

Abstract
We demonstrate the integration of DNA amplification and detection functionalities developed on a lab‐on‐a‐chip microdevice utilizing solid‐phase polymerase chain reaction (SP‐PCR) for point‐of‐need (PON) DNA analyses. First, the polycarbonate microdevice was fabricated by thermal bonding to contain microchambers as reservoirs for performing SP‐PCR. Next, the microchambers were subsequently modified with polyethyleneimine and glutaraldehyde for immobilizing amine‐modified forward primers. During SP‐PCR, the immobilized forward primers and freely diffusing fluorescence‐labeled reverse primers cooperated to generate target amplicons, which remained covalently attached to the microchambers for the fluorescence detection. The SP‐PCR microdevice was used for the direct identifications of two widely detected foodborne pathogens, namely Salmonella spp. and Staphylococcus aureus, and an alga causing harmful algal blooms annually in South Korea, Cochlodinium polykrikoides. The SP‐PCR microdevice would be versatilely applied in PON testing as a universal platform for the fast identification of foodborne pathogens and environmentally threatening biogenic targets.
Keywords: alga, foodborne pathogen, integrated microdevice, on‐chip detection, point‐of‐need (PON) testing, solid‐phase polymerase chain reaction (SP‐PCR)
A polycarbonate (PC) microdevice capable of performing solid‐phase polymerase chain reaction (SP‐PCR) was fabricated for the identification of pathogenic microorganisms. By immobilizing one of the primers on the chemically modified PC surface, Pham and coworkers have integrated DNA amplification and detection on a monolithic microdevice, which was used to detect two major foodborne pathogens and a harmful marine microalga.
1. INTRODUCTION
The early detection of foodborne microbial agents is a critical concern in many interdisciplinary research fields. Among diverse pathogenic microorganisms, foodborne pathogens have accounted for the largest number of domestic and global outbreaks and are responsible for high rates of morbidity and mortality worldwide (Foudeh, Fatanat Didar, Veres, & Tabrizian, 2012; Turner et al., 2016). Foodborne pathogens can also originate from the marine environment and include toxic algae, as well as harmful bacteria that could become a huge threat to human health (Visciano et al., 2016). The biotoxins that accumulate in contaminated seafood and the co‐occurring pathogens that proliferate during harmful algal blooms may be passed on to higher trophic levels in the marine food network and eventually to humans (Oh et al., 2016). Such pressures on human healthcare have stimulated the development of several diagnostic methods for the identification of microbial pathogens with high precision, short turnover time, and ease of operation (Foudeh et al., 2012; Law, Mutalib, Chan, & Lee, 2014).
Nucleic‐acid‐based testing has played a central role in the detection of harmful microorganisms at the molecular level (Clerc & Greub, 2010; Craw & Balachandran, 2012; Niemz, Ferguson, & Boyle, 2011). Attributed to the ability to quickly make several copies of genetic materials, the polymerase chain reaction (PCR) is widely used as a molecular diagnostic method in biomedical and biochemical research (Clerc and Greub, 2010; Craw and Balachandran, 2012; Law et al., 2014; Niemz et al., 2011; Oh et al., 2016; Turner et al., 2016; Visciano et al., 2016; Zhang & Ozdemir, 2009). Despite providing high sensitivity and selectivity of DNA amplification, the conventional PCR technique has limitations in realizing downstream detection or separation processes directly after PCR (Adessi et al., 2000; Lazcka, Campo, & Muñoz, 2007). Advances in lab‐on‐a‐chip (LOC) techniques have enabled the miniaturization and integration of several functional components of PCR, allowing timely detection of biomolecular targets with a better analytical performance. Among several variations of PCR, solid‐phase PCR (SP‐PCR) is a promising method for developing integrated LOC platforms because it enables amplification followed by immediate detection of DNA. This advantage allows the rapid on‐site detection of amplicons in a time‐saving manner by overcoming multiple manipulations required for post‐analyses of PCR products (Khodakov & Ellis, 2014; Mercier, Slater, & Mayer, 2003; Shin, Perera, Kim, & Park, 2013).
Since the invention of SP‐PCR, the technique has been developed in the form of microarray‐based approaches and further incorporated into LOC systems (Chin et al., 2017; J. Hoffmann, M. Trotter, Stetten, Zengerle, & Roth, 2012; J. Hoffmann, S. Hin, Stetten, Zengerle, & Roth, 2012; Kersting, Rausch, Bier, & von Nickisch‐Rosenegk, 2014). Most of the reported SP‐PCR LOC platforms have focused on the surface modification of solid substrates for the immobilization of solid‐support primers. Despite the noticeable improvements highlighted in these systems, disadvantages remain, which need to be overcome. Specifically, a microarray platform for the genotyping of human mutation genes was developed (Damin, Galbiati, Ferrari, & Chiari, 2016). However, that platform requires the synthesis of a copolymer coated onto the substrate surface for the immobilization of primers, which demands extensive and time‐consuming preparation steps. An organosilanization method was also introduced to immobilize primers (J. Hoffmann, S. Hin et al., 2012). Although this approach is compatible with SP‐PCR, the reproducibility of the coating chemistry depends on highly controlled operative conditions. Besides, the use of bulky apparatuses for the plasma activation of the substrate surface before the coating procedures used in both the abovementioned studies may not meet the conditions of resource‐poor laboratories. These shortcomings hamper the realization of an SP‐PCR microdevice.
To address these challenges, this study aims to fabricate a thermoplastic SP‐PCR microdevice using a chemically robust and thermostable surface modification for primer grafting. For the fabrication, the polycarbonate (PC) microdevice was hot embossed to contain microchambers. The amine groups of polyethyleneimine (PEI) were first coated onto PC via aminolysis to form the urethane linkages without requiring prior surface oxidation of PC. The PEI‐coated PC was then activated with glutaraldehyde (GA) to produce a reactive surface for the covalent tethering of amine‐modified primers. The microdevice was used for the simultaneous amplification and detection of two major foodborne pathogens, namely Salmonella spp. and Staphylococcus aureus (S. aureus). Furthermore, to examine the universal applicability of the microdevice, we applied the platform for the detection of a representative harmful marine microalga, Cochlodinium polykrikoides (C. polykrikoides). After on‐chip PCR, the amplicons were directly detected inside the microchambers by fluorescence imaging without having to take out the amplicons. The surface modifications of the PC substrates were characterized by water contact angle measurements, and the density of the coated amine functional groups was also assessed by fluorescence measurements. The feasibility of the SP‐PCR microdevice as a portable platform for simultaneous detection of multiple analytes was evaluated via fluorescence measurements directly inside the microchambers immediately after PCR in a consecutive manner.
2. MATERIALS AND METHODS
2.1. Materials and chemicals
PEI (Mw = 25,000), GA (25 wt% in water), sodium cyanoborohydride, bovine serum albumin (BSA, V fraction), sodium dodecyl sulfate (SDS), and fluorescein‐5‐isothiocyanate (FITC) were purchased from Sigma‐Aldrich (St. Louis, MO). Sodium phosphate buffer (pH 7.0) and 20× saline sodium citrate (SSC) were purchased from Biosesang (Seongnam, Korea). Commercial PC sheets with the thickness of 2 mm were obtained from Goodfellow (Huntingdon, England). Mueller Hinton Broth (MHB) was purchased from Becton Dickinson (Franklin Lakes, NJ), and nutrient broth (NB) was purchased from Neogen (Lansing, MI). Wizard genomic DNA purification kit was purchased from Promega (Madison, WI), and RNeasy Mini Kit was purchased from Qiagen (Hilden, Germany). PCR kit was purchased from BioFact (Daejeon, Korea). All PCR primers (Supporting Information Table S1) were synthesized by Integrated DNA Technologies (Hanam, Korea). Ethidium bromide dye was purchased from Dynebio (Seongnam, Korea). The 100‐bp (base pair) DNA ladder was purchased from Takara (Shiga, Japan). Agarose powder was purchased from BioShop (Burlington, ON, Canada).
2.2. Fabrication of microdevice
Figure 1a illustrates the fabrication of the microdevice and the functionalization of the PC surface for the immobilization of amine‐modified primers. Before surface modification, the microchambers were engraved on a PC substrate using a computer numerical control (CNC) machine. The engraved substrate was cleaned by sonicating and dried completely with compressed air. The PC substrate bearing the microchamber structures was then embossed with another PC substrate at 145°C under 0.1 MPa for 15 min to seal the microdevice.
Figure 1.

(a) Overall scheme of the fabrication and surface modification of the SP‐PCR microdevice for primer grafting. A PC substrate containing three microchambers was embossed with another flat one. Microchambers were successively modified with PEI and GA, and amine‐modified primers of corresponding microbe targets were directly immobilized onto the microchamber surfaces. (b) SP‐PCR procedures for the detection of foodborne pathogens and alga. PCR mixtures were introduced into the microchambers afterward to perform SP‐PCR. Synthesized amplicons remained covalently attached to the solid surface, and the amplified target signals were collected using fluorescence imaging. Green and red fluorescence indicate the signals of Alexa 488 and Alexa 647 dyes, respectively. GA, glutaraldehyde; PC, polycarbonate; PEI, polyethyleneimine; RT, room temperature; SP‐PCR, solid‐phase polymerase chain reaction [Color figure can be viewed at wileyonlinelibrary.com]
Surface modification was carried out inside the closed microchambers. First, the microchambers were incubated with an aqueous solution of 5 wt% PEI for 1 hr at room temperature. Afterward, the microchambers were washed with deionized water and completely dried with compressed air. The aminated surfaces of the microchambers were activated with freshly prepared 2.5% (v/v) GA solution diluted in phosphate buffer (pH 7.0) containing 10 mM sodium cyanoborohydride, which functions as a reducing agent, for 2 hr at room temperature. The activated microchamber surfaces were thoroughly rinsed with phosphate buffer (pH 7.0) to remove unreacted GA and then dried completely with compressed air. Finally, a solution of 0.5‐µM amine‐modified primers prepared in phosphate buffer (pH 7.0) was directly introduced into the microchambers to react with PEI‐coated PC modified with GA (PEI‐GA‐coated PC) for 1 hr at 50°C. After primer immobilization, the microchambers were washed with 0.1× SSC and 0.1% SDS solutions to remove the unreacted primers. The microchambers were then rinsed with deionized water and dried with compressed air.
As shown in Figure 1b, three microchambers immobilized with three types of amine‐modified primers were used for subsequent SP‐PCR to detect three microbial targets. Besides the PCR reagents, the PCR mixture contained an asymmetric concentration of floating forward primers and fluorescence‐modified primers in the ratio of 1:4. For SP‐PCR, the amplification undergoes two phases. First, liquid‐phase amplification occurs preferably to generate a number of double‐stranded and single‐stranded DNA that have the fluorescence dyes. When the limited number of forward primers was depleted, solid‐phase amplification becomes dominant, resulting in the formation of amplicons covalently attached to the surface of the microchamber, allowing the on‐site detection of target amplicons by collecting their fluorescence signals inside the microchambers.
2.3. Characterization of surface modification (1): Water contact angle measurement
Water contact angles were measured on the surfaces of pristine PC, PEI‐coated PC, and PEI‐GA‐coated PC to confirm the surface modification. Water contact angles were also used to evaluate the hydrolytic resistance of PEI‐coated PC under thermal cycling condition after 5, 10, 15, 20, 25, and 30 thermal cycles. A Phoenix 300 contact angle analyzer (Surface Electro Optics, Suwon, Korea) was used for the analyses with the sessile drop method. The obtained results were analyzed with Image Pro 300 software. Measurements were made five times and averaged.
2.4. Characterization of surface modification (2): Quantification of amine density coated on PC
To quantify the density of amine groups on the surface of PEI‐coated PC, a fluorescence measurement was performed using FITC, which can react with amine groups. A standard curve was established by spotting different concentrations of PEI (0–50 nmol) coupled with 0.1 mg ml–1 FITC and measuring the fluorescence signal. The actual density of amine groups coated on PC was quantified by staining the PEI‐coated PC substrates with FITC and comparing the fluorescence signal with the standard curve. A fluorescence signal was detected and analyzed using the Gel Doc EZ system (Bio‐Rad) and the Image Lab 4.0 software (Bio‐Rad). Differences in the fluorescence signals were statistically analyzed by one‐way analysis of variance followed by Tukey’s pairwise comparison with 95% confidence intervals using Minitab version 16 (Minitab, State College, PA).
2.5. Characterization of surface modification (3): Analysis of surface coating stability
To analyze the stability of the surface coatings on PC under thermal cycling condition, PEI‐GA‐coated microchambers were immobilized with PCR amplicons amplified off‐chip from 10 ng (3.25 × 106 copies) DNA template. The amplicons were produced to have both amine groups and Alexa 488 dyes at the 5′ and 3′ ends, respectively. The microchambers were then filled with the solutions of PCR buffer and water and were subjected to the thermal cycling conditions used for SP‐PCR. Fluorescence signals of the immobilized amplicons were measured before and after 45 cycles of thermal treatment using an Olympus IX71 inverted fluorescence microscope and were analyzed with ProgRes® Capture Pro 2.8 software (Jenoptik, Jena, Germany). The effect of temperature on the stability of the surface coatings was evaluated by analyzing the fluorescence signals before and after the thermal treatment using the Minitab version 16 (Minitab, State College, PA) with 95% confidence intervals, as mentioned above.
2.6. Genomic DNA extraction from bacteria and microalgae
For foodborne pathogens, liquid cultures of Salmonella spp. and S. aureus were grown in NB and MHB media, respectively, at 37°C for 16 hr. One milliliter of the overnight culture was then collected and centrifuged at 15,000g for DNA extraction. Isolation of genomic DNA (gDNA) was carried out using the Wizard Genomic DNA purification kit. Samples of the alga C. polykrikoides were directly collected from the ocean during its blooming season in 2013, and algal gDNA was extracted using the RNeasy Mini Kit. Extracted gDNA was stored at 4°C.
2.7. Temperature measurement
In this study, the flat heat block of a commercialized thermal cycler (Gene‐Touch TC‐E‐96GA, Bioer) was used to conduct SP‐PCR. A PC substrate was placed on the heat block, and the surface temperatures were measured using an infrared camera (FLIR Thermovision A320, Wilsonville, OR). To evaluate the temperature distribution, 10 spots were randomly selected within the microchamber area for the measurement at each temperature zone. The average temperature was analyzed using ThermaCAM Researcher 2.8 software.
2.8. SP‐PCR on the microdevice
After immobilizing amine‐modified primers, the surfaces of the microchambers were blocked with 2 mg ml–1 BSA for 1 hr at room temperature. The microchambers were then washed with deionized water and completely dried with compressed air. SP‐PCR was applied on the PC microdevice to amplify invA, nuc, and large subunit ribosomal RNA (LSU rRNA) genes of Salmonella spp., S. aureus, and C. polykrikoides, respectively. The amplification of each target was performed in a 20‐µl reaction mixture containing 10 ng of DNA template (3.25 × 106 copies of gDNA), 10× Taq reaction buffer, 10 mM of each deoxynucleotide, 1.5 mg ml–1 BSA, 100 nM of forward primer, 400 nM of fluorescence‐labeled reverse primer, and 1.25 U of Taq DNA polymerase. After introducing the PCR mixtures into the microchambers, the inlet and outlets were clamped, and the microdevice was placed on the heat block of the thermal cycler. SP‐PCR was performed with an initial denaturation at 100°C for 5 min followed by 30 thermal cycles at 100°C for 35 s, 63°C for 35 s, and 76°C for 35 s. In this study, amplification of three targets was performed at the same annealing temperature of 63°C. After SP‐PCR, the microchambers were washed, dried, and observed under the fluorescence filters.
As control experiments, DNA amplifications were also performed on pristine microchambers under the same temperature conditions of SP‐PCR but without the primer preimmobilization process. The obtained PCR amplicons were also cross checked by agarose gel electrophoresis. The amplicons were stained with ethidium bromide and detected using the Gel Doc EZ system (Bio‐Rad).
3. RESULTS AND DISCUSSION
3.1. Water contact angle measurement
Figure 2 shows the results of the water contact angle measurements of the pristine PC, PEI‐coated PC, and PEI‐GA‐coated PC substrates. The water contact angle of the pristine PC was approximately 80.1° ± 0.96°, which decreased significantly to 32.8° ± 4.55° after PEI treatment and then increased slightly to 53.5° ± 5.41° after GA activation. These results were in accordance with those reported in other studies (Gunda, Singh, Norman, Kaur, & Mitra, 2014; Jankowski, Ogończyk, Lisowski, & Garstecki, 2012; M. Jang, C. K. Park, & Lee, 2014; M. Jang, S. Park, & Lee, 2014). As shown in Figure 2, changes in the wettability of the substrate surface after each coating step confirmed the successful modification of PC with PEI and subsequently with GA. The PC surface was rendered hydrophilic after the PEI treatment due to the presence of positive charges of dense amine groups decorated on the surface of PC. Amine functional groups were previously reported to directly react with the carbonate backbones of PC chains to form stable urethane linkages (M. Jang, S. Park et al., 2014). This advantage simplifies the modification procedure by getting rid of prior surface oxidation by plasma, allowing the microchambers to be chemically modified even after sealing the microdevice. The next step of activating PEI‐coated PC with GA made the surface relatively hydrophobic compared with the earlier PEI treatment alone, probably due to the existence of hydrocarbon chains of GA. Sodium cyanoborohydride was used to reduce the Schiff base bonds formed between aldehyde and amine groups to stable and irreversible secondary amine bonds. The reducing agent targets Schiff bases while sparing free aldehyde groups for the conjugation of amine‐modified oligonucleotides (Migneault, Dartiguenave, Bertrand, & Waldron, 2004).
Figure 2.

Water contact angles measured on pristine PC, PEI‐coated PC, and PEI‐GA‐coated PC substrates. Data are presented as mean ± standard deviation (n = 5). GA, glutaraldehyde; PC, polycarbonate; PEI, polyethyleneimine; RT, room temperature; SP‐PCR, solid‐phase polymerase chain reaction [Color figure can be viewed at wileyonlinelibrary.com]
3.2. Quantification of amine density on PC
We evaluated the effects of PEI concentration (1%, 5%, and 10%) and the PEI modification time (30, 45, and 60 min) on the density of amine groups coated on PC. As shown in Supporting Information Figure S1, the surface amine density increased with the increase in the PEI concentration and the modification time but decreased after 45 and 60 min of coating with 10% PEI. This reduction probably occurred because a higher PEI concentration and longer modification time might have resulted in the stronger polymer entanglement of PEI chains, reducing the interactions between the amine groups of coated PEI and FITC, as well as the final amine density of PEI coated on the PC surface (Pan et al., 2014). The amine density reached 37.13 ± 0.91 nmol cm–2 after 60 min of modification with 5% PEI, which was the highest measured density and was significantly different from the other treatments (P < 0.05). Therefore, we finally selected this treatment condition for the modification of PC with PEI.
3.3. On‐chip detection: Effect of coated microchamber on capturing amine‐modified PCR amplicons
We further confirmed the surface modification of the microchamber with PEI and GA by determining the ability of the PEI‐GA‐coated microchamber to capture PCR amplicons. The amplicons were produced to have both amine groups and Alexa 488 fluorescence dyes, as mentioned previously. Figure 3a shows the schematic of immobilizing the amplicons on the PEI‐GA‐coated PC. Figure 3b shows a photograph of the microdevice. Figure 3c–e shows the fluorescence images of the PEI‐GA‐coated microchamber immobilized with PCR amplicons, the pristine microchamber only, and the PEI‐GA‐coated microchamber only, respectively. As shown in Figure 3c, the strong green fluorescence of the immobilized amplicons with a low signal background after washing indicated that the PC surface modified with PEI and GA could capture amine‐modified PCR amplicons. Control experiments were also performed to evaluate the inherent autofluorescence property of PC (Piruska et al., 2005). The pristine microchamber did not display noticeable fluorescence signals (Figure 3d) even after coating (Figure 3e). From those results, we could conclude that the amine‐modified PCR amplicons were successfully captured on the PEI‐GA‐coated microchambers, as demonstrated by the green fluorescence signal. The surface of PC was successfully modified with PEI and GA within approximately 3 hr without preactivation with plasma, which was not time‐consuming as well as did not require extensive coating steps or long‐time synthesis of chemicals used for the modification process.
Figure 3.
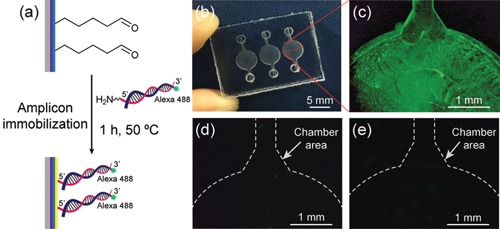
(a) Schematic of the immobilization of PCR amplicons on the PEI‐GA‐coated PC. (b) Image of the PC microdevice. Fluorescence images of (c) PEI‐GA‐coated microchamber decorated with PCR amplicons produced off‐chip to have both Alexa 488 fluorescence dyes and amine groups, (d) pristine microchamber only, and (e) PEI‐GA‐coated microchamber only. GA, glutaraldehyde; PC, polycarbonate; PEI, polyethyleneimine; PCR, polymerase chain reaction [Color figure can be viewed at wileyonlinelibrary.com]
The use of PEI to form stable urethane linkages on the PC surface before GA crosslinking was necessary to conjugate amine‐modified PCR amplicons because neither the pristine PC surface nor PC surface modified only with GA could capture PCR amplicons after washing, as shown in Supporting Information Figure S2a,b, respectively. Also, PEI was more effective than 3‐aminopropyltriethoxysilane (APTES) in functionalizing the PC surface with amine groups and forming more stable bonds against hydrolytic cleavage (Supporting Information Figure S2c). This was probably because PEI has much higher amine content with the ratio of primary, secondary, and tertiary amines being 1:2:1, as compared with primary amine‐bearing APTES. Since primary and secondary amines can potentially react with GA (Bai et al., 2006), PEI might have displayed a higher amplicon‐capturing efficiency compared with APTES.
3.4. Evaluation of coating stability under thermal cycling condition
An important criterion of SP‐PCR is that chemical linkages between immobilized primers and the surface coatings can withstand hydrolytic cleavage and thermal degradation under the thermal cycling conditions. Primary amines functionalized on PC were reported to suffer from hydrolytic cleavage during the amplification reaction (M. Jang, C. K. Park et al., 2014), which could return PC to its innate hydrophobic characteristic. However, this tendency was mediated in case of PEI, as demonstrated through the ignorable hydrophobic recovery of PEI‐coated PC after 30 PCR cycles (Supporting Information Figure S3). This was probably because interactions between PEI and PC take place at both primary and secondary amines (Lee & Ram, 2009), resulting in more stable urethane linkages between PEI and PC.
The surface coatings of PEI and GA and the grafted amine‐modified PCR amplicons were further evaluated under the thermal cycling conditions. Figure 4a,b displays the schematic of the thermal stability test of the surface coating and the immobilized amplicons, which was amplified off‐chip from 10 ng (3.25 × 106 copies) DNA template. As shown in the fluorescence images in Figure 4c,d, the fluorescence intensities were relatively identical before and after thermal treatment. On the basis of the statistical analyses (Supporting Information Figures S2–S4), the continuous changes in temperature during the thermal cycling program resulted in a nonsignificant reduction in the fluorescence intensity after thermal treatment (P > 0.05). Since 85.3% of the fluorescence signal remained after 45 thermal cycles, the linkages between the surface coatings and the immobilized amplicons were highly stable under the thermal cycling conditions. The thermal‐induced fluorescence decrease in this study was comparable or even lower to that reported from a study using a copolymer coated on silicon substrates (Damin et al., 2016). A greater loss of fluorescence (31.1%–55.6%) under thermal cycling conditions was also reported for the immobilization of DNA on cyclic olefin copolymer or polymer, polypropylene, polydimethylsiloxane, and glass using 1,4‐diphenylene diisothiocyanate, a homobifunctional cross‐linker (J. Hoffmann, S. Hin et al., 2012). Fluorescence decreases of 40%–56% were reported for DNA immobilization on glass using self‐synthesized linkers of benzene‐1,3,5‐triacetic acid and 1‐ethyl‐3‐(3‐dimethylaminopropyl)‐carbodiimide after 40 thermal cycles (Fedurco, Romieu, Williams, Lawrence, & Turcatti, 2006) and various sulfonated analogs, which are water‐soluble heterobifunctional linking molecules, after 50 thermal cycles (Adessi et al., 2000). The preliminary data obtained in this study showed that thermally stable bonds were established between the immobilized primers and the solid substrate, which confirms the capability of the surface modification strategy to realize SP‐PCR using our developed PC microfluidic platform.
Figure 4.
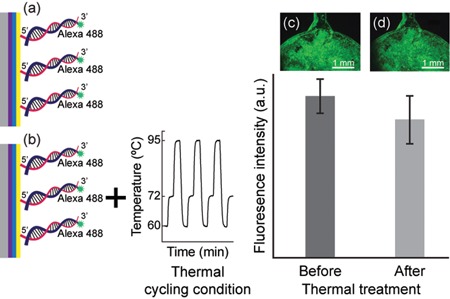
Thermal stability of the surface coating and the immobilized PCR amplicons under the influence of thermal cycling condition. The fluorescence intensities of the immobilized amplicons were compared before and after the thermal treatment. Data are presented as mean ± standard deviation (n = 3). (a) Schematic of PEI‐GA‐coated PC immobilized with amplicons and (c) the corresponding fluorescence image result. (b) The schematic of the thermal treatment and (d) the corresponding fluorescence image result. The amplicons were amplified off‐chip from 10 ng (3.25 × 106 copies) DNA template. GA, glutaraldehyde; PC, polycarbonate; PEI, polyethyleneimine; PCR, polymerase chain reaction [Color figure can be viewed at wileyonlinelibrary.com]
3.5. On‐chip PCR: The effects of surface coating on DNA amplification efficiency
To evaluate the compatibility of the surface coatings inside the microchamber with the DNA amplification efficiency, on‐chip amplifications were carried out without the primer preimmobilization step. A homogeneous temperature distribution was established on the surface of the microchamber during the denaturation, annealing, and extension steps used for SP‐PCR (Supporting Information Figure S4). Figure 5a shows the results for the amplifications of nuc gene (S. aureus), invA gene (Salmonella spp.), and LSU rRNA gene (C. polykrikoides) performed in both pristine and PEI‐GA‐coated microchambers. On the basis of the result of gel electrophoresis, the intensities of the target bands amplified from both pristine and coated microchambers were almost identical. The results were reproducible as indicated by the similar intensities of the amplicons in three targets, demonstrating that the surface coatings did not affect the DNA amplification efficiency.
Figure 5.
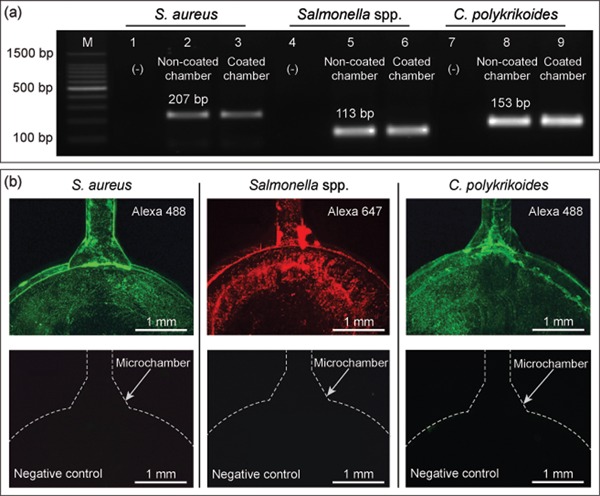
(a) On‐chip PCR results of three microbial targets performed in both pristine and coated microchambers. Lane M is the 100‐bp DNA ladder. Lanes 1–3, 4–6, and 7–9 show the results of amplified target genes of S. aureus, Salmonella spp., and C. polykrikoides, respectively. Lanes 1, 4, and 7 are negative controls. Lanes 2, 5, and 8 are target genes amplified with the pristine microchambers, whereas lanes 3, 6, and 9 are those amplified with the coated microchambers. (b) Fluorescence images of SP‐PCR results of S. aureus, Salmonella spp., and C. polykrikoides and their corresponding negative controls (without DNA templates). SP‐PCR, solid‐phase polymerase chain reaction [Color figure can be viewed at wileyonlinelibrary.com]
3.6. On‐chip SP‐PCR: Simultaneous amplification and detection of microbial targets
After demonstrating the stability of the surface coatings and realizing on‐chip PCR, simultaneous solid‐phase amplification and detection of three microbial targets was realized using the SP‐PCR microdevice. As shown in Figure 5b, the existence of immobilized amplicons was confirmed through the distinct fluorescence signals of Alexa 647 designed for Salmonella spp. and Alexa 488 designed for both S. aureus and C. polykrikoides. The signals indicated that all three targets were successfully amplified and remained covalently attached to the microchambers after the withdrawal of the solution and the subsequent washing steps. The negative controls of each target displayed no fluorescence signal, confirming the absence of nonspecific adsorption of fluorescence‐labeled reverse primers. The result also indicated the multiplexing ability of the microdevice, which was comparable to other multiplexing schemes, as presented in Table 1. In addition, the number of desired targets can be increased by fabricating more reaction chambers on the same substrate. As mentioned above, two typical reactions that take place during SP‐PCR are liquid‐phase and solid‐phase amplifications. Liquid‐phase amplification usually proceeds efficiently because of the free diffusion of the aqueous primers and outcompetes the solid priming reactions hampered by the steric hindrance of dense solid‐support primers (Adessi et al., 2000). To achieve a high efficiency of solid‐phase amplification performance, an asymmetric balance of the aqueous forward and fluorescence‐labeled reverse primers was adopted. The addition of floating forward primers at a low concentration is advantageous in reducing the steric constraint commonly associated with SP‐PCR (Adessi et al., 2000; Khan, Poetter, & Park, 2008) as well as minimizing the competition between the aqueous and surface‐grafted primers. In this study, different ratios of aqueous forward and fluorescence‐labeled reverse primers were examined, and the ratio of 1:4 was found to suit our experimental setup (Supporting Information Figure S5). Since fluorescence signals independently originate from the fluorescence dyes labeled on the reverse primers regardless of successful amplification of DNA, additional controls were performed to exclude the possibility of nonspecific physical adhesion of the fluorescence‐modified reverse primers to the engraved surface of the microchambers. The obtained results demonstrated that nonspecific adhesion of fluorescence‐modified primers did not occur either for the pristine and modified microchambers (Supporting Information Figure S6). Also, a procedure of step‐by‐step surface coating followed by the immobilization of amine‐modified primers is crucial for realizing on‐chip SP‐PCR (Supporting Information Figure S7).
Table 1.
Comparison of some nucleic‐acid‐based methods for multiplex assay
| Reference | Target analytes | Method | Amplification time | Detection limit |
|---|---|---|---|---|
| This work | Staphylococcus aureus |
|
~52 min | 32.5 gDNA copies |
| Salmonella spp. | ||||
| Cochlodinium polykrikoides | ||||
| Damin et al. (2016) | KRAS oncogenic mutations |
|
~75 min | 0.1 ng DNA |
| Song et al. (2017) | Schistosoma spp. |
|
≤40 min | 1 PFU of Zika virus |
| Plasmodium falciparum | ||||
| Salmonella spp. | ||||
| Brugia malayi | ||||
| Strongyloides stercoralis | ||||
| Zika virus | ||||
| HIV virus | ||||
| HPV virus | ||||
| Shao, Zhu, Jin, and Chen (2011) | Salmonella spp. |
|
~62 min | 10–4 ng gDNA |
| Shigella spp. | ||||
| Mollasalehi and Yazdanparast (2013) | Salmonella enteritidis |
|
~90 min | <10 CFU/ml |
| Salmonella typhimurium | ||||
| Mahony et al. (2007) | Influenza virus |
|
~100 min | 50–250 viral genome equivalent |
| Respiratory syncytial virus | ||||
| Rhinovirus | ||||
| Enterovirus | ||||
| Parainfluenza | ||||
| Coronavirus | ||||
| Adenovirus | ||||
| Silva et al. (2011) | Salmonella spp. |
|
~102 min | 103 CFU/ml |
| Salmonella enteritidis | ||||
| Reddington, Tuite, Minogue, and Barry (2014) | Salmonella spp. |
|
~5 hr | Not stated |
| Aeromonas spp. | ||||
| Shigella spp. | ||||
| Escherichia coli | ||||
| Campylobacter spp. | ||||
| Yersinia spp. | ||||
| Kawasaki et al. (2010) | Listeria monocytogenes |
|
~72 min | 2 × 102 CFU/ml |
| Escherichia coli O157:H7 | ||||
| Salmonella spp. | ||||
| Perandin et al. (2004) | Plasmodium falciparum |
|
~69 min | 0.7–1.5 parasites/µl |
| Plasmodium vivax | ||||
| Plasmodium ovale | ||||
| Suo et al. (2010) | Escherichia coli O157:H7 |
|
Not stated | 10−4 ng gDNA |
| Salmonella enterica | ||||
| Listeria monocytogenes | ||||
| Campylobacter jejuni |
Note. CFU, colony‐forming unit; gDNA, genomic DNA; HIV, human immunodeficiency virus; HPV, human papilloma virus; KRAS mutation, Kirsten Ras mutation; LAMP, loop‐mediated isothermal amplification; NASBA, nucleic acid sequence‐based amplification; PFU, plaque‐forming unit; RPA, recombinase polymerase amplification; RT‐PCR, reverse transcription polymerase chain reaction; SP‐PCR, solid‐phase polymerase chain reaction; TSPE, target‐specific primer extension.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Besides, the limit of detection of SP‐PCR microdevice was determined using 10‐fold dilution series of S. aureus gDNA varying from 3.25 to 3.25 × 106 copies. For comparison, homogeneous PCR, which was normal on‐chip PCR, was also performed similarly to evaluate the efficiency of SP‐PCR. The collected on‐chip PCR products were captured by the PEI‐GA‐coated microchambers, which were then washed to measure the fluorescence signals of the immobilized amplicons. To confirm the accuracy of dilutions, off‐chip PCR reactions were first carried out using a commercial thermal cycler, and the amplicons were analyzed using gel electrophoresis. As shown in Supporting Information Figure S8, the intensities of target bands decreased with decreasing concentration of DNA template, confirming the precision of the dilution series. Figure 6 shows the sensitivity comparison between SP‐PCR and homogeneous PCR and their corresponding fluorescence images. As shown in Figure 6a, the efficiency of SP‐PCR was generally lower than that of the homogeneous PCR probably due to the steric interactions between the freely diffusing primers and solid‐support primers in SP‐PCR, as mentioned above. Despite this lower efficiency, the fluorescence signals of the immobilized amplicons in SP‐PCR were clearly observed compared with homogeneous PCR. The lowest amount of DNA template at which fluorescence signal can still be observed was 32.5 copies of gDNA. The signals increased with increasing amount of DNA template and almost remained stable when the amount of DNA template was 3.25 × 105 copies, indicating the saturation of the surface coating with the amplicons.
Figure 6.

(a) Comparison of efficiency of SP‐PCR and homogeneous PCR using S. aureus as a model pathogen. Each experiment was repeated three times. The initial DNA template was varied with 10‐fold dilution series from 3.25 to 3.25 × 106 copies of gDNA. (b) Fluorescence images of SP‐PCR and homogeneous PCR at each concentration of gDNA (scale bar = 1 mm). gDNA, genomic DNA; SP‐PCR, solid‐phase polymerase chain reaction [Color figure can be viewed at wileyonlinelibrary.com]
4. CONCLUSION
In summary, we developed a LOC platform for the performance of SP‐PCR to detect two foodborne agents and a microalga. The simple and reliable immobilization of amine‐modified primers for SP‐PCR was successfully realized by a robust two‐step surface modification of PC with PEI and GA. The chemical coatings on the surface of the microchambers exhibited a high stability under thermal cycling condition with 85.3% of the immobilized PCR amplicons remaining after the thermal treatment. The obtained results showed the possibility of using a monolithic plastic platform for the simultaneous amplification and detection of not only prominent foodborne pathogens but also microalgae that annually cause harmful algal blooms. This versatility ensures the wide and universal applicability of the introduced platform as a portable PON testing device, while extending its ranges to other molecular analyses, including gene expression, mutation analysis, and genotyping.
CONFLICTS OF INTEREST
The authors declare that there is no conflict of interests.
Supporting information
Supporting information
ACKNOWLEDGMENTS
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP; No. NRF‐2017R1A2B4008179).
Pham QN, Trinh KTL, Jung SW, Lee NY. Microdevice‐based solid‐phase polymerase chain reaction for rapid detection of pathogenic microorganisms. Biotechnology and Bioengineering. 2018;115:2194–2204. 10.1002/bit.26734
References
REFERENCES
- Adessi, C. , Matton, G. , Ayala, G. , Turcatti, G. , Mermod, J. J. , Mayer, P. , & Kawashima, E. (2000). Solid phase DNA amplification: Characterisation of primer attachment and amplification mechanisms. Nucleic Acids Research, 28(20), e87–e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, Y. , Koh, C. G. , Boreman, M. , Juang, Y. J. , Tang, I. C. , Lee, L. J. , & Yang, S. T. (2006). Surface modification for enhancing antibody binding on polymer‐based microfluidic device for enzyme‐linked immunosorbent assay. Langmuir, 22(22), 9458–9467. [DOI] [PubMed] [Google Scholar]
- Chin, W. H. , Sun, Y. , Høgberg, J. , Hung, T. Q. , Wolff, A. , & Bang, D. D. (2017). Solid‐phase PCR for rapid multiplex detection of Salmonella spp. at the subspecies level, with amplification efficiency comparable to conventional PCR. Analytical and Bioanalytical Chemistry, 409(10), 2715–2726. [DOI] [PubMed] [Google Scholar]
- Clerc, O. , & Greub, G. (2010). Routine use of point‐of‐care tests: Usefulness and application in clinical microbiology. Clinical Microbiology and Infection, 16(8), 1054–1061. [DOI] [PubMed] [Google Scholar]
- Craw, P. , & Balachandran, W. (2012). Isothermal nucleic acid amplification technologies for point‐of‐care diagnostics: A critical review. Lab on a Chip, 12, 2469–2486. [DOI] [PubMed] [Google Scholar]
- Damin, F. , Galbiati, S. , Ferrari, M. , & Chiari, M. (2016). DNA microarray‐based solid‐phase PCR on copoly (DMA–NAS–MAPS) silicon coated slides: An example of relevant clinical application. Biosensors and Bioelectronics, 78, 367–373. [DOI] [PubMed] [Google Scholar]
- Fedurco, M. , Romieu, A. , Williams, S. , Lawrence, I. , & Turcatti, G. (2006). BTA, a novel reagent for DNA attachment on glass and efficient generation of solid‐phase amplified DNA colonies. Nucleic Acids Research, 34(3), e22–e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foudeh, A. M. , Fatanat Didar, T. , Veres, T. , & Tabrizian, M. (2012). Microfluidic designs and techniques using lab‐on‐a‐chip devices for pathogen detection for point‐of‐care diagnostics. Lab on a Chip, 12, 3249–3266. [DOI] [PubMed] [Google Scholar]
- Gunda, N. S. K. , Singh, M. , Norman, L. , Kaur, K. , & Mitra, S. K. (2014). Optimization and characterization of biomolecule immobilization on silicon substrates using (3‐aminopropyl)triethoxysilane (APTES) and glutaraldehyde linker. Applied Surface Science, 305, 522–530. [Google Scholar]
- Hoffmann, J. , Hin, S. , Stetten, F. , Zengerle, R. , & Roth, G. (2012). Universal protocol for grafting PCR primers onto various lab‐on‐a‐chip substrates for solid‐phase PCR. RSC Advances, 2, 3885–3889. [Google Scholar]
- Hoffmann, J. , Trotter, M. , Stetten, F. , Zengerle, R. , & Roth, G. (2012). Solid‐phase PCR in a picowell array for immobilizing and arraying 100000 PCR products to a microscope slide. Lab on a Chip, 12, 3049–3054. [DOI] [PubMed] [Google Scholar]
- Jang, M. , Park, C. K. , & Lee, N. Y. (2014). Modification of polycarbonate with hydrophilic/hydrophobic coatings for the fabrication of microdevices. Sensors and Actuators, B: Chemical, 193, 599–607. [Google Scholar]
- Jang, M. , Park, S. , & Lee, N. Y. (2014). Polycarbonate bonding assisted by surface chemical modification without plasma treatment and its application for the construction of plastic‐based cell arrays. Sensors and Actuators, A: Physical, 206, 57–66. [Google Scholar]
- Jankowski, P. , Ogończyk, D. , Lisowski, W. , & Garstecki, P. (2012). Polyethyleneimine coating renders polycarbonate resistant to organic solvents. Lab on a Chip, 12, 2580–2584. [DOI] [PubMed] [Google Scholar]
- Kawasaki, S. , Fratamico, P. M. , Horikoshi, N. , Okada, Y. , Takeshita, K. , Sameshima, T. , & Kawamoto, S. (2010). Multiplex real‐time polymerase chain reaction assay for simultaneous detection and quantification of Salmonella species, Listeria monocytogenes, and Escherichia coli O157:H7 in ground pork samples. Foodborne Pathogens and Disease, 7, 549–554. [DOI] [PubMed] [Google Scholar]
- Kersting, S. , Rausch, V. , Bier, F. F. , & von Nickisch‐Rosenegk, M. (2014). Multiplex isothermal solid‐phase recombinase polymerase amplification for the specific and fast DNA‐based detection of three bacterial pathogens. Microchimica Acta, 181(13–14), 1715–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, Z. , Poetter, K. , & Park, D. J. (2008). Enhanced solid phase PCR: Mechanisms to increase priming by solid support primers. Analytical Biochemistry, 375(2), 391–393. [DOI] [PubMed] [Google Scholar]
- Khodakov, D. A. , & Ellis, A. V. (2014). Recent developments in nucleic acid identification using solid‐phase enzymatic assays. Microchimica Acta, 181(13–14), 1633–1646. [Google Scholar]
- Law, J. W. F. , Mutalib, N. S. A. , Chan, K. G. , & Lee, L. H. (2014). Rapid methods for the detection of foodborne bacterial pathogens: Principles, applications, advantages and limitations. Frontiers in Microbiology, 5, 770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazcka, O. , Campo, F. J. D. , & Muñoz, F. X. (2007). Pathogen detection: A perspective of traditional methods and biosensors. Biosensors and Bioelectronics, 22(7), 1205–1217. [DOI] [PubMed] [Google Scholar]
- Lee, K. S. , & Ram, R. J. (2009). Plastic‐PDMS bonding for high pressure hydrolytically stable active microfluidics. Lab on a Chip, 9, 1618–1624. [DOI] [PubMed] [Google Scholar]
- Mahony, J. , Chong, S. , Merante, F. , Yaghoubian, S. , Sinha, T. , Lisle, C. , & Janeczko, R. (2007). Development of a respiratory virus panel test for detection of twenty human respiratory virus by use of multiplex PCR and fluid microbead‐based assay. Journal of Clinical Microbiology, 45, 2965–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier, J. F. , Slater, G. W. , & Mayer, P. (2003). Solid phase DNA amplification: A simple Monte Carlo lattice model. Biophysical Journal, 85(4), 2075–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migneault, I. , Dartiguenave, C. , Bertrand, M. J. , & Waldron, K. C. (2004). Glutaraldehyde: Behavior in aqueous solution, reaction with proteins, and application to enzyme crosslinking. Biotechniques, 37, 790–802. [DOI] [PubMed] [Google Scholar]
- Mollasalehi, H. , & Yazdanparast, R. (2013). Development and evaluation of a novel nucleic acid sequence‐based amplification method using one specific primer and one degenerate primer for simultaneous detection of Salmonella enteritidis and Salmonella typhimurium . Analytica Chimica Acta, 770, 169–174. [DOI] [PubMed] [Google Scholar]
- Niemz, A. , Ferguson, T. M. , & Boyle, D. S. (2011). Point‐of‐care nucleic acid testing for infectious diseases. Trends in Biotechnology, 29(5), 240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh, S. J. , Park, B. H. , Choi, G. , Seo, J. H. , Jung, J. H. , Choi, J. S. , … Seo, T. S. (2016). Fully automated and colorimetric foodborne pathogen detection on an integrated centrifugal microfluidic device. Lab on a Chip, 16, 1917–1926. [DOI] [PubMed] [Google Scholar]
- Pan, J. , Lyu, Z. , Jiang, W. , Wang, H. , Liu, Q. , Tan, M. , … Chen, H. (2014). Stimulation of gene transfection by silicon nanowire arrays modified with polyethyleneimine. ACS Applied Materials and Interfaces, 6(16), 14391–14398. [DOI] [PubMed] [Google Scholar]
- Perandin, F. , Manca, N. , Calderaro, A. , Piccolo, G. , Galati, L. , Ricci, L. , … Chezzi, C. (2004). Development of a real‐time PCR assay for detection of Plasmodium falciparum, Plasmodium vivax, and Plasmodium ovale for routine clinical diagnosis. Journal of Clinical Microbiology, 42, 1214–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piruska, A. , Nikcevic, I. , Lee, S. H. , Ahn, C. , Heineman, W. R. , Limbach, P. A. , & Seliskar, C. J. (2005). The autofluorescence of plastic materials and chips measured under laser irradiation. Lab on a Chip, 5, 1348–1354. [DOI] [PubMed] [Google Scholar]
- Reddington, K. , Tuite, N. , Minogue, E. , & Barry, T. (2014). A current overview of commercially available nucleic acid diagnostics approaches to detect and identify human gastroenteritis pathogens. Biomolecular Detection and Quantification, 1, 3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao, Y. , Zhu, S. , Jin, C. , & Chen, F. (2011). Development of multiplex loop‐mediated isothermal amplification‐RFLP (mLAMP‐RFLP) to detect Salmonella spp. and Shigella spp. in milk. International Journal of Food Microbiology, 148, 75–79. [DOI] [PubMed] [Google Scholar]
- Shin, Y. , Perera, A. P. , Kim, K. W. , & Park, M. K. (2013). Real‐time, label‐free isothermal solid‐phase amplification/detection (ISAD) device for rapid detection of genetic alteration in cancers. Lab on a Chip, 13, 2106–2114. [DOI] [PubMed] [Google Scholar]
- Silva, D. S. P. , Canato, T. , Magnani, M. , Alves, J. , Hirooka, E. Y. , & de Oliveira, T. C. R. M. (2011). Multiplex PCR for the simultaneous detection of Salmonella spp. and Salmonella enteritidis in food. International Journal of Food Science & Technology, 46, 1502–1507. [Google Scholar]
- Song, J. , Liu, C. , Mauk, M. G. , Rankin, S. C. , Lok, J. B. , Greenberg, R. M. , & Bau, H. H. (2017). Two‐stage isothermal enzymatic amplification for concurrent multiplex molecular detection. Clinical Chemistry, 63, 714–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suo, B. , He, Y. , Paoli, G. , Gehring, A. , Tu, S. I. , & Shi, X. (2010). Development of an oligonucleotide‐based microarray to detect multiple foodborne pathogens. Molecular and Cellular Probes, 24, 77–86. [DOI] [PubMed] [Google Scholar]
- Turner, L. M. , Alsterberg, C. , Turner, A. D. , Girisha, S. K. , Rai, A. , Havenhand, J. N. , … Godhe, A. (2016). Pathogenic marine microbes influence the effects of climate change on a commercially important tropical bivalve. Scientific Reports, 6, 32413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visciano, P. , Schirone, M. , Berti, M. , Milandri, A. , Tofalo, R. , & Suzzi, G. (2016). Marine biotoxins: Occurrence, toxicity, regulatory limits and reference methods. Frontiers in Microbiology, 7, 1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , & Ozdemir, P. (2009). Microfluidic DNA amplification—A review. Analytica Chimica Acta, 638(2), 115–125. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information