

Abstract
Case series
Patients: Male, 19-year-old • Female, 31-year-old
Final Diagnosis: TAFRO syndrome
Symptoms: Fever • splenomegaly • lymphadenopathies
Medication: —
Clinical Procedure: —
Specialty: Hematology
Objective:
Unusual clinical course
Background:
Thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly (TAFRO) syndrome is a variant of idiopathic multicentric Castleman disease. Adrenal hemorrhage has rarely been reported in TAFRO syndrome, and previous cases have mainly been Asian patients. This report is of two Caucasian patients with TAFRO syndrome presenting with acute adrenal insufficiency due to bilateral adrenal hemorrhage.
Case Reports:
Case 1 was a 19-year-old Caucasian man with no significant past medical history who was admitted with acute abdominal pain, vomiting, anorexia, and moderate weight loss. Case 2 was a 31-year-old Caucasian woman with no past medical history who was admitted to hospital with fever, dyspnea, thoracic and abdominal pain, polyarthralgia, and hypotension. Both patients had splenomegaly, mild lymphadenopathy, thrombocytopenia, acute kidney injury, and myelofibrosis. In both cases, lymph node biopsy histology showed mixed-type idiopathic multicentric Castleman disease. In both patients, a diagnosis of TAFRO was made, and they developed bilateral adrenal hemorrhage with adrenal insufficiency. Case 1 was treated with high-dose steroids, followed by tocilizumab infusion. Due to persistent thrombocytopenia, second-line treatment commenced with rituximab, but the patient relapsed two months later. Tocilizumab treatment was recommenced, which was followed by an immuno-allergic adverse event. He then had a good response to sirolimus. Case 2 died nine months after diagnosis due to acute respiratory distress.
Conclusions:
Two cases of TAFRO syndrome presented with acute adrenal insufficiency due to bilateral adrenal hemorrhage. The symptoms were only partially controlled with tocilizumab, rituximab, and tacrolimus. Adrenal hemorrhage may be a specific manifestation of TAFRO syndrome.
MeSH Keywords: Adrenal Insufficiency, Edema, Giant Lymph Node Hyperplasia, Primary Myelofibrosis, Renal Insufficiency, Thrombocytopenia
Background
Thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly (TAFRO) syndrome is a variant of idiopathic multicentric Castleman disease [1]. In 2010, Takai et al. described TAFRO syndrome as a new variant of human Herpesvirus 8 (HHV8)-negative idiopathic multicentric Castleman disease in Asian patients. The clinical features initially described included thrombocytopenia, anasarca (edema, pleural effusion, and ascites), fever, renal dysfunction, reticulin myelofibrosis, and organomegaly (hepatosplenomegaly and lymphadenopathy) [2]. The lymph node histology in patients with TAFRO syndrome is characterized by hyalinization of lymphoid follicles, capillary proliferation, and increased numbers of plasma cells, consistent with mixed type idiopathic multi-centric Castleman disease [3]. A central role for interleukin-6 (IL-6) has been proposed for the pathogenesis of multicentric Castleman disease [4].
Despite similar histopathology, TAFRO syndrome can be distinguished from multicentric Castleman disease by several clinical and histological findings. A normal immunoglobulin level, thrombocythemia (platelet count, <100×109/L), mild lymphadenopathy, with a lymph node diameter of ≤1.5 cm, hypoalbuminemia, and anasarca are more typical for TAFRO syndrome. Polyclonal hypergammaglobulinemia, thrombocytosis, and lymphadenopathy are characteristics of multicentric Castleman disease. A recently published retrospective study analyzed 220 patients from the Multicenter Collaborative Retrospective Study for Establishing the Concept of TAFRO Syndrome database that included 87 patients with idiopathic multicentric Castleman disease, not otherwise specified (iMCD-NOS), 63 patients with TAFRO-iMCD, and 19 patients with TAFRO alone [5]. The findings showed that the outcome for patients with TAFRO was poor, with a five-year survival rate of 65% [5]. However, while the pathogenesis of HHV-8-associated multicentric Castleman disease is better understood, idiopathic multicentric Castleman disease and TAFRO syndrome remain to be determined.
Adrenal involvement in multicentric Castleman disease has previously been described, and increased expression of IL-6 has been reported in these cases [6,7]. However, adrenal hemorrhage has only been reported in TAFRO syndrome in the idiopathic multicentric Castleman disease subtype. Also, the majority of reported cases were Asian patients, with few reported cases in Caucasians [8]. To our knowledge, hemorrhagic events in other organs during TAFRO syndrome had not been previously described. This report is of two Caucasian patients with TAFRO syndrome who presented with acute adrenal insufficiency due to bilateral adrenal hemorrhage.
Case Reports
Case 1
A 19-year-old French Caucasian man with no significant past medical history was admitted to hospital with acute abdominal pain and vomiting associated with anorexia and moderate weight loss. Physical examination showed splenomegaly, normal arterial blood pressure, and no fever. Blood tests initially showed a high serum C-reactive protein (CRP) level (27 mg/dL), hyperfibrinogenemia (5 g/L), thrombocytopenia (platelet count, 114×109/L), lymphopenia (1×109/L), hypoalbuminemia (29 g/L), hyponatremia, and hyperkalemia. The prothrombin time (PT) was 12.5 s (normal range, 9–14 s), and the partial thromboplastin time (PTT) was 31 s (normal range, 27–40 s). Blood cultures were negative. The serum cortisol level was low, and the adrenocorticotrophic hormone (ACTH) level was increased, in keeping with a diagnosis of acute adrenal insufficiency. Replacement therapy with hydrocortisone and fludrocortisone began with an excellent clinical response within a few days.
An abdominal computed tomography (CT) scan showed bilateral adrenal hemorrhage associated with a mild celiac, mesenteric, and axillary lymphadenopathy. He did not have splenomegaly, and there were no pleural, pericardial, or abdominal effusions. Adrenal hemorrhage was confirmed by magnetic resonance imaging (MRI) (Figure 1). The QuantiFERON-TB Gold (QFT) test for Mycobacterium tuberculosis (Qiagen, Hilden, Germany) and viral detection by polymerase chain reaction (PCR) were negative. Serum anti-adrenal antibodies, antibodies to beta2-glycoprotein I (anti-beta2-GPI), anti-cardiolipin antibodies, and antinuclear antibodies were negative. The Dilute Russell’s viper venom time (dRVVT) test for lupus anticoagulant (LA) was negative at 12 weeks. Gamma globulins were within the normal range (<14 g/L).
Figure 1.

Case 1. Magnetic resonance imaging (MRI) of the adrenal gland with thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly (TAFRO) syndrome, associated with adrenal hemorrhage and insufficiency. The adrenal gland axial volume acceleration-flexible (LAVA-Flex) magnetic resonance imaging (MRI) (blue arrow). The coronal T2-weighted sequence (yellow arrow) shows bilateral adrenal masses with a fluid collection (hemorrhage) and a hyperintense T1-weighted non-enhancing lesion, typical for adrenal hemorrhage.
Two weeks later, he developed fever (39°C), night sweats, limb edema, dyspnea, bilateral exudative pleural effusion, ascites, and purpura. All laboratory parameters worsened over time with normochromic anemia (6.3 g/dL), severe thrombocytopenia (platelet count, 26×109/L), high serum ferritin level (2294 µg/L), and hypoalbuminemia (23 g/L). Imaging by computed tomography (CT) showed anasarca, or generalized edema. A bone marrow biopsy showed grade 1 myelofibrosis. Right axillary and left inguinal lymph node biopsies were performed and showed histological changes of multicentric mixed-type Castleman disease, characterized by a hyaline vascular pattern (Figure 2), atrophic germinal centers (Figure 3) and mantle cell hyperplasia with polyclonal plasmacytosis (Figure 4). Immunohistochemistry staining for the latency-associated nuclear antigen (LANA-1) of human herpesvirus 8 (HHV-8) was negative. A slight increase in serum vascular endothelial growth factor (VEGF) was detected at 171 pg/mL (normal range, 0–115 pg/mL). He rapidly developed acute kidney injury with a rise in serum creatinine levels from 95 µmol/L at baseline to 155 µmol/L, without proteinuria or microscopic hematuria.
Figure 2.
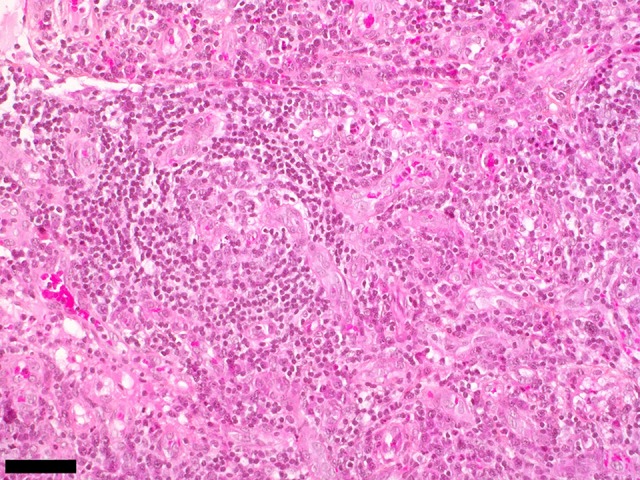
Case 1. Photomicrograph of the histology of the lymph node biopsy in a case of thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly (TAFRO) syndrome associated with adrenal hemorrhage and insufficiency. Lymph node histology shows a mixed subtype Castleman disease showing an onion skin pattern of fibrosis around an atrophic germinal center with prominent vascularity. The bar represents 60 μm. Hematoxylin and eosin (H&E).
Figure 3.
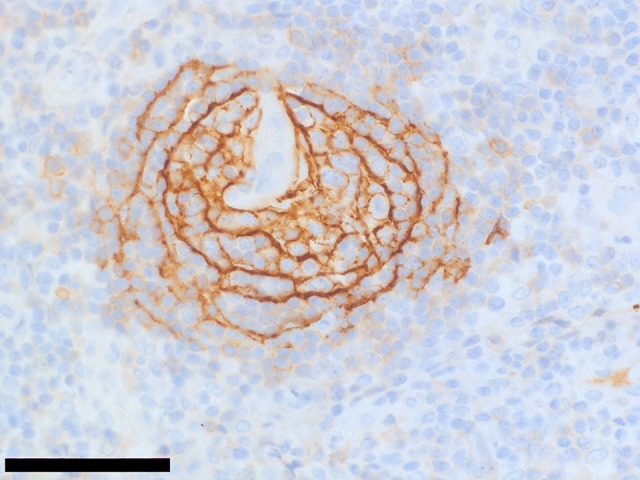
Case 1. Photomicrograph of the immunohistochemistry for CD23 in the lymph node biopsy in a case of thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly (TAFRO) syndrome associated with adrenal hemorrhage and insufficiency. Immunohistochemistry with a primary monoclonal antibody to CD23 for dendritic cells shows atrophic and concentric follicular dendritic cells distributed in the germinal center, which a histological pattern found in idiopathic multicentric Castleman disease. The bar represents 60 μm.
Figure 4.

Case 1. Photomicrograph of the immunohistochemistry for CD138 in the lymph node biopsy in a case of thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly (TAFRO) syndrome, associated with adrenal hemorrhage and insufficiency. Immunohistochemistry with a primary monoclonal antibody to CD138 (syndecan-1) shows lymph node infiltration by plasma cells. The bar represents 160 μm.
Based on the clinical manifestations, biochemical laboratory findings, and histopathology, the patient was diagnosed with thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly (TAFRO) syndrome, according to the 2015 diagnostic criteria [9]. He was treated with tocilizumab (8 mg/kg/15 days) and high-dose prednisone using 1 g for two days, followed by 1 mg/kg/day. A few days after starting tocilizumab treatment, his fever resolved, the CRP and creatinine levels decreased, but thrombocytopenia persisted, at <50×109/L. Treatment was suspended after three injections of tocilizumab because of severe hypotension. Second-line therapy with rituximab commenced using four once-weekly doses of 375 mg/m2. Two months later, the patient experienced disease relapse, with auto-immune hemolytic anemia (Hb, 3.9 g/dL), acute kidney injury, systemic inflammation, and anasarca, which was treated with tocilizumab. The patient developed a further immune drug-related reaction after the second infusion, and a third-line therapy with sirolimus began. Sirolimus treatment resulted in a hematologic response with the recovery of the platelet count to >100×109/L at six months after disease onset (Figure 5).
Figure 5.

Case 1. The clinical course in a patient with thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly (TAFRO) syndrome associated with adrenal hemorrhage and insufficiency. The clinical course in a 19-year-old Caucasian man included fever, platelet count, C-reactive protein (CRP), and creatinine levels. A partial response was obtained with tocilizumab injection. The patient was treated with high-dose steroids, followed by tocilizumab infusion. Due to persistent thrombocytopenia, second-line treatment commenced with rituximab, but the patient relapsed two months later. Tocilizumab treatment was recommenced, which was followed by an immuno-allergic adverse event. The patient then had a good response to sirolimus.
Case 2
A 31-year-old Caucasian woman with no significant past medical history was admitted to hospital with a fever of 39°C, dyspnea, thoracic and abdominal pain, polyarthralgia, and low blood pressure. Physical examination showed limb edema, pleuritis, mild lymphadenopathy, and bilaterally enlarged parotid glands. Blood tests showed an increased CRP level (11 mg/dL), normocytic anemia (Hb, 10 g/dL), thrombocytopenia (platelet count, 120×109/L), increased alkaline phosphatase (ALP) (142 g/L), hyponatremia, and hyperkalemia. Microbiology tests were negative. Thoracic and abdominal CT scans showed a large bilateral pleural effusion, mild ascites, mediastinal and subdiaphragmatic lymphadenopathy, mild hepatosplenomegaly, and bilateral adrenal hemorrhage, which supported the diagnosis of acute adrenal insufficiency. Pleural aspiration showed an exudative effusion containing neutrophils without bacterial growth. Because of respiratory failure and the worsening biochemical laboratory findings, she was admitted to the intensive care unit (ICU). Antinuclear antibodies (ANAs) were positive (1/1280) with a high titer of ANAs for Sjögren’s syndrome A (SSA) and SSB, a low titer of DNA antibodies, reduced levels of complement C3 and C4 factors, and a low titer of mixed type III cryoglobulins. Lupus anticoagulant, IgG anti-cardiolipin, and anti-beta2 glycoprotein 1 antibodies were initially positive. The partial thromboplastin time (PTT) was 31.6 s (normal range, 27–40 s), the prothrombin time (PT) was 13.6 s (normal range, 9–14 s), and the fibrinogen level was 7.14 g/L. She had moderate polyclonal hypergammaglobulinemia (15 g/L), and increased ferritin (2,000 μg/L), with a normal percentage of ferritin glycosylation.
A preliminary diagnosis was made of systemic lupus erythematosus (SLE) with articular, pleural, and hematologic manifestations associated with antiphospholipid syndrome. She was initially treated with three daily doses of 500 mg of methylprednisolone, then 1 mg/kg/day of prednisone, with anticoagulant therapy. However, there was no clinical or biochemical improvement. She then developed progressive subacute renal dysfunction and required hemodialysis. There was no glomerular proteinuria or microscopic hematuria. The serum VEGF level was increased (500 pg/mL). However, antiphospholipid antibodies were negative, three months after her initial diagnosis. A bone marrow biopsy showed a grade 1 reticulin pattern indicating myelofibrosis. A computed tomography (CT) scan confirmed anasarca with mild generalized lymphadenopathy and organomegaly. Two lymph node biopsies were successively performed, which showed a polyclonal population of lymphoplasmacytic cells and vascular hyalinization consistent with the histological features of multicentric Castleman disease. The patient was diagnosed with TAFRO syndrome complicated by bilateral adrenal hemorrhage.
This patient had a minor salivary gland biopsy that showed lymphocytic sialadenitis (focus score ≥1). The diagnosis of Sjögren syndrome was also considered, as the diagnosis of SLE and antiphospholipid syndrome were excluded. She received second-line treatment with tocilizumab (8 mg/kg/15 days), but developed toxic epidermal necrolysis after the first infusion, and tocilizumab was withdrawn. Third-line therapy commenced with rituximab (375 mg/m2 by intravenous infusion) using a four doses per week regimen, which resulted in clinical improvement and a partial response with reduced fever and systemic inflammation, and increased platelet levels. Her anasarca and renal dysfunction persisted, and tacrolimus treatment was initiated. However, the patient died suddenly with acute respiratory distress from an unknown cause nine months after the diagnosis of TAFRO syndrome, and 15 days after the tacrolimus treatment.
Discussion
Thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly (TAFRO) syndrome is a variant of idiopathic multicentric Castleman disease [9]. TAFRO syndrome is a systemic inflammatory disorder that is challenging to diagnose and treat. In 2015, Masaki et al. proposed a disease severity classification, diagnostic criteria, and recommendations for the treatment of TAFRO syndrome [9]. The two patients described in this report met all the major and minor diagnostic criteria for TAFRO syndrome [9]. The first case was a typical case of idiopathic multicentric Castleman disease or TAFRO subtype, without evidence of autoimmune diseases. The second case had clinical features of TAFRO syndrome and overlap with Sjögren syndrome, which is an association that has been previously described [10].
Auto-antibodies, such as antinuclear or anti-SS-A antibodies have been described in Caucasian patients with TAFRO syndrome [9]. However, the criteria used to define a connective tissue disease are usually not found in these patients [8]. A definitive diagnosis of connective tissue disease would exclude TAFRO syndrome [9]. However, all the clinical, biochemical, and histological findings from the second case could not be explained only by a diagnosis of Sjögren syndrome. Severe thrombocytopenia, reticulin myelofibrosis, mild organomegaly, anasarca, and acute kidney injury were consistent with a diagnosis of TAFRO syndrome. The optimal therapeutic strategy for idiopathic multicentric Castleman disease with TAFRO syndrome has not been clearly defined. Treatment with steroids has been suggested as first-line therapy, but a lack of response is common. A significant improvement of the symptoms can be achieved using interleukin-6 (IL-6) blockade with tocilizumab or siltuximab [9,11,12]. Previously published studies have shown that rituximab can be effective in controlling idiopathic multicentric Castleman disease with TAFRO syndrome [13,14].
The therapeutic use of inhibitors of mammalian target of rapamycin (mTOR), such as sirolimus, are a promising new approach for treating refractory idiopathic multicentric Castleman disease. A recent study analyzed the potential targeting pathways in three patients with multicentric Castleman disease with TAFRO syndrome refractory to IL-6 blockade. All the patients received sirolimus, and a clinical response included reduced T-cell activation and reduced levels of VEGF-A in all patients [15]. The two patients described in this report initially presented with acute adrenal insufficiency due to bilateral adrenal hemorrhage. The symptoms of TAFRO syndrome developed a few days for the first patient, and concomitantly for the second patient.
There have been three previously published cases of bilateral adrenal hemorrhage associated with TAFRO syndrome, which supports the possibility that adrenal hemorrhage may be a specific manifestation of TAFRO syndrome [16–18]. To our knowledge, there have been no previously reported cases of other subtypes of Castleman disease associated with adrenal hemorrhage. Nara et al. reported a case of a 48-year-old Japanese man with a two-month history of general fatigue with a diagnosis of TAFRO syndrome and adrenal hemorrhage [16]. Crump et al. reported a case of idiopathic multicentric Castleman disease associated with adrenal hemorrhage and acute adrenal insufficiency, 15 years before the first description of TAFRO syndrome [17]. In this previously published case report, the patient had an atypical presentation for idiopathic multicentric Castleman disease because of thrombocytopenia and myelofibrosis on bone marrow biopsy [17]. Currently, this patient would meet the diagnostic criteria of TAFRO syndrome and was probably the first description of this association [17]. A previous case reported by Fumiko and al. in 2017 was of a 48-year-old Japanese man who fulfilled all the diagnostic criteria for TAFRO syndrome with adrenal insufficiency due to bilateral adrenal hemorrhage on computed tomography (CT) scan, without evidence of antiphospholipid syndrome [18].
Antiphospholipid syndrome can be associated with unilateral or bilateral adrenal hemorrhage and is sometimes associated with Castleman disease [19,20]. Antiphospholipid antibodies were initially positive in the two patients presented in this report but were negative 12 weeks later. Also, the two patients did not experience episodes of thrombosis during follow-up, which excluded the diagnosis of antiphospholipid syndrome. Antiphospholipid antibodies were also negative in the three previously published cases of TAFRO syndrome associated with adrenal hemorrhage. Therefore, antiphospholipid syndrome does not seem to be the mechanism of adrenal hemorrhage in TAFRO syndrome. Adrenal involvement without hemorrhage has previously been reported in Castleman disease [6,7]. In these cases, Castleman disease presented as an adrenal mass without adrenal insufficiency [6,7].
The mechanisms involved in the development of adrenal hemorrhage are not well understood, but several theories have been proposed. An increase in levels of ACTH induced by stress leads to increased blood flow to the adrenal glands, which can lead to hemorrhage from the capillaries at the cortico-medullary junction due to the fragility of the capillary plexus [21]. Patients with TAFRO syndrome might experience ACTH release as a cellular stress response. Experimental studies have shown that IL-6 has a role in the activation of the hypothalamic-pituitary-adrenal axis in humans is a nonspecific stress-induced defense mechanism involving the innate immune system [22]. Mastorakos et al. showed a trophic effect of IL-6 on the adrenal gland in patients treated with a parenteral form of IL-6 [22]. However, evidence for the potential effect of IL-6 on adrenal hemorrhage is weak. Also, adrenal hemorrhage has not previously been described in association with high Il-6 levels, including inflammatory diseases or hemophagocytic lymphohistiocytosis, and inflammatory cytokines are also involved in TAFRO syndrome. A catecholamine surge from the adrenal gland may promote vasoconstriction and platelet aggregation, which may induce vasospasm and hemorrhage of the fragile adrenal capillary vessels. Fujiwara et al. identified a hypercoagulation state in TAFRO syndrome, but catecholamine levels were not studied [11]. Finally, profound thrombocytopenia as a common manifestation of TAFRO syndrome that may lead to adrenal hemorrhage.
Conclusions
This report described two patients with thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly (TAFRO) syndrome associated with bilateral adrenal hemorrhage with acute adrenal insufficiency. TAFRO) syndrome is recognized to be a variant of idiopathic multicentric Castleman disease. These two cases are distinctive in that they occurred in Caucasian patients, rather than previously described Asian patients. Although adrenal hemorrhage remains to be incorporated in the diagnostic criteria of TAFRO, adrenal insufficiency is now well described. These reports highlight that the presence of adrenal hemorrhage should be sought in the presence of abdominal pain in TAFRO syndrome with abdominal imaging and biochemical investigations. Adrenal hemorrhage in TAFRO syndrome may occur due to the hypercoagulation state and thrombocytopenia, but these predisposing factors require further investigation. As these cases have shown, adrenal hemorrhage may be a specific manifestation of TAFRO syndrome.
Footnotes
Conflict of interest
None.
References:
- 1.Sakashita K, Murata K, Takamori M. TAFRO syndrome: Current perspectives. J Blood Med. 2018;9:15–23. doi: 10.2147/JBM.S127822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takai K, Nikkuni K, Shibuya H, Hashidate H. [Thrombocytopenia with mild bone marrow fibrosis accompanied by fever, pleural effusion, ascites and hepatosplenomegaly] Rinsho Ketsueki. 2010;51:320–25. [in Japanese] [PubMed] [Google Scholar]
- 3.Fajgenbaum DC, Uldrick TS, Bagg A, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood. 2017;129:1646–57. doi: 10.1182/blood-2016-10-746933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang H-W, Pittaluga S, Jaffe ES. Multicentric Castleman disease: Where are we now? Semin Diagn Pathol. 2016;33:294–306. doi: 10.1053/j.semdp.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fujimoto S, Sakai T, Kawabata H, et al. Is TAFRO syndrome a subtype of idiopathic multicentric Castleman disease? Am J Hematol. 2019 doi: 10.1002/ajh.25554. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 6.Müssig K, Horger M, Wehrmann M. Adrenal Castleman’s disease. Ann Hematol. 2007;86:63–65. doi: 10.1007/s00277-006-0200-7. [DOI] [PubMed] [Google Scholar]
- 7.Santomauro M, Choe C, Heimbigner J, et al. Castleman’s disease in the left suprarenal region, mimicking an adrenal neoplasm. Urology. 2011;78:319. doi: 10.1016/j.urology.2010.12.031. [DOI] [PubMed] [Google Scholar]
- 8.Louis C, Vijgen S, Samii K, et al. TAFRO syndrome in caucasians: A case report and review of the literature. Front Med (Lausanne) 2017;4:149. doi: 10.3389/fmed.2017.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masaki Y, Kawabata H, Takai K, et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome, 2015 version. Int J Hematol. 2016;103:686–92. doi: 10.1007/s12185-016-1979-1. [DOI] [PubMed] [Google Scholar]
- 10.Fujimoto S, Kawabata H, Kurose N, et al. Sjögren’s syndrome manifesting as clinicopathological features of TAFRO syndrome: A case report. Medicine. 2017;96:e9220. doi: 10.1097/MD.0000000000009220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujiwara S, Mochinaga H, Nakata H, et al. Successful treatment of TAFRO syndrome, a variant type of multicentric Castleman disease with thrombotic microangiopathy, with anti-IL-6 receptor antibody and steroids. Int J Hematol. 2016;103:718–23. doi: 10.1007/s12185-016-1978-2. [DOI] [PubMed] [Google Scholar]
- 12.Sakai K, Maeda T, Kuriyama A, et al. TAFRO syndrome successfully treated with tocilizumab: A case report and systematic review. Mod Rheumatol. 2018;28:564–69. doi: 10.3109/14397595.2015.1120389. [DOI] [PubMed] [Google Scholar]
- 13.Jain P, Verstovsek S, Loghavi S, et al. Durable remission with rituximab in a patient with an unusual variant of Castleman’s disease with myelofibrosis – TAFRO syndrome. Am J Hematol. 2015;90:1091–92. doi: 10.1002/ajh.24015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hiramatsu S, Ohmura K, Tsuji H, et al. Successful treatment by rituximab in a patient with TAFRO syndrome with cardiomyopathy. Nihon Rinsho Meneki Gakkai Kaishi. 2016;39:64–71. doi: 10.2177/jsci.39.64. [DOI] [PubMed] [Google Scholar]
- 15.Fajgenbaum DC, Langan R-A, Japp AS, et al. Identifying and targeting pathogenic PI3K/AKT/mTOR signaling in IL-6 blockade – refractory idiopathic multicentric Castleman disease. J Clin Invest. 2019;129:4451–63. doi: 10.1172/JCI126091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nara M, Komatsuda A, Itoh F, et al. Two cases of thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly (TAFRO) syndrome with high serum procalcitonin levels, including the first case complicated with adrenal hemorrhaging. Intern Med. 2017;56:1247–52. doi: 10.2169/internalmedicine.56.7991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crump JA, Beard ME, Angus HB, et al. Acute adrenal insufficiency: A new presentation of Castleman’s disease. J Intern Med. 1995;238:81–84. doi: 10.1111/j.1365-2796.1995.tb00903.x. [DOI] [PubMed] [Google Scholar]
- 18.Ito F, Kameoka Y, Nara M, et al. [TAFRO Syndrome with Bilateral Adrenal Hemorrhage] Nippon Naika Gakkai Zasshi. 2017;106:288–94. [in Japanese] [PubMed] [Google Scholar]
- 19.Isilak Z, Uzun M, Incedayi M, et al. Coronary pseudoaneurysm and superior vena cava occlusion in a young patient with multicentric Castleman’s disease and antiphospholipid antibody positivity. Eur J Echocardiogr. 2011;12:E35. doi: 10.1093/ejechocard/jer092. [DOI] [PubMed] [Google Scholar]
- 20.Jakubíková M, Piťha J, Latta J, et al. Myasthenia gravis, Castleman disease, pemphigus, and anti-phospholipid syndrome. Muscle Nerve. 2013;47:447–51. doi: 10.1002/mus.23657. [DOI] [PubMed] [Google Scholar]
- 21.Vella A, Nippoldt TB, Morris JC. Adrenal hemorrhage: A 25-year experience at the Mayo Clinic. Mayo Clin Proc. 2001;76:161–68. doi: 10.1016/S0025-6196(11)63123-6. [DOI] [PubMed] [Google Scholar]
- 22.Mastorakos G, Chrousos GP, Weber JS. Recombinant interleukin-6 activates the hypothalamic-pituitary-adrenal axis in humans. J Clin Endocrinol Metab. 1993;77:1690–94. doi: 10.1210/jcem.77.6.8263159. [DOI] [PubMed] [Google Scholar]