

Abstract
Vaccination is one of the most effective interventions in global health. The worldwide vaccination programs significantly reduced the number of deaths caused by infectious agents. A successful example was the eradication of smallpox in 1979 after two centuries of vaccination campaigns. Since the first variolation administrations until today, the knowledge on immunology has increased substantially. This knowledge combined with the introduction of cell culture and DNA recombinant technologies revolutionized vaccine design. This review will focus on vaccines against human viral pathogens, recent developments on vaccine design and cell substrates used for their manufacture. While the production of attenuated and inactivated vaccines requires the use of the respective permissible cell substrates, the production of recombinant antigens, virus‐like particles, vectored vaccines and chimeric vaccines requires the use – and often the development – of specific cell lines. Indeed, the development of novel modern viral vaccine designs combined with, the stringent safety requirements for manufacture, and the better understanding on animal cell metabolism and physiology are increasing the awareness on the importance of cell line development and engineering areas. A new era of modern vaccinology is arriving, offering an extensive toolbox to materialize novel and creative ideas in vaccine design and its manufacture.
Keywords: Cell lines, Cell substrates, Chimeric vaccines, Viral vaccines, Virus‐like particles
In the past, animal cell culture and DNA recombinant technologies were decisive for vaccine progress, allowing the development of inactivated, attenuated and subunit protein based vaccines. Today, synthetic biology further expands vaccine design originating a myriad of different vaccines as chimeric ones, still under development but holding great expectations. This review focuses on vaccines against human viral pathogens, recent developments on vaccine design and cell substrates used for their manufacture. A new era of modern vaccinology is arriving, offering an extensive toolbox to materialize novel and creative ideas in vaccine design and its manufacture.
Abbreviations:
Ad, Adenovirus; APC, antigen presenting cell; BCG, Bacille Calmette–Guérin; BEVS, baculovirus expression system; CHO, Chinese hamster ovary; EMA, European Medicines Agency; FDA, Food and Drug Administration; HA, hemagglutinin; HBV, hepatitis B virus; HCV, hepatitis C virus; HEK, human embryonic kidney; HIV, human immunodeficiency virus; HPV, human papillomavirus; MDCK, Madin Darby canine kidney; NA, neuraminidase; NAb, neutralizing antibody; RCA, replication competent adenovirus; VLP, virus‐like particle
1 Introduction
1.1 Viral vaccines: A historical perspective
Since the first use of a vaccine by Edward Jenner, vaccination became an indispensable procedure. In the 20th century, diseases including poliomyelitis, measles, mumps, rubella and others caused an estimated 39 million infections in the United States of America alone; vaccines however rendered these infections uncommon in the 21st century [1]. Indeed, the past 50 years have witnessed tremendous advances in the development of viral vaccines. All in all, vaccination has reduced global mortality rates caused by infectious diseases by approximately three million people per year [2, 3].
Inoculation, or variolation in the case of smallpox virus, was the first systematic practice to protect humans against infectious agents (Box 1) [4, 5, 6]. This procedure became popular in Europe in the 18th century. But by the end of the 18th century Edward Jenner introduced the use of cowpox live viruses to inoculate humans and protect them against smallpox. This procedure was called vaccination and was considered a remarkable advance to variolation [5].
Only in the 20th century, in the 1930s, the way viral vaccines were produced would be revolutionized. The introduction of animal cell cultures allowed in vitro propagation of viruses [7] and paved the way for the development of several vaccines (Fig. 1). Virus attenuation or inactivation became standard practices being, in the 20th century, the most used form of vaccination against viral infections. Between 1950 and 1980 numerous attenuated vaccines for measles, poliovirus or rubella, were developed using cell culture passage [8].
Figure 1.
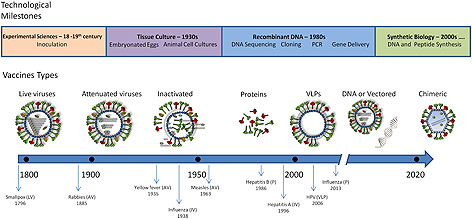
Schematic representation of the vaccine design evolution following the technological advancements. Inoculation was a standard procedure in the 18–19th centuries. The first vaccine against smallpox was introduced by Edward Jenner in 1796 and consisted in inoculating the live virus (LV) cowpox obtained from infected cattle. In the middle of the 20th century animal cell culture became a standard procedure to grow virus allowing the development of attenuated (AV) and inactivated (IV) vaccines. In the late 20th century the DNA recombinant technologies allowed to develop subunit vaccines based on presenting protein antigens (P and VLPs), coding for the antigens (DNA and vectored vaccines) or both. Further advances on genetic engineering originated a myriad of different vaccine designs such as chimeric vaccines still under development but holding great expectations.
Box 1
Inoculation, variolation and vaccination procedures
Inoculation, or variolation in the case of smallpox, was the first systematic practice to protect humans against infectious agents [1, 2]. Initially, live viruses were used through inoculation, i.e., subcutaneous administration of liquid taken from a pustule of a person showing mild symptoms into a healthy person to confer immune protection against the viral pathogen [3]. The term variolation refers solely to the inoculation with smallpox virus (from the Latin name of the virus “variola”). However, inoculation had its attendant risks: the recipients could develop the disease and spread it to others. Additionally the transmission of other diseases via bloodborne route, such as syphilis, was also a concern [2]. At the end of the 18th century, cowpox live viruses (the variant affecting cows) were inoculated in humans to protect against smallpox, the human variant. This procedure was called vaccination, the word is derived from the Latin vacca, meaning cow.
The introduction of recombinant DNA techniques from 1970s onwards enabled a further technological leap in vaccine development. The use of molecular biology and genetic engineering provided not only novel strategies for viral attenuation but also novel types of vaccines. The expression of antigenic proteins from its cDNA was now possible, resulting in the production of large amounts of highly pure antigens and thus, providing more doses. For example, if the first hepatitis B (HBV) vaccine was manufactured by purifying the plasma of infected patients, nowadays recombinant HBV vaccines are produced in CHO cells through genetic cell line development [9]. Advances in animal cell culture technology introduced novel cell lines, improving the production bioprocess by allowing scale up in suspension cultures and providing more competitive yields.
1.2 Viral vaccines: Progress on vaccine design and current challenges
In the beginning of the 21st century we witnessed the approval and commercialization of a novel type of viral vaccines: virus‐like particles (VLPs). VLPs structurally resemble to whole virus but lack genetic material. Gardasil® and Cervarix® are examples of marketed VLPs vaccines developed against human papillomavirus (HPV) [10, 11].
Currently, the toolbox for vaccine development is quite vast. The deeper comprehension of our immune system together with the remarkable technological innovations led to the design of a myriad of different viral vaccines (e.g. DNA vaccines, chimeric or hybrid viral particles, vectored vaccines, replication‐defective viruses, antigens obtained through DNA shuffling of different viral strains) (Fig. 1). Despite the technological progress and the promising prospects of disease control by vaccination, several challenges still stand. For many diseases there are no vaccines available and novel viruses are sure to emerge. If available, vaccines may not be 100% effective in preventing the disease, or their supply may still be insufficient, or their costs may be too high for worldwide distribution, particularly in developing countries [8]. For instance, traditional manufacturing methods for influenza vaccines are not able to rapidly respond to a pandemia. The use of eggs to propagate the virus is one of the bottlenecks, posing manufacturing problems that occasionally result in distribution delays and, in the case of a pandemia, the required number of quality fertile eggs and the capacity to process them, are unlikely to match worldwide needs [12]. Additionally, there is a growing demand for improved vaccine safety, fueled by the manufacturing requirements recommended by the regulatory entities (e.g. FDA or EMA) and by antivaccination groups [8]. Indeed some technologies are outdated and pose safety issues in already licensed vaccines [1]. For example, paralysis side effects were observed after oral vaccination against poliovirus [13] and disseminated infection after Bacille Calmette–Guérin (BCG) vaccination [14]. Hence, some of the older vaccines, or their manufacturing processes, need to be re‐examined and re‐addressed. Indeed, vaccines have been continuously evolving with the technology in order to provide: safer means of immune protection to the patients and reduce possible side effects. The newer molecular technologies can play a role in this demand improving the safety profile of a vaccine both through its design and manufacturing process. For example, recombinant vaccines can be designed to minimize or eliminate virulence reversion, a potential threat of traditional attenuated vaccines. In what concerns manufacturing, the production and purification processes should also be carefully established. At this level, one particular concern is the use and choice of the cell substrate used for viral vaccines production. A thorough characterization of cell substrates is essential to establish quality control systems that allow, for example, confirming that the final product is not contaminated with adventitious agents, oncogenic proteins or DNA from the host cell. Additionally, increasing efforts are being placed in the optimization of animal cell substrates to improve the production capacity and reducing the costs, aiming at worldwide distribution.
The triumphs of traditional viral vaccines are undeniable and inspiring. Notwithstanding, and as introduced previously, a variety of challenges remain for the development of novel vaccines: (i) obtaining efficient protection against pathogens for which no vaccine is yet available and for other viruses certain to emerge; (ii) reducing the costs of vaccine production; (iii) establishing manufacturing platforms allowing worldwide supply; and (iv) increasing vaccine safety to comply with stringent regulatory guidelines [8]. These will be discussed in view of novel molecular biology tools, available for the design of new viral vaccines for human use, and animal cell substrates for their manufacture.
2 Types of viral vaccines
There is a broad diversity of viral vaccines. Commercially available vaccines against viral pathogens can be classified into three general types: live attenuated vaccines; inactivated vaccines and subunit vaccines (Fig. 1) [15]. Within this generally accepted classification, different technological approaches can be applied, giving rise to the design of several sub‐types of vaccines.
2.1 Live attenuated vaccines
Live attenuated viral vaccines use live viruses and were the first type of vaccines to be used. Historically, live viruses were initially used through inoculation. Although inoculation procedures are still used in veterinary vaccination, for human use this is no longer the case. The only type of live viruses used for human vaccination are live attenuated (licensed) or vectored vaccines (under clinical development). There are several approaches to attenuate viruses. One of the most used procedures involves growing the viruses in foreign hosts, as animal cell cultures, where they replicate poorly. Several vaccines have been developed using this procedure including poliovirus, measles or rubella vaccines [8]. Today, live attenuated vaccines can be obtained through additional molecular strategies such as: reassortment (e.g. influenza and rotavirus) [16], mutation or deletion of viral genes (e.g. dengue virus) [17], or codon deoptimization (e.g. poliovirus) [18]. One concern related with live attenuated vaccines is the possibility of reversion into a virulent form, as seen for the first polio oral vaccine. In this context, inactivated vaccines provide safer alternatives to attenuated vaccines.
2.2 Inactivated vaccines
Inactivated vaccines are generated by killing or destroying the pathogen. This can be achieved by chemical means, using for example formalin or formaldehyde, or physically by heat or irradiation. Several inactivated viral vaccines are commercially available against, for example, influenza, rabies or poliovirus (Table 1). In addition to presenting a safer profile comparatively to attenuated viral vaccines, inactivated vaccines are also more stable. Although inactivated vaccines are quite effective for some pathogens, for others, they do not induce an effective and/or long‐lasting immunity since they do not give rise to cytotoxic T cells immune response, which is important to efficiently fight intracellular pathogens as viruses. Because of that, multiple doses and adjuvants are often used [19].
Table 1.
Examples of vaccines manufactured using animal cells
| Vaccine type | Description | Cells | Status | Ref. |
|---|---|---|---|---|
| Live/Attenuated | Oral polio vaccine (OPV) – Attenuated vaccine produced by the passage of the virus through non‐human cells | Vero | Licensed | [119] |
| RotaTeq® – Pentavalent vaccine containing (five) live attenuated reassortant rotaviruses | Vero | Licensed | [120] | |
| Varilix® – Lyophilized preparation of attenuated virus derived from the Oka strain, vaccine against varicella | MRC5 | Licensed | [121] | |
| smallpox vaccine ACAM2000 – Attenuated vaccinia‐based smallpox vaccine | Vero | Licensed | [122] | |
| Inactivated | Imovax® – Monovalent vaccine containing inactivated rabies | MRC5 | Licensed | [123] |
| Havrix® – Inactivated hepatitis A vaccine with formaldehyde | MRC5 | Licensed | [124] | |
| Inactivated polio vaccine (IPV) – Trivalent inactivated poliovirus | Vero | Licensed | [125] | |
| Optaflu® –Trivalent cell culture‐derived influenza vaccine | MDCK | Licensed | [90] | |
| Subunit Protein | Flublok® –Recombinant trivalent hemagglutinin (rHA) vaccine produced in insect cell culture using BEVs | expresSF+ | Licensed | [22] |
| Glycoprotein B – recombinant truncated secreted form of gB | CHO | Phase II | [126] | |
| DEN1‐80E – Adjuvanted recombinant envelope protein vaccine to protect against dengue virus | Drosophila S2 | Phase I | [127] | |
| Subunit VLP | GenHevacB® – Vaccine agents hepatitis B composed of PreS1 and preS2 of HBV S‐antigen assembled into HBV‐like particles | CHO | Licensed | [9] |
| Cervarix® – Vaccine against HPV infection composed of HPV L1 capsid protein from HPV16 and HPV18 assembled into a HPV‐like particle | High5 | Licensed | [11] | |
| NCT01596725 – Influenza backbone (M1 structural protein) displaying HA and NA of influenza H1N1 | Sf9 | Phase I | [128] | |
| Subunit Vectored Vaccine | Ad26.ENVA.01 – Ad26 vector for the expression of a modified HIV env glycoprotein (gp140HIV‐1Clade A) | HER.96 | Phase II | [129] |
| ChimeriVAX® – Chimeric YF17D/DEN vaccine against dengue using an attenuated yellow fever virus as a vector coding for PrM and E genes from dengue | Vero | Phase III | [130] | |
| AVX601 – Vaccine against HCMV it uses alphavirus as vector for the expression of HCMV gB/pp65/IE1 | Vero | Phase I | [131] |
2.3 Subunit vaccines
Subunit vaccines contain only parts of the pathogen. These vaccines use immunodominant antigens, specific parts of the virus (full proteins or peptides) known to stimulate the generation of neutralizing antibodies (Nabs). Subunit viral vaccines are a further development of inactivated viral vaccines but instead of generating antibodies against all of the pathogen antigens, only one or just a few antigens are used. The real challenge in the development of subunit vaccines is to identify which antigens induce protective immunity [20]. Examples of subunit viral vaccines available are the hepatitis B vaccine (e.g. GSK's Engerix® or Merck's RecombivaxHB®) or the recently approved influenza vaccine Flublok® (Protein Sciences). The subunit vaccines for hepatitis B are produced by recombinant technology using either yeast or CHO cells [9, 21]. The Flublok® vaccine is a recombinant trivalent hemagglutinin (rHA) vaccine produced in insect cell culture using the baculovirus expression system [22]. Similarly to inactivated vaccines, subunit vaccines are stable and are considered safer than live attenuated vaccines. Their main disadvantage is the lower immune response elicited, when compared to live attenuated, requiring co‐administration of adjuvants and often multiple doses [15].
Complex vaccine designs such as VLPs, DNA vaccines, vectored vaccines and vectored VLPs vaccines are often classified as subunit vaccines since they only provide a few antigens of the pathogen, either in the form of protein or genetic material. For the purpose of this review we will further describe these types of vaccines.
VLPs are particles that self‐assemble from virus‐derived structural antigens, mimicking the three dimensional structure of the pathogen. VLPs are non‐infectious, replication‐defective and are devoid of any genetic material. Because of that, VLPs are considered safer than live attenuated vaccines.
VLPs can be subcategorized in non‐enveloped or enveloped VLPs based on their structure. Non‐enveloped VLPs are typically composed of one or more proteins of the virus that self‐assemble into particles and do not incorporate any host proteins. Enveloped VLPs are more complex structures as they are wrapped in a lipid membrane (an envelope) derived from the producer cell, where target antigens are anchored [23].
The ordered and repetitive structure that VLPs exhibit has excellent self‐adjuvant properties capable of eliciting both innate and adaptive immune responses [24]. Indeed, VLPs can mount an effective immune protection without the use of co‐adjuvants molecules and requiring lower doses than soluble protein antigens (see [25] for a review). Additionally, the VLPs structure favors its uptake by antigen presenting cells (APCs) essential to elicit long‐lasting immune protection [11]. APCs display the foreign antigens complexed with major histocompatibility complexes (MHCs) on their surfaces. T‐cells can recognize these complexes through their T‐cell receptors (TCRs) starting an immune response by stimulating B and CD8+ lymphocytes [24].
The first VLP licensed for human use was the human papillomavirus (HPV) vaccine Gardasil® from Merck, produced in yeast [10] (Table 1). HPV Cervarix® was approved shortly after and is produced using the Baculovirus Expression System (BEVS) that uses insect cells [11]. Cervarix® is a bivalent HPV vaccine (L1 proteins of HPV‐16 and HPV‐18) indicated for the prevention of cervical cancer, was EMA approved in 2007 and FDA approved in 2009.
VLPs structural diversity and functional versatility forecasts the intensification in VLP research and future development of innovative vaccine designs.
DNA vaccines were developed as bacterial plasmids constructed to express an encoded antigen. They are administered in vivo, by transfecting the patient cells, eliciting both cellular and humoral immunity [26]. DNA vaccines have been approved for veterinary use (e.g. West Nile‐Innovator® vaccine for horses [27] and Apex®‐IHN for Salmonid fish [28]) but not yet for human use. One of the initial bottlenecks associated with DNA vaccines was the low potency which has been improved through the development of more efficient delivery systems in vivo and improved formulations with adjuvants (for a review on DNA vaccines see [26] ).
More complex subunit vaccines have been proposed. The engineering of modern vaccines based on different types of vectored vaccines and VLPs will be further discussed in the next section.
3 Modern viral vaccines and future trends
The increasing knowledge on immunology combined with the recent advances in molecular biology and engineering tools have provided tools for novel vaccine designs. Efforts have been made to design safer and more efficient vaccines. As a result, new and more complex vaccines are being developed, including chimeric VLPs, vectored VLPs and vectored vaccines.
3.1 Chimeric‐VLPs vaccines
The development of VLPs has revealed to be truly challenging for many viruses (e.g. rotavirus, hepatitis C virus (HCV) and influenza) [29, 30, 31, 32]. Therefore a subset of VLPs containing viral proteins from unrelated viruses, known as chimeric VLPs, has emerged. In non‐enveloped VLPs heterologous antigen presentation can be achieved through molecular fusion or chemical conjugation. [33, 34]. Fueled by the existence of an already licensed HBV vaccine, HBV‐based chimeric VLPs are one of the most used scaffolds for protein or peptide display. The engraftment of foreign sequences into HBV capsid protein can however, interfere with correct VLP assembly [35, 36]. A solution to maintain the VLP integrity is to combine two different proteins in hybrid VLPs. Strong and broad immune responses were generated using hybrid VLPs composed of L1 and L2 proteins from HPV and different HIV glycoproteins [37, 38].
Heterologous presentation of glycoproteins can also be achieved by pseudotyping enveloped VLPs with envelope proteins of unrelated virus [39]. Indeed, pseudotyping retroviral vectors has been widely explored in the field of gene therapy [40]. The most common examples of pseudotyping in vaccine development are the use of HCV or HIV envelopes to pseudotype murine leukeamia virus (MLV)‐derived VLPs [41, 42, 43, 44, 45]. Pseudotyping can also generate bivalent vaccine candidates when glycoproteins from different virus are incorporated in the same envelope, as Newcastle disease virus and influenza A virus [46], or rabies virus and ebolavirus [47]. Whole heterologous antigen presentation is also being explored as strategy to maintain structural authenticity of target antigens and thus, to meet quality criteria otherwise difficult to achieve when expressing the isolated target antigen. Indeed, expressing the HIV envelope proteins gp120 individually hinders the natural conformational arrangement into trimers, hampering its correct presentation. It has been shown that monomeric gp120 subunits fail to induce broad NAbs and do not prevent infection [48]. Conversely, Hammonds et al. demonstrated, in guinea pig models, enhanced neutralizing antibody response elicited by HIV VLPs when compared to the same HIV isolate subunit protein [49].
3.2 Vectored VLPs vaccines
VLPs can also incorporate small molecules or proteins; we will refer to these as vectored VLPs. For simplicity whenever genetic material is incorporated into a viral particle we will refer to them as vectored vaccines (section 3.3). VLPs (and vectored vaccines) can also be chimeric, coding and displaying the antigen required for the vaccination purpose, but having in their composition proteins from other viruses (serving as a display scaffold or providing gene delivery function).
The potential of some viral capsid proteins to encapsulate and deliver small molecules, nucleic acids or proteins has been largely explored by drug development and gene therapy companies to mediate cargo delivery to specific cells, tissues or organs. Cell and tissue specificity can be achieved using tissue‐specific viral envelope proteins [45, 50], embedded antibodies [51, 52] or cellular receptors [53]. Vaccine development has translated these strategies into a new class of VLP‐based vaccines known as vectored VLPs vaccines [41]. Encapsidation of small molecules like doxorubicin [54] in cell‐free assembled VLPs can potentiate the activity of anti‐cancer vaccines [55]. Full proteins or peptides can be fused with capsid proteins, with or without cleavable linkers, to be released upon cell entry. This can be applied to soluble proteins [56], specific antigen epitopes delivered as peptides [57], or to deliver functional immunomodulatory proteins as transcription factors, cytokines, TLR ligands or adjuvants [58, 59].
VLPs surface modification can be used for several purposes; polymers as polyethylenimine act as adjuvants [60] while polyethylene glycol (PEG) can increase VLPs stability [61, 62]. In addition VLPs can be covered with immunostimulatory agents. For example rabbit hemorrhagic fever VLPs covered with the glycolipid α‐galactosylceramide showed enhanced immunogenicity when compared to non‐coated VLPs [63].
3.3 Vectored vaccines
The challenge of achieving efficient transfections with DNA vaccines has led to the development of vectored vaccines. Vectored vaccines are herein defined as non‐pathogenic vehicles carrying genes from viruses, with the purpose of expressing them in the host cells to elicit an immune protective response. Vectored vaccines, like DNA vaccines, have the potential to elicit both B and T cell immunity. However, since vectored vaccines require live viruses, the vaccine is less stable than plasmid DNA vaccines and more difficult to manufacture. Several vaccine vectors coding viral genes from immunodeficiency virus, yellow fever virus or human cytomegalovirus have been tested in experimental candidate vaccines (Table 1).
DNA or RNA can be explored as cargos for gene delivery. All viruses harbor a DNA or RNA genome; therefore, several strategies for the transport of nucleic acids can be explored. Vaccines delivering nucleic acids often aim to promote the transient expression of the cargo encoded protein with immunostimulatory properties. Immunogens are often encoded to promote antigen presentation via MHC‐I [64]. Nevertheless these nucleic acids can also induce the expression of an immunomodulatory protein as IL10 [65], or block immunosupressor pathways with the delivery of small interfering RNA (siRNA) [66].
The concept of vectored vaccines has been extensively explored. Ideally, vectored vaccines for human use are replication‐defective viruses able to deliver the antigen(s) to the target cell in the form of genetic material. Nevertheless, replication competent vectored vaccines have been developed for veterinary use [67]. A myriad of viral vectors are available based on adenovirus, poxvirus, herpes simplex virus, cytomegalovirus, alphaviruses, or adeno‐associated virus. Adenoviruses have been perhaps the most used vaccine vector. Adenoviruses are easily produced in Human Embryonic Kidney 293 (HEK 293) or PER.C6® cells to high titers and express the transgene(s) to high levels activating the innate immune system and stimulating dendritic cell maturation [68]. Several candidate vaccines against HIV use adenovirus. Adenovirus type 5 vaccines expressing individual antigens of HIV such as the envelope protein or Gag were shown to induce CD8+ T cell responses. The clinical study from Merck evaluating recombinant adenovirus type 5 (rAd5) vectors expressing clade B Gag, Pol, and Nef in high‐risk individuals terminated however unsuccessfully. But yet another clinical trial provided the first signal of an HIV vaccine efficacy. In the latter a recombinant canarypox vector vaccine, ALVAC‐HIV, was evaluated on a prime envelope gp120 protein boost regimen and it demonstrated a 31% protective effect against acquisition of HIV infection in low‐risk subjects [69].
Ongoing efforts on the development of next generation recombinant adenovirus vector‐based vaccines for HIV include, the development of alternative serotypes of adenoviral vectors, the incorporation of adenoviral vectors into heterologous vector prime‐boost regimens, and the use of adenoviral vectors to express novel HIV antigens to improve immunologic coverage of circulating viruses [70].
3.4 Hybrid vaccine designs
A new concept that is also emerging is the in vivo expression of VLPs, or plasmoVLPs, using DNA vaccination technology. PlasmoVLPs combine DNA and VLP types of vaccines in one. Given the complementary potential of DNA and VLP as vaccines, Bellier and colleagues designed a single vector form that would be endowed with their respective advantageous properties, the plasmo‐retroVLPs [71]. These are plasmids administered as DNA vaccines that express all the components needed for retrovirus‐based VLPs formation. These retroVLPs are based on murine leukemia virus (MLV). Plasmo‐retroVLPs combine the straightforwardness, the stability, the large‐scale and low‐cost production of DNA vaccines with the immunostimulatory properties of VLP vaccines, circumventing laborious VLP vaccine production, purification and formulation processes. [72].
This technology was shown to produce correctly folded and assembled VLPs in vivo, both in mice and macaques, inducing earlier and significantly higher levels of NAbs than simply expressing the viral envelope proteins in a plasmid DNA. Also circulating NAbs were maintained over a longer period of time and could be boosted by additional plasmo‐retroVLP delivery [72]. Later studies, using animal models, showed that plasmo‐VLPs were able to prime APCs and to generate protective immune response for HCV, HIV and appear as a promising strategy for the treatment of HPV‐induced cancers [41, 56, 57, 73].
Other hybrid designs of vaccines are the cases of vectored vaccines combined with vectored VLPs, i.e. the display of the antigens in the form of protein is combined with the delivery of the antigen in the form of genetic material, either in DNA or RNA. This is the case of alphaviruses and flaviviruses that can be produced without a portion of its genome and further modified to code for a foreign viral antigen of interest. When used for immunization, these VLPs have only one replication cycle [74]. These types of vaccines are still in the development phase holding great potential.
3.5 Other trends in viral vaccine design
Another current interest is the development of universal viral vaccines. This would be of great interest for highly diverse viruses as influenza. A universal flu vaccine could provide protection regardless of the strain or subtype of the circulating virus, allowing the vaccine to be prepared in advance and be ready to use in the event of a pandemic [75, 76]. It would also have several advantages over currently available seasonal vaccines, requiring less frequent administration, ideally just one [77]. The development of universal vaccines relies on the utilization of highly conserved antigenic targets. The main difficulty lies on accessibility to conserved antigenic epitopes since these are usually less exposed and are generally weak immunogens. Therefore, in a universal vaccine the immunogenicity of the identified conserved antigens has to be increased to induce protective immunity. In the case of Influenza virus several promising candidate antigens have been proposed, including the hemagglutinin (HA), matrix protein (M1 and M2e), nucleoprotein and neuraminidase (NA) proteins (for an extensive revision see [77]).
The strategy to find conserved antigenic targets is not confined to influenza virus. The same concept is of interest for the also highly variable viruses HIV and HCV. The research efforts for finding a conserved and immunogenic epitope on HIV have been exhaustive. The envelope glycoprotein gp120 has been one of the primary targets as it is largely exposed on the virion surface and antibody response in an HIV infected individual is, to a great extent, directed against it [78]. The variability and extensive glycosylation of gp120 has however posed difficulties. HIV‐infected patients do generate a broad neutralization response [79], and a large number of broadly neutralizing antibodies (bNAbs) against HIV have been discovered in the last decade [80]. There are four major epitopes on the Env surface targeted by bNAbs and the very existence of such, indicates it is possible to elicit a neutralizing response and develop effective vaccines (for a review on the several strategies ongoing see [81]).
Similarly to HIV, the diversity of HCV virus also poses major challenges for vaccine development. Yet, broadly cross‐neutralizing monoclonal antibodies, directed against the E2 envelope glycoprotein and, efficiently blocking HCV infection of various genotypes have been recently isolated from HCV‐chronically infected patients and immunized animals [82, 83, 84]. Other studies also identified essential and conserved epitopes on E2 that may be used for vaccine design, thus renewing the hope on the development of a sterilizing antibody‐based vaccine for HCV [85, 86]. A major challenge hampering a faster progress on the development of HCV vaccines is still present: the inexistence of suitable animal models able to reproduce all stages of infection and immune response found in humans and chimpanzees [87].
In summary vaccination strategies aiming to deliver conserved and immunogenic antigens have generated the development of several vaccine types as, recombinant proteins, VLPs, vectored vaccines, and others. All of them having advantages and disadvantages as described above (sections 2.3 and 3.1 through 3.4). Common to most viral vaccines, traditional or recombinant, is the need of cell substrates – biological factories – enabling the production of the vaccine. In the next section, we will revisit the history of cell substrates use in viral vaccines manufacture and how the recent developments in animal cell culture development and engineering are leveraging the production of both traditional and modern vaccines.
4 Cell substrates for viral vaccine manufacture: A historical perspective
In 1796, Edward Jenner demonstrated the protective effect of cowpox against smallpox infection using matter from fresh cowpox lesions from a milkmaid for direct immunization of a healthy eight year‐old boy [5]. Human skin can, thus, be considered the first cell substrate for viral vaccine manufacture. But it was not until the late 1930s that cell culture was first used, when John Enders, Thomas Weller and Frederick Robbins, were able to propagate poliovirus in non‐nervous tissue, namely human embryonic skin and muscle tissue. This breakthrough would grant them the Nobel Prize in Physiology and Medicine in 1954 [88].
Until the 1990s, tissue and primary cultures were the privileged cell substrate choices for the production of viral vaccines. A few diploid cell lines (cell strains) derived from lung tissue were also used to grow virus for the production of: a rubella vaccine (WI‐38 cells); vaccines against poliovirus, hepatitis A virus, rabies virus and varicella virus (MRC‐5 cells); and rotavirus and rabies vaccines (FRhL‐2).
The last decade of the 20th century witnessed the use of the first continuous, non‐tumorigenic cell line – Vero – derived from the kidney of an African green monkey, for the manufacture of an inactivated poliomyelitis vaccine (IPV) and later a live‐attenuated oral poliomyelitis vaccine (OPV) [89]. The approval of Vero cell line for vaccine manufacture was a landmark as it opened an important precedent in the use of cell substrates by initiating the transition of cell strains to cell lines. It was also during those years that the use of tumorigenic cell lines for the production of vaccines against emerging viral pathogens started to be discussed. In the beginning of the 21st century, the first tumorigenic cell line was introduced in the production of a live viral vaccine. PER.C6®, a proprietary cell line from Crucell, was designed for the production of a replication‐defective adenovirus vectored HIV vaccine. This was also the first example of a “designer‐cell substrate”. Shortly after PER.C6®, Madin Darby canine kidney (MDCK) cell line was proposed for the production of inactivated influenza virus vaccines. MDCK cell line was isolated from the kidney of an apparently normal dog and was spontaneously immortalized. Some variants of MDCK cells have been described as highly tumorigenic, while others are not [90]. Optaflu® (EMA approved in 2009) and Flucelvax® (FDA approved in 2012) were the first human flu vaccines produced using the MDCK cell line, putting Novartis as the pioneer company transitioning from egg‐based to mammalian cell‐culture production of influenza vaccines [91].
The first decade of the 21st century also witnessed the approval of insect cell lines and the BEVS platform for the production of a human viral vaccine, Cervarix® (GSK). In 2013, the BEVS system gained further attention when the first recombinant vaccine for flu, solely based in protein, Flublok® (Protein Sciences) was approved [92].
By the end of 2012, the use of cell lines derived from human tumors started to be considered for vaccine manufacture, when the 'Vaccines and Related Biological Products Advisory Committee' (VRBPAC) recognized that “the current repertoire of cell substrates is inadequate for manufacture of certain types of new vaccines” and that “cell lines derived from tumors may be the optimal and in some cases the only cell substrate that can be used to propagate certain vaccine viruses” [93].
5 Cell line development and engineering: Current and future trends
It has been a long progress since the first cell substrates have been used for the production of viral vaccines until the more recent developments of “designer cells”. But this journey is likely to be just in the beginning. The complex and hypervariable organisms such as HIV or HCV, the high pathogenicity of ebolavirus and the emergence of new diseases as Middle East respiratory syndrome coronavirus (MERS‐CoV), first detected in 2012 [94], are examples that demonstrate that a continuous effort is required to develop new and more efficient vaccines. The availability of sophisticated technologies also leveraged the creation of modern vaccinology approaches, as described in section 3, aiming at the development of vaccines eliciting better humoral and cellular immune responses, safer and with lower costs. These approaches often require the development and engineering of new cell lines. For example, vectored vaccines rely on the production of replication‐defective virus requiring suitable transcomplementing cell lines that allow the production of these particles. Likewise, the production of complex enveloped VLPs will profit from the establishment of stable cell lines where a continuous production is enabled, facilitating bioreaction and purification processes (avoiding the need to produce plasmid DNA or baculovirus stocks and their subsequent removal from the final product). On the other hand, to increase the production yields and reduce the costs cell engineering strategies may be envisaged [95].
For simplicity purposes, we shall distinguish cell line development from cell line engineering, as illustrated in Fig. 2. In this section, we will discuss the main genetic approaches in cell line development and engineering for the manufacture of viral vaccines and vaccine candidates. Some examples on genetic approaches in cell line development and engineering are given in Tables 2 and 3, respectively.
Figure 2.

Genetic approaches in cell line development and engineering for the manufacture of vaccines. (A) Conceptually, cell line development encloses all the steps leading to a clonal culture with extended life‐span. At the end of development, some of the cell lines already support viral propagation (e.g. influenza virus propagates in MDCK, Vero, HEK293, etc). Spontaneous immortalization is usually based on chromosomal rearrangements – not externally induced – resulting in the loss of senescence‐related and/or activation of immortalizing genes; cell line development without external genetic manipulation is called simple cell line establishment. Induced immortalization can rely on chemical or physical agents (e.g. methylcholanthrene or UV) or on the integration of immortalizing genes (telomerase, SV40 large T antigen, adenovirus E1 genes, etc.). Depending on the immortalizing gene, the cell lines can also support the propagation of partially deleted viral vectors (such as HEK293 or PER.C6, immortalized with human adenovirus E1 gene being denominated as transcomplementing cell lines for E1‐deleted adenovirus). Cell line development also encloses genetic manipulation specifically conceived to support the production of a particular virus or viral components, resulting in stable (or inducible) cells lines that constitutively (or upon induction) express the viral components [33]. (B) Finally, cell line engineering can be defined as a genetic manipulation designed to improve the production performance of a pre‐existing cell line, mostly for increasing specific titers. Strategies to facilitate the production process or to provide the produced particles with specific traits can also be categorized as cell line engineering.
Table 2.
Examples of genetic manipulation in cell line development for vaccine manufacture
| Cell origin | Cell line name | Type of vaccines | Genetic manipulation in cell line development | Ref. |
|---|---|---|---|---|
| HER (Human embryonic retinoblasts) | HER.911 | Designed for vectored vaccines (and viral vectors) based on E1‐deleted replication deficient human adenovirus type 5 | Expression of the Ad serotype 5 (Ad5) sequence (nucleotides 79 – 5789) | [99] |
| HER | PER.C6 | Constitutive expression of Ad5 E1A‐ and E1B‐encoding sequences (nucleotides 459–3510) under the control of the human phosphoglycerate kinase (PGK) promoter. | [132] | |
| Primary human amniocytes | N52.E6 | Constitutive expression of Ad5 E1A‐ and E1B‐encoding sequences (nucleotides 505–3522) under the control of the mouse PGK promoter | [101] | |
| HeLa | GH329 | Constitutive expression of Ad5 E1A‐ and E1B‐encoding sequences (nucleotides 511–3924) under the control of the mouse PGK promoter | [100] | |
| Primary muscovy | AGE1.CR | Propagation of attenuated MVA | Constitutive expression of human Ad5 E1A | [105] |
| duck cells | AGE1.CA | under control of the human PGK promoter and | ||
| (retina (CR), somite (CS) and amnion membrane (CA)) | AGE1.CS | Propagation of other viruses has also been demonstrated | bovine growth hormone polyadenylation site and E1B is under control of herpes simplex virus thymidine kinase (HSV‐TK) promoter and polyadenylation site. | |
| CHO (Chinese hamster ovary cells) | CHO‐HBsAg | Hepatitis B surface antigen HBsAg | Constitutive expression of the HBsAg genes selected under methotrexate | [106] |
| HEK 293 (Human embryonic kidney cells) | HEK‐G | VLP‐based vaccine for rabies | Constitutive expression of the rabies virus G protein for continuous production of rabies virus VLPs | [107] |
| RK13 (Rabbit kidney cells) | J12#26 | VLP‐based vaccine for Japanese encephalitis (JEV) | centerConstitutive expression of C , prM, and E proteins from JEV, strain Beijing‐1 | [133] |
| BHK‐21 (Hamster kidney cells) | BJ‐ME | Constitutive expression of codon‐optimized cDNA encoding JEV prM and E protein, strain SA14‐14‐2 | [108] | |
| MDCK (Canine kidney cells) | MDCK SFS | Attenuated influenza vaccine (attenuated by NS1 deletion) | Establishment of two stable MDCK cell lines that show inducible expression of the allele B NS1 protein | [134] |
Table 3.
Examples of genetic manipulation in cell line engineering for vaccine manufacture
| Cell origin | Cell line name | Type of vaccines | Genetic manipulation in cell line engineering | Ref. |
|---|---|---|---|---|
| AGE1.CR® | AGE1.CR.pIX | Propagation of attenuated MVA | Stable expression of the structural gene pIX from human adenovirus to increase the titers | [105] |
| Propagation of other viruses has also been demonstrated | of poxvirus in AGE1® cell lines | |||
| MDCK (Canine kidney cells) | MDCK shIRF7 | Recombinant attenuated vaccine for seasonal flu | Stable down‐regulation of interferon regulatory factor 7 (IRF7), for increased viral titers (seven fold) | [116] |
| MDCK (Canine kidney cells) | MDCK siat7e | Recombinant attenuated vaccine for seasonal flu | Stable expression of siat7 to overcome anchorage dependency of MDCK cell line, also leading to increased titers (20‐fold). | [114] |
| HEK 293 (Human embryonic kidney cells) | 293‐SCARB2 | Wild type inactivated vaccine for hand‐foot‐mouth disease (HFMD) | Over‐expression of human scavenger receptor class B, member 2 (SCARB2) to increase human enterovirus type 71 (EV71) and coxsackievirus A group type 16 (CA16) titers (100‐ to 1000‐fold for 293 and RD and 10‐fold for Vero) | [117] |
| RD (Human rhabdomyosarcoma cells) | RD‐SCARB2 | |||
| Vero (Green monkey kidney cells) | Vero‐SCARB2 | |||
| 293 GP (Human embryonic kidney cells) | 293 GP siCD81 | RetroVLPs for foreign antigen display | Stable knock‐down of CD81 for the production of CD81‐free retroVLPs | [42] |
5.1 Cell line development
Cell line development can be defined as a genetic manipulation leading to a clonal culture with extended life‐span.
5.1.1 Cell line immortalization and designer cell lines
One of the major challenges in animal cell culture was the immortalization and establishment of continuous cell lines. Spontaneous immortalization is a rare event in human cells but it can be promoted by oncogenes. It was soon observed that some viruses had the ability to induce immortality. Viral genes or genomes are thus used to immortalize cells being the most classical example the HEK 293 cells, established in the early 1970s by transformation of cultures of normal human embryonic kidney cells with sheared adenovirus 5 DNA [96]. Currently several immortalizing viral genes or sequences have been identified, e.g. adenovirus E1 genes, simian virus 40 large T antigen, HPV E6 and E7 genes, Epstein‐Barr virus LMP, EBNA 1 and 2 genes, and human T‐cell leukemia virus sequence Tax. Cellular oncogenes, and mutant p53 gene can also immortalize cells [97].
HEK 293 cells are widely used to propagate vectored vaccines based on replication‐defective adenovirus lacking E1 genes (E1A and E1B) on their genome. A major issue when using HEK 293 cells for adenoviral vectored vaccines production is the formation of replication competent adenovirus (RCA) [98] through recombination of the virus genome with the adenovirus DNA used in the process of immortalization of HEK 293. E1‐only transcomplementing “designer cell lines” appeared then to respond to the need of safer cell lines than HEK 293 (Table 2). The overall strategy was based on reducing the Ad5 E1 sequences for cell immortalization and led to the development of the PER.C6® cell line. PER.C6® are human embryonic retinoblasts (HER) cells, transformed with Ad5 sequence 459 to 3510, comprising the E1A and E1B encoding sequences only. In addition E1A and E1B are under control of the human PGK promoter (that compensate for the removal of the 5' endogenous E1A promoter) [99]. A few years later, other E1‐transcomplementing, or 'designer', cell lines from HeLa cells [100] and human aminocytes [101] were established, to further reduce sequence homology, between E1 immortalizing DNA (integrated in the cells genome) and vectored vaccine viral genome, reducing the possibility of double homologous recombination and generation of RCAs. The cells derived from human aminocytes are the proprietary cell line CAP® (Cevec). Further details on the history of E1‐transcomplementing cell lines have been reviewed in [102].
Both PER.C6® and CAP® cell lines have already been tested and proven suitable for the propagation of other viruses than adenovirus, including Influenza [103, 104].
Cell line development comprises any genetic manipulation performed even when the gene stably expressed is not directly related to the virus to be produced. This is the case of AGE1® (Probiogen) cell lines, developed to obviate the dependence on primary chicken embryo fibroblasts (CEF) for the production of modified vaccinia Ankara virus (MVA) [105]. AGE1® cell lines are derived from muscovy duck – retina (CR), somite (CS) and amnion membrane (CA) cells – and were immortalized with E1A and E1B genes from human Ad5 (Table 2). Interestingly, CA cells were reported to be refractory to MVA replication, whereas evident differences for the anchorage‐dependence phenotype were observed for CR and CS cells: CR cells can be cultivated without anchorage whereas CS cells proliferate in monolayers [105]. This output highlights the importance of the cell background in the properties of the final cell line, some of which, very relevant to establish robust and cost‐effective vaccine production processes. AGE1.CR® cell line was also already subjected to cell line engineering (see section 5.2).
5.1.2 Stable cell line development for constitutive expression of vaccines
Another type of cell line development concerns the constitutive expression of viral components for stable production of vaccines based on viral proteins or viral particles (e.g. VLPs). An example on this type of cell line development is the establishment of CHO‐HBsAG for the production of the HBV vaccine [106] (Table 2). Cell line development for stable production of VLPs has been used when the viral proteins are not cytotoxic. Two very recent examples are HEK‐G cells developed for the production of rabies VLPs [107] and BJ‐BM cells for the production of Japanese encephalitis virus VLPs [108] (Table 2). When the proteins are cytotoxic, transient approaches are used instead.
Stable production of VLPs in animal cells has faced a difficult competition with the impressive yields obtained with the BEVS system [109]. However, BEVS system may pose a particularly challenging downstream processing difficulty, since the separation of the vaccine products from the baculovirus contaminants may be complicated [110]. This is the case when the VLPs are enveloped. Because of this, stable insect cell lines systems based on recombinase mediated cassette exchange have been developed [111]. Another example, still in the context of stable insect cell lines, reports stable double transfection with HIV Gag and HIV‐gp140 for the production of an enveloped HIV‐Gag VLP presenting trimeric gp140 on the surface [112]. Other groups are engineering the baculovirus in order to make them non‐replicative thus, reducing baculovirus contamination to minimal levels [113].
5.2 Cell line engineering
Cell line engineering comprises genetic manipulation to improve the production performance of a pre‐existing cell line mostly, but not exclusively, to increase the titers (Fig. 2). Reports on cell line engineering for viral vaccine manufacture are relatively scarce. In fact, gene engineering of animal cells is labor intensive and time‐consuming and may be perceived as low‐rewarding for a cell line that already supports vaccine production. Still, Table 3 shows that several efforts on cell line engineering for viral vaccine manufacture have been made, particularly in the last five years, highlighting the increasing interest in this field.
One of the first studies on cell line engineering was reported by Chu et al. (2009) [114]. After a transcriptome based study, comparing anchorage‐dependent and anchorage‐independent HeLa cells, siat7e (ST6GalNacV) was identified as one of the genes playing a role in controlling the cell adhesion [115] and chosen to be over expressed in MDCK cell line. MDCK‐siat7e cell line not only lost the anchorage dependent phenotype as it produced 20 times higher HA titers than the parental MDCK cells.
MDCK cell line has also been subjected to genetic engineering for the down‐regulation of pro‐apoptotic intracellular innate defenses, more specifically, interferon regulatory factor 7 (IRF7), for increased influenza titers [116]. The authors could not link the inhibition of IFN‐alpha/beta to titer increase, suggesting that RNA interference mediated knockdown of IRF7 in MDCK cells enhances virus propagation through other unknown mechanisms.
As previously mentioned, AGE1.CR® cell line was, after development, subjected to further cell line engineering. Stable expression of the structural gene pIX from human adenovirus increased the titers of poxvirus, the main viral product manufactured in AGE1® cells (Table 3). The authors suggested that the improvement in viral titers is mediated via induction of heat shock pathway [108].
More recently, the overexpression of human scavenger receptor class B member 2 (SCARB2), was reported to improve the production of human enterovirus type 71 (EV71) and coxsackievirus A group type 16 (CA16) in HEK 293, human rhabdomyosarcoma (RD) and Vero cell lines [117]. Indeed, SCARB2 had been previously identified as one of the receptors of EV71 and CA16, suggesting its overexpression could be a potential gene engineering approach to increase the susceptibility of expressing cells to these viruses.
Cell line engineering can also be used to provide specific features to a viral particle. For example, our group has used stable expression of short‐hairpin mediated RNA interference to knockdown CD81 expression in a HEK 293 derived cell line stably producing retroVLPs [42] (Table 3). This cell line is currently being used for the production of chimeric HCV vaccine candidates as it consists of E1/E2 HCV glycoproteins anchored on a retroviral core VLP. These particles assemble at the cellular membranes, naturally incorporating several host cell proteins including, and to a large extent, CD81 [118]. The later elicits an unwanted humoral immune response against the retroVLPs hampering an effective anti‐E1E2 immune response of the HCV candidate vaccine [43].
The field of cell line engineering has witnessed significant investment but it has been more focused on improving the production of recombinant proteins (mainly, monoclonal antibodies) with several pathways being targeted including: apoptosis, protein secretory pathways, glycosylation, and glycolysis. Leveraged by the growing knowledge on virus‐host interactions an intensification of cell engineering for vaccine production is also expected. A review on cell line engineering for the manufacture of vaccines and viral vectors can be found in Rodrigues et al. (2014) [95].
6 Concluding remarks: A new era – new cell lines and new engineering approaches for new vaccines
Replacing a cell line for an already marketed vaccine requires substantial investment. Hence, when it comes to changing to a new cell substrate, the manufacturing industry tends to be very conservative. However, the resistance is starting to break, being the shortage of vaccine supply one of the driving forces. For instance, Novartis already uses MDCK for the production of Optaflu® (or Flucelvax®), although maintaining the production of the oldest version, Fluvirin® which uses the traditional embryonated eggs manufacture process. New designs for old vaccines are also being pursued. Protein Sciences used insect cells for the production of Flublok®, exclusively based on protein (HA), the first vaccine for seasonal flu that avoids the use of replication‐competent influenza virus. The introduction of novel vaccine designs and cell lines should face less resistance in the case of diseases for which no vaccines or treatment are available, particularly those that are life threatening (or severely debilitating).
On one hand globalization is accelerating the spreading of many diseases that were contained to certain areas and countries; on the other hand, certain viruses have mechanisms of immune evasion for which some traditional vaccines no longer offer a solution. This pushes for further investment of the scientific and medical community, and flexibility in exploring advanced solutions. In addition, the available scientific knowledge and technological development is tremendous, and it is expected to continue to grow almost exponentially, offering an extensive toolbox to materialize novel, ambitious and creative ideas in vaccine design. Thus, when it comes to viral vaccine design and cell line development and engineering, we are certain to enter into a whole new era of modern vaccinology.
Biographical Information
Ana Sofia Coroadinha has a Degree in Biochemistry and a PhD in Chemical Engineering received from 'Instituto de Tecnologia Química e Biológica' (Universidade Nova de Lisboa). Since July 2009, she has been the Head of Cell Line Development and Molecular Biotechnology Lab at Animal Cell Technology Unit, iBET, Portugal.
Her current research interests focus on the manufacturing of viral vectors and vaccines using animal cells. It comprises the development of cell substrates, engineering recombinant virus and viral particles, and developing enabling tools for virus research.

Acknowledgements
The authors acknowledge Fundação Para a Ciência e a Tecnologia (FCT), Portugal, for financial support: PTDC/EBB‐BIO/118621/2010, PTDC/EBB‐BIO/118615/2010 and EXPL/BBB‐BIO/1129/2013). H. R. Soares and M. R. Guerreiro acknowledge FCT for the PhD grant (SFRH/BD/81598/2011 and SFRH/BD/90685/2012).
The authors declare no financial or commercial conflict of interest.
References
- 1. Nabel, G. J. , Designing tomorrow's vaccines. N. Engl. J. Med. 2013, 368, 551–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. World Health Organization, Global Alliance for Vaccines and Immunization (GAVI) Fact sheet 169, revised March 2001, http://www.who.int/mediacentre/factsheets/fs169/en.
- 3. Ehreth, J. , The value of vaccination: A global perspective. Vaccine 2003, 21, 4105–4117. [DOI] [PubMed] [Google Scholar]
- 4. McCollum, A. M. et al., Poxvirus viability and signatures in historical relics. Emerg. Infect. Dis. 2014, 20, 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Riedel, S. , Edward Jenner and the history of smallpox and vaccination. Proc. Bayl. Univ. Med. Cent. 2005, 18, 21–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bonanni P. S. J., Vaccine evolution, in: Garçon, N., Stern, P. L., Cunningham, A. L., Stanberry, L. R. (Eds.), Understanding Modern Vaccines: Perspectives in Vaccinology, Elsevier 2011, pp. 1–24.
- 7. Weller, T. H. , Robbins, F. C. , Enders, J. F. , Cultivation of poliomyelitis virus in cultures of human foreskin and embryonic tissues. Proc. Soc. Exp. Biol. Med. 1949, 72, 153–155. [DOI] [PubMed] [Google Scholar]
- 8. Plotkin, S. A. , Vaccines: Past, present and future. Nat. Med. 2005, 11, (Suppl.) S5–S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jungers, P. , Chauveau, P. , Courouce, A. M. , Devillier, P. et al., Immunogenicity of the recombinant GenHevac B Pasteur vaccine against hepatitis B in chronic uremic patients J. Infect. Dis. 1994, 169, 399–402. [DOI] [PubMed] [Google Scholar]
- 10. Garland, S. M. , Hernandez‐Avila, M. , Wheeler, C. M. , Perez, G. et al., Quadrivalent vaccine against human papillomavirus to prevent anogenital diseases. N. Engl. J. Med. 2007, 356, 1928–1943. [DOI] [PubMed] [Google Scholar]
- 11. Kirnbauer, R. , Booy, F. , Cheng, N. , Lowy, D. R. , Schiller, J. T. , Papillomavirus L1 major capsid protein self‐assembles into virus‐like particles that are highly immunogenic. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 12180–12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nicholson, K. G. , Influenza and vaccine development: A continued battle. Expert Rev. Vaccines 2009, 8, 373–374. [DOI] [PubMed] [Google Scholar]
- 13. World Health Oranisation, The relathionship between persisting spinal paralysis and poliomyelites vaccine – results of a ten‐year enquiry. Bull. World Health Organ. 1982, 60, 231–242. [PMC free article] [PubMed]
- 14. Hoft, D. F. , Leonardi, C. , Milligan, T. , Nahass, G. T. et al., Clinical reactogenicity of intradermal bacille Calmette‐Guerin vaccination. Clin. Infect. Dis. 1999, 28, 785–790. [DOI] [PubMed] [Google Scholar]
- 15. Strugnell, R., Zepp, F., Cunningham, A., Tantawichien, T., Vaccine antigens, in: Garçon, N., Stern, P. L., Cunningham, A. L., Stanberry, L. R. (Eds.), Understanding Modern Vaccines: Perspectives in Vaccinology, Elsevier 2011, pp. 61–88.
- 16. Clark, H. F. , Offit, P. A. , Plotkin, S. A. , Heaton, P. M. , The new pentavalent rotavirus vaccine composed of bovine (strain WC3)‐human rotavirus reassortants. Pediatr. Infect. Dis. J. 2006, 25, 577–583. [DOI] [PubMed] [Google Scholar]
- 17. Men, R. , Bray, M. , Clark, D. , Chanock, R. M. , Lai, C. J. , Dengue type 4 virus mutants containing deletions in the 3' noncoding region of the RNA genome: Analysis of growth restriction in cell culture and altered viremia pattern and immunogenicity in rhesus monkeys. J. Virol. 1996, 70, 3930–3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mueller, S. , Papamichail, D. , Coleman, J. R. , Skiena, S. , Wimmer, E. , Reduction of the rate of poliovirus protein synthesis through large‐scale codon deoptimization causes attenuation of viral virulence by lowering specific infectivity. J. Virol. 2006, 80, 9687–9696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baxter, D. , Active and passive immunity, vaccine types, excipients and licensing. Occup. Med. (London) 2007, 57, 552–556. [DOI] [PubMed] [Google Scholar]
- 20. Levine, M. M., Lagos, R., Esparza, J., Vaccines and vaccination in historical perspective, in: Levine, M. M. (Ed.), New Generation Vaccines, Marcel Dekker, Inc Ed. 1997, pp. 1–19.
- 21. Hepatitis B vaccines prepared from yeast by recombinant DNA techniques: Memorandum from a WHO meeting. Bull. WHO 1985, 63, 57–61. [PMC free article] [PubMed]
- 22. Cox, M. M. , Hollister, J. R. , FluBlok, a next generation influenza vaccine manufactured in insect cells. Biologicals 2009, 37, 182–189. [DOI] [PubMed] [Google Scholar]
- 23. Kushnir, N. , Streatfield, S. J. , Yusibov, V. , Virus‐like particles as a highly efficient vaccine platform: Diversity of targets and production systems and advances in clinical development. Vaccine 2012, 31, 58–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ludwig, C. , Wagner, R. , Virus‐like particles‐universal molecular toolboxes. Curr Opin Biotechnol 2007, 18, 537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grgacic, E. V. , Anderson, D. A. , Virus‐like particles: Passport to immune recognition. Methods 2006, 40, 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu, M. A. , DNA vaccines: An historical perspective and view to the future. Immunol. Rev. 2011, 239, 62–84. [DOI] [PubMed] [Google Scholar]
- 27. Davis, B. S. , Chang, G. J. , Cropp, B. , Roehrig, J. T. et al., West Nile virus recombinant DNA vaccine protects mouse and horse from virus challenge and expresses in vitro a noninfectious recombinant antigen that can be used in enzyme‐linked immunosorbent assays J. Virol. 2001, 75, 4040–4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Purcell, M. K. , Nichols, K. M. , Winton, J. R. , Kurath, G. et al., Comprehensive gene expression profiling following DNA vaccination of rainbow trout against infectious hematopoietic necrosis virus. Mol. Immunol. 2006, 43, 2089–2106. [DOI] [PubMed] [Google Scholar]
- 29. Bellier, B. , Klatzmann, D. , Virus‐like particle‐based vaccines against hepatitis C virus infection. Expert Rev. Vaccines 2013, 12, 143–154. [DOI] [PubMed] [Google Scholar]
- 30. Li, T. , Lin, H. , Zhang, Y. , Li, M. et al., Improved characteristics and protective efficacy in an animal model of E. coli‐derived recombinant double‐layered rotavirus virus‐like particles. Vaccine 2014, 32, 1921–1931. [DOI] [PubMed] [Google Scholar]
- 31. Subbarao, K. , Matsuoka, Y. , The prospects and challenges of universal vaccines for influenza. Trends Microbiol. 2013, 21, 350–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thompson, C. M. , Petiot, E. , Lennaertz, A. , Henry, O. , Kamen, A. A. , Analytical technologies for influenza virus‐like particle candidate vaccines: Challenges and emerging approaches. Virol. J. 2013, 10, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jariyapong, P. , Xing, L. , van Houten, N. E. , Li, T. C. et al., Chimeric hepatitis E virus‐like particle as a carrier for oral‐delivery. Vaccine 2013, 31, 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rivera‐Hernandez, T. , Hartas, J. , Wu, Y. , Chuan, Y. P. et al., Self‐adjuvanting modular virus‐like particles for mucosal vaccination against group A streptococcus (GAS). Vaccine 2013, 31, 1950–1955. [DOI] [PubMed] [Google Scholar]
- 35. Skrastina, D. , Petrovskis, I. , Petraityte, R. , Sominskaya, I. et al., Chimeric derivatives of hepatitis B virus core particles carrying major epitopes of the rubella virus E1 glycoprotein. Clin. Vaccine Immunol 2013, 20, 1719–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Billaud, J. N. , Peterson, D. , Barr, M. , Chen, A. et al., Combinatorial approach to hepadnavirus‐like particle vaccine design J. Virol. 2005, 79, 13656–13666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schellenbacher, C. , Kwak, K. , Fink, D. , Shafti‐Keramat, S. et al., Efficacy of RG1‐VLP vaccination against infections with genital and cutaneous human papillomaviruses. J. Invest. Dermatol. 2013, 133, 2706–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Visciano, M. L. , Diomede, L. , Tagliamonte, M. , Tornesello, M. L. et al., Generation of HIV‐1 virus‐like particles expressing different HIV‐1 glycoproteins. Vaccine 2011, 29, 4903–4912. [DOI] [PubMed] [Google Scholar]
- 39. Chua, A. J. , Vituret, C. , Tan, M. L. , Gonzalez, G. et al., A novel platform for virus‐like particle‐display of flaviviral envelope domain III: Induction of Dengue and West Nile virus neutralizing antibodies. Virol. J. 2013, 10, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Coroadinha, A. S. , Gama‐Norton, L. , Amaral, A. I. , Hauser, H. et al., Production of retroviral vectors: Review. Curr. Gene Ther. 2010, 10, 456–473. [DOI] [PubMed] [Google Scholar]
- 41. Garrone, P. , Fluckiger, A. C. , Mangeot, P. E. , Gauthier, E. et al., A prime‐boost strategy using virus‐like particles pseudotyped for HCV proteins triggers broadly neutralizing antibodies in macaques. Sci. Transl. Med. 2011, 3, 94ra71. [DOI] [PubMed] [Google Scholar]
- 42. Rodrigues, A. F. , Guerreiro, M. R. , Santiago, V. M. , Dalba, C. et al., Down‐regulation of CD81 tetraspanin in human cells producing retroviral‐based particles: Tailoring vector composition. Biotechnol. Bioeng. 2011, 108, 2623–2633. [DOI] [PubMed] [Google Scholar]
- 43. Rodrigues, A. F. , Guerreiro, M. R. , Castro, R. , Tomás, H. et al., Down‐regulation of CD81 in human cells producing HCV‐E1/E2 retroVLPs. BMC Proc. 2011, 5, 8 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang, W. , Chen, X. , Xue, C. , Du, Y. et al., Production and immunogenicity of chimeric virus‐like particles containing porcine reproductive and respiratory syndrome virus GP5 protein. Vaccine 2012, 30, 7072–7077. [DOI] [PubMed] [Google Scholar]
- 45. Watson, D. J. , Kobinger, G. P. , Passini, M. A. , Wilson, J. M. et al., Targeted transduction patterns in the mouse brain by lentivirus vectors pseudotyped with VSV, Ebola, Mokola, LCMV, or MuLV envelope proteins. Mol. Ther. 2002, 5, 528–537. [DOI] [PubMed] [Google Scholar]
- 46. Shen, H. , Xue, C. , Lv, L. , Wang, W. et al., Assembly and immunological properties of a bivalent virus‐like particle (VLP) for avian influenza and Newcastle disease. Virus Res. 2013, 178, 430–436. [DOI] [PubMed] [Google Scholar]
- 47. Blaney, J. E. , Marzi, A. , Willet, M. , Papaneri, A. B. et al., Antibody quality and protection from lethal Ebola virus challenge in nonhuman primates immunized with rabies virus based bivalent vaccine. PLoS Pathog. 2013, 9, e1003389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Flynn, N. M. , Forthal, D. N. , Harro, C. D. , Judson, F. N. et al., Placebo‐controlled phase 3 trial of a recombinant glycoprotein 120 vaccine to prevent HIV‐1 infection. J. Infect. Dis. 2005, 191, 654–665. [DOI] [PubMed] [Google Scholar]
- 49. Hammonds, J. , Chen, X. , Fouts, T. , DeVico, A. et al., Induction of neutralizing antibodies against human immunodeficiency virus type 1 primary isolates by Gag‐Env pseudovirion immunization. J. Virol. 2005, 79, 14804–14814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Horowitz, E. D. , Weinberg, M. S. , Asokan, A. , Glycated AAV vectors: Chemical redirection of viral tissue tropism. Bioconjug. Chem. 2011, 22, 529–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Goyvaerts, C. , De Groeve, K. , Dingemans, J. , Van Lint, S. et al., Development of the nanobody display technology to target lentiviral vectors to antigen‐presenting cells. Gene Ther. 2012, 19, 1133–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kaliberov, S. A. , Kaliberova, L. N. , Buggio, M. , Tremblay, J. M. et al., Adenoviral targeting using genetically incorporated camelid single variable domains. Lab. Invest. 2014, 94, 893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kitai, Y. , Fukuda, H. , Enomoto, T. , Asakawa, Y. et al., Cell selective targeting of a simian virus 40 virus‐like particle conjugated to epidermal growth factor. J. Biotechnol. 2011, 155, 251–256. [DOI] [PubMed] [Google Scholar]
- 54. Zhao, Q. , Chen, W. , Chen, Y. , Zhang, L. et al., Self‐assembled virus‐like particles from rotavirus structural protein VP6 for targeted drug delivery. Bioconjug. Chem. 2011, 22, 346–352. [DOI] [PubMed] [Google Scholar]
- 55. Shi, X., Li, C., Gao, S., Zhang, L. et al., Combination of doxorubicin‐based chemotherapy and polyethylenimine/p53 gene therapy for the treatment of lung cancer using porous PLGA microparticles. Colloids Surf., B 2014, 122, 498–504. [DOI] [PubMed]
- 56. Huret, C. , Desjardins, D. , Miyalou, M. , Levacher, B. et al., Recombinant retrovirus‐derived virus‐like particle‐based vaccines induce hepatitis C virus‐specific cellular and neutralizing immune responses in mice. Vaccine 2013, 31, 1540–1547. [DOI] [PubMed] [Google Scholar]
- 57. Jain, S. , Patrick, A. J. , Rosenthal, K. L. , Multiple tandem copies of conserved gp41 epitopes incorporated in gag virus‐like particles elicit systemic and mucosal antibodies in an optimized heterologous vector delivery regimen. Vaccine 2010, 28, 7070–7080. [DOI] [PubMed] [Google Scholar]
- 58. Wang, B. Z. , Xu, R. , Quan, F. S. , Kang, S. M. et al., Intranasal immunization with influenza VLPs incorporating membrane‐anchored flagellin induces strong heterosubtypic protection. PLoS One 2010, 5, e13972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wu, D. T. , Roth, M. J. , MLV based viral‐like‐particles for delivery of toxic proteins and nuclear transcription factors. Biomaterials 2014, 35, 8416–8426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Keswani, R. , Su, K. , Pack, D. W. , Efficient in vitro gene delivery by hybrid biopolymer/virus nanobiovectors. J. Control Release 2014, 192, 40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mendez, N. , Herrera, V. , Zhang, L. , Hedjran, F. et al., Encapsulation of adenovirus serotype 5 in anionic lecithin liposomes using a bead‐based immunoprecipitation technique enhances transfection efficiency. Biomaterials 2014, 35, 9554–9561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sheppard, N. C. , Brinckmann, S. A. , Gartlan, K. H. , Puthia, M. et al., Polyethyleneimine is a potent systemic adjuvant for glycoprotein antigens. Int. Immunol. 2014, 26, 531–538. [DOI] [PubMed] [Google Scholar]
- 63. McKee, S. J. , Young, V. L. , Clow, F. , Hayman, C. M. et al., Virus‐like particles and alpha‐galactosylceramide form a self‐adjuvanting composite particle that elicits anti‐tumor responses. J. Control Release 2012, 159, 338–345. [DOI] [PubMed] [Google Scholar]
- 64. Astray, R. M. , Ventini, D. C. , Boldorini, V. L. , Silva, F. G. et al., Rabies virus glycoprotein and immune response pattern using recombinant protein or recombinant RNA viral vectors. Vaccine 2014, 32, 2829–2832. [DOI] [PubMed] [Google Scholar]
- 65. Boettler, T. , Cunha‐Neto, E. , Kalil, J. , von Herrath, M. , Can an immune‐regulatory vaccine prevent HIV infection? Expert Rev. Anti Infect. Ther. 2012, 10, 299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gantier, M. P. , Tong, S. , Behlke, M. A. , Irving, A. T. et al., Rational design of immunostimulatory siRNAs. Mol. Ther. 2010, 18, 785–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Esaki, M. , Godoy, A. , Rosenberger, J. K. , Rosenberger, S. C. et al., Protection and antibody response caused by turkey herpesvirus vector Newcastle disease vaccine. Avian Dis. 2013, 57, 750–755. [DOI] [PubMed] [Google Scholar]
- 68. Morelli, A. E. , Larregina, A. T. , Ganster, R. W. , Zahorchak, A. F. et al., Recombinant adenovirus induces maturation of dendritic cells via an NF‐kappaB‐dependent pathway. J. Virol. 2000, 74, 9617–9628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rerks‐Ngarm, S. , Pitisuttithum, P. , Nitayaphan, S. , Kaewkungwal, J. et al., Vaccination with ALVAC and AIDSVAX to prevent HIV‐1 infection in Thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [DOI] [PubMed] [Google Scholar]
- 70. Barouch, D. H. , Novel adenovirus vector‐based vaccines for HIV‐1. Curr. Opin. HIV AIDS 2010, 5, 386–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bellier, B. , Dalba, C. , Clerc, B. , Desjardins, D. et al., DNA vaccines encoding retrovirus‐based virus‐like particles induce efficient immune responses without adjuvant. Vaccine 2006, 24, 2643–2655. [DOI] [PubMed] [Google Scholar]
- 72. Bellier, B. , Huret, C. , Miyalou, M. , Desjardins, D. et al., DNA vaccines expressing retrovirus‐like particles are efficient immunogens to induce neutralizing antibodies. Vaccine 2009, 27, 5772–5780. [DOI] [PubMed] [Google Scholar]
- 73. Lescaille, G. , Pitoiset, F. , Macedo, R. , Baillou, C. et al., Efficacy of DNA vaccines forming e7 recombinant retroviral virus‐like particles for the treatment of human papillomavirus‐induced cancers. Hum. Gene Ther. 2013, 24, 533–544. [DOI] [PubMed] [Google Scholar]
- 74. Ljungberg, K. , Whitmore, A. C. , Fluet, M. E. , Moran, T. P. et al., Increased immunogenicity of a DNA‐launched Venezuelan equine encephalitis virus‐based replicon DNA vaccine. J. Virol. 2007, 81, 13412–13423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Epstein, S. L. , Price, G. E. , Cross‐protective immunity to influenza A viruses. Expert Rev. Vaccines 2010, 9, 1325–1341. [DOI] [PubMed] [Google Scholar]
- 76. Kesik‐Brodacka, M. , Plucienniczak, G. , A universal flu vaccine. Acta Biochim. Pol. 2014, 61, 523–530. [PubMed] [Google Scholar]
- 77. Arinaminpathy, N. , Ratmann, O. , Koelle, K. , Epstein, S. L. et al., Impact of cross‐protective vaccines on epidemiological and evolutionary dynamics of influenza. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 3173–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kwong, P. D. , Wyatt, R. , Robinson, J. , Sweet, R. W. et al., Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. van Gils, M. J. , Euler, Z. , Schweighardt, B. , Wrin, T. , Schuitemaker, H. , Prevalence of cross‐reactive HIV‐1‐neutralizing activity in HIV‐1‐infected patients with rapid or slow disease progression. AIDS 2009, 23, 2405–2414. [DOI] [PubMed] [Google Scholar]
- 80. Klein, F. , Mouquet, H. , Dosenovic, P. , Scheid, J. F. et al., Antibodies in HIV‐1 vaccine development and therapy. Science 2013, 341, 1199–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rathore, U. , Kesavardhana, S. , Mallajosyula, V. V. , Varadarajan, R. et al., Immunogen design for HIV‐1 and influenza. Biochim. Biophys. Acta 2014, 1844, 1891–1906. [DOI] [PubMed] [Google Scholar]
- 82. Keck, Z. Y. , Li, T. K. , Xia, J. , Gal‐Tanamy, M. et al., Definition of a conserved immunodominant domain on hepatitis C virus E2 glycoprotein by neutralizing human monoclonal antibodies. J. Virol. 2008, 82, 6061–6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Law, M. , Maruyama, T. , Lewis, J. , Giang, E. et al., Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 2008, 14, 25–27. [DOI] [PubMed] [Google Scholar]
- 84. Tarr, A. W. , Owsianka, A. M. , Timms, J. M. , McClure, C. P. et al., Characterization of the hepatitis C virus E2 epitope defined by the broadly neutralizing monoclonal antibody AP33. Hepatology 2006, 43, 592–601. [DOI] [PubMed] [Google Scholar]
- 85. Khan, A. G. , Whidby, J. , Miller, M. T. , Scarborough, H. et al., Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 2014, 509, 381–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kong, L. , Giang, E. , Nieusma, T. , Kadam, R. U. et al., Hepatitis C virus E2 envelope glycoprotein core structure. Science 2013, 342, 1090–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Couto, L. B., Kolykhalov, A. A., Animal models for HCV study, in: Tan, S. L. (Ed.) Hepatitis C Viruses: Genomes and Molecular Biology, Norfolk (UK) 2006, pp. 353–372.
- 88. Norrby, E. , Prusiner, S. B. , Polio and Nobel prizes: Looking back 50 years. Ann. Neurol. 2007, 61, 385–395. [DOI] [PubMed] [Google Scholar]
- 89. Montagnon, B. J., Polio and rabies vaccines produced in continuous cell lines: A reality for Vero cell line. Dev. Biol. Stand. 1989, 70, 27–47. [PubMed]
- 90. Omeir, R. L. , Teferedegne, B. , Foseh, G. S. , Beren, J. J. et al., Heterogeneity of the tumorigenic phenotype expressed by Madin‐Darby canine kidney cells. Comp. Med. 2011, 61, 243–250. [PMC free article] [PubMed] [Google Scholar]
- 91. Doroshenko, A. , Halperin, S. A. , Trivalent MDCK cell culture‐derived influenza vaccine Optaflu (Novartis Vaccines). Expert Rev. Vaccines 2009, 8, 679–688. [DOI] [PubMed] [Google Scholar]
- 92. Yang, L. P. , Recombinant trivalent influenza vaccine (flublok®): A review of its use in the prevention of seasonal influenza in adults. Drugs 2013, 73, 1357–1366. [DOI] [PubMed] [Google Scholar]
- 93. Cell lines derived from human tumors for vaccine manufacture in: FDA Briefing Document – Vaccines and Related Biological Products Advisory Committee Meeting. 2012, http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/BloodVaccinesandOtherBiologics/VaccinesandRelatedBiologicalProductsAdvisoryCommittee/UCM319573.pdf.
- 94. de Groot, R. J. , Baker, S. C. , Baric, R. S. , Brown, C. S. et al., Middle East respiratory syndrome coronavirus (MERS‐CoV): Announcement of the Coronavirus Study Group. J. Virol. 2013, 87, 7790–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Rodrigues, A. F. , Carrondo, M. , Alves, P. M. , Coroadinha, A. S. , Cellular targets for improved manufacturing of virus‐based‐biopharmaceuticals. Trends Biotechnol. 2014, 32, 602–607. [DOI] [PubMed] [Google Scholar]
- 96. Graham, F. L. , Smiley, J. , Russell, W. C. , Nairn, R. et al., Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977, 36, 59–74. [DOI] [PubMed] [Google Scholar]
- 97. Katakura, Y. , Alam, S. , Shirahata, S. , Immortalization by gene transfection. Methods Cell Biol. 1998, 57, 69–91. [DOI] [PubMed] [Google Scholar]
- 98. Hehir, K. M. , Armentano, D. , Cardoza, L. M. , Choquette, T. L. et al., Molecular characterization of replication‐competent variants of adenovirus vectors and genome modifications to prevent their occurrence. J. Virol. 1996, 70, 8459–8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Fallaux, F. J. , Kranenburg, O. , Cramer, S. J. , Houweling, A. et al., Characterization of 911: A new helper cell line for the titration and propagation of early region 1‐deleted adenoviral vectors. Hum. Gene Ther. 1996, 7, 215–222. [DOI] [PubMed] [Google Scholar]
- 100. Gao, G. P. , Engdahl, R. K. , Wilson, J. M. , A cell line for high‐yield production of E1‐deleted adenovirus vectors without the emergence of replication‐competent virus. Hum. Gene Ther. 2000, 11, 213–219. [DOI] [PubMed] [Google Scholar]
- 101. Schiedner, G. , Hertel, S. , Kochanek, S. , Efficient transformation of primary human amniocytes by E1 functions of Ad5: Generation of new cell lines for adenoviral vector production. Hum. Gene Ther. 2000, 11, 2105–2116. [DOI] [PubMed] [Google Scholar]
- 102. Kovesdi, I. , Hedley, S. J. , Adenoviral producer cells. Viruses 2010, 2, 1681–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Genzel, Y. , Behrendt, I. , Rodig, J. , Rapp, E. et al., CAP, a new human suspension cell line for influenza virus production Appl. Microbiol. Biotechnol. 2013, 97, 111–122. [DOI] [PubMed] [Google Scholar]
- 104. Koudstaal, W. , Hartgroves, L. , Havenga, M. , Legastelois, I. et al., Suitability of PER.C6 cells to generate epidemic and pandemic influenza vaccine strains by reverse genetics. Vaccine 2009, 27, 2588–2593. [DOI] [PubMed] [Google Scholar]
- 105. Jordan, I. , Vos, A. , Beilfuss, S. , Neubert, A. et al., An avian cell line designed for production of highly attenuated viruses. Vaccine 2009, 27, 748–756. [DOI] [PubMed] [Google Scholar]
- 106. Diminsky, D. , Schirmbeck, R. , Reimann, J. , Barenholz, Y. , Comparison between hepatitis B surface antigen (HBsAg) particles derived from mammalian cells (CHO) and yeast cells (Hansenula polymorpha): Composition, structure and immunogenicity. Vaccine 1997, 15, 637–647. [DOI] [PubMed] [Google Scholar]
- 107. Fontana, D. , Kratje, R. , Etcheverrigaray, M. , Prieto, C. , Rabies virus‐like particles expressed in HEK293 cells. Vaccine 2014, 32, 2799–2804. [DOI] [PubMed] [Google Scholar]
- 108. Hua, R. H. , Li, Y. N. , Chen, Z. S. , Liu, L. K. et al., Generation and characterization of a new mammalian cell line continuously expressing virus‐like particles of Japanese encephalitis virus for a subunit vaccine candidate. BMC Biotechnol. 2014, 14, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Cox, M. M. , Recombinant protein vaccines produced in insect cells. Vaccine 2012, 30, 1759–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. van Oers, M. M. , Pijlman, G. P. , Vlak, J. M. , Thirty years of baculovirus‐insect cell protein expression: From dark horse to mainstream technology. J. Gen. Virol. 2015, 96(Pt1), 6–23. [DOI] [PubMed] [Google Scholar]
- 111. Fernandes, F. , Vidigal, J. , Dias, M. M. , Prather, K. L. et al., Flipase‐mediated cassette exchange in Sf9 insect cells for stable gene expression. Biotechnol. Bioeng. 2012, 109, 2836–2844. [DOI] [PubMed] [Google Scholar]
- 112. Tagliamonte, M. , Visciano, M. L. , Tornesello, M. L. , De Stradis, A. et al., HIV‐Gag VLPs presenting trimeric HIV‐1 gp140 spikes constitutively expressed in stable double transfected insect cell line. Vaccine 2011, 29, 4913–4922. [DOI] [PubMed] [Google Scholar]
- 113. Marek, M. , van Oers, M. M. , Devaraj, F. F. , Vlak, J. M. , Merten, O. W. , Engineering of baculovirus vectors for the manufacture of virion‐free biopharmaceuticals. Biotechnol. Bioeng. 2011, 108, 1056–1067. [DOI] [PubMed] [Google Scholar]
- 114. Chu, C. , Lugovtsev, V. , Golding, H. , Betenbaugh, M. , Shiloach, J. , Conversion of MDCK cell line to suspension culture by transfecting with human siat7e gene and its application for influenza virus production. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 14802–14807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Jaluria, P. , Betenbaugh, M. , Konstantopoulos, K. , Frank, B. , Shiloach, J. , Application of microarrays to identify and characterize genes involved in attachment dependence in HeLa cells. Metab. Eng. 2007, 9, 241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Hamamoto, I. , Takaku, H. , Tashiro, M. , Yamamoto, N. , High yield production of influenza virus in Madin Darby canine kidney (MDCK) cells with stable knockdown of IRF7. PLoS One 2013, 8, e59892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Li, X. , Fan, P. , Jin, J. , Su, W. et al., Establishment of cell lines with increased susceptibility to EV71/CA16 by stable overexpression of SCARB2. Virol. J. 2013, 10, 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Segura, M. M. , Garnier, A. , Di Falco, M. R. , Whissell, G. et al., Identification of host proteins associated with retroviral vector particles by proteomic analysis of highly purified vector preparations. J. Virol. 2008, 82, 1107–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Chezzi, C. , Dommann, C. J. , Blackburn, N. K. , Maselesele, E. et al., Genetic stability of oral polio vaccine prepared on primary monkey kidney cells or Vero cells – effects of passage in cell culture and the human gastrointestinal tract. Vaccine 1998, 16, 2031–2038. [DOI] [PubMed] [Google Scholar]
- 120. El Khoury, A. C. , Mast, T. C. , Ciarlet, M. , Markson, L. et al., Projecting the effectiveness of RotaTeq® against rotavirus‐related hospitalizations and deaths in six Asian countries. Hum Vaccin. 2011, 7, 506–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Papaloukas, O. , Giannouli, G. , Papaevangelou, V. , Successes and challenges in varicella vaccine. Ther. Adv. Vaccines 2014, 2, 39–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Greenberg, R. N. , Kennedy, J. S. , ACAM2000: A newly licensed cell culture‐based live vaccinia smallpox vaccine. Expert Opin. Investig. Drugs 2008, 17, 555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Briggs, D. J. , The role of vaccination in rabies prevention. Curr. Opin. Virol. 2012, 2, 309–314. [DOI] [PubMed] [Google Scholar]
- 124. Hens, N. , Habteab Ghebretinsae, A. , Hardt, K. , Van Damme, P. , Van Herck, K. , Model based estimates of long‐term persistence of inactivated hepatitis A vaccine‐induced antibodies in adults. Vaccine 2014, 32, 1507–1513. [DOI] [PubMed] [Google Scholar]
- 125. Bakker, W. A. , Thomassen, Y. E. , van't Oever, A. G. , Westdijk, J. et al., Inactivated polio vaccine development for technology transfer using attenuated Sabin poliovirus strains to shift from Salk‐IPV to Sabin‐IPV. Vaccine 2011, 29, 7188–7196. [DOI] [PubMed] [Google Scholar]
- 126. Zhang, C. , Buchanan, H. , Andrews, W. , Evans, A. , Pass, R. F. , Detection of cytomegalovirus infection during a vaccine clinical trial in healthy young women: Seroconversion and viral shedding. J. Clin. Virol. 2006, 35, 338–342. [DOI] [PubMed] [Google Scholar]
- 127. Coller, B. A. , Clements, D. E. , Bett, A. J. , Sagar, S. L. , Ter Meulen, J. H. , The development of recombinant subunit envelope‐based vaccines to protect against dengue virus induced disease. Vaccine 2011, 29, 7267–7275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Nolan, T. , Roy‐Ghanta, S. , Montellano, M. , Weckx, L. et al., Relative efficacy of AS03‐adjuvanted pandemic influenza A(H1N1) vaccine in children: Results of a controlled, randomized efficacy trial J. Infect. Dis. 2014, 210, 545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Barouch, D. H. , Liu, J. , Peter, L. , Abbink, P. et al., Characterization of humoral and cellular immune responses elicited by a recombinant adenovirus serotype 26 HIV‐1 Env vaccine in healthy adults (IPCAVD 001). J. Infect. Dis. 2013, 207, 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Guy, B. , Barrere, B. , Malinowski, C. , Saville, M. et al., From research to phase III: Preclinical, industrial and clinical development of the Sanofi Pasteur tetravalent dengue vaccine. Vaccine 2011, 29, 7229–7241. [DOI] [PubMed] [Google Scholar]
- 131. Bernstein, D. I. , Reap, E. A. , Katen, K. , Watson, A. et al., Randomized, double‐blind, Phase 1 trial of an alphavirus replicon vaccine for cytomegalovirus in CMV seronegative adult volunteers. Vaccine 2009, 28, 484–493. [DOI] [PubMed] [Google Scholar]
- 132. Fallaux, F. J. , Bout, A. , van der Velde, I. , van den Wollenberg, D. J. et al., New helper cells and matched early region 1‐deleted adenovirus vectors prevent generation of replication‐competent adenoviruses. Hum. Gene Ther. 1998, 9, 1909–1917. [DOI] [PubMed] [Google Scholar]
- 133. Kojima, A. , Yasuda, A. , Asanuma, H. , Ishikawa, T. et al., Stable high‐producer cell clone expressing virus‐like particles of the Japanese encephalitis virus e protein for a second‐generation subunit vaccine. J. Virol. 2003, 77, 8745–8755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. van Wielink, R. , Harmsen, M. M. , Martens, D. E. , Peeters, B. P. et al., MDCK cell line with inducible allele B NS1 expression propagates delNS1 influenza virus to high titres. Vaccine 2011, 29, 6976–6985. [DOI] [PubMed] [Google Scholar]