusion. The AEC‐I are flanked by cuboidal alveolar type‐II cells (AEC‐II), which are indispensable for the regulation of alveolar homeostasis by producing and secreting surfactant,3 other bioactive mediators, and contributing to lung tissue repair.4
During every breath, this extensive and intricate air–blood barrier is exposed to mechanical stress5 and inhaled air (around 10.000 L day−1) containing particles ranging from pollutants to allergens or infectious agents. The activation level of the resident alveolar immune cells such as macrophages6 and dendritic cells, in concert with the lung epithelium,7 decide if and how such inhaled agents induce a systemic or just a local immune response (“keep calm”). In cases of systemic activation, recruitment of further immune cells including polymorphonuclear granulocytes,8 monocytes,9 or T lymphocytes10 contribute to combat the perturbing agents. Notably, certain circumstances, such as severe pneumonia, may lead to inadequate or overwhelming activation of such resident lung cells triggering a deleterious pro‐inflammatory cascade significantly contributing to the development of life‐threatening acute lung injury.11 As there are major physiological differences between most animal lungs and human lungs, and animal models are unable to completely recapitulate human disease, ex vivo three‐dimensional (3D) human lung models are required and helpful for understanding the underlying molecular and cellular events and mechanisms of such diseases in a functional, spatial, and timely manner.
1.1. Why Investigate Living Human Lung Tissue and Primary Human Cells of the Lung?
There is a high social and medical need for a better understanding of human lung diseases. This is because lower respiratory tract infections, chronic obstructive pulmonary disease (COPD), lung cancer, and tuberculosis belong to the ten most common causes of death worldwide.12, 13 A study analyzing the global burden of pneumonia for 2010 estimated about 120 million episodes occurring in children younger than 5 years. Of those, around 14 million progressed to severe pneumonia, causing about 1.3 million deaths.14 Furthermore, a study estimating the global burden of COPD for the same year identified more than 230 million cases among urban dwellers, and 153.7 million among rural dwellers worldwide, thereby providing an overview of the high prevalence of lung diseases in the population.15 Additionally, diseases such as asthma also affect millions of people worldwide and show likewise an increasing prevalence. Although mortality due to asthma has decreased over the last decades, no curative therapy is available and personal constraints in daily life activities persist.16, 17 Lung cancer is still the leading cause of cancer‐related deaths world‐wide.18 Models, which more accurately reflect human lung cancer could help aid in our understanding of the deranged signaling networks and help identify and validate new potential therapies. Taken together, there is a high global burden of lung diseases ranging from the economic to the individual level19 and a prerequisite to understand these diseases and to identify novel treatment options is the creation and validation of more complex models with multiple readout options that better mimic physiologic tissues and accurately reflect the clinical situation.
One could argue that the use of immortalized cell lines and animal models with rodents should reveal the essential information to understand and treat human lung disease. While there is no doubt that these techniques and approaches have helped to make major contributions to the field. The scientific community is increasingly recognizing that these models and their outcomes suffer from serious limitations. It is estimated that over 80% of the potential therapeutics deemed to be safe and effective in animals fail in humans due to non‐rigorous study design, which raises ethical concerns with regard to the sacrificed animals as well as the patients, who receive ineffective agents with potential side‐effects.20 Moreover, lung anatomy, cellular composition, or molecular responses in man significantly differ from widely used animal models, such as rodents.21, 22, 23 For instance, chronic bronchitis and COPD are characterized both by excessive mucus production. However, in contrast to humans, bronchial glands of mice and rats anatomically localize only to the proximal trachea making it difficult to reproduce this particular disease aspect.24 In parallel to these challenges, current attempts to simulate the effect of cigarette smoke, air pollution, and lung cancer induction in animal models are limited in multiple ways by such basic animal‐human differences.22, 25, 26, 27, 28 Therefore, a vigorous debate has arisen in the last decade regarding the predictive value of mouse models in inflammatory diseases, across many diseases and organs.29, 30
In that context, another intriguing example for the need to use primary human lung samples are emerging zoonotic infections such as Severe Acute Respiratory Syndrome (SARS), Middle East Respiratory Syndrome (MERS), or new influenza A virus (IAV) pandemics. Several animal models are naturally not permissive to these severe infections or need virus adaption to the animal host by serial passaging, thereby affecting viral pathogenicity. Animal models often need transgenic expression of human receptors (e.g., human angiotensin I converting enzyme 2 (ACE2) for SARS coronavirus (CoV), or human dipeptidyl peptidase 4 (DPP4) for MERS CoV), or simply do not reflect the clinical course of the disease in humans.31, 32 Furthermore, an inherent characteristic of a zoonosis is its specific occurrence between a distinct animal species and the human host. Species specific factors, but mostly unknown factors from both, the pathogen and the host, determine the zoonotic axis. Therefore, the investigation of fundamental factors defining how a pathogen crosses the species barrier must include material and infection models from these “zoonotic partner species” itself, meaning in the local context such as the human lung.33 Although such complex human lung tissue models suffer from physiological limitations, it is obviously purposeful to make efforts toward analyzing the fundamentals of lung biology in the original tissue of interest. This tissue can be used to mimic the various diseases and even to elaborately analyze diseased tissue biopsies, which can serve as models themselves. In addition, the currently established human in vitro models require further improvement. For example, culture medium‐submerged bronchial lung epithelia cells lack cilia and hardly form the tight junctions typically expressed in vivo and observed in air–liquid interface cultures.34 There is thus a need to recapitulate the complex 3D structure and matrix of the lung tissue with a suitable cell mixture35 to establish an appropriate intercellular network.36 Besides those integrated models, human clonal or primary cells are useful for dissecting fundamental aspects of the biology of a single lung cell population in vitro, which is indispensable to complement the understanding of species specific signaling pathways.
In conclusion, the high medical need together with fundamental methodological considerations substantiate the necessity for development of human‐based disease models for acute and chronic lung diseases. For this purpose, recently developed techniques now allow for innovative and more meaningful investigation of 3D human lung tissue. This review will outline some examples of the models, applications developed to date, and will discuss further directions of improvement.
2. Existing Co‐Cultures and 3D Models For Pulmonary Research
Some major aspects of advantages as well as disadvantages of the different models presented below are summarized in Table 1.
Table 1.
Advantages and disadvantages of 3D models
| Model | Advantages | Disadvantages | Ref. |
|---|---|---|---|
| Air‐liquid‐interface | Self‐made: | Self‐made: | 42, 43, 44, 51, 52, 53, 54, 55, 56 |
| • Long‐term cultivation (weeks to months) | • Lack of complexity | ||
| • Opportunity for direct application of substances | Commercially available: | ||
| • Perfusion possible | • Lack of modifiability | ||
| • Great variety of functional studies | |||
| • Simulating breathing motions of the lung | |||
| Commercially available: | |||
| • High data reproducibility | |||
| • Low batch‐to‐batch variabilities | |||
| • Well characterized | |||
| • Can be provided mimicking several pathologies | |||
| • Long‐term cultivation (up to 12 months) | |||
| Spheroids | • Simple | • High shear force | 45, 47, 48 |
| • Allows co‐culture with different cell types | • Long term culture difficult (hours to days) | ||
| Lung tissue explants | • Model cellular and molecular interplay | • Short term cultivation (up to 96 h) | 71, 72, 74 |
| • Characterization of resident innate immune events | • No immune cell recruitment | ||
| • Live cell imaging possible | • No systemic perfusion | ||
| • Genetic modifications become possible | • No ventilation | ||
| • Genetic modifications still difficult | |||
| Precision cut lung slices | • Long‐term cultivation (up to 1 week) | • Flushing with low melting point agarose necessary | 75, 76, 136, 158 |
| • Retain cellular and structural organization of the lung | • Others as above in lung tissue explants | ||
| • Generation of PCLS from diseased tissue is challenging | |||
| Bronchial rings | • Direct investigation of bronchial physiological responses‐, e.g., contraction‐pharmacological controllability | • Short term cultivation (hours to days) | 77, 78, 79, 80, 81, 83, 84, 85, 86 |
| • Technically cultivation circuits necessary | |||
| Ex vivo perfused and ventilated human lungs | • Investigation of lung edema formation, oxygenation capacity, vascular reactivity, bacterial infection, and stem cell therapy | • Limited available | 87, 88, 89, 90, 91, 92, 93, 94 |
| • Technically elaborated | |||
| • Cultivated so far for only several hours | |||
| Scaffold based models | • Maintain characteristics of their respective disease pathologies | • Cells are seeded in two or three dimensions | 95, 96, 97, 98, 99, 100, 105 |
| • Physiologic seeding of cells into either the airway or vascular compartments with an artificial pleura | • Initial cell seeding is stochastic | ||
| • Able to recapitulate the heterogeneity of human disease | • Limited access to nutrients and oxygen in the inner portions of the scaffold | ||
| • Cultivation for up to 1 month |
2.1. In Vitro Models Originating From Primary Cells and Immortalized Cell Lines
The use of human lung tissue shown in this review was approved by the ethics committee of the Charite Universitätsmedizin Berlin (projects: EA2/050/08 and EA2/023/07); written informed consent from all patients was obtained. Over the last decades, tissue engineering approaches have made tremendous progress. Several in vitro models for studying inhalation toxicity or diseases have been established, which aim to improve our understanding of (patho‐)physiological processes and to provide novel and more reliant experimental systems for pharmacological and toxicological studies. Currently, in vitro lung models of all important segments of the respiratory system are available, starting from the nasal cavity and trachea down to the proximal and distal airways.37 Cell culture models of human lung epithelium are based either on primary cells including normal human (trachea) bronchial epithelial cells (NH(T)BEC)), AEC, induced pluripotent stem cell (iPSC) derived epithelial cells,38, 39 or are generated using cell lines. iPSC derived epithelial cell organoids from patients with lung diseases, such as cystic fibrosis, or cells manipulated in vitro through gene editing have been recently shown to recapitulate functions known to be deranged in human lung disease. iPSC derived lung bud organoids have also been recently used for studying respiratory syncytial viral infection.39 These models thus provide an opportunity to identify new treatments or to further understand the underlying biology of human disease. The most commonly used lung epithelial cell lines are Calu‐3, H441, 16HBE14o‐, and A549. These cell lines offer several advantages over primary cells such as almost unlimited cell source and passaging. However, they suffer from distinct limitations such as the lack of mucus production for 16HBE14o‐or absent tight junction formation for A549.40 To induce cell differentiation, cells are typically cultured on biocompatible scaffolds or matrices to mimic the natural environment. The matrices can consist of natural (e.g., collagen, gelatin, elastin, alginate, silk, Matrigel®) or synthetic compounds (e.g., polyglycolic acid, polylactic acid, polyethylene glycol, polyvinyl alcohol, polyacrylic acid).41 An important step for improving the physiological relevance of these models is the culture of lung epithelial cells at the air–liquid‐interface (ALI), since increased oxygen and air exposure play crucial roles for the development of a well‐differentiated barrier.42 Bronchial epithelial cells cultured under ALI conditions develop a three‐dimensional, multi‐cellular epithelium comprised of basal cells as well as ciliated surface cells and goblet cells, whereas submerged cultures express substantially fewer ciliated cells.43 Additionally, ALI cultures offer the opportunity for direct application of aerosols or solid particles onto semi‐dry cell surfaces, more closely resemble the in vivo situation and can be kept in culture for several month.44
In addition to the use of primary and immortalized cells for modeling normal cell biology, tumor models using human cells have become important tools for cancer research. Their complexity is dependent on the objectives and research questions. Tumor models have been developed to provide insight into, e.g., tumor growth, invasion, matrix remodeling, or drug delivery. Many lung tumor studies use spheroid models for therapeutic screening where clusters of cells undergo self‐assembly to form viable, 3D tumor‐like structures. Spheroids can be generated using low attachment U‐bottom plates,45 extracellular matrices (MatrigelTM),46 rotatory cell culture systems or the hanging drop method.47 Hanging drops can be used to produce 3D mono‐ and co‐culture spheroids for a better reflection of in vivo conditions of cancer cells to investigate, e.g., tumor‐stroma interactions.48 The culture time depends on the cell type and the experimental question. Small spheroids have been used for drug testing and large to recapitulate oxygen gradients with hypoxic regions or proliferation gradients. More complex models of lung cancer and tumors have been established to serve as tools to identify new drug targets. Three‐dimensional decellularized tissue matrix derived from rodent lungs have been recellularized with human lung cancer cell lines to provide a perfusable tumor model.49 Alternatively, a 3D collagen gel based model has been described which contains human lung adenocarcinoma cells, human lung fibroblast cells, and macrophages.50
Several research groups have established 3D airway models for pulmonary research consisting of undifferentiated NHTB/biliary epithelial cells (BEC).51, 52 Besides the use of in‐house‐models, well characterized, fully differentiated and standardized 3D human airway models are commercially available offering the major advantage of higher data reproducibility due to lower batch‐to‐batch variabilities. These models are used for a wide range of applications including safety and risk assessment and research on, e.g., new anti‐viral compounds, drug delivery systems, or common lung diseases such as inflammation and fibrosis. EpiAirway® (MatTek Corp., MA, USA) and MucilAir™ (Epithelix Sàrl, Suisse) are composed of normal NHTB/BEC from non‐smokers, but epithelium mimicking several pathologies like asthma, COPD, or cystic fibrosis can also be obtained. OncoCilAir™ (Epithelix Sàrl, Suisse) is a 3D human lung cancer model combining functional reconstituted human airway epithelium, human lung fibroblasts, and lung adenocarcinoma cells which can be used for drug efficacy testing, toxicity effects, analyzing off‐target‐effects, and tumor recurrence. A major advantage of these models is their ability to be cultured for long‐term. For example, OncoCilAir™ and EpiAirway® have a useful lifetime for up to 3 months, whereas MucilAir™ can be cultivated up to 12 months, allowing long‐term and repeat toxicity and efficacy studies. The main disadvantages of commercial models are the lack of complexity and ability to be customized for particular applications. In this regard, in‐house models offer more flexibility, particularly with regard to cell selection or cell manipulation like genetic modifications via gene knockdown or knockout.53 Moreover, endothelial cells can be implemented to mimic the alveolar‐capillary barrier54, 55 or the models can be co‐cultured with dendritic cells and/or macrophages to generate immunocompetent tissues allowing more detailed studies of lung (patho‐)physiology.35, 56, 57 The implementation of immune cells, however, is a challenging approach and requires extensive research efforts to develop reliable and predictive models closely resembling the in vivo situation.
Besides the above‐mentioned modification of including more cell populations in those models, a further area of improvement is the application of physical forces and motion to the culture systems. Benam et al.58 presented a system in which human lung bronchial epithelial cells (from healthy individuals or asthma and COPD patients), differentiated in an ALI culture and grown on an extracellular matrix (ECM) coated dimethylsiloxane membrane, were integrated in a microfluidic system with cultured human endothelial cells on the lower side. The endothelial channel is perfused by culture medium. The culture of patient‐derived airway cells in combination with the application of stimulating agents via the airflow or the perfusion channel (so via the vascular route) allowed for a great variety of functional studies including, e.g., the recruitment of immune cells from the bloodstream into the airways. The same group used this experimental setup to model the alveolar region of the lung.59, 60, 61 By integrating a flexible membrane and a two side hollow chamber they induce stretching of both the epithelial cells grown on the upper side and the endothelial cells grown on the bottom side of the membrane, thereby simulating breathing motions of the lung. Notably, in this model, human alveolar cells were exposed to an airstream and thus cultured in ALI conditions (alias “lung‐on‐chip”). These systems are now commercialized (Emulate, Inc., Boston, MA, USA) and thus may be available in the future for a range of applications. To this end, varieties of human 3D models of different complexity based on in vitro culture of lung cells are now available.
2.2. Ex Vivo Models
Beside the development of various in vitro 3D models mimicking the upper and lower human respiratory tract, the ex vivo culturing of human lung tissue enables a broad spectrum of investigation in the dynamic context of the native organ‐specific 3D structure (especially in the alveolar compartment) benefiting from controlled experimental conditions as well as the representation of natural patient heterogeneity. Early studies used direct post‐surgical snap frozen or homogenized human lung tissue specimens for the analytical determination of molecular markers,62 antibiotic metabolism,63 or pathogen detection.64 However, these experimental approaches are excluded from ex vivo lung modeling discussed here, due to the lack of ex vivo cultivation and stimulation. The majority of tissue explants studied in the literature are based on tissue pieces obtained from thoracic surgery, mostly due to different types of lung carcinoma. In addition, complete lobectomy or pneumonectomy explants as well as whole lungs unsuitable for transplantation are used.
Due to limited experience and techniques available for cell culture, initial studies investigated short‐term human lung tissue cultures (for several hours) in saline media, e.g., the identification of mediator release such as colony stimulating activities.65 Although limited by the short time of tissue culture possible in the 1970s, the group of Austen et al. were already able to investigate the role of histamine in anaphylaxis in a series of elegant studies, thereby indicating the power of this experimental approach.66, 67, 68, 69, 70
Currently, following pathological examination, the tissue is typically transferred into the research units within a few hours, where manual preparation into smaller pieces differing in size (1 mm3–10 cm3) or weight (0.5 μg–2 g) can occur. The culture time for lung tissue explants is up to 96 h.71, 72, 73, 74 For generation of precision cut lung slices (PCLS) the obtained tissue is gently inflated with lung slice medium, composed of cultivation medium, certain supplements, and low melting point agarose; the latter allows production of lung sections of defined thickness (100–500 μm) using a microtome (e.g., Krumdieck tissue slicer or a vibratome).75, 76 Depending on size or weight, generated specimens are cultivated in tissue culture plates varying in size or tissue culture flasks, with the appropriate amount of classical cell culture medium (such as RPMI‐1640, CMRL‐166, or MEM) for up to one week. Culture media and its supplementation differs considerably between research groups; and varying amounts of antibiotics, antifungal drugs, glutamine, fetal calf serum, or bovine serum albumin are used in the studies. Typically, tissue is washed for several times in culture media followed by an overnight incubation before stimulation with, e.g., drugs, pathogens, or other stimuli of interest.
Some laboratories are particularly interested in the responses of larger airways and thus explore bronchial rings, prepared from lung tissue specimens, by applying similar culture conditions as for peripheral lung parenchyma (e.g.,77, 78, 79, 80). However, in most studies investigators used shorter periods of bronchial ring cultivation (e.g., 1 day) than in studies testing peripheral alveolar lung tissue. They cultured the human bronchial rings in special organ chambers coupled to mechanical transducers81 or electric field stimulation82 allowing for the investigation of “classical” physiological responses of the bronchi, such as bronchoconstriction. Consequently, many studies focused on, e.g., pharmacological interventions or the identification of bronchial contractility‐mediating substances (e.g.,81, 82, 83, 84, 85, 86).
A small number of groups investigated isolated, ex vivo perfused, and ventilated human lungs or lobes.87, 88, 89, 90, 91, 92, 93, 94 In these models, lung tissue was perfused via the cannulated pulmonary artery with cell free perfusion media, with cell‐containing (e.g., diluted whole human blood preparations) solutions,90, 91, 92 or solutions for lung preservation during transport before transplantation.87, 93, 94 The lungs were ventilated via the natural airways for up to 24 h. Such limited available and technically elaborated models allow for the investigation of lung edema formation,89, 90, 91, 92 oxygenation capacity, vascular reactivity, bacterial infection,91 and innovative therapeutic approaches such as stem cell therapies.90, 91, 92 These examples demonstrate that there are multiple, scalable approaches of different complexity to use living human lung tissue in its original tissue architecture for experimental research and testing of new clinical interventions.
2.3. De‐Re‐Cellularization Models
Recently, the use of acellular lung tissue (also referred to as decellularized) has emerged as a novel in vitro/ex vivo model system to study ECM interactions in the context of normal and diseased repair and regeneration. Acellular patient‐derived lung scaffolds were shown to retain unique, individual protein compositions as well as disease‐specific differences, as most comprehensively assessed by mass spectrometry proteomic analysis.95, 96, 97 In addition to normal lungs, lungs derived from scleroderma, idiopathic pulmonary fibrosis (IPF), and COPD patients have all been decellularized to date and have been shown to maintain characteristics (composition and structure) of their respective disease pathologies for up to 1 month.95, 96, 98
Acellular lung scaffolds can be generated from entire lungs, individual lobes, or small‐dissected segments and modifications might be necessary for decellularizing diseased scaffolds.95, 97, 99, 100, 101 For both entire lungs and individual lobes, decellularization has been achieved via perfusion of decellularization solutions such as detergents, hypo/hypertonic solutions, and DNase through either the vasculature or both the vasculature and airways. Once decellularized, scaffolds can be recellularized with either primary stem and progenitor cells,96, 97, 102, 103 induced pluripotent stem cells (iPS),104 or immortalized cell lines.
Since the recellularization of entire lobes is costly, there are generally three techniques that are used to re‐introduce cells into acellular lung scaffolds. The most common method is the generation of thin slices of decellularized tissue, followed by direct application of cells.95, 98, 99 This method is arguably the simplest to perform and allows for high throughput studies from a single acellular scaffold. However, it suffers from the fact that cells are seeded in two dimensions and the initial cell seeding is thought to be stochastic (e.g., cells randomly attach to the upper surface of the slice with no documented preference for adhering to the physiologic compartment corresponding to the cells in vivo location). Alternatively, cell suspensions can be injected into dissected decellularized tissue segments (∼3 cm3), and then further processed into thin slices.105 If segments are not further sliced, cells in the inner portions of the scaffold will have limited access to nutrients and oxygen. The third high throughput method of recellularization allows for physiologic seeding of cells into either the airway or vascular compartments with an artificial pleura.96, 97, 103
Primary human fibroblasts seeded on acellular lung scaffolds derived from patients with IPF were found to obtain a myofibroblast phenotype, as assessed by α‐smooth muscle actin (α‐SMA) expression.95 In a follow up study, it was determined that the matrix, in which cells were seeded, played a more significant role in determining cell fate than the source of the cells (i.e., normal versus IPF).106 Immortalized cells such as human fibroblasts (MRC5) have also been seeded onto normal acellular human scaffolds and stimulated with recombinant CHI3L (a prototypic chitinase like protein) to induce α‐SMA expression and a contractile phenotype.107 In addition to their usage in IPF research, acellular lung scaffolds derived from COPD patients have also been used. Cells seeded onto COPD scaffolds were unable to survive as long as they did on scaffolds derived from normal patients.96 These cells were all grown under conditions known to stimulate proliferation in two dimensions on tissue culture plastic. This indicates that either the matrix itself or a component in the matrix/scaffold suppressed proliferation or enhanced cell death. In both instances, the findings in vitro match known hallmarks of human disease. However, it is important to note that mass spectrometry proteomics consistently detects a large number of non‐ECM components in acellular human lung scaffolds from both normal and diseased origin, many of which are growth factors known to be involved in disease onset and pathogenesis.95 Therefore, acellular scaffolds and results from these experiments should not only be viewed as models of cell‐ECM interactions but should be interpreted as being models of the extracellular environment of normal or diseased lungs, including matrix sequestered growth factors. Their ability to recapitulate the heterogeneity of human disease in an in vivo like environment makes acellular human lung scaffolds a powerful in vitro tool for studying human disease.
3. Modeled Diseases
Most of the above‐mentioned human‐based ex vivo models are characterized by a relatively short culture period. Researchers thus concentrate their use of these systems on the investigation of rapid (seconds to hours) or short‐term (within days) responses. In asthma research, PCLS are used extensively for the investigation of new drugs or interventions interfering with bronchial constriction.108, 109, 110, 111, 112 Noteworthy, carefully done studies point to remarkable differences between mice and man113 with respect to the regulation of bronchial constriction.114, 115 Although very costly, work‐intensive, and of enormous importance for the estimation of the potential translational impact of studies using mice as a model in asthma research, scientific journals often do not adequately honor such work using human material or models alone.
Many studies use ALI cultures116, 117, 118, 119, 120, 121 and explanted human lung tissue72, 74, 122, 123, 124, 125 for the investigation of lung tissue infection and inflammation. For example, our own work has assessed viral replication, cellular tropism of viruses as well as the induction of mediator release by the host tissue. The use of original human lung tissue is of particular importance for the estimation of the actual zoonotic potential of an emerging virus.72, 122, 125 Furthermore, in some cases such as MERS coronavirus, no meaningful animal model is available31, 32 or (as for pneumococci) humans are the sole natural host of a pathogen.126, 127 To this end, original patient‐derived viral and bacterial strains and thereon based mutants are indispensible models to assess the impact of different wild‐type strains and specific virulence factors on pathogen replication and tissue infection as well as its resulting affection.71, 72, 122, 125 New pharmacological interventions, ranging from antivirals to antibiotics or host tissue‐based manipulations have been tested as well.
Recurrent infections are considered as an important disease‐accelerating factor in COPD, and thus researchers have compared the effect of infection on ALI cultures derived from healthy humans or COPD patients.128, 129, 130, 131 The alteration of lung epithelial function in PCLS or ALI cultures by toxic inhaled pollutants such as cigarette smoke or diesel exhaust is also an expanding area of research on original human material in the COPD field.130, 132, 133 Studies range from the observation of acute effects to the investigation of epithelial dedifferentiation and mesenchymal transition.134
These studies relate to those that focus on the investigation of the toxic potential of inhaled substances in human lung tissue per se.135, 136 Again, the comparison of results obtained in original human material with results in animal models is of great importance to estimate the validity of data from the respective models for the human population. Fewer studies based on human material and not on animals have investigated lung cancer137 and in vitro models of solid tumors vary in complexity. Furthermore, in silico models and computer‐based systems to analyze CT scans to improve cancer diagnosis138, 139 may be used with 3D models in the future for drug efficacy testing or to mimic, e.g., tumor invasion related processes or matrix remodeling. These have only been investigated in a handful of studies using original human tissue or patient tumor–derived cells140 thus far.
Overall, these briefly reviewed examples already indicate that these models are of great value for human‐related lung research.
4. Current Limitations of the Ex Vivo/In Vitro Models
Despite several advantages, which have been highlighted before, the proposed ex vivo/in vitro approaches have clear limitations, which need to be considered carefully. For example, reproducing experiments in ex vivo models is not always straightforward, the access to lung tissue ex vivo is highly limited and the tissue sections are mostly obtained from individuals with some other underlying disease, which raises the challenge of studying potential alterations at baseline due to primary diseases.
Major drawbacks of 3D models are their poor standardization, which hampers inter‐laboratory reproducibility as well as the lack of complexity. However, while challenging to standardize, the different approaches may also yield complementary information. Numerous publications report on novel 3D tissue models, each of them relying on differing protocols, of which the most are not published with sufficient information to completely and independently reproduce results. Moreover, varying cells/cell lines, scaffolds, and cell culture media are used which further complicates the inter‐laboratory transfer. Another major obstacle is the lack of complexity of the organ models. Although the current state of the art includes two or even more cells in a model, the complexity of a living tissue in vivo cannot yet be completely emulated ex vivo. Similarly, vascularization, systemic distribution, immune responses as well as tissue regeneration are not yet possible in all models. Considering these aspects, animal models remain useful as a complementary approach, although their predictive power is limited by distinct interspecies‐related differences and their use raises more and more ethical concerns.
Notably, preclinical research exclusively relies on model systems, all of which have advantages and limitations. Considering these carefully and choosing complementary approaches could help to improve the predictive power of preclinical research and significantly reduce the number of animal testing at the same time.
5. Further Developments – Where Should We Go From Here?
Although these models based on original human material have already considerably contributed to furthering our understanding of human lung biology and disease, it is clear that several aspects need improvement in the near future.
Most of these models are based on the experience of single laboratories and hamper from standardization. Although this applies to many areas in biomedical research, it is of particular importance when the undertaken investigations endeavor to be of clinical and translational relevance. Regulatory authorities ask for clear descriptions of robust models and standard operation procedures to make such models acceptable beyond the use in basic science. In addition to this, the usability of a specific model compared to alternative (animal) models needs to be investigated in detail. Once accepted by the competent authorities and the pharmaceutical industry, these experimental approaches will considerably contribute to speed up translational processes and to reduce the use of animals in biomedical science. Therefore, third‐party donors and scientific journals need to be encouraged by the scientific community to support such methodological improvements and standardization projects.
Beyond standardization, there are specific scientific challenges. For example, ways to extend the culture time of explanted lung tissue cultures and PCLS are needed to allow for the investigation of longer lasting processes in the human lung. To this end, the implementation of lung explant cultures into microfluidic models141, 142, 143, 144 might improve tissue nutrition and culture duration. Microfluidic systems are also instrumental to investigate the interaction of the lung with other organs (e.g., gut‐lung axis, gut‐liver‐axis, human‐on‐a‐chip145) and to induce physical forces (“breathing lung on chip”141, 143). Of particular importance is the addition of an – at least‐minimal functional immune system in the microfluidic systems.
One of the biggest advantages of these models is accompanied by a great challenge: researchers are faced with the problem to gain cell‐specific mechanistic answers out of a 3D structure built up from different cell types over time. One key method is therefore the further development of high‐end live tissue microscopy techniques. Unfortunately, classical 2‐photon microscopy has no significant advantage in living lung tissues compared to confocal imaging.146 This is due to the strong light scattering of the chambered alveolar system, which limits both techniques. Next, lung tissue has very strong auto‐fluorescence hampering specific fluorescence signal separation. Therefore, such methods must be combined with spectral microscopy,147 and developed further with the fast (lattice) light sheet approach.148, 149 To show the advantage of spectral imaging we demonstrate two examples, where we have illustrated that spectral microscopy and linear unmixing provides the possibility to overcome the high auto‐fluorescence of the tissue and allows even for subcellular resolution of the complex cellular composition of the human alveolus. This kind of imaging techniques will drastically improve the methodological potential to visualize different cellular and structural aspects in living (lung) tissue and 3D models in general (Figure 1). Innovative live cell labeling combined with the use of viral transduction systems for gene editing will allow for mechanistic analysis of such lung tissue models in the future.150 The rapid development of stem cell based technologies151, 152, 153; decellularization procedures and tissue printing154, 155 make the building of human‐lung related tissue pieces for use in basic science more realistic within the next years.
Figure 1.
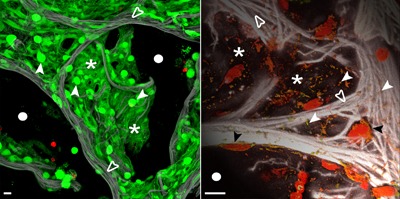
High‐end spectral confocal microscopy of living human lung tissue to illustrate the complex cellular composition of the human alveolus and the methodological potential. The tissue in the left panel was loaded with live/dead stain (calcein, green/life; ethidium bromide, red/dead). Green stained cells constitute the intact alveolar wall, bright ones are AEC‐II (white arrow heads), whereas dim ones reflect flat AEC‐I (asterisk). A tight intertwined network of elastic fibers (collagen/elastin, gray) serves as scaffold for the AEC (which is highly auto‐fluorescent) and the hidden capillary system (open arrowheads). The dark black areas indicate the air space of the alveoli (white circles). The right panel shows that live tissue microscopy can reach a resolution at the organelle level. Mitochondria stained for their DNA (Syto82, red) and membrane potential (MitoTrackerOrange, green) are depicted in orange (white arrow heads) demonstrating their distribution in the alveolar septae (asterisk). The strong elastic scaffold (gray, open arrow heads) and nuclear DNA of AEC (Syto82, red) shape the alveolar wall, black areas show the transition into the air space (white circles). All images have been acquired using a LSM 780 spectral microscope (37 °C, 5% CO2), Objective LCI 40xW C‐Apochromat NA/1.2 (Carl Zeiss, Jena, Germany). Spectral confocal imaging and linear unmixing of fluorescence dyes and tissue auto‐fluorescence was performed for z‐stacks up to 50 μm tissue depth. Display adjustment for clear visualization of structures was performed. Left image is a maximum intensity projection and right panel a 3D surface rendering. Bar 10 μm.
Overall, although complex developmental steps need to be overcome, there is already a fascinating outlook for the future of human pulmonary 3D models in experimental and translational research, which we have summarized in a multi‐level matrix (Figure 2). This matrix illustrates the different requirements for different types of 3D human pulmonary models. It also gives an overview over techniques which come into focus and which need to be improved to adequately analyze such models. It is one central goal to make the human lung “stand alone” models more integrative to allow for more physiologic models, which are on par or exceed the classical and widely accepted in vivo animal models. Such efforts combined with high quality cryo‐preservation and culture techniques of lung tissue156, 157 will substantially increase the expansion of those methods into the scientific community and the pharmaceutical industry. An inter‐laboratory exchange of experiences and a detailed publication of protocols is a first step to improve future work with 3D models.
Figure 2.
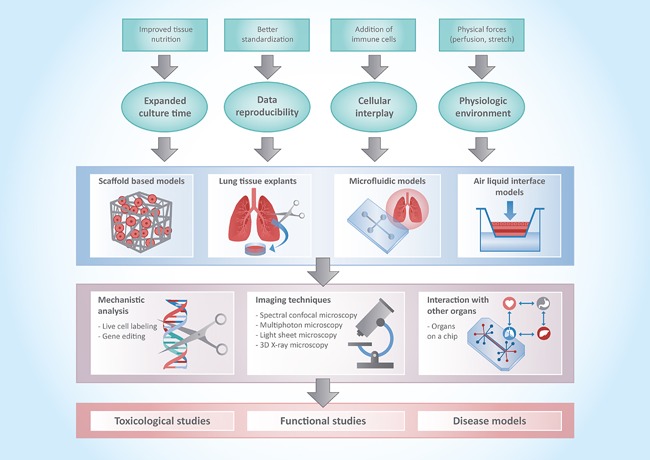
Illustration of the different requirements and achievements (first and second level) for different types of 3D human pulmonary models (third level), which then can be applied in different methodological modalities and read‐out systems (fourth level) to give insight in toxicological, functional, or disease related aspects (fifth level).
Abbreviations
3D, 3‐dimensional; AEC‐I, Alveolar epithelial type I cells; AEC‐II, alveolar epithelial type II cells; COPD, chronic obstructive pulmonary disease; SARS, Severe Acute Respiratory Syndrome; MERS, Middle East Respiratory Syndrome; ACE2, Angiotensin I converting enzyme 2; DPP4, Dipeptidyl peptidase 4; IAV, influenza A virus; CoV, coronavirus; NHBEC, normal human bronchial epithelial cells; NHTBC, normal human tracheobronchial cells; AEC, alveolar epithelial cells; ALI, air‐liquid‐interface; BEC, biliary epithelial cells; ECM, extracellular matrix; PCLS, precision cut lung slices; IPF, idiopathic pulmonary fibrosis; iPS, induced pluripotent stem cells; α‐SMA, α‐smooth muscle actin; CT, Computed Tomography.
Acknowledgements
This study was supported by the Transregional Collaborative Research Center SFB‐TR84 of the Deutsche Forschungsgemeinschaft (grants B6 and TF1 to A.C. Hocke and S. Hippenstiel, Z1a to A.C. Hocke, and B1 to N. Suttorp). We thank all the human material donors and the clinical partners for their generous support making human material‐based research possible. The authors thank the current and former members of our laboratory for their contributions. The authors apologize to authors whose relevant contributions could not be cited because of space limitations. Illustration of human 3D models (Figure 2) was carried out by Domino‐Illustration, Berlin Germany.
Conflict of Interest
The authors declare no financial or commercial conflict of interest.
Biographies
Katja Zscheppang studied Biotechnology at the University of Applied Sciences in Senftenberg and did her PhD at the Hannover Medical School. After a post‐doctoral position at the Tufts Medical Center in Boston and the OncoRay – National Center for Radiation Research in Oncology in Dresden, she joined 2014 the Molecular Imaging of Immunoregulation group at the Charité Berlin. Her research focusses on inflammatory and immunological analysis of the cellular and molecular host‐pathogen interactions in the human alveolar compartment.

Andreas Hocke studied Human Medicine at the University of Gießen and specialized during his PhD in light microscopy and pathogen‐host interaction of the lung. During his post‐doctoral time at the Charité – Universitätsmedizin Berlin/Dept. of Infectious and Respiratory Diseases he established a new model of living human lung tissue, which was awarded in 2011 from the Berlin government for innovative alternatives in animal testing. In 2016, he became full professor of the group “Molecular Imaging of Immunoregulation,” which dedicates its research for molecular and functional high‐end microscopy in human lung immunology and pneumonia.

References
- 1. Fahy J. V., Dickey B. F., N. Engl. J. Med. 2010, 363, 2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ochs M., Nyengaard J. R., Jung A., Knudsen L., Voigt M., Wahlers T., Richter J., Gundersen H. J., Am. J. Respir. Crit. Care Med. 2004, 169, 120. [DOI] [PubMed] [Google Scholar]
- 3. Whitsett J. A., Wert S. E., Weaver T. E., Annu. Rev. Med. 2010, 61, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guillot L., Nathan N., Tabary O., Thouvenin G., Le Rouzic P., Corvol H., Amselem S., Clement A., Int. J. Biochem. Cell Biol. 2013, 45, 2568. [DOI] [PubMed] [Google Scholar]
- 5. Roan E., Waters C. M., Am. J. Physiol. 2011, 301, L625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Byrne A. J., Mathie S. A., Gregory L. G., Lloyd C. M., Thorax 2015, 70, 1189. [DOI] [PubMed] [Google Scholar]
- 7. Hippenstiel S., Opitz B., Schmeck B., Suttorp N., Respir. Res. 2006, 7, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hoenderdos K., Condliffe A., Am. J. Respir. Cell Mol. Biol. 2013, 48, 531. [DOI] [PubMed] [Google Scholar]
- 9. Morales‐Nebreda L., Misharin A. V., Perlman H., Budinger G. R. S., Eur. Respir. Rev. 2015, 24, 505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen K., Kolls J. K., Annu. Rev. Immunol. 2013, 31, 605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Matthay M. A., Ware L. B., Zimmerman G. A., J. Clin. Invest. 2012, 122, 2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Global Burden of Disease Study, C., Lancet 2015, 386, 743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mokdad A. H., Forouzanfar M. H., Daoud F., Mokdad A. A., El Bcheraoui C., Moradi‐Lakeh M., Kyu H., Barber R., Wagner J., Cercy K., Kravitz H., Coggeshall M., Chew A., O'Rourke K., Steiner C., Tuffaha M., Charara R., Al‐Ghamdi E., Adi Y., Afifi R., Alahmadi H., AlBuhairan F., Allen N., AlMazroa M., Al‐Nehmi A., AlRayess Z., Arora M., Azzopardi P., Barroso C., Basulaiman M., Bhutta Z., Bonell C., Breinbauer C., Degenhardt L., Denno D., Fang J., Fatusi A., Feigl A., Kakuma R., Karam N., Kennedy E., Khoja T., Maalouf F., Obermeyer C., Mattoo A., McGovern T., Memish Z., Mensah G., Patel V., Petroni S., Reavley N., Zertuche D., Saeedi M., Santelli J., Sawyer S., Ssewamala F., Taiwo K., Tantawy M., Viner R., Waldfogel J., Zuñiga M., Naghavi M., Wang H., Vos T., Lopez A., Al Rabeeah A., Patton G., Murray C., Lancet 2016, 387, 2383. [DOI] [PubMed] [Google Scholar]
- 14. Walker C. L., Rudan I., Liu L., Nair H., Theodoratou E., Bhutta Z. A., O'Brien K. L., Campbell H., Black R. E., Lancet 2013, 381, 1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Adeloye D., Chua S., Lee C., Basquill C., Papana A., Theodoratou E., Nair H., Gasevic D., Sridhar D., Campbell H., Chan K., Sheikh A., Rudan I., J. Global Health 2015, 5, 020415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Beasley R., Semprini A., Mitchell E. A., Lancet 2015, 386, 1075. [DOI] [PubMed] [Google Scholar]
- 17. Martinez F. D., Vercelli D., Lancet 2013, 382, 1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ferlay J., Shin H. R., Bray F., Forman D., Mathers C., Parkin D. l., Int. J. Cancer 2010, 127, 2893. [DOI] [PubMed] [Google Scholar]
- 19. Ehteshami‐Afshar S., FitzGerald J. M., Doyle‐Waters M. M., Sadatsafavi M., Int. J. Tuberc. Lung Dis. 2016, 20, 11. [DOI] [PubMed] [Google Scholar]
- 20. Perrin S., Nature 2014, 507, 423. [DOI] [PubMed] [Google Scholar]
- 21. Boers J., Ambergen A., Thunnissen F. J. M., Am. J. Respir. Crit. Care Med. 1999, 159, 1585. [DOI] [PubMed] [Google Scholar]
- 22. Churg A., Sin D. D., Wright J. L., Am. J. Respir. Cell Mol. Biol. 2011, 45, 1111. [DOI] [PubMed] [Google Scholar]
- 23. Irvin C. G., Bates J. H. T., Respir. Res. 2003, 4, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Churg A., Cosio M., Wright J. L., Am. J. Physiol. 2008, 294, L612. [DOI] [PubMed] [Google Scholar]
- 25. Coggins C. R. E., Inhal. Toxicol. 2002, 14, 991. [DOI] [PubMed] [Google Scholar]
- 26. Coggins C. R. E., Inhal. Toxicol. 2010, 22, 974. [DOI] [PubMed] [Google Scholar]
- 27. Kellar A., Egan C., Morris D., BioMed Res. Int. 2015, 2015, 621324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leberl M., Kratzer A., Taraseviciene‐Stewart L., Front. Physiol. 2013, 4, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Seok J., Warren H. S., Cuenca A. G., Mindrinos M. N., Baker H. V., Xu W., Richards D. R., McDonald‐Smith G. P., Gao H., Hennessy L., Finnerty C. C., Lopez C. M., Honari S., Moore E. E., Minei J. P., Cuschieri J., Bankey P. E., Johnson J. L., Sperry J., Nathens A. B., Billiar T. R., West M. A., Jeschke M. G., Klein M. B., Gamelli R. L., Gibran N. S., Brownstein B. H., Miller‐Graziano C., Calvano S. E., Mason P. H., Cobb J. P., Rahme L. G., Lowry S. F., Maier R. V., Moldawer L. L., Herndon D. N., Davis R. W., Xiao W., Tompkins R. G., Proc. Natl. Acad. Sci. USA 2013, 110, 3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Takao K., Miyakawa T., Proc. Natl. Acad. Sci. USA 2015, 112, 1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gretebeck L. M., Subbarao K., Curr. Opin. Virol. 2015, 13, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sutton T. C., Subbarao K., Virology 2015, 479–480, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bean A. G., Baker M. L., Stewart C. R., Cowled C., Deffrasnes C., Wang L. F., Lowenthal J. W., Nat. Rev. Immunol. 2013, 13, 851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cao X., Lin H., Muskhelishvili L., Latendresse J., Richter P., Heflich R. H., Respir. Res. 2015, 16, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harrington H., Cato P., Salazar F., Wilkinson M., Knox A., Haycock J. W., Rose F., Aylott J. W., Ghaemmaghami A. M., Mol. Pharm. 2014, 11, 2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bhowmick R., Gappa‐Fahlenkamp H., Lung 2016, 194, 419. [DOI] [PubMed] [Google Scholar]
- 37. BéruBé K., Aufderheide M., Breheny D, Clothier R, Combes R, Duffin R, Forbes B, Gaça M, Gray A, Hall I, Kelly M, Lethem M, Liebsch M, Merolla L, Morin JP, Seagrave J, Swartz MA, Tetley T. D., Umachandran M., Altern. Lab. Anim. 2009, 37, 89. [PubMed] [Google Scholar]
- 38. Chen Y.‐W., Huang S. X., de Carvalho A. L. R. T., Ho S.‐H., Islam M. N., Volpi S., Notarangelo L. D., Ciancanelli M., Casanova J. L., Bhattacharya J., Liang A. F., Palermo L. M., Porotto M., Moscona A., Snoeck H. W, Nat. Cell Biol. 2017, 19, 542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McCauley K. B., Hawkins F., Serra M., Thomas D. C., Jacob A., Kotton D. N., Cell Stem Cell 2017, 20, 844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Braakhuis H. M., Kloet S. K., Kezic S., Kuper F., Park M. V. D. Z., Bellmann S., van der Zande M., Le Gac S., Krystek P., Peters R. J. B., Rietjens I. M. C. M., Bouwmeester H., Arch. Toxicol. 2015, 89, 1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee J., Cuddihy M. J., Kotov N. A., Tissue Eng. Part B Rev. 2008, 14, 61. [DOI] [PubMed] [Google Scholar]
- 42. Adler K. B., Holden‐Stauffer W. J., Repine J. E., J. Clin. Invest. 1990, 85, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sakagami M., Adv. Drug Deliv. Rev. 2006, 58, 1030. [DOI] [PubMed] [Google Scholar]
- 44. BéruBé K., Prytherch Z., Job C., Hughes T., Toxicology 2010, 278, 311. [DOI] [PubMed] [Google Scholar]
- 45. Ekert J. E., Johnson K., Strake B., Pardinas J., Jarantow S., Perkinson R., Colter D. C., PLoS ONE 2014, 9, e92248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Storch K., Dickreuter E., Artati A., Adamski J., Cordes N., PLoS ONE 2016, 11, e0167931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zanoni M., Piccinini F., Arienti C., Zamagni A., Santi S., Polico R., Bevilacqua A., Tesei A., Sci. Rep. 2016, 6, 19103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Amann A., Zwierzina M., Gamerith G., Bitsche M., Huber J. M., Vogel G. F., Blumer M., Koeck S., Pechriggl E. J., Kelm J. M., Hilbe W., Zwierzina H., PLoS ONE 2014, 9, e92511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stratmann A. T., Fecher D., Wangorsch G., Gottlich C., Walles T., Walles H., Dandekar T., Dandekar G., Nietzer S. L., Mol. Oncol. 2014, 8, 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu X. Q., Kiefl R., Roskopf C., Tian F., Huber R. M., PLoS ONE 2016, 11, e0156268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Choe M. M., Tomei A. A., Swartz M. A., Nat. Protocols 2006, 1, 357. [DOI] [PubMed] [Google Scholar]
- 52. Marrazzo P., Maccari S., Taddei A., Bevan L., Telford J., Soriani M., Pezzicoli A., PLoS ONE 2016, 11, e0153985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Klein S. G., Hennen J., Serchi T., Blömeke B., Gutleb A. C., Toxicol. In Vitro 2011, 25, 1516. [DOI] [PubMed] [Google Scholar]
- 54. Chowdhury F., Howat W. J., Phillips G. J., Lackie P. M., Exp. Lung Res. 2010, 36, 1. [DOI] [PubMed] [Google Scholar]
- 55. Papritz M., Pohl C., Wübbeke C., Moisch M., Hofmann H., Hermanns M. I., Thiermann H., Kirkpatrick C. J., Kehe K., Toxicol. Appl. Pharmacol. 2010, 245, 361. [DOI] [PubMed] [Google Scholar]
- 56. Brandenberger C., Rothen‐Rutishauser B., Mühlfeld C., Schmid O., Ferron G. A., Maier K. L., Gehr P., Lenz A. G., Toxicol. Appl. Pharmacol. 2010, 242, 56. [DOI] [PubMed] [Google Scholar]
- 57. Rothen‐Rutishauser B. M., Kiama S. G., Gehr P., Am. J. Respir. Cell Mol. Biol. 2005, 32, 281. [DOI] [PubMed] [Google Scholar]
- 58. Benam K. H., Novak R., Nawroth J., Hirano‐Kobayashi M., Ferrante T. C., Choe Y., Prantil‐Baun R., Weaver J. C., Bahinski A., Parker K. K., Ingber D. E., Cell Syst. 2016, 3, 456. [DOI] [PubMed] [Google Scholar]
- 59. Huh D., Ann. Am. Thorac. Soc. 2015, 12, S42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Huh D., Leslie D. C., Matthews B. D., Fraser J. P., Jurek S., Hamilton G. A., Thorneloe K. S., McAlexander M. A., Ingber D. E., Sci. Transl. Med. 2012, 4, 159ra147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Huh D., Matthews B. D., Mammoto A., Montoya‐Zavala M., Hsin H. Y., Ingber D. E., Science 2010, 328, 1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Perkoff G. T., Arch. Intern. Med. 1968, 122, 326. [PubMed] [Google Scholar]
- 63. Hansen I., Lykkegaard Nielsen M., Heerfordt L., Henriksen B., Bertelsen S., Chemotherapy 1973, 19, 221. [DOI] [PubMed] [Google Scholar]
- 64. Meade G. M., W. Steenken, Jr. , Am. Rev. Tuberc. 1949, 59, 429. [DOI] [PubMed] [Google Scholar]
- 65. Hinterberger W., Paukovits W. R., Kinast H., Moritz E., Zwinz O., Blut 1978, 37, 69. [DOI] [PubMed] [Google Scholar]
- 66. Kaliner M., Austen K. F., J. Exp. Med. 1973, 138, 1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kaliner M., Orange R. P., Austen K. F., J. Exp. Med. 1972, 136, 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lewis R. A., Wasserman S. I., Goetzi E. J., Austen K. F., Formation of slow‐reacting substance of anaphylaxis in human lung tissue and cells before release. J. Exp. Med. 1974, 140, 1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Orange R. P., Austen W. G., Austen K. F., J. Exp. Med. 1971, 134, 136. [PMC free article] [PubMed] [Google Scholar]
- 70. Tauber A. I., Kaliner M., Stechschulte D. J., Austen K. F., J. Immunol. 1973, 111, 27. [PubMed] [Google Scholar]
- 71. Fatykhova D., Rabes A., Machnik C., Guruprasad K., Pache F., Berg J., Toennies M., Bauer T. T., Schneider P., Schimek M., Eggeling S., Mitchell T. J., Mitchell A. M., Hilker R., Hain T., Suttorp N., Hippenstiel S., Hocke A. C., Opitz B., PLoS ONE 2015, 10, e0137108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hocke A. C., Becher A., Knepper J., Peter A., Holland G., Tonnies M., Bauer T. T., Schneider P., Neudecker J., Muth D., Wendtner C. M., Ruckert J. C., Drosten C., Gruber A. D., Laue M., Suttorp N., Hippenstiel S., Wolff T., Am. J. Respir. Crit. Care Med. 2013, 188, 882. [DOI] [PubMed] [Google Scholar]
- 73. Szymanski K. V., Toennies M., Becher A., Fatykhova D., N'Guessan P. D., Gutbier B., Klauschen F., Neuschaefer‐Rube F., Schneider P., Rueckert J., Neudecker J., Bauer T. T., Dalhoff K., Dromann D., Gruber A. D., Kershaw O., Temmesfeld‐Wollbrueck B., Suttorp N., Hippenstiel S., Hocke A. C., Eur. Respir. J. 2012, 40, 1458. [DOI] [PubMed] [Google Scholar]
- 74. Weinheimer V. K., Becher A., Tonnies M., Holland G., Knepper J., Bauer T. T., Schneider P., Neudecker J., Ruckert J. C., Szymanski K., Temmesfeld‐Wollbrueck B., Gruber A. D., Bannert N., Suttorp N., Hippenstiel S., Wolff T., Hocke A. C., J. Infect. Dis. 2012, 206, 1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Uhl F. E., Vierkotten S., Wagner D. E., Burgstaller G., Costa R., Koch I., Lindner M., Meiners S., Eickelberg O., Konigshoff M., Eur. Respir. J. 2015, 46, 1150. [DOI] [PubMed] [Google Scholar]
- 76. Alsafadi H. N., Staab‐Weijnitz C. A., Lehmann M., Lindner M., Peschel B., Konigshoff M., Wagner D. E., Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L896. [DOI] [PubMed] [Google Scholar]
- 77. Morin C., Sirois M., Echave V., Gomes M. M., Rousseau E., Am. J. Physiol. 2007, 293, L1037. [DOI] [PubMed] [Google Scholar]
- 78. Morin C., Sirois M., Echave V., Gomes M. M., Rousseau E., Am. J. Respir. Cell Mol. Biol. 2008, 38, 192. [DOI] [PubMed] [Google Scholar]
- 79. Morin C., Sirois M., Echave V., Rousseau E., Am. J. Respir. Cell Mol. Biol. 2008, 39, 638. [DOI] [PubMed] [Google Scholar]
- 80. Tabet Y., Sirois M., Sirois C., Rizcallah E., Rousseau É., Am. J. Physiol. 2013, 304, L562. [DOI] [PubMed] [Google Scholar]
- 81. Le Guen M., Naline E., Grassin‐Delyle S., Devillier P., Faisy C., PLoS ONE 2015, 10, e0127765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Calzetta L., Luongo L., Cazzola M., Page C., Rogliani P., Facciolo F., Maione S., Capuano A., Rinaldi B., Matera M. G., Life Sci. 2015, 131, 44. [DOI] [PubMed] [Google Scholar]
- 83. Calzetta L., Spina D., Cazzola M., Page C. P., Facciolo F., Rendina E. A., Matera M. G., Am. J. Respir. Cell Mol. Biol. 2011, 45, 1222. [DOI] [PubMed] [Google Scholar]
- 84. Cazzola M., Calzetta L., Page C. P., Rogliani P., Facciolo F., Gavalda A., Matera M. G., Eur. J. Pharmacol. 2014, 745, 135. [DOI] [PubMed] [Google Scholar]
- 85. Faisy C., Grassin‐Delyle S., Blouquit‐Laye S., Brollo M., Naline E., Chapelier A., Devillier P., PLoS ONE 2014, 9, e111350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Faisy C., Pinto F. M., Le Guen M., Naline E., Grassin Delyle S., Sage E., Candenas M. L., Devillier P., Crit. Care 2011, 15, R208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Fernandes L. M., Mariani A. W., Medeiros I. L., Samano M. N., Abdalla L. G., Correia A. T., Nepomuceno N. A., Canzian M., Pego‐Fernandes P. M., Acta Cir. Bras. 2015, 30, 359. [DOI] [PubMed] [Google Scholar]
- 88. Frank J. A., Briot R., Lee J. W., Ishizaka A., Uchida T., Matthay M. A., Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L52. 17351061 [Google Scholar]
- 89. Gnadt M., Kardziev B., Schmidt M., Hogger P., Lung 2012, 190, 431. [DOI] [PubMed] [Google Scholar]
- 90. Lee J. W., Fang X., Gupta N., Serikov V., Matthay M. A., Proc. Natl. Acad. Sci. USA 2009, 106, 16357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lee J. W., Krasnodembskaya A., McKenna D. H., Song Y., Abbott J., Matthay M. A., Am. J. Respir. Crit. Care Med. 2013, 187, 751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. McAuley D. F., Curley G. F., Hamid U. I., Laffey J. G., Abbott J., McKenna D. H., Fang X., Matthay M. A., Lee J. W., Lung Cell. Mol. Physiol. 2014, 306, L809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Medeiros I. L., Pego‐Fernandes P. M., Mariani A. W., Fernandes F. G., Unterpertinger F. V., Canzian M., Jatene F. B., Clinics 2012, 67, 1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Sadaria M. R., Smith P. D., Fullerton D. A., Justison G. A., Lee J. H., Puskas F., Grover F. L., J. C. Cleveland, Jr. , Reece T. B., Weyant M. J., Ann. Thorac. Surg. 2011, 92, 478. [DOI] [PubMed] [Google Scholar]
- 95. Booth A. J., Hadley R., Cornett A. M., Dreffs A. A., Matthes S. A., Tsui J. L., Weiss K., Horowitz J. C., Fiore V. F., Barker T. H., Moore B. B., Martinez F. J., Niklason L. E., White E. S., Am. J. Respir. Crit. Care Med. 2012, 186, 866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wagner D. E., Bonenfant N. R., Parsons C. S., Sokocevic D., Brooks E. M., Borg Z. D., Lathrop M. J., Wallis J. D., Daly A. B., Lam Y. W., Deng B., DeSarno M. J., Ashikaga T., Loi R., Weiss D. J., Biomaterials 2014, 35, 3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wagner D. E., Bonenfant N. R., Sokocevic D., DeSarno M. J., Borg Z. D., Parsons C. S., Brooks E. M., Platz J. J., Khalpey Z. I., Hoganson D. M., Deng B., Lam Y. W., Oldinski R. A., Ashikaga T., Weiss D. J., Biomaterials 2014, 35, 2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sun H., Zhu Y., Pan H., Chen X., Balestrini J. L., Lam T. T., Kanyo J. E., Eichmann A., Gulati M., Fares W. H., Bai H., Feghali‐Bostwick C. A., Gan Y. Peng, Moore X., White M. W., Sava E. S., Gonzalez P., Cheng A. L., Niklason Y., Arthritis Rheumatol. 2016, 68, 1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Gilpin S. E., Guyette J. P., Gonzalez G., Ren X., Asara J. M., Mathisen D. J., Vacanti J. P., Ott H. C., J. Heart Lung Transplant. 2014, 33, 298. [DOI] [PubMed] [Google Scholar]
- 100. O'Neill J. D., Anfang R., Anandappa A., Costa J., Javidfar J., Wobma H. M., Singh G., Freytes D. O., Bacchetta M. D., Sonett J. R., Vunjak‐Novakovic G., Ann. Thorac. Surg. 2013, 96, 1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zvarova B., Uhl F., Uriarte J., Borg Z., Coffey A. L., Bonenfant N. R., Weiss D. J., Wagner D. E., Tissue Eng. Part C Methods 2016, 22, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Gilpin S. E., Charest J. M., Ren X., Tapias L. F., Wu T., Evangelista‐Leite D., Mathisen D. J., Ott H. C., Biomaterials 2016, 108, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Wagner D. E., Fenn S. L., Bonenfant N. R., Marks E. R., Borg Z., Saunders P., Oldinski R. A., Weiss D. J., Cell. Mol. Bioeng. 2014, 7, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Huang S. X. L., Islam M. N., O'Neill J., Hu Z., Yang Y. G., Chen Y. W., Mumau M., Green M. D., Vunjak‐Novakovic G., Bhattacharya J., Snoeck H. W., Nat. Biotech. 2014, 32, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Nichols J. E., Niles J., Riddle M., Vargas G., Schilagard T., Ma L., Edward K., La Francesca S., Sakamoto J., Vega S., Ogadegbe M., Mlcak R., Deyo D., Woodson L, McQuitty C., Lick S., Beckles D., Melo E., Cortiella J., Tissue Eng. Part A 2013, 19, 2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Parker M. W., Rossi D., Peterson M., Smith K., Sikstrom K., White E. S., Connett J. E., Henke C. A., Larsson O., Bitterman P. B., J. Clin. Invest. 2014, 124, 1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Zhou Y., Peng H., Sun H., Peng X., Tang C., Gan Y., Chen X., Mathur A., Hu B., Slade M. D., Montgomery R. R., Shaw A. C., Homer R. J., White E. S., Lee C. M., Moore M. W., Gulati M., Lee C. G., Elias J. A., Herzog E. L., Sci. Transl. Med. 2014, 6, 240ra276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Brown S. M., Koarai A., Sturton R. G., Nicholson A. G., Barnes P. J., Donnelly L. E., Eur. J. Pharmacol. 2013, 702, 109. [DOI] [PubMed] [Google Scholar]
- 109. Koziol‐White C. J., Yoo E. J., Cao G., Zhang J., Papanikolaou E., Pushkarsky I., Andrews A., Himes B. E., Damoiseaux R. D., Liggett S. B., Di Carlo D., Kurten R. C., R. A. Panettieri, Jr , Br. J. Pharmacol. 2016, 173, 2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Banerjee A., Trivedi C. M., Damera G., Jiang M., Jester W., Hoshi T., Epstein J. A., R. A. Panettieri, Jr , Am. J. Respir. Cell Mol. Biol. 2012, 46, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Jiang H., Abel P. W., Toews M. L., Deng C., Casale T. B., Xie Y., Tu Y., J. Pharmacol. Exp. Ther. 2010, 334, 703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Cooper P. R., R. A. Panettieri, Jr. , J. Allergy Clin. Immunol. 2008, 122, 734. [DOI] [PubMed] [Google Scholar]
- 113. Mullane K., Williams M., Biochem. Pharmacol. 2014, 87, 131. [DOI] [PubMed] [Google Scholar]
- 114. Schleputz M., Rieg A. D., Seehase S., Spillner J., Perez‐Bouza A., Braunschweig T., Schroeder T., Bernau M., Lambermont V., Schlumbohm C., Sewald K., Autschbach R., Braun A., Kramer B. W., Uhlig S., Marti C., PLoS ONE 2012, 7, e47344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Ressmeyer A. R., Larsson A. K., Vollmer E., Dahlen S. E., Uhlig S., Martin C., Eur. Respir. J. 2006, 28, 603. [DOI] [PubMed] [Google Scholar]
- 116. Kindler E., Jonsdottir H. R., Muth D., Hamming O. J., Hartmann R., Rodriguez R., Geffers R., Fouchier R. A., Drosten C., Muller M. A., Dijkman R., Thiel V., mBio 2013, 4, e00611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Dijkman R., Jebbink M. F., Koekkoek S. M., Deijs M., Jonsdottir H. R., Molenkamp R., Ieven M., Goossens H., Thiel V., van der Hoek L., J. Virol. 2013, 87, 6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Raj V. S., Mou H., Smits S. L., Dekkers D. H., Muller M. A., Dijkman R., Muth D., Demmers J. A., Zaki A., Fouchier R. A., Thiel V., Drosten C., Rottier P. J., Osterhaus A. D., Bosch B. J., Haagmans B. L., Nature 2013, 495, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Bertram S., Dijkman R., Habjan M., Heurich A., Gierer S., Glowacka I., Welsch K., Winkler M., Schneider H., Hofmann‐Winkler H., Thiel V., Pohlmann S., J. Virol. 2013, 87, 6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Lundin A., Dijkman R., Bergstrom T., Kann N., Adamiak B., Hannoun C., Kindler E., Jonsdottir H. R., Muth D., Kint J., Forlenza M., Muller M. A., Drosten C., Thiel V., Trybala E., PLoS Pathog. 2014, 10, e1004166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Kolesnikova L., Heck S., Matrosovich T., Klenk H. D., Becker S., Matrosovich M., J. Gen. Virol. 2013, 94, 971. [DOI] [PubMed] [Google Scholar]
- 122. Knepper J., Schierhorn K. L., Becher A., Budt M., Tonnies M., Bauer T. T., Schneider P., Neudecker J., Ruckert J. C., Gruber A. D., Suttorp N., Schweiger B., ippenstiel S., Hocke A. C., Wolff T., mBio 2013, 4, e00601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wu W., Booth J. L., Duggan E. S., Patel K. B., Duggan E. S., Patel K. B., Coggeshall K. M., Metcalf J. P., J. Gen. Virol. 2010, 91, 1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Wu W., Zhang W., Booth J. L., Metcalf J. P., PLoS ONE 2012, 7, e49856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Zhang J., Zhang Z., Fan X., Liu Y., Wang J., Zheng Z., Chen R., Wang P., Song W., Chen H., Guan Y., J. Infect. Dis. 2010, 201, 1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Chi F., Leider M., Leendertz F., Bergmann C., Boesch C., Schenk S., Pauli G., Ellerbrok H., Hakenbeck R., J. Bacteriol. 2007, 189, 6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Mehr S., Wood N., Paediatr. Respir. Rev. 2012, 13, 258. [DOI] [PubMed] [Google Scholar]
- 128. Jiang D., Berman R., Wu Q., Stevenson C., Chu H. W., J. Clin. Cell. Immunol. 2016, 7, pii: 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Amatngalim G. D., Schrumpf J. A., Henic A., Dronkers E., Verhoosel R. M., Ordonez S. R., Haagsman H. P., Fuentes M. E., Sridhar S., Aarbiou J., Janssen R. A. J., Lekkerkerker A. N., Hiemstra P. S., J. Innate. Immun. 2017, 9, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Schamberger A. C., Staab‐Weijnitz C. A., Mise‐Racek N., Eickelberg O., Sci. Rep. 2015, 5, 8163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Gohy S. T., Detry B. R., Lecocq M., Bouzin C., Weynand B. A., Amatngalim G. D., Sibille Y. M., Pilette C., Am. J. Respir. Crit. Care Med. 2014, 190, 509. [DOI] [PubMed] [Google Scholar]
- 132. Zarcone M. C., van Schadewijk A., Duistermaat E., Hiemstra P. S., Kooter I. M., Respir. Res. 2017, 18, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Leclercq B., Happillon M., Antherieu S., Hardy E. M., Alleman L. Y., Grova N., Perdrix E., Appenzeller B. M., Lo Guidice J. M., Coddeville P., Garcon G., Environ. Pollut. 2016, 218, 1074. [DOI] [PubMed] [Google Scholar]
- 134. Gohy S. T., Hupin C., Fregimilicka C., Detry B. R., Bouzin C., Gaide Chevronay H., Lecocq M., Weynand B., Ladjemi M. Z., Pierreux C. E., Birembaut P., Polette M., Pilette C., Eur. Respir. J. 2015, 45, 1258. [DOI] [PubMed] [Google Scholar]
- 135. Sewald K., Braun A., Xenobiotica 2013, 43, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Morin J. P., Baste J. M., Gay A., Crochemore C., Corbiere C., Monteil C., Xenobiotica 2013, 43, 63. [DOI] [PubMed] [Google Scholar]
- 137. Emura M., Aufderheide M., Exp. Toxicol. Pathol. 2016, 68, 255. [DOI] [PubMed] [Google Scholar]
- 138. El‐Baz A., Beache G. M., Gimel'farb G., Suzuki K., Okada K., Elnakib A., Soliman A., Abdollahi B., Int. J. Biomed. Imaging 2013, 2013, 942353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Soliman A., Khalifa F., Elnakib A., Abou El‐Ghar M., Dunlap N., Wang B., Gimel'farb G., Keynton R., El‐Baz A., IEEE Trans. Med. Imaging 2017, 36, 263. [DOI] [PubMed] [Google Scholar]
- 140. Onion D., Argent R. H., Reece‐Smith A. M., Craze M. L., Pineda R. G., Clarke P. A., Ratan H. L., Parsons S. L., Lobo D. N., Duffy J. P., Atherton J. C., McKenzie A. J., Kumari R., King P., Hall B. M., Grabowska A. M., Mol. Cancer Ther. 2016, 15, 753. [DOI] [PubMed] [Google Scholar]
- 141. Huh D., Leslie D. C., Matthews B. D., Fraser J. P., Jurek S., Hamilton G. A., Thorneloe K. S., McAlexander M. A., Ingber D.E, Sci. Transl. Med. 2012, 4, 159ra147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Huh D., Matthews B. D., Mammoto A., Montoya‐Zavala M., Hsin H. Y., Ingber D. E., Science 2010, 328, 1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Huh D. D., Ann. Am. Thorac. Soc. 2015, 12, S42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Ingber D. E., Cell 2016, 164, 1105. [DOI] [PubMed] [Google Scholar]
- 145. Esch E. W., Bahinski A., Huh D., Nat. Rev. Drug Discov. 2015, 14, 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Schiessl I. M., Castrop H., Pflugers Arch. 2016, 468, 1505. [DOI] [PubMed] [Google Scholar]
- 147. DaCosta R. S., Wilson B. C., Marcon N. E., ScientificWorldJournal 2007, 7, 2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Lim J., Lee H. K., Yu W., Ahmed S., Analyst 2014, 139, 4758. [DOI] [PubMed] [Google Scholar]
- 149. Chen B. C., Legant W. R., Wang K., Shao L., Milkie D. E., Davidson M. W., Janetopoulos C., Wu X. S., J. A. Hammer, 3rd , Liu Z., English B. P., Mimori‐Kiyosue Y., Romero D. P., Ritter A. T., Lippincott‐Schwartz J., Fritz‐Laylin L., Mullins R. D., Mitchell D. M., Bembenek J. N., Reymann A. C., Bohme R., Grill S. W., Wang J. T., Seydoux G., Tulu U. S., Kiehart D. P., Betzig E., Science 2014, 346, 1257998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Burgstaller G., Vierkotten S., Lindner M., Konigshoff M., Eickelberg O., Am. J. Physiol. 2015, 309, L323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Wilkinson D. C., Alva‐Ornelas J. A., Sucre J. M., Vijayaraj P., Durra A., Richardson W., Jonas S. J., Paul M. K., Karumbayaram S., Dunn B., Gomperts B. N., Stem Cells Transl. Med. 2016, 6, 622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Dye B. R., Miller A. J., Spence J. R., Curr. Pathobiol. Rep. 2016, 4, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Choi J., Iich E., Lee J. H., Dev. Biol. 2016, 420, 278. [DOI] [PubMed] [Google Scholar]
- 154. Li J., Chen M., Fan X., Zhou H., J. Transl. Med. 2016, 14, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Jung J. P., Bhuiyan D. B., Ogle B. M., Biomater. Res. 2016, 20, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Baatz J. E., Newton D. A., Riemer E. C., Denlinger C. E., Jones E. E., Drake R. R., Spyropoulos D. D., In Vivo (Athens, Greece). 2014, 28, 411. [PMC free article] [PubMed] [Google Scholar]
- 157. Rosner S. R., Ram‐Mohan S., Paez‐Cortez J. R., Lavoie T. L., Dowell M. L., Yuan L., Ai X., Fine A., Aird W. C., Solway J., Fredberg J. J., Krishnan R., Am. J. Respir. Cell Mol. Biol. 2014, 50, 876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Bai Y., Krishnamoorthy N., Patel K. R., Rosas I., Sanderson M. J., Ai X., Am. J. Respir. Cell Mol. Biol. 2016, 54, 656. [DOI] [PMC free article] [PubMed] [Google Scholar]