

Abstract
The rapid growth of harmful pathogens and their multidrug‐resistance poses a severe challenge for health professionals and for the development of new healthcare products. Various strategies are exploited for the development of effective antimicrobial agents, and nanoparticles are a particularly promising class of materials in this respect. This review summarizes recent advances in antimicrobial metallic, polymeric, and lipid‐based nanoparticles such as liposomes, solid lipid nanoparticles, and nanostructured lipid carriers. The latter materials in particular are engineered for antimicrobial agent delivery and act by encapsulation, receptor‐based binding, and disruption of microbial adherence to cellular substrates. Potential strategies for the design of multifunctional antimicrobial nanocarriers, combining material chemistry and biological interface science, are also discussed.
Keywords: antimicrobial, formulations, nanoparticles, pathogens, resistant
The rapid growth of harmful pathogens and their multidrug‐resistance poses severe challenges in infectious diseases. One of the most promising materials used in antimicrobial treatments are nanoparticles, including metallic nanoparticles, polymeric nanoparticles, and lipid‐based nanoparticles. This review summarizes the most recent developments in research on these sophisticated materials, with diverse functions oriented toward therapeutic antimicrobial agent deliveries.
1. Introduction
The spread of antibiotic‐resistant pathogens is increasing with potentially catastrophic consequences.1 The emergence of clinically significant resistance is occurring within only a few years after the deployment of any new antibiotic. It has been estimated that about 70% of the hospital‐acquired infections are resistant to one or more current antibiotic treatments.2 These infections have significant impact on treatment failures, longer hospital stays, and health care costs. A significant number of newly developed antibiotics represent minor alterations to well‐known chemical scaffolds and are therefore particularly prone to resistance.3 The relentless increase in the number of resistant pathogens has threatened 70 years of medical advancement and led to the suggestion that we are reentering the pre‐antibiotic era.4 This is happening at a time when large pharmaceutical companies have ended or down‐sized their antibiotic discovery programs because they are too expensive.
Selective pressure imposed by the widespread use of antibiotics is responsible for this antibiotic resistance. Microbial pathogens have developed resistance mechanisms including: (i) decreased uptake and increased efflux of the drug through transmembrane efflux pumps; (ii) genetic alteration of the antibiotic targets with reduced affinity compared to the unaltered targets, (iii) production of enzymes which covalently modify or degrade the key structural motifs important for activity; and (iv) modification of membrane charge or sterol structure/composition.5, 6, 7 The induction of antibiotic resistance by pathogens is complex and governed by factors such as the mode of antibiotic action, whether or not the antibiotics is natural or synthetic and whether the antibiotic action is time‐ or concentration‐dependent.8 A high level of ingenuity is therefore needed to develop antibiotics with novel modes of action.
Prior to the discovery of penicillin by Sir Alexander Fleming, metals, metallic oxides, and metallic salts were used to treat bacterial and fungal infections.9 Their medicinal utility was diminished with the antibiotic era. The dearth of new antibiotics in the pipeline and the emergence of antibiotic resistance have led to the revival of these ancient antimicrobial agents. Nanoscience and nanotechnology offer potential opportunities in many fields, such as medical devices and implants, processing of food, water treatment, and agriculture, and therapeutics for cancer, pain, and infectious diseases.10 Figure 1 illustrates that the focus of research in the past decade on antimicrobial nanoparticles (NPs) has increased exponentially in the hope of a solution for the rising mutated strains of microorganisms. NPs have been used as carriers of antibiotics so that a localized high concentration of the drug can be generated at the site of infection. Alternatively, the NPs themselves, or in combination with a light source, act as antimicrobial agents against resistant pathogens.
Figure 1.
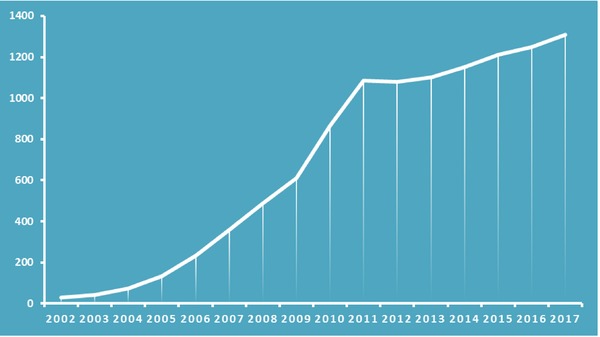
Number of publications on antimicrobial nanoparticles over the period 2002–2017. Data extracted from PubMed.
Antimicrobial NPs carriers should be able to protect the drugs from degradation and improve drug bioavailability for treatment.11, 12, 13 They may control the release of loaded‐antimicrobial drugs, which is useful to maintain an optimum level of drug concentration in the bloodstream for a period of time. These NPs may also provide a platform for surface modification that allows specific targeting of diseased sites. The size of NPs should be small enough to penetrate anatomical or cellular barriers.14 In addition, they should produce minimal toxicity and aspire to decrease the emergence of drug‐resistant strains of microorganisms.15, 16 Producing NPs at a low cost and on a large‐scale level is necessary for commercially viability, particularly in less‐developed countries. In summary, the traits of an ideal antimicrobial NP carrier includes: drug protection from degradation, improved bioavailability, controlled release for a sustained concentration, specific targeting of diseased site, overcoming anatomical or cellular barriers, minimal toxicity, decreased emergence of drug‐resistant, low cost, and scalable production.17, 18, 19
The purpose of this review is to summarize the published work on antimicrobial NPs and their therapeutic potential for combating resistant pathogens. With the increase emergence of mutated strains of microbes, such as the recently discovered coronavirus H7N9, coating of face masks with NPs may be a useful preventive measure for health care workers and civilians. In a study carried out by Li et al., ten treated and untreated masks with a mixture of Ag NPs and TiO2 NPs were tested against E. coli and S. aureus.20 No growth was reported on the treated masks after 48 h incubation while the untreated masks showed an increase in bacterial numbers. Wound dressing is another important area of investigation. Bacterial cellulose fibers do not possess antimicrobial activity. However, upon incorporation of Ag NPs into the fibers, they exhibited a strong antimicrobial response against E. coli and S. aureus.21 NPs have proven to overcome some of the shortcomings of existing antibiotics include having relatively short half‐lives, low bioavailability, and possible toxic side effects.22 For example, gentamicin was previously studied in different NP drug delivery system such as poly (lactide‐co‐glycolide) (PLGA) and chitosan.23, 24 It was observed that NPs carrying gentamicin aided in the drug delivery to specific sites and with sustained release of the drug.25 In addition, the encapsulation of antibiotics within NPs was observed to reduce their toxic side effects relative to conventional administration.26
2. Classification of Antimicrobial Nanoparticles
Antimicrobial nanoparticles can be classified into (1) metallic nanoparticles and (2) nonmetallic nanoparticles, including polymeric nanoparticles and lipid‐based nanoparticles.
2.1. Antimicrobial Metallic Nanoparticles
Metallic nanoparticles including silver, gold, and copper are reported to kill different types of bacteria. Each of these nanometals has its own specific properties and mechanism of action. The general antimicrobial mechanism of nanometals is proposed to involve disruption of cell membrane metabolism.27 Metallic nanoparticles may penetrate the cell membrane and disrupt associated enzymes leading to microbial death (Figure 2 ). Moreover, metal nanoparticles can generate reactive oxygen species (ROS) which can cause DNA damage and bacterial death. Attachment of nanoparticle to the cell membrane also reduces bacterial replication. Important parameters for antimicrobial NPs include their nanometer dimension, shape, and concentration.
Figure 2.
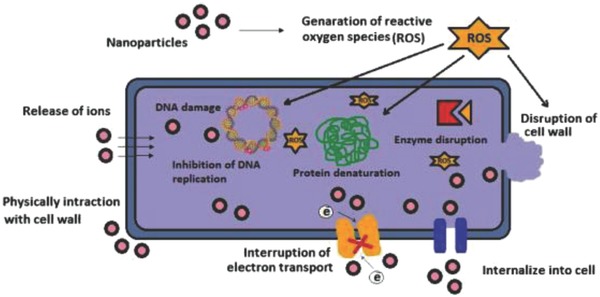
Proposed antimicrobial mechanisms of metallic nanoparticles by disrupting cell membrane metabolism. Reproduced with permission.27 Copyright 2014, Elsevier.
Of all the metals investigated as antibacterial agents, silver (Ag) NPs have perhaps proved the most useful against a wide spectrum of microorganisms.28 Silver and its compounds can possess potent bactericidal activity and have been developed into NPs for potential drug treatments.29 The antimicrobial efficacy of silver NPs is determined by size, shape, and nature of the capping agents. A linear correlation has been observed between size of the silver NPs and the minimum inhibitory concentration for both Gram‐positive and Gram‐negative strains.30 Ag NPs have been shown to kill yeast by disrupting its plasma membrane and thus affecting the membrane potential of the cells. Amphotericin B (AmB) was used as a positive control and the plasma membrane DPH fluorescence anisotropy of Candida albicans displayed a significant decrease as the concentrations of Ag NPs and AmB increased.31, 32 This result was consistent with the disruption of the plasma membrane by Ag NPs and AmB. In the same study, Ag NPs exhibited a minimum inhibitory concentration (MIC) of 2 µg mL−1, which is similar to that exhibited by AmB (2.5–5 µg mL−1).32 Moreover, nano‐Ag caused less hemolysis of erythrocytes (6% lysis) compared to AmB (10% lysis) at the same concentration.32 Ag NPs display average MIC values of 4.86 µg mL−1 compared to 500 µg mL−1 for ZnO NPs and 197 µg mL−1 for Au NPs against Streptococcus mutans.33
Ag NPs are also found to possess antiviral properties.34 This may prove to be an important finding in view of the rising numbers of virus‐related deaths worldwide. Viruses are highly adaptable and able to switch to a new host to overcome antiviral mechanisms.35 ≈2.5 million people were newly infected with human immunodeficiency virus infection (HIV) in 2011 (WHO), bringing the number of people living with HIV to a total of 34 million in 2011. About 1.7 million acquired immunodeficiency syndrome (AIDS) deaths were reported in the same year. Ag NPs are viricidal, not only to cell‐free HIV‐1 but also to cell‐associated HIV‐1, based on viral infectivity assays.36 These NPs appear to block viral entry through gp120‐CD4 interactions in the pre‐infection stage. In another study, poly(N‐vinyl‐2‐pyrrolidone) (PVP)‐coated Ag NPs were investigated as a potential topical vaginal microbicide against HIV‐1 infection.37 Cervical tissues were exposed to the nonspermicidal gel with PVP‐coated Ag NPs for 48 h and did not display signs of toxicity. A 1 min pretreatment of the gel prevented the transmission of HIV‐1 isolates, and a 20 min treatment followed by a thorough washing of the drug protected the tissues from HIV‐1 infection for 48 h.37
The emergence of multidrug‐resistant (MDR) bacteria has become a major threat to public health. A recent study showed that coating Au nanoparticles with cationic and hydrophobic functional groups could suppress the growth of both Gram‐negative and Gram‐positive uropathogens, including MDR pathogens (Figure 3 ).38 This result revealed that surface chemistry plays an important role in Au NP antimicrobial activity, providing a rational design element to develop new antibiotic NPs. Taken together with the high biocompatibility and slow development of resistance through generations, the use of functionalized Au NPs reported in this study provides a promising approach for t combating MDR infections.
Figure 3.

Schematic illustration showing cationic and hydrophobic functionalized Au NPs used for combating of MDR bacteria. Reproduced with permission.38 Copyright 2014, American Chemical Society.
Copper nanoparticles (Cu NPs) also exhibit a strong ability to inhibit microbes. Cu NPs are easy to develop and eco‐friendly, and their effectiveness against pathogenic bacteria, algae, and viruses has been demonstrated (Figure 4 ).39 It has been proposed that Cu NPs act against a wide spectrum of bacterial species by interactions with —SH groups, which results in protein denaturation. They also exhibit a high affinity with and amines and carboxyl groups present on the cell membrane. After penetration, the Cu NPs can disturb the structure of DNA by cross‐linking nucleic acid strands, which also results in bacterial cell death. Table 1 summarizes some of the most studied metallic NPs and their antimicrobial mechanism of action.
Figure 4.
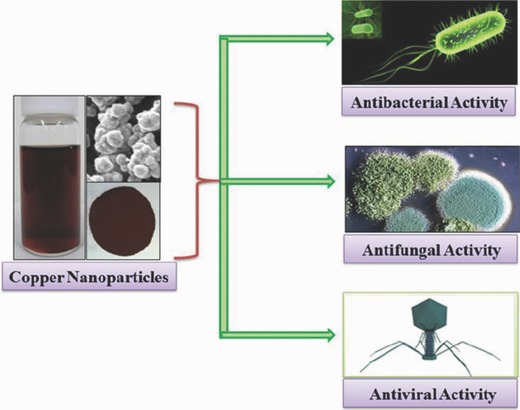
Schematic representation illustrating antimicrobial activity of copper nanoparticles against bacteria, fungi, and viruses. Reproduced with permission.39 Copyright 2014, Springer.
Table 1.
The most studied antimicrobial metallic nanoparticles and their mechanism of action
| Nanoparticles | Targeted microorganisms | Antimicrobial mechanism | Ref. |
|---|---|---|---|
| Ag | E. coli, B. subtilis, S. aureus |
|
40 |
| C. albicans |
|
41 | |
|
|||
| Au | HIV‐1 |
|
42 |
| P. aeruginosa, E. coli |
|
43 | |
| C. albicans |
|
44 | |
| HSV‐1 |
|
45 | |
| ZnO | E. coli, S. aureus |
|
46, 47, 48 |
| B. cinerea, P. expansum |
|
||
|
|||
|
|||
| TiO2 | E. coli, B. megaterium |
|
49 |
|
|||
| C. albicans |
|
50 | |
| Cu | E. coli, B. subtilis |
|
51 |
| Issatchenkia orientalis |
|
||
|
|||
| MgO | E. coli, S. aureus, B. subtilis, B. megaterium |
|
52 |
| S. cerevisiae, C. albicans |
|
53 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Metallic NPs can also be functionalized with antimicrobial surface molecules. The advantages of these NPs include producing longer lasting antimicrobial effectiveness and ensuring that microbes encountering the antimicrobials drugs are exposed to only the high surface concentrations. Silica nanoparticles (SNP) have been functionalized with AmB, for example. SNPs were chosen for their low toxicity, high stability, durability, and ease of modification to allow the incorporation of an array of different functional molecules.54, 55 SNPs incorporating DexOxAmB were found to totally eliminate C. albicans.55 AmB is known to cause a degree of hemolysis. However, when it is immobilized, no significant release of hemoglobin was observed.55 In the same study, SNPs functionalized with AmB were found to be more effective against C. albicans than silver nanoparticles.55 It was observed that the silver NPs were not fungistatic at the concentrations observed for SNPs functionalized with AmB.
The increased effectiveness of antimicrobial molecules tethered to NPs is further supported by the work of Gajbhiye et al., in which fluconazole displayed increased antifungal activity in the presence of Ag NPs.56 The zone of inhibition was increased 3.69 times when tested against C. albicans, but showed little difference when tested against F. semitectum and P. herbarum (increase of 0.15‐fold in area).56 However, adverse biological effects of nanomaterials have been reported over recent years. In one study, single‐wall carbon nanotubes were shown to produce free radicals, resulting in oxidative stress and cellular toxicity. This caused the loss of dermal cell viability after 18 h of the carbon nanotubes exposure.57 Another study reported that TiO2 nanoparticles in surface water may be a risk to aquatic life.58 Further development and research is necessary to determine the potential threat that NPs may pose.
2.2. Nonmetallic Nanoparticles Carrying Antimicrobials
2.2.1. Polymeric Nanoparticles
Polymeric NPs are very useful for antimicrobial drug delivery because: (i) they are structurally stable, and thus can be synthesized with a narrow size distribution; (ii) appropriate selection of polymer lengths, surfactants, and organic solvents can generate a precise set of particle properties; and (iii) the functional groups on the surfaces of polymeric NPs can be chemically modified for more effective targeting of microbes.59, 60
Polymeric NPs include poly‐lactic acid (PLA), polyhydroxyalkanoates (PHA), poly‐d‐l‐glycolide (PLG), PLGA, polycaprolactone (PCL), and poly‐cyanoacrylate (PCA), which are biodegradable.61, 62, 63, 64, 65 These NPs are in general cleared rapidly from the blood system by phagocytic cells, thereby minimizing bioaccumulation and associated side effects.66 Hence, it is also necessary for surface modification to escape or delay phagocytic clearance.67 Polymeric NPs can be modified with polyethylene glycol (PEG) to improve blood circulation half‐life.68, 69 Yuan et al. have reported the synthesis and antibacterial properties of biodegradable poly(ethylene oxide)‐poly(ε‐caprolactone)‐poly[(2‐tert‐butylaminoethyl) methacrylate] (PEO‐PCL‐PTBAM) (Figure 5 ).70 In this study, the desired triblock copolymers were synthesized by a two‐step reaction process, including ring opening polymerization (ROP) to prepare PEO‐PCL diblock copolymer, followed by a chain extension of PTBAM via atom transfer radical polymerization (ATRP). The presence of hydrophobic PCL segments provided the driving force for the copolymer to self‐assemble into micelles, and also imparted biodegradability, whereas PEGylation afforded better biocompatibility and colloidal stability. PEO‐PCL‐PTBAM micelles are inherently antibacterial and membrane‐active despite the absence of quaternary ammonium groups. This is because of the presence of secondary amines in the PTBAM block, which are cationic under physiological conditions. In addition, the utilization of PTBAM can circumvent the significant hemolytic effects of quaternary ammonium‐based antimicrobial materials. Antimicrobial testing showed that PEO‐PCL‐PTBAM copolymer with the highest PTBAM content exhibited the most potent effect against E. coli and S. aureus, with MIC values of 0.19 and 0.06 × 10−3 m, respectively. More importantly, the optimal MIC values were much higher than its critical micellization concentration (CMC), indicating self‐assembly is a prerequisite for the polymer to exhibit antimicrobial efficacy.70
Figure 5.

Schematic illustration showing the self‐assembly of PEO‐PCL‐PTBAM block copolymer into micelles in aqueous solution, postulated to interact with bacterial membranes through electrostatic interactions. Reproduced with permission.70 Copyright 2012, Royal Society of Chemistry.
Many researches have also demonstrated the potential role of polymeric NPs for antimicrobial drug delivery. PLGA has been loaded with gentamicin, which is an aminoglycoside antibiotic used in the treatment of Pseudomonoas infections.71 Data collected from this study showed that PLGA nanoparticles carrying gentamicin performed better than free gentamicin. The latter reduced P. aeruginosa infection after 24 h but the level of infection returned to levels comparable to saline‐treated controls after 96 h. However, antimicrobial activity remained high even after 96 h when an equal concentration of gentamicin‐loaded PLGA nanoparticles was used.71 This result was further supported by the work of Peng et al. investigating the effectiveness of free voriconazole versus PLGA NPs loaded with voriconazole. It was found that PLGA NPs loaded with voriconazole showed greater potency in vivo and extended fungicidal times than free voriconazole in vitro.72
Research on other microorganisms has similarly supported the advantages of polymeric NPs in drug delivery systems. Cavalli et al. investigated the efficacy of antiviral NPs loaded with acyclovir. Acyclovir is used to treat herpes simplex virus (HSV) infections.73 However, acyclovir needs to be taken orally five times daily due to its short half‐life (≈2 h) and low bioavailability (about 15–30%). Polymeric NPs were prepared using a β‐cyclodextrin‐poly(a‐acryloylmorpholine) monoconjugate (β‐CD‐PACM). Free acyclovir and acyclovir‐loaded β‐CD‐PACM NPs were incubated with vero cells separately and the intracellular drug concentration was determined. It was found that the intracellular drug concentration was much higher in cells incubated with acyclovir‐loaded β‐CD‐PACM NPs than for cells incubated with free acyclovir.73 Likewise, HIV protease inhibitor saquinavir and nucleoside analog zalcitabine were loaded into polyhexycyanocrylate NPs. Free saquinavir displayed little antiviral activity at <10 × 10−9 m against acutely infected human monocytes/macrophages cells, while saquinavir‐loaded NPs showed good antiviral activity at 1 × 10−9 m. However, zalcitabine loaded into NPs did not appear to enhance antiviral activity. Hence, it appears that protease inhibitor drugs may gain more from NP‐loading than nucleoside analogs. Table 2 summarizes some typical examples of polymeric NPs and their antimicrobial activities.
Table 2.
Polymeric nanoparticles for antimicrobial drug delivery
| Function | Nanoparticle | Drug | Targeted microorganisms | Activity and features | Ref. |
|---|---|---|---|---|---|
| Antibacterial | Poly‐lactic acid (PLA) | Arjunglucoside | Leishmania donovani |
|
74 |
| Poly‐d‐l‐lactide‐co‐glycolide (PLGA) | Gentamicin | P. aeruginosa |
|
71 | |
| Poly‐d‐l‐glycolide (PLG) | Econazole Moxifloxacin | Mycobacterium tuberculosis |
|
75 | |
| Polyethylene glycol (PEG)‐PLA | Halofantrine | Plasmodiumberghe |
|
76 | |
| Alginate | Rifampicin Pyrazinamide | Mycobacterium tuberculosis |
|
77 | |
| Antifungal | Poloxamer 188 coated poly(epsilon‐caprolactone)78 | Amphotericin B | Candida albicans |
|
79 |
| Antiviral | β‐cyclodextrin‐poly(a‐acryloylmorpholine) | Acyclovir | HSV‐1 and HSV‐2 |
|
73 |
| Polyhexycyanocrylate | Saquinavir | HIV |
|
80 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
2.2.2. Lipid‐Based Nanoparticles
Liposomes: Liposomes are composed of a bilayer membrane consisting of amphipathic phospholipids enclosing an interior space and have been researched intensively for antimicrobial drug delivery. In the 1990s, an anticancer drug, Doxil, was produced by Ortho‐Biotech and was used in the treatment of AIDS‐associated Kaposi's sarcoma. It became the first liposomal delivery system to be approved by the Food and Drug Administration (FDA).59
Liposomes can exhibit antimicrobial properties, either alone or through encapsulation to enhance the antiviral efficacy of drugs. The bilayer membrane of liposomes mimics cell membranes and possesses the ability to fuse directly with the microbe. This allows the drug payloads of liposomes to be released into the microbe. In addition, liposome surfaces can be easily modified to enhance their in vivo stability, or for targeting ligands to enable more specific drug delivery (Figure 6 ).59, 81 PEG is commonly used on the liposome surfaces to create a “protective” layer that increases the blood circulation half‐life.
Figure 6.
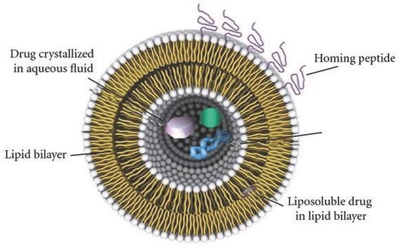
Liposomes are efficient carriers for drug delivery. Reproduced with permission.81 Copyright 2012, Hindawi.
Polyunsaturated ER liposomes (PERLs) significantly reduce viral secretion and infection for the treatment of HBV, HCV, and HIV infections. They act by the reduction of cholesterol which is essential for virus infectivity. Mareuil et al. have compared the antiviral efficacy of SPC3 (a synthetic polymeric peptide) encapsulated in liposomes relative to free SPC3.82 Liposomal encapsulated SPC3 exhibited greater antiviral efficacy by more than ten and fivefold in HIV‐infected C8166 T‐cells and human peripheral blood lymphotcytes (PBLs), respectively. Table 3 summarizes the most studied examples of liposomes that have been used for antimicrobial drug delivery.
Table 3.
Representative examples of liposomes for antimicrobial drug delivery
| Function | Encapsulant | Drug | Targeted microorganism | Activity and features | Ref. | ||
|---|---|---|---|---|---|---|---|
| Natural | Synthetic | Natural | Synthetic | ||||
| Antibacterial | Partially hydrogenated egg phosphatidylcholine (PHEPC), cholesterol | 1,2‐distearoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐(polyethylene glycol‐2000) (PEGDSPE) | Gentamicin | – | Klebsiella pneumonia |
|
83 |
| Soybean phosphatidylcholine, cholesterol | – | – | Ampicillin | Micrococcus lluterus, Salmonella tphimurium |
|
84 | |
| Phosphatidylcholine, cholesterol, phosphatidylinositol | – | – | Netilmicin | B. subtilis, E. coli |
|
85 | |
| Egg phosphatidylcholine, diacetylphosphate, cholesterol | – | Vancomycin, teicoplanin | – | Methicillin‐resistant S. aureus |
|
86 | |
| Antifungal | Hydrogenated soybean phosphatidylcholine, cholesterol | Distearolyphophatidylglycerol, monoethoxypolyethylene‐glycol 1900 succinimidyl succinate (activated PEG) | – | Amphotericin B | C. albicans |
|
87 |
| Phosphatidylcholine, cholesterol | Cardiolipin | – | Nystatin, amphotericin B | Fusarium oxysporum |
|
88 | |
| Antiviral | Phosphatidylcholine, stearylamine | Dioleoyl phosphatidylethanolamine | – | Phosphorothioate antisense oligodeoxynucleotides (PS‐ODN) | Duck hepatitis B virus (DHBV) |
|
89 |
| – | 1,2‐Didocosahexaenoyl‐sn‐glycero‐3‐phosphoethanolamine, 1,2‐didocosahexaenoyl‐sn‐glycero‐3‐phosphocholine, l‐α‐phosphatidylserine | – | – | HBV, HCV, and HIV |
|
90 | |
| Egg phosphatidylcholine, egg l‐α‐phosphatidylglycerol, cholesterol, tocopherol | – | – | SPC3 | HIV‐1 |
|
82 | |
| Stearylamine (SA) | Dicetylphosphate | – | Zidovudine | HIV |
|
91 | |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Solid Lipid Nanoparticles (SLNs) and Nanostructured Lipid Carriers (NLCs): SLNs are made from lipids that are solid at room temperature and they encompass a solid lipid core. Drug mobility is lower in the solid lipid state compared to lipids in the oily phase, allowing better controlled release of drugs.92 Surfactants are used for emulsification to further enhance SLN stability.92
SLNs have been used in cosmetic and pharmaceutical products for the skin care industry. SLNs are found to have occlusive excipients that form a thin film when applied to skin. This reduces evaporation, helping to retain skin moisture, and enhance molecule penetration.93 Another advantage of SLNs is their stability in water and dermal creams, which makes it easier to incorporate them into cosmetic and skin care products.94 SLNs are used topically.95, 96 Their small size can prolong the drug residence time in the skin and aid drug penetration through the skin.97 It has been found that SLNs loaded with econazole nitrate (particle diameter of about 150 nm) showed increased drug diffusion rates into deeper skin layers.98 This is further supported by a study of Lv et al., who reported a more than twofold increase (130% increase) in the cumulative amount of penciclovir penetrating through excised rat skins from penciclovir‐loaded SLNs, relative to that penetrating from a commercial cream (control) 12 h after administration.
The improved drug targeting to specific sites is especially useful for drug delivery to the central nervous system (CNS), as the blood–brain barrier (BBB) and blood–cerebrospinal fluid barrier obstruct the effective transportation of drugs. SLNs may provide a solution to this problem. Chattopadhyay et al. showed a higher drug uptake in the human brain endothelial cells (a representative of BBB) in vitro when atazanavir was delivered via SLNs. SLNs can be used for oral administration by producing them in powder form for pellets, capsules, or tablets.99 Tobramycin‐loaded SLNs displayed greater effectiveness against Pseudomonas aeruginosa infections in the gastrointestinal tracts of patients with cystic fibrosis.100 SLNs consist of natural components and do not require high‐pressure homogenization and sophisticated machinery,92 thereby allowing large‐scale, inexpensive manufacturing. However, SLN formulations may be limited by unwanted “burst release” of the drug due to particle growth by agglomeration or coagulation.92, 101
NLCs encompass an imperfect crystal or amorphous particle, which allows drug loading in the molecular form and in clustered aggregates. This enhances drug loading and gives rise to less pronounced “burst release” of the drug because of the reduced proportion of crystalline structures.102 Table 4 gives a summary of lipid‐based NPs for antimicrobial drug delivery. Cai et al. reported a new lipid polymer nanoparticle formulation with rhamnolipid and phospholipids as the outer mixed lipids layer (RHL‐PC‐LPN) (Figure 7 ).109 Amoxicillin (AMX) (10.2%) and an anti H. pylori adhesion material pectin sulfate (PECS) were loaded into the LPNs to facilitate the disruption of H. pylori biofilms and further enhanced their susceptibility to antibacterial agents. This study showed that RHL‐PC‐LPN could significantly disrupt H. pylori biofilm and exerted stronger potency than the control nanoparticles with phospholipids alone as the outer layer. In addition, the strategy of using combined PECS with amoxicillin exhibited stronger antimicrobial activity than amoxicillin alone. A lower inhibition concentration of amoxicillin‐loaded nanoparticles to biofilm was detected, demonstrating that the nanoparticles could reduce the infection resistance. More importantly, RHL‐PC‐LPN could also provide protection to AGS cells against H. pylori infection.
Table 4.
Solid lipid nanoparticles for antimicrobial drug delivery
| Function | Encapsulant | Drug | Targeted microorganism | Activity and features | Ref. | ||
|---|---|---|---|---|---|---|---|
| Natural | Synthetic | Natural | Synthetic | ||||
| Antibacterial | Chitosan | Myristyl myristate | Tretinonin | – | Propionibacterium acnes, S. aureus |
|
103 |
| Stearic acid, soya phosphatidyl‐choline | – | – | Tobramycin | P. aeruginosa |
|
104 | |
| Antifungal | Glyceryl tripalmitate | Tyloxapol | – | Clotrimazole | Fungi |
|
105 |
| Glycerol palmitostearate | – | – | Miconazole nitrate | Fungi |
|
106 | |
| Antiviral | Stearic acid | – | – | Atazanavir | HIV |
|
107 |
| Egg phosphotidylcholine, glyceryl monostearate | – | – | Penciclovir | – |
|
108 | |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Figure 7.

The design strategy for lipid polymer nanoparticles to eradicate bacterial biofilms. Reproduced with permission.103 Copyright 2015, Elsevier.
3. Methods for Preparation of Nonmetallic Antimicrobial Nanoparticles
Polymeric NPs can be formed via spontaneous self‐assembly of hydrophilic and hydrophobic segments into diblock copolymers.59 Both polymers and drugs are first dissolved in a water‐miscible organic solvent such as acetone or acetonitrile. The mixture is then added to an aqueous solution. As the organic solvent evaporates, the polymers and drugs undergo nanoprecipitation via rapid solvent diffusion, to produce NPs.110 Alternatively, polymeric NPs can be prepared from linear polymers via an emulsion polymerization method. Monomers are first dissolved in polymerization media with surfactants, followed by the addition of polymerization initiators, resulting in the formation of nanocapsules. Antimicrobials are either absorbed into the NPs during this process or they can be covalently attached to the surface of the NPs after they are formed. Hydrophobic drugs are typically loaded via absorption while hydrophilic drugs are attached via covalent conjugation.111 Table 5 summarizes the reported methods for preparing polymeric NPs. Different preparation methods and different compositions of liposomes contribute to their efficacy as antimicrobial carriers. Electrospray can produce a broad array of NPs. This technique generates monodisperse droplets in sizes ranging from nanometers to micrometers (depending on the processing parameters). The electric field induces free charge which concentrates on the surface of the liquid and this tends not to affect sensitive biomolecules such as nucleic acids. This method provides better control with high drug or nucleic acid encapsulation efficiency (Table 6 ).
Table 5.
Methods for preparation of polymeric nanoparticles used for antimicrobial applications
| Method | Polymer | Solvent | Stabilizer | Size [nm] | Ref. |
|---|---|---|---|---|---|
| Solvent diffusion | PLGA | Acetone | Pluronic F127 | 200 | 68 |
| Solvent displacement | PLA | Acetone/methylene chloride | Pluronic F68 | 100–146 | 112 |
| Nanoprecipitation | PLGA/PLA/PCL | Acetone | Pluronic 68 | 110–208 | 113 |
| Solvent evaporation | PLA‐PEG‐PLA | Dichloromethane | – | 193–335 | 114 |
| Multiple emulsion | PLGA | Ethyl acetate | – | >200 | 115 |
| Salting out | PLA | Acetone | PVA | 300–700 | 116 |
| Ionic gelation | Chitosan | Sodium tripolyphosphate | – | 275–281 | 112 |
| Polymerization | Polyethylcyanoacrylate | – | Pluronic F68 | 308–332 | 66 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Table 6.
Summary of methods for antimicrobial liposome preparation
| Method | Formulation | Solvent | Size | Ref. |
|---|---|---|---|---|
| Dehydration‐rehydration vesicles | Egg phosphatidylcholine,78 phosphatidic acid (PA), stearylamine (SA), distearoyl phosphatidylcholine (DSPC), cholesterol | – | – | 117 |
| Reverse‐phase evaporation vesicles | Dipalmitoyl‐DL‐α‐phospatidyl‐l‐serine (PS), cholesterol, egg phosphatidylcholine (EPC), 1,2‐dipalmitoyl‐sn‐glycerophosphocholine monohydrate, 1,2‐dimyrostoyl‐sn‐glycerophosphoethanolamine (PE),1,2‐dipalmitoyl‐sn‐glycerophosphatidic acid disodium salt (PA), dihexadecyl hydrogen phosphate (DP) | Diethyl ether, isotonic buffer | 207–265 | 118 |
| Freeze‐thaw multilamellar vesicles | Dipalmitoyl‐DL‐α‐phospatidyl‐l‐serine (PS), cholesterol, egg phosphatidylcholine (EPC), 1,2‐dipalmitoyl‐sn‐glycerophosphocholine monohydrate, 1,2‐dimyrostoyl‐sn‐glycerophosphoethanolamine (PE), 1,2‐dipalmitoyl‐sn‐glycerophosphatidic acid disodium salt (PA), dihexadecyl hydrogen phosphate (DP) | Phosphate‐buffered saline (PBS) | 945–1307 | 78 |
| Stable plurilamellar vesicles | Egg phosphatidylcholine, CHL, dicetylphosphate (DP), stearylamine (ST), bovine HDL | Diethyl ether, HEPES buffer | – | 119 |
| Electrospray | Lecithin | Polyethylene glycol, poly(allylamine hydrochloride) | 1300–4600 | 120 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
In a study by Wu et al., the effectiveness of antisense oligodeoxynucleotides (ODN) encapsulated lipoplexes generated by coaxial electrospray was compared to those produced via the standard ethanol dilution method.121 It was found that coaxial electrospray is a simpler, continuous one‐step method that requires less time and effort to produce the lipoplexes. A good balance of lipoplex productivity, size, and surface charge can be achieved by adjusting processing parameters. The lipoplexes produced can be used for intravenous injection or as aerosol for inhalation therapy. SLNs and NLCs can be produced using several methods which are summarized in Table 7 . High‐pressure homogenization (HPH) is arguably the most effective technique yielding a narrow particle size distribution, avoiding the use of organic solvents and is scalable.132 However, HPH involves melted lipids and high energy input, making it unsuitable for temperature‐ and mechanically sensitive biomolecules. Wu et al. demonstrated that electrospray is an efficient method to produce highly monodisperse cholesterol NPs. The cholesterol concentration in the final solution was 150 ± 8 µg mL−1 after filtration, which is 100 times higher than the cholesterol solubility limit.
Table 7.
Summary of methods for preparation of lipid‐based nanoparticles
| Nanoparticles | Method | Formulation | Solvent | Stabilizer | Size [nm] | Ref. |
|---|---|---|---|---|---|---|
| Microemulsion | Stearic acid | Methanol | Pluronic F68 | 167–224 | 122 | |
| High pressure homogenization | Myristyl myristate | – | Pluronic F68 | 162.7 ± 1.4 | 123 | |
| Solid‐lipid NPs | Solvent emulsification‐evaporation | Para‐dodecanoyl‐calix[4]arene | Acetone, methanol, ethanol, glycerol | Pluronic F68 | <60 | 124 |
| Solvent injection | Triglycerides | Acetone, methanol, ethanol, glycerol, isopropanol | Phosphatidylcholine, polysorbate 80, poloxamer 188 | 80–300 | 125 | |
| Water‐in‐oil‐in‐water double emulsion | Stearic acid, dioctyl sodium sulfosuccinate, egg lecithin | – | – | – | 126 | |
| High shear homogenization | Benzyl nicotinate, hydrogenated soybean lecithin, cholesterol | – | Poloxamer 188, glycerol | 240 | 127 | |
| Membrane contactor | Propanol, glyceryl behenate | Sodium hydroxide | – | 70–215 | 128 | |
| Electrospray | Cholesterol | Ethanol | – | ≈150 | ||
| Nanostructured lipid carriers (NLC) | Solvent diffusion | Monostearic acid, caprylic triglycerides | Acetone, ethanol | Poloxamer 188 | 300–400 | 129 |
| Stearic acid, oleic acid | Acetone, ethanol | Poloxamer 188 | 160–430 | 130 | ||
| Melt‐emulsification and ultrasonication | Glyceryl behenate, polyoxyglycerides, Solutol HS‐15, C8‐C12 triglyceride, chitosan oligosaccharides | – | – | 55–170 | 131 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
4. Novel Strategies for the Delivery of Antimicrobial Nanoparticles
The normal method for achieving a large antimicrobial effect is to simply increase the dosage. However, high dosage will increase the likelihood of cytotoxicity and drug resistance. Thus, new strategies for specifically delivering the antimicrobial agents are needed.
4.1. Encapsulation
Encapsulation is one of the best strategies for passive targeting and exploits the enhanced permeability and retention effect for drug delivery. The interior aqueous spaces within liposomes allow loading space for antimicrobials. Polymyxin B, for example, is effective against P. aeruginosa and is used in the treatment of pneumonias and chronic bronchio‐pneumonia of cystic fibrosis. Its usage is, however, limited by toxic side effects and this toxicity was reduced by encapsulating the drug in liposomes. In addition, the antimicrobial activity against resistant strains of P. aeruginosa was found to be improved.133 Polymyxin B‐loaded liposomes undergo membrane fusion with that of P. aeruginosa, allowing a high dose of drug to be delivered into the bacteria, overwhelming any resistance by efflux mechanisms of the bacteria.133 In another example, anticancer drug Doxil (doxorubicin‐encapsulated PEGlyated liposome) was observed to have better drug retention and circulation time relative to free doxorubicin.134, 135
4.2. Receptor‐Based Binding
Receptor‐based binding is an active targeting method using conjugated receptor specific ligands for site specific targeting. Antimicrobials can be conjugated with a cell or tissue‐specific ligand, allowing accumulation at the targeted site. Choi et al. conjugated PEGlyated gold NPs with human transferrin and observed greater intracellular delivery to cancer cells than was possible with nontargeted NPs.136 This observation was further supported by results from Kocbek et al., who modified PLGA NPs with monoclonal antibodies (mAb) and found that the modified PLGA NPs showed enhanced binding to targeted cancer cells than was possible with nontargeted NPs.137 Site‐specific targeting via receptors allows NPs carrying therapeutic drugs to reach the diseased site more accurately and rapidly.
This strategy has been applied to antimicrobial drugs. Liposomes containing aminoglycoside demonstrated direct delivery of antibiotics, thus enhancing the intraphagocytic killing of bacteria.138
4.3. Disruption of Microbial Adherence to Cellular Substrates
The first step to infection is the adherence of microorganisms to the epithelial cells.139, 140, 141 Research has generally been focused on the efficiency and effectiveness of drug loading capacity in NPs and subsequent drug delivery. However, using chemotherapeutic agents against microorganisms has unfortunately led to the rise of resistant strains.139, 140 The prevention of infections may serve as an alternative strategy to treatment. In a study conducted by McCarron et al., polymeric NPs were adsorbed onto the surface of blastospores, thereby significantly reducing the adhesion to buccal epithelial cells (BEC) in vitro.142 Although complete inhibition of microbial adherence was not achieved, the extent of reduction was substantial relative to untreated blastospores.142 This may prove to be useful for the prophylaxis of candidosis of the oral cavity.
5. Conclusions and Future Prospects
Increasing antibiotic resistance has become a critical issue for the health care industry, and is responsible for the failure of conventional antimicrobial therapies. Metallic nanoparticles display potent antimicrobial activity with rapid‐time‐kill and avoidance of antibiotic resistance based on their specific properties and mechanisms of action involving disruption of membrane metabolism and membrane‐related processes. Nonmetallic nanoparticles including polymer and lipid‐based nanoparticles have been used as carriers to deliver antimicrobial drugs to targets, enhancing efficacy by increasing the payload of drugs, and reducing cytotoxicity.
The biocidal mechanisms of action of different classes of nanoparticles are still not clear. Many investigations ascribe antibacterial activity to either oxidative stress or reactive oxygen species (ROS), while for some other nanoparticles (e.g., MgO) antibacterial activity may not be related to bacterial metabolism. Thus, more work in this area is required in the quest for rational antimicrobial therapies.
Another limitation of the current research is the lack of standardization. Different investigations employ a variety of bacterial strains with different reaction times using varied nanoparticle formulations, which makes comparing antimicrobial efficacy difficult. Likewise, researchers employ a variety of in vivo and in vitro models to measure antimicrobial activity and mammalian cytotoxicity drawing quite different conclusions from each model. Nevertheless, it is clear that nanoparticles are useful vaccine and drug delivery systems, either applied topically or accessing specific diseased sites via the blood circulatory system. Greater efficiency in particle engineering, minimizing costs, and reducing environmental harm will lead to improved commercial viability of this technology.
The long‐term toxic effects of NPs remain unknown. Again, understanding the mechanism by which each NP functions is crucial for avoiding possible adverse side effects at the cellular level. The half‐life of NPs in the body is also fundamental knowledge in this respect.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
R.L and E.Y. contributed equally to this work.
Biographies
Zibiao Li obtained his Ph.D. in 2014 from the National University of Singapore (NUS). Currently, he is working as a research scientist at IMRE, A*STAR, Singapore. His research interests are focused on biodegradable and functional polymeric materials design, structure–properties correlations and their formulations for healthcare and consumer care applications.

Xian Jun Loh is the head of the Soft Materials Department in the Institute of Materials Research and Engineering (IMRE), A*STAR and an assistant professor at the National University of Singapore (NUS). His main research interests are in the design of supramolecular and stimuli‐responsive polymers and hydrogels for biomedical and personal care applications.

Lakshminarayanan R., Ye E., Young D. J., Li Z., Loh X. J., Adv. Healthcare Mater. 2018, 7, 1701400 10.1002/adhm.201701400
Contributor Information
Rajamani Lakshminarayanan, Email: lakshminarayanan.rajamani@seri.com.sg.
Zibiao Li, Email: lizb@imre.a-star.edu.sg.
Xian Jun Loh, Email: lohxj@imre.a-star.edu.sg.
References
- 1. Millan B., Park H., Hotte N., Mathieu O., Burguiere P., Tompkins T. A., Kao D., Madsen K. L., Clin. Infect. Dis. 2016, 62, 1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tang Y., Fang L., Xu C., Zhang Q., Anim. Health Res. Rev. 2017, 10.1017/S1466252317000135. [DOI] [PubMed] [Google Scholar]
- 3. Lam S. J., Wong E. H., Boyer C., Qiao G. G., Prog. Polym. Sci. 2017, 76, 46. [Google Scholar]
- 4. Yousefi M., Dadashpour M., Hejazi M., Hasanzadeh M., Behnam B., de la Guardia M., Shadjou N., Mokhtarzadeh A., Mater. Sci. Eng., C 2017, 74, 568. [DOI] [PubMed] [Google Scholar]
- 5. Matai I., Sachdev A., Dubey P., Kumar S. U., Bhushan B., Gopinath P., J. Colloids Surf., B 2014, 115, 359. [DOI] [PubMed] [Google Scholar]
- 6. Liu H., Pei H., Han Z., Feng G., Li D., Food Control 2015, 47, 444. [Google Scholar]
- 7. Asri L. A., Crismaru M., Roest S., Chen Y., Ivashenko O., Rudolf P., Tiller J. C., van der Mei H. C., Loontjens T. J., Busscher H. J., Adv. Funct. Mater. 2014, 24, 346. [Google Scholar]
- 8. Zou X., Zhang L., Wang Z., Luo Y., J. Am. Chem. Soc. 2016, 138, 2064. [DOI] [PubMed] [Google Scholar]
- 9. Perelshtein I., Lipovsky A., Perkas N., Gedanken A., Moschini E., Mantecca P., Nano Res. 2015, 8, 695. [Google Scholar]
- 10. Beyth N., Houri‐Haddad Y., Domb A., Khan W., Hazan R., J. Evidence‐Based Complementary Altern. Med. 2015, 2015, 246012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee S. J., Heo D. N., Moon J.‐H., Ko W.‐K., Lee J. B., Bae M. S., Park S. W., Kim J. E., Lee D. H., Kim E.‐C., Carbohydr. Polym. 2014, 111, 530. [DOI] [PubMed] [Google Scholar]
- 12. Gao W., Fang R. H., Thamphiwatana S., Luk B. T., Li J., Angsantikul P., Zhang Q., Hu C.‐M. J., Zhang L., Nano Lett. 2015, 15, 1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Armentano I., Arciola C. R., Fortunati E., Ferrari D., Mattioli S., Amoroso C. F., Rizzo J., Kenny J. M., Imbriani M., Visai L., Sci. World J. 2014, 2014, 410423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barua S., Mitragotri S., Nano Today 2014, 9, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arami H., Khandhar A., Liggitt D., Krishnan K. M., Chem. Soc. Rev. 2015, 44, 8576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sirelkhatim A., Mahmud S., Seeni A., Kaus N. H. M., Ann L. C., Bakhori S. K. M., Hasan H., Mohamad D., Nano‐Micro Lett. 2015, 7, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dizaj S. M., Lotfipour F., Barzegar‐Jalali M., Zarrintan M. H., Adibkia K., Mater. Sci. Eng., C 2014, 44, 278. [DOI] [PubMed] [Google Scholar]
- 18. Samiei M., Farjami A., Dizaj S. M., Lotfipour F., Mater. Sci. Eng., C 2016, 58, 1269. [DOI] [PubMed] [Google Scholar]
- 19. Allaker R. P., Memarzadeh K., Int. J. Antimicrob. Agents 2014, 43, 95. [DOI] [PubMed] [Google Scholar]
- 20. Li Y., Leung P., Yao L., Song Q. W., Newton E., J. Hosp. Infect. 2006, 62, 58. [DOI] [PubMed] [Google Scholar]
- 21. Maneerung T., Tokura S., Rujiravanit R., Carbohydr. Polym. 2008, 72, 43. [Google Scholar]
- 22. Tange R. A., Dreschler W. A., Prins J. M., Buller H. R., Kuijper E. J., Speelman P., Clin. Otolaryngol. Allied Sci. 1995, 20, 118. [DOI] [PubMed] [Google Scholar]
- 23. Lecaroz C., Blanco‐Prieto M. J., Burrell M. A., Gamazo C., J. Antimicrob. Chemother. 2006, 58, 549. [DOI] [PubMed] [Google Scholar]
- 24. Soliman G. M., Szychowski J., Hanessian S., Winnik F. M., Soft Matter 2010, 6, 4504. [Google Scholar]
- 25. Gao P., Nie X., Zou M., Shi Y., Cheng G., J. Antibiot. 2011, 64, 625. [DOI] [PubMed] [Google Scholar]
- 26. Seleem M. N., Munusamy P., Ranjan A., Alqublan H., Pickrell G., Sriranganathan N., Antimicrob. Agents Chemother. 2009, 53, 4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dizaj S. M., Lotfipour F., Barzegar‐Jalali M., Zarrintan M. H., Adibkia K., Mater. Sci. Eng., C 2014, 44, 278. [DOI] [PubMed] [Google Scholar]
- 28. Surmeneva M. A., Sharonova A. A., Chernousova S., Prymak O., Loza K., Tkachev M. S., Shulepov I. A., Epple M., Surmenev R. A., Colloids Surf., B 2017, 156, 104. [DOI] [PubMed] [Google Scholar]
- 29. Ertem E., Gutt B., Zuber F., Allegri S., Le Ouay B., Mefti S., Formentin K., Stellacci F., Ren Q., ACS Appl. Mater. Interfaces 2017, 9, 34762. [DOI] [PubMed] [Google Scholar]
- 30. Gomez‐Carretero S., Nybom R., Richter‐Dahlfors A., Adv. Healthcare Mater. 2017, 6, 1700435. [DOI] [PubMed] [Google Scholar]
- 31. Hartsel S., Bolard J., Trends Pharmacol. Sci. 1996, 17, 445. [DOI] [PubMed] [Google Scholar]
- 32. Kim K.‐J., Sung W., Suh B., Moon S.‐K., Choi J.‐S., Kim J., Lee D., Biometals 2009, 22, 235. [DOI] [PubMed] [Google Scholar]
- 33. Hernández‐Sierra J. F., Ruiz F., Cruz Pena D. C., Martínez‐Gutiérrez F., Martínez A. E., de Jesús Pozos Guillén A., Tapia‐Pérez H., Martínez Castañón G., Nanomed.: NBM 2008, 4, 237. [DOI] [PubMed] [Google Scholar]
- 34. Sharma R. K., Cwiklinski K., Aalinkeel R., Reynolds J. L., Sykes D. E., Quaye E., Oh J., Mahajan S. D., Schwartz S. A., Immunol. Invest. 2017, 46, 833. [DOI] [PubMed] [Google Scholar]
- 35. Sun B., Barnard A. S., Nanoscale 2017, 9, 12698. [DOI] [PubMed] [Google Scholar]
- 36. Kumar S. D., Singaravelu G., Ajithkumar S., Murugan K., Nicoletti M., Benelli G., J. Cluster Sci. 2017, 28, 359. [Google Scholar]
- 37. Milovanovic M., Arsenijevic A., Milovanovic J., Kanjevac T., Arsenijevic N., Antimicrobial Nanoarchitectonics, Elsevier, Amsterdam: 2017, p. 383. [Google Scholar]
- 38. Li X., Robinson S. M., Gupta A., Saha K., Jiang Z., Moyano D. F., Sahar A., Riley M. A., Rotello V. M., ACS Nano 2014, 8, 10682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ingle A. P., Duran N., Rai M., Appl. Microbiol. Biotechnol. 2014, 98, 1001. [DOI] [PubMed] [Google Scholar]
- 40. Jung W. K., Koo H. C., Kim K. W., Shin S., Kim S. H., Park Y. H., Appl. Environ. Microbiol. 2008, 74, 2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vazquez‐Muñoz R., Avalos‐Borja M., Castro‐Longoria E., PLoS One 2014, 9, e108876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bowman M.‐C., Ballard T. E., Ackerson C. J., Feldheim D. L., Margolis D. M., Melander C., J. Am. Chem. Soc. 2008, 130, 6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhao Y., Tian Y., Cui Y., Liu W., Ma W., Jiang X., J. Am. Chem. Soc. 2010, 132, 12349. [DOI] [PubMed] [Google Scholar]
- 44. Bankar A., Joshi B., Kumar A. R., Zinjarde S., Colloids Surf., B 2010, 80, 45. [DOI] [PubMed] [Google Scholar]
- 45. Baram‐Pinto D., Shukla S., Gedanken A., Sarid R., Small 2010, 6, 1044. [DOI] [PubMed] [Google Scholar]
- 46. Jones N., Ray B., Ranjit K. T., Manna A. C., FEMS Microbiol. Lett. 2008, 279, 71. [DOI] [PubMed] [Google Scholar]
- 47. Liu Y., He L., Mustapha A., Li H., Hu Z., Lin M., J. Appl. Microbiol. 2009, 107, 1193. [DOI] [PubMed] [Google Scholar]
- 48. He L., Liu Y., Mustapha A., Lin M., Microbiol. Res. 2011, 166, 207. [DOI] [PubMed] [Google Scholar]
- 49. Darbari S., Abdi Y., Haghighi F., Mohajerzadeh S., Haghighi N., J. Phys. D: Appl. Phys. 2011, 44, 245401. [Google Scholar]
- 50. Karunakaran G., Suriyaprabha R., Manivasakan P., Yuvakkumar R., Rajendran V., Kannan N., J. Nanosci. Nanotechnol. 2013, 13, 678. [DOI] [PubMed] [Google Scholar]
- 51. Esteban‐Tejeda L., Malpartida F., Esteban‐Cubillo A., Pecharromán C., Moya J., Nanotechnology 2009, 20, 505701. [DOI] [PubMed] [Google Scholar]
- 52. Tang Z.‐X., Lv B.‐F., Braz. J. Chem. Eng. 2014, 31, 591. [Google Scholar]
- 53. Sawai J., Yoshikawa T., J. Appl. Microbiol. 2004, 96, 803. [DOI] [PubMed] [Google Scholar]
- 54. Slowing I. I., Trewyn B. G., Giri S., Lin V. S. Y., Adv. Funct. Mater. 2007, 17, 1225. [Google Scholar]
- 55. Paulo C. S. O., Vidal M., Ferreira L. S., Biomacromolecules 2010, 11, 2810. [DOI] [PubMed] [Google Scholar]
- 56. Gajbhiye M., Kesharwani J., Ingle A., Gade A., Rai M., Nanomed.: NBM 2009, 5, 382. [DOI] [PubMed] [Google Scholar]
- 57. Shvedova A., Castranova V., Kisin E., Schwegler‐Berry D., Murray A., Gandelsman V., Maynard A., Baron P., J. Toxicol. Environ. Health, Part A 2003, 66, 1909. [DOI] [PubMed] [Google Scholar]
- 58. Kaegi R., Ulrich A., Sinnet B., Vonbank R., Wichser A., Zuleeg S., Simmler H., Brunner S., Vonmont H., Burkhardt M., Boller M., Environ. Pollut. 2008, 156, 233. [DOI] [PubMed] [Google Scholar]
- 59. Zhang L., Pornpattananangkul D., Hu C.‐M., Huang C.‐M., Curr. Med. Chem. 2010, 17, 585. [DOI] [PubMed] [Google Scholar]
- 60. Lam S. J., Wong E. H. H., Boyer C., Qiao G. G., Prog. Polym. Sci. 2017. [Google Scholar]
- 61. Wu Y. L., Wang H., Qiu Y. K., Liow S. S., Li Z., Loh X. J., Adv. Healthcare Mater. 2016, 5, 2679. [DOI] [PubMed] [Google Scholar]
- 62. Liu Q., Cheng S., Li Z., Xu K., Chen G. Q., J. Biomed. Mater. Res., Part A 2009, 90, 1162. [DOI] [PubMed] [Google Scholar]
- 63. Li Z., Cheng S., Li S., Liu Q., Xu K., Chen G. Q., Polym. Int. 2008, 57, 887. [Google Scholar]
- 64. Fan X., Jiang S., Li Z., Loh X. J., Mater. Sci. Eng., C 2017, 73, 275. [DOI] [PubMed] [Google Scholar]
- 65. Tan B. H., Muiruri J. K., Li Z., He C., ACS Sustainable Chem. Eng. 2016, 4, 5370. [Google Scholar]
- 66. Mahapatro A., Singh D. K., J. Nanobiotechnol. 2011, 9, 55. [Google Scholar]
- 67. Hans M. L., Lowman A. M., Curr. Opin. Solid State Mater. Sci. 2002, 6, 319. [Google Scholar]
- 68. Gref R., Lück M., Quellec P., Marchand M., Dellacherie E., Harnisch S., Blunk T., Müller R., Colloids Surf., B 2000, 18, 301. [DOI] [PubMed] [Google Scholar]
- 69. Li Z., Chee P. L., Owh C., Lakshminarayanan R., Loh X. J., RSC Adv. 2016, 6, 28947. [Google Scholar]
- 70. Yuan W., Wei J., Lu H., Fan L., Du J., Chem. Commun. 2012, 48, 6857. [DOI] [PubMed] [Google Scholar]
- 71. Abdelghany S. M., Quinn D. J., Ingram R. J., Gilmore B. F., Donnelly R. F., Taggart C. C., Scott C. J., Int. J. Nanomed. 2012, 7, 4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Peng H.‐S., Liu X.‐J., Lv G.‐X., Sun B., Kong Q.‐F., Zhai D.‐X., Wang Q., Zhao W., Wang G.‐Y., Wang D.‐D., Li H.‐L., Jin L.‐H., Kostulas N., Int. J. Pharm. 2008, 352, 29. [DOI] [PubMed] [Google Scholar]
- 73. Cavalli R., Donalisio M., Civra A., Ferruti P., Ranucci E., Trotta F., Lembo D., J. Controlled Release 2009, 137, 116. [DOI] [PubMed] [Google Scholar]
- 74. Tyagi R., Lala S., Verma A. K., Nandy A., Mahato S. B., Maitra A., Basu M. K., J. Drug Targeting 2005, 13, 161. [DOI] [PubMed] [Google Scholar]
- 75. Ahmad Z., Pandey R., Sharma S., Khuller G., Int. J. Antimicrob. Agents 2008, 31, 142. [DOI] [PubMed] [Google Scholar]
- 76. Mosqueira V. C., Loiseau P. M., Bories C., Legrand P., Devissaguet J.‐P., Barratt G., Antimicrob. Agents Chemother. 2004, 48, 1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zahoor A., Sharma S., Khuller G., Int. J. Antimicrob. Agents 2005, 26, 298. [DOI] [PubMed] [Google Scholar]
- 78. Dalgleish A., Beverley P., Clapham P., Crawford D., Greaves M., Weiss R., Nature 1984, 312, 763. [DOI] [PubMed] [Google Scholar]
- 79. Espuelas M., Legrand P., Loiseau P., Bories C., Barratt G., Irache J., J. Drug Targeting 2002, 10, 593. [DOI] [PubMed] [Google Scholar]
- 80. Duchêne D., Ponchel G., J. Inclusion Phenom. Macrocyclic Chem. 2002, 44, 15. [Google Scholar]
- 81. Bitounis D., Fanciullino R., Iliadis A., Ciccolini J., ISRN Pharm. 2012, 2012, 738432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. de Mareuil J., Mabrouk K., Doria E., Moulard M., de Chasteigner S., Oughideni R., van Rietschoten J., Rochat H., De Waard M., Sabatier J.‐M., Antiviral Res. 2002, 54, 175. [DOI] [PubMed] [Google Scholar]
- 83. Schiffelers R. M., Storm G., Marian T., Bakker‐Woudenberg I. A., Antimicrob. Agents Chemother. 2001, 45, 464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Schumacher I., Margalit R., J. Pharm. Sci. 1997, 86, 635. [DOI] [PubMed] [Google Scholar]
- 85. Mimoso I. M., Francisco A. P. G., Cruz M. E. M., Int. J. Pharm. 1997, 147, 109. [Google Scholar]
- 86. Onyeji C., Nightingale C., Marangos M., Infection 1994, 22, 338. [DOI] [PubMed] [Google Scholar]
- 87. Van Etten E., Ten Kate M., Stearne L., Bakker‐Woudenberg I. A., Antimicrob. Agents Chemother. 1995, 39, 1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Iamskov I., Kuskov A., Babievskiĭ K., Berezin B., Kraiukhina M., Samoıˇlova N., Tikhonov V., Shtil'man M., Prikl. Biokhim. Mikrobiol. 2008, 44, 688. [PubMed] [Google Scholar]
- 89. Soni P. N., Brown D., Saffie R., Savage K., Moore D., Gregoriadis G., Dusheiko G. M., Hepatology 1998, 28, 1402. [DOI] [PubMed] [Google Scholar]
- 90. Pollock S., Nichita N. B., Böhmer A., Radulescu C., Dwek R. A., Zitzmann N., Proc. Natl. Acad. Sci. USA 2010, 107, 17176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kaur C. D., Nahar M., Jain N. K., J. Drug Targeting 2008, 16, 798. [DOI] [PubMed] [Google Scholar]
- 92. Martins S., Sarmento B., Ferreira D. C., Souto E. B., Int. J. Nanomed. 2007, 2, 595. [PMC free article] [PubMed] [Google Scholar]
- 93. Müller R., Radtke M., Wissing S., Adv. Drug Delivery Rev. 2002, 54, S131. [DOI] [PubMed] [Google Scholar]
- 94. Wissing S. A., Müller R. H., Int. J. Pharm. 2003, 254, 65. [DOI] [PubMed] [Google Scholar]
- 95. Müller R. H., Mäder K., Gohla S., Eur. J. Pharm. Biopharm. 2000, 50, 161. [DOI] [PubMed] [Google Scholar]
- 96. Wissing S., Lippacher A., Müller R., J. Cosmet. Sci. 2001, 52, 313. [PubMed] [Google Scholar]
- 97. Yang W., Wiederhold N. P., Williams R. O. III, Expert Opin. Drug Delivery 2008, 5, 1199. [DOI] [PubMed] [Google Scholar]
- 98. Sanna V., Gavini E., Cossu M., Rassu G., Giunchedi P., J. Pharm. Pharmacol. 2007, 59, 1057. [DOI] [PubMed] [Google Scholar]
- 99. Pouton C. W., Eur. J. Pharm. Sci. 2000, 11, S93. [DOI] [PubMed] [Google Scholar]
- 100. Gilligan P. H., Clin. Microbiol. Rev. 1991, 4, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wissing S., Kayser O., Müller R., Adv. Drug Delivery Rev. 2004, 56, 1257. [DOI] [PubMed] [Google Scholar]
- 102. Puri A., Loomis K., Smith B., Lee J.‐H., Yavlovich A., Heldman E., Blumenthal R., Crit. Rev. Ther. Drug. 2009, 26, 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ridolfi D. M., Marcato P. D., Justo G. Z., Cordi L., Machado D., Duran N., Colloids Surf., B 2012, 93, 36. [DOI] [PubMed] [Google Scholar]
- 104. Cavalli R., Gasco M. R., Chetoni P., Burgalassi S., Saettone M. F., Int. J. Pharm. 2002, 238, 241. [DOI] [PubMed] [Google Scholar]
- 105. Souto E., Wissing S., Barbosa C., Müller R., Int. J. Pharm. 2004, 278, 71. [DOI] [PubMed] [Google Scholar]
- 106. Bhalekar M. R., Pokharkar V., Madgulkar A., Patil N., Patil N., AAPS PharmSciTech 2009, 10, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Chattopadhyay N., Zastre J., Wong H.‐L., Wu X. Y., Bendayan R., Pharm. Res. 2008, 25, 2262. [DOI] [PubMed] [Google Scholar]
- 108. Lv Q., Yu A., Xi Y., Li H., Song Z., Cui J., Cao F., Zhai G., Int. J. Pharm. 2009, 372, 191. [DOI] [PubMed] [Google Scholar]
- 109. Cai J., Huang H., Song W., Hu H., Chen J., Zhang L., Li P., Wu R., Wu C., Int. J. Pharm. 2015, 495, 728. [DOI] [PubMed] [Google Scholar]
- 110. Cheng J., Teply B. A., Sherifi I., Sung J., Luther G., Gu F. X., Levy‐Nissenbaum E., Radovic‐Moreno A. F., Langer R., Farokhzad O. C., Biomaterials 2007, 28, 869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Abeylath S. C., Turos E., Dickey S., Lim D. V., Bioorg. Med. Chem. 2008, 16, 2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Shenoy D. B., Amiji M. M., Int. J. Pharm. 2005, 293, 261. [DOI] [PubMed] [Google Scholar]
- 113. Kwok K. K., Groves M. J., Burgess D. J., Pharm. Res. 1991, 8, 341. [DOI] [PubMed] [Google Scholar]
- 114. Pandey R., Ahmad Z., Sharma S., Khuller G., Int. J. Pharm. 2005, 301, 268. [DOI] [PubMed] [Google Scholar]
- 115. Vila A., Sanchez A., Tobıo M., Calvo P., Alonso M., J. Controlled Release 2002, 78, 15. [DOI] [PubMed] [Google Scholar]
- 116. Aslani P., Kennedy R. A., J. Controlled Release 1996, 42, 75. [Google Scholar]
- 117. Kirby C., Gregoriadis G., Nat. Biotechnol. 1984, 2, 979. [Google Scholar]
- 118. Puglisi G., Fresta M., Mazzone G., Furneri P. M., Tempera G., Int. J. Pharm. 1995, 118, 65. [Google Scholar]
- 119. Vitas A. I., Diaz R., Gamazo C., Antimicrob. Agents Chemother. 1996, 40, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Maruyama T., Fukui Y., Tsuchiya E., Fujii A., RSC Adv. 2012, 2, 11672. [Google Scholar]
- 121. Wu Y., Yu B., Jackson A., Zha W., Lee L. J., Wyslouzil B. E., Mol. Pharm. 2009, 6, 1371. [DOI] [PubMed] [Google Scholar]
- 122. Chattopadhyay N., Zastre J., Wong H.‐L., Wu X., Bendayan R., Pharm. Res. 2008, 25, 2262. [DOI] [PubMed] [Google Scholar]
- 123. Ridolfi D. M., Marcato P. D., Justo G. Z., Cordi L., Machado D., Durán N., Colloids Surf., B 2012, 93, 36. [DOI] [PubMed] [Google Scholar]
- 124. Shahgaldian P., Da Silva E., Coleman A. W., Rather B., Zaworotko M. J., Int. J. Pharm. 2003, 253, 23. [DOI] [PubMed] [Google Scholar]
- 125. Schubert M. A., Müller‐Goymann C. C., Eur. J. Pharm. Biopharm. 2003, 55, 125. [DOI] [PubMed] [Google Scholar]
- 126. Morel S., Terreno E., Ugazio E., Aime S., Gasco M. R., Eur. J. Pharm. Biopharm. 1998, 45, 157. [DOI] [PubMed] [Google Scholar]
- 127. Kržicˇ M., Šentjurc M., Kristl J., J. Controlled Release 2001, 70, 203. [DOI] [PubMed] [Google Scholar]
- 128. Charcosset C., El‐Harati A., Fessi H., J. Controlled Release 2005, 108, 112. [DOI] [PubMed] [Google Scholar]
- 129. Hu F.‐Q., Jiang S.‐P., Du Y.‐Z., Yuan H., Ye Y.‐Q., Zeng S., Int. J. Pharm. 2006, 314, 83. [DOI] [PubMed] [Google Scholar]
- 130. Hu F.‐Q., Jiang S.‐P., Du Y.‐Z., Yuan H., Ye Y.‐Q., Zeng S., Colloids Surf., B 2005, 45, 167. [DOI] [PubMed] [Google Scholar]
- 131. Luo Q., Zhao J., Zhang X., Pan W., Int. J. Pharm. 2011, 403, 185. [DOI] [PubMed] [Google Scholar]
- 132. Uner M., Yener G., Int. J. Nanomed. 2007, 2, 289. [PMC free article] [PubMed] [Google Scholar]
- 133. Alipour M., Halwani M., Omri A., Suntres Z. E., Int. J. Pharm. 2008, 355, 293. [DOI] [PubMed] [Google Scholar]
- 134. Gullotti E., Yeo Y., Mol. Pharm. 2009, 6, 1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Parveen S., Misra R., Sahoo S. K., Nanomed.: NBM 2012, 8, 147. [DOI] [PubMed] [Google Scholar]
- 136. Parveen S., Mitra M., Krishnakumar S., Sahoo S. K., Acta Biomater. 2010, 6, 3120. [DOI] [PubMed] [Google Scholar]
- 137. Kocbek P., Obermajer N., Cegnar M., Kos J., Kristl J., J. Controlled Release 2007, 120, 18. [DOI] [PubMed] [Google Scholar]
- 138. Dees C., Schultz R., Vet. Immunol. Immunopathol. 1990, 24, 135. [DOI] [PubMed] [Google Scholar]
- 139. Jones D. S., McGovern J. G., Woolfson A. D., Gorman S. P., Pharm. Res. 1997, 14, 1765. [DOI] [PubMed] [Google Scholar]
- 140. Jones D. S., Schep L. J., Shepherd M. G., Pharm. Res. 1995, 12, 1896. [DOI] [PubMed] [Google Scholar]
- 141. Douglas L. J., Crit. Rev. Ther. Drug 1987, 15, 27. [DOI] [PubMed] [Google Scholar]
- 142. McCarron P. A., Donnelly R. F., Canning P. E., McGovern J. G., Jones D. S., Biomaterials 2004, 25, 2399. [DOI] [PubMed] [Google Scholar]